

Abstract
Sphingolipids that contain a sphingoid base are composed of hundreds to thousands of distinct compounds, many of which serve as lipid regulators of biological functions. The global analysis of the large number of low-abundance sphingolipid molecular species has been hampered in many cases by the sphingolipid molecular species being overwhelmed by the quantity of other classes of lipid (e.g., glycerophospholipid) molecular species present, thereby imposing severe restrictions on the dynamic range of their measurement using shotgun lipidomics. Herein, we developed a facile approach in which the sphingolipids of cellular extracts were dramatically enriched by direct alkaline methanolysis of lipid extracts followed by extraction to remove the large majority of other endogenous lipid classes. Through direct infusion of the resultant enriched solution, we identified and quantitated a variety of very-low-abundance sphingolipid classes (e.g., sphingosine, psychosine, and lysosphingomyelin) and molecular species (e.g., sphingomyelin) using electrospray ionization mass spectrometry (i.e., shotgun sphingolipidomics). Accordingly, through utilization of these facile enrichment techniques, direct penetrance into the sphingolipidomes has been greatly extended, facilitating new insights into their metabolism and signaling functions in biological systems.
Keywords: Electrospray ionization mass spectrometry, Shotgun lipidomics, Shotgun sphingolipidomics, Sphingolipidome, Lysosphingomyelin, Psychosine, Sphingomyelin, Sphingosine
Lipidomics, the large-scale study of the pathways and networks of cellular lipids, is an emerging and rapidly expanding research field [1–3]. Currently, lipidomics research has been focused on identifying alterations in lipid metabolic pathways and networks induced by a disease state, a gene mutation (knockout or overexpression), a therapeutic treatment, or other perturbations. In the future, research in lipidomics will expand to include the dynamics of lipidomes, subcellular organizations of lipidomes, and interactions of lipids with lipids, proteins, and other cellular moieties. Although lipidomics has emerged as a distinct field only within the past few years [1–3], numerous new discoveries and/or advances have already been made [3–12]. Its essential roles in identifying the biochemical mechanisms of lipid metabolism, investigating the functions of an individual gene of interest, identifying novel biomarkers, and evaluating drug efficacy, among others, are becoming increasingly recognized.
One of the major analytical platforms in current lipidomics practice is multidimensional mass-spectrometry (MS)1 -based shotgun lipidomics [13–15]. This platform has now evolved into a mature technology that includes a series of simple steps such as multiplexed extractions, intrasource separation, identification of individual lipid molecular species using multidimensional MS, and quantitation of the identified lipid molecular species using a two-step procedure [4,15,16]. The principle of shotgun lipidomics is to maximally exploit differences in the physical and chemical properties of the classes of lipids present in biological samples to maximize the penetrance and dynamic range of analyses.
Therefore, the differential solubility of the lipid classes in various solvents under varying pH conditions is an initial critical step to maximally separate and enrich the lipid class(es) of interest. For example, many lipid classes (e.g., sphingosine-1-phosphate, lysophosphatidic acid, acylcarnitine, etc.) can be efficiently extracted under acidic conditions [4]. Gangliosides and acylCoAs are highly soluble in polar solvents and are partitioned into the aqueous phase during chloroform extraction [17–19], which can be reverse-extracted by using butanol under acidic conditions. Moreover, very hydrophobic lipid classes (e.g., cholesterol and its esters, triacylglycerol, free fatty acid, etc.) can be extracted and enriched with hexane.
Next, in shotgun lipidomics, the differential acidic or basic properties of lipid classes in an extracted lipid solution are exploited to selectively ionize different lipid classes in the positive- or negative-ion modes (i.e., intrasource separation) and achieve a maximal ionization sensitivity [20]. Therefore, lipid classes containing phosphate, sulfate, and carboxylate (e.g., anionic phospholipids, acyl-CoA, sulfatide, gangliosides, free fatty acid, and sphingosine-1-phosphate) can be selectively ionized in the negative-ion mode, particularly under basic conditions (e.g., in the presence of NH4OH or LiOH), whereas lipid classes containing amine groups (e.g., acylcarnitine) can be readily ionized in the positive-ion mode under acidic conditions [4]. Molecular species of other lipid classes can be ionized as either alkaline or anion (e.g., chloride, acetate, or formate) adducts in the positive- or negative-ion mode, respectively.
Finding sensitive and unique fragments produced after collision-induced dissociation (CID) specific to a class or a group of lipids of interest to scan is the third key step for successfully profiling and quantifying individual molecular species in the class or group. Either neutral-loss (NL) scanning or precursor-ion (PI) scanning at the mass or m/z ratio of a fragment, respectively, can be performed to “isolate” a given class or group of lipids from which individual lipid molecular species can be identified through multidimensional array analysis. Each of these fragments represents a building block of the class or the group of lipids [13].
Finally, quantitation by shotgun lipidomics is performed in a two-step procedure [13,21,22]. First, the abundant and nonoverlapping molecular species of a class are quantified by comparison with a preselected internal standard of the class after 13C deisotoping [4,14] from a survey scan. Second, some or all of these determined molecular species of the class are used as standards to determine the contents of other low-abundance or overlapping molecular species using one or multiple NL and/or PI scans as described above. Through this second step in the quantitation process, the linear dynamic range of quantitation can be dramatically extended by eliminating background noise and by filtering the overlapping molecular species through a multidimensional MS approach [4]. However, due to signal overlaps of low-abundance molecular species of a class of interest (e.g., sphingomyelin (SM)) with potential isomeric lipid molecular species in other lipid class(es) (e.g., choline glycerophospholipid (GPCho)) in both first and second steps of quantitation in shotgun lipidomics, identification and quantitation of these molecular species was previously inaccessible. Furthermore, the linear dynamic range of quantitation of sphingolipid molecular species by comparison of ion peak intensities with a preselected internal standard was compromised by the presence abundance of other choline-containing phospholipids and other lipid classes (see discussion below for details).
Although sphingolipids that contain a sphingosine (i.e., trans-4-sphingenine) backbone or its analogs (Scheme 1) are highly bioactive compounds which serve as components of biological structures and regulators (see [23] for recent review), the majority of these compounds are generally in extremely low abundance (e.g., <0.1% of cellular lipids) which renders quantitation of these compounds difficult by conventional methods. To this end, analysis of sphingolipids by shotgun lipidomics is challenging due to the hundreds of molecular species present, with many present in diminutive amounts. The majority of established methods for the large-scale analysis of sphingolipids are developed based on LC and LC-MS [24–30].
Scheme 1.
