

Abstract
While the specificity and timing of membrane fusion in diverse physiological reactions, including virus–cell fusion, is determined by proteins, fusion always involves the merger of membrane lipid bilayers. We have isolated a lipid-dependent stage of cell–cell fusion mediated by influenza hemagglutinin and triggered by cell exposure to mildly acidic pH. This stage preceded actual membrane merger and fusion pore formation but was subsequent to a low pH–induced change in hemagglutinin conformation that is required for fusion. A low pH conformation of hemagglutinin was required to achieve this lipid-dependent stage and also, downstream of it, to drive fusion to completion. The lower the pH of the medium applied to trigger fusion and, thus, the more hemagglutinin molecules activated, the less profound was the dependence of fusion on lipids. Membrane-incorporated lipids affected fusion in a manner that correlated with their dynamic molecular shape, a characteristic that determines a lipid monolayer's propensity to bend in different directions. The lipid sensitivity of this stage, i.e., inhibition of fusion by inverted cone–shaped lysophosphatidylcholine and promotion by cone-shaped oleic acid, was consistent with the stalk hypothesis of fusion, suggesting that fusion proteins begin membrane merger by promoting the formation of a bent, lipid-involving, stalk intermediate.
Protein-mediated membrane fusion is a ubiquitous process in living systems. By far the best characterized example is fusion mediated by the influenza virus hemagglutinin (HA)1 (White, 1992; White, 1996). The influenza virus enters cells by receptor-mediated endocytosis (Yoshimura et al., 1982). The low pH environment in the endosome induces a conformational change in the viral hemagglutinin (HA), through which this protein mediates fusion between the viral and endosomal membranes to deliver the viral nucleocapsid into host cell cytosol. Two posttranslational cleavage products of the HA0 precursor (HA1 and HA2) are responsible for viral binding to sialic acids of receptors on the surface of host cells and for membrane fusion, respectively (Wiley and Skehel, 1987). Both the structure of the nonfusogenic form of HA at neutral pH and the conformational changes this glycoprotein undergoes at low pH have been studied (Wilson et al., 1981; Carr and Kim, 1993; Bullough et al., 1994; White, 1996). We know that this conformational change results in a lengthening of a coiled-coil helix in the center of the HA trimer. At low pH, a highly conserved apolar NH2-terminal HA2 segment, the fusion peptide, is exposed, which then binds hydrophobically to both the target bilayer and viral membrane and interacts with both of them (Stegmann et al., 1991; Tsurudome et al., 1992; Weber et al., 1994; Tatulian et al., 1995). These interactions are thought to trigger membrane fusion in a process that can be affected both by specific mutations of the fusion protein (Daniels et al., 1985; Gething et al., 1986; Schoch and Blumenthal, 1993; Kemble et al., 1994) and by the lipid composition of membranes (White et al., 1982; Stegmann et al., 1985; van Meer et al., 1985). The specific mechanisms of the membrane rearrangements in fusion remain unknown.
Since the first detectable event in HA-mediated fusion is the opening of the fusion pore (Tse et al., 1993; Zimmerberg et al., 1994), it is natural to imagine that a low pH conformation of HA (activated HA), spanning both bilayers, starts fusion by promoting the formation of a proteinaceous pore (Fig. 1 c) similar to gap junction channels and, only later, the expansion of this fusion pore allows mixing of lipids and actual merger of two fusing membrane bilayers (Tse et al., 1993; Lindau and Almers, 1995). In agreement with this model, simultaneous measurements of lipid dye flux and fusion pores in HA-mediated cell–cell fusion showed that fusion pore formation precedes lipid dye redistribution (Tse et al., 1993; Zimmerberg et al., 1994). However, lipid flow from membrane to membrane before the expansion of an initial fusion pore can be hindered by the complexes of integral membrane fusion proteins in the fusion site (Tse et al., 1993; Zimmerberg et al., 1993; Kemble et al., 1994).
Figure 1.

Two hypothetical pathways in HA-mediated fusion. (a) HA-expressing membrane in contact with the target membrane. (b) Low pH induces dramatic changes in HA conformation including insertion of the HA fusion peptide into the target membrane. (c) Formation of a proteinaceous pore. (d) Completion of the fusion process. Dashed lines show the boundaries of the hydrophobic surfaces of monolayers. (e) Local merger of membrane outer monolayers. According to the stalk hypothesis, a transient and local connection between membranes (stalk) has a net negative curvature and its formation should be facilitated and hindered by cone-shaped lipids (e.g., OA), and by inverted cone–shaped lipids (e.g., LPC), respectively. Dashed lines show the boundaries of the hydrophobic surfaces of two monolayers.
An alternative class of models assumes that activated fusion proteins bring membrane lipid bilayers into close contact and decrease the energy barrier for lipids to mix (Monck and Fernandez, 1992; Zimmerberg et al., 1993; Chernomordik et al., 1995b ; Melikyan et al., 1995c ). In this scenario, local mixing of membranes' lipids occurs before fusion pore opening (Fig. 1 e). Recent findings confirm that membrane merger is not necessarily accompanied by fusion pore formation. A modification of HA, engineered so that the ectoviral domains are anchored in the outer leaflet of cell membranes via a glycosylphosphatidylinositol (GPI) tail, rather than via its normal transmembrane domain, promotes low pH–triggered mixing of lipids but not water-soluble probes (Kemble et al., 1994; Melikyan et al., 1995c ). These results suggest that the ectodomains of HA mediate hemifusion (i.e., fusion of contacting membrane monolayers without merger of the inner monolayers) and that the transmembrane HA domains then merge the inner membrane leaflets to open the fusion pore.
In contrast to the model of a proteinaceous fusion pore, early fusion intermediates in the alternative models already involve membrane lipids, so the lipid composition of membranes may affect early stages of membrane fusion. To determine which class of models fits HA-mediated membrane fusion, we decided to systematically study the effect of exogenous lipids on the fusion of human red blood cells (RBC) with cells expressing influenza HA. We were guided in our choice of lipids by a specific model, the “stalk” hypothesis, which suggests that membrane fusion proceeds via the formation of stalk intermediates, which are local lipidic connections between contacting monolayers of fusing membranes (Fig. 1 e; Kozlov and Markin, 1983; Siegel, 1993a ,b; Chernomordik et al., 1995b ), and gives specific predictions for the effect of lipids on fusion.
Stalks are highly bent local hemifusion intermediates, and thus the elastic energy of monolayer bending is considered in the stalk hypothesis as a very important component of the stalk's energy. Different lipids bend lipid monolayers in different directions reflecting the dynamic molecular shape of the lipids (spontaneous curvature of the monolayer), which in turn depends on the conformation of individual lipid molecules and on the interactions between lipid molecules in a monolayer. Inverted cone–shaped lysophosphatidylcholine, LPC, promotes a micellar positive spontaneous curvature (Epand, 1985), and cone-shaped cis-unsaturated fatty acids and phosphatidylethanolamine promote negative spontaneous curvature, as in the inverted hexagonal HII phase (Epand et al., 1991). The net curvature of a stalk is negative (Chernomordik et al., 1995b ). Thus, one may expect LPC present in the contacting monolayers of membranes to inhibit, and phosphatidylethanolamine or cis-unsaturated fatty acids to promote stalk formation and membrane merger (Fig. 1 e).
In the present study, we found a lipid-sensitive stage common to fusion mediated by HA of different strains of influenza (Japan/305/57 and X:31 strains) and for hemifusion mediated by GPI-anchored HA. This lipid-sensitive stage was subsequent to the low pH–induced change in HA conformation but preceded or coincided with lipid mixing as assayed by membrane dye redistribution from labeled to unlabeled cells upon their fusion. This stage was also upstream of fusion pore formation assayed both as aqueous dye transfer between cells and by electrophysiological recording. The lipid sensitivity of fusion, i.e., its inhibition by LPC and promotion by oleic acid (OA), is similar to that reported for some other examples of biological fusion and for fusion of purely lipid bilayers and is consistent with the stalk mechanism of membrane fusion.
Materials and Methods
Preparation of Cells
HAb2 cells, a line of stably transfected NIH-3T3 fibroblasts expressing the A/Japan/305/57 strain of influenza virus HA (Doxsey et al., 1985), were cultured as described (Melikyan et al., 1995a ). CHO-K1 cells expressing the X:31 strain of influenza virus HA (referred to as HA300a cells) or expressing the GPI-anchored X:31 HA (BHA-PI cells) (Kemble et al., 1993, 1994) were grown as described (Kemble et al., 1994).
Human RBC, freshly isolated from whole blood obtained from the National Institutes of Health blood bank (Bethesda, MD), were labeled with fluorescent dyes. To label RBC membranes with R18 (Hoekstra et al., 1984), we rapidly added 15 μl of R18 solution in ethanol (1 mg/ml) to 10 ml of a RBC suspension (1% hematocrit) in PBS. Unbound R18 was removed by washing RBC with complete medium followed by four washings with PBS. Under these conditions, R18 incorporated into RBC membranes to a high enough concentration to cause significant self-quenching of fluorescence. Because of slow fusion–independent redistribution of R18 between contacting membranes observed in experiments when HAexpressing cells with uncleaved HA were incubated at neutral pH with prebound R18-labeled RBCs for more than 1 h, in longer experiments (see Figs. 6 and 8 b) we also used another fluorescent membrane dye PKH26 (Sigma Chemical Co., St. Louis, MO). Cell labeling with PKH26 was performed as described (Dimitrov and Blumenthal, 1994).
Figure 6.

The inhibition of pore formation by LPC. Fusion of a HAb2 cell with a single RBC. The arrows indicate the beginning of the application of pH 4.9 solution. Note that these experiments were performed at 33°C rather than at room temperature to accelerate fusion pore formation upon low pH application (Spruce et al., 1989). (a) No LPC added. The increase in capacitance reflects the fusion event. The waiting time between the beginning of low pH application and the onset of the capacitance increase was 16.8 ± 8.9 s (n = 10). (b) Capacitance trace in the presence of 25 μM lauroyl LPC.
Figure 8.
