

Abstract
Multiple sclerosis (MS) is the most common human demyelinating disease of the central nervous system where oxidative stress has been proposed to play an important role in oligodendroglial death. However, molecular mechanisms that couple oxidative stress to the loss of oligodendrocytes are poorly understood. This study underlines the importance of neutral sphingomyelinase–ceramide pathway in mediating oxidative stress-induced apoptosis and cell death of human primary oligodendrocytes. Various oxidative stress-inducing agents, such as, superoxide radical produced by hypoxanthine and xanthine oxidase, hydrogen peroxide, aminotriazole capable of inhibiting catalase and increasing intracellular level of H2O2, or reduced glutathione-depleting diamide induced the activation of neutral sphingomyelinase and the production of ceramide. It is interesting to note that antisense knockdown of neutral but not acidic sphingomyelinase ablated oxidative stress-induced apoptosis and cell death in human primary oligodendrocytes. This study identifies neutral but not acidic sphingomyelinase as a target for possible therapeutic intervention in MS.
Keywords: oligodendrocytes, cell death, oxidative stress, ceramide, neutral sphingomyelinase, antisense knockdown
Introduction
Reactive oxygen species (ROS) accumulate and lead to oxidative stress when there is an imbalance in the redox status of a cell (Smith et al. 1999). One of the hallmarks of various neurodegenerative and neuroinflammatory disorders is oxidative stress-induced central nervous system (CNS) damage. Such oxidative stress can damage lipids, proteins, and nucleic acids of cells and power-house mitochondria causing cell death in various cell types including oligodendroglia, myelin-synthesizing cells in the CNS. There is a growing body of evidence indicating that oligodendroglial death might contribute to the pathology of multiple sclerosis (MS) (Boccaccio and Steinman 1996; Lucchinetti et al. 1996). Being the predominant iron containing cells of the brain (Connor et al. 1995) and having reduced level of glutathione (GSH), glutathione peroxidase and mitochondrial manganese superoxide dismutase, oligodendrocytes are highly susceptible to oxidative stress-induced damage (Bernhard et al. 1998). Furthermore, myelin sheaths contain polyunsaturated fatty acids that react with peroxide and hydroxyl radical, triggering a cascade of oxidative damage (Halliwell 1992). Consistently, several lines of evidence support the hypothesis that oligodendrocytes are vulnerable to intracellular glutathione depletion in vitro and in vivo (Back et al. 1998). These include the findings that N-acetyl cysteine (NAC), a precursor of GSH, prevented oligodendrocyte death evoked by tumor necrosis factor-α (TNF-α) (Back et al. 1998). According to Oka et al. (1993), oligodendrocytes die in response to GSH depletion caused by buthione sulfoxamine, and this sensitivity to GSH depletion is ameliorated by free radical scavengers, such as vitamin E and idebenone.
Although at higher concentrations, ROS may induce necrotic cell death, there are also several reports that suggest apoptotic cell death in oligodendrocytes under oxidative stress conditions. Mronga et al. (2004) have demonstrated that H2O2-induced apoptosis in oligodendrocytes involves mitochondrial damage and cytochrome c release accompanied by the activation of the death-related caspases 3 and 9. They have also reported up-regulation of death protein Bax in oligodendrocytes under oxidative stress insult. Mature rat brain oligodendrocytes exposed to oxidative stress also undergo chromatin segmentation via mechanisms involving transcriptional activation of the immediate early genes c-fos and c-jun (Richter-Landsberg and Vollgraf 1998). Consistently, TUNEL-positive cells bearing the myelin marker has been demonstrated in fresh frozen MS cerebrum by in situ TUNEL technique (Dowling et al. 1997).
However, mechanisms that trigger oligodendroglial apoptosis under oxidative stress condition remain poorly understood. Although sphingomyelin was initially considered as an inert structural component of plasma membrane, several investigations so far have established its role in signal transduction pathways associated with cell regulation, growth arrest, and apoptosis (Hannun 1996; Pettus et al. 2002). Because ceramide, a putative lipid second messenger, produced from the degradation of sphingomyelin by sphingomyelinases is a prototypic inducer of apoptosis in various cell types, including glial and neuronal cells (Hannun 1996; Pettus et al. 2002; Singh et al. 1998; Testai et al. 2004), we were prompted to explore whether ceramide was involved in the apoptosis of primary human oligodendrocytes. In this study, we report that various ROS-producing molecules or ROS itself induce the activation of sphingomyelinase and the production of ceramide in human primary oligodendrocytes. Of particular interest is the finding that the neutral but not the acidic sphingomyelinase is involved in oxidative stress-induced oligodendroglial apoptosis and cell death. The current results underscores a scenario in which attenuation of neutral sphingomyelinase may constitute a crucial step in preventing oligodendroglial loss in demyelinating disorders such as MS.
Materials and methods
Reagents
Fetal bovine serum, Hank’s balanced salt solution (HBSS), trypsin, and DMEM/F-12 were from Mediatech (USA). Phosphorothioate-labeled antisense and scrambled oligodeoxynucleotides were synthesized in the DNA-synthesizing facility of Invitrogen. Diamide, aminotriazole, and H2O2 were purchased from Sigma.
