

Abstract
Kappa opioid receptor (KOR) desensitization was previously shown to follow agonist-dependent phosphorylation of serine 369 by G-protein receptor kinase (GRK) and β-arrestin binding in transfected cells. To study the in vivo effects induced by phosphorylation of KOR(S369), C57Bl/6 mice were administered single or repeated doses of the KOR agonist, U50,488, and isolated brain glycoprotein was probed with an antibody, KOR-P, that specifically recognized phosphoserine 369 KOR. Western blot analysis using KOR-P antibody showed that labeling intensity increased after either single or repeated treatment of mice with U50,488 by 59 ± 22% and 101 ± 29%, respectively. In contrast, there was no change in labeling intensity by nonphosphoselective KOR antibodies following acute or chronic in vivo treatment with kappa agonist. Moreover, mice lacking GRK3 showed no increase in KOR-P labeling and developed significantly less analgesic tolerance following treatment with kappa agonist. The result suggests that tolerance to kappa agonists includes phosphorylation of serine 369 within KOR by GRK3. Recovery of analgesic potency and reduction of elevated KOR-P labeling in wild-type mice both required 2 weeks to return to base line. Consistent with these results, in vitro phosphorylation by GRK3 of KOR isolated from tolerant mice resulted in 46 ± 7% less 32P incorporation than in KOR isolated from untreated mice. In addition, in vitro 32P incorporation returned to base line levels only in KOR isolated from tolerant mice allowed to recover for 2 weeks. The coincident reversal of analgesic tolerance and slow return to a basal phosphorylation state matched the regeneration rate of functional kappa receptors following irreversible antagonism and suggested that receptor replacement rather than dephosphorylation was required to restore sensitivity.
Opioid agonists are valuable clinical tools in the management of pain, but continued administration produces a pronounced tolerance, and chronic use may lead to addiction (1, 2). The mechanisms underlying opioid tolerance are likely to include molecular changes resulting in opioid receptor desensitization (3). Opioid receptors are members of the family of seven transmembrane G-protein-coupled receptors (GPCR)1 that are desensitized following phosphorylation by G-protein receptor kinases (GRKs) and subsequent arrestin binding (see Ref. 4). Most of the insights concerning GPCR receptor desensitization mechanisms are derived from in vitro studies using transfected cells. In these heterologous gene expression models, GRKs rapidly phosphorylate the agonist-activated conformation of the GPCR (5–9). Site-directed mutagenesis of the GPCR has been used to identify the specific serine and threonine residues in the mu, kappa, and delta opioid receptors required for GRK/arrestin-dependent desensitization (see Ref. 10). For example, mutation of serine 369 to alanine in the C-terminal domain of the kappa opioid receptor (KOR) blocked GRK3-mediated desensitization (11).
These results suggest that GRK/arrestin-dependent mechanisms normally regulate KOR activity in the nervous system; however, this hypothesis has not been directly tested. Studies measuring phosphorylation have typically involved the agonist-induced incorporation of 32P into proteins matching the approximate weight of the opioid receptor after isolation from in vitro model systems, such as cell cultures (5, 6, 12). In one ex vivo study, immunoprecipitation of the KOR from guinea pig hippocampal slices preincubated in [32P]orthophosphoric acid revealed basal phosphorylation of a 53-kDa protein that increased 50% after a 75-min pretreatment with the KOR agonist U50,488 (7). The kinase responsible and the phosphorylation sites affected were not defined. In addition, the relationship between these molecular events and opioid tolerance measured behaviorally was not established.
To address these questions, we generated an antibody able to distinguish the phosphorylated state of the KOR at serine 369 (13). Using the KOR-P antibody with confocal microscopy of transfected cells, we previously found that kappa agonist-induced stimulation and internalization of KOR-GFP was correlated with phosphorylation of the kappa opioid receptor on serine 369 (13). Using this reagent and GRK3 knockout mice in the present study, we found a direct correlation between opioid receptor phosphorylation by GRK3 at this site and analgesic tolerance.
EXPERIMENTAL PROCEDURES
Chemicals and Antibodies
A phosphorylated peptide corresponding to the C-terminal domain of KOR designated KOR-P (KRVRNT-VQDPAS(PO4)MRD, matching amino acids 359–372 within the rodent kappa opioid receptor sequence) was used to produce an antibody selective for the phosphorylated state of the receptor, KOR-P, as described previously (13). The two antibodies KT-2 (raised against the C-terminal tail of the KOR) and KE-4 (raised against the predicted fourth extra-cellular loop of the rat KOR300–312: GSTSHSTAALSSY) were produced as previously described (Refs. 14 and 7, respectively). Both KT-2 and KE-4 were used interchangeably to detect kappa opioid receptors by Western analysis in this study, because they gave identical results. All chemicals, unless otherwise specified, were from Sigma (St. Louis, MO).
Preparation of Cell Lysates and Immunoprecipitation of KOR-GFP
HEK293 cells in 35-mm plates were transiently transfected with 10 μg of KOR-GFP cDNA as described previously (13). After 24–48 h of expression, cells were pretreated for 30 min with 10 μM U50,488 at 37 °C, then rinsed once with ice-cold phosphate-buffered saline. Cells were incubated for 5 min with 0.5 ml of ice-cold cell lysis buffer (20 mM Tris, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, 2 mM Na3VO4, and 1 μg/ml leupeptin (pH 7.5)), removed from plates, and sonicated briefly on ice. Cell lysates were centrifuged for 10 min at 4 °C, and 400 μl of supernatant were then incubated with 20 μg of KE-4 antibody overnight with gentle rocking at 4 °C. Protein A-agarose beads (10 μl of 50% bead slurry, Sigma) were added to the lysate/antibody incubation with gentle rocking for 2.5 h at 4 °C. Tubes were microcentrifuged for 30 s at 4 °C, and the pellets were washed four times with 500 μl of cell lysis buffer. Precipitated protein was eluted from Protein A-agarose beads with the addition of 35 μl of SDS-sample buffer (187.5 mM Tris-HCl (pH 6.8), 6% w/v SDS, 30% glycerol, 150 mM dithiothreitol, and 0.03% w/v bromphenol blue) and boiling for 10 min. Samples were briefly centrifuged, and the supernatant was analyzed by Western immunoblotting (see below).
Wheat Germ Agglutinin Purification of Mouse Brain Membrane Proteins
Mice were sacrificed, and brain tissue lacking cerebellum was removed, weighed, and homogenized in 9× volume (w/v) of homogenizing buffer (25 mM Tris and 250 mM sucrose, pH 7.4, with protease inhibitors 1 mM phenylmethylsulfonyl fluoride, 2 μg/ml leupeptin, and 2 μg/ml aprotinin) using a Dounce tissue homogenizer. Homogenized tissue was centrifuged at 3000 × g for 20 min at 4 °C. The supernatant was diluted with 100 mM NaCl/0.2 mM MgSO4, followed by ultracentrifugation at 100,000 × g for 15 min. The membrane pellet was solubilized in 4 volumes of solubilization buffer (50 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, and 1% Triton X-100, with 2 μg/ml each of leupeptin and aprotinin added as protease inhibitors) for 1 h on ice, and again ultracentrifuged at 100,000 × g for 15 min to remove unsolubilized material. Solubilized brain membrane proteins were diluted 4× in WGA buffer (10 mM Tris (pH 7.4), 50 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, and 0.1% Triton X-100, with 2 μg/ml each of protease inhibitors leupeptin and aprotinin), and glycosylated protein was affinity-purified using a WGA-Sepharose column (Sigma). Bound protein was eluted with 0.25 M N-acetylglucosamine in WGA buffer. Protein concentration was estimated by UV absorbance at 280 nm, plotted against measured bovine serum albumin protein standard curves.
Immunoblotting
WGA-purified solubilized mouse brain protein (90 μg/lane unless otherwise noted) was resolved by SDS-PAGE, and proteins were transferred to nitrocellulose and incubated for 1 h in 10% nonfat dry milk (NFDM) in Tris-buffered saline (TBS, 50 mM Tris base and 150 mM NaCl, pH 7.4). Note that some experiments were performed with precast 10% PAGE gels (Invitrogen) under nonreducing conditions where mentioned in the figure legends. All nitrocellulose membranes were then incubated overnight at 4 °C under gentle agitation in 5% NFDM/TBS containing 5 μg/ml KOR-P antibody (to measure KOR phosphorylation) or 15 μg/ml KT-2 antibody or 20 μg/ml KE-4 antibody (as controls to verify consistent loading of KOR between conditions). Membranes were then washed three times in 5% NFDM/TBS and incubated in horseradish peroxidase-conjugated goat antirabbit IgG (diluted 1:4000 in 5% NFDM/TBS) for 90 min at room temperature. The membranes were again washed three times in 5% NFDM/TBS, then once in TBS, and antibody labeling of protein was visualized with an enhanced chemiluminescence (ECL, Amersham Biosciences) detection kit on Kodak X-OMAT film.