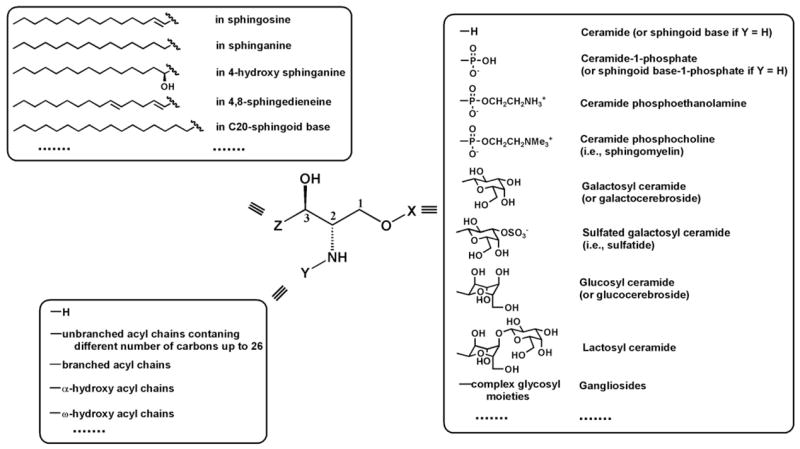
General structure of sphingoid-based lipids. The building block X represents a different polar moiety (linked to the oxygen at the C1 position of the sphingoid base). The building block Y represents fatty acyl chains (acylated to the primary amine at the C2 position of the sphingoid base) with or without the presence of a hydroxyl group which is usually located at the α or ω position. The building block Z represents the aliphatic chains in all possible sphingoid bases, which are carbon–carbon linked to the C3 position of sphingoid bases and vary with the aliphatic chain length, the degree of unsaturation, the presence of a branch, and the presence of an additional hydroxyl group. This illustration has been modified from [7] with permission.
In this report, we exploited two distinct chemical characteristics of sphingolipids to extend the current shotgun lipidomics platform for the analysis of low-abundance sphingolipids. First, we exploited the base-resistant character of all sphingolipids under mildly basic conditions. Second, we dramatically enriched the sphingolipid content of the sample by liquid–liquid phase portioning of resultant reaction products by washing with hexane (removal of nonpolar lipids) followed by a Bligh and Dyer [31] extraction to further remove the aqueous-phase-soluble components produced during alkaline methanolysis. Through these simple procedures, the depth of penetrance and dynamic range of shotgun sphingolipidomics has been greatly extended.
Materials and methods
Materials
Synthetic sphingolipids including N-lauroryl sphingomyelin (N12:0 SM), sphingosine analogs (C14 to C18), C17:0 sphinganine analog, N-heptadecanoyl ceramide (N17:0 Cer), and most of the synthetic phospholipids used for internal standards were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). Semisynthetic N-palmitoyl sulfatide (N16:0 sulfatide), perdeuterated N-stearoyl galactosylceramide (d35-N18:0 GalCer), and N-pentadecanoyl galactosylceramide (N15:0 GalCer) were obtained from Matreya, Inc. (Pleasant Gap, PA). All the solvents were obtained from Burdick and Jackson (Honeywell International Inc., Burdick and Jackson, Muskegon, MI). All other chemicals were purchased from Sigma–Aldrich (St. Louis, MO). It should be noted that the prefix “N” denotes the fatty acyl amide chain and “C” represents the sphingoid base.
Preparation of lipid extracts from biological samples
Mice (male, C57 BL/J background, 4 months of age) were purchased from The Jackson Laboratory (Bar Harbor, ME). All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals and were approved by the Animals Studies Committee at Washington University. Mice were killed by asphyxiation with CO2. Mouse brain tissues (including cortex, cerebellum, brain stem, and spinal cord) were dissected quickly. The collected brain tissues were immediately freeze-clamped at the temperature of liquid nitrogen. The tissue wafers were pulverized into a fine powder with a stainless steel mortar and pestle. Mouse plasma was also collected in the presence of a small amount of heparin (approximately 5 units) followed by centrifugation at 2500 rpm for 10 min to remove red blood cells, platelets, and other particles.
A sample (approximately 10 mg) from each dissected brain tissue was weighed and homogenized in 0.3 ml of ice-cooled diluted (10×) phosphate-buffered saline with a Potter–Elvehjem tissue grinder. Protein assays on each individual homogenate were performed using a bicinchoninic acid protein assay kit (Pierce, Rockford, IL) with bovine serum albumin as a standard. After homogenate (~0.1 ml) from each sample was transferred to a disposable culture borosilicate glass tube (16 × 100 mm), 4 ml of CHCl3/MeOH (1/1, v/v) and the proper volume of LiCl solution to make up 1.8 ml of the final LiCl solution (50 mM) were added to each individual test tube. Internal standards in a premixed solution for global lipid analysis including N12:0 SM (2.5 nmol/mg protein), N17:0 Cer (0.8 nmol/mg protein), N16:0 sulfatide (5.0 nmol/mg protein), C16:0 sphingosine analog (0.12 nmol/mg protein), and d35-N18:0 GalCer (10 nmol/mg protein) and necessary internal standards for quantitation of individual molecular species of other lipid classes were also added to each brain tissue sample based on protein concentration prior to extraction of lipids. Thus, the lipid content could be normalized to the protein content and quantified directly. These internal standards were selected because they represent ≪1% of endogenous cellular lipid molecular species present as demonstrated by electrospray ionization mass spectrometry (ESI/MS) lipid analysis without addition of these internal standards. A similar procedure was also used to prepare lipid extracts from 100 μl of mouse plasma with a new set of internal standards including N17:0 SM (15 nmol/mg protein), N17:0 Cer (1 nmol/mg protein), and C16:0 sphingosine analog (0.12 nmol/mg protein) and necessary internal standards for quantitation of individual molecular species of other lipid classes. Lipids were extracted by the modified method of Bligh and Dyer [31] as described previously [16,32]. Each individual lipid extract was reconstituted with 500 μl/mg of tissue protein or 20 ml/ml of plasma in 1:1 CHCl3/MeOH. The lipid extracts were finally flushed with nitrogen, capped, and stored at −20 °C for ESI/MS (typically analyzed within 1 week) as previously described [4].
Preparation of lipid samples for shotgun sphingolipidomics
The procedures of preparing lipid samples for shotgun sphingolipidomics is illustrated in Scheme 2, which was modified from the commonly used procedure of alkaline hydrolysis of lipids reported previously [33,34]. Briefly, a small portion of each individual crude lipid extract equivalent to the lipid extract from a tissue sample containing 0.1 mg protein was transferred to a conical centrifuge glass test tube and the solvent was evaporated under a stream of nitrogen. A small volume of ice-cooled LiOMe solution (1M, 50 μl) in MeOH was added to the test tube at 0 °C. The reaction mixture was vortexed for 15 s, placed in an ice bath for 1 h, and quenched with 2 ml of 0.4% acetic acid solution. The pH of the quenched reaction solution should be adjusted to 4–5 by addition of acetic acid if necessary. The aqueous phase was washed with hexane (2 ml × 3 times) and the hexane solution was saved for other purposes. The lipids in the aqueous phase were extracted by the modified Bligh and Dyer method as described above. The combined CHCl3 phase was dried under a stream of nitrogen. The residue was reextracted against a 10 mM aqueous LiCl solution. Each individual extract was reconstituted in 100 μl of CHCl3/MeOH (1/1, v/v) after filtering through a 0.2-μm polytetrafluoroethylene membrane filter, flushed with nitrogen, capped, and stored at −20 °C for ESI/MS analysis.