Isolation of human primary oligodendrocytes
Human fetal tissue was obtained from the Human Embryology Laboratory, University of Washington, Seattle. All of the experimental protocols were reviewed and approved by the Institutional Review Board (IRB Number 224-01-FB) of the University of Nebraska Medical Center. Primary oligodendrocytes were isolated from fetal brain tissues as described earlier (Jana and Pahan 2005). Briefly, 11- to 17-week-old fetal brains obtained from the Human Embryology Laboratory (University of Washington, Seattle, WA, USA) were dissociated by trituration and trypsinization (0.25% trypsin in PBS at 37°C for 15 min). The trypsin was inactivated with 10% heat-inactivated FBS (Mediatech, Washington, DC, USA). The dissociated cells were filtered successively through 380- and 140-μm meshes (Sigma, St. Louis, MO, USA) and pelleted by centrifugation. The resulting suspension was centrifuged for 10 min at 1,500 rpm and then resuspended in DMEM supplemented with 20% heat-inactivated FBS. Cells were plated on poly-D-lysine-coated 75 cm2 flasks and incubated at 37°C with 5% CO2 in air. Culture medium was changed after every 3 days. The initial mixed glial cultures, grown for 9 days, were placed on a rotary shaker at 240 rpm at 37°C for 2 h to remove loosely attached microglia. The oligodendrocytes were detached after shaking for 18 h at 200 rpm at 11 days. To purify oligodendrocytes from astrocytes and microglia, the detached cell suspension was plated in tissue culture dishes (2 × 106 cells/100 mm) for 60 min at 37°C. This step was repeated twice for nonadherent cells to minimize the contamination. The nonadhering cells, mostly oligodendrocytes, were seeded into poly-D-lysine-coated 6-well culture plates in complete medium at 37°C with 5% CO2 in air.
Assay of neutral and acidic sphingomyelinases
Activities of SMase(s) were assayed as described previously (Jana and Pahan 2004a, b). Briefly, after stimulation, the cells were washed with PBS, harvested, and resuspended in buffer A (100 mM Tris–HCl, 0.1% Triton X-100, 1 mM EDTA, and protease inhibitors, pH 7.4). The cell suspension was sonicated and centrifuged at 500×g at 4°C for 5 min. The supernatant was used as the enzyme source. For N-SMase, the reaction mixture contained enzyme preparation in 100 mM Tris–HCl, 5 nmol of [14C]sphingomyelin, 5 nmol of phosphatidylserine, 5 mM DTT, 0.1% Triton X-100 (1.54 mM), and 5 mM MgCl2, pH 7.4, in a final volume of 100 μl. A-SMase activity was measured in a 100 μl reaction mixture consisting of enzyme preparation in 100 mM sodium acetate, 5 nmol of [14C]sphingomyelin, and 0.1% Triton X-100, pH 5.0. The enzyme reaction was initiated by the addition of 50 μl of substrate and stopped by the addition of 1.5 ml of chloroform/methanol (2:1, v/v) and 0.2 ml of water. After vortexing and phase separation, aqueous phase was removed for counting.
Lipid extraction
Approximately 5 × 105 cells were exposed to different oxidizing agents for different periods of time, and lipids were extracted according to the methods described previously (Pahan et al. 1998, 2000).
Quantification of ceramide levels by diacylglycerol kinase assay
Ceramide content was quantified using diacylglycerol (DAG) kinase as described previously (Pahan et al. 1998, 2000). Briefly, dried lipids were solubilized in 20 μl of an octyl β-D-glucoside-cardiolipin solution (7.5% octyl β-D-glucoside and 5 mM cardiolipin in 1 mM diethylene-triaminepentaacetic acid) by sonication in a sonicator bath. The reaction was then performed in a final volume of 100 μl containing the 20 μl sample solution, 50 mM imidazole HCl, 50 mM NaCl, 12.5 mM MgCl2, 1 mM EGTA, 2 mM dithiothreitol, 6.6 μg of DAG kinase, and 1 mM [γ-32P]ATP (specific activity of 1–5 × 105 cpm/nmol), pH 6.6, for 30 min at room temperature. The labeled ceramide-1-phosphate was resolved with a solvent system consisting of methyl acetate/n-propanol/chloroform/methanol/0.25% KCl in water/acetic acid (100:100:100:40:36:2). A standard sample of ceramide was phosphorylated under identical conditions and developed in parallel. Both standard and experimental samples had identical RF values (0.46). Quantification of ceramide-1-phosphate was performed by autoradiography and densitometric scanning by a FluorChem 8800 Imaging System (Alpha Innotech, San Leandro, CA, USA). Values are expressed as either arbitrary units (absorbance) or percentage change.