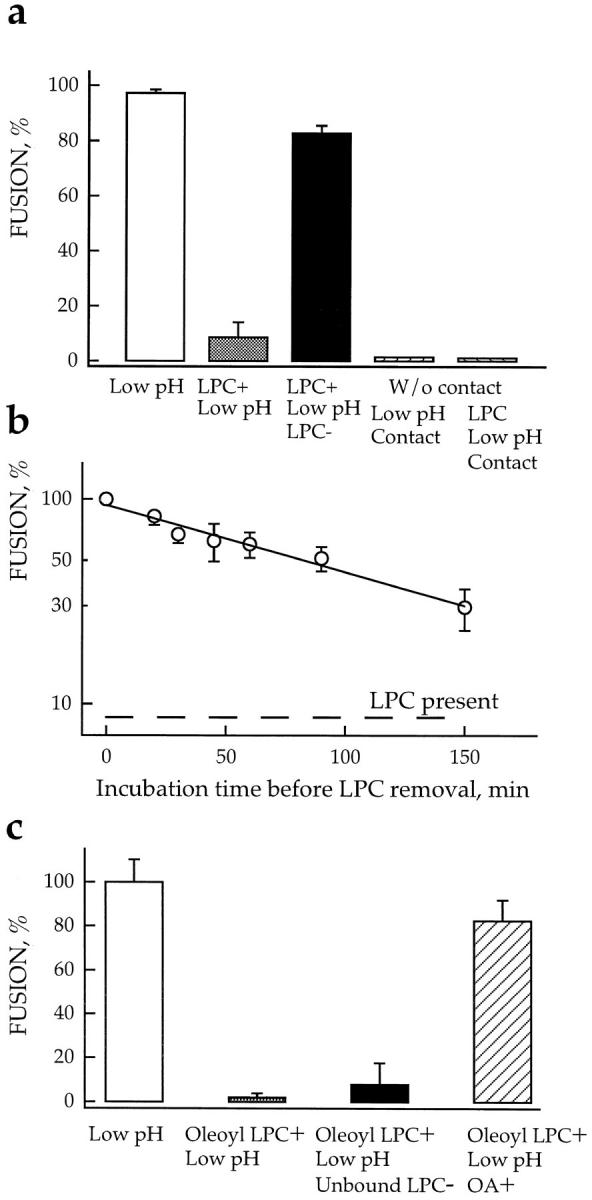
Lipids affect fusion downstream of the triggering step. (a) LPC reversibly arrested fusion downstream low pH–dependent stages. Fusion was triggered by a 2-min application of pH 4.9 medium and assayed by fluorescence microscopy as R18 redistribution. □, fusion of HAb2 cells with prebound RBCs, no lipids added; ▒⃞ , starting 5 min before the low pH pulse, cells were incubated in the presence of 150 μM lauroyl LPC; ▪, LPC was washed out by LPC-free PBS 20 min after the end of low pH application; ▨ , the same pulse of low pH was applied to a suspension of HAb2 cells in the absence of RBCs. Then, back at neutral pH , R18-labeled RBCs were added to the low pH–treated HAb2 cells. ▨ , similar to the previous experiment, but low pH was applied to HAb2 cells in the presence of 150 μM lauroyl LPC, then R18-labeled RBCs were added at neutral pH but still in the presence of LPC, and, finally, HAb2 cells with bound RBCs were washed out with LPC-free PBS to remove lysolipid. Fusion was assayed 20 min later. (b) Slow inactivation of the committed fusion stage arrested by LPC. Fusion of HAb2 cells with prebound PKH26-labeled RBCs was triggered in the presence of 150 μM lauroyl LPC by a 2-min application of pH 4.9 medium. At different time intervals after low pH application, LPC was removed by washing cells with LPC-free PBS. t = 0 corresponds to the end of the low pH pulse. The extent of fusion observed at t = 2 h in the experiment when LPC was not removed is shown by dashed line. Fusion extents assayed by fluorescence microscopy as PKH26 redistribution were normalized to those in control experiments with no lipids added. Each point is mean ± SEM, n = 3. (c) Adding OA released cells from the LPC-arrested fusion stage. Cells were triggered to fuse by a 2-min treatment with pH 4.95 medium in the absence or in the presence of 9.6 μM oleoyl LPC and then returned to the LPC-free neutral pH medium. The recovery of fusion previously arrested by LPC reached an extent close to that of the control experiment (no lipids added) upon addition of 5 μM OA at neutral pH , 5 min after the end of low pH pulse. No fusion was observed in control experiments when OA was added to cells not treated by low pH . Thus, OA canceled the LPC block to low pH–triggered fusion rather than having induced fusion on its own. Each point is mean ± SEM, n = 3.
To label RBC ghosts with the fluorescent water-soluble dyes carboxyfluorescein (CF) or ethidium bromide, we used mild hypotonic lysis followed by resealing as described (Ellens et al., 1989; Melikyan et al., 1995a ). NBD-taurine was loaded into RBC as described (Morris et al., 1993).
Fusion Experiments
Expressed HA0 was cleaved into its fusion-competent HA1-S-S-HA2 form with 5 μg/ml trypsin (Fluka AG, Buchs, Switzerland) in the presence of 250 μg/ml neuraminidase (Sigma Chemical Co.) for 10 min at room temperature. The reaction was terminated by washing cells twice with MEM containing 10% fetal serum. Cells were washed twice by PBS and then incubated for 10 min with a 1 ml suspension of RBCs or RBC ghosts (0.05% hematocrit) at room temperature (20–22°C). The unbound RBCs were removed by three washings with PBS. HA-expressing cells with bound RBCs (∼0–2 erythrocytes per cell) were then used for experiments. In some experiments, we evaluated the effects of exogenous lipids and some enzymatic treatments on the RBC binding to cells. We randomly selected several areas of the dish and screened at least 200 cells to find the average number of RBCs bound to one HA-expressing cell.
All fusion experiments were performed at room temperature (20– 22°C). Fusion was triggered by replacing PBS with an isoosmotic buffer titrated by citrate to acidic pH. After incubation of cells at low pH for 2 min (if not stated otherwise), the acidic solution was replaced by PBS.
The extent of fusion, as defined by dye redistribution from RBCs (or RBC ghosts) to HA-expressing cells, was quantified using fluorescence microscopy as the ratio of dye-redistributed bound RBCs to the total number of the bound RBCs. Extent of fusion was always measured >20 min after low pH application. Longer incubations (up to 1 h) did not increase the extent of fusion. Because extent of fusion and kinetics varied from day to day, apparently because of variation in the level of HA expression, number of attached cells, differences in blood samples, and temperature, we routinely started the experiments by choosing the precise conditions of the low pH treatment and concentrations of exogenous lipids to use. Each experiment presented here was repeated at least three times, and all functional dependences reported were observed in each experiment. If not stated otherwise, all data presented in one figure correspond to the same experiment performed in triplicate.
In some experiments, HAb2 cells with bound RBCs were treated by neuraminidase (0.25 mg/ml, 20 min) or proteinase K (Sigma Chemical Co.; 0.2 mg/ml, 20 min), or both enzymes together in PBS at room temperature before or after low pH application. Reactions were terminated by washing cells twice with PBS.
Spectrofluorometric Measurements of Membrane Fusion and Lipid Incorporation
An SLM Aminco Luminescence Spectrometer (Urbana, IL) was used for all experiments. Excitation and emission wavelengths were 550 and 590 nm for R18 and 473 and 515 nm for NBD-taurine. The standard fusion assay was performed as follows: 2 ml of suspension of HA-expressing cells with prebound RBCs in PBS were placed into a fluorescence cuvette and stirred with a teflon-coated flea. The pH in the cuvette was changed to 4.9 by injecting citric acid. The increase in fluorescence was normalized to the maximum dequenching signal obtained at infinite dilution of the probe by lysing cells with 0.06% SDS.
Incorporation of exogenously added lipids into R18-labeled RBC membranes decreased the membrane concentration of R18 and thus caused some relief of dye self-quenching and an increase in fluorescence. A larger fluorescence increase corresponds to more incorporation. To evaluate how change in R18 concentration affects the extent of R18 selfquenching, we measured the fluorescence of RBCs labeled with different amounts of R18 before (F ) and after (F 0) SDS addition. These data were used as a calibration curve to roughly estimate the incorporation of different LPCs and OA into cell membranes from the decrease in R18 quenching (F 0 /F ratio) caused by adding different lipids to HAb2 cells with prebound R18-labeled RBCs. This experimental approach is based on the assumption that both R18 and exogenously added lipids are homogeneously and independently distributed in the same monolayers of RBC membranes, i.e., all these lipids are present only in the outer monolayer or in both membrane monolayers. Because characteristic times for flip-flop of all LPCs used (∼hours; Mohandas et al., 1982; Bhamidipati and Hamilton, 1995) are orders of magnitude slower than those of OA and R18 (∼seconds; Broring et al., 1989; Leenhouts and De Kruijff, 1995; Melikyan et al., 1996), this assumption can cause some underestimation of LPC incorporation. In contrast to the binding measurements (Weltzien, 1979; Mohandas et al., 1982; Chernomordik et al., 1993), this assay gave us an estimate, although only approximate, of how much of the exogenous lipid was incorporated, rather than just bound to membranes.
This approach allowed us to evaluate in the same experiment, first, incorporation of exogenous lipids into RBC membranes and then, after low pH application, fusion of RBCs with HA-expressing cells. Fusion efficiencies found by the R18 dequenching assay were not affected by decreasing the initial extent of the dye self-quenching (F 0 /F) from 3.3 to 2.45 by lowering the amount of R18 solution used to label RBCs from 15 to 10 μl (data not shown). Thus, the smaller decreases in the extent of R18 selfquenching before low pH application, caused by lipid incorporation, did not interfere with the fusion assay.
In control experiments using RBCs without HA-expressing cells or with HAb2 cells that were fusion incompetent (not trypsin treated), we found that low pH application did not affect lipid incorporation. In some experiments, HAb2 cells with prebound R18-labeled RBCs were incubated with stearoyl LPC and then pelleted. Because all R18 fluorescence was found to stay associated with cells upon their pelleting, dequenching of R18 measured after LPC addition appears to be caused by LPC incorporation into cell membranes rather than by extraction of R18 into lysolipid micelles.
Treatment of Cells with Exogenous Lipids
Stock solutions of lysolipids, purchased from Avanti Polar Lipids (Birmingham, AL), were freshly prepared as a 0.5% (wt/wt) aqueous dispersion and vortexed until clear. Stock solutions of oleic and arachidonic acids (Sigma Chemical Co.), monoolein, and diolein (Nu Chek Prep, Elysian, MN) were freshly prepared as 25–50 mM ethanolic solutions. In spectrofluorometry experiments, exogenous lipids were added directly to the cell suspension by rapid injection of a few microliters of the corresponding stock solutions into the cuvette under constant stirring. In fluorescence microscopy experiments, the cell medium bathing the plastic or glass attached HA-expressing cells with prebound RBCs was replaced by 0.5 ml of medium supplemented with exogenous lipids 5 min before low pH application to the cell medium, unless stated otherwise. Low pH medium (used to trigger fusion) and “normal” pH medium (used to terminate the low pH treatment) were supplemented with the same concentration of lipid. Note that since exogenous lipids were added in different ways in the spectrofluorometry and fluorescence microscopy experiments, conditions of lipid partition into membranes would differ between these two assays. Added lipids did not change the pH of the medium. Ethanol at the concentrations used to add the exogenous lipids had no effect on fusion.
Some of the lipids tested in this study were toxic for cells at some concentrations. However, no correlation was found between the toxicity of these compounds, assayed using trypan blue exclusion test, and their ability to affect low pH–induced cell–cell fusion (see also Chernomordik et al., 1993). Even dead cells were able to fuse upon exposure to low pH (see also Sarkar et al., 1989). All data presented in the paper were obtained at nontoxic concentrations of tested compounds.
In some experiments, treatment of cells with exogenous lipids was followed by removing unbound lipids. Cells were washed by 2 ml PBS three times. To extract long-chain lipids such as stearoyl and oleoyl LPC or OA, we washed cells with PBS supplemented by 10 mg/ml of fatty acid–free BSA (Sigma Chemical Co.).
