

Abstract
To measure rate constants for discrete steps of single-round transcription (preinitiation complex formation, promoter escape, and transcript elongation), kinetic studies were performed in a well defined human RNA polymerase II transcription system. These experiments revealed that promoter escape limits the rate of transcription from the adenovirus major late promoter (AdMLP) contained on negatively supercoiled DNA. TFIIE and TFIIH were found to significantly increase fractional template usage during a single round of transcription in an ATP-dependent reaction. The observed rate constant for promoter escape, however, was not greatly affected by TFIIE and TFIIH. Our results are explained by a model in which transcription branches into at least two pathways: one that results in functional promoter escape and full-length RNA synthesis, and another in which preinitiation complexes abort during promoter escape and do not produce full-length RNA transcripts. These results with negatively supercoiled templates agree with our earlier conclusion that TFIIE, TFIIH, and ATP direct promoter escape and support a model in which the TFIIH helicases stimulate promoter escape in an ATP-dependent reaction.
Eukaryotic transcription is catalyzed by RNA polymerase II and facilitated by the concerted action of auxiliary proteins called general transcription factors (TFIIA, -B, -D, -E, -F, and -H; as reviewed in ref. 1). The transcription cycle consists of multiple steps, including preinitiation complex formation, open complex formation, promoter escape, transcript elongation, termination, and reinitiation, all of which have the potential to be regulated by promoter-specific transcriptional activators and repressors. Biochemical studies have established that the first step in basal (or unregulated) transcription is the binding of TFIID to core promoter sequences. After TFIID binding, RNA polymerase II and the other general transcription factors assemble on promoter DNA to form stable preinitiation complexes that become open complexes upon melting of the DNA in the region of the transcription start site (2–6). Upon the addition of nucleoside triphosphates, RNA polymerase II enters promoter escape, during which open complexes transform into elongation complexes as the first phosphodiester bonds are synthesized and the melted region of DNA moves away from the start site. Transcripts then are elongated and finally mRNA synthesis terminates as the RNA polymerase II is released from the template to reinitiate transcription.
Not all of the general transcription factors are required for transcription at TATA-containing promoters in vitro. For example, the human TFIID complex consisting of the TATA-binding protein (TBP) and at least eight associated factors (TAFs) can be replaced by the single subunit human TBP in basal transcription at promoters containing TATA boxes (7, 8). In addition, negative supercoiling of the template DNA alleviates the requirement for TFIIE and TFIIH in basal transcription at multiple promoters (9–12). Two of the subunits of TFIIH are helicases that hydrolyze rATP or dATP to melt DNA (13, 14). It has been hypothesized that negative superhelicity facilitates promoter melting during open complex formation and/or promoter escape (11, 12, 15), thereby overcoming the need for the ATP-dependent, TFIIH-associated helicase activities.
The mechanisms by which the TFIIH helicases function in transcription remain unresolved. It is evident that on linear DNA templates TFIIE, TFIIH, and ATP are required for promoter escape (12). It is also clear that these factors facilitate melting of linear promoter DNA, as detected by permanganate footprinting (6, 16). In contrast, the role of TFIIE, TFIIH, and ATP in abortive initiation, which detects functional initiation complexes capable of synthesizing short (3-nt), aborted RNA products, is controversial. We and others have reported that TFIIE, TFIIH, and ATP are not required for abortive initiation at promoters contained on linear DNA templates (6, 12, 17). Alternatively, in another report all three of these factors were required for abortive initiation from linear templates (18). This discrepancy could be explained by the recent finding that an excess of some general transcription factors in reactions imposes a requirement for TFIIE, TFIIH, and ATP in abortive initiation (D. Reinberg, personal communication).
An approach to understanding mechanisms of transcription, including the function of the TFIIH helicases, is to determine the rate constant for each step in the transcription cycle (19, 20). Previous data indicate functional open complexes are not capable of escaping the promoter on linear templates in the absence of TFIIE, TFIIH, and ATP (12). More recent studies have shown that TFIIE, TFIIH, and ATP can suppress promoter proximal pausing by RNA polymerase II under conditions of low nucleotide concentrations (21, 22). Together these studies suggest that promoter escape is rate limiting for transcription under some conditions and might represent a key point for regulating levels of transcription.
