

Abstract
MRG15 is a core component of the NuA4/Tip60 histone acetyltransferase complex that modifies chromatin structure. We here demonstrate that Mrg15 null and heterozygous mouse embryonic fibroblasts exhibit an impaired DNA damage response post gamma irradiation, when compared to wild-type cells. Defects in DNA repair and cell growth, and delayed recruitment of repair proteins to sites of damage were observed. Formation of phosphorylated H2AX and 53BP1 foci was delayed in Mrg15 mutant versus wild-type cells following irradiation. These data implicate a novel role for MRG15 in DNA damage repair in mammalian cells.
Keywords: MORF4, NuA4, Sin3-HDAC, ATM, 53BP1
1. Introduction
Normal cells have a finite ability to divide in culture, a phenomenon known as replicative senescence. Cell fusion of normal with immortal tumor cells demonstrated that senescence is a dominant phenotype and provided the first evidence that senescence is a mechanism of tumor suppression [1]. These studies resulted in the isolation of MORF4 as a senescence inducing gene. MORF4 is a member of a family of transcription factors including the MORF4-Related Gene on human chromosome 15 (MRG15) [2,3].
MRG15 has a 96% similarity to MORF4 in amino acid sequence but fails to induce senescence upon introduction into immortal cells. The most striking structural difference between the two proteins is the presence of an N-terminal extension in MRG15, which includes a chromodomain. Proteins containing a chromodomain, characterized to date, have been found to be chromatin remodeling factors involved in causing conformational changes in chromatin by ATP-dependent movement of nucleosomes and modification of histones [4-6]. Histone modifying enzymes, the histone acetyltransferases and deacetylases (HATs, HDACs), are present in complexes involved in transcription and, recently, HAT complexes have been implicated in DNA-damage detection and repair [7]. MRG15 is present in both the NuA4/Tip60-HAT and Sin3-HDAC chromatin modifying complexes [8].
We have shown that MRG15 is important for cell proliferation in primary mouse embryonic fibroblasts (MEFs), and that deletion of the gene in mice results in gross developmental defects leading to embryonic lethality [9]. We here demonstrate that MRG15 is required for effective DNA damage repair post exposure to ionizing radiation (IR) in MEFs and is important for efficient recruitment of DNA repair proteins at sites of damage and acetylation of H2A and H2AX. Loss of a single copy of MRG15 in MEFs delays repair of DNA-damage post irradiation, indicating that even a modest decrease in MRG15 levels affects the function of associated complexes. This suggests a novel and critical role for MRG15 in DNA repair in mammalian cells.
2. Methods
2.1 Cell culture and gamma irradiation conditions
Generation of Mrg15 null and heterozygous (het) MEFs and conditions for cell culture have been described previously [9]. To determine the optimal dose of IR, MEFs were exposed to 0, 2, 3, 5 and 10Gy from a 137Cs source and seeded at 2500 cells per 60-mm tissue culture dish in triplicate. Cells were incubated for 10 days, fixed and stained and total colony number and cell numbers per colony scored [9]. Cloning efficiency was equivalent in cells exposed to 3-5Gy and doses in this range were used in all experiments, except for detection of H2AX and 53BP1.
2.2 Colony formation and growth assays
Long-term (10 days) colony formation assays were performed as described above, following 3Gy exposure. For cell attachment/short term cloning efficiency and cell growth assays, γ-irradiated (3Gy) or untreated MEFs were seeded at 100 cells per 35-mm tissue culture dish in triplicate or 3 × 104 cells per well in 24 well plates, respectively. Mass cell growth was measured by the MTT assay [10] and cell number determined from 1 to 5 days after irradiation, at 24 h intervals.
2.3 Strand break detection using the alkaline comet assay
Strand break repair was analyzed by single-cell agarose gel electrophoresis under alkaline conditions as described previously [11]. Cells were irradiated (4Gy), and harvested immediately or at 30 and 120 min post exposure to IR.