Immunostaining of myelin proteins and GFAP
Immunostaining was performed as described earlier (Jana and Pahan 2004a, b). Briefly, coverslips containing 200–300 cells/mm2 were fixed with 4% paraformaldehyde for 15 min, followed by treatment with cold ethanol (−20°C) for 5 min and two rinses in PBS. Samples were blocked with 3% BSA in PBS containing Tween 20 (PBST) for 30 min and incubated in PBST containing 1% BSA and goat anti-GalC (1:50), rabbit anti-GFAP (1:50), rabbit anti-CNPase (1:50), rabbit anti-MOG (1:50), and rabbit anti-PLP (1:50). After three washes in PBST (15 min each), slides were further incubated with Cy5 and Cy2 (Jackson ImmunoResearch, West Grove, PA, USA). For negative controls, a set of culture slides was incubated under similar conditions without the primary antibodies. The samples were mounted and observed under a Bio-Rad (Hercules, CA, USA) MRC1024ES confocal laser-scanning microscope (Jana and Pahan 2004a, b).
Fragment end labeling of DNA
Fragmented DNA was detected in situ by the terminal deoxynucleotidyl transferase (TdT)-mediated binding of 3′-OH ends of DNA fragments generated in response to apoptotic signals, using a commercially available kit (TdT FragEL) from Calbiochem (La Jolla, CA, USA) as described earlier (Jana and Pahan 2004a, b). Briefly, coverslips were treated with 20 μg/ml proteinase K for 15 min at room temperature and washed before TdT staining.
Cell viability measurement
Mitochondrial activity was measured with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma).
Lactate dehydrogenase measurement
The activity of lactate dehydrogenase (LDH) was measured using the direct spectrophotometric assay using an assay kit from Sigma.
Results
Identification of human primary oligodendrocytes
Among different brain cells, the development of myelin-producing cells oligodendrocytes has been well-characterized. Oligodendrocytes arise from oligodendrocytes progenitor cells (OPC) through a series of developmental stages, e.g., proliferative prooligodendrocyte stage, nonproliferative immature oligodendrocyte stage, and finally nonproliferative mature oligodendrocyte stage (Pfeiffer et al. 1993; Miller 2002). These developmental stages are marked by sequential expression of developmental markers, such as immature oligodendrocytes express galactocerebroside (GalC) and 2′, 3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) while only mature oligodendrocytes express myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG), and proteolipid protein (PLP) (Pfeiffer et al. 1993; Miller 2002). To investigate if primary oligodendrocytes isolated from 11- to 17-week-old fetal brains were mature and expressed different myelin-specific markers, purified oligodendrocytes were immunostained with antibodies against GalC, MOG, PLP, and CNPase. To examine purity, cells were also double-labeled with GalC and GFAP (a specific marker of astrocytes). Immunofluorescence analysis in Fig. 1a shows that human primary oligodendrocytes readily expressed MOG, PLP, CNPase (Fig. 1b), and MBP (data not shown). As shown in Fig. 1b, these cells also expressed GalC and the expression of GFAP was rarely observed (one astrocyte out of 50 oligodendroglia) in this oligodendroglial preparation. By semiquantitative RT-PCR, we were also unable to detect any GFAP mRNA in purified oligodendrocytes (data not shown). Taken together, these studies suggest that human primary oligodendrocytes were mature and about 98% pure. These purified oligodendrocytes were used for subsequent experiments.
Fig. 1.

Immunofluorescence analysis of human primary oligodendrocytes for the expression of GalC, CNPase, MOG, and PLP. (a) Cells were immunostained with antibodies against MOG, CNPase and PLP. (b) To assess the purity of oligodendrocytes, cells were double-immunolabeled with antibodies against GalC and astroglial marker GFAP (left panel, GalC; middle panel, GFAP; right panel, both).
Oxidative stress caused by H2O2 leads to cell death and apoptosis in human primary oligodendrocytes
H2O2 is a widely accepted reagent for studying oxidative stress-induced apoptosis in many cell types. To examine if H2O2 is also capable of inducing apoptosis and cell death in human primary oligodendrocytes, we challenged these cells with 300 μM H2O2. Chromatin condensation and DNA fragmentation are common nuclear changes seen in apoptotic cells. TUNEL test for DNA fragmentation revealed marked induction of TdT-mediated labeling of DNA fragments after 6 h treatment of oligodendrocytes with H2O2 (Fig. 2a, b). It is known that mitochondrial dehydrogenase in active mitochondria of living cells cleave the MTT to formazan crystals and its amount is directly proportional to the number of living cells. On the other hand, LDH is a stable cytoplasmic enzyme present in all cells. It is released in the supernatant when the plasma membrane gets damaged. An increase in LDH activity in the supernatant reflects an increase in the number of plasma membrane damaged cells. Therefore, cell viability was determined by the MTT metabolism assay in cells and LDH release assay in supernatants. After 9 h treatment, H2O2 reduced cell viability in human primary oligodendrocytes as evidenced by decrease in MTT metabolism and increase in LDH release (Fig. 2b).
Fig. 2.