32P Incorporation Experiments
GRK3 was produced using a bacu-lovirus expression system (generous gift of Dr. Jeffrey Benovic) as described previously (15). WGA-purified solubilized mouse brain protein (90 μg/tube) was incubated with 10 μM U50,488, isolated GRK3, and [γ-32P]ATP according to the method detailed previously (15) for 2 h at 37 °C. Samples were then incubated with 10 μg of KT-2 antibody and protein A-agarose beads (30 μl of 50% bead slurry, Sigma) with gentle rocking for 2 h at 25 °C to immunoprecipitate the KOR. Tubes were microcentrifuged, the pellets washed, and precipitated protein was eluted with the addition of 35 μl of SDS-sample buffer as described above. Samples were briefly centrifuged, and the supernatant was analyzed by separation across precast 10% PAGE gels (Invitrogen) and exposure to Kodak X-OMAT film for 1–48 h.
Breeding and Genotyping of GRK3 and KOR Knockout Mice
Homozygous GRK3(−/−) knockout mice were prepared by homologous recombination as described previously (16) and provided for this study. Animals were backcrossed for seven generations with C57Bl/6 mice. Then breeding pairs of heterozygotes were used to generate GRK3(−/−) mice and paired littermate GRK3(+/+) controls for this study. Individual mice were genotyped by extracting DNA from tail samples that was then used as template in a pair of PCR reactions. A common primer (5′-CAGGGCTAGGTGTGACTGTCATGT) recognizing a sequence present in both wild-type and knockout mice, a wild-type (wt) primer based on a sequence within the GRK3 gene (5′-CTTCCACAGCTGAGCATGAACGAC), and a neomycin primer (5′-CTGACTAGGGGAGGAGTAGAAGGT) were used for genotyping. Homozygous KOR(−/−) and wild-type littermate mice were generated as described elsewhere (17), and isolated brains provided by Dr. John Pintar were used in this study.
Chronic U50,488 Treatment
To induce kappa opioid tolerance, adult C57Bl/6 mice (15–25 g) were administered U50,488 (intraperitoneally, Sigma) twice a day in an ascending dosage schedule of 10 and 15, 25 and 30, 50 and 60, and 70 and 75 mg/kg body weight (adapted from Ref. 18). On day 5, the animals were injected with a test dose of 25 mg/kg U50,488 and then sacrificed 35 min later. All injections were performed with the same bolus volume (0.3 ml/100 g of body weight). As a control for nonspecific effects of either animal handling, injections, or learned tolerance, “naïve” animals received vehicle (sterile water) injections for days 1–4 on the same schedule before testing with U50,488 on day 5. The development of tolerance was monitored by measuring the antinociceptive effect of U50,488 in the mouse 55 °C warm water, tail immersion assay 30 min after the morning injection each day (see below).
Antinociceptive Testing: the 55 °C Warm Water, Tail Immersion Assay
The nociceptive stimulus was 55 °C water, with the latency to withdraw the tail taken as the end point (19). After determining baseline latencies before the day of testing, the mice received a single intraperitoneal dose of the kappa agonist U50,488, and the antinoci-ceptive effect was measured 30 min after injection. A cut-off time of 15 s was used throughout the study for analgesic testing. If the mouse failed to display a tail withdrawal in that time, the tail was removed from the water and the animal was assigned a maximal antinociceptive score. Mice that showed no response within 5 s in the initial predrug latency test would have been excluded from the experiment; however, no mice were excluded by this criterion.
Data Analysis
Signal intensity in Western blots and 32P autoradio-grams were quantified with Image 1.62 software (National Institutes of Health) on an Apple G4 computer. Statistical significance throughout the study was defined by p < 0.05 as determined by Student’s t test, performed with Excel 97 software (Microsoft, Redmond, WA).
RESULTS
Mice Chronically Treated with Kappa Agonist Demonstrate Prolonged Analgesic Tolerance
The antinociceptive effect of an acute dose of 30 mg/kg U50,488 was measured using the 55 °C warm water, tail withdrawal assay. Antinociception reached a peak 30 min after U50,488 injection, and tail withdrawal latency increased 4.7-fold to 11.0 ± 1.6 s (n = 9). Consistent with previous results demonstrating the kappa receptor selectivity of U50,488 (20), nor-BNI (10 mg/kg, intraperitoneally) significantly (p < 0.01) blocked the effects of U50,488; nor-BNI-pretreated mice displayed a tail withdrawal latency of 3.2 ± 0.3 s 30 min after 30 mg/kg U50,488 (n = 4). To induce opioid tolerance, we used a standard protocol in which escalating doses of U50,488 were given twice daily for 4 days (18). On the fifth day, a test dose of U50,488 (25 mg/kg, intraperitoneal) produced a tail withdrawal latency of 2.0 ± 0.1 s that was not significantly greater than base-line or repeated saline values and was significantly (p < 0.01) less than the response to an acute injection of U50,488 (Fig. 1B). Naïve animals, defined as mice receiving vehicle injections for 4 days on the same schedule as the U50,488-treated set, showed no change in latency over the course of injections and had a normal antinociceptive response to 25 mg/kg U50,488 on the fifth day of injection (9.3 ± 1.3 s, p > 0.05). The lack of change in response latency following vehicle injections and in response to U50,488 on day 5 suggests that neither handling stress nor repeated testing produced the tolerance observed in this study (Fig. 1A).
Fig. 1. Reversal of analgesic tolerance following chronic kappa opioid agonist treatment is slow.
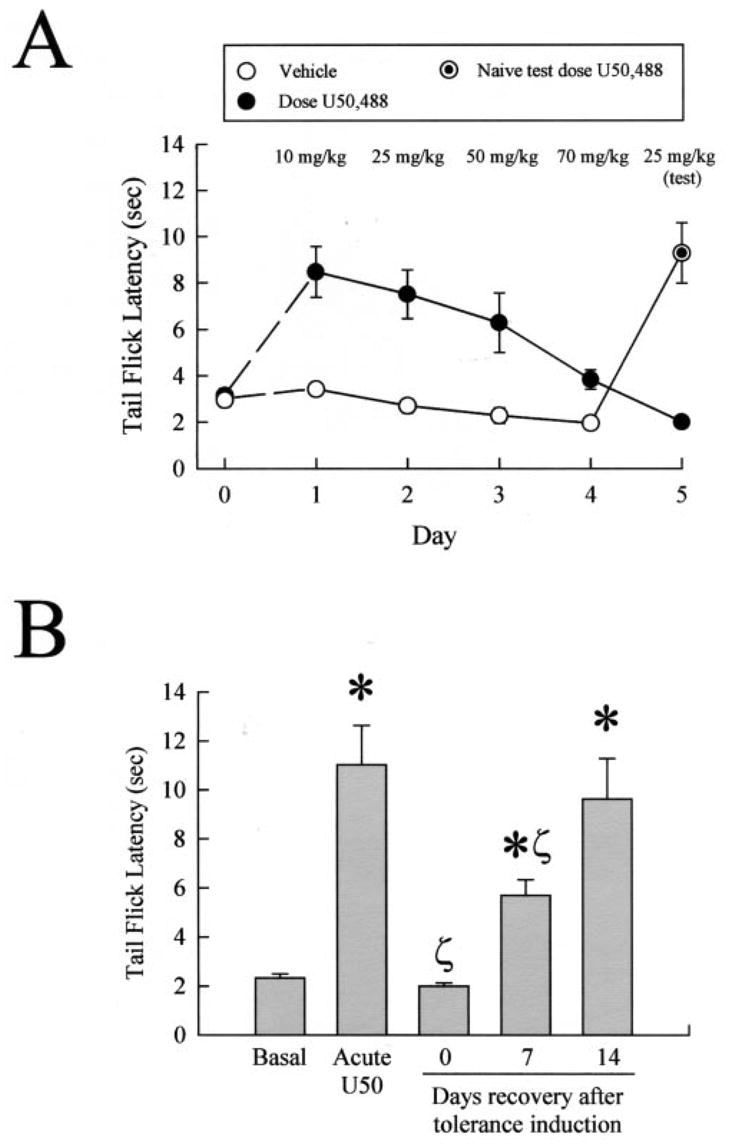
A, mice developed tolerance to the antinociceptive effects of U50,488 following repeated administration. Latency of tail withdrawal from the 55 °C warm water was measured in untreated animals, which were then injected over 4 days with increasing doses of U50,488 (●) or vehicle (○) as detailed under “Experimental Procedures.” Antinociceptive response to morning injection of U50,488 was measured each day, 30 min after administration, to monitor the generation of tolerance. Animals pretreated for 5 days with U50,488 developed pronounced antinociceptive tolerance (day 5, ●), whereas naïve animals receiving only vehicle prior to the test dose showed a normal antinociceptive effect from 25 mg/kg U50,488 (⊙). Data points represent the means ± S.E. of 10 mice from two separate experiments. In some cases, symbol size exceeds error bar width. B, mice made tolerant to U50,488 recover sensitivity to U50,488 after 2 weeks being drug-free. Mice made tolerant to U50,488 as above were then allowed to recover 1 or 2 weeks (12 or 19 days after initial drug administration) without drug. After the recovery period, animals were tested by measuring the tail withdrawal latency response 30 min after intraperitoneal 25 mg/kg U50,488. Data points represent the means ± S.E. of 7–8 mice from two separate experiments. *, p < 0.05 compared with basal group;ζ, p < 0.05 compared with acute U50,488.