Here we have used a highly purified in vitro transcription system to study the kinetics of three steps of RNA polymerase II transcription at the adenovirus major late promoter (AdMLP) contained on a negatively supercoiled DNA template: preinitiation complex assembly, promoter escape, and transcript elongation. We measured the rate constants for these three steps and studied the effect of TFIIE, TFIIH, and ATP on transcription. Our results indicate that promoter escape limits the rate of transcription from negatively supercoiled templates, and that TFIIE, TFIIH, and ATP increase the fraction of preinitiation complexes that escape the promoter to produce full-length RNA.
MATERIALS AND METHODS
Transcription Factors.
Recombinant proteins were expressed and purified as described: human TBP (7), human TFIIB (23), and human TFIIE-34/TFIIE-56 (24). Human RAP30 and human RAP 74 were expressed in Escherichia coli as described (25–27) and purified separately by ion-exchange chromatography in TGED buffer (20 mM Tris⋅HCl, pH 7.9/10% glycerol/1 mM EDTA/1 mM DTT/0.2 mM phenylmethylsulfonyl fluoride) containing 6 M urea and NaCl. Purified RAP30 and RAP74 were combined in a 3:1 molar ratio, at a final protein concentration of 0.2 mg/ml in TGED containing 6 M urea and 100 mM NaCl, and subjected to stepwise dialysis to remove urea: (i) TGED, 1 M urea, 1 M NaCl; (ii) TGED, 1 M NaCl; (iii) TGED, 0.1 M NaCl. The TFIIF complex was purified on a Poros-Q column (PerSeptive Biosystems, Framingham, MA) with a linear gradient of 100–500 mM NaCl in TGED. Human RNA polymerase II was purified from HeLa nuclear pellets through the DEAE-5PW step as described (28). Human TFIIH was purified from HeLa cytoplasmic extracts as described (4). All basal factors, except RNA polymerase II, were dialyzed against buffer DB100 (20 mM Tris⋅HCl, pH 7.9/20% glycerol/1 mM DTT/0.1 mM EDTA/100 mM KCl). RNA polymerase II was dialyzed against a similar buffer containing 100 mM ammonium sulfate and 0.05% Nonidet P-40 in place of KCl.
In Vitro Transcription.
Transcription reactions were performed in buffer A containing 10 mM Tris⋅HCl, pH 7.9/10 mM Hepes. pH 8.0/10% glycerol/1 mM DTT/4 mM MgCl2/50 mM KCl/100 μg/ml BSA/15 units of RNAsin (Promega). Reactions contained the following amounts of protein factors: 5 ng TBP, 4 ng TFIIB, 6 ng TFIIF, 50 ng RNA polymerase II, 15 ng TFIIE-34, 6 ng TFIIE-56, and ≈14 ng TFIIH. The DNA template (0.8 nM) was negatively supercoiled plasmid DNA containing the AdMLP (−53 to +10) fused to a 380-bp G-less cassette (12). Calf thymus DNA (ctDNA, Sigma) was sonicated and purified by phenol extraction and ethanol precipitation. Transcription factors were preincubated in buffer A (10 μl per time point) for 2 min at 30°C, promoter DNA in buffer A (10 μl per time point) was added, and the incubation was continued for variable times as indicated in the figures. Nucleotides and ctDNA (2 μl of an 11× mixture per time point) were added to initiate transcription at final concentrations of 325 μg/ml ctDNA, 625 μM ATP, 625 μM UTP, 25 μM [α-32P]CTP (5 μCi per reaction) unless otherwise indicated in the figure legends. Transcription was allowed to proceed for 20 min at 30°C unless otherwise indicated. Reactions (22 μl per time point) were stopped with 100 μl of stop mix containing 3.1 M ammonium acetate/10 μg of carrier yeast RNA/15 μg of proteinase K. After ethanol precipitation, the samples were resolved by 6% denaturing PAGE.
Rate Constant Calculations.
The amount of RNA produced during in vitro transcription reactions was quantitated with a Molecular Dynamics PhosphorImager. After subtracting background, PhosphorImager units from full-length RNA at each time point were divided by the average PhosphorImager units produced at the longest time points (in the plateau region) to obtain the fractional completion at each time point. These values were plotted and fit to the equation Fc = 1 − e−kt, where Fc is the fractional completion, k is the observed rate constant, and t is time in seconds. The reported errors are one standard deviation from the mean rate constants calculated from three separate experiments. Observed rate constants for preinitiation complex formation and transcript elongation were estimated to be greater than the inverse of the fastest time point taken.
Template Usage Calculations.