2.4 Immunoblot analysis for detection of histone acetyl-K5-H2A (Ac-H2A) and phosphorylated H2AX (γ-H2AX)
Histone proteins were acid extracted from trichostatin A (TSA)-treated cells (0.4 μM, 16 h) or irradiated cells (10Gy) at 0, 30,45, 60, 90 and 180 min post exposure according to manufacturer's instructions (Upstate Biotechnology, Charlottesville, VA). Acid extracted histones from the same number of cells were loaded onto 15% SDS-polyacrylamide gels and western blotted using anti-acetyl-K5-H2A (abcam, ab1764), anti-phosphorylated H2AX (Ser139) (Upstate, #05-636), or anti-histone H2A (Santa Cruz, sc-10807) antibodies, as described previously [12].
2.5 Indirect immunofluorescence to detect γ-H2AX and 53BP1 foci
Cells were fixed at 0, 30 and 60 min post irradiation (10Gy) with cold 70% ethanol for 30 min at 4°C. Nonspecific binding was saturated for 5 min at room temperature in block solution (1% bovine serum albumin and 10% horse serum in PBS). After incubation with anti- γ-H2AX or 53BP1 antibodies, Fluorescein and Texas Red-conjugated secondary antibodies were added. Staining with 0.5 μg/ml DAPI was done for 5 min. A Zeiss AxioVert 200M optical sectioning microscope equipped with a Zeiss AxioCam B&W CCD camera was used to collect digital images and three-dimensional deconvolution performed with the Zeiss software to resolve foci.
3. Results
3.1. Comet assays demonstrate Mrg15 null and het MEFs are defective in repair of IR induced DNA damage
MRG15 is an essential component of the NuA4/Tip60-HAT complex that has been shown to promote accessibility to chromatin and, thereby, facilitate recruitment of DNA-repair machinery to sites of DNA-damage in Drosophila and mammalian cells [13]. Post DNA repair, other complexes, such as the Sin3-HDAC complex, in which MRG15 is also a component, have been postulated to restore condensed chromatin at sites of damage to maintain genome integrity. In this study, we analyzed Mrg15 null and heterozygous (het) MEFs to determine if they were defective in DNA-repair in response to IR. We initially quantified DNA damage using alkaline single-cell agarose gel electrophoresis (comet assay).
MEFs derived from E13.5 wild-type, Mrg15 null and het embryos [9], were either mock treated (-IR) or exposed to 4Gy IR (+IR) and harvested at various times post treatment. DNA damage in -IR was low and no major differences were observed between wild-type, Mrg15 null and het cells. At 10 min following exposure to IR, wild-type MEFs exhibited a high percentage of DNA in the comet tail, representing damaged DNA. However, by 120 min post exposure the cells had efficiently repaired damaged DNA to levels comparable to -IR controls (Fig. 1). In contrast, the Mrg15 null and het MEFs had un-repaired DNA in the tail at 120 min. At least two independent clones of MEF cell lines were analyzed for each genetic background and decreased DNA-repair at 120 min was observed in the Mrg15 null and het MEF clones tested. These results demonstrate that loss of even one copy of MRG15 is sufficient to affect efficient repair of DNA-damage post IR.
Fig. 1. MRG15 is important for DNA-repair.

Wild-type, Mrg15 het and null MEFs were untreated (−IR) or γ-irradiated at 4Gy (+IR) and harvested at various times post exposure for comet analysis. Two clones of each genotype were tested. Distributions of percent cells with damaged DNA in tails are shown.
3.2 Long- and short-term clonal and growth assays confirm that Mrg15 null and het MEFs have impaired growth, not increased apoptosis, following IR
Based on the results of the comet assay, we determined whether cell growth of Mrg15 null and het cells was affected following DNA-damage using a clonogenic assay. Irradiated Mrg15 null and het MEFs cloned poorly and the percentage of large colonies were much fewer than in unirradiated controls (Fig. 2A and B). Irradiated wild-type cells, in contrast, cloned as well as unirradiated cells. This result was obvious by viewing the dishes, and was confirmed when we counted the number of colonies with over 100 cells in treated and untreated MEFs of all genotypes. In contrast to wild-type MEFs, which had the same number of colonies in both conditions, Mrg15 het and null MEFs showed reduced cell growth after IR (Fig. 2C). Due to clonal heterogeneity, and to demonstrate that the result was not unique to a single clonal isolate, we used a minimum of two independently isolated wild-type, null and het clones in these studies. The results demonstrate that loss of one copy of MRG15 is sufficient to affect MEF cell growth post exposure to IR.