Scheme 2.
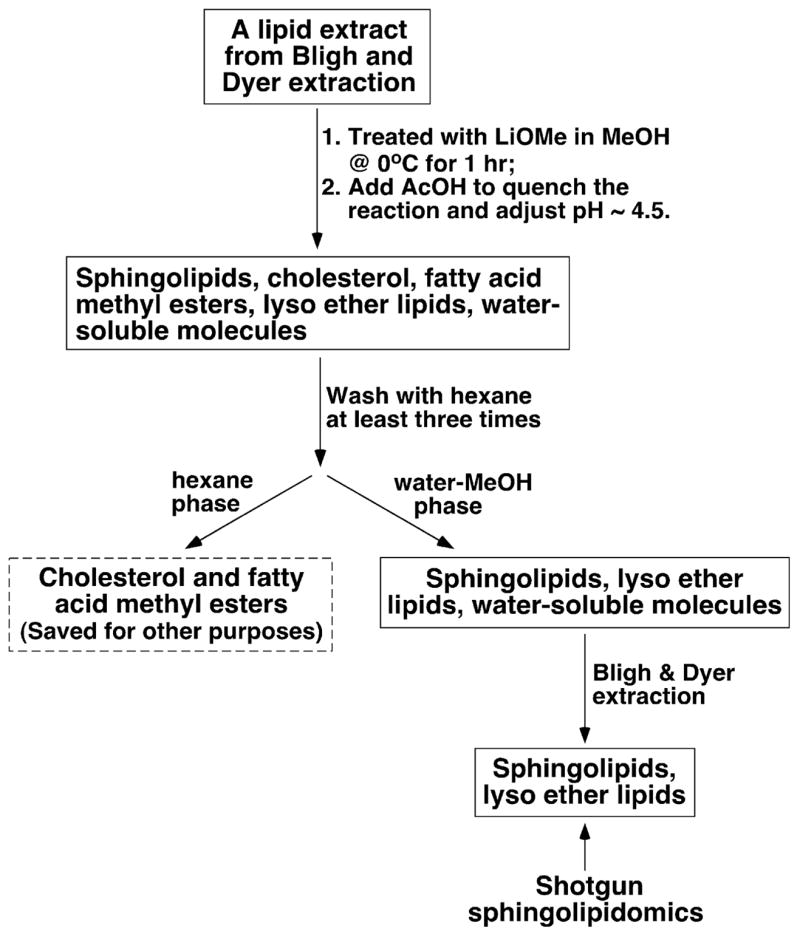
Schematic illustration of sample preparation for shotgun sphingolipidomics.
Mass spectrometric analysis of sphingolipids
A triple-quadrupole mass spectrometer (Thermo Electron TSQ Quantum Ultra Plus; San Jose, CA) equipped with an electrospray ion source and Xcalibur system software was utilized in the study under conditions as previously described [15]. The first and third quadrupoles serve as independent mass analyzers using a mass resolution setting of peak width 0.7 Th while the second quadrupole serves as a collision cell for tandem mass spectrometry. The diluted lipid extract was directly infused into the ESI source at a flow rate of 4 μl/min with a syringe pump. Typically, a 1-min period of signal averaging in the profile mode was employed for each MS spectrum. For tandem mass spectrometry, the collision gas pressure was set at 1.0 mTorr but the collision energy varied with the classes of lipids as described previously [4,15]. Typically, a 2- to 5-min period of signal averaging in the profile mode was employed for each tandem MS spectrum. All MS spectra and tandem MS spectra were automatically acquired by a customized sequence subroutine operated under Xcalibur software. Data processing of two-dimensional MS analyses, including ion peak selection, baseline correction, data transfer, peak intensity comparison, 13C deisotoping, and quantitation, was conducted using custom-programmed Microsoft Excel macros as outlined previously [15].
Results and discussion
Treatment of lipid extracts for shotgun sphingolipidomics
Sample preparation is a key step in shotgun lipidomics. We exploited two special chemical features of cellular lipids to maximize their suitability for shotgun sphingolipidomics. These features include the base resistance of sphingolipids and the facile removal of the resultant nonpolar lipids after alkaline methanolysis through liquid–liquid phase partitioning (Scheme 2). Briefly, when a lipid extract was treated with LiOMe solution in methanol, all ester-linked fatty acyl moieties were converted into fatty acid methyl esters which, along with cholesterol and free fatty acids if they were present in the lipid extract, were readily removed from the reaction mixture by extraction with hexane. Next, the sphingolipidome present in the reaction mixture could be recovered by Bligh and Dyer extraction through which the majority of polar compounds generated by methanolysis could be removed. Although alkaline hydrolysis followed by chromatographic separation for sphingolipid analysis has been employed previously (see [27,30,33–35] for examples), to the best of our knowledge, this is the first time the unique chemical features of sphingolipids were used to enrich the sphingolipidome for direct chemical analysis without the utilization of chromatographic separation.
To achieve the best experimental results for shotgun sphingolipidomics analyses, we examined a variety of the reaction conditions including the base used for cleavage of esters, temperature, and reaction time in the study. We tested the reaction under different base conditions by using LiOH, NaOH, KOH, NaOMe, or LiOMe at various concentrations. We found that the free fatty acids generated from the treatment of LiOH, NaOH, or KOH could not be efficiently removed by washing with hexane due to a limited solubility of free fatty acids in hexane and that the quality of lipid analysis achieved using shotgun lipidomics might be affected when bases other than those containing lithium were used to treat lipid extracts [4]. The recovery of sphingosine was affected if ether was used to remove the free fatty acids generated. However, abundant free fatty acids present in the lipid extract from mouse plasma (100 μl) had to be removed by at least a single wash with ether.
In this study, we also examined the effects of reaction temperature (tested at −20, 0, 25, and 50 °C) and reaction time (tested for 0.5, 1, and 2 h) to optimize penetrance into the sphingolipidome. We found that the treatment of crude lipid extracts with LiOMe at 0 °C for 1 h represented the best experimental conditions for the preparation of samples for shotgun sphingolipidomics. Furthermore, under these experimental conditions, the cleavage of amide, sulfate, phosphate, and ether bonds of sphingolipids was not detected as examined by using synthetic sphingolipids before and after the LiOMe treatment.
The recoveries of sphingolipids after treatment were determined by comparing the content levels of each individual sphingolipid class obtained from a sample with an internal standard added posttreatment with those from the identical sample with the internal standard added prior to the base treatment. The recoveries of sulfatide, SM, and GalCer molecular species were in the range of 80–95%. It should be emphasized that, even if the content of sphingolipids after treatment was not completely recovered, this would be only a secondary effect on the quantitation of the sphingolipid molecular species because an internal standard for each individual sphingolipid class was added to the lipid extracts prior to treatment.
