

Abstract
The medical utility of proteins, e.g. therapeutic enzymes, is greatly restricted by their liable nature and inadequate delivery. Most therapeutic enzymes do not accumulate in their targets and are inactivated by proteases. Targeting of enzymes encapsulated into substrate-permeable Polymeric Nano-Carriers (PNC) impermeable for proteases might overcome these limitations. To test this hypothesis, we designed endothelial targeted PNC loaded with catalase, the H2O2-detoxifying enzyme, and tested if this approach protects against vascular oxidative stress, a pathological process implicated in ischemia-reperfusion and other disease conditions. Encapsulation of catalase (MW 240KD), peroxidase (MW 42kD) and xanthine oxidase (XO, MW 300 kD) into ~300nm diameter PNC composed of co-polymers of PEG-PLGA (polyethylene glycol and poly-lactic/poly-glycolic acid) was in the range ~10% for all enzymes. PNC/catalase and PNC/peroxidase were protected from external proteolysis and exerted the enzymatic activity on their PNC diffusible substrates, H2O2 and ortho-phenylendiamine, whereas activity of encapsulated XO was negligible due to polymer impermeability to the substrate. PNC targeted to platelet-endothelial cell adhesion molecule-1 delivered active encapsulated catalase to endothelial cells and protected the endothelium against oxidative stress in cell culture and animal studies. Vascular targeting of PNC-loaded detoxifying enzymes may find wide medical applications including management of oxidative stress and other toxicities.
Introduction
High specificity and potency are the key strengths of protein therapeutics (e.g., enzymes), which have seen an exponential increase in the rate of Food and Drug Administration (FDA) approval over the past decade [1]. Unfortunately, sub-optimal stability and delivery to therapeutic sites impede the medical use of proteins. Strategies including stealth technologies (i.e., coupling of hydrophilic polyethylene glycol, PEG, that protects cargoes from recognition by host defense systems), designer stabilized enzymes, loading proteins into synthetic depots with controlled release rates [2], synthesis of targeted protein conjugates, fusion and recombinant mutant proteins have all been pursued in an effort to overcome these hurdles [3–6].
The key role and daunting challenges of optimal delivery of therapeutic enzymes is illustrated by the three decades worth of efforts aimed at the important, yet still elusive goal of vascular oxidative stress containment. This pathological condition, induced by reactive oxygen species including H2O2 produced by leukocytes and vascular cells themselves, is implicated in acute lung injury, ischemia-reperfusion, stroke, myocardial infarction, inflammation and other maladies [7]. Antioxidant inducers and high doses of non-enzymatic antioxidants alleviate some forms of chronic oxidative stress, but afford no protection against acute severe insults [8]. Unfortunately, the more potent antioxidant enzymes (e.g., catalase which reduces H2O2 to water) have no utility in the treatment of vascular oxidative stress due to poor stability and inadequate delivery to endothelial cells (EC) lining the vascular lumen [7]. EC represent both a source and a critically important, vulnerable target of oxidants [9–12].
In order to improve endothelial delivery, diverse means have been designed [13, 14]. In particular, endothelial cell adhesion molecules (CAMs, e.g., Platelet-Endothelial Cell Adhesion Molecule-1, PECAM-1) represent a good target determinant for delivery of antioxidants and other protein therapeutics for the treatment of vascular maladies [15, 16]. PECAM-1 is stably expressed or up-regulated on the surface of endothelium during inflammation and its blockade attenuates leukocyte transmigration [17]. Targeting of catalase conjugated with antibodies against PECAM-1 alleviates acute severe oxidative stress in cell cultures[18, 19], perfused organs[20], lung transplantation in rats[13] and oxidative lung injury in mice[21].
Although promising, this approach offers a relatively short (<3 hours) duration of protection, due to endothelial proteolysis of conjugates [22], which is sub-optimal for applications requiring more prolonged therapy. In theory, catalase loaded into targeted Polymeric Nano-Carriers (PNC) could be more resistant to inactivation. The immobilization of enzymes inside macro- and micro-scale polymeric matrixes protects them from thermal and proteolytic degradation [23, 24]. Conceivably, enzymes encapsulated within nanocarriers permeable for their substrates but impermeable to proteases could result in a prolonged therapeutic effect. Previous work of other labs with enzyme reactors designed for industrial use [23, 25] that provides high stability of encapsulated proteins [24] lends indirect support for this hypothetical concept.
Catalase is active at the acidic milieu typical of lysosomes and ischemic pathological foci (pH 4–6) and decomposes the highly permeable small oxidant, H2O2. Thus, catalase is an attractive candidate for PNC encapsulation. We previously loaded enzymatically active catalase into ~300–400 nm diameter PNC composed of biodegradable di-block copolymers PEG-poly(lactide co glycolide) (PEG-PLGA) permeable for H2O2 and showed that PNC-encapsulated catalase (PNC/catalase) was resistant to proteases and degraded H2O2 diffusing through the PNC shell [26]. Based on this initial success, we hypothesized that targeting of PNC/catalase to PECAM would both serve to deliver the antioxidant enzyme to the endothelium and provide sustained protection against vascular oxidative stress. Furthermore, to analyze this platform for vascular detoxification systematically, we loaded a series of other enzymes into PNC and tested their protection against external proteolysis and ability to convert substrates of varying size and hydrophobicity.
METHODS
Reagents
Methoxypoly(ethylene glycol) MW 5000 (mPEG) was obtained from Polysciences (Warrington, PA). Poly(lactic-co-glycolic acid)(50:50) in the free acid (38,000 MW) form was from Alkermes, Inc (Cincinnati, OH). Bovine liver catalase (242,000 MW) was obtained from Calbiochem (EMD Biosciences, San Diego, CA). 10-acetyl-3,7-dihydroxyphenoxazine, Amplex red ®, and Alexa Flour-488-labeled goat anti-mouse antibodies were from Molecular Probes (Eugene, OR). Succinimidyl 4-[N-maleimidomethyl]cyclohexane-1-carboxylate, N-Succinimidyl-S-acetylthioacetate and N-succinimidyl-biotin (SMCC, SATA, NHS-biotin, respectively) were from Pierce Biotechnology (Rockford, IL). Na125I and Na251CrO4 were from Perkin Elmer (Boston, MA). Pronase, protease cocktail derived from Streptomyces griseus, and all other reagents and solvents were from Sigma-Aldrich (St. Louis, MO).
Protein Iodination
All proteins were radiolabeled with Na125I using the Iodogen (Pierce Biotech., Rockford, IL) method following manufacturer’s recommendations, and unbound iodine was removed using gel permeation chromatography (Bio-spin 6 Columns, Bio-Rad Labs, Hercules, CA).
Copolymer Synthesis
Two separate methods were used to prepare copolymers for this study. For all polymers, molecular weight and polydispersity index were determined using proton-nuclear magnetic resonance spectroscopy (1H-NMR) and gel permeation chromatography (GPC).
mPEG-PLA
D,L-lactide was recrystalized twice in anhydrous ether, and then mixed with mPEG in weight ratios predetermining its molecular weight (30KDa). The bulk material was raised to 140°C for 2 hours under a purged nitrogen atmosphere, then the temperature was reduced to 120°C, 1 wt% stannous 2-ethyl-hexanoate was added, and the polymerization proceeded for 6 hours. The resulting polymer was dissolved in dichloromethane (DCM), and precipitated twice in cold diethyl ether. The final product was serially dried in a rotovap (Safety Vap 205, Buchi, Switzerland) followed by a freeze dryer (RCT 60, Jouan Inc, Winchester, VA) to remove any residual solvent.
Biotin-PEG-PLGA
PLGA (50:50) (38kDa) polymer containing a carboxylate end group and PEG-diamine (10,000 MW) were freeze dried overnight to remove bound water. The polymers were mixed in a 6:1 molar ratio (PEG:PLGA) in anhydrous DCM to a final polymer concentration of 2 wt%. Next, 2,2-dicyclocarbodiimide (DCC) was added to the polymer solution at the molar ratio of 1.2:1, DCC:PLGA. Conjugation was carried out under a N2 atmosphere at room temperature for 18 hours. The resulting precipitate, dicyclohexylurea, was filtered out, and the polymer was precipitated twice in anhydrous ether. The filtrate was then dried, dissolved in acetone and precipitated in deionized water. This precipitate was filtered and freeze dried. Next, Biotin-N′-Succinimidyl ester was added (1.2:1 molar ratio) with the polymer in DCM. After 4 hours, the polymer was precipitated twice more in ether. Fourier transform infrared spectroscopy (FTIR) was used to verify biotin conjugation by monitoring absorbance at 1630 cm−1, the characteristic absorbance of the biotin urea bond.
PNC synthesis
Two types of PNC syntheses were employed in the course of this study, unloaded solid core PNC and catalase-loaded PNC. Particle sizes were determined by dynamic light scattering (DLS, 90PLUS Particle Sizer, Brookhaven Instruments, Holtsville, NY).