Electrophysiological Recording
To prepare HAb2 cells for patch clamp experiments, we followed the protocol described above but lifted cells using EGTA/Trypsin (1 mM EGTA; 0.25% trypsin; GIBCO BRL, Gaithersburg, MD) and diluted them in the external solution: 155 mM N-methyl-glucamine glutamate, 5 mM MgCl2, 2 mM Cs-Hepes, pH 7.4 (Tse et al., 1993). Then cells were kept on ice until the experiment (1–3 h). Whole-cell recordings were made on HAb2 fibroblasts with prebound RBC using glass micropipettes (Microcaps, 75 μl; Drummond Sci. Co., Broomall, PA) coated with Sylgard (World Precision Instruments, Sarasota, FL). Patch pipettes with resistances of 1–3 MΩ contained (in mM): 155 Cs-glutamate, 5 MgCl2, 5 BAPTA, 10 Cs-Hepes, pH 7.4 (Tse et al., 1993). Capacitance measurements were made by applying 250-, 500-, and 750-Hz sine waves simultaneously (Melikyan et al., 1995b ). The sine waves were superimposed over a −29 mV holding potential using an in-house software lock-in amplifier (program available upon request). Partially attached cells were patched at 10 ± 2°C. After wholecell configuration (access resistance: 4.1 ± 1.3 MΩ [control cells, n = 10], 4.7 ± 1.9 MΩ [cells in the presence of LPC, n = 15]; capacitances: 20.8 ± 5.3 pF [control cells, n = 10], 12.1 ± 7.3 pF [cells in the presence of LPC, n = 15]), cells were pulled off the glass cover slip and held in suspension while the bath temperature was raised to accelerate fusion pore development upon low pH application (Spruce et al., 1989). We found that this experimental procedure increased the probability of maintaining a stable whole-cell configuration. Once the cell warmed up to 33 ± 2°C, a pipette (same size as patch pipette) containing pH 4.9 medium (same as the external solution but with 20 mM succinic acid replacing 2 mM Hepes) was positioned ∼40 μm from the cell and a pulse of positive pressure (<10 p.s.i. for 5 min) from a pneumatic picopump (model PV830; World Precision Instruments) forced a flow of pH 4.9 solution around the patched cell. When LPC was present in the external medium, it was also added to the pH pipette to avoid washing lysolipids out of the cell by the flow of the low pH medium. The concentration of LPC in the external solution was determined for each experiment by finding the concentration providing maximum inhibition of PKH26 redistribution without significant cell lysis. During electrical recording, we followed PKH26 redistribution using an inverted microscope with an intensified camera (model VE1000SIT; Dage MTI, Inc., Michigan City, IN) connected to a tape recorder (Sony Electronics, Lanham, MD) (Zimmerberg, 1993).
Results
As expected, brief application of mildly acidic pH to HAexpressing cells with bound RBCs caused both lipid mixing (assayed as the transfer of the membrane marker, R18, from RBCs to HA-expressing cells) and mixing of aqueous contents (assayed as the redistribution of the content probe CF from RBC ghosts to HA-expressing cells) (Sarkar et al., 1989; Kemble et al., 1994; Melikyan et al., 1995c ). In contrast, application of low pH to GPI-HA–expressing cells (BHA-PI cells) with bound RBC resulted in hemifusion, i.e., R18 redistribution with no CF transfer (Kemble et al., 1994; Melikyan et al., 1995c ).
Because the net curvature of the lipid monolayer in the hypothetical stalk intermediate has the same sign as that of the inverted HII hexagonal phase, stalk formation should be inhibited by inverted cone–shaped LPC and promoted by cone-shaped OA. Thus, if HA-mediated fusion proceeds via stalk intermediates, LPC should inhibit and OA should promote merger of membrane lipid bilayers. In the following experiments, we have studied low pH–triggered hemifusion and fusion mediated by different HA forms to verify these predictions of the stalk hypothesis of membrane fusion.
Lysolipids Inhibit, and cis-Unsaturated Fatty Acids Promote HA-Mediated Fusion
Altering membrane lipid composition by adding exogenous lipids to the medium affected the extent of low pH– triggered fusion, assayed by spectrofluorometry, in a manner consistent with the predictions of the stalk hypothesis. LPCs, with saturated hydrocarbon chains of lengths varying from 12 to 18 hydrocarbon groups, all decreased the extent of fusion, although at rather different lysolipid concentrations in the medium—the longer the hydrocarbon chain of the lysolipid, the lower the inhibiting concentration (Fig. 2 a). Because the length of the hydrocarbon chains of the lipid affects its partitioning into RBC membrane considerably (Weltzien, 1979), we evaluated the membrane concentrations of exogenously added lipids into cell membranes using R18 dequenching. Incorporation of lipids into RBC membrane labeled by self-quenching concentration of R18 resulted in membrane dye dilution and, consequently, in an increase in the fluorescence. Membrane concentrations of added lipids were estimated using the calibration curve of R18 self-quenching vs. dye concentration.
Figure 2.
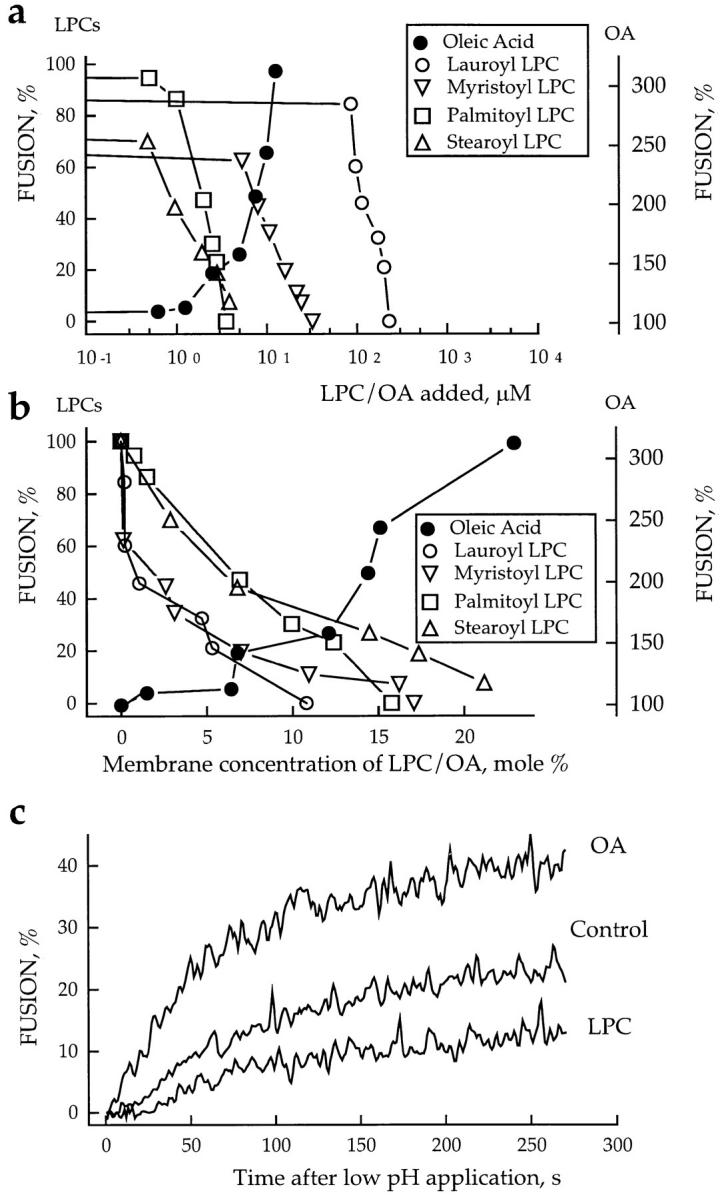
Inhibition and promotion of fusion by lipids. (a and b) The extent of R18-labeled RBC fusion to HAb2 cells in the presence of OA or LPCs with different hydrocarbon chains. In this representative experiment, fusion was triggered by lowering the pH to 4.9. Fusion, assayed by spectrofluorometry, was plotted vs. concentration of exogenous lipids in the medium (a) or in RBC membranes (b). Membrane concentration of exogenous lipids was estimated from R18 dequenching as described in Materials and Methods. (c) Effects of oleoyl LPC (2.5 μM) and OA (10 μM) on the kinetics of membrane mixing. The increase of fluorescence with time after lowering the pH to 5.0 reflects dilution of self-quenched R18 upon lipid mixing between labeled RBCs and unlabeled HAb2 cells upon their fusion.
The dependence of fusion extent on membrane concentrations of different LPCs is presented in Fig. 2 b. While the aqueous concentration of stearoyl LPC required to cause twofold inhibition of fusion was ∼125 times lower than that of lauroyl LPC (∼0.8 vs. ∼100 μM) (Fig. 2 a), the membrane concentration of stearoyl LPC required for equivalent (∼50%) inhibition of fusion was approximately six times higher than that of lauroyl LPC (∼6 vs. ∼1 mol %). Thus, the membrane concentrations of different lysolipids required to cause fusion inhibition vary significantly less than their corresponding aqueous concentrations. Judging from membrane concentrations, shortchained lauroyl and myristoyl LPCs, having more profound inverted cone–shape, appear to be stronger inhibitors than the longer-chained palmitoyl and stearoyl LPCs.
In contrast, cone-shaped molecules OA (Fig. 2 b), arachidonic acid (AA), monoolein, and diolein (not shown) promoted fusion in a dose-dependent manner. In control experiments, no fusion was observed in the presence of 50 μM OA without application of low pH or with application of low pH to cells expressing the uncleaved HA0 (data not shown). The R18 dequenching assay was also used to compare the concentrations at which different lipid promoters enhanced fusion at pH 5.3 by ∼50%: from 29 ± 4% (no lipids added, n = 9) to 43 ± 7% (1.25 μM diolein, n = 3), 45 ± 6% (4.2 μM monoolein, n = 3), and 45 ± 6% (2.5 μM OA, n = 3), respectively. The corresponding membrane concentrations were 2 ± 0.7, 4.5 ± 1.4, and 9.4 ± 3 for diolein, monoolein, and OA, respectively. While the differences between aqueous concentrations of these lipids apparently characterize the differences between their partition into the membrane under our experimental conditions, the membrane concentrations required for fusion promotion correlated with dynamic molecular shape of these lipids judged by their relative effects on inverted hexagonal phase formation. Diolein is a more potent promoter of HII phase than monoolein, which is in turn a better promoter of HII phase than OA (Epand et al., 1988, 1991). These data indicate that the more profound the cone shape of the lipid, the lower is its membrane concentration required for fusion promotion.
Altering membrane lipid composition affected not only the extent of fusion but also the apparent fusion kinetics (Fig. 2 c). Oleoyl LPC increased and OA decreased the delay time between low pH application and the onset of R18 dequenching due to cell–cell fusion. Also, LPC decreased and OA increased the rate of R18 dequenching.
The lipid effects on fusion inferred from the R18 dequenching assay were verified by direct counting of the fused cells with fluorescence microscopy. The spectrofluorometry and fluorescence microscopy assays significantly differ in ways of introducing exogenous lipids and in total numbers of suspended cells (spectrofluorometry) and attached cells (microscopy) used. Thus, quantitative relationships between aqueous and membrane concentrations of the exogenous lipids found with R18 dequenching assay (Fig. 2) cannot be used to directly estimate membrane concentrations of lipids in fluorescence microscopy experiments.