Shotgun sphingolipidomics reveals the presence of many low-abundance molecular species of SM and the efficient ionization of HexCer molecular species in the positive-ion mode
One of the advantages of the developed approach is to allow shotgun sphingolipidomics to reveal the presence of many low-abundance molecular species of SM, which overlap with the abundant GPCho molecular species and/or are buried in the baseline noise in our original shotgun lipidomics approach. For example, Fig. 1 shows the comparisons of MS analyses of a lipid extract from mouse cortex before (Fig. 1A) and after (Fig. 1B) the treatment of the lipid extract with LiOMe along with the mass spectra acquired by using neutral loss scanning of 183.1 u (i.e., phosphocholine) which is specific for GPCho and SM molecular species in this mass region. The mass spectra clearly demonstrate that many additional SM molecular species in low abundance could be readily identified and quantified by shotgun sphingolipidomics (Fig. 1B2).
Fig. 1.

Shotgun lipidomics analyses of sphingolipid molecular species before and after treatment of a mouse cortex lipid extract with LiOMe in the positive-ion mode in the presence of a small amount of LiOH. The mass spectra were acquired directly from a lipid extract of mouse cortex before (left) and after (right) treatment with LiOMe as illustrated in Scheme 2, respectively. Mass spectra A1 and B1 are the survey scans and mass spectra A2 and B2 were acquired from the neutral loss of 183.1 u (i.e., phosphocholine). IS denotes internal standard. The ion peaks in mass spectrum B2 represent lithiated SM molecular species. Each spectrum displayed is normalized to the base peak in the spectrum.
Specifically, in the original descriptions of shotgun lipidomics technology, we were able to assess the content of lithiated N18:0 SM at m/z 737.5 in comparisons with that of N12:0 SM at m/z 653.5 after 13C deisotoping with an acceptable error. Limitations resulted mainly from the high background noise in the region of interest in particular cases (Fig. 1A1). By exploiting multidimensional mass spectrometry in the second step of shotgun lipidomics, the content of N16:0 SM at m/z 709.5 in the lipid extract could be assessed with minimal background noise through ratiometric comparisons with molecular ions quantified directly (Fig. 1A2). In shotgun sphingolipidomics, the quantities of lithiated N18:1 SM at m/z 735.5, N18:0 SM at m/z 737.5, and N24:1 SM at m/z 819.6 were accurately determined in comparisons with that of N12:0 SM at m/z 653.5 after 13C deisotoping (Fig. 1B1). The contents of all other low-abundance molecular species of SM (see inset in Fig. 1B2) were determined through the second step of quantitation by using N12:0, N18:0, and N24:1 SM as internal standards. These quantities of SM molecular species along with those from other mouse brain tissues and mouse plasma are listed in Table 1. The sphingoid base of major SM molecular species was determined as previously described [36]. The acyl amide moieties were derived based on the sphingoid base and m/z of each individual molecular species.
Table 1.
Sphingomyelin molecular species and content in lipid extracts of mouse cerebellum, cortex, spinal cord, brain stem, and plasma
| Molecular species | [M+Li]+ | Cerebellum | Cortex | Spinal cord | Brain stem | Plasma |
|---|---|---|---|---|---|---|
| N14:0 | 681.55 | — | — | — | — | 0.44 ± 0.02 |
| N15:0 | 695.57 | — | — | — | — | 1.49 ± 0.17 |
| N16:1 | 707.57 | 0.02 ± 0.00 | 0.02 ± 0.01 | 0.07 ± 0.04 | 0.03 ± 0.01 | 4.08 ± 0.94 |
| N16:0 | 709.58 | 0.20 ± 0.05 | 0.15 ± 0.05 | 0.41 ± 0.08 | 0.29 ± 0.08 | 20.49 ± 0.77 |
| N18:2 | 733.58 | — | — | — | — | 0.15 ± 0.03 |
| N18:1 | 735.60 | 0.09 ± 0.02 | 0.63 ± 0.03 | 0.18 ± 0.05 | 0.57 ± 0.06 | 0.74 ± 0.08 |
| N18:0 | 737.61 | 2.88 ± 0.41 | 3.86 ± 0.59 | 3.66 ± 0.60 | 5.63 ± 0.98 | 1.64 ± 0.22 |
| N19:0 | 751.63 | — | 0.05 ± 0.01 | — | — | 0.15 ± 0.03 |
| N20:1 | 763.63 | 0.04 ± 0.01 | — | — | — | 0.35 ± 0.07 |
| N20:0 | 765.65 | 0.23 ± 0.07 | 0.16 ± 0.05 | 0.67 ± 0.15 | 0.55 ± 0.14 | 0.51 ± 0.16 |
| N22:2 | 789.65 | — | — | — | — | 0.38 ± 0.09 |
| N22:1 | 791.66 | 0.08 ± 0.01 | 0.06 ± 0.02 | 0.80 ± 0.15 | 0.20 ± 0.05 | 2.63 ± 0.51 |
| N22:0 | 793.68 | 0.12 ± 0.04 | 0.10 ± 0.04 | 1.42 ± 0.24 | 0.49 ± 0.14 | 4.39 ± 0.95 |
| N23:2 | 803.66 | — | — | — | — | 0.23 ± 0.06 |
| N23:1 | 805.68 | — | — | 0.19 ± 0.04 | — | 1.51 ± 0.17 |
| N23:0 | 807.69 | — | 0.05 ± 0.01 | 0.34 ± 0.02 | 0.17 ± 0.03 | 1.79 ± 0.11 |
| N24:3 | 815.66 | — | — | — | — | 0.52 ± 0.05 |
| N24:2 | 817.68 | — | — | 0.26 ± 0.03 | 0.17 ± 0.02 | 4.89 ± 0.81 |
| N24:1 | 819.69 | 0.62 ± 0.02 | 0.49 ± 0.10 | 6.62 ± 0.63 | 2.86 ± 0.25 | 14.55 ± 2.62 |
| N24:0 | 821.71 | 0.16 ± 0.03 | 0.09 ± 0.02 | 1.70 ± 0.13 | 0.68 ± 0.23 | 3.16 ± 0.48 |
| N25:1 | 833.71 | — | — | 0.11 ± 0.02 | 0.07 ± 0.02 | 0.43 ± 0.13 |
| N25:0 | 835.72 | — | 0.05 ± 0.00 | — | 0.29 ± 0.08 | 0.90 ± 0.19 |
| Total | 4.53 ± 0.57 | 5.74 ± 0.89 | 16.50 ± 1.47 | 12.03 ± 1.79 | 65.43 ± 3.80 |
Lipid samples were prepared as illustrated in Scheme 2. The contents of SM molecular species were quantitated in a two-step procedure as described in the text. The results are expressed in nmol/mg protein (except that in plasma which represents nmol/ml) and represent X ± SD of five animals. The acyl amide chains were derived after determining the sphingoid base as sphingosine.