After allowing the C57Bl/6 mice to recover for 7 days following the 5-day induction protocol, mice showed only a partial return of U50,488 sensitivity (Fig. 1B). Tolerant mice allowed to recover for 7 days and then treated with 25 mg/kg U50,488 had tail withdrawal latencies of 5.7 ± 0.6 s, significantly less than that produced in drug naïve mice (p < 0.05, n = 7). Complete recovery was only evident 14 days after tolerance induction, with mice demonstrating a tail withdrawal latency of 9.6 ± 1.9 s after 25 mg/kg U50,488. These results demonstrate that the tolerance induced by repeated agonist exposure was reversible, but required a prolonged drug-free interval. The mechanisms responsible for the induction of analgesic tolerance and the slow reversal were not clear, but prior in vitro studies suggest opioid receptor phosphorylation and dephosphorylation may have been involved (3).
Phosphorylation of KOR Is Elevated in Mice Acutely or Chronically Treated with Kappa Agonist
To test the hypothesis that the analgesic tolerance observed resulted from kappa receptor phosphorylation, we utilized an antibody raised against phosphoserine 369 within the KOR (13). This sequence was previously shown by site-directed mutagenesis to be required for in vitro receptor desensitization (11). The resulting affinity-purified anti-phosphopeptide antibody (KOR-P) was previously shown to recognize the phosphorylated KOR-(359–372) but not the matching nonphosphorylated peptide in enzyme-linked immunosorbent assay (13). The KOR-P antibody used in Western analysis detected an ~85-kDa mass protein band expressed in HEK293 cells transfected with GFP-tagged KOR but not untransfected cells (Fig. 2A, left panel). Additional confocal imaging studies using the KOR-P antibody to selectively label phosphorylated kappa receptors in agonist-treated transfected cells previously confirmed the specificity of this reagent (13). Specificity of the KOR-P antibody for the kappa opioid receptor was further demonstrated with Western blot analysis of glycoproteins isolated from KOR wild-type (+/+) and knockout (−/−) mouse brains (17) by WGA purification. The KOR-P antibody labeled protein in the KOR(+/+) samples, but no signal was detected in protein from KOR(−/−) brains (Fig. 2A, right panel). In this nondenaturing gel, the KOR-P immunoreactivity moved with an apparent molecular mass of ~85 kDa as predicted (Fig. 2A, right panel). To assess the utility of this antibody for in vivo studies, C57Bl/6 wild-type mice were injected with U50,488 and sacrificed 30 min later, and brain membrane glycoproteins were isolated by WGA purification. In Western blot analyses of the isolated protein both a previously characterized antibody directed against a nonphosphorylated domain of the C-terminal region of KOR (7) and the phosphoselective KOR-P antibody recognized a brain membrane protein of molecular mass ~55 kDa under reducing conditions (Fig. 2B). The observed molecular mass is consistent with a glycosylated form of the mouse KOR (7, 21). Western blot analysis using the KOR-P antibody demonstrated a 59 ± 22% increase in the labeling intensity of KOR protein isolated from mice acutely treated with U50,488 over KOR protein from untreated, control mice (Fig. 2, B and C). Western blots comparing isolated KOR protein from both untreated and acute-U50,488-treated mouse brains showed no significant change in KOR labeling measured by the nonphosphoselective antibody (Fig. 2B). WGA-purified protein from the brains of nor-BNI, then U50,488, pretreated mice displayed no significant difference in KOR-P labeling compared with untreated controls (p > 0.05, n = 16). The results confirm that the change in KOR-P labeling required kappa receptor activation and further suggest that opioid receptor phosphorylation may be initiated during the earliest exposure to agonist and not require chronic drug administration.
Fig. 2. Opioid agonist-induced tolerance corresponds to an increase in kappa opioid receptor phosphorylation detected by the KOR-phosphoselective antibody, KOR-P.
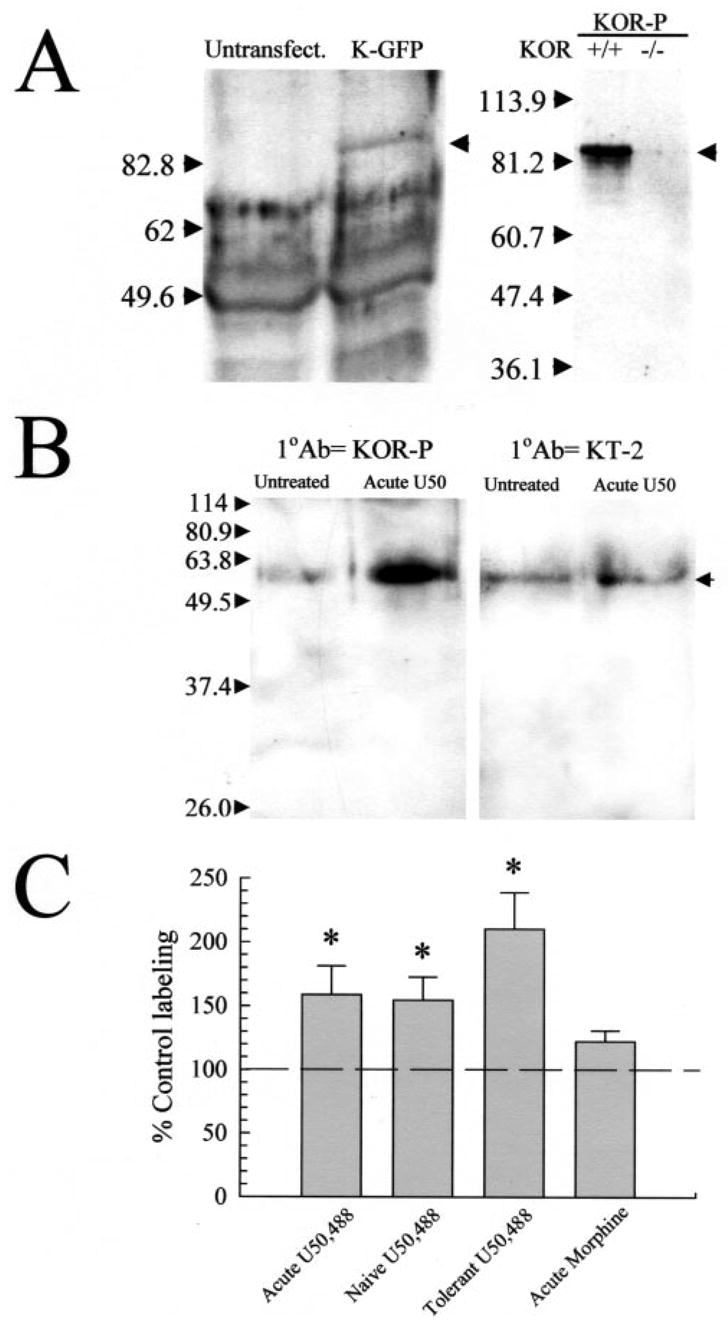
A, kappa opioid receptor selective binding by the KOR-P antibody. Left panel: representative gel shows immunoprecipitated KOR-GFP labeled by KOR-P. Lysates from untransfected HEK293 cells or cells transiently transfected with KOR-GFP cDNA were immunoprecipitated with 20 μg of KE-4 and then analyzed by Western blot with 5 μg of KOR-P. Cells were pre-treated with 10 μM U50,488 for 30 min at 37 °C. The arrow on the right side highlights an ~85-kDa band corresponding to the predicted molecular mass of the KOR-GFP fusion protein which was labeled in the KOR-GFP-transfected cell lysate but not in the untransfected cell lysates. Right panel: representative Western blot of isolated mouse brain membrane protein (40 μg/lane) taken from either KOR wild-type (WT) or knockout (KO) mice, and incubated with KOR-P. For these experiments, brain protein was resolved on nonreducing PAGE gels. The KOR-P antibody labeled protein in the WT, but not KO, brain protein samples. Identical specificity results were obtained with the KOR-selective antibody, KT-2, labeling of KOR(+/+) and KOR(−/−) brain membrane proteins (data not shown). B, representative Western blots of isolated mouse brain membrane protein (90 μg/lane), incubated with KOR-P (left) or KT-2 (right). For these and subsequent experiments, brain protein was resolved on reducing PAGE gels containing 0.01% SDS. Both antibodies recognized a protein of ~55 kDa corresponding to the KOR in samples from untreated control animals (Untreated). Pre-treatment with 30 mg/kg intraperitoneal U50,488 (Acute) increased blotting intensity with KOR-P compared with untreated controls. This increase in blotting intensity indicates an increase in KOR phosphorylation, as the nonphosphospecific KOR antibodies KT-2 (shown) or KE-4 (not shown) detected no significant change. C, phosphorylation state of the KOR is increased in animals following agonist treatment. A single administration of the kappa-selective agonist (Acute U50,488 group) increased KOR phosphorylation ~60%, whereas chronic administration more than doubles basal phosphorylation of the KOR (Tolerant). Mice treated with vehicle for 5 days then tested with a single injection of 25 mg/kg U50,488 on day 5 (Naïve U50,488 group) demonstrated a 63 ± 16% increase in KOR-P labeling, comparable to that produced by a single U50,488 injection. In contrast, while acute morphine (30 mg/kg, intraperitoneally) produced a tail withdrawal latency at the 15 s cutoff in the 55 °C warm water tail withdrawal assay 30 min after administration, it did not significantly increase the phosphorylation of KOR measured in subsequent Western blot analysis using KOR-P antibody. The lack of effect of morphine further demonstrates that specific kappa receptor activation was required for increased KOR-P labeling. The graph summarizes the analysis results of protein blots using NIH densitometry software of the ECL image, standardized against matching, untreated control protein from the same experiment. Data points represent the means ± S.E. of 26–33 independent experiments. Statistical significance was determined with Student’s paired t test. *, p < 0.05 against matching KE-4 antibody-labeled band.