For each experiment a standard curve of [α-32P]CTP spotted on Whatman 3-mm paper was quantitated by PhosphorImager analysis and used to determine the moles of [α-32P]CTP per PhosphorImager unit. Using the standard curve, the moles of RNA produced in each reaction were calculated and divided by the moles of DNA template to obtain a value for template usage.
RESULTS
To study the kinetic parameters of individual steps in transcription, we took advantage of competitor DNA. Our minimal transcription system consisted of TBP, TFIIB, TFIIF, RNA polymerase II, and a negatively supercoiled DNA template containing the AdMLP. By sequestering free TBP and RNA polymerase II, competitor DNA was used to divide the transcription cycle into two steps: (i) stable preinitiation complex formation and (ii) RNA synthesis (Fig. 1A). Stable preinitiation complex formation encompasses those steps that occur before the addition of nucleotides and competitor DNA. RNA synthesis includes all steps that occur after preinitiation complexes are provided with nucleotides and competitor DNA.
Figure 1.

Competitor DNA divides transcription into two steps and limits it to a single round. (A) Competitor DNA divides transcription into preinitiation complex formation and RNA synthesis by sequestering free TBP and RNA polymerase II. When competitor DNA is added at point 1 it prevents preinitiation complexes from forming on the promoter. When competitor DNA is added at point 2, after preinitiation complex formation is complete, the competitor prevents further preinitiation complexes from forming during RNA synthesis. (B) ctDNA was titrated into in vitro transcription reactions. When added to reactions before addition of the proteins (point 1 in schematic), ctDNA completely inhibited transcription (dashed line). When added to reactions with nucleotides (point 2 in schematic), ctDNA decreased the level of transcription approximately 8-fold (solid line).
Sonicated calf thymus DNA when added to transcription reactions as a competitor limits transcription to a single round of RNA synthesis. When ctDNA is titrated into reactions before the addition of proteins (point 1 of Fig. 1A), transcription is abolished by 200 μg/ml ctDNA (Fig. 1B, dashed line). Conversely, when ctDNA is titrated into transcription reactions with the nucleotides, after preinitiation complexes have formed at the promoter (point 2 of Fig. 1A), the level of transcription decreases 8-fold and plateaus by 200 μg/ml of ctDNA (Fig. 1B, solid line). Subsequent experiments all contained 325 μg/ml ctDNA that, when added at point 2, limited transcription to a single round. Control experiments confirmed that under these conditions TBP and RNA polymerase II were sequestered rapidly by ctDNA (data not shown).
The Rate of RNA Synthesis Limits Transcription from a Negatively Supercoiled Template.
We first measured the rate of stable preinitiation complex formation, as diagrammed in Fig. 2A. Promoter DNA was limiting with respect to general transcription factors in all reactions. The data show that preinitiation complexes assemble on the promoter within 30 s. In an attempt to detect a measurable rate constant for preinitiation complex formation, subsequent experiments were done with shorter time points. As shown in Fig. 2B, preinitiation complexes formed within 10 s. It was not technically possible to measure rates faster than 10 s using these experimental techniques; therefore, we conclude that under our experimental conditions the observed rate constant for stable preinitiation complex formation is greater than 0.1 s−1.
Figure 2.

Stable preinitiation complex formation is rapid. (A) Transcription reactions were assembled, initiated, and stopped according to the procedure diagrammed at the top of the figure. At varying time points after the addition of AdMLP, 20-μl aliquots were removed and added to ctDNA and nucleotides to produce a 390-nt G-less transcript. The star indicates an internal start site within the G-less cassette. (B) Stable preinitiation complexes form within 10 s. Reactions were carried out as in A.
We next measured the rate of RNA synthesis (moles of 390-nt transcript produced per second) from preformed stable complexes. RNA synthesis encompasses all steps that occur after preinitiation complex formation upon addition of nucleotides and ctDNA, i.e., promoter escape and transcript elongation. The protocol used to measure the rate constant for RNA synthesis is diagrammed in Fig. 3. The results show that RNA synthesis occurs at a relatively slow rate, with an observed rate constant of 1.9 ± 0.4 × 10−3 s−1 (derived from three independent experiments). The rate constant measured for RNA synthesis is at least 50 times less than that measured for preinitiation complex formation; hence, the rate-limiting step in a single transcription cycle occurs after the addition of nucleotides and within the events encompassed by RNA synthesis.
Figure 3.

RNA synthesis is slower than preinitiation complex formation. Reactions were assembled and initiated as depicted. Transcription was stopped at varying time points after the addition of nucleotides and ctDNA by removing 20-μl aliquots and adding them to stop solution.