Shotgun sphingolipidomics analysis also demonstrates the abundant ion peaks other than SM in the mass spectrum acquired in the positive-ion mode in the presence of a small amount of LiOH (Fig. 1B1). Multidimensional MS analysis (spectra not shown) as described previously [37] identified these molecular species as lithiated monohexose cerebroside (HexCer) molecular species that were barely identifiable in the mass spectrum in the survey scanning mode acquired from the crude lipid extracts prior to the LiOMe treatment (Fig. 1A1). Furthermore, quantitation of HexCer molecular species was more accurate than that from untreated samples due to the presence of abundant and nonoverlapping HexCer ion peaks in the MS survey scan (Fig. 1B1). Thus, a greater number of HexCer molecular species could be quantified in the first step of the quantitation procedure by using shotgun sphingolipidomics without heavily relying on the second step which was conducted by using neutral loss of 162.1 u, corresponding to the loss of a hexose (spectrum not shown) as previously described [37].
Through application of mild alkaline hydrolysis in conjunction with shotgun sphingolipidomics, sulfatide molecular species can be analyzed in the negative-ion mode with greater accuracy
Sulfatide molecular species can be analyzed by using shotgun lipidomics in the negative ion mode. However, due to the so-called “ion suppression” and overlapping of sulfatide molecular species with either GPIns isotopologues at the high mass end or HexCer-chloride adducts at the low mass region (Fig. 2A), the accuracy of the assessment of the quantities of low abundant sulfatide molecular species is questionable in some cases [38,39]. By using shotgun sphingolipidomics, the overlapping of sulfatide molecular species with GPIns isotopologues was cleaned up and negative-ion ESI mass spectrum in the m/z range 600–1000 shows only deprotonated sulfatides and HexCer-chloride adducts (Fig. 2B1). Therefore, the contents of many sulfatide molecular species (e.g., all the ions higher than m/z 876.6) were determined in the MS survey scan in the first step of quantitation (Fig. 2B1). The contents of other low-abundance sulfatide molecular species at m/z 804.6, 806.6, 820.6, 822.6, 832.6, 834.6, 848.6, 850.6, 860.6, 862.6, and 874.6 (which were overlapped with chlorinated HexCer molecular species) were assessed through the second step of quantitation by using precursor-ion (PI) scanning of 97 Th (i.e., sulfate) (Fig. 2B2) and using the determined abundant sulfatide molecular species as internal standards. Moreover, neutral loss of 36 u (i.e., HCl) in shotgun sphingolipidomics under the experimental conditions (Fig. 2B3) allows us to readily identify the HexCer molecular species containing an hydroxy moiety at the α positions as previously described [37]. If necessary, the identity of the hexose moiety could be identified by product ion analysis of chlorinated HexCer after CID as described previously [37].
Fig. 2.
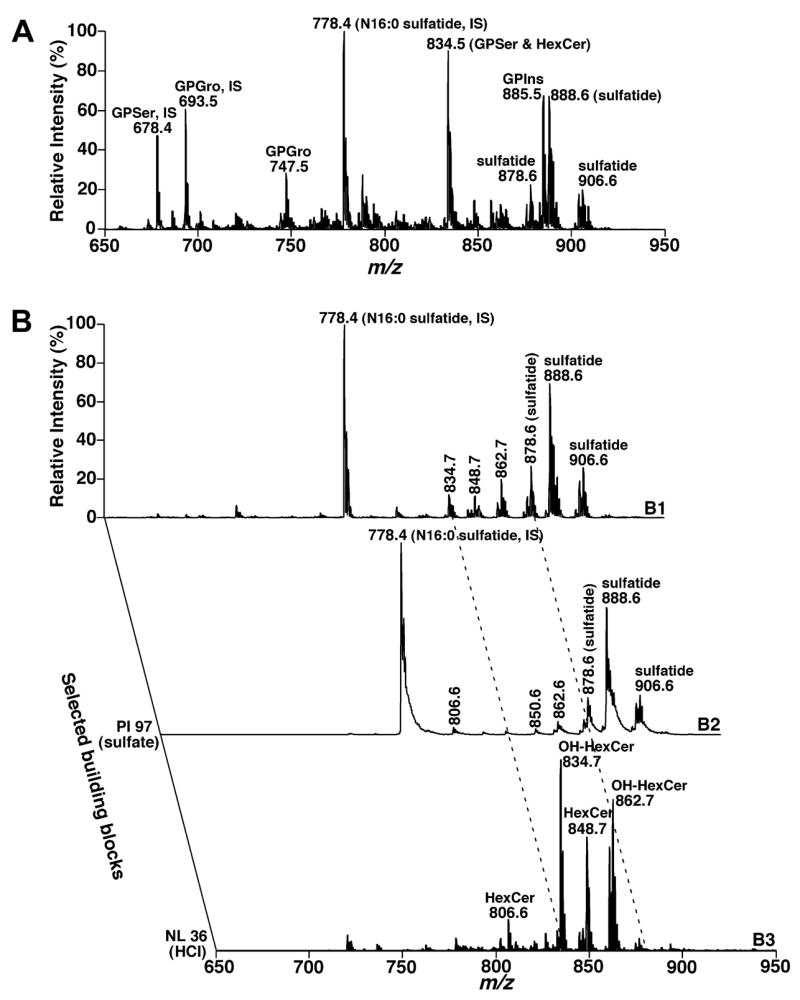
Shotgun lipidomics analyses of sphingolipid molecular species before and after treatment of a mouse cortex lipid extract with LiOMe in the negative-ion mode. The mass spectra in A and B1 were acquired directly from a lipid extract of mouse cortex before and after treatment with LiOMe as illustrated in Scheme 2, respectively. Tandem mass spectrometric analyses of the lipid extract after treatment with LiOMe were acquired by using precursor-ion scanning of 97 Th (i.e., sulfate) (B2) and neutral loss scanning of 36 u (loss of HCl from chlorinated HexCer molecular species) (B3), respectively. IS denotes internal standard. Each spectrum displayed is normalized to the base peak in the spectrum.
Shotgun sphingolipidomics enables identification and quantification of very minor sphingolipid classes without enrichment by using chromatography
The total lipid concentration in the infused solution used in shotgun lipidomics is limited to approximately 10 and 100 pmol/μl in 1:2 and 1:1 (v/v) CHCl3/MeOH, respectively, to avoid the formation of aggregates which causes complications for the analysis of molecular species in each individual lipid class and their quantitation [4,7,13]. Therefore, in addition to removing potential overlapping nonsphingolipid molecular species, treatment of lipid extracts with LiOMe also provides two other advantages. First, these procedures can enrich sphingolipids without the use of column chromatography(s). Second, this approach dramatically increases the effective dynamic range of quantitation for sphingolipid molecular species because many of the coexisting lipids (glycerolipids, cholesterol, etc.) have been eliminated. Therefore, the advantages of this shotgun sphingolipidomics approach allow us to establish a foundation to analyze many sphingolipid classes (e.g., sphingosine, lysosphingomyelin, psychosine, etc.) in the very-low-abundance regime without prior enrichment by column chromatography. Under the experimental conditions where the concentration of infused solution was kept below 100 pmol of total lipids/μl in 1:1 CHCl3/MeOH to avoid lipid aggregation, a 10- to 50-fold enrichment after base treatment could be easily achieved because the composition of sphingolipids in crude lipid extracts is commonly only a small percentage of total lipids.