Solid core
PEG-PLGA with or without 15 mol% Biotin-PEG-PLGA was dissolved into acetone (10mg/ml, 2.5 ml). This solution was then slowly pipetted into 20 ml PBS under mild agitation. Acetone was stripped off under vacuum using a dry nitrogen stream. PNC were collected by centrifuging at 30,000g for 30 minutes, and resuspended into 1 ml of PBS. PNC mass concentrations were determined by a previously published chemical and enzymatic assay [26]. Briefly, A 50 μl aliquot of the concentrated nanoparticle prep was saponified by adding 200 μl of 5 M NaOH and reacting overnight at 80 °C. This solution was then neutralized with 200 μl of 5 M HCl. PEG concentration was determined by a colorimetric assay based off of the PEG–Barium Iodide complex. Absorbance of the color product was measured at 550 nm using a microplate reader (Model 2550-UV, Bio-Rad Labs, Hercules, CA).
To measure PLA concentration, an enzymatic assay for L-lactic acid was used. 5 μl of sample was added to 45 μl of 50 mM PBS in a microplate well. To this well, 50 μl of assay buffer was added. The assay buffer consisted of 2 U ml 1 horseradish peroxidase, 20 mU lactate oxidase and 1 μg ml 1 of Amplex Red. After 10 min of incubation at room temperature, the product concentration was determined by UV absorbance at 550 nm. Concentrations were measured in triplicate for each individual particle preparation.
Protein Loaded PNC
Nanocarrier synthesis was based upon the previously published double emulsion, freeze-thaw strategy. The primary emulsion was formed by homogenizing at 15,000 rpm for 1 minute (−80 °C, dry ice/acetone bath) a 100 μl aqueous drug solution (1–25 mg/ml protein in PBS) in a 1ml organic polymer solution (25 mg/ml of mPEG-PLA with 15 mol% Biotin-PEG-PLA in DCM), using a 7mm blade homogenizer (Kinemetica Polytron 3100 equipped with a PTDA3007/2 generator, Brinkmann Instruments, Westbury, NY). This primary solution was immediately pipetted into a secondary aqueous phase (5 ml) containing 2 wt% of the PEG-poly(propylene glycol) triblock copolymer surfactant, Pluronic F68 (Sigma, MO), and homogenized at 15,000 rpm for 1 minute. This second homogenization was added to 10 more ml of the same surfactant solution, and stirred overnight under mild agitation to remove residual solvent. PNC were then washed twice more (centrifuged at 20,000g for 30 minutes, resuspended and centrifuged again). PNC were finally resuspended in 1ml PBS and stored at 4°C prior to use.
Antibody-streptavidin conjugate preparation
The heterobifunctional cross-linker, SMCC, was used to introduce stable maleimide reactive groups onto streptavidin molecule (SA). The reaction was performed at a 40-fold molar excess of SMCC at room temperature for 1 h. In parallel, sulfhydryls were introduced onto the antibody through primary amine directed chemistry using SATA. Preliminary experiments showed that the yield of the reaction was about 20%. Thus, to introduce 1 sulfhydryl per IgG molecule, antibody was incubated with 5-fold molar excess of SATA at room temperature for 30 min. This extent of modification prevented potential cross-linking of SA and subsequent protein polymerization. Acetylated sulfhydryls were deprotected using hydroxylamine (50mM final concentration) and antibody was conjugated with activated SA at 2:1 IgG to SA molar ratio. At each step unreacted components were removed using desalting columns (G-25 Sephadex, Roche Applied Science, Indianapolis, IN).
Binding of anti-PECAM/SA or IgG/SA conjugates to PNC
PNC (250 μg) and conjugates (200 μg) were mixed on vortex for 1 minute in 200μl PBS. After subsequent 1 hour incubation at 20°C, samples were diluted to 1ml with 1% BSA in PBS, centrifuged for 10 minutes at 16,000g, and resuspended in 1 ml of 1% BSA-PBS. Particle size was measured before and after preparation, and extent of conjugate binding was monitored by 125I-labeled conjugate using a Wizard 1470 gamma counter (Wallac Oy, Turku, Finland). Particle concentration was obtained by dividing the total polymer mass concentration by the mass of a single particle, as calculated by assuming the volume of a sphere and assuming a polymer density of 1.08. PNC diameter was obtained by DLS.
Determination of substrate permeabilities in PLGA
Substrate permeability through PLGA were determined by using a two chamber diffusion apparatus. Polymer films of esterified PLGA (34,000 MW) were prepared via a solvent casting procedure. PLGA was dissolved in DCM (50 mg/ml solution) and poured onto a glass plate. The solvent was allowed to evaporate off at ambient conditions for 1 hour. At this time, the polymer was placed into a vacuum over (40°C) for 1 hr. The remaining polymer film was then subsequently pealed from the glass surface. Film thickness was measured by digital calipers before and after permeability studies (typical thickness varied from 100–500μm). For the permeability studies, the film was then compression fitted between the two chambers(1ml) and checked for leaks. The donor cell contained either a 5 mM H2O2, 5 mM o-phenylenediamine (OPD) or 0.1 mM hypoxanthine in phosphate buffered saline (PBS) (10 mM, 7.4 pH), and the receptor cell contained pure PBS buffer. At specific time intervals (15 minutes and 30 minutes), the receptor cell contents were removed and replaced with fresh buffer. The concentration of the H2O2 in the receptor cell was determined by UV absorbance at 242 nm. OPD concentration was determined by a horseradish peroxidase (HRP) mediated oxidation reaction with 1mM H2O2 and measuring absorbance at 450nm. Hypoxanthine concentration was determined by its oxidation with xanthine oxidase (XO) and monitoring H2O2 production using an OPD/HRP chemical assay (Cary 50 UV-Vis, Varian, Palo Alto, CA). Permeability studies were performed in triplicate for two independently cast polymer films.
Proteolytic degradation of enzyme-loaded PNC
125I-labeled enzymes encapsulated in PNC were incubated for 4 hours at 37°C in a PBS solution containing 0.2 wt% pronase. Total enzyme activity and mass of enzyme associated with PNC were determined at 0 hr and 4 hr. Mass of enzyme was calculated by centrifuging the particles at 30,000 g for 10 minutes and measuring the amount of radioactivity in the supernatant and the PNC pellet. For enzyme activities, catalase activity was measured using a direct kinetic spectrophotometric analysis of H2O2 degradation at 242nm [26]. The rate of decrease in absorbance as a function of time is proportional to catalase activity. HRP and XO activity was obtained by a microplate assay. HRP activity was determined by measuring the kinetics of OPD oxidation (working solution contained 2mM OPD and 5mM H2O2 in PBS). XO activity was determined by the oxidation of hypoxanthine by measuring H2O2 production using OPD oxidation (working solution, 0.3 mM Hypoxanthine, 10Units/ml HRP and 2mM OPD in PBS). 50μl of PNC solution was added to 150μl of the working solutions and reference cells contained 150μl PBS. Absorbance was measured at 0, 5, 10, 15, 30, 45 and 60 minutes using a microplate reader (450nm filter). Scattering as a result of particles was subtracted out from the measurements.
Cell culture
Pooled human umbilical vein endothelial cells, HUVEC (Clonetics, San Diego, CA), were cultured at 37°C, 5% CO2, and 95% relative humidity in M199 medium (GIBCO, Grand Island, NY), 10 % fetal calf serum (FCS, GibcoBRL, Grand Island, NY) supplemented with 100 μg/ml heparin (Sigma, MO), 2 mM L-glutamine (Gibco), 15 μg/ml endothelial cell growth supplement (Upstate, Lake Placid, NY), 100 U/ml penicillin, and 100 μg/ml streptomycin and used at passage 4–5. REN cells (human mesothelioma) were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, supplemented with 2 mM -glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. REN cells stably transfected with human PECAM (REN/PECAM cells, which express PECAM-1 at levels and cellular localization similar to those found in human endothelial cells were maintained in the same growth medium supplemented with 0.5 mg/ml G418-sulfate. G418-sulfate was omitted from the medium during experiments. For microscopy studies, cells were seeded onto gelatinized 12mm coverslips in 24 well plates. For all other studies, cells were coated directly onto Gelatinized 24 well plates.