To examine the hypothesis that phosphorylation of KOR serine 369 coincides with agonist desensitization and tolerance, Western blot analyses using KOR-P or nonphosphoselective antibodies were performed with protein isolated from mice made tolerant to U50,488. The amount of protein detected by the nonphosphoselective antibody showed no significant change after chronic kappa receptor activation (Fig. 3, A and B). In contrast, KOR isolated from mice made tolerant to U50,488 showed a 101 ± 29% increase in KOR-P labeling intensity compared with untreated controls (Fig. 2C). The increase in KOR-P labeling following acute or chronic agonist treatment without a change in KE-4 or KT-2 labeling supports the conclusion that in vivo exposure to U50,488 results in kappa receptor phosphorylation on serine 369.
Fig. 3. Kappa receptor number is unchanged by acute or chronic U50,488 treatment.
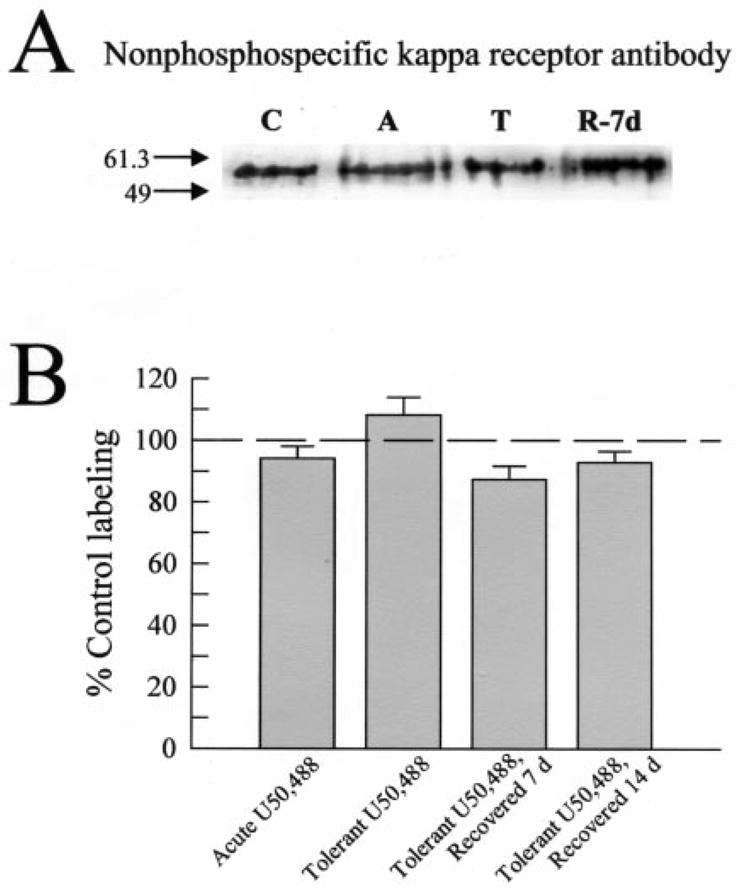
A, representative Western blot analysis with KE-4 antibody and 90 μg/lane mouse brain membrane protein isolated from untreated control mice (“C”), mice pretreated acutely with U50,488 (25 mg/kg intraperitoneal, “A”), mice made tolerant to U50,488 and sacrificed immediately (“T”), or after a recovery period of 7 days (“R-7d”). KE-4 antibody labeling intensity was consistent across all protein samples, suggesting the total number of KOR remained unchanged despite the different pretreatments with U50,488 and recovery time. B, summary graph of densitometry analysis of Western blots shows KE-4 antibody labeling of KOR from each set of pretreated mice. No significant differences were found in KE-4 antibody labeling across the tissue sets tested. Data points represent the means ± S.E. of 18 –24 independent Western blot experiments.
GRK3 Knockout Mice Do Not Phosphorylate KOR Serine 369 after Acute Kappa Agonist Treatment
If the behavioral tolerance and elevation of KOR-P labeling observed were both caused by kappa receptor phosphorylation, kinase inhibition would be predicted to block both changes. The kinase responsible is not known, but a plausible candidate is the G-protein receptor kinase 3 (GRK3), previously shown to be particularly effective in inducing β-arrestin-dependent KOR desensitization in vitro (11). To examine the hypothesis that GRK3 mediates agonist-induced phosphorylation of KOR, mice either expressing or lacking the GRK3 protein (16) were administered acute or chronic doses of U50,488. The GRK3 knockout (−/−) mice and wild-type (+/+) littermates demonstrated equivalent base-line latencies in the tail flick assay (3.2 ± 0.2 s and 2.8 ± 0.3 s, respectively, n = 8). Both GRK3(+/+) and GRK3(−/−) mice showed strong and equivalent antinociceptive responses to U50,488 30 min after acute administration (10 mg/kg, intra-peritoneally, Fig. 4A). Acute U50,488 at 30 mg/kg gave a maximal tail withdrawal latency for both the GRK3(+/+) and GRK3(−/−) mice. KOR-P labeling doubled in intensity after 30 mg/kg U50,488-treatment of GRK3(+/+) mice (Fig. 4, B and C). In contrast, GRK3(−/−) mice did not significantly change in KOR-P labeling intensity following agonist treatment (Fig. 4, B and C). These results suggest that GRK3 may be uniquely responsible for the phosphorylation of KOR serine 369 detected by KOR-P labeling.
Fig. 4. Kappa agonist administration induces KOR phosphorylation in mice mediated by GRK3.