Fig. 2. Mrg15 null and het MEFs are impaired in growth post exposure to IR.

(A) Representative wild-type and Mrg15 null MEF clones exposed to 3Gy and seeded at 2500 cells per dish. Cells were grown for 10 days and stained with crystal violet. (B) Independent clonal isolates of wild-type, Mrg15 null and het MEFs treated as in A. (C) Quantitation of colony formation efficiency of Mrg15 mutant MEFs (from 2B) after IR. Colonies with >100 cells were counted. The percentage of untreated cells was set at 100%. (This is a representative result for at least two independent clones for each genotype.) Error bars represent standard error (SE) for triplicate samples.
We then determined the short-term survival of Mrg15 mutant MEFs after IR using colony formation and cell growth assays. We counted attached cells (1 day) and number of cells per colony (4 days) post-treatment. At day one, cell numbers were no different among genotypes or between untreated and treated cells (data not shown). This indicates that apoptosis does not contribute to the differential survival of Mrg15 mutant versus wild-type MEFs observed in the long-term clonogenic assay, after IR (Fig 2). Data obtained at 4 days, confirmed this as wild-type MEFs showed the same pattern of colony formation in untreated and treated conditions (Fig. 3), whereas the percentage of large clones in irradiated null and het MEFs was decreased relative to untreated cells. Conversely there were more small clones following treatment in these cells. This was further confirmed by mass cell culture growth measurements done at 24 h intervals using the MTT assay, from days one to five after IR; wild-type cell growth unaffected by IR, mutant MEF growth inhibited (data not shown).
Fig. 3. Colony size distribution assay of MEFs.

Untreated and γ-irradiated MEFs (100 cells) were plated in 60-mm dishes and incubated for 4 days. The cell number in each colony was determined.
These data suggest that reduced colony numbers after long-term culture of Mrg15 mutant MEFs in response to IR, reflect defects in growth and not increased apoptosis, and that wild-type MEFs have efficiently repaired DNA damage at this dose.
3.3. Histone acetylation is delayed in Mrg15 null and het MEFs
The NuA4/Tip60-HAT complex acetylates H2A at lysine 5 (K5) and TRRAP (another component of the Tip60 complex) deficient MEFs exhibit impairment of acetylation of H2A post IR that results in defective accessibility and recruitment of additional repair proteins, such as 53BP1, to the damage sites [14]. In D. melanogaster, dTip60 and dMRG15 proteins have been shown to be necessary for the exchange of γ-H2Av for unmodified H2Av at sites of repair [15]. We therefore determined whether MRG15 might be required for proper acetylation of H2A in response to DNA damage.
Acid extracted histones were analyzed by Western blot to assess whether there were any differences in the levels of acetyl-H2A and γ-H2AX in Mrg15 wild-type and null MEFs following IR. To determine the specificity of the anti-Ac-K5-H2A antibody, we prepared acid extracted histones from wild-type and Mrg15 null MEFs treated with or without trichostatin A (TSA), an inhibitor of class I HDACs, and performed Western analyses using anti-Ac-K5-H2A or anti-H2A antibodies (Fig. 4A). The major band recognized by the anti-Ac-K5-H2A antibody was the size expected for H2A and the intensity of this band increased significantly following TSA treatment. We designated this band H2A. An additional protein, of a slightly higher molecular weight, also exhibited increased acetylation following TSA treatment. This band most likely is Ac-H2AX because the size was the same as that of γ-H2AX, as discussed below. There was no difference in the response to wild-type or null cells following TSA treatment.
Fig. 4. Mrg15 null MEFs show delayed H2A-K5 acetylation and γ-H2AX foci formation in response to IR.