Promoter Escape Limits the Rate of Transcription.
Upon determining that RNA synthesis events are slow, we next asked which step within RNA synthesis dictates the rate observed. Because promoter escape and elongation are the primary steps that occur after the addition of nucleotides, we hypothesized that one of these steps is rate limiting. The rate of transcript elongation was measured by monitoring the synthesis of full-length RNA from a paused 16-nt transcript (Fig. 4). A mutant template in which the +1 site on the nontemplate strand of the AdMLP was changed from A to G was used. In this promoter the first A in the nontemplate strand is at +16. Preinitiation complexes were formed, limited nucleotides (CpG, CTP, and UTP) and ctDNA were added, and transcription to +15 was allowed to occur. Next, ATP was added to elongate the paused 16-nt RNA into full-length RNA, reactions were stopped at varying time points, and the amount of full-length RNA at each time point was quantitated. The data in Fig. 4B show that the 16-nt RNA is elongated into full-length RNA within 30 s. Therefore, the rate constant for elongation to full-length RNA is greater than 0.03 s−1. This is much faster than the rate measured for the entirety of RNA synthesis. Therefore, we conclude that the rate-limiting step in transcription is not elongation, but rather promoter escape, which includes those events occurring after nucleotides are added to stable preinitiation complexes and before synthesis of RNA to position +15 is complete.
Figure 4.

Transcript elongation is rapid. (A) The schematic depicts the method used to measure the rate at which a short RNA transcript elongates into a full-length RNA. The promoter template contained a mutation in which the A/T base pair at +1 was mutated to G/C (12). As diagrammed, the limited nucleotide substrates CpG, CTP, UTP, and ctDNA allowed the synthesis of a 16-nt transcript. When given ATP the 16-nt RNA elongates into the full-length 86-nt RNA. (B) Elongation of the 16-nt RNA to a full-length RNA occurs within 30 s. Nucleotides were at final concentrations of 1 mM CpG/625 μM UTP/25 μM [α-32P]CTP (5 μCi per reaction). Elongation occurred after the addition of 625 μM ATP. The RNA products were resolved by 8% denaturing PAGE, and different exposures were used to show the full-length and 16-nt RNAs.
Although we could detect the 16-nt RNA paused at this promoter, we were not able to accurately measure the rate of synthesis of this short RNA because the signal-to-background ratio at short time points was too low. It is possible to measure the rate of promoter escape directly by decreasing the concentration of CTP to increase the specific activity of [α-32P]CTP labeling. We found, however, that decreasing the CTP concentration caused increased promoter proximal pausing of RNA polymerase II. This is not surprising because the rate of RNA synthesis is intimately tied to the concentration of nucleotides, and decreasing nucleotide concentrations is predicted to decrease the rate of RNA synthesis and stall RNA polymerase II. Although this approach provides a useful tool for examining characteristics of promoter escape (21, 22), it does not allow us to determine the rate-limiting step under standard in vitro transcription conditions. We were able to directly measure the rate of promoter escape in the presence of TFIIE, TFIIH, and dATP, conditions that give rise to a significantly better signal (see below).
TFIIE and TFIIH Increase the Extent of Promoter Escape.
Upon determining that promoter escape is rate limiting from a negatively supercoiled template, we asked whether TFIIE, TFIIH, and ATP could enhance this rate. We hypothesized that the TFIIH helicases might stimulate promoter escape in an ATP-dependent manner. The rate of RNA synthesis was measured in the presence of TFIIE and TFIIH (Fig. 5A). In this experiment TFIIE and TFIIH were added along with the other general transcription factors during preinitiation complex assembly; however, neither rATP nor dATP was included during preinitiation complex formation. ATP, CTP, and UTP were added at a later time point with ctDNA to activate the TFIIH helicases and initiate RNA synthesis. RNA synthesis was relatively slow in the presence of TFIIE and TFIIH with a rate constant of 2.3 ± 0.5 × 10−3 s−1. Hence, the rate constant for RNA synthesis is approximately the same in the presence and absence of TFIIE and TFIIH. Including dATP during preinitiation complex formation did not increase the observed rate constant for RNA synthesis (data not shown). In the presence of TFIIE and TFIIH the rate of RNA synthesis is still much slower than the rates of preinitiation complex formation and elongation. Therefore, promoter escape is rate limiting for transcription in the presence of TFIIE, TFIIH, and dATP.