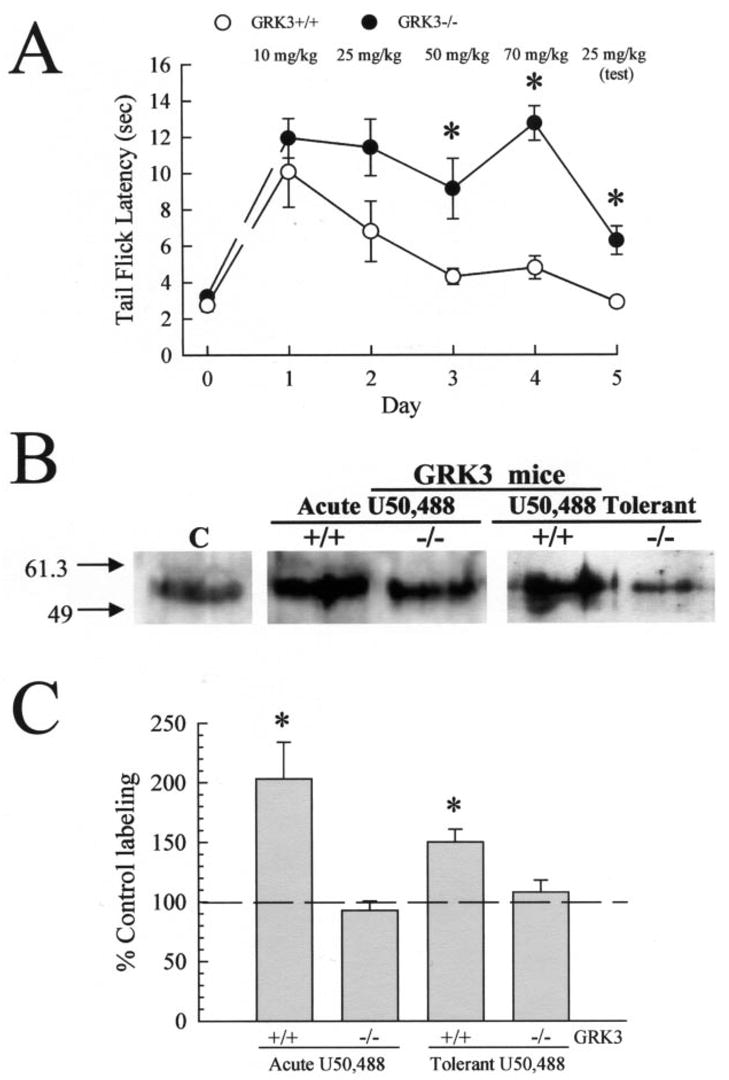
A, onset of U50,488 antinociceptive tolerance was impaired in GRK3(−/−) mice in the mouse 55 °C warm water, tail withdrawal assay. Latency to tail flick from a warm water stimulus was measured in untreated animals, and then increasing doses of U50,488 were administered over 4 days to GRK3(+/+) (○) or (−/−) (●) mice as detailed under “Experimental Procedures.” Antinociceptive response to morning injection of U50,488 was measured each day, 30 min after administration, to monitor the progress of antinociceptive tolerance. Development of antinociceptive tolerance was tested on day 5 with a test dose of 25 mg/kg U50,488. GRK3(+/+) animals pretreated for the week with U50,488 developed pronounced antinociceptive tolerance (day 5, ○), whereas GRK3(−/−) animals showed a reduced U50,488 antinociceptive tolerance with delayed onset of effect (●). Data points represent the means ± S.E. of 8 mice from two separate determinations. Both GRK3(+/+) and (−/−) mice were also injected acutely with 30 mg/kg intraperitoneal U50,488 and sacrificed 30 min later. B, representative Western blot analysis of 90 μg/lane brain membrane protein isolated from untreated C57Bl/6 mice (Control, C) or GRK3(+/+) or GRK3(−/−) mice treated acutely (center) or chronically (right) with U50,488 and blotted with 5 μg/ml KOR-P. KOR protein isolated from U50,488-pre-treated GRK(+/+), but not GRK(−/−), mice showed increased labeling by KOR-P. KE-4 showed no change in blotting intensity between the protein sets tested. A summary graph is shown in C, with data representing the means ± S.E. of 19–20 independent experiments analyzed with NIH Image software of the ECL image and standardized against matching, untreated control C57Bl/6 mouse brain protein from the same experiment. Brain membrane protein was isolated by WGA column purification in all cases as detailed under “Experimental Procedures.” Statistical significance was determined with Student’s paired t test. *, p < 0.05 against matching KE-4 antibody-labeled band.
Because the GRK3(+/+) and knockout mice showed equivalent sensitivities to U50,488, it was possible to next investigate the involvement of GRK3 in the induction of kappa opioid analgesic tolerance. GRK3 knockout (−/−) mice and wild-type (+/+) littermates were administered escalating doses of U50,488 twice daily for 4 days as in the previous experiment. The development of analgesic tolerance was monitored daily using the tail immersion test each morning after administration of U50,488 (Fig. 4A). As expected, the progression of analgesic tolerance for wild-type GRK3(+/+) littermate mice over 4 days was comparable to C57Bl/6 mice as previously described (Fig. 1A). In contrast, GRK3(−/−) knockout mice showed significantly less tolerance to U50,488 on days 3–5 than matched, wild-type littermates (Fig. 4A). Tolerant GRK3(+/+) mice showed a 50 ± 11% increase in KOR-P labeling; however, KOR-P labeling did not increase significantly in GRK3(−/−) mice (Fig. 4, B and C). Thus, GRK3 phosphorylation of serine 369 is likely to have a key role in the development of antinociceptive tolerance to kappa opioids. However, GRK3 gene deletion did not completely block kappa tolerance. Although the response of the GRK3(−/−) mice to 25 mg/kg U50,488 on day 5 (6.3 ± 0.8 s) was significantly greater than the response of GRK3(+/+) mice (2.9 ± 0.3 s, p < 0.01), the GRK3(−/−) response was still significantly less than the wild-type response to an acute 25 mg/kg dose of U50,488 (11.0 ± 1.6 s, p < 0.05); thus other mechanisms may also contribute to kappa opioid analgesic tolerance.
Tolerance Reversal Rate
If the receptor phosphorylation detected by the KOR-P antibody was responsible for a substantial portion of the analgesic tolerance observed, then the slow reversal of tolerance should be associated with slow dephosphorylation. To test this prediction, we next measured the rate of decline in KOR-P labeling following acute or chronic agonist exposure. Surprisingly, mice tested 7 days after a single dose of U50,488 (30 mg/kg, intraperitoneally) showed a modest but significant degree of antinociceptive tolerance (6.6 ± 2.0 s latency, n = 8, p < 0.01). Only after 14 days of recovery did mice acutely treated with U50,488 show sensitivity to U50,488 similar to the initial, acute response (11.1 ± 2.0 s, n = 5, p > 0.05). Consistent with the behavioral result, KOR-P labeling remained significantly elevated (34.2 ± 9.1%) over basal labeling (p < 0.05, n = 11–15) in samples isolated from mice administered a single acute dose of U50,488 1 week previously. Following chronic U50,488 administration, the recovery to basal KOR-P labeling was similarly slow. KOR-P labeling was significantly elevated 1-week following tolerance induction and only returned to base line 14-days after the last U50,488 dose (Fig. 5, A and B). During the recovery period, the levels of kappa receptor did not significantly change as determined by Western blot analysis using the nonphosphoselective antibody (Fig. 3).
Fig. 5. Return to basal KOR phosphorylation state in U50,488-tolerant mice follows recovery from antinociceptive tolerance after 14 days.
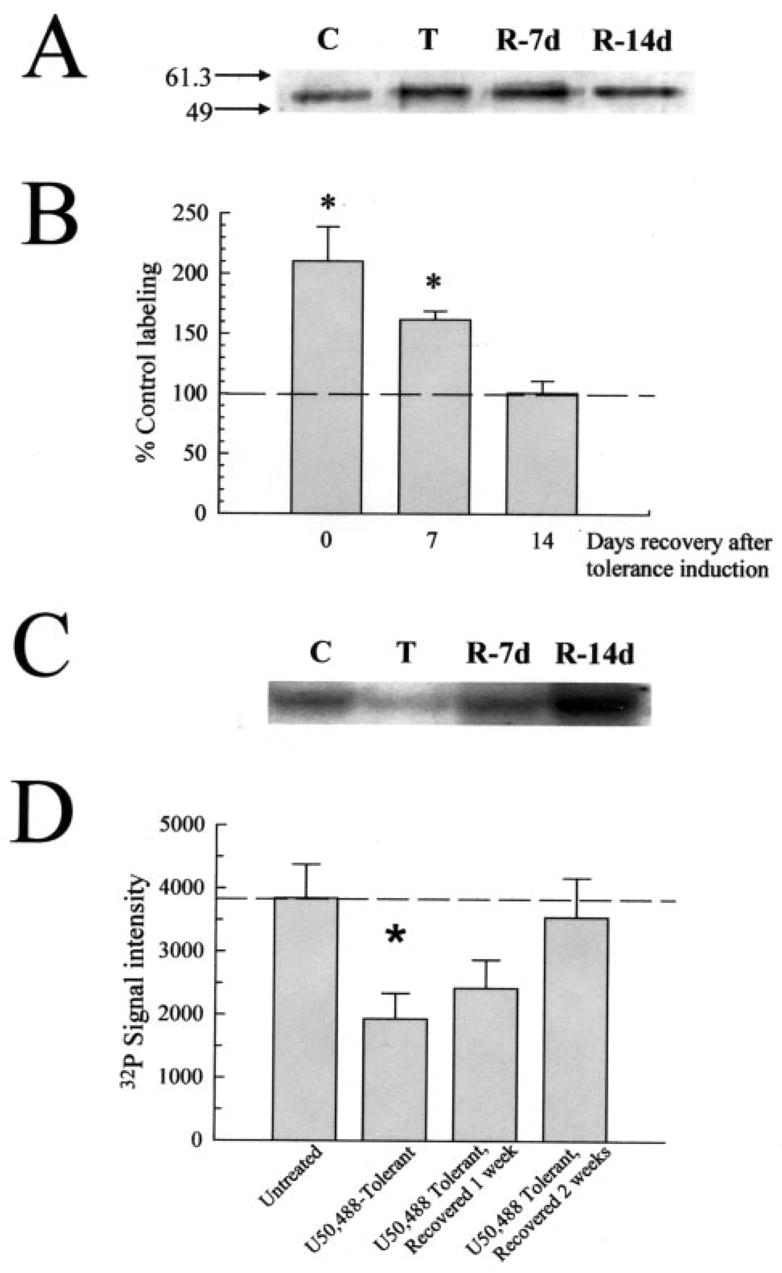
A, representative Western blot analysis with KOR-P and 90 μg/lane mouse brain membrane protein isolated from untreated control mice (“C”), or mice made tolerant to U50,488 and sacrificed immediately (“T”), or after a recovery period of 7 or 14 days (“R-7d” and “R-14d,” respectively). KOR-P labeling intensity was elevated in protein samples from tolerant and 1-week recovered animals, suggestive of prolonged KOR phosphorylation matching the demonstration of antinociceptive tolerance. B, summary graph of densitometry analysis of Western blots shows that the phosphorylation intensity of KOR from tolerant mice requires 2 weeks to return to basal values. Data points represent the means ± S.E. of 6 –24 independent experiments. *, p < 0.01 against matching KE-4-labeled band. C, representative PAGE gel with 90 mg/lane mouse brain protein isolated from untreated control mice (“C”), or mice made tolerant to U50,488 and sacrificed immediately (“T”), or after a recovery period of 7 or 14 days (“R-7d” and “R-14d,” respectively) following 32P incorporation with isolated GRK3 enzyme. Following GRK3/32P incubation, all samples were immunoprecipitated with the KOR-selective antibody, KT-2 as described under “Experimental Procedures.” Adjusting for background, 32P labeling intensity was decreased in protein samples from tolerant and 1-week recovered animals, suggestive of prolonged KOR phosphorylation. These results match the changes KOR-P labeling and antinociceptive tolerance, indicating that GRK3 phosphorylation sites were occupied by nonradioactive phosphate. D, summary graph of GRK3/32P incorporation experiments shows 32P signal intensity level of KOR from tolerant mice requires 2 weeks to return to basal values. Data points represent the means ± S.E. of five independent experiments. *, p < 0.05 against matching untreated control brain protein value.