Inhibition and promotion of fusion by LPC and OA, respectively, were seen with fluorescence microscopy in all three molecular species of HA studied (Fig. 3). Lipid inhibitors and promoters of fusion were additive and canceled the effects of each other when added together. LPC and OA added one after another did not affect the incorporation of each other into membranes as evaluated by R18 dequenching measurements (data not shown).
Figure 3.
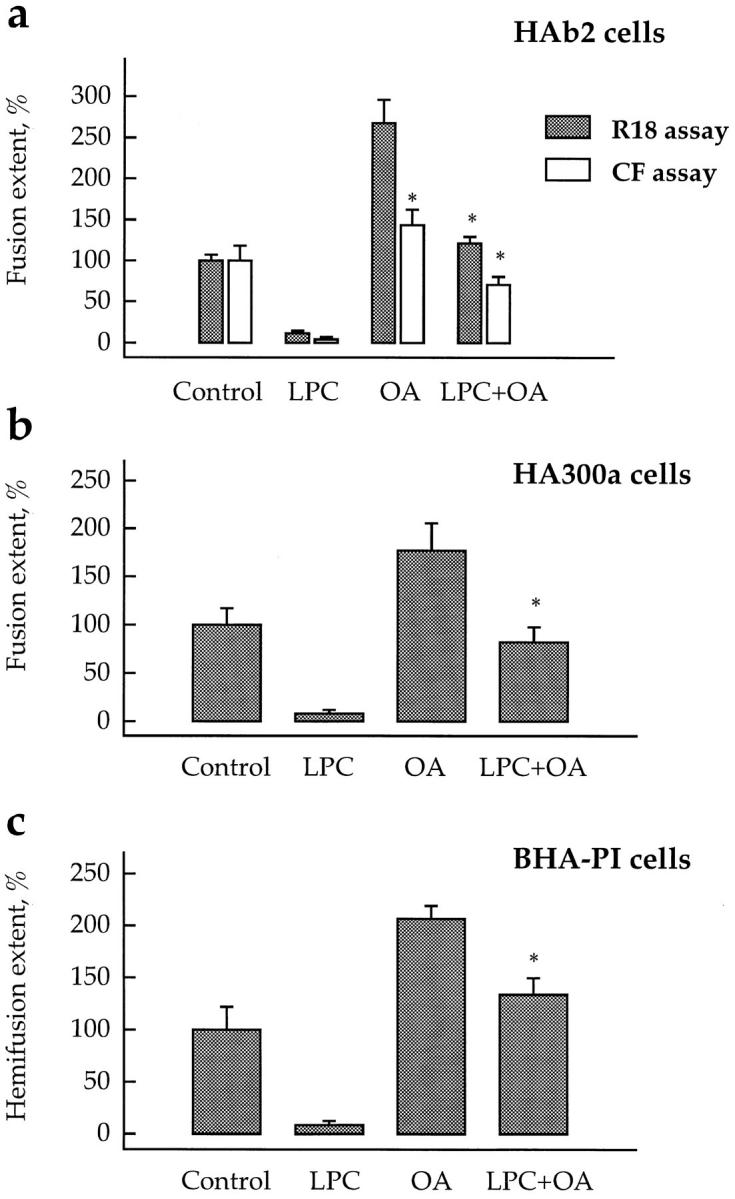
LPC and OA added together compensate the effects of each other on lipid mixing and content mixing mediated by different forms of HA. Fusion of HA-expressing cells (HAb2 [a] and HA300a [b]) and hemifusion of GPI-HA–expressing cells (BHA-PI [c]) with prebound RBCs labeled by either membrane dye R18 or aqueous content marker CF was studied with no added lipids (Control) or in the presence of oleoyl LPC (50 μM), OA (50 μM), or both LPC and OA (same concentrations). Fusion was triggered by a 2-min application of low pH medium (pH 5.2 for HAb2 and HA300a cells, and pH 5.25 for BHA-PI cells). Fusion extents, assayed by fluorescence microscopy as R18 redistribution (hatched bars) or as CF redistribution (open bars), were normalized to those in control experiments: 36.1, 26.8, 13.2, and 17.7% for HAb2, HA300a and BHA-PI (all -R18 assay), and HAb2- CF assay, respectively. Each bar is mean ± SEM, n = 3. Bars which were not statistically different from the corresponding controls are labeled by asterisks. In a, fusion extent for HAb2 (CF assay) in the presence of OA alone was significantly higher than that in the presence of both OA and LPC (P < 0.05).
The more acidic the pH applied to cells was, and thus, the more fusion proteins activated, the less profound were the effects of exogenous lipids (Fig. 4). Fusion inhibition by 19 μM LPC increased from ∼1.2 to ∼20 times, when the pH applied to trigger fusion was increased from 4.9 to 5.3. Similarly, the fusion promotion by 25 μM OA increased from ∼1.8 to ∼8 times when the pH applied was increased from 5.15 to 5.35. These results suggest that activated HA molecules more strongly affect the spontaneous curvature of membrane monolayers (and thus the energetics of the bent stalk intermediates) than nonbilayer lipids such as LPC and OA. This may explain the recent findings (Stegmann, 1993; Alford et al., 1994; Gunther-Ausborn et al., 1995; Shangguan et al., 1996) that, although nonbilayer lipids affect HA-mediated fusion of lipid vesicles or RBCs to influenza virus particles, which presumably have very high surface density of HA molecules at the acidic pH applied, these effects appear to be less profound than those found in the present study.
Figure 4.
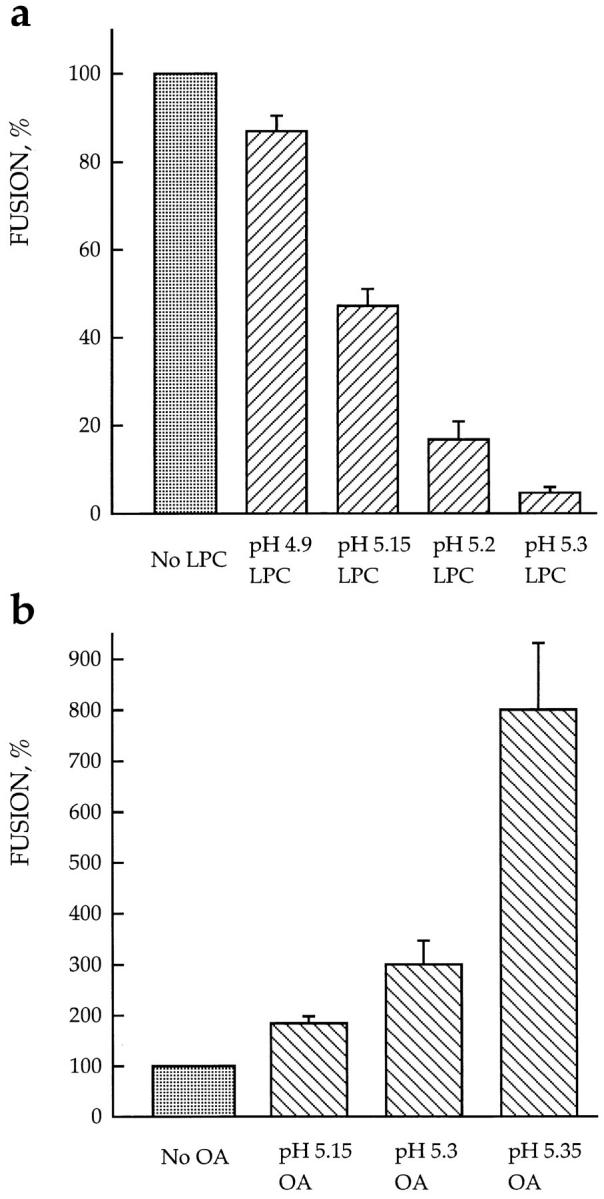
The lower the pH applied to trigger fusion, the less profound is the dependence of fusion on lipids. Fusion of HAb2 cells with prebound RBCs was triggered by a 2-min application of low pH medium in the presence of 19 μM stearoyl LPC (a) or 25 μM OA (b). The extent of fusion assayed by fluorescence microscopy as R18 redistribution was normalized to those in the corresponding control experiments with no lipids added: (a) 98 ± 0.5%, 68 ± 3%, 53 ± 4%, and 36 ± 4.5% for pH 4.9, 5.15, 5.2, and 5.3, respectively; (b) 39 ± 3%, 15 ± 3%, and 3 ± 0.5% for pH 5.15, 5.3, and 5.35, respectively. Each point is mean ± SEM, n = 3. Note that because of day-to-day variability (see Materials and Methods), fusion efficiency observed for the same suboptimal pH values with no exogenous lipids added was somehow different in two independent experiments presented in a and b.
Both inhibition and promotion of cell–cell fusion by exogenous lipids were reversible. For all three lines of HAexpressing cells studied, the fusion competence of membranes treated with short-chain lysolipids (lauroyl LPC and myristoyl LPC) could be completely restored before low pH application by merely washing the cells with PBS (data not shown). Thus, the effects of lipids on fusion cannot be mediated by any irreversible processes such as cell lysis or solubilization of membrane components (e.g., cholesterol depletion; Golan et al., 1988).
Lipids with longer hydrocarbon chains (e.g., stearoyl LPC, oleoyl LPC, OA, or AA) remained in the cell membrane even when cells were washed with fresh medium (Fig. 5 a). To extract these lipids from membranes and to restore the fusion competence, we had to wash the cells with solutions containing fatty acid–free BSA. The low solubility of stearoyl LPC in water allowed us to add LPC exclusively to only one of the fusion partners, HA-expressing cells (HAb2) or RBC. We treated the given type of cells with stearoyl LPC, washed the unbound LPC out, brought treated cells in contact with untreated ones, and applied low pH. LPC treatment of only one of the two bound membranes (either HA-bearing membrane or RBC membrane) inhibited fusion (Fig. 5 b). This experiment allows us to exclude the possibility that LPC was transferred from treated to untreated cells via the aqueous phase. Even if all of the LPC present in the treated cells was released into the medium, the corresponding aqueous concentration of the lysolipid would be two orders of magnitude lower than that required for inhibition of fusion. It is still possible that some LPC had been directly transferred from treated to untreated membrane in the contact region.
Figure 5.

LPC incorporated into either one or both of two fusing membranes reversibly inhibits fusion. Fusion was triggered by a 10-min application of pH 4.9 medium and assayed by fluorescence microscopy as R18 redistribution. Each bar is mean ± SEM, n = 3. (a) Fusion inhibition is caused by membrane-incorporated rather than unbound LPC. □, fusion of HAb2 cells with prebound RBCs, no lipids added; ▒⃞ , fusion in the presence of 36 μM stearoyl LPC; ▪, unbound LPC was removed before low pH application. Cells were incubated with LPC (same concentration as above) for 10 min and then washed out with LPC-free PBS; ▨ , LPC was extracted from membranes by BSA before low pH application. Cells were incubated with LPC as above and then washed out with LPC-free PBS containing BSA. Each point is mean ± SEM, n = 3. (b) LPC inhibits fusion when present either in HA-expressing membrane or in RBC membrane. □, fusion of HAb2 cells with RBCs, no lipids added, ▒⃞ , only HAb2 cells were treated by stearoyl LPC (42 μM); ▪, only RBCs were treated by stearoyl LPC (1 μM/5 × 106 RBCs); ▨ , both RBCs and HAb2 cells were separately treated with stearoyl LPC (same concentrations as above).
