

Abstract
The effect of laminin on the distribution of dystroglycan (DG) and other surface proteins was examined by fluorescent staining in cultures of muscle cells derived from Xenopus embryos. Western blotting confirmed that previously characterized antibodies are reactive in Xenopus. In control cultures, αDG, βDG, and laminin binding sites were distributed as microclusters (<1 μm2 in area) over the entire dorsal surface of the muscle cells. Treatment with laminin induced the formation of macroclusters (1–20 μm2), accompanied by a corresponding decline in the density of the microclusters. With 6 nM laminin, clustering was apparent within 150 min and near maximal within 1 d. Laminin was effective at 30 pM, the lowest concentration tested. The laminin fragment E3, which competes with laminin for binding to αDG, inhibited laminin-induced clustering but did not itself cluster DG, thereby indicating that other portions of the laminin molecule in addition to its αDG binding domain are required for its clustering activity. Laminin-induced clusters also contained dystrophin, but unlike agrin-induced clusters, they did not contain acetylcholine receptors, utrophin, or phosphotyrosine, and their formation was not inhibited by a tyrosine kinase inhibitor. The results reinforce the notion that unclustered DG is mobile on the surface of embryonic muscle cells and suggest that this mobile DG can be trapped by at least two different sets of molecular interactions. Laminin self binding may be the basis for the laminin-induced clustering.
αdystroglycan (αDG)1 and βDG are present in a variety of tissues including brain but have been studied most intensively in skeletal muscle, where they are thought to play a role in maintaining the structural integrity of the sarcolemma and in clustering acetylcholine receptors (AChRs) at the neuromuscular junction (Fallon and Hall, 1994; Tinsley et al., 1994; Campbell, 1995; Carbonetto and Lindenbaum, 1995; Ozawa et al., 1995). These dystrophin associated proteins (DAPs) exist as a complex, bound tightly to each other, and are derived from the same precursor protein (Ibraghimov-Beskrovnaya et al., 1992, 1993; Bowe et al., 1994; Yoshida et al., 1994). αDG is a highly glycosylated extracellular peripheral membrane protein that binds laminin and agrin and can thereby establish a link with the extracellular matrix in extrajunctional regions as well as the neuromuscular junction (IbraghimovBeskrovnaya et al., 1992; Ervasti and Campbell, 1993; Gee et al., 1993, 1994; Campanelli et al., 1994). Its glycosylated transmembrane partner βDG can bind dystrophin (Suzuki et al., 1994; Jung et al., 1995). Since dystrophin can interact with cytoskeletal actin (Hemmings et al., 1992), this complex of molecules is thought to provide a link from the extracellular matrix to the cytoskeleton. The dystrophinrelated protein utrophin substitutes for dystrophin at sites where AChRs are clustered (Fallon and Hall, 1994; Tinsley et al., 1994; Carbonetto and Lindenbaum, 1995) and is also complexed with DG in nonmuscle cells which lack dystrophin (Matsumura et al., 1993, James et al., 1996). Other transmembrane DAPs such as the muscle-specific sarcoglycan complex as well as intracellular peripheral membrane DAPs such as the syntrophins are also associated with this extracellular matrix–cytoskeleton link (Kramarcy et al., 1994; Suzuki et al., 1994). The notion that this complex linkage is an important structural element is supported by the association of different forms of muscular dystrophy with genetic mutations in dystrophin, the sarcoglycans, and the laminin α2 chain (Campbell, 1995; Ozawa et al., 1995; Worton, 1995).
That DG also plays a role in clustering AChRs at the neuromuscular junction is based on the following evidence. In addition to being concentrated at the neuromuscular junction together with other DAPs and utrophin, αDG is the major agrin-binding protein on the surface of skeletal muscle cells (Bowe et al., 1994; Campanelli et al., 1994; Gee et al., 1994). Agrin is deposited by growing nerve fibers at newly forming synapses on muscle cells (Cohen and Godfrey, 1992) and is the major neural agent involved in triggering the aggregation of AChRs at these sites in culture (Reist et al., 1992) as well as in vivo (Gautam et al., 1996). αDG accumulates very early together with AChRs and neurally released agrin at newly forming synapses, within as little as 1 h after the establishment of nerve–muscle contact (Cohen et al., 1995a ). Clusters of AChRs induced by the addition of agrin to the culture medium of embryonic muscle cells likewise contain an accumulation of αDG as well as other DAPs and utrophin (Campanelli et al., 1994; Gee et al., 1994). Tyrosine phosphorylation has been strongly implicated in agrin's clustering activity (Wallace, 1994, 1995; Ferns et al., 1996), and recent data indicate that this phosphorylation depends upon activation of the transmembrane receptor tyrosine kinase MuSK by agrin (DeChiara et al., 1996; Glass et al., 1996). Glass et al. suggest that αDG could play a role in this activation by helping to present agrin to MuSK.