The elevated KOR-P labeling detected in brain protein isolated from U50,488-tolerant mice was unexpected because receptor dephosphorylation required less than 1 h in vitro as measured by the loss of KOR-P labeling of agonist-treated, KOR-transfected cells (13). To verify this result, 32P incorporation experiments were performed with isolated GRK3 and WGA-purified protein isolated from the brains of untreated mice or mice made tolerant to U50,488 and allowed to recover up to 2 weeks. KOR protein immunoprecipitated with the KT-2 antibody was separated by PAGE and exposed to x-ray film to demonstrate incorporation of the 32P label. In these experiments, KOR phosphorylated at serine 369 would be expected to incorporate less 32P than KOR from untreated mouse brain, resulting in less 32P signal intensity in these samples. Consistent with Western blots using the KOR-P antibody, 32P labeling of KOR immunoprecipitated from tolerant mice was significantly reduced 46 ± 7% from untreated control protein (Fig. 5, C and D). Moreover, 32P labeling was likewise reduced 34 ± 10% from base line in KOR immunoprecipitated from the brains of mice allowed to recover 1 week after the last U50,488 dose and returned to values not significantly different from untreated mouse brain protein 14 days after the induction of U50,488 tolerance (Fig. 5, C and D). Because KOR protein levels did not significantly change in the recovery period (Fig. 3), and the same protein concentration was used across incubations, these data support the conclusions drawn from the Western blot analysis with the KOR-P antibody.
The surprisingly slow return to basal phosphorylation of KOR might have resulted from a slow pharmacokinetic clearance of U50,488. To test this possibility, mice made tolerant to U50,488 were subsequently injected with 10 mg/kg nor-BNI immediately following the 5-day induction protocol and again 3 days later. This treatment with nor-BNI was shown to fully block the kappa opioid receptors for a week (22), and in vivo nor-BNI treatment alone did not affect KOR-P labeling (data not shown). The intensity of KOR-P labeling of protein isolated 1 week after the tolerance/nor-BNI protocol was still 85 ± 18% greater than untreated control levels (p < 0.01, n = 10). Thus, recovery to basal KOR phosphorylation state in U50,488-tolerant mice could not be accelerated with antagonist treatment. Together, these data suggest that the recovering opioid-tolerant mice experienced prolonged elevation of kappa receptor phosphorylation that declined at the same rate as the loss of analgesic tolerance.
Prolonged Recovery of KOR- but Not MOR-mediated Antinociception following Irreversible Antagonist Treatment
As noted above, the slow rate of recovery was unexpected, because receptor dephosphorylation required less than 1 h in vitro as measured by the loss of KOR-P labeling of agonist-treated, KOR-transfected cells (13). Furthermore, a 2-week recovery time seemed slower than the predicted rate of new receptor synthesis. Although the kappa receptor synthesis rate in vivo is not known, mu opioid receptor synthesis rates in vivo have been estimated by measuring the recovery rate of morphine sensitivity following irreversible antagonism (23). In that study, morphine effectiveness was 90 –95% restored within 3 days following β-chlornaltrexamine (β-CNA) treatment. If the rate of new kappa receptor synthesis was faster than the rate of KOR-P labeling decay, the result would support an active rephosphorylation mechanism rather than a failure of phos-phatase activity. A hypothetical example of an active rephos-phorylation mechanism would be if the kinase remained active and continually rephosphorylated previously desensitized receptors long after agonist was removed. To distinguish these alternative mechanisms, mice were pretreated with 2.4 μg of β-CNA, intracerebroventricularly, then tested for sensitivity to U50,488 or morphine (Fig. 6). Prior studies have established that β-CNA specifically antagonizes opiate effects by covalently binding to mu, delta, and kappa opioid receptors (24). Following β-CNA treatment, the analgesic response to U50,488 required 2 weeks to recover (Fig. 6A). In contrast, morphine effects recovered within 3 days, as expected (Fig. 6B). Thus, these results do not support an active rephosphorylation mechanism and instead suggest that kappa receptors phosphorylated on serine 369 fail to dephosphorylate in vivo. The basis for the sustained phosphorylation is not clear from these studies, but the results suggest that recovery of response following opioid tolerance may require new receptor synthesis, transport to active site, and assembly of a functional signaling complex rather than the readily reversible phosphorylation event previously assumed.
Fig. 6. Mice pretreated with β-CNA require prolonged recovery for kappa, but not mu, opioid-induced antinociception.
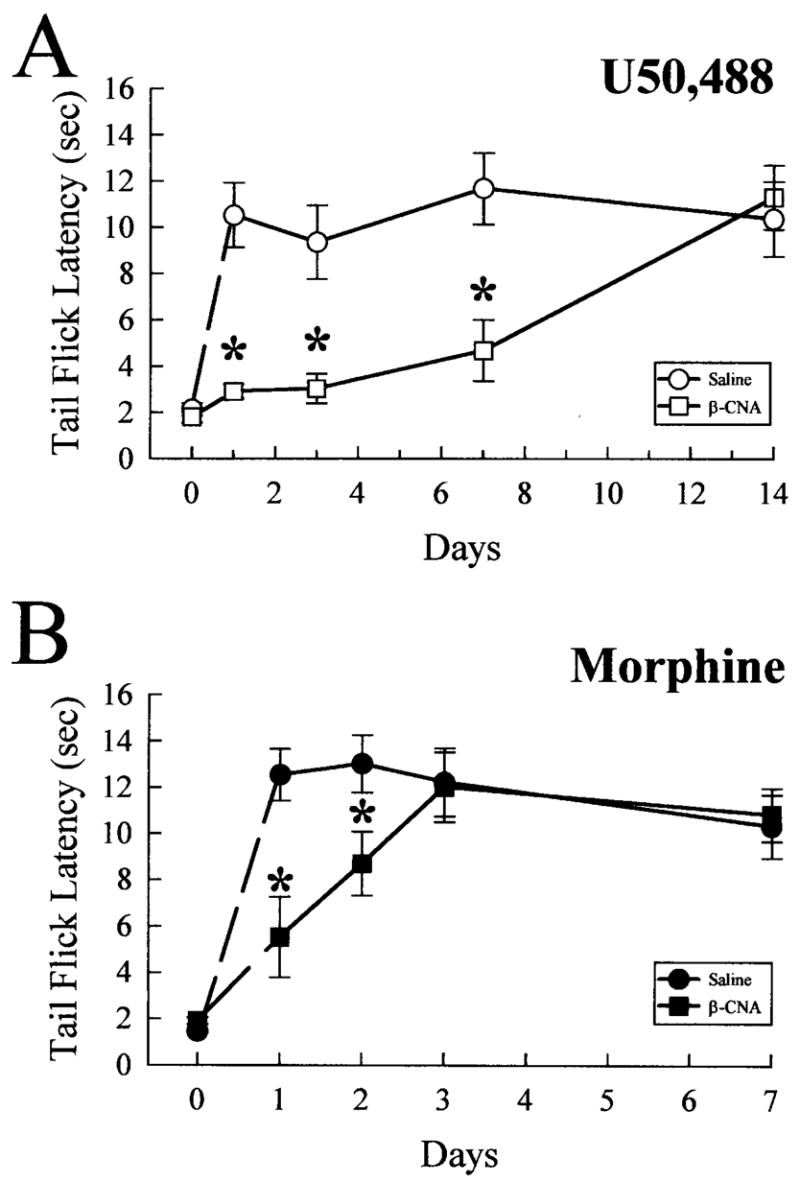
C57Bl/6 mice were treated with saline (0.9% intraperitoneally, circles) or β-CNA (2.4 nmol intracerebroventricularly, squares) and returned to their home cages for 1–14 days prior to use in analgesic testing with opioid agonists. A, antinociception induced by the kappa-selective agonist, U50,488, is blocked by pretreatment with opioid antagonists up to 2 weeks. After recovering from antagonist pretreatment for 1–14 days, mice were administered U50,488 (25 mg/kg, intraperitoneally), and the antinociceptive effect was measured 30 min later. Pretreatment with the irreversible opioid antagonist β-CNA (open squares) produced significant antagonism of U50,488-induced antinociception up to 7 days after antagonist administration, with recovery observed after 14 days. In contrast, similar β-CNA pre-treatment (closed squares, B) significantly reduced the antinociception induced by subsequent administration of morphine (30 mg/kg, intraperitoneally, closed circles; B) only up to 2 days, with recovery observed after 3 days. Each animal received only a single dose of agonist (U50,488 or morphine). Data points represent the means ± S.E. of 9 –14 mice. *, p < 0.05 against matching saline pre-treated time point.