Although both the specific fluorescence of membrane dyes and their lateral mobility can be changed by altering membrane composition (Golan et al., 1988; MacDonald, 1990), the effects presented cannot be explained by some lipid-dependent changes in the properties of our probes. Fusion inhibition by LPC was observed with a variety of different assays including R18 dequenching (Fig. 2), fluorescence microscopy with different membrane dyes (R18 and PKH26, Figs. 3 and 8 B, respectively) and water-soluble probes (e.g., CF; Fig. 3 A), and electrophysiological recording (Fig. 6). The additivity of effects of LPC and OA observed in the fluorescence microscopy experiments with R18 (Fig. 3) was also observed with PKH26 (not shown).
Lipid Action Is Upstream of Membrane Merger and Fusion Pore Formation
What stage of the fusion reaction is lipid-sensitive? LPC inhibited and OA promoted mixing of the membrane lipids in fusion mediated by HA (Figs. 2 and 3, a and b) and in hemifusion mediated by GPI-HA (Fig. 3 c). LPC also strongly inhibited the transfer of water-soluble dyes such as CF (Fig. 3 a) and ethidium bromide (not shown) from RBC ghosts or NBD-taurine (data not shown) from RBCs into the cytoplasm of HA-expressing cells (HAb2 and HA300a cells) (Fig. 3 a), indicating that LPC blocked the mixing of the aqueous contents of fusing cells. These results proved that lipids affect fusion before membrane merger, including hemifusion, and suggested that the lipidsensitive stage precedes fusion pore formation. Because the diffusion of some water-soluble dyes is restricted in the smallest fusion pores (Tse et al., 1993; Zimmerberg et al., 1994), we have applied the most sensitive assay for fusion pore formation, electrophysiological recording, which allows one to detect a pore as soon as ions can pass through it.
Patch clamp studies of HAb2 cell fusion to RBCs confirmed that LPC inhibits fusion pore formation at concentrations that inhibit membrane dye redistribution in the same pair of cells. The HAb2 cell with RBCs, labeled by fluorescent membrane marker PKH26, was patch-clamped in the whole-cell configuration in the absence or in the presence of lauroyl LPC in the external medium, low pH was applied, and both the capacitance of the membrane of HAb2 cell and the PKH26 transfer into this membrane were measured using simultaneous electrical and video recording. In 10 out of 12 control experiments with no LPC added, we observed changes in capacitance corresponding to the development of a fusion pore (Fig. 6) and accompanied by PKH26 transfer. The capacitance change was 1.2 ± 0.1 pF (n = 7), which is in agreement with the values of RBC capacitance reported before (Tse et al., 1993; Zimmerberg et al., 1994). In 10 out of 15 experiments performed in the presence of LPC, we observed neither membrane dye redistribution nor formation of fusion pores larger than our resolution limit of 100 pS. In five remaining experiments, we observed both dye redistribution and pore formation. Thus, when LPC inhibited fusion, it did so upstream of both membrane merger and fusion pore formation.
Lipids Affect Fusion Downstream of HA's Conformational Change
The lipid-sensitive stage of fusion was downstream of membrane binding. Both inhibition of fusion by oleoyl LPC and promotion of fusion by OA were accomplished without concomitant changes in the RBC's adhesion to HAb2 and HA300a cells, assayed by direct counting of the bound R18-labeled RBCs (data not shown).
The extent of fusion depended on the duration of the low pH pulse (Fig. 7). For short pH pulses, the longer the pulse, the higher the fraction of RBC-HAb2 contacts wherein cells were committed to fuse by completion of the low pH–dependent stages, and thus the higher the fusion extent. The same dependence of fusion extent on low pH pulse duration was observed when the low pH medium was supplemented by lauroyl LPC at concentrations completely inhibiting fusion. The LPC-free, neutral pH medium (used to terminate the pulse) washed LPC out and allowed fusion of cells previously committed to fuse in the presence of LPC. The results of this experiment indicated that LPC had not affected the effectiveness of the low pH– dependent conformational changes of HA required for fusion.
Figure 7.

LPC does not affect the low pH–dependent conformational changes of HA required for fusion. HAb2 cells with prebound R18-labeled RBCs were incubated in low pH medium (pH 5.2), LPC-free, or supplemented with 150 μM lauroyl LPC for different times. At the end of the low pH pulse, the low pH medium was replaced by LPC-free PBS (pH 7.4) to both terminate the fusion-triggering pulse and to wash out LPC. Fusion was assayed by fluorescence microscopy as R18 redistribution. Each point is mean ± SEM, n = 3. In the experiments when the PBS used to terminate the low pH pulse was supplemented with 150 μM lauroyl LPC, and thus LPC was not washed out, the extent of fusion was 0.2 ± 0.16% (n = 3).
Additional indications that the lipid-sensitive stage was downstream of the triggering step came from the following experiments. If cells were exposed to a 2-min pulse of low pH and then returned to normal pH, fusion occurred (Fig. 8 a). In the presence of LPC, there was no fusion. However, if LPC was removed up to 30 min after giving this short pulse of low pH, almost the full extent of fusion was observed. There was a reduction in the extent of fusion seen upon LPC removal if cells were kept in the LPCarrested fusion stage for longer times (Fig. 8 b). This decrease of the extent of fusion may be caused by slow inactivation or internalization of activated HA. Interestingly, compensating for LPC by adding OA, instead of washing out LPC, also led to complete fusion. Thus, adding OA to LPC-arrested cells removed the block that keeps membranes from merger (Fig. 8 c).
Both low pH application and membrane contact were required concomitantly to reach the LPC-arrested fusion stage. We first applied a low pH pulse to HAb2 cells for 1 min and then 10 min later, at neutral pH, added RBCs to establish cell–cell contact. No fusion was observed in these experiments (Fig. 8 a). Likewise, no fusion was observed when this experiment was conducted in the presence of LPC, even though it was removed after the establishment of cell–cell contact. This eliminated the possibility that lysolipid stabilized an activated state in the absence of membrane contact.
Additional evidence that LPC arrests fusion at a stage after the low pH–triggered conformational change of HA came from experiments with neuraminidase. Neuraminidase treatment of HAb2 or HA300a cells with prebound RBC inhibits both HA-mediated fusion (Fig. 9 a) and RBCs binding to HA-expressing cells (Fig. 9 b), presumably by cleaving the sialic acids required for HA1-receptor binding (White et al., 1983). In contrast, the LPC-arrested stage was insensitive to neuraminidase. In these experiments, LPC was present during a 2-min application of low pH. Then cells were incubated with neuraminidase at neutral pH, and LPC was withdrawn. Both fusion and binding were unaffected by neuraminidase treatment.
Figure 9.
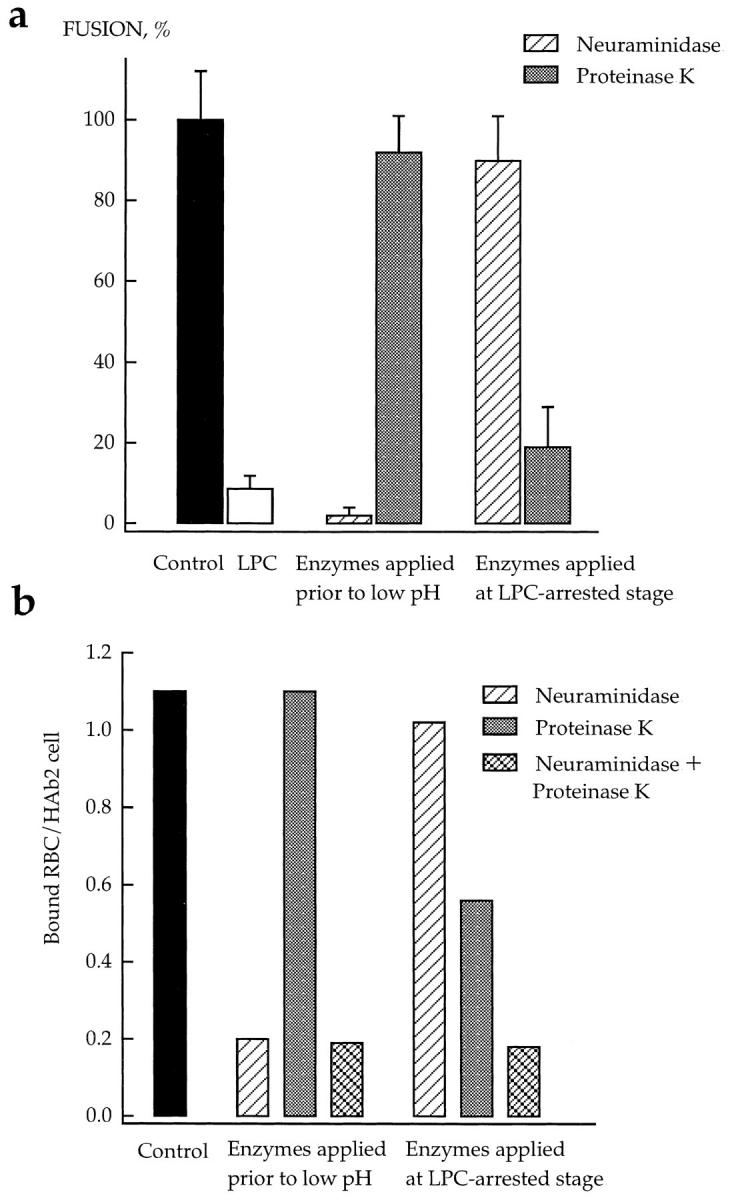
Effects of neuraminidase and proteinase K on the LPCarrested stage of HA-mediated fusion. Fusion of HAb2 cells with prebound R18-labeled RBCs was triggered by a 3-min pH 4.9 pulse applied in the presence of 50 μM lauroyl LPC. After a 20min incubation of cells at neutral pH in LPC-containing PBS, lysolipid was removed by washing cells with LPC-free PBS. Cells were treated by neuraminidase or proteinase K before low pH application or at the LPC-arrested fusion stage in the time interval between the end of low pH and removal of LPC. (a) Fusion. The extent of fusion after removal of LPC was measured as R18 redistribution using fluorescence microscopy, and normalized to those in the control experiments (▪, neither LPC nor enzymes applied). □, the extent of fusion observed at t = 2 hours in the experiments when LPC was not removed. ▨ and ▒⃞ , cells were treated with neuraminidase or proteinase K, respectively. Each point is mean ± SEM, n = 3. (b) Binding. In this representative experiment, the effects of neuraminidase (▨ ) or proteinase K (▒⃞ ), or the combination of both enzymes (▩ ) on binding of R18labeled RBCs to HAb2 cells before low pH application and during the LPC-arrested fusion stage were evaluated with fluorescence microscopy by counting RBCs bound to ∼100 HAb2 cells. ▪, An average number of RBCs bound per one HAb2 cell in the control experiments with neither LPC nor enzymes applied.