Figure 5.

TFIIE and TFIIH enhance the extent of promoter escape. (A) TFIIE and TFIIH have a minimal effect on the rate of RNA synthesis. Transcription reactions were assembled and initiated in a large volume as indicated in the schematic. (B) TFIIE and TFIIH greatly enhance the extent of RNA synthesis. Transcription reactions were performed as in A, both in the presence and absence of TFIIE and TFIIH. The RNA products in lanes 1 and 3 result from 20 min of transcription, whereas those in lanes 2 and 4 result from 30 min of transcription. (C) TFIIE, TFIIH, and dATP enhance the extent of transcription by affecting promoter escape. Reactions were performed according to the procedure diagrammed beside the figure. Note that dATP was added with the nucleotides rather than with the proteins. AMP (1 mM) was added as the initiating nucleotide along with UTP (625 μM), CTP (25 μM [α-32P]CTP, 5 μCi per reaction), and, where indicated, dATP (5 μM). The 15-nt RNA product was resolved by 14% denaturing PAGE.
The results of Fig. 5A prompted us to ask whether the extent rather than the rate of RNA synthesis is affected by TFIIE and TFIIH. To address this question we monitored single-round transcription in the absence and presence of TFIIE and TFIIH. The amount of RNA produced was increased 11-fold by the addition of TFIIE and TFIIH (Fig. 5B). Thus, TFIIE and TFIIH increase the extent but not the observed rate constant for RNA synthesis.
The results of Fig. 5 A and B raise the following questions: which step in a single round of transcription is influenced by TFIIE and TFIIH and is ATP required for this event? To address these questions we measured the amount of a 15-nt RNA produced in the absence and presence of TFIIE, TFIIH, and ATP. To limit transcription to a 15-nt RNA we used the nucleotides AMP, CTP, and UTP and the wild-type AdMLP. AMP can be used only as the initiating nucleotide and cannot be incorporated into other positions of an elongating RNA. TFIIE and TFIIH were either included or omitted from the protein mixture, whereas dATP was either included or omitted from the nucleotide mixture. It is important to note that dATP was not present during preinitiation complex formation, hence, any effect TFIIE and TFIIH have on this step would be independent of dATP. As shown in Fig. 5C, the amount of 15-nt RNA increased greatly when TFIIE and TFIIH were added (lane 1 versus lane 2). Importantly, this effect depended on the presence of dATP (lane 1 versus lane 3). Thus, TFIIE and TFIIH increased the amount of 15-nt transcript in a dATP-dependent manner. We took advantage of the increased signal in the presence of TFIIE, TFIIH, and dATP to determine directly the rate constant for promoter escape in the presence of these factors (data not shown). We found that the rate constant for production of a 15-nt RNA was 5.1 ± 1.4 × 10−3 s−1. From these results, we conclude that TFIIE and TFIIH use ATP to increase the fraction of preinitiation complexes that functionally escape the promoter and synthesize a 15-nt RNA.
DISCUSSION
Here we have investigated the rate-limiting step in transcription at the AdMLP on a negatively supercoiled template. In doing so, we developed a system to divide transcription into discrete steps and to limit transcription to a single round, thereby facilitating kinetic measurements. Our results indicate that under the conditions described, promoter escape is the rate-limiting step for transcription from the AdMLP. In our experiments, stable preinitiation complex formation and transcript elongation were rapid. Although we expected transcript elongation to be rapid, we were surprised that preinitiation complex formation occurred within 10 s. This could be because of the way in which the general transcription factors were preincubated separately from the promoter DNA (unpublished results). Previous experimental evidence indicated that TFIIE, TFIIH, and ATP are intimately involved in promoter escape from linear templates (12, 21, 22). The results presented here extend these earlier observations to negatively supercoiled DNA templates.
A Kinetic Model for Promoter Escape.
Our results show that TFIIE and TFIIH do not greatly affect the observed rate constant for promoter escape; however, they cause an 11-fold increase in the fraction of preinitiation complexes that produce a 15-nt RNA in an ATP-dependent reaction. It is not possible to explain these observations if the process of transcription is a linear series of steps. If, however, the transcription reaction branches during promoter escape, then it is feasible that TFIIE and TFIIH could increase the extent of transcription without affecting the rate-limiting step. This is possible if one of the branches results in functional promoter escape while the other branch(es) end in nonfunctional or abortive complexes. Descriptions of the kinetics of branched transcription reactions have been explained in detail elsewhere (29, 30).