DISCUSSION
Using a novel phosphospecific antibody against the kappa opioid receptor, we monitored the phosphorylation of a specific residue in KOR suggested by prior studies to regulate homologous receptor desensitization. The principal findings of this study were that 1) agonist activation of the kappa receptor in mice resulted in phosphorylation of serine 369 within the C-terminal domain of the receptor; 2) kappa opioid receptor phosphorylation at serine 369 was strongly dependent on GRK3 expression; and 3) the rate of analgesic tolerance development and the prolonged recovery of opiate sensitivity were correlated with serine 369 phosphorylation.
The selectivity of the KOR-P antibody was defined by several means. Previously, KOR-P reacted with the synthetic phosphopeptide used as the immunogen, but not the unphosphoryl-ated version of the same peptide as measured by enzyme-linked immunosorbent assay (13). KOR-P reacted with membrane protein expressed in agonist-treated HEK293 cells transfected with KOR, but not with untransfected cells or cells transfected with KOR(S369A) in confocal microscopy experiments (13). In the present study, KOR-P detected a band at the predicted molecular mass from transfected cells first immunoprecipitated by a different antibody raised against a separate portion of the kappa opioid receptor. KOR-P also reacted with membrane protein isolated from the brains of KOR wild-type mice, but not knockout littermates. Moreover, KOR-P selectively recognized a solubilized brain membrane protein of the same apparent mass (~55 kDa) as was recognized by two different antibodies raised against separate portions of the kappa opioid receptor. Agonist treatment in mice additionally increased KOR-P antibody labeling, and this increase was blocked by a kappa receptor antagonist and was not evident in the absence of GRK3. These results strongly suggest that the KOR-P antibody selectively recognized the phosphorylated serine 369 form of KOR.
We were surprised to find that mice lacking GRK3 failed to significantly increase KOR(S369) phosphorylation after agonist treatment. The lack of redundancy suggests that GRK3 may preferentially phosphorylate serine 369 in KOR in vivo. Because the conclusion is based on a gene knockout model, indirect effects of GRK3 loss on neuronal development have not been excluded; however, the potential association between GRK3 and KOR is intriguing. The apparent selectivity might result from a colocalization of GRK3 and KOR in a subcellular compartment within neurons. Evidence for a physical association of signaling receptors and regulatory proteins by preassembled complexes is common in other systems (25) and has been suggested for GPCRs (26). Alternatively, substrate specificity might result from the presence of other ancillary proteins or GRK regulators expressed in fully differentiated neurons as opposed to transfected cell systems. Although further work is required, the results suggest that a selective GRK3 antagonist might specifically reduce tolerance to kappa opioid agonists.
Knockout of GRK3 significantly reduced the magnitude and delayed the onset of antinociceptive tolerance. No significant phosphorylation of KOR at serine 369 was detected in these animals, suggesting the tolerance seen in these animals after chronic U50,488 administration did not result from serine 369 phosphorylation. Phosphorylations at other KOR sites or other types of compensatory changes are possible (see Refs. 2 and 3), but these mechanisms were not further characterized in this study. Consistent with previously characterized G-protein-coupled receptor desensitization mechanisms, KOR desensitization was shown to require both GRK-mediated phosphorylation and β-arrestin binding (11). Results in the present study extend prior work using β-arrestin gene deletion. Mice lacking β-arrestin-2 did not desensitize the mu opioid receptor after chronic morphine treatment and failed to develop antinociceptive tolerance (27). In the present study, tolerance to kappa agonist treatment of GRK3(−/−) mice was delayed, supporting a role for GRK/β-arrestin-mediated desensitization in opioid analgesic tolerance. The delayed tolerance in the absence of GRK3 may have been produced by a PKC-mediated mechanism as demonstrated previously for morphine tolerance (28). Thus, deletion of either GRK3 or β-arrestin-2 affects opioid receptor desensitization and analgesic tolerance. Interestingly, mice lacking the β-arrestin 2 gene showed potentiation and prolongation of the analgesic effect of morphine (29), whereas GRK3 gene deletion did not affect the acute response to kappa agonist in the present study. The basis for this difference is not clear, but the difference may be consistent with the additional role of β-arrestin in G-protein-coupled receptor scaffolding (26).
The third major finding in this study was that the kinetics of phosphorylation followed the changes in the behavioral response. Acute exposure to a kappa agonist initiated both a behavioral response and KOR-P labeling. Assuming that KOR-P labeling is a valid marker of receptor desensitization, the result indicates that the onset of tolerance begins with the initial exposure. Behavioral tolerance to opiates consistently increases during several days of exposure, and that was confirmed in this study. As predicted, KOR labeling was greater after 5 days of exposure than after 30 min. Tolerance did not result in a change in receptor density as measured by KOR labeling by a nonphosphoselective antibody, and this result is consistent with prior studies showing that opioid receptor numbers do not change in the tolerant animal (see Ref. 30). The slow recovery of behavioral sensitivity following the cessation of opiate administration has been observed previously (31). The novel observation in this study is that the decrement in KOR-P labeling was similarly slow. The data suggest that the slow return of behavioral sensitivity was caused by the continued presence of phosphorylated KOR. This conclusion was verified by back-phosphorylation experiments incorporating 32P into the KOR by incubation with GRK3.
The basis for the sustained receptor phosphorylation and slow recovery of KOR sensitivity is not clear. Studies using transfected opiate receptors indicate that in cell lines, dephosphorylation occurs quickly following cessation of agonist stimulation (13, 32, 33). In the present study, either KOR dephosphorylation occurs slowly or mechanisms exist allowing continued rephosphorylation. Without selective kinase inhibitors, these possibilities cannot be resolved. Alternatively, the recovery of KOR sensitivity in vivo may involve new receptor synthesis rather than dephosphorylation. Notably, U50,488-induced antinociception was antagonized by β-CNA pretreatment in the present study and was restored only after 14 days, a recovery time course matching the duration of kappa agonist-induced antinociceptive tolerance. Thus, surprisingly, new receptor synthesis could account for the recovery without requiring receptor dephosphorylation.
An important implication of the results of this study is that assembly of a functional kappa receptor signaling complex in neurons appears to be irreversibly disrupted by the events initiated by receptor phosphorylation. Once phosphorylated by GRK3, KOR is presumably sequestered by a β-arrestin-mediated process, but it is neither degraded (there is no loss of KOR protein) nor dephosphorylated (there is no loss of KOR-P signal). This outcome is clearly different from that observed in transfected cells where signaling is quickly restored by receptor recycling initiated by okadaic acid-sensitive phosphatases (13). The simplest explanation is that phosphorylated KOR is sequestered in vivo at a site inaccessible to intrinsic phosphatases, but the nature of that compartment is presently unknown. Liu-Chen and colleagues (34, 35) have shown that the rodent kappa receptors are not readily internalized following activation with low (≤1 μM) concentrations of synthetic kappa agonists, although strong agonist activation (10 μM U50,488) was shown to induce clustering and internalization of transfected rodent KOR (13), and incubation of R1.1 mouse thymoma cells with 100 nM U50,488 for 24 h resulted in a 50% reduction of KOR number as measured in radioligand binding assays (36). If KOR naturally expressed in mouse brain is resistant to internalization, receptor dephosphorylation and recycling might also be impaired. A test of this prediction requires further immunocytochemical analysis of receptor trafficking in vivo following agonist administration.