Since agrin is a ligand for αDG and can cluster it, it seemed of interest to explore whether laminin, the more ubiquitous ligand, would also cluster DG. In fact, laminininduced changes in the distribution of its binding sites were briefly reported in a study on cultured rat myotubes (Vogel et al., 1983). In the present study we have examined this action of laminin in more detail in cultures of embryonic Xenopus muscle cells. We report that DG is clustered by laminin, that these clusters contain dystrophin, and that this laminin-induced clustering involves a corresponding depletion of DG in regions surrounding the clusters. The laminin fragment E3 has no clustering activity but inhibits laminin's. Unlike nerve and agrin-induced clustering, laminin-induced clustering of DG is not accompanied by an accumulation of AChRs or utrophin and occurs in the absence of tyrosine phosphorylation. The findings support the notion that unclustered DG is mobile on the surface of embryonic muscle cells and suggest that this mobile DG can be trapped by at least two different sets of molecular interactions. Since laminin, a t-shaped molecule, binds to αDG through its G domain at the end of its long arm (Ibraghimov-Beskrovnaya et al., 1992; Ervasti and Campbell, 1993; Gee et al., 1993) and can cross link with itself through the terminal regions of its short arms (Yurchenco and Cheng, 1993; Colognato-Pyke et al., 1995), we speculate that this self binding property of laminin causes mobile DG to cluster.
Materials and Methods
Membrane Protein Isolation and Western Blot Analysis
For tests on βDG, membrane proteins were isolated from leg muscle of the frog Xenopus laevis and from leg and back muscles of rabbit as described previously (Cohen et al., 1995a ). Briefly, muscle was homogenized in a Polytron mixer (Brinkmann Instruments Inc., Rexdale, ON, Canada) in 7.5 vol of homogenization buffer (20 mM sodium pyrophosphate, 20 mM sodium phosphate monohydrate, 1 mM MgCl2, 0.303 M sucrose, 0.5 mM EDTA, pH 7.0) in the presence of the following protease inhibitors: aprotinin (1 μM), leupeptin (1 μM), pepstatin A (1 μM), benzamidine (1 mM), iodoacetamide (1 mM), and PMSF (1 mM). The homogenate was centrifuged at 14,000 g at 4°C for 15 min. The supernatant was retained and the pellet re-extracted (and recentrifuged) in 75% of the original buffer volume. The ensuing supernatants were pooled and centrifuged at 30,000 g for 30 min at 4°C to pellet the heavy microsome fraction. The pellet was then resuspended in 0.6 M KCl, 0.303 M sucrose, 50 mM Tris-HCl, pH 7.4, with protease inhibitors and incubated on ice. After 30 min this suspension was centrifuged at 142,000 g for 30 min at 4°C. The KCl-washed heavy microsomes (pellet) were solubilized on ice for 30 min in 1% digitonin, 50 mM Tris-HCl, pH 7.4, with protease inhibitors. This material was subjected to centrifugation at 85,000 g for 30 min at 4°C and the supernatant retained.
Equal amounts (20 μg) of solubilized heavy microsomal membranes from rabbit and Xenopus muscle were electrophoretically separated on 7.5% SDS-PAGE gels (Laemmli, 1970) and the protein subsequently transferred to nitrocellulose membranes. The blots were blocked in 10 mM Tris-HCl, pH 7.4, 0.15 M NaCl, 0.1% Tween-20 with 5% skim milk. The anti-βDG mAb 43DAG1/8D5 (Novocastra Laboratories Ltd., Newcastle upon Tyne, UK) was equilibrated with the membrane in blocking buffer for 30 min at room temperature. The blot was washed with repeated buffer changes for 1 hour in the above buffer without skim milk and then incubated with secondary antibody conjugated to horseradish peroxidase (Sigma Chemical Co., St. Louis, MO). Excess secondary antibody was removed by washing for 2 h. Bound antibody was visualized using chemiluminescence (DuPont-New England Nuclear, Boston, MA).
For tests on dystrophin and utrophin, human and Xenopus tissues were homogenized in 4 ml/g of extraction buffer (2% SDS, 5% 2-mercaptoethanol, 5% sucrose, 62.5 mM Tris-HCl, pH 6.8) using a Polytron homogenizer as previously described (Nguyen thi Man et al., 1990). The homogenates were boiled for 2 min and clarified by centrifugation at 100,000 g for 20 min. The extracts were subjected to SDS-PAGE on 3–12.5% gradient gels, together with prestained molecular mass markers, and electrophoretically transferred to nitrocellulose membranes for 16 h at 100 mA in 25 mM Tris, 192 mM glycine, 0.003% SDS. The Western blots were developed with 1:100 dilutions of mAb culture supernatants followed by a biotinylated anti–mouse Ig and avidin–peroxidase detection system (Vectastain; Vector Labs Inc., Peterborough, UK) as previously described (Nguyen thi Man et al., 1991).
Cultures
Myotomal muscle cells derived from 1-d-old Xenopus embryos were plated in glass culture chambers as described previously (Cohen et al., 1994). The coverslip which formed the floor of the chamber was coated with rat tail collagen (Upstate Biotechnology, Inc., Lake Placid, NY) and bovine plasma fibronectin (Sigma Chemical Co.). The culture medium consisted of 67% (vol/vol) L15 and 0.08% (wt/vol) bovine albumin (fraction V; GIBCO BRL, Burlington, ON, Canada). Treatment with mouse laminin-1, isolated from the EHS sarcoma (GIBCO BRL), with the E3 fragment of laminin (prepared as described by Yurchenco and Cheng, 1993), or with an extract of Torpedo agrin (a generous gift from E.W. Godfrey, Medical College of Wisconsin, Milwaukee, WI) was begun after the cells had been in culture for 1–3 d. The agrin extract was used at a dilution of 1:100–1:200. In cases in which the tyrphostin RG50864 (Rhône-Pulenc Rover Pharmaceuticals, Inc., Collegeville, PA) was included with laminin or agrin, it was added to the culture up to 4 h earlier. Unless indicated otherwise the cultures were maintained at room temperature (22–24°C).