The insensitivity of the LPC-arrested stage to neuraminidase treatment suggests that LPC blocks fusion at a “committed state” where the fusion peptides of HA are already inserted into the target membrane. This means that the HA1–RBC sialic acid complex is not required for completion of fusion from the LPC-arrested stage and that the HA-receptor binding energy does not contribute to the completion of fusion (see also van Meer et al., 1985; Schoch et al., 1992).
The same experiments with proteinase K gave opposite results. Only HA that has been exposed to low pH is cleaved by proteinase K (Doms et al., 1985), so the finding of normal fusion and binding when cells were treated by proteinase K before low pH application was expected. However, the LPC-arrested stage was found to be sensitive to proteinase K, as removal of LPC after both a low pH pulse and proteinase K treatment resulted in a considerably reduced extent of fusion (Fig. 9 a). RBCs binding to HAb2 cells at LPC-arrested stage was also susceptible to proteinase K applied alone and, in particular, in combination with neuraminidase (Fig. 9 b), indicating that this binding is mediated by insertion of HA fusion peptides into RBC membrane and by HA1 interaction with RBC's sialic acid.
These data confirm that LPC blocks fusion at a stage subsequent to low pH–induced changes in HA conformation and control for the neuraminidase experiments by showing that the relevant HA molecules at a fusion site are apparently accessible to external macromolecules. We also conclude that activated fusion proteins are still needed for the fusion reaction when the system is released from the LPC-arrested stage.
Discussion
The existing hypotheses on the mechanisms of membrane fusion differ fundamentally in their predictions on whether a lipidic connection precedes or follows the establishment of aqueous continuity (fusion pore formation) (Monck and Fernandez, 1992; Zimmerberg et al., 1993; Tse et al., 1993; Lindau and Almers, 1995). We have now found that membrane lipids modulate cell–cell fusion mediated by influenza HA at a stage preceding both the merger of membrane lipid bilayers and fusion pore formation but subsequent to the low pH–induced conformational change in HA. Hemifusion mediated by GPI-anchored HA had the same dependence on membrane lipids as that observed for membrane merger in fusion mediated by wild-type HA (Fig. 3). These data are consistent with the hypothesis that both extended hemifusion and membrane merger in complete fusion involve similar membrane rearrangements such as local hemifusion (Zimmerberg et al., 1993; Kemble et al, 1994; Melikyan et al., 1995c ).
Our data demonstrate that lipids are involved in the very early fusion intermediates, which is inconsistent with models where the first stage of fusion is the lipid-independent opening of a proteinaceous fusion pore. The data also argue against the possibility that while lipids are not directly involved into the initial proteinaceous fusion pore, they affect the formation of this pore by altering the function of membrane proteins, such as HA. For instance, the composition of HA-expressing membranes can influence the association of membrane proteins into some multimolecular complex required to drive membrane merger (Gutman et al., 1993; Danieli et al., 1996). Alternatively, the lipid composition of the target membrane can affect the orientation of the HA fusion peptide inserted into this membrane (Martin et al., 1993). Our finding that LPC inhibited fusion regardless of the membrane in which it was located (HA-expressing membrane or target membrane, Fig. 5 b) argues against both of these hypotheses.
Nonbilayer lipids may modulate fusion by direct interaction with HA. An increase in the hydrophobicity of the HA fusion peptide upon HA activation manifests itself by a significant increase in the binding of nonionic detergent and lipid to the extramembraneous protein domain (Doms et al., 1985). Perhaps blocking the incorporation of the HA fusion peptide into the target membrane by direct binding of monomeric or micellar LPC to the fusion peptide may account for LPC inhibition of influenza virus–liposome fusion (Gunther-Ausborn et al., 1995; Shangguan et al., 1996). It appears, however, that this mechanism is not valid here. The LPC-arrested stage was already insensitive to neuraminidase, indicating that LPC inhibits fusion downstream of fusion peptide insertion into the target membrane (see below). The effects of lipids on fusion were dependent on membrane-incorporated lipids rather than on monomeric or micellar lipids. In addition, the interaction of the fusion peptide with amphiphilic compounds appears to be fairly nonspecific (Doms et al., 1985), but cone-shaped lipids promoted fusion instead of inhibiting it. Finally, the similarity of the lipid dependencies between HA-mediated fusion and other examples of biological fusion and even fusion of purely lipid bilayers (see below) argues against the hypothesis that lipids inhibit or promote formation of a proteinaceous fusion pore via direct lipid–HA interactions.
In contrast, our results corroborate the hypothesis that HA-mediated membrane fusion includes an early stage of local hemifusion, when a specific lipid-involving intermediate, the fusion stalk, is formed (Kozlov and Markin, 1983; Chernomordik et al., 1987; Siegel, 1993a ,b; Zimmerberg et al., 1993). The elastic energy of a strongly bent monolayer in the stalk depends on the dynamic molecular shape of the lipids in contacting monolayers. Lipid effects on fusion found in the present study are consistent with the specific predictions of this model.
Fusion Is Affected by Membrane-incorporated Lipids in Correlation with Their Dynamic Molecular Shape
It is the membrane-incorporated lipid that affects fusion. Stearoyl LPC inhibited fusion, and OA promoted fusion, even when cells treated by LPC or OA were thoroughly washed by fresh medium to remove unbound lipids (monomers, micelles, and mixed micelles) before low pH application (Fig. 5 a). Our finding that inhibition by LPC can be reversed by adding OA (Figs. 3 and 8), and the lack of any direct correlation between the critical micelle concentration, CMC, of different lipids and their effects on fusion also argue against the involvement of monomers and micelles. Lauroyl and stearoyl LPCs inhibited fusion below and above their CMCs of ∼430 and 0.4 μM (Kramp et al., 1984), respectively. While the CMC of palmitoyl LPC is about 10 times higher than that of stearoyl LPC, these two lysolipids incorporated into biological membranes and inhibited fusion in the same range of concentrations (Fig. 2, a and b).
Asymmetric intercalation of lysolipids can affect fusion by causing compression and buckling of membrane monolayers. However, while different LPCs induce echinocytosis at the same membrane concentrations (Fujii and Tamura, 1984), membrane concentrations of lauroyl LPC and stearoyl LPC required for inhibition of fusion differed significantly (Fig. 2 b).
The net curvature of a strongly bent monolayer in the stalk has the same sign as in inverted HII phase (Chernomordik et al., 1995b ), and thus stalk formation should be promoted by the inclusion of cone-shaped lipids into contacting membrane monolayers. Cone-shaped lipids such as OA, AA, monoolein, diolein (this work), and unsaturated phosphatidylethanolamine (White et al., 1982; van Meer et al., 1985; Stegmann, 1993; Alford et al., 1994) did promote fusion. Moreover, the fusion-promoting activity of OA, monoolein, and diolein correlated with their dynamic molecular shape: the more profound the cone shape of the lipid judging by its effects on the inverted HII phase formation, the lower the membrane concentration of the lipid required for fusion promotion.
In contrast, inverted cone–shaped LPCs inhibited fusion (this work and Gunther-Ausborn et al., 1995; Shangguan et al., 1996). The fusion-inhibiting activity of different LPCs was higher for shorter hydrocarbon tails (lauroyl and myristoyl) and, therefore, for compounds having more profound inverted cone shapes (Fig. 2 b). Lipids of complementary dynamic shapes (LPC and OA) were found to cancel the effects of each other with respect to membrane fusion (Fig. 3), in agreement with the known additivity of lipid effects on the spontaneous curvature of lipid monolayer (Madden and Cullis, 1982).
Lipids Affect Fusion at a Stage Downstream of HA Activation
The stalk model suggests that nonbilayer lipids should affect the initial stage of actual membrane merger. By adding LPC and altering the membrane lipid composition to be nonpermissive for fusion, we have isolated a lipid-sensitive stage of fusion. This stage is upstream of lipid mixing and fusion pore formation, but downstream of a low pH– induced conformational change in HA. The presence of LPC did not affect the low pH–dependent stages of HA activation (Fig. 7). Release from the LPC-arrested fusion stage occurred at neutral pH upon altering membrane lipid composition either by washing LPC out or by adding OA (Fig. 8), so the inhibited stage was after the event in which pH triggered fusion. Establishment of this “activated” stage was accompanied by drastic changes in the system's sensitivity to neuraminidase and proteinase K (Fig. 9). The initial RBC binding to HA-expressing cells is mediated by HA1 binding to sialic acids and is sensitive to neuraminidase (White et al., 1983). This binding mechanism was apparently superseded at an LPC-arrested stage by mechanisms based on the low pH–induced insertion of the HA fusion peptide into RBC membranes (Stegmann et al., 1991; Tsurudome et al., 1992), and thus fusion was insensitive to neuraminidase. In contrast, both membrane binding and fusion upon release from the LPC-arrested stage were inhibited by proteinase K treatment (Fig. 9), indicating that activated HA molecules are still required at this stage to keep membranes together and to drive fusion to completion. These data indicate that nonbilayer lipids affect the merger of membrane lipids rather than any other stage of the fusion process.
HA-mediated Fusion and Many Other Fusion Reactions Can Involve Formation of Common, Stalk Intermediates
Lysolipids added between fusing membranes inhibit and cis-unsaturated fatty acids promote not only diverse biological fusion reactions (this paper and Creutz, 1981; Glick and Rothman, 1987; Chernomordik et al., 1993; Paiement et al., 1994; Yeagle et al., 1994; Chernomordik et al., 1995c ; Gunther-Ausborn et al., 1995; but see Nagao et al., 1995; Coorssen, 1996), but also fusion of purely lipid bilayers (for review see Chernomordik et al., 1995b ). Importantly, LPC inhibits HA-mediated fusion at membrane concentrations similar to those found to inhibit syncytia formation mediated by the Sendai virus F protein (Yeagle et al., 1994) and baculovirus gp64 (Chernomordik et al., 1995c ), as well as for microsome-microsome fusion (Chernomordik et al., 1993) and vesicle-planar bilayer fusion (Chernomordik et al., 1995a). We suggest that fusion mediated by HA and other proteins and fusion of purely lipid bilayers proceed via a common lipid-involving intermediate—a stalk structure, producing local and transient hemifusion of membranes. Nonbilayer lipids may influence the energy required for stalk formation by altering some properties of membrane lipid bilayers and, in particular, the propensity of lipid monolayers to bend into nonbilayer fusion intermediates (Chernomordik et al., 1995b ), the hydrophobicity of membrane surfaces (Ohki, 1988), and the repulsive pressures and the distances between the contacting monolayers (Rand and Parsegian, 1989; McIntosh et al., 1995).