Endothelial targeting of anti-PECAM/PNC/catalase in cell culture
Binding studies were carried out using both radiotracing and fluorescent microscopy. Fluorescence studies were carried out using REN and REN/PECAM cells growing onto glass coverslips. Cells were incubated with solid core PNC and solid core anti-PECAM/PNC for 1 hour at 37°C. Cells were then washed 5 times using PBS and fixed (2%paraformaldyhde, 15 minutes ambient conditions). After fixation cells were washed, and then labeled with an Alexa Four-488-labeled secondary goat antibody to mouse IgG. Coverslips were then mounted and imaged using an Olympus IX70 inverted fluorescence microscope, 40x or 60x PlanApo objectives. Images were acquired using a Hamamatsu Orca-1 CCD camera and analyzed with ImagePro 3.0 imaging software (Media Cybernetics, Silver Spring, MD). For radiotracing studies, IgG/PNC and anti-PECAM/PNC were labeled with 125I-IgG-SA conjugate (5% of total conjugate surface coating). Cells were incubated for 1 hour at 37°C, at which time cells were washed 5 times and then lysed using 1% Triton X-100 in 1N NaOH. Cell supernatants and lysates were collected and CPM measured on a gamma counter to determine extent of binding.
Injection of anti-PECAM/PNC/catalase in mice
Anesthetized C57BL/6 male mice were injected intravenously with 10 mg/kg 125I-labeled anti-PECAM/PNC/catalase and IgG/PNC/catalase. Blood samples were collected from the retro-orbital plexus at 1 min, 15 min and 30 min post-injection, and organs (heart, kidneys, liver, spleen, and lungs) were collected at 30 min post-injection. The radioactivity and weight of the samples was determined to calculate nanocarrier targeting parameters, including percent of injected dose per gram (%ID/g), organ-to-blood ratio (localization ratio), and immunospecificity index [27].
Antioxidant protection studies in cell culture
Cell death was determined by the specific release of 51Cr [13, 19]. To label the cells, 51Cr isotope (200,000 cpm/well) was added 24 h prior to the experiment. The cells were then washed and incubated with anti-PECAM/PNC/catalase or IgG/PNC/catalase for 1 hour. Cells were then washed 5 times with RPMI 1640 without phenol red. 5mM H2O2 was added to the cells, and then returned to incubation at 37°C with 5% CO2. After 5 hours incubation, total radioactivity in the supernatant and in the cell lysates was determined. Release of 51Cr was used as a marker of cell death.
At the indicated times, H2O2 remaining in the supernatant medium was measured by H2O2-dependent oxidation of OPD (15 mM final concentration) in the presence of HRP (5 μg/ml final concentration) as determined by absorbance at 490 nm in a BioRad 3550 Microtiter Plate Reader [28]. The stability of catalase loaded into PNC was determined by incubating PNC with cells for various times prior to hydrogen peroxide insult. During the 24 period of 51Cr incubation, PNC were introduced into the chromium containing medium at specific times for 1 hour, washed 5 times with HUVEC medium, and then incubated with 51Cr containing medium for the remainder of the 24 hour period. After this time, cells were washed and the injury experiment was conducted as previously stated.
Isolated Perfused Mouse Lung Studies
Either anti-PECAM/PNC/catalase or IgG/PNC/catalase (10 mg/kg) was injected into anesthetized C57/BL6 mice via jugular vein. Thirty min after injection, lungs were isolated and perfused in a warm chamber via the pulmonary artery with 40 ml of recirculating filtered Krebs-Ringer buffer (KRB) (pH 7.4), containing 10 mM glucose and 3% BSA, under constant ventilation with a humidified gas mixture containing 5% CO2 and 95% air as described (5, 6). After 10 minutes perfusion, H2O2 was injected into the perfusate to a final concentration of 5 mM. Lungs were further perfused for 1 hour, washed with fresh buffer for 10 min and flash frozen. Frozen lungs were then analyzed for lipid peroxides using a previously published thiobarbituric acid-reactive substances (TBARS) assay [29].
For 2-photon imaging studies, lungs were perfused for 15 min using 2μM solution of 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (DCF, Molecular Invitrogen, Eugene, WA) in PBS. After this single pass perfusion, lungs were perfused with either PBS or 1mM H2O2 in PBS for 15 minutes.
2-Photon imaging of H2O2 in intact lung
Isolated, perfused lungs were placed in a Plexiglas chamber, and a nylon mesh was used to hold the tissue securely against the glass bottom of the chamber. Images were acquired using the Prairie Technologies Ultima multiphoton system attached to an Olympus BX-61 fixed stage upright microscope. Excitation light was provided by a diode-pumped broadband mode-locked titanium:sapphire femtosecond laser system (Spectraphysics MaiTai system; 720nm–920nm, <140fs, 80MHZ). The laser beam was raster-scanned and focused onto the specimen by a 40x water-immersion objective (NA 0.80, LUMPlanFl/IR, Olympus). An excitation wavelength of 850 nm was used to maximize the dynamic range of the DCF emission. The excitation beam was attenuated with neutral density filters to minimize phototoxic effects. Emitted fluorescence was collected with the same objective lens, reflected by a dichroic filter (660LP) passing through an IR cut filter (650SP) (Chroma Technology) and a filter set, where the fluorescence signal is divided and detected with two photomultipliers (R3896, Hamamatsu Photonics). Images were acquired using Prairie View software and image stacks were analyzed using Metamorph software.
Results
Design of anti-PECAM/PNC/catalase
For coupling antibodies to PNC composed of PEG-PLGA containing a biotin residue on the PEG amino-terminus, we employed a modular streptavidin (SA) cross-linker widely used for conjugation of drugs [6], liposomal [30] and polymeric materials [31, 32]. To solve the typical problem of aggregation of biotinylated molecules and carriers that results from SA possessing four biotin binding sites, we designed a series of covalent anti-PECAM/SA and IgG/SA conjugates, in which the anti-PECAM and IgG moiety mask ~50% of biotin-binding sites in SA. These conjugates offer a single step non-aggregating coupling to biotin-PNC. For example, 40% of added IgG/SA was coupled to solid-core (no encapsulated protein) biotin-PEG-PLGA PNC with mean diameter ~140 nm, i.e., 177.5±8.6 125I-IgG/SA molecules bound per biotin-PEG-PLGA PNC (Fig. 1A,B). This corresponds to coating by IgG/SA of ~50% of theoretical maximum of surface of PNC with diameter ~150 nm. DLS analysis showed that the coupling of this amount of IgG/SA increased PNC diameter by ~25 nm (Fig. 1B) reflecting uniform monomolecular coating by IgG/SA (conjugate size ~10 nm). Binding of IgG/SA to biotin-free PEG-PLGA PNC, as well as binding of non-conjugated IgG to biotin-PEG-PLGA PNC was negligible (Fig. 1B,C). Unless specified otherwise, in the subsequent in vitro and in vivo studies, we employed enzyme loaded PNC with mean diameter of ~450 nm formed using 10 mol% biotin-PEG-PLGA, carrying 1250±60 molecules of 125I-anti-PECAM/SA conjugate per PNC (anti-PECAM/PNC), which corresponds to coating of ~25% the PNC surface (Fig. 1C).
Figure 1. Antibody coupling to PNC using modular streptavidin-biotin system.

(A) 125I-IgG/SA was incubated with solid core PNC and centrifuged to remove unbound protein. One centrifugation was sufficient to remove most of unattached IgG/SA from non-biotinylated PNC used as a control (closed bars), while subsequent centrifugations did not remove IgG/SA coupled to biotinylated PNC (gray bars). (B) Size (left scale, closed bars) and amount of IgG/SA bound per PNC (right scale, grey bars) after coupling of 125I-IgG/SA to biotinylated (left) or non-biotinylated (right) solid PNC with initial diameter ~140 nm. (C) 125I-IgG/SA, but not 125I-IgG coupling to catalase loaded Biotin-PEG-PLGA PNC (initial diameter 420nm). (N=3, M±SD).
Targeting of anti-PECAM/PNC/catalase in cell cultures
Targeting of anti-PECAM/PNC was initially characterized in cell culture studies. First, fluorescent microscopy was used to test binding of fluorescently labeled solid-core PLGA anti-PECAM/PNC (~170 nm diameter) vs non-targeted PNC to either PECAM-transfected or control PECAM-negative REN cells (Fig. 2). This cell line forms an endothelium-like monolayer in vitro and provides an ideal cell-type matched model system for testing specific PECAM targeting [18]. Fluorescent imaging analysis shows binding of 117±11 vs 17.5±6.2 anti-PECAM/PNC per PECAM-transfected vs control REN cell, respectively (Fig. 3A). Very few non-targeted PNC (3.1±0.7 per cell) bound to either the PECAM positive or negative REN cells, likely due to the highly adhesion resistant nature of a non-conjugated PEG corona.
Figure 2. Fluorescent microscopy of anti-PECAM/PNC targeting to PECAM transfected or control REN cells.