Sphingosine (and sphinganine) were demonstrated to be readily ionized in the positive-ion mode in the presence of a small amount of formic acid (e.g., 0.1%) in 1:1 CHCl3/MeOH (Fig. 3A). The ionization efficiencies of the protonated sphingosine and its analogs (e.g., C14 to C18) were essentially identical (within experimental error) after 13C deisotoping as examined using a variety of mixtures of these compounds with various concentrations at different molar ratios. Fig. 3A shows an example of the mass spectra for the analysis of an equimolar mixture of these compounds (0.1 pmol/μl each in 1:1 CHCl3/MeOH with 0.1% formic acid).
Fig. 3.

ESI/MS and MS/MS analyses of sphingosine and its analogs. ESI/MS analysis (A) of an equimolar mixture of sphingosine analogs (0.1 pmol/μl each in 1:1 CHCl3/MeOH in the presence of 0.1% formic acid) was performed in the positive-ion mode. Product ion analysis of sphingosine (B) was performed after collision-induced dissociation. Tandem MS analyses of the equimolar mixture and a lipid extract from mouse spinal cord were also performed by using neutral-loss (NL) scanning of 48 u (i.e., loss of a H2O and a CH2O) (C and D, respectively). IS denotes internal standard.
Moreover, sphingosine and its analogs could be readily fragmented after CID (Fig. 3B). The CID mass spectra of this class of sphingolipids acquired by use of a low collision energy (e.g., 10 to 15 eV) display an abundant, common fragment which is very specific for sphingosine and its analogs and which corresponds to the neutral loss of 48.0 u (i.e., a formaldehyde molecule and a water molecule) (e.g., Fig. 3B). In addition, two predominant, non-specific water-loss fragments which are commonly seen in the CID analysis of some sphingolipids such as Cer in the positive-ion mode under acidic conditions are also present in these CID mass spectra (e.g., Fig. 3B). A fragmentation pattern of sphinganine quite similar to that of sphingosine was obtained under the experimental conditions (spectra not shown). Again, the mass spectrum acquired by using neutral-loss (NL) scanning of 48 u from an equimolar mixture of sphingosine and its analogs (0.1 pmol/μl each in 1:1 CHCl3/MeOH with 0.1% formic acid) shows a profile (Fig. 3C) essentially identical to that acquired by the survey scan of the mixture (Fig. 3A) except for the C17 sphinganine analog. The differential intensities in NL48 spectra of sphingosine and sphinganine analogs represent the different fragmentation kinetic rates of these two subclasses in generation of this identical fragment.
Further studies with other molar ratios at various concentrations of sphingosine and its analogs showed that the fragmentation kinetics of these compounds under such a low collision energy condition (i.e., 10 to 15 eV) are quite similar. Therefore, quantitation of sphingosine through direct ratiometric comparison with one of their analogs after 13C deisotoping was accurate within experimental error. A linear dynamic range over 1000-fold from 1 fmol/μl to 1 pmol/μl for quantitation of sphingosine by using C16 sphingosine analog as an internal standard and a detection limit of 0.1 fmol/μl were determined by using NL48 spectra. A correction factor of 1.30 ± 0.05 for quantitation of sphinganine by using C16 sphingosine analog as an internal standard was determined with a correlation coefficient (γ2) of 0.9994. In some cases, a background peak at m/z 288.3 (overlapping with C17 sphinganine analog) in NL48 spectra was observed. Therefore, a correction factor relative to C16 sphingosine analog as an internal standard rather than direct using C17 sphinganine analog as an internal standard for quantitation of sphinganine by using NL48 spectra is recommended. It should be emphasized that the CID mass spectra of sphingosine and its analogs acquired using a collision energy >20 eV show predominant fragments related to the cleavage of the aliphatic chain (i.e., ions at m/z 50 to 150; spectra not shown), which not only affected the detection limit but also changed the fragmentation kinetics. Direct profiling of sphingolipid extracts from mouse cortex, cerebellum, brain stem, spinal cord, and plasma using shotgun sphingolipidomics through NL scanning of 48 u demonstrated abundant ion peaks at m/z 300.3 and a minor ion peak at m/z 302.3, corresponding to sphingosine and sphinganine, respectively, along with the internal standards at m/z 272.2 (Fig. 3D). The contents of sphingosine (and sphinganine) in the determined mouse brain and plasma samples are compiled in Table 2. These results are within the range of the values published previously [40,41].
Table 2.
Sphingosine and sphinganine contents in mouse brain tissue and plasma samples
| Sample | Sphingosine | Sphinganine |
|---|---|---|
| Spinal cord | 107.7 ± 8.8 | 32.5 ± 9.9 |
| Brain stem | 100.1 ± 16.6 | 30.0 ± 4.8 |
| Cerebellum | 67.9 ± 2.7 | 23.9 ± 1.8 |
| Cortex | 96.8 ± 7.7 | 33.2 ± 3.4 |
| Plasma | 33.7 ± 1.7 | 29.5 ± 1.4 |
Lipid samples were prepared as shown in Scheme 2. The contents of sphingosine and sphinganine were quantitated by ratiometric comparison to the internal standard (i.e., C16:0 sphingosine analog) using tandem mass spectra after neutral loss of 48 u. The results are expressed in pmol/mg protein (except that in plasma which represents pmol/ml) and represent X ± SD of five animals.
Similar to other choline-containing phospholipids, product ion analysis of protonated lysoSM shows a predominant fragment at m/z 184.1 (Fig. 4A). Using shotgun sphingolipidomics, lysoSM molecular species were readily detected in the lipid extracts of mouse brain tissues and mouse plasma samples using precursor-ion scanning of m/z 184.1 (Fig. 4B). Therefore, lysoSM could also be quantitated using shotgun sphingolipidomics similar to that described above for quantitation of sphingosine utilizing proper internal standard(s).
Fig. 4.

Tandem mass spectrometric analyses of lysosphingomyelin. Product ion analysis of lysosphingomyelin (0.1 pmol/μl in 1:1 CHCl3/MeOH in the presence of 0.1% formic acid) was performed after collision-induced dissociation with a collision gas (nitrogen) pressure at 1.0 mTorr and a collision energy of 22 eV (A). Precursor-ion scanning of 184.1 Th (i.e., protonated phosphocholine) from a lipid extract of mouse spinal cord after treatment with LiOMe was conducted under experimental conditions as described (B).
Psychosine was also characterized under acidic conditions (e.g., in the presence of 0.1% formic acid in the infused solution). It was found that protonated psychosine was readily ionizable under acidic conditions and a detection limit of 1 fmol/μl was achieved. CID of protonated psychosine (0.1 pmol/μl) yielded multiple informative fragments as indicated (Fig. 5A). The abundant product ions at m/z 282.2 and 264.2 generated after CID correspond to the NL of a galactose (180 u) and a galactose plus a water molecule (198 u), respectively, from the protonated psychosine (Fig. 5A). Using shotgun sphingolipidomics, psychosine was readily detected from the lipid extracts of mouse brain tissue and plasma samples using NL of both 180 and 198 u (Figs. 5B and 5C). Therefore, psychosine could also be quantitated using shotgun sphingolipidomics with proper internal standard(s) after determination of a linear dynamic range for quantitation.