Fluorescent Staining
For extracellular epitopes, cultures were stained alive at 6–8°C and then fixed. To stain intracellular epitopes, it was necessary to fix and permeabilize the cells (see Fig. 5, A and B) either with precooled (−16°C) 95% ethanol for 5–10 min or with 4% (wt/vol) formaldehyde for 10–15 min, followed by 1% (vol/vol) Triton X-100 for 20 min. After fixation, all steps of the staining protocol were carried out at 6–8°C. Permeabilized cultures were exposed to 10% goat serum (in 67% L15) for 10 min before staining them. Cultures were exposed to primary antibodies and fluorescent reagents for 30–60 min and rinsed with 1% goat serum. Stained cultures were stored in 4% formaldehyde in the refrigerator and subsequently processed for fluorescence microscopy (Cohen and Godfrey, 1992).
Figure 5.

Laminin-induced clusters of βDG and LBS. (A1 and A2) βDG and LBS immunofluorescence after treatment with 2.4 nM laminin for 3 d, revealing large numbers of macroclusters and a greatly reduced density of microclusters (compare with Fig. 3 B). (B1 and B2) βDG immunofluorescence in the absence of permeabilization and LBS immunofluorescence after the same laminin treatment as in A. Only some slight “bleedthrough” of the bright LBS immunofluorescence is seen in B1, thereby confirming that the immunofluorescence in A1 involved the binding of the antiβDG mAb to an intracellular epitope. (C) βDG immunofluorescence on the ventral surface of a cell after the same laminin treatment as in A. (D) βDG immunofluorescence on the ventral surface of a cell in a culture which was not treated with laminin. Comparison of C and D reveals that the laminin treatment induced an extensive accumulation of βDG at the cell periphery but did not induce clustering over the rest of the ventral surface which is inaccessible to laminin. The laminin-induced clustering at the cell periphery is out of focus in A and B2. Bar, 5 μm.
AChRs were stained with rhodamine-conjugated α-bungarotoxin (Molecular Probes, Inc., Eugene, OR) at 2–4 μg/ml. The following primary antibodies were used: 1:100 mAb IIH6 directed against αDG (Ervasti and Campbell, 1993; Cohen et al., 1995a ); 1:500 anti-laminin, a polyclonal antibody (GIBCO BRL) which also binds to the laminin fragment E3; 1:20–1:40 anti-βDG mAb 43DAG1/8D5; 5 μg/ml mAb 4G10, directed against phosphotyrosine (Upstate Biotechnology, Inc.); and anti-utrophin (MANCHO 3; Nguyen thi Man et al., 1991) and anti-dystrophin mAbs (MANEX 737410A11; Morris et al., 1995) at 1:5–1:20. Appropriate affinity purified secondary antibodies conjugated with fluorescein or rhodamine (Organon Teknika, Inc., Scarborough, ON, Canada; Molecular Probes, Inc.) were used at 10 μg/ml. For some experiments primary antibodies were followed by biotin-conjugated secondary antibodies and fluorescent streptavidins (Molecular Probes, Inc.). To stain for laminin binding sites (LBS) without inducing clustering, cultures were cooled to 6–8°C and then exposed to 0.6–6 nM laminin for 10–30 min, followed by anti-laminin and secondary antibody. The latter protocol was also used when muscle cells were cultured for a day or more in the presence of a lower concentration of laminin.
The widespread distribution of αDG microclusters seen in control cultures of the present study was not observed consistently in a previous study (Cohen et al., 1995a ). Methodological differences probably account for this discrepancy. In the latter study some of the cultures were stained with a more dilute solution of mAb IIH6, and some were fixed before staining. In addition, the culture substrate contained laminin rather than fibronectin. Laminin competes with mAb IIH6 for binding to αDG (Ervasti and Campbell, 1993) and reduces the intensity of mAb IIH6 immunofluorescence (see Fig. 3 D).
Figure 3.
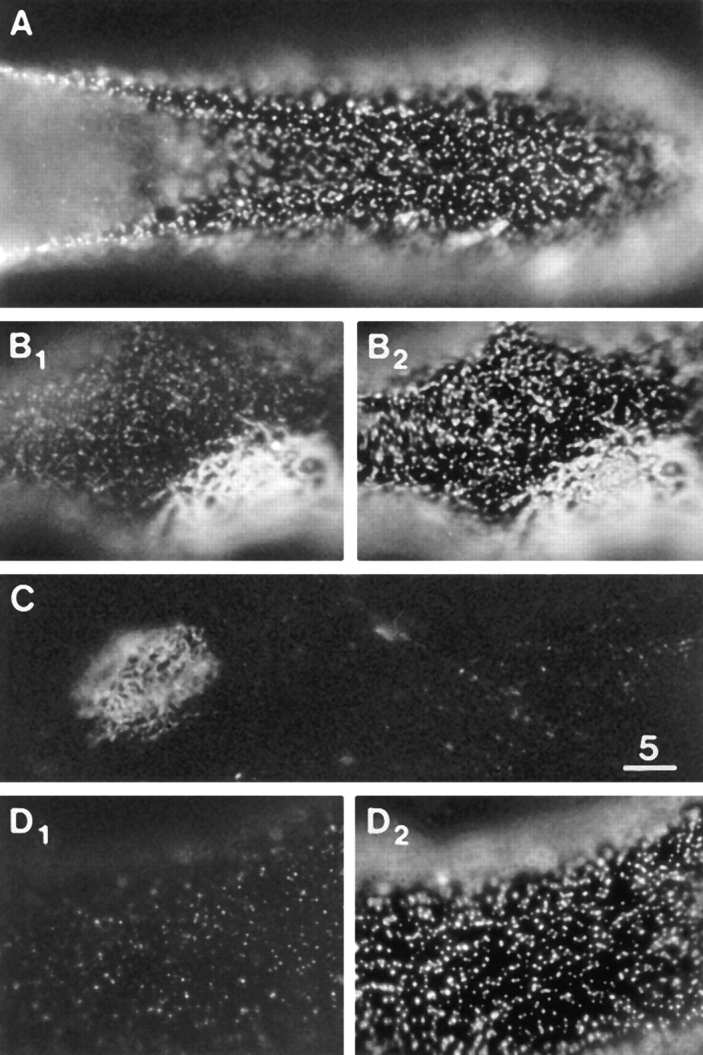
Distribution of DG and LBS on the dorsal surface of muscle cells in control cultures. (A) αDG immunofluorescence, revealing microclusters and a few macroclusters. (B1) βDG immunofluorescence. Part of a megacluster is seen. Microclusters are also apparent but are relatively faint. (B2) LBS immunofluorescence in same field as B1. Microclusters, as well as the megacluster, are well resolved. (C) Laminin immunofluorescence in the absence of pretreatment with laminin, revealing a megacluster but very few microclusters. (D1 and D2) αDG and LBS immunofluorescence, obtained by exposing the culture to 6 nM laminin for 10 min at low temperature, followed by staining for αDG and laminin. The brief pretreatment with laminin inhibited the staining for αDG (compare D1 with A). Bar, 5 μm.