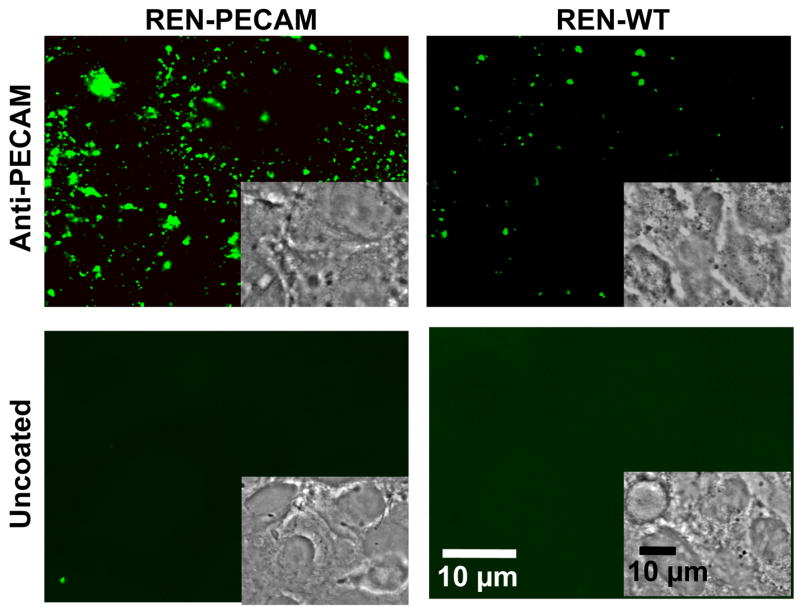
Anti-PECAM/PNC or uncoated PNC were incubated with REN cells or REN/PECAM cells for 1 hour, washed, fixed and permeabilized and stained with an Alexaflour® 488-fluorescent secondary goat antibody against mouse IgG.
Figure 3. Endothelial targeting of catalase encapsulated into PECAM-targeted stealth polymer nanocarriers (anti-PECAM/PNC).

(A) Solid core PNC, uncoated vs coated with anti-PECAM, were incubated with REN cells or REN/PECAM cells for 1 hour. Cells were then washed, fixed, permeabilized, and stained with a FITC-labeled secondary antibody. Quantification was obtained by averaging 10 fields each from 2 separate slides. Unless specified otherwise, data in this and other figures is shown as M±SD. (B). Binding to HUVEC of anti-PECAM/PNC (closed symbols) vs IgG/PNC (open symbols) labeled by a 5 wt% fraction of 125I-IgG-SA as a tracer coupled to solid PNC with 180 nm diameter. N=4. (C) Binding of 125I-catalase encapsulated into anti-PECAM vs IgG coated PNC with 450 nm diameter to endothelial cells. N=4. (D) Biodistribution of anti-PECAM/PNC (closed bars) vs control IgG/PNC (gray bars) traced by 5mol% 125I-IgG-SA 30 min after IV injection in naïve anesthetized mice. (N=3, M±SE).
We also tested binding of solid-core anti-PECAM/PNC and catalase-loaded anti-PECAM/PNC to human endothelial cells in culture using radioisotope tracing. In order to avoid potentially misleading tracing of radiolabeled anti-PECAM/SA (that may or may not reflect targeting of the PNC or its cargo), in this and the following experiments including animal studies we used anti-PECAM/PNC containing either surface-coupled 125I-IgG/SA tracer (mixed at 10% to anti-PECAM/SA), or encapsulated 125I-catalase. Figures 3 B and C show that both solid-core anti-PECAM/PNC and catalase-loaded anti-PECAM/PNC, but not IgG/PNC counterpart, specifically bind to endothelial cells (maximum level of 193±16.2 and 330±101 vs 15.6±0.8 PNC per cell). The higher targeting capacity of catalase-loaded anti-PECAM/PNC vs smaller solid core anti-PECAM/PNC can be explained by more effective multivalent binding of the catalase loaded preparation, which carries almost ten times more anti-PECAM molecules(see above).
Pulmonary targeting of anti-PECAM/PNC/catalase circulating in naïve animals
To test endothelial targeting in vivo, 125I-catalase encapsulated into anti-PECAM/PNC vs non-targeted PNC was injected intravenously in mice. At 30 minutes, as much as 30.9±2.2 and 21.2±2.3 % of injected dose (%ID/g) of the PNC/catalase and the anti-PECAM/PNC/catalase was circulating in the bloodstream, likely due to stealth function of PEG-corona, yet only 19±4 vs. 122±8 %ID/g was found in the lungs, respectively (Fig. 3D). This result was consistent with previous studies showing that after intravascular injection, anti-PECAM conjugates accumulate in the lungs of naïve, but not PECAM-deficient animals due to specific PECAM-mediated targeting to endothelial cells in the pulmonary vasculature [33–35]. This is due to the fact that the lungs, representing ~30% of total endothelial surface in the vascular system, are the only organ that receives the whole cardiac blood output and that pulmonary blood perfusion is relatively slow, favoring binding to endothelium [36]. The total difference in absolute values of pulmonary uptake (1.9±0.4 vs. 12.2±0.8 %ID) practically matches the difference in the circulating fraction of IgG/PNC/catalase vs. anti-PECAM/PNC/catalase (54.0±3.8 and 37.1±4.0 %ID, respectively), suggesting that the reduction of the circulating pool of the latter formulation is directly due to endothelial binding.
Anti-PECAM/PNC/catalase protects endothelium against oxidative stress
To test if catalase delivered by anti-PECAM/PNC protects from oxidative injury, we employed a model system that we developed previously for testing protective effects of protein anti-PECAM/SA/catalase conjugates[6, 18, 19]. 51Cr-labeled endothelial cells (HUVEC) were pre-incubated for 1 hour with either anti-PECAM/PNC/125I-catalase or IgG/PNC/125I-catalase, washed and exposed to 5 mM of H2O2. Cell death was detected by 51Cr release from cells reflecting irreversible damage to cell membrane. Anti-PECAM/PNC/catalase provided statistically greater catalase delivery (12.8±4.4 vs. 0.4±1.5 ng/well, p<0.01) and protection against H2O2-indced cell death (100±8.6 vs. 14.1±4.3 % protection, p<0.01) than IgG/PNC/catalase (Fig. 4A).
Figure 4. Endothelial targeting of catalase encapsulated into anti-PECAM coated stealth polymer nanocarriers protects against oxidative stress.

Panel A: cell culture studies. Human endothelial cells were incubated with 125I-catalase loaded IgG/PNC (black bars) or anti-PECAM/PNC (grey bars) for 1 hour, washed and treated with 5mM H2O2. The amount of catalase delivered was determined by iodine tracing. Cell death was measured by 51Cr-release after 5 hours (56.1±2.1% 51Cr was released from control cells treated with 5mM H2O2) and percent of protection was calculated. N=4. Panels B–E: animal studies. Anesthetized mice were injected with either anti-PECAM/PNC/catalase or catalase-free anti-PECAM/PNC and lungs were isolated from the animals 30 min later to test their capacity to decompose perfused H2O2 (see Methods). Anti-PECAM/PNC loaded with catalase suppressed both lipid peroxidation in the lung determined by TBARs level (B: N=4, M±SE) and DCF-fluorescence (C–E) induced by H2O2. Typical images of 2-photon fluorescent microscopy show DCF fluorescence in the vasculature in control lungs without and with H2O2 infusion (C and D, respectively), as well as in the lungs obtained from mice injected anti-PECAM/PNC/catalase and infused with H2O2 (E). Scale bar = 54μm. Asterisks denote statistical significance (*p<=0.5, **p<=0.01).
To determine whether this drug delivery system alleviates oxidative stress in the pulmonary vasculature, anesthetized mice were injected intravenously with anti-PECAM/PNC/catalase or control formulations. Thirty minutes later, lungs were removed, loaded with H2O2-sensitive probe DCF and perfused with a buffer containing H2O2, which in control lungs resulted in a two-fold elevation of the products of lipid peroxidation (p<0.01, Fig. 4B) and increased green fluorescence, indicating DCF oxidation (Fig. 4C–E). Injection of anti-PECAM/PNC/catalase in mice prior to isolation of lungs significantly (p<0.01) suppressed lipid peroxidation to practically normal control level (56.7±2.9 vs 101.9±3.8 pmol TBARS/mg of protein in control lungs). This result was confirmed by the visible suppression of H2O2-induced oxidation of DCF in the lungs of mice injected with anti-PECAM/PNC/catalase (Fig. 4E). Unloaded anti-PECAM/PNC did not prevent tissue oxidation (87.8±7.1 pmol TBARS/mg of protein). Thus PECAM-targeted polymer nanocarriers deliver encapsulated active catalase to the pulmonary endothelium, which resulted in alleviation of vascular oxidative stress in the lungs.