H2O2 induces apoptosis and cell death in human primary oligodendrocytes. Differentiated oligodendrocytes were treated with 300 μM H2O2. Control cells received only vehicle (PBS). (a) After 6 h of insult, apoptotic events were detected by TUNEL. Digital images were collected under bright-field setting using a 40× objective. (b) TUNEL-positive cells were counted manually in four different images of each of three coverslips by three individuals blinded to the experiment. Values obtained from the control group served as 100%, and data obtained in other groups were calculated as percent of control accordingly. After 9 h of insult, cells were also subjected to MTT reduction activity assay and culture medium was collected for LDH release assay. Results are expressed as mean ± SD of three different experiments. ap<0.001 vs control.
Extracellular and intracellular ROS induce the production of ceramide in human primary oligodendrocytes
Because ceramide is a well-known inducer of apoptosis, we next examined whether an increase in cellular ceramide content is detrimental to oligodendrocytes. As demonstrated in other studies (Jana and Pahan 2004a), cell-permeable C2-ceramide markedly induced apoptosis in primary oligodendrocytes as revealed by TUNEL staining (data not shown). This result instigated us to examine if ceramide is playing a role in oxidative stress-induced apoptosis in human fetal oligodendrocytes. Several reagents were used to model the oxidative stress condition. As for example, the combination of hypoxanthine and xanthine oxidase was used to mimic the effect of superoxide. Aminotriazole (ATZ) is an inhibitor of catalase; therefore, it was used to examine the effect of endogenously produced H2O2. Similarly, diamide is a prooxidant that converts reduced glutathione (GSH) to oxidized glutathione (GSSG). Therefore, this thiol-depleting agent was used to facilitate the elevation of endogenous oxidants in a cell. Human primary oligodendrocytes were treated with the combination of 10 μM hypoxanthine and 1 mU/ml xanthine oxidase, 300 μM H2O2, 100 μM diamide, and 3 mM aminotriazole. The level of ceramide was measured at different time points using the DAG kinase assay. As revealed from Fig. 3a, all the oxidants and prooxidants increased the level of ceramide significantly within 1 h of challenge, and the level of ceramide increased further with the increase in time of incubation. As for example, after 6 h of treatment, H2O2 increased the level of ceramide by about eightfold compared to about fourfold increase after 1 h of treatment (Fig. 3a). Among four different treatment groups, H2O2 was the most potent one in increasing the level of ceramide (Fig. 3a). Because DAG kinase phosphorylates both the DAG and ceramide using [γ-32P] ATP as substrate, both lipids can be quantified in the same assay. Although the DAG content was much higher than the ceramide content, in contrast to a time-dependent increase in the production of ceramide, the level of DAG was unchanged at different time points of stimulation (Fig. 3b) suggesting that various oxidative stress-causing agents increase the level of ceramide but not DAG in human primary oligodendrocytes.
Fig. 3.

Effect of extracellular and intercellular ROS on ceramide level in human primary oligodendrocytes. Differentiated oligodendrocytes were treated with 300 μM H2O2, the combination of 10 μM hypoxanthine and 1 mU/ml xanthine oxidase, 100 μM diamide, and 3 mM aminotriazole (ATZ). After 1 and 6 h of oxidative insults, total lipids were extracted and levels of ceramide (a) and DAG (b) were measured using the DAG kinase assay as mentioned under “Materials and methods”. Control group served as 100%, and data from other groups were expressed as percentage of control. Results are mean ± SD of three different experiments. ap<0.001 vs control.
Extracellular and intracellular ROS induce the activation of sphingomyelinases in human primary oligodendrocytes
To date five distinct sphingomyelinases have been identified based upon their pH optima, cellular localization, and cation dependence (Hannun 1996; Ruvolo 2003). However, the neutral membrane-bound Mg2+-independent sphingomyelinase (NSMase) and the lysosomal acid pH optima sphingomyelinase (ASMase) have been the best studied for their roles in ceramide generation (Hannun 1996; Ruvolo 2003). To test the potential link between accumulation of ceramide and activation of sphingomyelinase in response to oxidative stress, we investigated whether the above oxidants/prooxidants were capable of inducing the activation of neutral (N-SMase) and acidic sphingomyelinases (A-SMase). Significant increments in N-SMase activity were detected in human primary oligodendrocytes as early as 5 min in response to H2O2 (Fig. 4a) and hypoxanthine/xanthine oxidase (H + XO) (Fig. 4b) that peaked at 30 min and decreased afterwards. On the other hand, the induction of N-SMase in response to diamide began at 15 min and peaked at 30 min of stimulation (Fig. 4c). As aminotriazole enters into the cell, inhibits catalase and favors an increase in intracellular H2O2 level, this prooxidant induced the activation N-SMase at later time points, such as 30 and 60 min (Fig. 4d). In contrast, under same conditions, all the oxidants/prooxidants were unable to induce the activation of A-SMase in human primary oligodendrocytes (data not shown). These findings strongly support that oxidative stress results in exclusive activation of N-SMase without engagement of the acidic one in human primary oligodendrocytes.
Fig. 4.

Effect of extracellular and intercellular ROS on the activation of N-SMase in human primary oligodendrocytes. Differentiated oligodendrocytes were treated with 300 ìM H2O2(a), the combination of 10 μM hypoxanthine and 1 mU/ml xanthine oxidase (b), 100 μM diamide (c), and 3 mM ATZ (d). At different minute intervals, cellular lysates were prepared and activity of N-SMase was assayed using radiolabeled 14C sphingomyelin as described under “Materials and methods”. Control group served as 100%, and data from other groups were expressed as percentage of control. Results are mean ± SD of three different experiments. ap<0.001 and bp<0.05 vs control.