Analysis
Because of their large size, laminin and mAb IIH6 (an IgM) have little if any access to the ventral muscle cell surface which is in contact with the culture substrate. Accordingly, LBS and αDG immunofluorescence as well as laminin-induced clustering were restricted to the dorsal surface. For photography and subsequent analysis, it was therefore necessary to locate regions of the dorsal surface which were in a single focal plane. All cultures were examined and photographed using a 100× oil immersion objective.
Negatives on 35-mm film (Kodak T-MAX 3200) were analyzed using the MCID imaging system (Imaging Research Inc., St. Catharines, ON, Canada) at a magnification of 208 pixels/μm2. For each frame, a rectangular region of interest (ROI) was chosen where all the clusters were in focus. ROIs were typically 200–400 μm2. The area of each discrete cluster within the ROI was recorded, thereby revealing the size distribution of the clusters, which were arbitrarily divided into microclusters (<1 μm2 in area) and macroclusters (1–20 μm2 in area). The percentage of the ROI's area occupied by macroclusters was used as a measure of laminin-induced clustering. Another measure was the decline in the density of microclusters. This density was calculated as the number of microclusters within the ROI divided by the difference between the ROI area and the total macrocluster area.
Since microclusters and laminin-induced macroclusters were both readily resolved after staining for LBS, photographs of this immunofluorescence were used for most of the quantitative analysis. One problem, however, was that the intensity of the immunofluorescence of the laminin-induced macroclusters was considerably greater than that of the microclusters. Accordingly, photographic exposure times that were optimal for the microclusters resulted in overexposure and some “blooming” of the macroclusters. Conversely, shorter photographic exposure times which were optimal for the macroclusters resulted in underexposure and some apparent loss of the microclusters. To avoid exaggerating the laminin-induced decline in the density of microclusters we opted for the longer exposure times (5–10 s). Subsequently, we adjusted the intensity of the light table on which the negative was placed to exclude from the measurements as much of the “blooming” as possible without the loss of microclusters.
In cultures stained with two different probes, colocalization was assessed on pairs of photographs printed at a magnification of 1,600×. A transparency was placed over one of the photographs, and the position of the fluorescein (or rhodamine) fluorescence was marked on the transparency. The transparency was then repositioned over the companion photograph, thereby revealing the location of the fluorescein fluorescence with respect to the rhodamine fluorescence.
Counts of cells with AChR-containing megaclusters were made directly on the microscope using an occular graticule to assess cluster size. Clusters whose diameters were at least 5 μm were categorized as megaclusters.
Results
Reactivity of Anti-βDG, -Dystrophin, and -Utrophin Antibodies in Xenopus
Based on previous Western blot analysis, the anti-αDG mAb IIH6 recognized Xenopus αDG (Cohen et al., 1995a ). The results of similar tests made for the anti-βDG mAb 43DAG1/8D5 are shown in Fig. 1. This mAb recognizes a single protein band on Western blots of adult rabbit and Xenopus skeletal muscle membrane extracts. In comparison to rabbit βDG, the Xenopus protein migrates a little more slowly on SDS-PAGE. This small difference was also noted previously in tests with another anti-DG antibody (Cohen et al., 1995a ).
Figure 1.