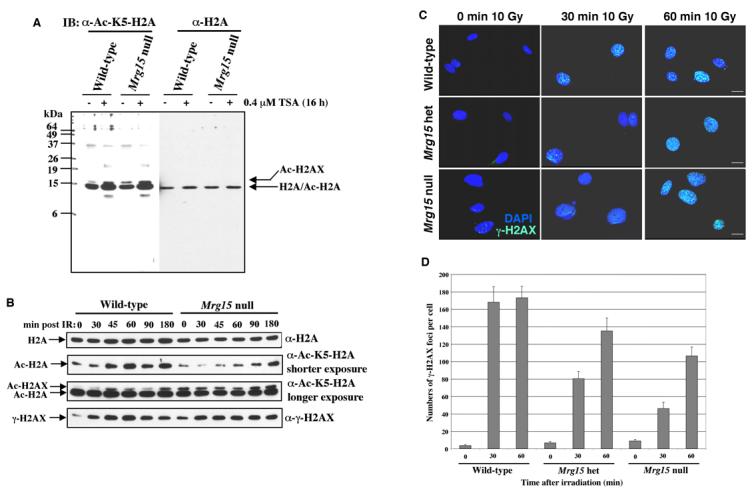
(A) Histones were acid extracted from wild-type and Mrg15 null MEFs treated without (−) or with (+) 0.4 μM trichostatin A for 16 h. Immunoblots were probed with anti-acetyl-K5-H2A and anti-H2A antibodies. (B) Histones were acid extracted from wild-type and Mrg15 null MEFs at various time points post treatment with 10Gy. Immunoblots were probed with anti-acetyl-K5-H2A antibodies (Second and third panels), stripped and probed with anti-γ-Ser139-H2AX (fourth panel) and, finally, stripped and probed with anti-H2A antibodies (top panel) as loading control. (C) Cells from wild-type, het, and null MEFs were fixed and stained with anti-γ-H2AX (FITC) antibodies at 0, 30 and 60 minutes post exposure to 10Gy. Representative cells from three experiments are shown. Scale bars, 10 μm. (D) The number of γ-H2AX foci per cell after treatment with IR, from two independent experiments.
The total amount of H2A did not change after IR in both cell types (Fig. 4B upper panel). Acetylation of H2A in wild-type MEFs increased significantly at 45 min after IR and these levels were maintained at later time points. In contrast, acetylation of H2A in Mrg15 null MEFs did not increase until 180 min after IR (Fig. 4B second panel). This impaired pattern of acetylation of H2A was very similar to that observed in TRRAP deficient MEFs following IR [14]. When the blot was developed for a longer time, an additional slightly higher molecular weight band than Ac-H2A was observed (Fig. 4B third panel). The size of this band was the same as that of γ-H2AX. The anti-Ac-K5-H2A antibody should recognize Ac-K5-H2AX as well as Ac-H2A due to sequence similarity. We therefore conclude that the upper band is indeed Ac-H2AX.
Acetylation of H2AX in wild-type MEFs increased after IR and the kinetics were similar to that of H2A (Fig. 4B third panel). In contrast, acetylation of H2AX in Mrg15 null MEFs did not increase until 180 min after IR, although basal levels were higher than wild-type (Fig. 4B third panel). Thus, MRG15 is important for efficient acetylation of both H2A and H2AX in mammalian cells, presumably through its association with the NuA4/Tip60-HAT complex.
The phosphorylation of H2AX (γ-H2AX), a mammalian histone variant, is another modification that occurs on chromatin flanking strand breaks and is required for proper recruitment of DNA-repair proteins to sites of damage [16]. However, Mrg15 null MEFs did not exhibit defects in this phosphorylation process post exposure to IR (Fig. 4B bottom panel).
We further analyzed the formation of γ-H2AX foci at different times following IR in various Mrg15 MEF clones by microscopy, using deconvolution algorithms. Western analysis indicated phosphorylation levels of H2AX were increased upon exposure to damage in Mrg15 null MEFs similar to what was observed in wild-type MEFs. Immunostaining of wild-type MEFs showed maximal foci formation as early as 30 min post exposure to IR (Fig. 4C and D). In null as well as het cells some γ-H2AX foci were observed at 30 min but did not reach maximum levels until 60 min post IR (Fig. 4C and D). These data suggest that even hemizygous expression of MRG15 influences efficient formation of γ-H2AX foci at sites of damage in mammalian cells.