Overall, these findings emphasize the significance of serine 369 phosphorylation as an initial step in KOR desensitization and internalization and strengthen the connection between the results obtained using transfected cells and those from in vivo tolerance studies. In conclusion, the mechanisms responsible for tolerance to opiate drugs in brain following sustained exposure are likely to include phosphorylation by G-protein receptor kinase.
Acknowledgments
KOR(−/−) mice were generated and maintained with grant support from RO1 DA-09040 from NIDA to Dr. Pintar. Dr. Suzanne Appleyard (Vollum Institute) arranged the phosphopep-tide synthesis and initial rabbit immunizations while a graduate student in the laboratory. We thank Dr. Jeffrey Benovic (Jefferson Medical College) for the GRK3-baculovirus. Dr. Terri Gilbert isolated GRK3 from Sf9 lysates used in the 32P back-phosphorylation experiments. Sumit Sud and Joe Novak genotyped and maintained the GRK3 mouse colony.
Footnotes
The work was supported in part by USPHS Grants RO1 DA11672 and T32 DA07278 from NIDA, National Institutes of Health (NIH). KOR(−/−) mice were generated and maintained with grant support from RO1 DA-09040 from NIDA, NIH (to J. E. P.).
The abbreviations used are: GPCR, G-protein-coupled receptor; KOR, kappa opioid receptor (following IUPHAR convention, this is the Kappa Opioid Peptide (KOP) receptor); KOR-P, affinity purified phos-phospecific rabbit anti rat/mouse kappa opioid receptor polyclonal antibody; MOR, mu opioid receptor; GRK3, G-protein receptor kinase 3; nor-BNI, nor-binaltorphimine; U50,488, (trans)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)-cyclohexyl]benzeneacetamide methanesulfonate hydrate; WGA, wheat germ agglutinin; β-CNA, β-chlornaltrexamine; GFP, green fluorescent protein; NFDM, nonfat dry milk.
References
- 1.Gutstein HB, Akil H. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. Vol. 10. McGraw-Hill Co.; New York, NY: 2001. pp. 569–619. [Google Scholar]
- 2.Nestler EJ. Curr Opin Neurobiol. 1997;7:713–719. doi: 10.1016/s0959-4388(97)80094-3. [DOI] [PubMed] [Google Scholar]
- 3.Law PY, Wong YH, Loh HH. Annu Rev Pharmacol Toxicol. 2000;40:389 –430. doi: 10.1146/annurev.pharmtox.40.1.389. [DOI] [PubMed] [Google Scholar]
- 4.Lefkowitz RJ. J Biol Chem. 1998;273:18677–18680. doi: 10.1074/jbc.273.30.18677. [DOI] [PubMed] [Google Scholar]
- 5.Pei G, Kieffer BL, Lefkowitz RJ, Freedman NJ. Mol Pharmacol. 1995;48:173–177. [PubMed] [Google Scholar]
- 6.Arden JR, Segredo V, Wang A, Lameh J, Sadeé W. J Neurochem. 1995;65:1636 –1645. doi: 10.1046/j.1471-4159.1995.65041636.x. [DOI] [PubMed] [Google Scholar]
- 7.Appleyard SM, Patterson TA, Jin W, Chavkin C. J Neurochem. 1997;69:2405–2412. doi: 10.1046/j.1471-4159.1997.69062405.x. [DOI] [PubMed] [Google Scholar]
- 8.Kovoor A, Nappey V, Kieffer BL, Chavkin C. J Biol Chem. 1997;272:27605–27611. doi: 10.1074/jbc.272.44.27605. [DOI] [PubMed] [Google Scholar]
- 9.Hasbi A, Polastron J, Allouche S, Stanasila L, Massotte D, Jauzac P. J Neurochem. 1998;70:2129 –2138. doi: 10.1046/j.1471-4159.1998.70052129.x. [DOI] [PubMed] [Google Scholar]
- 10.Chavkin C, McLaughlin JP, Celver JP. Mol Pharmacol. 2001;60:20–25. doi: 10.1124/mol.60.1.20. [DOI] [PubMed] [Google Scholar]
- 11.Appleyard SM, Celver J, Pineda V, Kovoor A, Wayman GA, Chavkin C. J Biol Chem. 1999;274:23802–23807. doi: 10.1074/jbc.274.34.23802. [DOI] [PubMed] [Google Scholar]
- 12.Deng HB, Yu Y, Wang H, Guang W, Wang JB. Brain Res. 2001;898:204 –214. doi: 10.1016/s0006-8993(01)02179-5. [DOI] [PubMed] [Google Scholar]
- 13.McLaughlin JP, Xu M, Mackie K, Chavkin C. J Biol Chem. 2003;278:34631–34640. doi: 10.1074/jbc.M304022200. [DOI] [PubMed] [Google Scholar]
- 14.Drake CT, Patterson TA, Simmons ML, Chavkin C, Milner TA. J Comp Neurol. 1996;370:377–395. doi: 10.1002/(SICI)1096-9861(19960701)370:3<377::AID-CNE8>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 15.Richardson RM, Kim C, Benovic JL, Hosey MM. J Biol Chem. 1993;268:13650 –13656. [PubMed] [Google Scholar]
- 16.Peppel K, Boekhoff I, McDonald P, Breer H, Caron MG, Lefkowitz RJ. J Biol Chem. 1997;272:25425–25428. doi: 10.1074/jbc.272.41.25425. [DOI] [PubMed] [Google Scholar]
- 17.Clarke S, Czyzyk T, Ansonoff M, Nitsche JF, Hsu MS, Nilsson L, Larsson K, Borsodi A, Toth G, Hill R, Kitchen I, Pintar JE. Eur J Pharmacol. 2002;16:1705–1712. doi: 10.1046/j.1460-9568.2002.02239.x. [DOI] [PubMed] [Google Scholar]
- 18.Jin W, Terman GW, Chavkin C. J Pharmacol Exp Ther. 1997;281:123–128. [PubMed] [Google Scholar]
- 19.Vaught JL, Takemori AE. J Pharmacol Exp Ther. 1979;208:86 –90. [PubMed] [Google Scholar]
- 20.Simonin F, Valverde O, Smadja C, Slowe S, Kitchen I, Dierich A, Le Meur M, Roques BP, Maldonado R, Kieffer BL. EMBO J. 1998;17:886 –897. doi: 10.1093/emboj/17.4.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie GX, Meng F, Mansour A, Thompson RC, Hoversten MT, Goldstein A, Watson WJ, Akil H. Proc Natl Acad Sci U S A. 1993;91:3779 –3783. doi: 10.1073/pnas.91.9.3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horan P, Taylor J, Yamamura HI, Porreca F. J Pharmacol Exp Ther. 1992;260:1237–1243. [PubMed] [Google Scholar]
- 23.Portoghese PS, Larson DL, Jiang JB, Takemori AE, Caruso TP. J Med Chem. 1978;21:598 –599. doi: 10.1021/jm00205a002. [DOI] [PubMed] [Google Scholar]
- 24.James IF, Chavkin C, Goldstein A. Proc Natl Acad Sci U S A. 1982;79:7570 –7574. doi: 10.1073/pnas.79.23.7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schlessinger J. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 26.Miller WE, Lefkowitz RJ. Curr Opin Cell Biol. 2001;13:139 –145. doi: 10.1016/s0955-0674(00)00190-3. [DOI] [PubMed] [Google Scholar]
- 27.Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Nature. 2000;408:720 –723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- 28.Bohn LM, Lefkowitz RJ, Caron MG. J Neurosci. 2002;23:10494 –10500. doi: 10.1523/JNEUROSCI.22-23-10494.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 30.Cox BM. Opioids I: Handbook of Experimental Pharmacology. I. Vol. 104. Springer-Verlag; Berlin: 1993. [Google Scholar]
- 31.Villar VM, Bhargava HN. Pharmacology. 1992;45:319 –328. doi: 10.1159/000139017. [DOI] [PubMed] [Google Scholar]
- 32.Hasbi A, Allouche S, Sichel F, Stanasila L, Massotte D, Landemore G, Polastron J, Jauzac P. J Pharmacol Exp Ther. 2000;293:237–247. [PubMed] [Google Scholar]
- 33.Koch T, Schulz S, Pfeiffer M, Klutzny M, Schroder H, Kahl E, Höllt V. J Biol Chem. 2001;276:31408–31414. doi: 10.1074/jbc.M100305200. [DOI] [PubMed] [Google Scholar]
- 34.Li JG, Luo LY, Krupnick JG, Benovic JL, Liu-Chen LY. J Biol Chem. 1999;274:12087–12094. doi: 10.1074/jbc.274.17.12087. [DOI] [PubMed] [Google Scholar]
- 35.Li JG, Benovic JL, Liu-Chen LY. Mol Pharmacol. 2000;58:795–801. [PubMed] [Google Scholar]
- 36.Joseph DB, Bidlack JM. J Pharmacol Exp Ther. 1995;272:970–976. [PubMed] [Google Scholar]