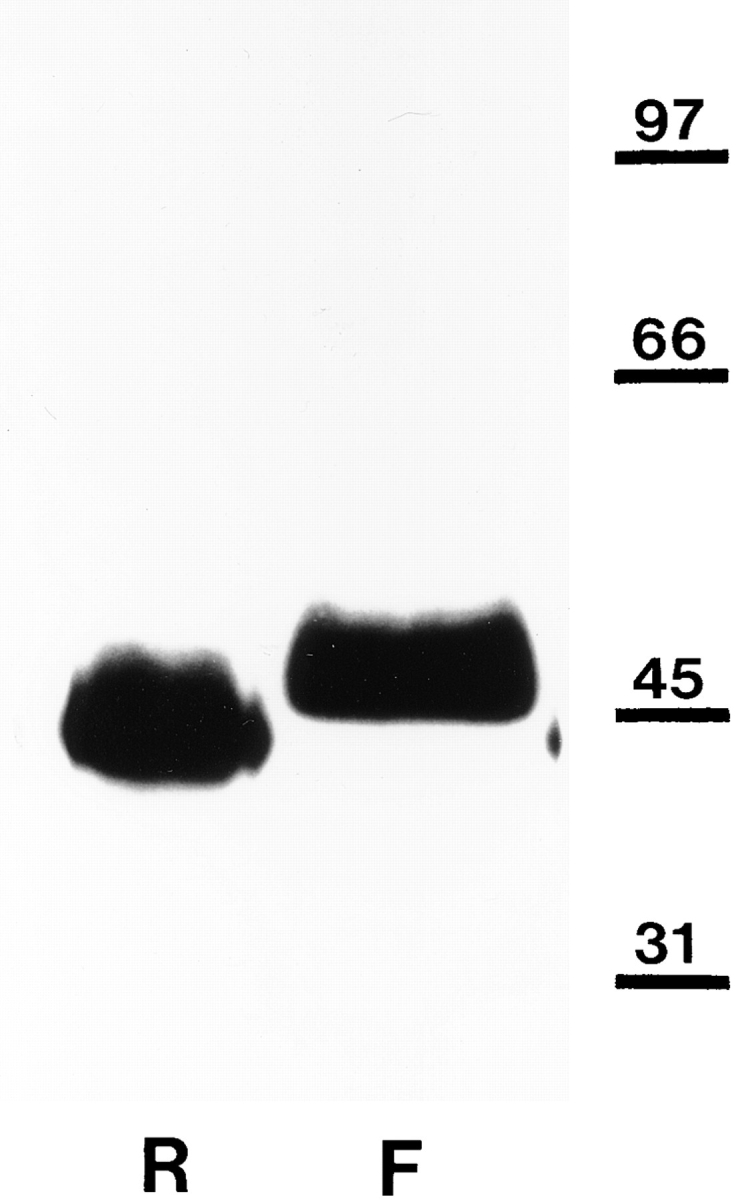
Western blot analysis of rabbit and Xenopus βDG. Nitrocellulose transfers of heavy microsomes from rabbit (R) and Xenopus (frog, F) muscle, which had been separated by SDSPAGE, were stained with mAb 43DAG1/8D5 raised to 15 of the last 16 amino acids at the extreme COOH terminus of the human dystroglycan sequence. It recognizes a band of roughly 43 kD in rabbit corresponding to the size of βDG (Ibraghimov-Beskrovnaya et al., 1992). A similar though somewhat larger band of 44–46 kD is visible in Xenopus. Molecular mass markers (in kD) are shown to the right.
Fig. 2 shows tests for the anti-dystrophin mAb MANEX 7374-10A11 and for the anti-utrophin mAb MANCHO 3. In human tissues, dystrophin is present in muscle but undetectable in lung (Fig. 2 B). By contrast, utrophin is present in most human tissues, including lung (Fig. 2 A), but is barely detectable in normal human muscle where it is confined to neuromuscular junctions, blood vessels, and nerve fibers (Nguyen thi Man et al., 1991). The anti-utrophin mAb also appears to show a high degree of specificity for Xenopus utrophin; it gives a strong band in the heart but not in skeletal muscle (Fig. 2 A), although dystrophin is abundant in Xenopus skeletal muscle (Fig. 2 B). Conversely, the anti-dystrophin mAb clearly recognizes Xenopus dystrophin in skeletal muscle, but does not cross react with the abundant utrophin in Xenopus heart (Fig. 2 B). We therefore used these mAbs for immunofluorescent staining of embryonic Xenopus muscle cell cultures.
Figure 2.

Western blot analysis of human and Xenopus utrophin (A) and dystrophin (B). The Xenopus (frog, F) tissues analyzed were skeletal muscle (m), liver (lr), and heart (h), and the human (H) tissues were skeletal muscle and lung (lg). Positions of molecular mass markers at 180, 116, and 84 kD are shown, while utrophin and dystrophin migrate at ∼400 kD. Lower molecular mass bands at ∼50 and 120 kD are nonspecific and due to cross reactions of the second antibody system.
Clusters in Control Cultures
Based on fluorescent staining for αDG, βDG, LBS, and AChRs, clusters on the dorsal surface of embryonic Xenopus myotomal muscle cells in culture can be classified into three size categories termed here megaclusters, macroclusters, and microclusters.
Megaclusters are >20 μm2 in area and often larger than 40 μm2. When viewed at high magnification some of them appear uniform (see Fig. 10 D), but most have a complex, patterned appearance (Fig. 3, B and C). Megaclusters contain similar distributions of αDG, βDG, LBS, and AChRs although at their periphery, αDG, βDG, and LBS can extend beyond the limits of the AChRs (see Fig. 3 A in Cohen et al., 1995a ). The overall incidence of megaclusters on the dorsal surface is low. Many cells have none and usually have instead, one or two megaclusters on their ventral surface (see Fig. 5, C and D and Fig. 11, A and C). The remaining cells rarely have more than one megacluster on their dorsal surface. Megaclusters were not included in the quantitative analysis shown in Figs. 4, 6, and 7.
Figure 10.