3.4. Recruitment of 53BP1 to nuclear foci is delayed in Mrg15 mutant MEFs
We assessed recruitment of the DNA-repair factor 53BP1 to strand breaks in Mrg15 mutants by immunofluorescence. Consistent with the histone H2A and H2AX acetylation data (Fig. 4B), formation of nuclear 53BP1 foci in Mrg15 null and het MEFs, at various times post exposure to IR, was markedly delayed when compared with wild-type cells (Fig. 5A). Foci were observed at 30 min post IR in wild-type MEFs and this number increased at 60 min post treatment. Relatively few to no 53BP1 foci were present at 30 min post exposure in het and null MEFs and, though the number of foci increased at 60 min post exposure, this was consistently lower when compared to wild-type (Fig 5B). Thus, in Mrg15 null and het MEFs recruitment of the initial DNA-repair machinery is less robust and delayed when compared with wild-type cells.
Fig. 5. 53BP1 foci formation is delayed in Mrg15 null MEFs post exposure to IR.

(A) Cells from wild-type, het, and null MEFs were fixed and stained with anti-53BP1 (Texas red) antibodies at various times post exposure to 10Gy. Representative cells from three experiments are shown. Scale bars, 10 μm. (B) The number of 53BP1 foci per cell after treatment with IR, from two independent experiments.
4. Discussion
In this study, we demonstrate a role for MRG15 in repair of DNA-damage and cell growth in response to IR in mammalian cells, even at hemizygous levels of expression. This sensitivity to MRG15 depletion most likely reflects its participation as a cofactor in multiple chromatin modifying complexes, including the NuA4/Tip60-HAT and Sin3-HDAC1 complexes, that have been implicated in DNA-repair. In fact, deletion of other components of the NuA4/Tip60-HAT complex has been shown to result in defects in histone H4 and H2A acetylation and, as a consequence, impaired recruitment of DNA-repair machinery to sites of damage [17].
It has been shown that, in D. melanogaster, the NuA4/Tip60-HAT complex is important for the exchange of γ-H2Av for unmodified H2Av at sites of DNA-repair [15]. We have found that Mrg15 null MEFs are delayed in H2A and H2AX acetylation and γ-H2AX foci formation after IR. The MRG15 protein does not itself have HAT activity and, therefore, Mrg15 null cell defects in H2A and H2AX acetylation are most likely due to improper targeting of the NuA4/Tip60-HAT complex to sites of damage in the absence of this protein. Another possibility is that deletion of MRG15 disrupts the integrity of the NuA4/Tip60-HAT complex in mammalian cells. The fact that the yeast NuA4 complex is intact in cells deleted for the yeast MRG15 homolog, EAF3 [18], does not eliminate the possibility that this process might differ across species. In support of this, deletion of the EAF3 gene in yeast does not affect cell viability, whereas deletion of MRG15 has many detrimental effects on mouse development [9]. We observed delayed acetylation in Mrg15 mutant MEFs and subsequently delayed 53BP1 foci formation in response to IR. MRG15 appears to be more important for γ-H2AX foci formation in contrast to observations made in Drosophila [15]. These results further support the idea that the function of MRG15 varies with cell type and origin, despite the high degree of conservation of the protein across species.
ATM kinase is a key player in the activation of cell cycle checkpoints in response to radiation-induced DNA damage [19] and Tip60 acetylates and activates ATM after DNA damage [20]. MRG15, which associates with the Tip60 complex, when decreased could potentially modify the action of this complex and thereby affect down stream pathway(s) of ATM. UV irradiation of cells also results in rapid phosphorylation of H2AX, but different kinases and pathways than those involved in DNA double-strand break repair are activated [21]. Thus, Mrg15 null and het cells may incorrectly activate such kinases or pathways as a compensatory mechanism for the decreased levels of MRG15 and thereby decreased activity of the Tip60 complex. We have observed that the kinetics of γ-H2AX levels in Mrg15 null MEFs following IR were similar to those of wild-type MEFs. However, maximal foci formation was delayed in Mrg15 null and het MEFs. This may result from (i) activation of phosphatases which dephosphorylate γ-H2AX outside foci after initiation of γ-H2AX foci formation, (ii) redistribution of γ-H2AX to DNA damage sites through unknown mechanisms. Our preliminary proteomic analysis of MRG15 indicates that MRG15 interacts with many other proteins and is associated with multiple protein complexes. The genotype of Mrg15 het or null most likely affects different processes than wild-type and results in the phenotypes we have observed. Details regarding this remain to be determined.