Antisense oligonucleotides against human N-SMase protect oligodendrocytes against oxidative stress-induced apoptosis and cell death
Because oxidative stress induced apoptotic cell death and activation of N-SMase in human primary oligodendrocytes, we were prompted to investigate whether knocking down of N-SMase would rescue these cells from oxidative insults. Earlier, we (Jana and Pahan 2004a, b) have shown that following antisense (ASO) but not scrambled (ScO) oligonucleotides against human N-SMase and A-SMase inhibit the activation of respective SMase in human primary neurons.
N-SMase
ASO: 5′- CAGCGAGCCCGTCCACCAGCC-3′
ScO: 5′- CACGCGTCCGACGCCGCACGA-3′
A-SMase
ASO: 5′- GACATCTCGGAGCCGGGGCA-3′
ScO: 5′- GGAAACCCGGTTAGGCCCGG-3′
We investigated the effect of these ASO and ScO against N-SMase and A-SMase on H2O2-induced apoptosis and cell death in human primary oligodendrocytes. Under the experimental condition of ASO/ScO treatment, control oligodendrocytes showed a few apoptotic bodies but treatment with H2O2 resulted in marked increase in apoptosis (Fig 5a, b). However, the ASO, but not the ScO against N-SMase markedly blocked H2O2-induced oligodendroglial apoptosis (Fig. 5a, b). On the other hand, ASO against A-SMase had no effect on H2O2-induced apoptosis (Fig. 5b).
Fig. 5.
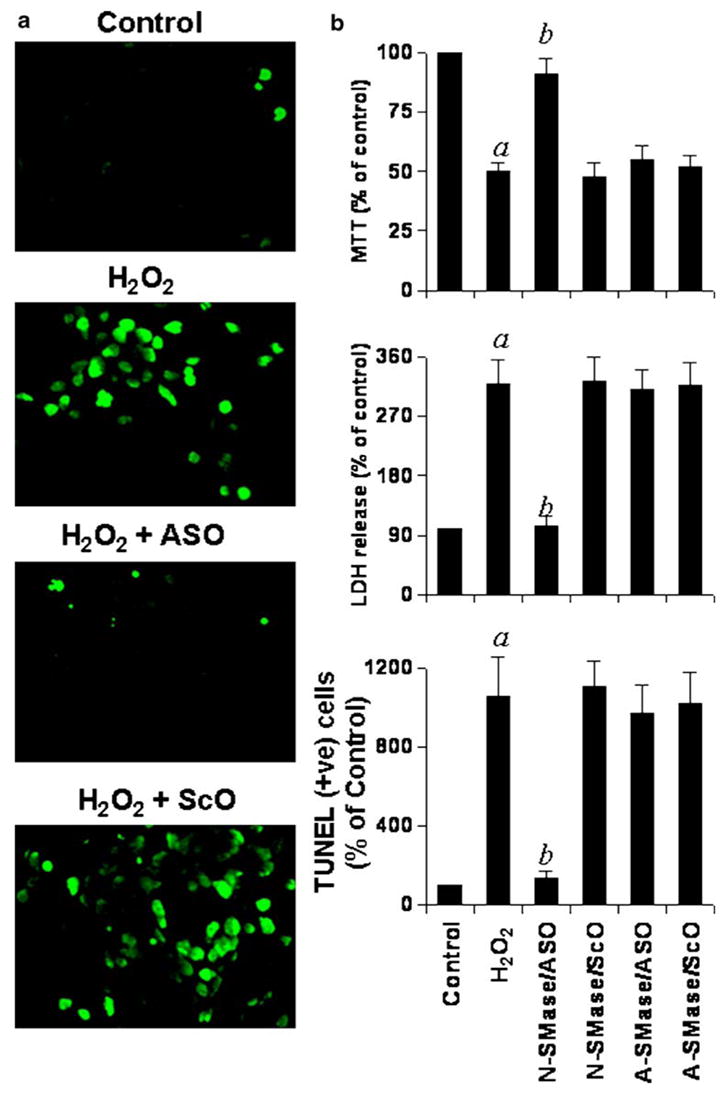
Effect of antisense oligonucleotides against N-SMase and A-SMase on H2O2-induced apoptosis and cell death in human primary oligodendrocytes. (a) Cells plated on poly-D-lysine-coated coverslips were treated with either 1 μM anti-sense (ASO) or scrambled (ScO) oligonucleotides against N-SMase. After 40 h of incubation, cells were treated with 300 μM H2O2 for 6 h, and apoptotic events were detected by TUNEL. Control cells received only vehicle (PBS). (b) TUNEL-positive cells were counted manually in four different images of each of three coverslips by three blinded individuals. Similarly, cells pre-incubated with 1 μM ASO or ScO against N-SMase and A-SMase for 40 h were challenged with 300 μM H2O2. After 9 h of insult, cells were subjected to MTT reduction activity assay and culture medium was collected for LDH release assay. Values obtained from the control group served as 100%, and data obtained in other groups were calculated as percentage of control accordingly. Results are mean ± SD of three different experiments. ap<0.001 vs control; bp<0.001 vs H2O2.