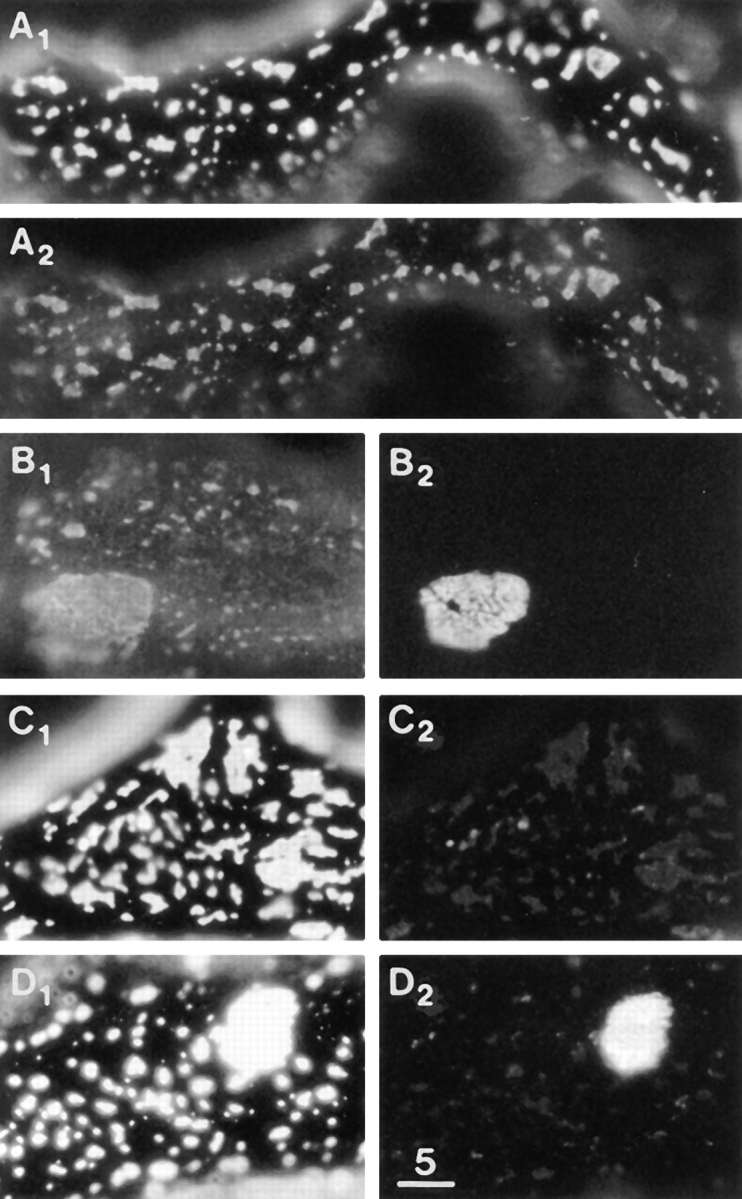
Laminin-induced clusters contain dystrophin but lack AChRs, utrophin, and PY. (A1 and A2) LBS and dystrophin immunofluorescence, revealing excellent colocalization. (B1 and B2) Dystrophin immunofluorescence and AChR stain, revealing AChRs at a megacluster but not at laminin-induced macroclusters. (C1 and C2) LBS and utrophin immunofluorescence. Utrophin was not detected at laminin-induced clusters (but was always observed wherever AChRs were clustered). (D1 and D2) LBS and PY immunofluorescence, revealing PY at a megacluster but not at the laminin-induced macroclusters. The laminin treatment was 2.4 nM for 3 d in A and C, 6 nM for 2 d in B, and 6 nM for 1 d in D. Some slight bleedthrough of the bright LBS immunofluorescence is seen in C2 and D2. Bar, 5 μm.
Figure 11.

Effect of agrin and laminin on megaclusters. Cultures were stained for AChRs and photographed at low magnification. (A) In control cultures each muscle cell typically has one or two megaclusters. (B) After treatment with agrin for 1 d, the muscle cells have many agrin-induced macroclusters but often lack megaclusters. (C) After treatment with 6 nM laminin for 1 d, each muscle cell still has one or two megaclusters. Variable numbers of autofluorescent yolk granules are apparent in the central regions of the cells. The faint outlining of some of the cells in C is due to bleedthrough of the very bright LBS immunofluorescence (not shown) at cell peripheries. Bar, 25 μm.
Figure 4.