Fig. 6 shows a proposed reaction pathway for RNA polymerase II transcription that includes the steps we have investigated here. Preinitiation complexes form rapidly at the AdMLP, with an observed rate constant k1(obs) greater than 0.1 s−1. When preinitiation complexes are provided with nucleotides they enter promoter escape. Only a fraction of the preinitiation complexes actually proceed through promoter escape and produce a 15-nt RNA. Most of the preinitiation complexes that attempt to escape the promoter abort and never produce a 15-nt RNA. By monitoring transcription from the preinitiation complexes that functionally escape the promoter we measured an observed rate constant for RNA synthesis (i.e., the combination of promoter escape and transcript elongation) of 0.0019 s−1. These complexes rapidly elongate a 15-nt RNA with an apparent rate constant (k3) greater than 0.03 s−1; therefore, promoter escape limits the rate of RNA synthesis with a rate constant, k2(obs), of 0.002 s−1.
Figure 6.

A model to describe the mechanism of single-round transcription. The rate constant for preinitiation complex formation, k1(obs), is greater than 0.1 s−1, and the rate constant for transcript elongation, k3, is greater than 0.03 s−1 under our standard in vitro transcription conditions. Promoter escape limits the rate of transcription with an observed rate constant, k2(obs), of 2 × 10−3 s−1. In the model, there are two pathways for promoter escape, functional and abortive, with rate constants k2f and k2a, respectively. TFIIE and TFIIH, in an ATP-dependent reaction, increase k2f and, in doing so, increase the fraction of preinitiation complexes that functionally escape the promoter. This is observed experimentally as an increase in the extent of transcription with no significant change in k2(obs) (explained in detail in Discussion).
If the pathway of transcription splits at promoter escape into functional and abortive branches, then the rate constant measured for promoter escape, k2(obs), is equal to the sum of the rate constants for functional promoter escape (k2f) and abortive escape (k2a).
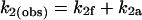 |
1 |
The rate constants for the two branches can be calculated from the following equations:
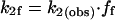 |
2 |
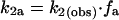 |
3 |
where ff is the fraction of the preinitiation complexes that escape the promoter to produce full-length RNA and fa is the fraction of the preinitiation complexes that abort during promoter escape. We can calculate the microscopic rate constants (k2f and k2a) from the template usage in our single-round transcription reactions. On average, in the absence of TFIIE and TFIIH only 0.2% of the templates in our reactions are used to produce full-length RNA (ff = 0.002). Assuming all promoter DNA forms preinitiation complexes, then 99.8% of the preinitiation complexes do not produce an RNA (fa = 0.998), and the calculated rate constants k2f and k2a are 4 × 10−6 s−1 and 2 × 10−3 s−1, respectively. Thus, in the absence of TFIIE and TFIIH, k2a ≫ k2f resulting in k2(obs) being equal to k2a.
In the presence of TFIIE and TFIIH, k2(obs) is 5.1 × 10−3 s−1 and ff is 0.022; hence, the calculated k2f and k2a are 1 × 10−4 s−1 and 5 × 10−3 s−1, respectively. Therefore, we propose that TFIIE and TFIIH in an ATP-dependent reaction increase k2f, which is observed as an increase in the extent of promoter escape. In the presence of TFIIE and TFIIH, k2a is still much greater than k2f; thus, the measured rate of promoter escape is still dominated by the rate of abortive complex formation (i.e., k2(obs) = k2a).
What is the nature of the abortive complexes that limit functional promoter escape? Answering this question will require further studies; however, we do have some clues from other experiments. First, we found that abortive initiation at the AdMLP ceases when the missing nucleotides are added to reactions (J.G., unpublished data). Although this is to be expected under conditions where full-length RNA can be observed, abortive initiation also stops with linear templates in the absence of TFIIE and TFIIH where full-length RNA is not produced. Thus, complexes that are functional for abortively synthesizing 3-nt RNA products become nonfunctional instead of escaping the promoter successfully. Second, Dvir et al. (21) observed that TFIIH could rescue promoter-proximal paused complexes containing short RNAs (5–7 nt). This indicates that TFIIH has the ability to suppress promoter-proximal pausing in an ATP-dependent reaction. Third, transcription by E. coli RNA polymerase can result in dead-end complexes during transcript elongation in the presence of all four nucleoside triphosphates (31). Fourth, Kubori and Shimamoto (32) have found that only 25% of the open complexes formed by E. coli RNA polymerase at the phage λ PR promoter synthesize a full-length RNA under single-round transcription conditions. The other 75% of the open complexes proceed through a branch in the pathway and are only capable of abortive RNA synthesis. We hypothesize that abortive complexes form upon addition of nucleotides to RNA polymerase II preinitiation complexes at the AdMLP. Timmers and colleagues (6) have elegantly shown that TFIIE, TFIIH, and ATP facilitate melting of the AdMLP contained on linear templates under conditions very similar to those used here. Perhaps the TFIIH helicases can act both on open complexes before the initiation of transcription and on complexes as they are escaping the promoter.