To further confirm this observation, we examined H2O2-induced cell death in ASO- and ScO-treated oligodendrocytes. Cell viability was determined by the MTT metabolism assay. After 9 h of treatment, H2O2 reduced cell viability as evidenced by a decrease in MTT metabolism (Fig. 5b). It is interesting to note that ASO against N-SMase, but not A-SMase, effectively prevented H2O2-induced loss of MTT metabolism (Fig. 5b). Similarly, H2O2 also induced an increase in LDH release and treatment of cells with ASO against N-SMase but not A-SMase resulted in significant reduction in LDH release (Fig. 5b). On the other hand, ScO against N-SMase had no effect on H2O2-induced loss of MTT metabolism and increase in LDH release (Fig. 5b). Similar to H2O2, other oxidizing agents, such diamide, (H + XO) and ATZ also induced cell death in primary human oligodendrocytes as evidenced by decrease in MTT metabolism (Fig. 6a) and increase in LDH release (Fig. 6b). Consistent to the effect of antisense knockdown of N-SMase on H2O2-mediated cell death (Fig. 5), ASO but not ScO against N-SMase protected oligodendrocytes from oxidative stress created by (H + XO), diamide and ATZ (Fig. 6). These results suggest that activation of N-SMase but not A-SMase plays the key role in oxidative stress-mediated apoptosis and cell death in human primary oligodendrocytes.
Fig. 6.

Effect of antisense oligonucleotides against N-SMase and A-SMase on superoxide-, diamide-, and aminotriazole-induced cell death in human primary oligodendrocytes. Cells preincubated with 1 μM ASO or ScO against N-SMase and A-SMase for 40 h were challenged with the combination of 10 μM hypoxanthine and 1 mU/ml xanthine oxidase, 100 μM diamide, and 3 mM ATZ. After 9 h of insult for all except 24 h for ATZ, cells were subjected to MTT reduction assay (a) and culture medium was collected for LDH release assay (b). Values obtained from the control group served as 100%, and data obtained in other groups were calculated as percentage of control accordingly. Results are mean ± SD of three different experiments. ap<0.001 vs H + XO; bp<0.001 vs diamide; cp<0.001 vs ATZ.
Discussion
Multiple sclerosis (MS) is a chronic inflammatory and demyelinating disease of the CNS in which oxidative stress plays a pathogenic role. Reactive oxygen species (ROS) generated by activated macrophages and microglial cells are thought to play a major role in damaging myelin and myelin-producing cells oligodendrocytes in MS (Smith et al. 1999). Therefore, antioxidant supplementation has been suggested as a possible primary or adjunct therapeutic approach for MS and other neurodegenerative disorders. However, apart from antioxidants, mechanisms that might protect oligodendrocytes from oxidative stress are poorly understood. Ceramide, a lipid second messenger molecule, is a prototype inducer of apoptosis in various cell types including glial and neuronal cells (Brugg et al. 1996; Weisner and Dawson 1996). Once ceramide is generated, different molecules such as Bcl-2, SAPK, PKB/Akt, and KSR become involved downstream of ceramide in the apoptotic pathway (Hannun 1996; Ruvolo 2003). Earlier, it has been shown that oxidative stress modulates the activation of neutral sphingomyelinase (N-SMase) in airway epithelial cells (Levy et al. 2006) and mammary carcinoma cells (Liu et al. 1998). Several lines of evidence presented in this study clearly demonstrate that oxidative stress kills human primary oligodendrocytes via the activation of N-SMase-ceramide pathway. First, H2O2 as well as ATZ, an inhibitor of catalase, that increases intercellular concentration of H2O2, markedly induced the activation of N-SMase and cell death in oligodendrocytes. However, antisense knockdown of N-SMase protected oligodendrocytes from H2O2- and ATZ-induced cells death. Second, diamide, a thiol depleting agent that modulates intracellular redox status by depleting GSH, induced cell death in oligodendrocytes that was rescued by antisense oligonucleotides against N-SMase. Third, antisense oligonucleotides against N-SMase saved oligodendrocytes from death induced by exogenously produced superoxide by a reaction involving hypoxanthine and xanthine oxidase. On the other hand, knockdown of acidic sphingomyelinase did not abrogate oligodendroglial cell death under any of the experimental conditions. These results clearly implicate the central role of N-SMase in endangering oligodendrocytes to oxidative stress-induced cell death.