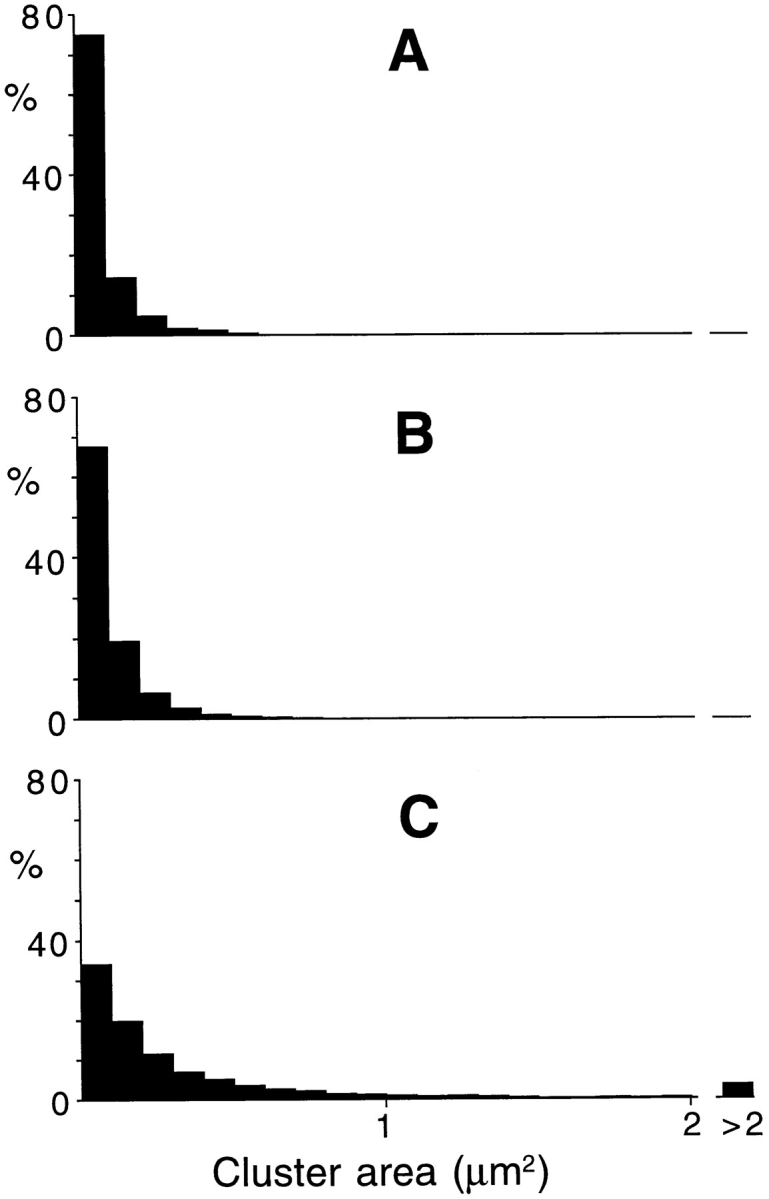
Sizes of individual αDG and LBS clusters. (A) αDG clusters (n = 4,989) in control cultures not exposed to laminin. (B) LBS clusters (n = 7,930) in control cultures exposed briefly (10–30 min) to laminin (0.6–6 nM) at low temperature immediately before staining. (C) LBS clusters (n = 3,621) after treatment with 6 mM laminin for 1 d at room temperature. In control cultures (A and B) there were very few macroclusters (>1 μm2), and most of the microclusters were <0.1 μm2. After treatment with laminin for 1 d (C) the incidence of macroclusters was increased whereas the incidence of the smallest microclusters was decreased.
Figure 6.

Effect of laminin treatment on (A) the percentage of surface area occupied by LBS macroclusters and (B) the density of LBS microclusters. Laminin concentrations (in nM) and exposure times are indicated at the bottom of B. The cultures were treated with laminin either at low temperature (open columns) or at room temperature (shaded columns). In some cases the laminin treatment was carried out in the presence of RG50864 (striped columns). On average the means and standard errors are based on 26 cells (range, 9–79).
Figure 7.

Relationship between the density of microclusters and the percentage of surface area occupied by macroclusters. From the data of Fig. 6.
Macroclusters, defined as 1–20 μm2 in area, are usually 1–3 μm2 and rarely as large as 10 μm2. They typically occur near the ends of some of the muscle cells in control cultures and can be seen after staining for αDG, βDG, or LBS. However, unlike megaclusters, the great majority of macroclusters do not contain AChRs.
Microclusters, defined as discrete sites of immunofluorescence having an area of <1 μm2, are distributed in a random fashion over the entire dorsal surface of the muscle cells. They were readily observed after staining for αDG (Fig. 3 A). Immunofluorescent staining for βDG, known from biochemical studies to be bound tightly to αDG (Yoshida et al., 1994), also revealed the microclusters (Fig. 3 B1) but not as consistently and with less resolution, presumably because of technical limitations such as reduced immunoreactivity associated with fixation and permeabilization (see Materials and Methods). A widespread distribution of microclusters was also seen after staining for LBS (Fig. 3 B2). Virtually all microclusters that were resolved with βDG immunofluorescence also contained LBS (Fig. 3 B). Such colocalization is in line with biochemical studies showing that laminin binds to αDG which is tightly complexed to βDG (Ibraghimov-Beskrovnaya et al., 1992; Ervasti and Campbell, 1993; Gee et al., 1993). In the absence of pretreatment with exogenous laminin, laminin immunofluorescence revealed megaclusters but very few microclusters (Fig. 3 C). A lack of endogenous laminin immunofluorescence over most of the cell surface, except at AChR-containing clusters, was also reported for cultures of fetal rat muscle cells (Vogel et al., 1983).
The density of microclusters stained for αDG was 1.33 ± 0.09/μm2 (mean ± SEM; n = 17) whereas the density of the LBS microclusters was 1.58 ± 0.04/μm2 (n = 95). Since the latter value is only slightly greater than that obtained for αDG microclusters and since laminin is known to bind to αDG, it follows that most if not all of the LBS microclusters contained αDG. The slight difference between the two densities may be related to the fact that the LBS immunofluorescence was generally brighter than the αDG immunofluorescence. Furthermore, in agreement with previous findings that laminin competes with mAb IIH6 for binding to biochemically isolated αDG (Ervasti and Campbell, 1993), laminin reduced the αDG immunofluorescence in a dose-dependent manner and even a 10 min pretreatment with 6 nM blocked much of the microcluster type of αDG immunofluorescence (Fig. 3 D). Overall then, the findings complement previous biochemical data and suggest that αDG functions as a laminin receptor in situ.