Anti-PECAM/PNC/catalase provides durable antioxidant protection of endothelial cells
To test the duration of the protective effect of delivered anti-PECAM/PNC/catalase, endothelial cells were exposed to H2O2 insult after different time intervals following delivery and elimination of non-bound anti-PECAM/PNC/catalase (Fig. 5). Even 21 hours after delivery, anti-PECAM/PNC/catalase greatly accelerated decomposition of H2O2 added to the cells (Fig. 5A). Thus, H2O2 degrading activity of catalase delivered to cells by anti-PECAM/PNC was more durable than that delivered by the catalase directly conjugated with anti-PECAM. In accord with previous studies of kinetics of degradation of protein conjugates targeted to endothelium [37, 38], the anti-PECAM/SA/catalase conjugate lost 90% of activity within <3 hours after delivery, while anti-PECAM/PNC/catalase was inactivated gradually by 60% during first five hours and retained a significant ~20% residual activity even after 21 hours after delivery (Fig. 5B). This bi-phase inactivation kinetics of anti-PECAM/PNC/catalase correlated well with the time course of inactivation of PNC-encapsulated catalase by external proteases (Fig. 5B, dash line) characterized in the previous study [26]. In theory, the first phase can be attributed to fast degradation of catalase loaded on the PNC surface and/or into PNC cavities accessible to external proteolysis.
Figure 5. Anti-PECAM/PNC/catalase provides durable antioxidant protection.

51Cr-labeled endothelial cells were incubated with anti-PECAM/PNC/catalase at the indicated time interval, washed 5 times, and exposed to a 5 mM H2O2 insult for 1 hour. Rate of H2O2 decay in the cell medium was monitored (A) to calculate the activity of catalase by fitting a first order decay equation to the data (B). The kinetics of catalase inactivation after delivery to the cells (B) has a profile similar to previously published kinetics of proteolytic inactivation of catalase encapsulated into PNC (dashed line). The duration of residual enzymatic activity of catalase encapsulated into anti-PECAM/PNC bound to cells greatly exceeded that of protein anti-PECAM/catalase conjugate bound to cells (B). Analysis of 51Cr release from endothelial cells showed that anti-PECAM/PNC/catalase provided significant (p<0.01) protection against H2O2-induced injury even 21 hours after delivery. N=4 in all panels.
However, cells exposed to H2O2 insult 3 hours after anti-PECAM/PNC/catalase delivery completely degraded H2O2 within <20 min (Fig. 5A, squares), which provided practically complete protection against H2O2 toxicity (Fig. 5C). Even in the case of 21 hour interval between delivery and insult, H2O2 was degraded within an hour (Fig. 5A, diamonds), which resulted in a statistically significant (56.7±8.6%, p<0.01) level of protection compared to the control unprotected cells (Fig. 5C).
Substrate permeability controls apparent activity of enzymes loaded into PNC
To evaluate the robustness and general utility of this carrier strategy and define role of substrate permeability through polymer matrix in the function of the enzymes encapsulated into PNC, two other enzymes, HRP and XO, were loaded into PNC. Catalase, HRP and XO have quite different molecular size ranging from 38 kD for HRP to 240 kD for catalase and 300 kD for XO, as well as diversity in pH optimum, co-substrates (HRP and XO require H2O2 and O2, respectively, to oxidize their substrates, ortho-phenylenediamine and xanthine/hypoxanthine), water solubility and sub-unit features (Table 1). Despite these differences, tracing of isotope-labeled proteins revealed no statistically significant difference in the loading of catalase, HRP and XO (10.2±1.2%, 8.2±1.7% and 7.8±2.5%, respectively) into PNC under identical formulation conditions (Fig. 6A). Further, the residual amount of radiolabeled proteins retained in the PNC after external proteolysis was similar (~30%) for all three proteins (Fig. 6B), indicating that comparable fractions of all three enzymes are encapsulated within PNC interior. Yet, catalase and HRP showed similar residual catalytic activity after external proteolysis (24.8±8.3% and 18.9±4.9%, respectively), whereas no remaining activity was detected in PNC/XO (0.1±2.5%).
Table 1. Summary of Enzyme Properties.
Enzymes tested for loading were selected based upon variations in size and enzyme function. Differences in enzyme molecular weight (MW), hydrodynamic radius (rh), and isoelectric point (IP) and tertiary structure prove the general applicability of the loading procedure used. Xanthine oxidase’s hydrodynamic radius was approximated using empirical correlations of proteins of known MW with rh determined by the Stokes-Einstein equation.[50]
| Protein | Function | MW (kDa) | Structure | rh (nm) | IP | Ref |
|---|---|---|---|---|---|---|
| Catalase | Reduces H2O2 to water and O2 | 240 | 4 identical subunits with HEME cofactors | 5.2 | 5.4 | [51, 52] |
| Horseradish Peroxidase | Oxidative conjugation of phenolic compounds | 42 | 1 glycolated subunit (18wt% carbohydrate) | 2.9 | 3–9 | [53, 54] |
| Xanthine Oxidase | Purine metabolism into uric acid and superoxide anion | 300 | 2 subunits each containing Mb, Fe and FAD | 6.8 | 6.9–7.6 | [55, 56] |
Figure 6. Encapsulation of diverse enzymes into PNC: substrate permeability via polymer control residual activity of the enzymes protected against external proteolysis.

(A) 125I-labeled catalase, horseradish peroxidase (HRP), and xanthine oxidase (XO) showed similar efficiency of encapsulation into PNC. (B). Residual enzymatic activity (closed bars) and load of radiolabeled enzyme (gray bars) after a 4 hour incubation of PNC loaded with catalase, HRP or XO in a 0.2% pronase solution. (C) Analysis of substrate permeability (a product of partitioning and diffusivity) via PLGA film showed that H2O2 and OPD have ~100-times higher permeability then xanthine. (C, inset) There was poor agreement between the experimental data and the Renkin diffusion model that describes the sieving effects of membrane pore size on substrate permeability. This result suggests that other chemical properties (e.g., polarity, electrostatic interactions, aqueous solubility) of the substrates also greatly impacted the observed rates of diffusion.
In theory, this result could be due to the different sensitivities of encapsulated enzymes to inactivation (a formidable obstacle to loading enzymes into sub-micron polymer carriers) or/and different permeability of their substrates via the PNC shell. Prior to incubation with proteases, measurable activities were noted for all PNC loaded proteins, which suggests that sensitivity of enzymes to our encapsulation procedure is not a concern. However, permeability measurements of PLGA films (Fig 6C) showed that the permeability of substrates of catalase, HRP and XO, i.e., H2O2, OPD and hypoxanthine is 3.3±0.4×10−7, 7.0±1.7×10−7 and 3.2±1.2×10−9 cm2/s, respectively. This result suggested that low substrate permeability via PLGA is the likely reason for undetectable activity of encapsulated XO.
DISCUSSION
The concept of targeted protein delivery using polymers [39] and PNC dates to at least early 1980s [40], yet advance in this area is relatively modest and slow compared to that of PNC delivery of small therapeutics like doxorubicin [41, 42] and oligo-nucleic acids[43]. The primary stumbling block has been that the available formulation methods (e.g., phase separation, micellization and mechanical or ultrasound homogenization) typically result in low loading levels and/or protein inactivation. Recently, we have designed a PNC synthesis using a freeze-thaw cycle that aides in the encapsulation of catalase without loss of enzymatic activity (24). In theory, this result could be explained by the transient gelation of the polymer phase, yet it was not clear if it could be generalized to the loading of other proteins. To probe the general utility of the proposed strategy of a protective polymer carrier (or rather cage) for enzyme delivery, horse radish peroxidase and xanthine oxidase were also encapsulated into PEG-PLGA PNC. Interestingly, it was found that degree of enzyme loading was similar for all three proteins, demonstrating that the freeze-thaw encapsulation was more dependent upon the synthesis conditions than the features of actual protein being loaded, a result not seen when equilibrium partitioning is a driving mechanism [44]. It is conceivable, therefore, that a wide variety of proteins can be loaded within PEG-PLGA PNC using this approach.
However, when exposed to external proteolysis, PNC-loaded xanthine oxidase lost its activity despite the fact that the amount of encapsulated protein cargo protected from proteolysis was similar to that in PNC/catalase and PNC/HRP, which both showed high residual activity that was not affected by protease treatment (Fig. 6C). It was found that permeability of H2O2 via PLGA polymer is 10 fold greater than its permeability via cellular membranes [45]. In contrast to H2O2 that, therefore, has a physiologically relevant diffusion rate, hypoxanthine’s permeability across the polymer was 100-fold lower, which helps to explain undetectable activity of XO encapsulated into the PNC (Fig. 6C). Oxygen permeability via PLGA may also be a limiting factor for enzymatic XO-catalyzed reaction, although oxygen size and water solubility (Table 2) argue against this notion (see below).
Table 2. Substrate Chemical Properties.