Since the discovery of the sphingomyelin cycle, several inducers have been shown to be coupled to N-SMase, including 1α,25-dihydroxyvitamin D3, radiation, antibody cross-linking, TNF-α, IL-1β, Fas, nerve growth factor, brefeldin A, and serum-deprivation (Hannun 1996; Pettus et al. 2002; Ruvolo 2003). The mechanisms involved in the activation of N-SMase in response to TNF-α are becoming clear. For example, TNF-α initiates the pathway through TNFR1 (55-kDa receptor) leading to phospholipase A2 activation, generation of arachidonic acid, and subsequent activation of N-SMase (Hannun 1996). While binding of Fan to the N-SMase activating domain (NSD) of TNFR1 is required for the activation of N-SMase, binding of TRADD to the death domain (DD) of the same receptor induces the activation of A-SMase (Hannun 1996; Ruvulo 2003). In addition, proteases have also been implicated in the pathway leading from TNF-α to the activation of N-SMase (Hannun 1996; Pettus et al. 2002). However, mechanisms involved in ROS-mediated activation of N-SMase in primary oligodendrocytes are not known. There is escalating evidence that ROS modulate both PKC (Konishi et al. 1997) and tyrosine kinase activities or inhibit tyrosine phosphatase activities (Garg and Aggarwal 2002). Therefore, it can be speculated that ROS may induce the activation of N-SMase by either influencing the subcellular localization of N-SMase or by activating one or several protein kinases or inhibiting tyrosine phosphatase activities. Alternatively, it could be hypothesized that ROS triggers the generation of second messengers which in turn activate N-SMase.
At low concentrations, ROS are believed to exert beneficial physiologic effects, such as control of gene expression and mitogenesis. However, at higher concentrations, ROS are reactive, and disintegrate biomolecules such as DNA, lipids and proteins, lead to deterioration of cellular functions. Recent studies show that ROS are also capable of transducing death signal by activating stress-related kinases (JNK/SAPK), mobilizing Ca+2, stimulating apoptotic proteases (caspases), and up-regulating a number of death proteins (Bax, Bad) (Hannun 1996; Ruvolo 2003). However, knocking out of the single molecule (N-SMase) is capable of disabling ROS in human primary oligodendrocytes suggesting that probably most of the death-inducing pathways stimulated by ROS converge on the activation of N-SMase. Recently, we have also demonstrated that antisense knockdown of human N-SMase protects human primary neurons from fibrillar Aβ- and HIV-1 gp120-mediated apoptosis and cell death (Jana and Pahan 2004a, b). Taken together, these findings suggest N-SMase may turn out to be a common therapeutic target for different neuroinflammatory and neurodegenerative disorders.
In summary, we have demonstrated that antisense oligonucleotides against human N-SMase protect human primary oligodendrocytes from oxidative insults caused by various oxidants. Although the local concentration of ROS present in the brain microenvironment of MS patients may differ from the concentration we used in primary oligodendrocytes, and the in vitro situation of human fetal oligodendrocytes in culture may not truly resemble the in vivo situation of oligodendrocytes in the brain of MS patients, our results clearly point out N-SMase as a possible therapeutic target to halt oxidative stress-induced oligodendroglial damage in MS and other demyelinating diseases.
Acknowledgments
This study was supported by grants from National Multiple Sclerosis Society (RG3422A1/1) and NIH (NS39940).
References
- Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccaccio GL, Steinman L. Multiple sclerosis: from a myelin point of view. J Neurosci Res. 1996;45:647–654. doi: 10.1002/(SICI)1097-4547(19960915)45:6<647::AID-JNR1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Brugg B, Mitchel PP, Agid Y, Ruberg M. Ceramide induces apoptosis in cultured mesencephalic neurons. J Neurochem. 1996;66:733–739. doi: 10.1046/j.1471-4159.1996.66020733.x. [DOI] [PubMed] [Google Scholar]
- Connor JR, Pavlick G, Karli D, Menzies SL, Palmer C. A histochemical study of iron-positive cells in the developing rat brain. J Comp Neurol. 1995;355:111–123. doi: 10.1002/cne.903550112. [DOI] [PubMed] [Google Scholar]
- Dowling P, Husar W, Menonna J, Donnenfeld H, Cook S, Sidhu M. Cell death and birth in multiple sclerosis brain. J Neurol Sci. 1997;149:1–11. doi: 10.1016/s0022-510x(97)05213-1. [DOI] [PubMed] [Google Scholar]