The finding that promoter escape can be rate-limiting for RNA polymerase II transcription suggests that this step is likely to be targeted by transcriptional activators. It has been shown that transcriptional activators can stimulate promoter escape (33). It has also been found that transcriptional activators can bind to TFIIH, suggesting that these activators may also influence promoter escape (34–36). We expect that other transcriptional activators will be found to target promoter escape as more mechanistic studies are undertaken in the future.
Acknowledgments
We thank L. J. Kim, S. D. Klube, R. D. Kuchta, T. N. Lively, O. C. Uhlenbeck, and H. Vodermaier for helpful discussions and for comments on the manuscript. We are grateful to M. E. Maxon for TFIIE. This research was supported in part by grants from The Council for Tobacco Research, USA, and The University of Colorado Council on Research and Creative Work. J.A.G. is a Special Fellow of the Leukemia Society of America.
ABBREVIATIONS
- AdMLP
adenovirus major late promoter
- ctDNA
calf thymus DNA
- TFII
transcription factor of RNA polymerase II
- TBP
TATA-binding protein
References
- 1.Zawel L, Reinberg D. Annu Rev Biochem. 1995;64:533–561. doi: 10.1146/annurev.bi.64.070195.002533. [DOI] [PubMed] [Google Scholar]
- 2.Reinberg D, Horikoshi M, Roeder R. J Biol Chem. 1987;262:3322–3330. [PubMed] [Google Scholar]
- 3.Buratowski S, Hahn S, Guarente L, Sharp P A. Cell. 1989;56:549–561. doi: 10.1016/0092-8674(89)90578-3. [DOI] [PubMed] [Google Scholar]
- 4.Flores O, Lu H, Reinberg D. J Biol Chem. 1992;267:2786–2793. [PubMed] [Google Scholar]
- 5.Jiang Y, Yan M, Gralla J D. Mol Cell Biol. 1996;16:1614–1621. doi: 10.1128/mcb.16.4.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holstege F C P, van der Vliet P C, Timmers H T M. EMBO J. 1996;15:1666–1677. [PMC free article] [PubMed] [Google Scholar]
- 7.Peterson M G, Tanese N, Pugh B F, Tjian R. Science. 1990;248:1625–1630. doi: 10.1126/science.2363050. [DOI] [PubMed] [Google Scholar]
- 8.Kao C C, Lieberman P M, Schmidt M C, Zhou Q, Pei R, Berk A J. Science. 1990;248:1646–1650. doi: 10.1126/science.2194289. [DOI] [PubMed] [Google Scholar]
- 9.Parvin J D, Timmers H T M, Sharp P A. Cell. 1992;68:1135–1144. doi: 10.1016/0092-8674(92)90084-p. [DOI] [PubMed] [Google Scholar]
- 10.Tyree C M, George C P, Lira-DeVito L M, Wampler S L, Dahmus M E, Zawel L, Kadonaga J T. Genes Dev. 1993;7:1254–1265. doi: 10.1101/gad.7.7a.1254. [DOI] [PubMed] [Google Scholar]
- 11.Parvin J D, Sharp P A. Cell. 1993;73:533–540. doi: 10.1016/0092-8674(93)90140-l. [DOI] [PubMed] [Google Scholar]
- 12.Goodrich J A, Tjian R. Cell. 1994;77:145–156. doi: 10.1016/0092-8674(94)90242-9. [DOI] [PubMed] [Google Scholar]
- 13.Schaeffer L, Roy R, Humbert S, Moncolli V, Vermeulen W, Hoeijmakers J H J, Chambon P, Egly J-M. Science. 1993;260:58–63. doi: 10.1126/science.8465201. [DOI] [PubMed] [Google Scholar]
- 14.Drapkin R, Reardon J T, Ansari A, Huang J-C, Zawel L, Ahn K, Sancar A, Reinberg D. Nature (London) 1994;368:769–772. doi: 10.1038/368769a0. [DOI] [PubMed] [Google Scholar]