Octanol/water partition coefficient, Log(P), provides an indication of relative hydrophobicity with positive values relating to hydrophobic substrates. It is expected that more hydrophobic molecules would partition better into the polymer layer, enhancing permeation. A more analytic representation of molecular polarity is provided by the total polar surface area (PSA), which is defined as the sum of surfaces of polar atoms in a molecule. Solubilities and Log(P) values were obtained from the Physical Properties Database (PhysProp) by Syracuse Research Corporation[57]. PSA values were calculated using a group contribution method [48].
| Substrate | MW | Aqueous Solubility | Log(P) | PSA (A2) |
|---|---|---|---|---|
| O2 | 32 | 6.7mg/L (37°C) | 0.65 | 34.1 |
| H2O2 | 34 | 1 kg/L | −1.5 | 40.5 |
| o-Phenylenediamine | 108 | 40g/L | 0.15 | 52.0 |
| Hypoxanthine | 136 | 0.7g/L | −1.11 | 74.4 |
Permeability is a function of both substrate diffusivity and partitioning. Diffusivity describes the relative ease of which a solute can move through a medium with substrate size being a primary factor controlling its diffusional capacity. Inset in Fig. 6C shows that permeability is minimal for the largest substrate. Partitioning describes the relative affinity of the solute for either the polymer phase or the exterior aqueous phase; hence, aqueous solubility of the substrate may also control permeability of its molecules via polymers. Therefore, it is expected that the PEG layer that provides a hydrophilic steric barrier for protein absorption will participate negligibly in solute diffusion, whereas the hydrophobic PLA, which creates an aqueous lean domain that structurally separates enzyme from the continuous phase, will be the primary isolating permeation barrier.
This isolating polymer layer can control the flux of solutes through physicochemical effects. Physically, the organization of polymer chains within this hydrophobic domain results in pockets which allow for the transport of solute across the polymer layer. In a setting where this effect dominates the diffusion, the size of these pockets in relation to the size of the solute directly determines the rate of diffusion and can be described by the equation:
where Da,layer is the diffusivity of substrate through the polymer film, D∞ is the diffusivity of the substrate in pure solvent (water) and λ is the ratio of solute to pore size. This model is a simplification of the Renkin model made by Anderson and Quinn [46, 47] for membranes containing cylindrical pores, yet it has been shown to fit with random polymer films with the gap size between polymer chains used as the pore diameter. When this model is fitted using a least squares method to the three substrates by allowing variation in the pore diameter as a control parameter, the best fit was obtained with a pore diameter of 1.2 nm, which is within reason for a solid polymer layer. The MW of pronase is 15–27kDa, which results in a globular protein size of ~4–5nm, further indicating that it is much too large to penetrate the PNC polymer.
In Fig. 6C inset, the curve fit for the Renkin model with a pore diameter of 1.2 nm is given. While the model predicts a continuous trend with an inverse relationship between MW and permeability, the experimental data did not possess this gradual change. Indeed, there was a two orders of magnitude step change in permeability between 108 and 136 MW (OPD and hypoxanthine, respectively), resulting in poor agreement with the pore model. This result suggests that MW of the substrate is not the only factor that controls the selective diffusion via polymer and that other chemical properties (e.g., polarity, electrostatic interactions, aqueous solubility) are likely to contribute to the outcome permeability.
A comparison of chemical properties of substrates is provided in Table 2. Classically, Octanol/water partitioning is used to describe relative hydrophobicity of chemicals and their permeability across lipid layers. A more accurate picture of molecular hydrophobicity can be obtained by the polar charge surface area, a measure of the surface polarity of a molecule, which was determined through a group contribution analysis [48]. Since hypoxanthine possesses such a high polar surface area, it is expected that its partitioning into the hydrophobic polymer layer would be significantly reduced, explaining the disproportionately (based on the MW solely) low permeability.
This analysis indicates that selectivity of substrate diffusion may be driven by molecular sieving effects and also the natural chemical affinity of the substrate for the polymer shell. This opens unique opportunities of functional control of targeted enzyme therapies. Many enzymes (e.g., the cytochrome P450s, peroxidases, peptidases) can catalyze conversion of variety of molecules. By designing polymer shells with selective diffusivities (e.g., through molecular imprinting or channel formation) it may be possible to control the apparent specificity of these multifunctional enzymes. This approach provides an opportunity to design degradable carriers that function on whole classes of molecules rather than just single substrate, for example, for detoxification of xenobiotics and endogenous toxins including oxidants, the elusive and harmful agents inducing and mediating a plethora of human maladies involving oxidative stress.
Our results indicate that detoxifying therapeutic enzymes can be encapsulated into polymer nanocarriers, which: i) can be targeted in the vasculature by affinity moieties coupled to their surface; ii) be stealth (PEG-coated) and small enough to circulate and provide intraendothelial delivery (i.e., <500 nm, according to our studies using prototype spherical polymer beads coated with anti-CAM [27]); and, iii) protect enzymatic cargo from proteolysis, while permitting their activity towards diffusible toxic substrates. An advantage of this drug delivery system is that substrate-diffusible PNC serves rather as a protective protein cage, hence no need for drug release. This paradigm may find medical utility including targeting of antioxidant enzymes.
Several factors control amplitude and duration of the local activity of the PNC encapsulated enzymes, including: i) Specificity and amplitude of targeting; ii) Activity of encapsulated cargo; iii) Permeability of the substrate via PNC; and, iv) Durability of the cargo and PNC itself. Targeting features of anti-PECAM/PNC/catalase match those of the previous protein anti-PECAM conjugates and fusion proteins [16, 18, 21, 33, 49], yet the duration of the protective effect afforded by anti-PECAM/PNC/catalase greatly exceeds that of protein conjugates, which is no longer than 2–3 hours (Fig. 5). In fact, despite loss of a large fraction of the activity of loaded catalase during the first, relatively rapid phase of inactivation (Fig. 5A), residual activity that apparently lasts for a prolonged time was sufficient to provide substantial protective benefit (Fig. 5C).
In theory, the two-phase kinetics of decrease in PNC/catalase activity can be attributed to i) PNC degradation; ii) enzyme release; and, iii) inactivation of the surface accessible enzyme fraction. In vitro stability studies showed that these formulations of PNC are stable for at least 1 week in acidic and protease environment with undetectable molecular weight change (not shown); hence PNC degradation does not seem the primary mechanism for activity loss. Since release of 240kD protein encapsulated into PEG-PLGA would be expected due to the carrier degradation, proteolysis or another cause of inactivation of surface-accessible cargo (e.g., in small cavities opened to the milieu) seems the most plausible explanation (Fig. 6, right panel).
Results of animal studies shown in this paper indicate that PECAM-targeted polymer nanoparticles loaded with encapsulated therapeutic enzymes have sufficient affinity to endothelial cells to accumulate in the pulmonary vasculature (Fig. 3D) and that the load of the catalase cargo is sufficient to provide anti-oxidant effect in the lungs (Fig. 4, B and C-E). This result is consistent with our previous publications that documented that the pulmonary vasculature is a preferable site of accumulation of intravenously injected protein conjugates, recombinant fusion constructs and model drug carriers targeted to PECAM-1 [13, 18, 21]. Low level of the pulmonary uptake of IgG/PNC of the same size as anti-PECAM/PNC confirms the specificity of targeting that is due to affinity to endothelial cells, not mechanical retention in the pulmonary microvasculature (Fig. 3D). Pulmonary vasculature represents a privileged target for agents circulating in the bloodstream due to its extreme extension (~30% of the total vascular surface in the body), slow rate of perfusion and the fact that this is the only organ in the body that receives the whole cardiac blood output [36]. On the other hand, this compartment in the body is extremely vulnerable to pro-inflammatory, oxidative and thrombotic insults, due to its proximity to the external atmosphere and function of filtering mechanical filtering of activated leukocytes, thrombi and debris from the venous blood [11, 12]. Therefore, drug delivery strategy proposed in this paper, targeting of durable detoxifying enzymes to the pulmonary vasculature, might find applications for treatment of diverse human pathological conditions including Acute Lung Injury (ALI), hyperoxia, sepsis and shock.
Conclusions
In summary, this work has demonstrated that polymer nanocarriers can indeed provide a means of enhancing and extending the benefit of therapeutic proteins, particularly the antioxidant enzyme, catalase. This effect was a result of both the capacity to target PNC to the vascular endothelium and the ability to extend therapeutic duration by reducing proteolytic inactivation. Long term enzyme activity was only observed for enzymes whose substrates were readily permeable across the polymer layer. Future studies in designing molecularly imprinted PNC shells will provide a means of controlling substrate diffusion rates, providing platforms for localized enzymatic therapies directed towards diverse substrates with controlled selectivity, adjustable to fit a wider variety of medical needs ranging from the treatment of oxidative stress evaluated here to new avenues in xenobiotic detoxification.
Acknowledgments
Authors thank Drs. Dennis Discher and Silvia Muro for stimulating discussions and helpful advice and Dr. Philip G Haydon for support in experiments involving 2-photon confocal microscope.
Grant support: National Institutes of Health Grants NHLBI RO1 HL71175, HL078785 and HL73940 and Department of Defense Grant PR 012262 (VRM).