An example of the complexity of MRG15 function(s) is the fact that it interacts with a number of proteins that are not associated with the NuA4/Tip60-HAT and Sin3-HDAC complexes, such as the hMOF-HAT and the retinoblastoma protein [22,23]. It has recently been shown that the hMOF-HAT acetylates the ATM kinase thereby modulating phosphorylation of its downstream DNA-repair targets in response to IR [24]. Thus, MRG15 function(s) in DNA-repair could be mediated through such protein associations, that occur independent of NuA4 and HDAC interactions. Many of these MRG15-interacting proteins are not conserved across species, suggesting that MRG15 will likely function in DNA-repair through a number of distinct mechanisms that are cell type and species dependent.
In summary, we have demonstrated a requirement for MRG15 in DNA-repair and cell growth in response to IR and this is dependent on protein levels, as hemizygous expression of MRG15 results in the same effect on DNA-repair as in null cells. Additionally, MRG15 is important for efficient acetylation of both H2A and H2AX, formation of γ-H2AX foci and as a consequence the recruitment of DNA-repair machinery to sites of damage. These results underscore the sensitivity of precise amounts of functional DNA-repair proteins for maintenance of an intact genome, and emphasize the importance of analyzing protein function in different species. Over- or under-expression of key proteins can disrupt a delicate balance resulting in a decrease in the efficacy of DNA-repair and related processes including cell growth, apoptosis, cell senescence and tumorigenicity.
Acknowledgements
We thank Christina Hawks for assistance with immunofluorescence microscopy, Natalia Podlutskaja for comet analysis and Emiko Tominaga for MEF culture. We also thank James R. Smith, Christi Walter and James Jackson for comments on the manuscript. This work was supported by NIA grants, T32AG021890 (SNG) and KO7AG25063 (AP), the Ellison Medical Foundation (OMP-S), and the American Federation for Aging Research (AP and KT).
Abbreviations
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- IR
ionizing radiation
- MEFs
mouse embryonic fibroblasts
- MORF4
mortality factor on chromosome 4
- MRG15
MORF4-related gene on chromosome 15
- TSA
trichostatin A
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pereira-Smith OM, Smith JR. Evidence for the recessive nature of cellular immortality. Science. 1983;221:964–966. doi: 10.1126/science.6879195. [DOI] [PubMed] [Google Scholar]
- 2.Bertram MJ, Berube NG, Hang-Swanson X, Ran Q, Leung JK, Bryce S, Spurgers K, Bick RJ, Baldini A, Ning Y, Clark LJ, Parkinson EK, Barrett JC, Smith JR, Pereira-Smith OM. Identification of a gene that reverses the immortal phenotype of a subset of cells and is a member of a novel family of transcription factor-like genes. Mol. Cell. Biol. 1999;19:1479–1485. doi: 10.1128/mcb.19.2.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pereira-Smith OM, Smith JR. Genetic analysis of indefinite division in human cells: identification of four complementation groups. Proc. Natl. Acad. Sci. USA. 1988;85:6042–6046. doi: 10.1073/pnas.85.16.6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ball LJ, Murzina NV, Broadhurst RW, Raine AR, Archer SJ, Stott FJ, Murzin AG, Singh PB, Domaille PJ, Laue ED. Structure of the chromatin binding (chromo) domain from mouse modifier protein 1. EMBO J. 1997;16:2473–2481. doi: 10.1093/emboj/16.9.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cowell IG, Austin CA. Self-association of chromo domain peptides. Biochem. Biophys. Acta. 1997;1337:198–206. doi: 10.1016/s0167-4838(96)00165-3. [DOI] [PubMed] [Google Scholar]