Fig. 5.

Tandem mass spectrometric analyses of psychosine. Product ion analysis of psychosine (0.1 pmol/μl in 1:1 CHCl3/MeOH in the presence of 0.1% formic acid) was performed after collision-induced dissociation with a collision gas (nitrogen) pressure at 1.0 mTorr and a collision energy of 24 eV (A). Neutral loss scanning of 180 u (i.e., loss of a galactose) (B) and 198 u (i.e., loss of a galactose and a water molecule) (C) from a lipid extract of mouse spinal cord after treatment with LiOMe was conducted under experimental conditions as described.
Quantitative analyses of Cer and sphingoid base-1-phosphate molecular species in shotgun sphingolipidomics were similarly performed as previously described [42,43] (data not shown). It should be pointed out that, owing to the quite nonpolar nature of ceramide, the hexane wash alters the profile of ceramide molecular species. Therefore, it is advised that this class of sphingolipid should be analyzed directly from the lipid extracts without mild alkaline treatment as previously described [7,42]. Lactosylceramide was poorly recovered by the current extraction method and an improved extraction method is making progress. Gangliosides which are essentially present in the aqueous phase during the Bligh and Dyer extraction were not covered by the described method. Collectively, identification of alterations in metabolic pathways and networks of sphingolipidome induced by any biological perturbation shall be greatly facilitated with our current progress in sphingolipidomics.
Conclusion
We developed a new shotgun lipidomics approach for the analysis of the sphingolipidome of biological samples by exploiting specific chemical properties of sphingolipids in conjunction with liquid–liquid phase portioning to markedly enrich biological samples for shotgun sphingolipidomics. Through this new development, individual molecular species of many abundant sphingolipid classes (e.g., SM, sulfatide, and cerebroside) and multiple minor sphingolipid classes (e.g., sphingosine, lysosphingomyelin, psychosine, and sphingoid base-1-phosphate) were accurately analyzed. Moreover, many sphingolipid molecular species present in either low abundance or those which overlapped with other lipid molecular species that were difficult to profile by our original shotgun lipidomics procedure could now be readily accessed by the addition of new multiplexed extraction conditions, thereby facilitating new horizons in shotgun sphingolipidomics.
Acknowledgments
This work was supported by NIA Grant R01 AG23168, NIA Grant R01 AG31675, NIH Grant P01 HL57278, and the Neurosciences Education and Research Foundation.
References
- 1.Han X, Gross RW. Global analyses of cellular lipidomes directly from crude extracts of biological samples by ESI mass spectrometry: a bridge to lipidomics. J Lipid Res. 2003;44:1071–1079. doi: 10.1194/jlr.R300004-JLR200. [DOI] [PubMed] [Google Scholar]
- 2.Lagarde M, Geloen A, Record M, Vance D, Spener F. Lipidomics is emerging. Biochim Biophys Acta. 2003;1634:61. doi: 10.1016/j.bbalip.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Wenk MR. The emerging field of lipidomics. Nat Rev Drug Discov. 2005;4:594–610. doi: 10.1038/nrd1776. [DOI] [PubMed] [Google Scholar]
- 4.Han X, Gross RW. Shotgun lipidomics: Electrospray ionization mass spectrometric analysis and quantitation of the cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom Rev. 2005;24:367–412. doi: 10.1002/mas.20023. [DOI] [PubMed] [Google Scholar]
- 5.Walker JM, Krey JF, Chen JS, Vefring E, Jahnsen JA, Bradshaw H, Huang SM. Targeted lipidomics: fatty acid amides and pain modulation. Prostaglandins Other Lipid Mediat. 2005;77:35–45. doi: 10.1016/j.prostaglandins.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 6.Serhan CN. Mediator lipidomics. Prostaglandins Other Lipid Mediat. 2005;77:4–14. doi: 10.1016/j.prostaglandins.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 7.Han X. Neurolipidomics: challenges and developments. Front Biosci. 2007;12:2601–2615. doi: 10.2741/2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ivanova PT, Milne SB, Forrester JS, Brown HA. LIPID arrays: New tools in the understanding of membrane dynamics and lipid signaling. Mol Interv. 2004;4:86–96. doi: 10.1124/mi.4.2.6. [DOI] [PubMed] [Google Scholar]
- 9.Welti R, Shah J, Li W, Li M, Chen J, Burke JJ, Fauconnier ML, Chapman K, Chye ML, Wang X. Plant lipidomics: discerning biological function by profiling plant complex lipids using mass spectrometry. Front Biosci. 2007;12:2494–2506. doi: 10.2741/2250. [DOI] [PubMed] [Google Scholar]
- 10.Schiller J, Suss R, Fuchs B, Muller M, Zschornig O, Arnold K. MALDI-TOF MS in lipidomics. Front Biosci. 2007;12:2568–2579. doi: 10.2741/2255. [DOI] [PubMed] [Google Scholar]
- 11.Albert CJ, Anbukumar DS, Monda JK, Eckelkamp JT, Ford DA. Myocardial lipidomics Developments in myocardial nuclear lipidomics. Front Biosci. 2007;12:2750–2760. doi: 10.2741/2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ejsing CS, Duchoslav E, Sampaio J, Simons K, Bonner R, Thiele C, Ekroos K, Shevchenko A. Automated identification and quantification of glycerophospholipid molecular species by multiple precursor ion scanning. Anal Chem. 2006;78:6202–6214. doi: 10.1021/ac060545x. [DOI] [PubMed] [Google Scholar]
- 13.Han X, Gross RW. Shotgun lipidomics: multi-dimensional mass spectrometric analysis of cellular lipidomes. Expert Rev Proteomics. 2005;2:253–264. doi: 10.1586/14789450.2.2.253. [DOI] [PubMed] [Google Scholar]
- 14.Han X, Gross RW. Quantitative analysis and molecular species fingerprinting of triacylglyceride molecular species directly from lipid extracts of biological samples by electrospray ionization tandem mass spectrometry. Anal Biochem. 2001;295:88–100. doi: 10.1006/abio.2001.5178. [DOI] [PubMed] [Google Scholar]
- 15.Han X, Yang J, Cheng H, Ye H, Gross RW. Towards fingerprinting cellular lipidomes directly from biological samples by two-dimensional electrospray ionization mass spectrometry. Anal Biochem. 2004;330:317–331. doi: 10.1016/j.ab.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 16.Cheng H, Jiang X, Han X. Alterations in lipid homeostasis of mouse dorsal root ganglia induced by apolipoprotein E deficiency: A shotgun lipidomics study. J Neurochem. 2007;101:57–76. doi: 10.1111/j.1471-4159.2006.04342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsui ZC, Chen QR, Thomas MJ, Samuel M, Cui Z. A method for profiling gangliosides in animal tissues using electrospray ionization-tandem mass spectrometry. Anal Biochem. 2005;341:251–258. doi: 10.1016/j.ab.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 18.Kalderon B, Sheena V, Shachrur S, Hertz R, Bar-Tana J. Modulation by nutrients and drugs of liver acyl-CoAs analyzed by mass spectrometry. J Lipid Res. 2002;43:1125–1132. doi: 10.1194/jlr.m200060-jlr200. [DOI] [PubMed] [Google Scholar]