Abbreviations
- ALI
acute lung injury
- BSA
bovine serum albumin
- CAM
cell adhesion molecule
- DCC
2,2-dicyclocarbodiimide
- DCF
5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate
- DCM
dichloromethane
- DLS
dynamic light scattering
- EC
endothelial cell
- FCS
fetal calf serum
- FTIR
fourier transform infrared spectroscopy
- GPC
gel permeation chromatography
- HRP
horseradish peroxidase
- HUVEC
human umbilical vein endothelial cells
- ID
injected dose
- IP
isoelectric point
- KRB
Krebs-Ringer buffer
- mPEG
methoxy-poly(ethylene glycol)
- MW
molecular weight
- M±SE
mean plus minus standard error
- NHS-biotin
N-succinimidyl-biotin
- NMR
nuclear magnetic resonance
- OPD
o-phenylenediamine
- PBS
phosphate buffered saline
- PECAM
platelet endothelial cell adhesion molecules
- PEG
poly(ethylene glycol)
- PhysProp
physical properties database
- PLA
poly(lactic acid)
- PLGA
poly(lactic co glycolic acid)
- PNC
polymer nanocarriers
- PSA
polar surface area
- REN
human mesothelioma cell line
- RPMI
Roswell Park Memorial Institute buffered media
- SA
streptavidin
- SATA
N-succinimidyl-S-acetylthioacetate
- SMCC
succinimidyl 4-[N-maleimidomethyl]cyclohexane-1-carboxylate
- TBARS
thiobarbituric acid-reactive substances
- XO
xanthine oxidase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pavlou AK, Reichert JM. Recombinant protein therapeutics--success rates, market trends and values to 2010. Nature biotechnology. 2004;22(12):1513–9. doi: 10.1038/nbt1204-1513. [DOI] [PubMed] [Google Scholar]
- 2.Langer R. Drug delivery. Drugs on target. Science. 2001;293(5527):58–9. doi: 10.1126/science.1063273. [DOI] [PubMed] [Google Scholar]
- 3.Frokjaer S, Otzen DE. Protein drug stability: a formulation challenge. Nat Rev Drug Discov. 2005;4(4):298–306. doi: 10.1038/nrd1695. [DOI] [PubMed] [Google Scholar]
- 4.Putney SD, Burke PA. Improving protein therapeutics with sustained-release formulations. Nature biotechnology. 1998;16(2):153–7. doi: 10.1038/nbt0298-153. [DOI] [PubMed] [Google Scholar]
- 5.Schwendeman SP. Recent advances in the stabilization of proteins encapsulated in injectable PLGA delivery systems. Crit Rev Ther Drug Carrier Syst. 2002;19(1):73–98. doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.20. [DOI] [PubMed] [Google Scholar]
- 6.Shuvaev VV, Dziubla T, Wiewrodt R, Muzykantov VR. Streptavidin-biotin crosslinking of therapeutic enzymes with carrier antibodies: nanoconjugates for protection against endothelial oxidative stress. Methods Mol Biol. 2004;283:3–19. doi: 10.1385/1-59259-813-7:003. [DOI] [PubMed] [Google Scholar]
- 7.Muzykantov VR. Delivery of antioxidant enzyme proteins to the lung. Antioxid Redox Signal. 2001;3(1):39–62. doi: 10.1089/152308601750100489. [DOI] [PubMed] [Google Scholar]
- 8.Christofidou-Solomidou M, Muzykantov VR. Antioxidant strategies in respiratory medicine. Treat Respir Med. 2006;5(1):47–78. doi: 10.2165/00151829-200605010-00004. [DOI] [PubMed] [Google Scholar]
- 9.Freeman BA, Young SL, Crapo JD. Liposome-mediated augmentation of superoxide dismutase in endothelial cells prevents oxygen injury. J Biol Chem. 1983;258(20):12534–42. [PubMed] [Google Scholar]
- 10.McCord JM, Edeas MA. SOD, oxidative stress and human pathologies: a brief history and a future vision. Biomed Pharmacother. 2005;59(4):139–42. doi: 10.1016/j.biopha.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 11.Varani J, Ward PA. Mechanisms of endothelial cell injury in acute inflammation. Shock. 1994;2(5):311–9. doi: 10.1097/00024382-199411000-00001. [DOI] [PubMed] [Google Scholar]
- 12.Heffner JE, Repine JE. Pulmonary strategies of antioxidant defense. Am Rev Respir Dis. 1989;140(2):531–54. doi: 10.1164/ajrccm/140.2.531. [DOI] [PubMed] [Google Scholar]
- 13.Kozower BD, Christofidou-Solomidou M, Sweitzer TD, Muro S, Buerk DG, Solomides CC, et al. Immunotargeting of catalase to the pulmonary endothelium alleviates oxidative stress and reduces acute lung transplantation injury. Nature biotechnology. 2003;21(4):392–8. doi: 10.1038/nbt806. [DOI] [PubMed] [Google Scholar]
- 14.McCord JM. Superoxide dismutase in aging and disease: an overview. Methods Enzymol. 2002;349:331–41. doi: 10.1016/s0076-6879(02)49348-2. [DOI] [PubMed] [Google Scholar]
- 15.Sakhalkar HS, Hanes J, Fu J, Benavides U, Malgor R, Borruso CL, et al. Enhanced adhesion of ligand-conjugated biodegradable particles to colitic venules. Faseb J. 2005;19(7):792–4. doi: 10.1096/fj.04-2668fje. [DOI] [PubMed] [Google Scholar]
- 16.Ding BS, Dziubla T, Shuvaev VV, Muro S, Muzykantov VR. Advanced drug delivery systems that target the vascular endothelium. Molecular interventions. 2006;6(2):98–112. doi: 10.1124/mi.6.2.7. [DOI] [PubMed] [Google Scholar]
- 17.Muro S, Muzykantov VR. Targeting of antioxidant and anti-thrombotic drugs to endothelial cell adhesion molecules. Current pharmaceutical design. 2005;11(18):2383–401. doi: 10.2174/1381612054367274. [DOI] [PubMed] [Google Scholar]
- 18.Muzykantov VR, Christofidou-Solomidou M, Balyasnikova I, Harshaw DW, Schultz L, Fisher AB, et al. Streptavidin facilitates internalization and pulmonary targeting of an anti-endothelial cell antibody (platelet-endothelial cell adhesion molecule 1): a strategy for vascular immunotargeting of drugs. Proc Natl Acad Sci U S A. 1999;96(5):2379–84. doi: 10.1073/pnas.96.5.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sweitzer TD, Thomas AP, Wiewrodt R, Nakada MT, Branco F, Muzykantov VR. PECAM-directed immunotargeting of catalase: specific, rapid and transient protection against hydrogen peroxide. Free radical biology & medicine. 2003;34(8):1035–46. doi: 10.1016/s0891-5849(03)00029-7. [DOI] [PubMed] [Google Scholar]
- 20.Atochina EN, Balyasnikova IV, Danilov SM, Granger DN, Fisher AB, Muzykantov VR. Immunotargeting of catalase to ACE or ICAM-1 protects perfused rat lungs against oxidative stress. Am J Physiol. 1998;275(4 Pt 1):L806–17. doi: 10.1152/ajplung.1998.275.4.L806. [DOI] [PubMed] [Google Scholar]
- 21.Christofidou-Solomidou M, Scherpereel A, Wiewrodt R, Ng K, Sweitzer T, Arguiri E, et al. PECAM-directed delivery of catalase to endothelium protects against pulmonary vascular oxidative stress. Am J Physiol Lung Cell Mol Physiol. 2003;285(2):L283–92. doi: 10.1152/ajplung.00021.2003. [DOI] [PubMed] [Google Scholar]
- 22.Muro S, Cui X, Gajewski C, Murciano JC, Muzykantov VR, Koval M. Slow intracellular trafficking of catalase nanoparticles targeted to ICAM-1 protects endothelial cells from oxidative stress. Am J Physiol Cell Physiol. 2003;285(5):C1339–47. doi: 10.1152/ajpcell.00099.2003. [DOI] [PubMed] [Google Scholar]
- 23.Torchilin VP, Tischenko EG, Smirnov VN, Chazov EI. Immobilization of enzymes on slowly soluble carriers. Journal of biomedical materials research. 1977;11(2):223–35. doi: 10.1002/jbm.820110208. [DOI] [PubMed] [Google Scholar]
- 24.Klibanov AM. Enzyme stabilization by immobilization. Anal Biochem. 1979;93(1):1–25. [PubMed] [Google Scholar]
- 25.Chang TM, Langer R, Sparks RE, Reach G. Drug delivery systems in biotechnology. Artif Organs. 1988;12(3):248–51. doi: 10.1111/j.1525-1594.1988.tb02766.x. [DOI] [PubMed] [Google Scholar]
- 26.Dziubla TD, Karim A, Muzykantov VR. Polymer nanocarriers protecting active enzyme cargo against proteolysis. J Control Release. 2005;102(2):427–39. doi: 10.1016/j.jconrel.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 27.Muro S, Dziubla T, Qiu W, Leferovich J, Cui X, Berk E, et al. Endothelial targeting of high-affinity multivalent polymer nanocarriers directed to intercellular adhesion molecule 1. The Journal of pharmacology and experimental therapeutics. 2006;317(3):1161–9. doi: 10.1124/jpet.105.098970. [DOI] [PubMed] [Google Scholar]