- 6.Nielsen AL, Oulad-Abdelghani M, Ortiz JA, Remboutsika E, Chambon P, Losson R. Heterochromatin formation in mammalian cells: interaction between histone and HP1 proteins. Mol. Cell. 2001;7:729–739. doi: 10.1016/s1097-2765(01)00218-0. [DOI] [PubMed] [Google Scholar]
- 7.Morrison AJ, Shen X. Chromatin modifications in DNA repair. Results Probl. Cell Differ. 2006;41:109–125. doi: 10.1007/400_008. [DOI] [PubMed] [Google Scholar]
- 8.Doyon Y, Selleck W, Lane WS, Tan S, Cote J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol. Cell. Biol. 2004;24:1884–1896. doi: 10.1128/MCB.24.5.1884-1896.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tominaga K, Kirtane B, Jackson JG, Ikeno Y, Ikeda T, Hawks C, Smith JR, Matzuk MM, Pereira-Smith OM. MRG15 regulates embryonic development and cell proliferation. Mol. Cell. Biol. 2005;25:2924–2937. doi: 10.1128/MCB.25.8.2924-2937.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 11.Olive PL, Banath JP, Durand RE. Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the “comet” assay. Radiat. Res. 1990;122:86–94. [PubMed] [Google Scholar]
- 12.Tominaga K, Magee DM, Matzuk MM, Pereira-Smith OM. PAM14, a novel MRG- and Rb-associated protein, is not required for development and T-cell function in mice. Mol. Cell. Biol. 2004;24:8366–8373. doi: 10.1128/MCB.24.19.8366-8373.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Downs JA, Cote J. Dynamics of chromatin during the repair of DNA double-strand breaks. Cell Cycle. 2005;4:1373–1376. doi: 10.4161/cc.4.10.2108. [DOI] [PubMed] [Google Scholar]
- 14.Murr R, Loizou JI, Yang Y-G, Cuenin C, Li H, Wang Z-Q, Herceg Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 2006;8:91–99. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- 15.Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates JR, 3rd, Abmayr SM, Washburn MP, Workman JL. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science. 2004;306:2084–2087. doi: 10.1126/science.1103455. [DOI] [PubMed] [Google Scholar]
- 16.Tsukuda T, Fleming AB, Nickoloff JA, Osley MA. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 2005;438:379–383. doi: 10.1038/nature04148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Squatrito M, Gorrini C, Amati B. Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol. 2006;16:433–42. doi: 10.1016/j.tcb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 18.Eisen A, Utley RT, Nourani A, Allard S, Schmidt P, Lane WS, Lucchesi JC, Cote J. The yeast NuA4 and Drosophila MSL complexes contain homologous subunits important for transcription regulation. J. Biol. Chem. 2001;276:3484–3491. doi: 10.1074/jbc.M008159200. [DOI] [PubMed] [Google Scholar]
- 19.Lavin FM, Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6:931–942. doi: 10.4161/cc.6.8.4180. [DOI] [PubMed] [Google Scholar]
- 20.Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA. 2005;102:13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc. Natl. Acad. Sci. USA. 2006;103:9891–9896. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neal KC, Pannuti A, Smith ER, Lucchesi JC. A new human member of the MYST family of histone acetyl transferases with high sequence similarity to Drosophila MOF. Biochem. Biophys. Acta. 2000;1490:170–174. doi: 10.1016/s0167-4781(99)00211-0. [DOI] [PubMed] [Google Scholar]
- 23.Pardo PS, Leung JK, Lucchesi JC, Pereira-Smith OM. MRG15 a novel chromodomain protein is present in two distinct multiprotein complexes involved in transcriptional activation. J. Biol. Chem. 2002;277:50860–50866. doi: 10.1074/jbc.M203839200. [DOI] [PubMed] [Google Scholar]
- 24.Gupta A, Sharma GG, Young CS, Agarwal M, Smith ER, Paull TT, Lucchesi JC, Khanna KK, Ludwig T, Pandita KT. Involvement of human MOF in ATM function. Mol. Cell. Biol. 2005;25:5292–5305. doi: 10.1128/MCB.25.12.5292-5305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]