Hypothetical stalk intermediates, if formed, can evolve in different directions. The connection between membranes can succumb, returning the system to its original state of two separate membranes. Alternatively, local hemifusion can rapidly develop into complete fusion via fusion pore formation. Finally, a local hemifusion intermediate can be stabilized or expand to form an extended hemifusion structure. Such an extended and long-lasting hemifusion structure has been reported for cell–cell fusion mediated by GPI-anchored HA (Melikyan et al., 1995c ). The wall of an initial fusion pore can be formed at least partially by lipids (Monck and Fernandez, 1992; Zimmerberg et al., 1991, 1993). In this case, as in the case of a stalk, the energy of the pore will be dependent on lipid composition. However fusion pore formation involves the bending of different membrane monolayers, and the direction of lipid monolayer bending in a pore and in a stalk will be opposite (Chernomordik et al., 1995b ). Thus, to study the lipid effects on fusion, one needs to know which lipids are added to which membrane monolayers and how fast the transmembrane redistribution of lipids is. For instance, OA quickly redistributes between outer and inner membrane monolayers (Broring et al., 1989). This may explain why in Fig. 3 a the enhancement of fusion by OA was found to be more profound when fusion was measured with lipid-mixing assay, which does not distinguish between hemifusion and complete fusion, than with contentmixing assay, which characterizes only complete fusion. Cone-shaped OA in contacting membrane monolayers is expected to promote stalk formation and hemifusion. The same lipid in the inner membrane monolayers should inhibit fusion pore development and thus can partially compensate the hemifusion and fusion promotion by OA present in outer monolayers.
To conclude, we have identified an early stage of HAmediated fusion that is dependent on membrane lipids in agreement with the predictions of the stalk hypothesis of membrane fusion. This hypothesis suggests that fusion involves local bending deformations of lipid monolayers (Fig. 1 e). The interaction of activated HA molecules with membrane lipid bilayers may dramatically decrease the energy of stalk intermediates and, thus, allow spontaneous fusion of membrane lipid bilayers by altering the geometry of fusion sites and/or the spontaneous curvature of membrane monolayers. The domineering role of fusion proteins in membrane bilayer rearrangement is substantiated by our findings that the more HA molecules activated, the less profound is the dependence of fusion on lipids and that the presence of activated HA was imperative for the completion of fusion downstream of the lipid-dependent fusion stage. However, since the stalk would be formed by bent lipid monolayers, nonbilayer lipids that aid or oppose the monolayer curvature dominating in the stalk, respectively, promoted and inhibited fusion when added to contacting monolayers of fusing membranes. Altering the lipid composition of membranes to be nonpermissive for fusion allowed us to isolate the long-living activated fusion state in which the fusion proteins are frozen in a fusioncompetent conformation. The fact that this state can last for hours suggests that our approach may help to find out whether the low pH conformation of HA determined by x-ray crystallography is the fusogenic conformation.
Acknowledgments
We are grateful to Drs. M. Kozlov, G. Melikyan, and J. White for valuable discussions. Special gratitude is due to Dr. J. White for providing us with HA300a and BHA-PI cells. We thank Drs. J. Coorssen and T. Whalley for critical reading of the manuscript.
Footnotes
Address all correspondence to Leonid V. Chernomordik, The Laboratory of Cellular and Molecular Biophysics, National Institute of Child Health and Human Development, National Institutes of Health, Bldg. 10, Room 10D04, 10 Center Drive, Bethesda, MD 20892-1885. Tel.: (301) 594-1128. Fax: (301) 594-0813. E-mail: lchern@helix.nih.gov
Abbreviations used in this paper: AA, arachidonic acid; CF, 6-carboxyfluorescein; NBD-taurine, N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)taurine; GPI, glycosylphosphatidylinositol; GPI–HA, HA ectodomain linked to GPI; HA, influenza virus hemagglutinin; LPC, lysophosphatidylcholine; OA, oleic acid; RBC, red blood cells.
References
- Alford D, Ellens H, Bentz J. Fusion of influenza virus with sialic acid-bearing target membranes. Biochemistry. 1994;33:1977–1987. doi: 10.1021/bi00174a002. [DOI] [PubMed] [Google Scholar]
- Bhamidipati SP, Hamilton JA. Interactions of lyso 1-palmitoylphosphatidylcholine with phospholipids: a 13C and 31P NMR study. Biochemistry. 1995;34:5666–5677. doi: 10.1021/bi00016a043. [DOI] [PubMed] [Google Scholar]
- Broring K, Haest CW, Deuticke B. Translocation of oleic acid across the erythrocyte membrane. Evidence for a fast process. Biochim Biophys Acta. 1989;986:321–331. doi: 10.1016/0005-2736(89)90484-7. [DOI] [PubMed] [Google Scholar]
- Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature (Lond) 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- Carr CM, Kim PS. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell. 1993;73:823–832. doi: 10.1016/0092-8674(93)90260-w. [DOI] [PubMed] [Google Scholar]
- Chernomordik LV, Melikyan GB, Chizmadzhev YA. Biomembrane fusion: a new concept derived from model studies using two interacting planar lipid bilayers. Biochim Biophys Acta. 1987;906:309–352. doi: 10.1016/0304-4157(87)90016-5. [DOI] [PubMed] [Google Scholar]
- Chernomordik LV, Vogel SS, Sokoloff A, Onaran HO, Leikina EA, Zimmerberg J. Lysolipids reversibly inhibit Ca(2+)-, GTP- and pHdependent fusion of biological membranes. FEBS Lett. 1993;318:71–76. doi: 10.1016/0014-5793(93)81330-3. [DOI] [PubMed] [Google Scholar]
- Chernomordik L, Chanturiya A, Green J, Zimmerberg J. The hemifusion intermediate and its conversion to complete fusion: regulation by membrane composition. Biophys J. 1995a;69:922–929. doi: 10.1016/S0006-3495(95)79966-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik L, Kozlov M, Zimmerberg J. Lipids in biological membrane fusion. J Membr Biol. 1995b;146:1–14. doi: 10.1007/BF00232676. [DOI] [PubMed] [Google Scholar]
- Chernomordik L, Leikina E, Zimmerberg J. Control of baculovirus gp64-induced syncytia formation by membrane lipid composition. J Virol. 1995c;69:3049–3058. doi: 10.1128/jvi.69.5.3049-3058.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coorssen JR. Phospholipase activation and secretion: evidence that PLA2, PLC, and PLD are not essential to exocytosis. Am J Physiol. 1996;270:C1153–C1163. doi: 10.1152/ajpcell.1996.270.4.C1153. [DOI] [PubMed] [Google Scholar]
- Creutz CE. cis-Unsaturated fatty acids induce the fusion of chromaffin granules aggregated by synexin. J Cell Biol. 1981;91:247–256. doi: 10.1083/jcb.91.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danieli T, Pelletier SL, Henis YI, White JM. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J Cell Biol. 1996;133:559–569. doi: 10.1083/jcb.133.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RS, Downie JC, Hay AJ, Knossow M, Skehel JJ, Wang ML, Wiley DC. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell. 1985;40:431–439. doi: 10.1016/0092-8674(85)90157-6. [DOI] [PubMed] [Google Scholar]
- Dimitrov DS, Blumenthal R. Photoinactivation and kinetics of membrane fusion mediated by the human immunodeficiency virus type 1 envelope glycoprotein. J Virol. 1994;68:1956–1961. doi: 10.1128/jvi.68.3.1956-1961.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doms RW, Helenius A, White J. Membrane fusion activity of the influenza virus hemagglutinin. The low pH-induced conformational change. J Biol Chem. 1985;260:2973–2981. [PubMed] [Google Scholar]
- Doxsey SJ, Sambrook J, Helenius A, White J. An efficient method for introducing macromolecules into living cells. J Cell Biol. 1985;101:19–27. doi: 10.1083/jcb.101.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellens H, Doxsey S, Glenn JS, White JM. Delivery of macromolecules into cells expressing a viral membrane fusion protein. Methods Cell Biol. 1989;31:155–178. doi: 10.1016/s0091-679x(08)61607-6. [DOI] [PubMed] [Google Scholar]
- Epand RM. Diacylglycerols, lysolecithin, or hydrocarbons markedly alter the bilayer to hexagonal phase transition temperature of phosphatidylethanolamines. Biochemistry. 1985;24:7092–7095. doi: 10.1021/bi00346a011. [DOI] [PubMed] [Google Scholar]
- Epand RM, Epand RF, Lancaster CR. Modulation of the bilayer to hexagonal phase transition of phosphatidylethanolamines by acylglycerols. Biochim Biophys Acta. 1988;945:161–166. doi: 10.1016/0005-2736(88)90478-6. [DOI] [PubMed] [Google Scholar]
- Epand RM, Epand RF, Ahmed N, Chen R. Promotion of hexagonal phase formation and lipid mixing by fatty acids with varying degrees of unsaturation. Chem Phys Lipids. 1991;57:75–80. doi: 10.1016/0009-3084(91)90051-c. [DOI] [PubMed] [Google Scholar]
- Fujii T, Tamura A. Shape change of human erythrocytes induced by phosphatidylcholine and lysophosphatidylcholine species with various acyl chain lengths. Cell Biochem Funct. 1984;2:171–176. doi: 10.1002/cbf.290020311. [DOI] [PubMed] [Google Scholar]
- Gething MJ, Doms RW, York D, White J. Studies on the mechanism of membrane fusion: site-specific mutagenesis of the hemagglutinin of influenza virus. J Cell Biol. 1986;102:11–23. doi: 10.1083/jcb.102.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick BS, Rothman JE. Possible role for fatty acyl-coenzyme A in intracellular protein transport. Nature (Lond) 1987;326:309–312. doi: 10.1038/326309a0. [DOI] [PubMed] [Google Scholar]
- Golan DE, Furlong ST, Brown CS, Caulfield JP. Monopalmitoylphosphatidylcholine incorporation into human erythrocyte ghost membranes causes protein and lipid immobilization and cholesterol depletion. Biochemistry. 1988;27:2661–2667. doi: 10.1021/bi00408a005. [DOI] [PubMed] [Google Scholar]
- Gunther-Ausborn S, Praetor A, Stegmann T. Inhibition of influenza-induced membrane fusion by lysophosphatidylcholine. J Biol Chem. 1995;270:29279–29285. doi: 10.1074/jbc.270.49.29279. [DOI] [PubMed] [Google Scholar]
- Gutman O, Danieli T, White JM, Henis YI. Effects of exposure to low pH on the lateral mobility of influenza hemagglutinin expressed at the cell surface: correlation between mobility inhibition and inactivation. Biochemistry. 1993;32:101–106. doi: 10.1021/bi00052a014. [DOI] [PubMed] [Google Scholar]
- Hoekstra D, de Boer T, Klappe K, Wilschut J. Fluorescence method for measuring the kinetics of fusion between biological membranes. Biochemistry. 1984;23:5675–5681. doi: 10.1021/bi00319a002. [DOI] [PubMed] [Google Scholar]