- Garg AK, Aggarwal BB. Reactive oxygen intermediates in TNF signaling. Mol Immunol. 2002;39:509–517. doi: 10.1016/s0161-5890(02)00207-9. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- Hannun YA. Functions of ceramide in coordinating cellular responses to stress. Science. 1996;274:1855–1859. doi: 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- Jana A, Pahan K. Fibrillar amyloid-beta peptides kill human primary neurons via NADPH oxidase-mediated activation of neutral sphingomyelinase. Implications for Alzheimer’s disease. J Biol Chem. 2004a;279:51451–51459. doi: 10.1074/jbc.M404635200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana A, Pahan K. Human immunodeficiency virus type 1 gp120 induces apoptosis in human primary neurons through redox-regulated activation of neutral sphingomyelinase. J Neurosci. 2004b;24:9531–9540. doi: 10.1523/JNEUROSCI.3085-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana M, Pahan K. Redox regulation of cytokine-mediated inhibition of myelin gene expression in human primary oligodendrocytes. Free Radic Biol Med. 2005;39:823–831. doi: 10.1016/j.freeradbiomed.2005.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juurlink BHJ, Thorburne SK, Hertz L. Peroxide-scavenging deficit underlies oligodendrocyte susceptibility to oxidative stress. Glia. 1998;22:371–378. doi: 10.1002/(sici)1098-1136(199804)22:4<371::aid-glia6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Konishi H, Tanaka M, Takemura Y, Matsuzaki H, Ono Y, Kikkawa U, Nishizuka Y. Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc Natl Acad Sci U S A. 1997;94:11233–11237. doi: 10.1073/pnas.94.21.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy M, Castillo SS, Goldcorn T. nSMase2 activation and trafficking are modulated by oxidative stress to induce apoptosis. Biochem Biophys Res Commun. 2006;344:900–905. doi: 10.1016/j.bbrc.2006.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Andrieu-Abadie N, Levade T, Zhang P, Obeid LM, Hannun YA. Glutathione regulation of neutral sphingomyelinase in tumor necrosis factor-alpha-induced cell death. J Biol Chem. 1998;273:11313–11320. doi: 10.1074/jbc.273.18.11313. [DOI] [PubMed] [Google Scholar]
- Lucchinetti CF, Bruck W, Rodriguez M, Lassmann H. Distinct patterns of multiple sclerosis pathology indicates heterogeneity on pathogenesis. Brain Pathol. 1996;6:259–274. doi: 10.1111/j.1750-3639.1996.tb00854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RH. Regulation of oligodendrocyte development in the vertebrate CNS. Prog Neurobiol. 2002;67:451–467. doi: 10.1016/s0301-0082(02)00058-8. [DOI] [PubMed] [Google Scholar]
- Mronga T, Stahnke T, Goldbaum O, Richter-Landsberg C. Mitochondrial pathway is involved in hydrogen-peroxide-induced apoptotic cell death of oligodendrocytes. Glia. 2004;46:446–455. doi: 10.1002/glia.20022. [DOI] [PubMed] [Google Scholar]
- Ohmori S, Shirai Y, Sakai N, Fujii M, Konishi H, Kikkawa U, Saito N. Three distinct mechanisms for translocation and activation of the delta subspecies of protein kinase C. Mol Cell Biol. 1998;18:5263–5271. doi: 10.1128/mcb.18.9.5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ. Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J Neurosci. 1993;13:1441–1453. doi: 10.1523/JNEUROSCI.13-04-01441.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahan K, Khan M, Singh I. Interleukin-10 and interleukin-13 inhibit proinflammatory cytokine-induced ceramide production through the activation of phosphatidylinositol 3-kinase. J Neurochem. 2000;75:576–582. doi: 10.1046/j.1471-4159.2000.0750576.x. [DOI] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Khan M, Namboodiri AM, Singh I. Sphingomyelinase and ceramide stimulate the expression of inducible nitric-oxide synthase in rat primary astrocytes. J Biol Chem. 1998;273:2591–2600. doi: 10.1074/jbc.273.5.2591. [DOI] [PubMed] [Google Scholar]
- Pettus BJ, Chalfant CE, Hannun YA. Ceramide in apoptosis: an overview and current perspectives. Biochim Biophys Acta. 2002;1585:114–125. doi: 10.1016/s1388-1981(02)00331-1. [DOI] [PubMed] [Google Scholar]
- Pfeiffer SE, Warrington AE, Bansal R. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 1993;3:191–197. doi: 10.1016/0962-8924(93)90213-k. [DOI] [PubMed] [Google Scholar]
- Richter-Landsberg C, Vollgraf U. Mode of cell injury and death after hydrogen peroxide exposure in cultured oligodendroglia cells. Exp Cell Res. 1998;244:218–229. doi: 10.1006/excr.1998.4188. [DOI] [PubMed] [Google Scholar]
- Ruvolo PP. Intracellular signal transduction pathways activated by ceramide and its metabolites. Pharmacol Res. 2003;47:383–392. doi: 10.1016/s1043-6618(03)00050-1. [DOI] [PubMed] [Google Scholar]
- Singh I, Pahan K, Khan M, Singh AK. Cytokine-mediated induction of ceramide production is redox-sensitive. Implications to proinflammatory cytokine-mediated apoptosis in demyelinating diseases. J Biol Chem. 1998;273:20354–20362. doi: 10.1074/jbc.273.32.20354. [DOI] [PubMed] [Google Scholar]
- Smith KJ, Kapoor R, Felts PA. Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathol. 1999;9:69–92. doi: 10.1111/j.1750-3639.1999.tb00212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testai FD, Landek MA, Dawson G. Regulation of sphingo-myelinases in cells of the oligodendrocyte lineage. J Neurosci Res. 2004;75:66–74. doi: 10.1002/jnr.10816. [DOI] [PubMed] [Google Scholar]
- Wiesner DA, Dawson G. Staurosporine induces programmed cell death in embryonic neurons and activation of the ceramide pathway. J Neurochem. 1996;66:1418–1425. doi: 10.1046/j.1471-4159.1996.66041418.x. [DOI] [PubMed] [Google Scholar]