- 15.Timmers H T M. EMBO J. 1994;13:391–399. doi: 10.1002/j.1460-2075.1994.tb06273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holstege F C P, Fiedler U, Timmers H T M. EMBO J. 1997;16:7468–7480. doi: 10.1093/emboj/16.24.7468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan G, Greenblatt J. J Biol Chem. 1994;269:30101–30104. [PubMed] [Google Scholar]
- 18.Dvir A, Garrett K P, Chalut C, Egly J-M, Conaway J W, Conaway R C. J Biol Chem. 1996;271:7245–7248. doi: 10.1074/jbc.271.13.7245. [DOI] [PubMed] [Google Scholar]
- 19.Hawley D K, Roeder R G. J Biol Chem. 1985;260:8163–8172. [PubMed] [Google Scholar]
- 20.Hawley D K, Roeder R G. J Biol Chem. 1987;262:3452–3461. [PubMed] [Google Scholar]
- 21.Dvir A, Conaway R C, Conaway J W. Proc Natl Acad Sci USA. 1997;94:9006–9010. doi: 10.1073/pnas.94.17.9006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dvir A, Conaway R C, Conaway J W. J Biol Chem. 1996;271:23352–23356. doi: 10.1074/jbc.271.38.23352. [DOI] [PubMed] [Google Scholar]
- 23.Ha I, Lane W S, Reinberg D. Nature (London) 1991;352:689–695. doi: 10.1038/352689a0. [DOI] [PubMed] [Google Scholar]
- 24.Peterson M G, Inostroza J, Maxon M E, Flores O, Admon A, Reinberg D, Tjian R. Nature (London) 1991;354:369–373. doi: 10.1038/354369a0. [DOI] [PubMed] [Google Scholar]
- 25.Aso T, Vasavada H A, Kawaguchi T, Germino F J, Ganguly S, Kitajima S, Weissman S M, Yasukochi Y. Nature (London) 1992;355:461–464. doi: 10.1038/355461a0. [DOI] [PubMed] [Google Scholar]
- 26.Finkelstein A, Kostrub C F, Li J, Chavez D P, Wang B Q, Fang S M, Greenblatt J, Burton Z F. Nature (London) 1992;355:464–467. doi: 10.1038/355464a0. [DOI] [PubMed] [Google Scholar]
- 27.Sopta M, Burton Z, Greenblatt J. Nature (London) 1989;341:410–414. doi: 10.1038/341410a0. [DOI] [PubMed] [Google Scholar]
- 28.Lu H, Flores O, Weinmann R, Reinberg D. Proc Natl Acad Sci USA. 1991;88:10004–10008. doi: 10.1073/pnas.88.22.10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goodrich J A, McClure W R. J Mol Biol. 1992;224:15–29. doi: 10.1016/0022-2836(92)90573-3. [DOI] [PubMed] [Google Scholar]
- 30.Herschlag D, Johnson F B. Genes Dev. 1993;7:173–179. doi: 10.1101/gad.7.2.173. [DOI] [PubMed] [Google Scholar]
- 31.Erie D A, Hajiseyedjavadi O, Young M C, von Hippel P H. Science. 1993;262:867–873. doi: 10.1126/science.8235608. [DOI] [PubMed] [Google Scholar]
- 32.Kubori T, Shimamoto N. J Mol Biol. 1996;256:449–457. doi: 10.1006/jmbi.1996.0100. [DOI] [PubMed] [Google Scholar]
- 33.Narayan S, Widen S G, Beard W A, Wilson S H. J Biol Chem. 1994;269:12755–12763. [PubMed] [Google Scholar]
- 34.Xiao H, Pearson A, Coulombe B, Truant R, Zhang S, Regier J L, Triezenberg S J, Reinberg D, Flores O, Ingles C J, et al. Mol Cell Biol. 1994;14:7013–7024. doi: 10.1128/mcb.14.10.7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blau J, Xiao H, McCracken S, O’Hare P, Greenblatt J, Bentley D. Mol Cell Biol. 1996;16:2044–2055. doi: 10.1128/mcb.16.5.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rochette-Egly C, Adam S, Rossignol M, Egly J M, Chambon P. Cell. 1997;90:97–107. doi: 10.1016/s0092-8674(00)80317-7. [DOI] [PubMed] [Google Scholar]