- Kemble GW, Henis YI, White JM. GPI- and transmembrane- anchored influenza hemagglutinin differ in structure and receptor binding activity. J Cell Biol. 1993;122:1253–1265. doi: 10.1083/jcb.122.6.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemble GW, Danieli T, White JM. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell. 1994;76:383–391. doi: 10.1016/0092-8674(94)90344-1. [DOI] [PubMed] [Google Scholar]
- Kozlov MM, Markin VS. Possible mechanism of membrane fusion. Biofizika. 1983;28:255–261. [PubMed] [Google Scholar]
- Kramp W, Pieroni G, Pinckard RN, Hanahan DJ. Observations on the critical micellar concentration of 1-O-alkyl-2-acetyl-sn-glycero-3phosphocholine and a series of its homologs and analogs. Chem Phys Lipids. 1984;35:49–62. doi: 10.1016/0009-3084(84)90032-x. [DOI] [PubMed] [Google Scholar]
- Leenhouts JM, De Kruijff B. Membrane potential-driven translocation of a lipid-conjugated rhodamine. Biochim Biophys Acta. 1995;1237:121–126. doi: 10.1016/0005-2736(95)00093-i. [DOI] [PubMed] [Google Scholar]
- Lindau M, Almers W. Structure and function of fusion pores in exocytosis and ectoplasmic membrane fusion. Curr Opin Cell Biol. 1995;7:509–517. doi: 10.1016/0955-0674(95)80007-7. [DOI] [PubMed] [Google Scholar]
- MacDonald RL. Characteristics of self-quenching of the fluorescence of lipid-conjugated rhodamine in membranes. J Biol Chem. 1990;265:13533–13539. [PubMed] [Google Scholar]
- Madden TD, Cullis PR. Stabilization of bilayer structure for unsaturated phosphatidylethanolamines by detergents. Biochim Biophys Acta. 1982;684:149–153. doi: 10.1016/0005-2736(82)90061-x. [DOI] [PubMed] [Google Scholar]
- Martin I, Dubois MC, Saermark T, Epand RM, Ruysschaert JM. Lysophosphatidylcholine mediates the mode of insertion of the NH2-terminal SIV fusion peptide into the lipid bilayer. FEBS Lett. 1993;333:325–330. doi: 10.1016/0014-5793(93)80680-s. [DOI] [PubMed] [Google Scholar]
- McIntosh TJ, Advani S, Burton RE, Zhelev DV, Needham D, Simon SA. Experimental tests for protrusion and undulation pressures in phospholipid bilayers. Biochemistry. 1995;34:8520–8532. doi: 10.1021/bi00027a002. [DOI] [PubMed] [Google Scholar]
- Melikyan GB, Niles WD, Cohen FS. The fusion kinetics of influenza hemagglutinin expressing cells to planar bilayer membranes is affected by HA density and host cell surface. J Gen Physiol. 1995a;106:783–802. doi: 10.1085/jgp.106.5.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan GB, Niles WD, Ratinov VA, Karhanek M, Zimmerberg J, Cohen FS. Comparison of transient and successful fusion pores connecting influenza hemagglutinin expressing cells to planar membranes. J Gen Physiol. 1995b;106:803–819. doi: 10.1085/jgp.106.5.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan GB, White JM, Cohen FS. GPI-anchored influenza hemagglutinin induces hemifusion to both red blood cell and planar bilayer membranes. J Cell Biol. 1995c;131:679–691. doi: 10.1083/jcb.131.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan GB, Deriy BN, Ok DC, Cohen FS. Voltage-dependent translocation of R18 and Dil across lipid bilayers leads to fluorescence changes. Biophys J. 1996;71:2680–2691. doi: 10.1016/S0006-3495(96)79459-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohandas N, Wyatt J, Mel SF, Rossi ME, Shohet SB. Lipid translocation across the human erythrocyte membrane. Regulatory factors. J Biol Chem. 1982;257:6537–6543. [PubMed] [Google Scholar]
- Monck JR, Fernandez JM. The exocytotic fusion pore. J Cell Biol. 1992;119:1395–1404. doi: 10.1083/jcb.119.6.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SJ, Zimmerberg J, Sarkar DP, Blumenthal R. Kinetics of cell fusion mediated by viral spike glycoproteins. Methods Enzymol. 1993;221:42–58. doi: 10.1016/0076-6879(93)21006-t. [DOI] [PubMed] [Google Scholar]
- Nagao T, Kubo T, Fujimoto R, Nishio H, Takeuchi T, Hata F. Ca(2+)-independent fusion of secretory granules with phospholipase A2treated plasma membranes in vitro. Biochem J. 1995;307:563–569. doi: 10.1042/bj3070563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohki, S. 1988. Surface tension, hydration energy and membrane fusion. In Molecular Mechanisms of Membrane Fusion. S. Ohki, D. Doyle, T.D. Flanagan, S.W. Hui, and M.E., editors. Plenum Press, New York. 123–139.
- Paiement J, Lavoie C, Gavino GR, Gavino VC. Modulation of GTP-dependent fusion by linoleic and arachidonic acid in derivatives of rough endoplasmic reticulum from rat liver. Biochim Biophys Acta. 1994;1190:199–212. doi: 10.1016/0005-2736(94)90075-2. [DOI] [PubMed] [Google Scholar]
- Rand RP, Parsegian VA. Hydration forces between phospholipid bilayers. Biochim Biophys Acta. 1989;988:351–376. [Google Scholar]
- Sarkar DP, Morris SJ, Eidelman O, Zimmerberg J, Blumenthal R. Initial stages of influenza hemagglutinin–induced cell fusion monitored simultaneously by two fluorescent events: cytoplasmic continuity and lipid mixing. J Cell Biol. 1989;109:113–122. doi: 10.1083/jcb.109.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch C, Blumenthal R. Role of the fusion peptide sequence in initial stages of influenza hemagglutinin-induced cell fusion. J Biol Chem. 1993;268:9267–9274. [PubMed] [Google Scholar]
- Schoch C, Blumenthal R, Clague MJ. A long-lived state for influenza virus-erythrocyte complexes committed to fusion at neutral pH. FEBS Lett. 1992;311:221–225. doi: 10.1016/0014-5793(92)81107-w. [DOI] [PubMed] [Google Scholar]
- Shangguan T, Alford D, Bentz J. Influenza-virus lipid mixing is leaky and largely insensitive to the material properties of the target membrane. Biochemistry. 1996;35:4956–4965. doi: 10.1021/bi9526903. [DOI] [PubMed] [Google Scholar]
- Siegel DP. Energetics of intermediates in membrane fusion: comparison of stalk and inverted micellar intermediate mechanisms. Biophys J. 1993a;65:2124–2140. doi: 10.1016/S0006-3495(93)81256-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, D.P. 1993b. Modeling protein-induced fusion mechanisms: insights from the relative stability of lipidic structures. In Viral Fusion. J. Bentz, editor. CRS Press, Boca Raton, FL. 477–512.
- Spruce AE, Iwata A, White JM, Almers W. Patch clamp studies of single cell-fusion events mediated by a viral fusion protein. Nature (Lond) 1989;342:555–558. doi: 10.1038/342555a0. [DOI] [PubMed] [Google Scholar]
- Stegmann T. Influenza hemagglutinin-mediated membrane fusion does not involve inverted phase lipid intermediates. J Biol Chem. 1993;268:1716–1722. [PubMed] [Google Scholar]
- Stegmann T, Hoekstra D, Scherphof G, Wilschut J. Kinetics of pH- dependent fusion between influenza virus and liposomes. Biochemistry. 1985;24:3107–3113. doi: 10.1021/bi00334a006. [DOI] [PubMed] [Google Scholar]
- Stegmann T, Delfino JM, Richards FM, Helenius A. The HA2 subunit of influenza hemagglutinin inserts into the target membrane before fusion. J Biol Chem. 1991;266:18404–18410. [PubMed] [Google Scholar]
- Tatulian SA, Hinterdorfer P, Baber G, Tamm LK. Influenza hemagglutinin assumes a tilted conformation during membrane fusion as determined by attenuated total reflection FTIR spectroscopy. EMBO (Eur Mol Biol Organ) J. 1995;14:5514–5523. doi: 10.1002/j.1460-2075.1995.tb00238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse FW, Iwata A, Almers W. Membrane flux through the pore formed by a fusogenic viral envelope protein during cell fusion. J Cell Biol. 1993;121:543–552. doi: 10.1083/jcb.121.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurudome M, Gluck R, Graf R, Falchetto R, Schaller U, Brunner J. Lipid interactions of the hemagglutinin HA2 NH2-terminal segment during influenza virus–induced membrane fusion. J Biol Chem. 1992;267:20225–20232. [PubMed] [Google Scholar]
- van Meer G, Davoust J, Simons K. Parameters affecting low-pHmediated fusion of liposomes with the plasma membrane of cells infected with influenza virus. Biochemistry. 1985;24:3593–3602. doi: 10.1021/bi00335a030. [DOI] [PubMed] [Google Scholar]
- Weber T, Paesold G, Galli C, Mischler R, Semenza G, Brunner J. Evidence for H(+)-induced insertion of influenza hemagglutinin HA2 N-terminal segment into viral membrane. J Biol Chem. 1994;269:18353–18358. [PubMed] [Google Scholar]
- Weltzien HU. Cytolytic and membrane-perturbing properties of lysophosphatidylcholine. Biochim Biophys Acta. 1979;559:259–287. doi: 10.1016/0304-4157(79)90004-2. [DOI] [PubMed] [Google Scholar]
- White J. Membrane fusion: the influenza paradigm. Cold Spring Harbor Symp Quant Biol. 1996;60:581–588. doi: 10.1101/sqb.1995.060.01.062. [DOI] [PubMed] [Google Scholar]
- White J, Kartenbeck J, Helenius A. Membrane fusion activity of influenza virus. EMBO (Eur Mol Biol Organ) J. 1982;1:217–222. doi: 10.1002/j.1460-2075.1982.tb01150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J, Kielian M, Helenius A. Membrane fusion proteins of enveloped animal viruses. Q Rev Biophys. 1983;16:151–195. doi: 10.1017/s0033583500005072. [DOI] [PubMed] [Google Scholar]
- White JM. Membrane fusion. Science (Wash DC) 1992;258:917–924. doi: 10.1126/science.1439803. [DOI] [PubMed] [Google Scholar]
- Wiley DC, Skehel JJ. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu Rev Biochem. 1987;56:365–394. doi: 10.1146/annurev.bi.56.070187.002053. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature (Lond) 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- Yeagle PL, Smith FT, Young JE, Flanagan TD. Inhibition of membrane fusion by lysophosphatidylcholine. Biochemistry. 1994;33:1820–1827. doi: 10.1021/bi00173a027. [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Kuroda K, Kawasaki K, Yamashina S, Maeda T, Ohnishi S. Infectious cell entry mechanism of influenza virus. J Virol. 1982;43:284–293. doi: 10.1128/jvi.43.1.284-293.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerberg J. Simultaneous electrical and optical measurements of individual membrane fusion events during exocytosis. Methods Enzymol. 1993;221:99–112. doi: 10.1016/0076-6879(93)21010-6. [DOI] [PubMed] [Google Scholar]
- Zimmerberg J, Curran M, Cohen FS. A lipid/protein complex hypothesis for exocytotic fusion pore formation. Annu NY Acad Sci. 1991;635:307–317. doi: 10.1111/j.1749-6632.1991.tb36501.x. [DOI] [PubMed] [Google Scholar]
- Zimmerberg J, Vogel SS, Chernomordik LV. Mechanisms of membrane fusion. Annu Rev Biophys Biomol Struct. 1993;22:433–466. doi: 10.1146/annurev.bb.22.060193.002245. [DOI] [PubMed] [Google Scholar]
- Zimmerberg J, Blumenthal R, Curran M, Sarkar D, Morris S. Restricted movement of lipid and aqueous dyes through pores formed by influenza hemagglutinin during cell fusion. J Cell Biol. 1994;127:1885–1894. doi: 10.1083/jcb.127.6.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]