- 28.Sweitzer TD, Thomas AP, Wiewrodt R, Nakada MT, Branco F, Muzykantov VR. Pecam-directed immunotargeting of catalase: specific, rapid and transient protection against hydrogen peroxide. Free Radical Biology & Medicine. 2003;34(8):1035–46. doi: 10.1016/s0891-5849(03)00029-7. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Feinstein SI, Manevich Y, Ho YS, Fisher AB. Lung injury and mortality with hyperoxia are increased in peroxiredoxin 6 gene-targeted mice. Free Radic Biol Med. 2004;37(11):1736–43. doi: 10.1016/j.freeradbiomed.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 30.Lee HY, Jung HS, Fujikawa K, Park JW, Kim JM, Yukimasa T, et al. New antibody immobilization method via functional liposome layer for specific protein assays. Biosensors & bioelectronics. 2005;21(5):833–8. doi: 10.1016/j.bios.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 31.Vinogradov SV, Batrakova EV, Kabanov AV. Nanogels for oligonucleotide delivery to the brain. Bioconjugate chemistry. 2004;15(1):50–60. doi: 10.1021/bc034164r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lackey CA, Press OW, Hoffman AS, Stayton PS. A biomimetic pH-responsive polymer directs endosomal release and intracellular delivery of an endocytosed antibody complex. Bioconjugate chemistry. 2002;13(5):996–1001. doi: 10.1021/bc010053l. [DOI] [PubMed] [Google Scholar]
- 33.Ding BS, Gottstein C, Grunow A, Kuo A, Ganguly K, Albelda SM, et al. Endothelial targeting of a recombinant construct fusing a PECAM-1 single-chain variable antibody fragment (scFv) with prourokinase facilitates prophylactic thrombolysis in the pulmonary vasculature. Blood. 2005;106(13):4191–8. doi: 10.1182/blood-2005-05-2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kozower BD, Christofidou-Solomidou M, Sweitzer TD, Muro S, Buerk DG, Solomides CC, et al. Immunotargeting of catalase to the pulmonary endothelium alleviates oxidative stress and reduces acute lung transplantation injury. Nature biotechnology. 2003;21(4):392–8. doi: 10.1038/nbt806. [DOI] [PubMed] [Google Scholar]
- 35.Muzykantov VR. Targeting pulmonary endothelium. Biomedical Aspects of Drug Targeting. 2002:129–48. [Google Scholar]
- 36.Muzykantov VR. Biomedical aspects of targeted delivery of drugs to pulmonary endothelium. Expert opinion on drug delivery. 2005;2(5):909–26. doi: 10.1517/17425247.2.5.909. [DOI] [PubMed] [Google Scholar]
- 37.Muro S, Gajewski C, Koval M, Muzykantov VR. ICAM-1 recycling in endothelial cells: a novel pathway for sustained intracellular delivery and prolonged effects of drugs. Blood. 2005;105(2):650–8. doi: 10.1182/blood-2004-05-1714. [DOI] [PubMed] [Google Scholar]
- 38.Muro S, Koval M, Muzykantov V. Endothelial endocytic pathways: gates for vascular drug delivery. Curr Vasc Pharmacol. 2004;2(3):281–99. doi: 10.2174/1570161043385736. [DOI] [PubMed] [Google Scholar]
- 39.Flanagan PA, Kopeckova P, Kopecek J, Duncan R. Evaluation of protein-N-(2-hydroxypropyl)methacrylamide copolymer conjugates as targetable drug carriers. 1. Binding, pinocytic uptake and intracellular distribution of transferrin and anti-transferrin receptor antibody conjugates. Biochimica et biophysica acta. 1989;993(1):83–91. doi: 10.1016/0304-4165(89)90146-3. [DOI] [PubMed] [Google Scholar]
- 40.Rhine WD, Hsieh DS, Langer R. Polymers for sustained macromolecule release: procedures to fabricate reproducible delivery systems and control release kinetics. J Pharm Sci. 1980;69(3):265–70. doi: 10.1002/jps.2600690305. [DOI] [PubMed] [Google Scholar]
- 41.Lee CC, Gillies ER, Fox ME, Guillaudeu SJ, Frechet JM, Dy EE, et al. A single dose of doxorubicin-functionalized bow-tie dendrimer cures mice bearing C-26 colon carcinomas. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(45):16649–54. doi: 10.1073/pnas.0607705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khandare JJ, Jayant S, Singh A, Chandna P, Wang Y, Vorsa N, et al. Dendrimer versus linear conjugate: Influence of polymeric architecture on the delivery and anticancer effect of paclitaxel. Bioconjugate chemistry. 2006;17(6):1464–72. doi: 10.1021/bc060240p. [DOI] [PubMed] [Google Scholar]
- 43.Bartlett DW, Davis ME. Physicochemical and Biological Characterization of Targeted, Nucleic Acid-Containing Nanoparticles. Bioconjugate chemistry. 2007 doi: 10.1021/bc0603539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JC, Bermudez H, Discher BM, Sheehan MA, Won YY, Bates FS, et al. Preparation, stability, and in vitro performance of vesicles made with diblock copolymers. Biotechnology and bioengineering. 2001;73(2):135–45. doi: 10.1002/bit.1045. [DOI] [PubMed] [Google Scholar]
- 45.Seaver LC, Imlay JA. Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. Journal of bacteriology. 2001;183(24):7182–9. doi: 10.1128/JB.183.24.7182-7189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deen WM. Hindered Transport of Large Molecules in Liquid Filled Pores. AICHE Journal. 1987;33(9):1409–34. [Google Scholar]
- 47.Anderson JL, Quinn JA. Restricted Transport in Small Pores - Model for Steric Exclusion and Hindered Particle Motion. Biophysical Journal. 1974;14(2):130–50. doi: 10.1016/S0006-3495(74)70005-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. Journal of medicinal chemistry. 2000;43(20):3714–7. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- 49.Danilov SM, Gavrilyuk VD, Franke FE, Pauls K, Harshaw DW, McDonald TD, et al. Lung uptake of antibodies to endothelial antigens: key determinants of vascular immunotargeting. Am J Physiol Lung Cell Mol Physiol. 2001;280(6):L1335–47. doi: 10.1152/ajplung.2001.280.6.L1335. [DOI] [PubMed] [Google Scholar]
- 50.Saltzman WM. Drug Delivery - Engineering Principles for Drug Therapy. New York, NY: Oxford Press; 2001. [Google Scholar]
- 51.Diesseroth A, Dounce AL. Catalase: Physical and Chemical Properties, Mechanism of catalysis, and Physiological Role. Physiological reviews. 1970;50(3):319–75. doi: 10.1152/physrev.1970.50.3.319. [DOI] [PubMed] [Google Scholar]
- 52.Samejima T, Kamata M, Shibata K. Dissociation of Bovine Liver Catalase at Low pH. J Biochem (Tokyo) 1962;51(181) doi: 10.1093/oxfordjournals.jbchem.a127518. [DOI] [PubMed] [Google Scholar]
- 53.Smith AT, Santama N, Dacey S, Edwards M, Bray RC, Thorneley RN, et al. Expression of a synthetic gene for horseradish peroxidase C in Escherichia coli and folding and activation of the recombinant enzyme with Ca2+ and heme. The Journal of biological chemistry. 1990;265(22):13335–43. [PubMed] [Google Scholar]
- 54.Rodriguez-Lopez JN, Hernández-Ruiz J, Garcia-Cánovas F, Thorneley RNF, Acosta M, Arnao MB. The Inactivation and Catalytic Pathways of Horseradish Peroxidase with m-Chloroperoxybenzoic Acid. A Spectrophotometric and Kinetic Study. J Biol Chem. 1997;272:5469–76. doi: 10.1074/jbc.272.9.5469. [DOI] [PubMed] [Google Scholar]
- 55.Sullivan CH, Mather IH, Greenwalt DE, Madara PJ. Purification of xanthine oxidase from the fat-globule membrane of bovine milk by electrofocusing. Mol Cell Biochem. 1982;44(1):13–22. doi: 10.1007/BF00573841. [DOI] [PubMed] [Google Scholar]
- 56.Brondino CD, Romao MJ, Moura I, Moura JJ. Molybdenum and tungsten enzymes: the xanthine oxidase family. Curr Opin Chem Biol. 2006;10(2):109–14. doi: 10.1016/j.cbpa.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 57.Syracuse Research Corporation. PhysProp-Physical Properties Database. 2006 http://www.syrres.com/esc/physdemo.htm.