- 19.Golovko MY, Murphy EJ. An improved method for tissue long-chain acyl-CoA extraction and analysis. J Lipid Res. 2004;45:1777–1782. doi: 10.1194/jlr.D400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Han X, Yang K, Yang J, Fikes KN, Cheng H, Gross RW. Factors influencing the electrospray intrasource separation and selective ionization of glycerophospholipids. J Am Soc Mass Spectrom. 2006;17:264–274. doi: 10.1016/j.jasms.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Han X, Cheng H, Mancuso DJ, Gross RW. Caloric restriction results in phospholipid depletion membrane remodeling and triacylglycerol accumulation in murine myocardium. Biochemistry. 2004;43:15584–15594. doi: 10.1021/bi048307o. [DOI] [PubMed] [Google Scholar]
- 22.Schwudke D, Oegema J, Burton L, Entchev E, Hannich JT, Ejsing CS, Kurzchalia T, Shevchenko A. Lipid profiling by multiple precursor and neutral loss scanning driven by the data-dependent acquisition. Anal Chem. 2006;78:585–595. doi: 10.1021/ac051605m. [DOI] [PubMed] [Google Scholar]
- 23.Zheng W, Kollmeyer J, Symolon H, Momin A, Munter E, Wang E, Kelly S, Allegood JC, Liu Y, Peng Q, Ramaraju H, Sullards MC, Cabot M, Merrill AH., Jr Ceramides and other bioactive sphingolipid backbones in health and disease Lipidomic analysis metabolism and roles in membrane structure dynamics signaling and autophagy. Biochim Biophys Acta. 2006;1758:1864–1884. doi: 10.1016/j.bbamem.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 24.Merrill AH, Caligan TB, Wang E, Peters K, Ou J. Analysis of sphingoid bases and sphingoid base 1-phosphates by high-performance liquid chromatography. Methods Enzymol. 2000;312:3–9. doi: 10.1016/s0076-6879(00)12894-0. [DOI] [PubMed] [Google Scholar]
- 25.Merrill AH, Jr, Sullards MC, Allegood JC, Kelly S, Wang E. Sphingolipidomics: high-throughput structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods. 2005;36:207–224. doi: 10.1016/j.ymeth.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 26.Sullards MC. Analysis of sphingomyelin glucosylceramide ceramide sphingosine and sphingosine 1-phosphate by tandem mass spectrometry. Methods Enzymol. 2000;312:32–45. doi: 10.1016/s0076-6879(00)12898-8. [DOI] [PubMed] [Google Scholar]
- 27.Sullards MC, Merrill AH., Jr Analysis of sphingosine 1-phosphate, ceramides, and other bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Sci STKE. 2001;2001:PL1. doi: 10.1126/stke.2001.67.pl1. [DOI] [PubMed] [Google Scholar]
- 28.Levery SB. Glycosphingolipid structural analysis and glycosphingolipidomics. Methods Enzymol. 2005;405:300–369. doi: 10.1016/S0076-6879(05)05012-3. [DOI] [PubMed] [Google Scholar]
- 29.Markham JE, Li J, Cahoon EB, Jaworski JG. Separation and identification of major plant sphingolipid classes from leaves. J Biol Chem. 2006;281:22684–22694. doi: 10.1074/jbc.M604050200. [DOI] [PubMed] [Google Scholar]
- 30.Bielawski J, Szulc ZM, Hannun YA, Bielawska A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods. 2006;39:82–91. doi: 10.1016/j.ymeth.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 31.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 32.Cheng H, Guan S, Han X. Abundance of triacylglycerols in ganglia and their depletion in diabetic mice: Implications for the role of altered triacylglycerols in diabetic neuropathy. J Neurochem. 2006;97:1288–1300. doi: 10.1111/j.1471-4159.2006.03794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gatt S. Enzymatic hydrolysis of sphingolipids .I. Hydrolysis and synthesis of ceramides by an enzyme from rat brain. J Biol Chem. 1966;241:3724–3730. [PubMed] [Google Scholar]
- 34.Portman OW, Alexander M. Metabolism of sphingolipids by normal and atherosclerotic aorta of squirrel monkeys. J Lipid Res. 1970;11:23–30. [PubMed] [Google Scholar]
- 35.Guan XL, He X, Ong WY, Yeo WK, Shui G, Wenk MR. Non-targeted profiling of lipids during kainate-induced neuronal injury. FASEB J. 2006;20:1152–1161. doi: 10.1096/fj.05-5362com. [DOI] [PubMed] [Google Scholar]
- 36.Hsu FF, Turk J. In: The encyclopedia of mass spectrometry. Caprioli RM, editor. Vol. 3. Elsevier; New York: 2005. pp. 430–447. [Google Scholar]
- 37.Han X, Cheng H. Characterization and direct quantitation of cerebroside molecular species from lipid extracts by shotgun lipidomics. J Lipid Res. 2005;46:163–175. doi: 10.1194/jlr.D400022-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Han X, Cheng H, Fryer JD, Fagan AM, Holtzman DM. Novel role for apolipoprotein E in the central nervous system: Modulation of sulfatide content. J Biol Chem. 2003;278:8043–8051. doi: 10.1074/jbc.M212340200. [DOI] [PubMed] [Google Scholar]
- 39.Cheng H, Xu J, McKeel DW, Jr, Han X. Specificity and potential mechanism of sulfatide deficiency in Alzheimer’s disease: An electrospray ionization mass spectrometric study. Cell Mol Biol. 2003;49:809–818. [PubMed] [Google Scholar]
- 40.te Vruchte D, Lloyd-Evans E, Veldman RJ, Neville DC, Dwek RA, Platt FM, van Blitterswijk WJ, Sillence DJ. Accumulation of glycosphingolipids in Niemann-Pick C disease disrupts endosomal transport. J Biol Chem. 2004;279:26167–26175. doi: 10.1074/jbc.M311591200. [DOI] [PubMed] [Google Scholar]
- 41.He X, Dagan A, Gatt S, Schuchman EH. Simultaneous quantitative analysis of ceramide and sphingosine in mouse blood by naphthalene-2,3-dicarboxyaldehyde derivatization after hydrolysis with ceramidase. Anal Biochem. 2005;340:113–122. doi: 10.1016/j.ab.2005.01.058. [DOI] [PubMed] [Google Scholar]
- 42.Han X. Characterization and direct quantitation of ceramide molecular species from lipid extracts of biological samples by electrospray ionization tandem mass spectrometry. Anal Biochem. 2002;302:199–212. doi: 10.1006/abio.2001.5536. [DOI] [PubMed] [Google Scholar]
- 43.Jiang X, Han X. Characterization and direct quantitation of sphingoid base-1-phosphates from lipid extracts: A shotgun lipidomics approach. J Lipid Res. 2006;47:1865–1873. doi: 10.1194/jlr.D600012-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]