As shown in Fig. 4, A and B, the size distributions of the clusters stained for αDG and LBS were essentially the same, with the microclusters greatly outnumbering the macroclusters. Interestingly, most of the microclusters were <0.1 μm2. This area corresponds to a diameter of <0.4 μm and encroaches on the limit of resolution of the light microscope. Thus if the immunofluorescence emanating from a single αDG molecule were bright enough to be photographed, its size would be similar to that of the smallest microclusters photographed in this study. Alternatively, the smallest microclusters may have contained several αDG molecules.
Laminin-induced Clustering
When cultures were treated with laminin at room temperature, macroclusters of a variety of shapes and sizes formed over the entire dorsal surface of the muscle cells. Invariably, the regions surrounding these laminin-induced macroclusters had a reduced density of microclusters (Fig. 5 A). As shown in Fig. 4 C, the size distribution of the clusters in laminin-treated cultures formed a continuum, with no demarcation between microclusters and macroclusters. Rather, the laminin treatment reduced the percentage of smallest microclusters and increased the percentage of macroclusters. In the example of Fig. 4 C, the microclusters outnumbered the macroclusters by about 10-fold, but because of size differences, the macroclusters actually occupied a greater proportion of the surface area.
The laminin-induced macroclusters appeared to be randomly distributed, were readily resolved with LBS and βDG immunofluorescence, but were considerably fainter with αDG immunofluorescence, further reflecting the competition noted between laminin and mAb IIH6 for binding to αDG. A pronounced laminin-induced clustering was also apparent at cell peripheries where the dorsal and ventral surfaces meet (Fig. 5 C). Combined staining for LBS and βDG revealed that laminin was bound at all sites of laminin-induced clustering of DG (Fig. 5 A). On the other hand the laminin treatment produced no apparent change in the pattern of βDG immunofluorescence on the ventral surface which is inaccessible to laminin (Fig. 5, C and D), thus indicating that the laminin-induced clustering was restricted only to those sites where laminin binding occurred.
The time and concentration dependence of the laminininduced changes are summarized in Fig. 6 A, which shows the percentage of surface area occupied by the LBS macroclusters, and Fig. 6 B, which shows the density of the LBS microclusters. In control cultures which were exposed to 0.6–6 nM laminin for 10–150 min at 6–8°C (Fig. 6, open columns), the macroclusters occupied only 0.2–1.2% of the surface area, and the density of the microclusters varied from 1.34–1.77/μm2. When cultures were exposed to 6 nM laminin at 22–24°C (Fig. 6, shaded columns) an increase in macrocluster formation was apparent within 150 min, and this was accompanied by a decrease in the density of the microclusters. Larger, temperature-dependent changes occurred with more prolonged treatment. After 1 d in 6 nM laminin the macroclusters occupied 10.2% of the surface area, and the density of microclusters was only 0.26/μm2. Significant laminin-induced clustering occurred within 1 d even at a concentration of 0.03 nM, the lowest concentration tested. Intermediate concentrations of laminin induced intermediate amounts of clustering. Extending the laminin treatment beyond 1 d resulted in some additional clustering, especially at the lower concentrations of laminin.
Taken together the findings indicate that low concentrations of laminin bind to αDG and induce an apparently random clustering of it and its transmembrane partner βDG. This clustering is accompanied by a depletion of DG in the surrounding regions. The decline in the density of microclusters that accompanied the formation of macroclusters is shown in more detail in Fig. 7. The inverse relationship suggests that the macroclusters were derived from the microclusters and can be readily explained by a clustering mechanism whereby the binding of laminin to mobile DG promotes aggregation of colliding laminin–DG complexes (see Discussion). That the decline in microcluster density was most-pronounced at the onset of macrocluster formation is also readily explained by this mechanism. As pointed out earlier, the immunofluorescence emitted from each of the smallest microclusters might be due to a much smaller source (and perhaps even a single laminin–DG complex). If pairs of these microclusters aggregated, their density would be reduced by half, but the size of the immunofluorescence associated with these newly formed pairs might not be changed at all. Nor would the area of the macroclusters be changed. In addition, since the smallest microclusters were the most numerous (Fig. 4), these would require considerable recruitment of surrounding mobile laminin–DG complexes to attain macrocluster dimensions. Based on these considerations, it is not unexpected that the decline in microcluster density was steepest at the onset of the laminin-induced clustering.
Effects of the Laminin Fragment E3
E3, one of the fragments obtained when laminin is cleaved by elastase, consists of the last two G repeats at the end of the long arm of the laminin molecule. In agreement with previous findings that this portion of the molecule binds to αDG (Gee et al., 1993), acute exposure of cultures to E3, like acute exposure to laminin, resulted in widespread binding in the form of microclusters and inhibited the widespread binding of mAbIIH6. The latter inhibition was 41% in the presence of 33 nM E3 and virtually complete (94%) in the presence of 3.3 μM E3. Unlike laminin, however, E3 did not exhibit any clustering activity. When cultures were treated for up to 2 d with 3.3 nM–3.3 μM E3 there was no detectable clustering as assessed by staining for E3 binding sites (Fig. 8 A) or for βDG. The density of microclusters after exposure to 3.3 μM E3 for 2 d was 1.326 ± 0.105/μm2, and macroclusters occupied only 0.2 ± 0.1% of the surface area (n = 13). Essentially similar values (1.271 ± 0.053/μm2 and 0.5 ± 0.2%, respectively; n = 15) were obtained for acute (30 min) exposure to 3.3 μM E3.
Figure 8.
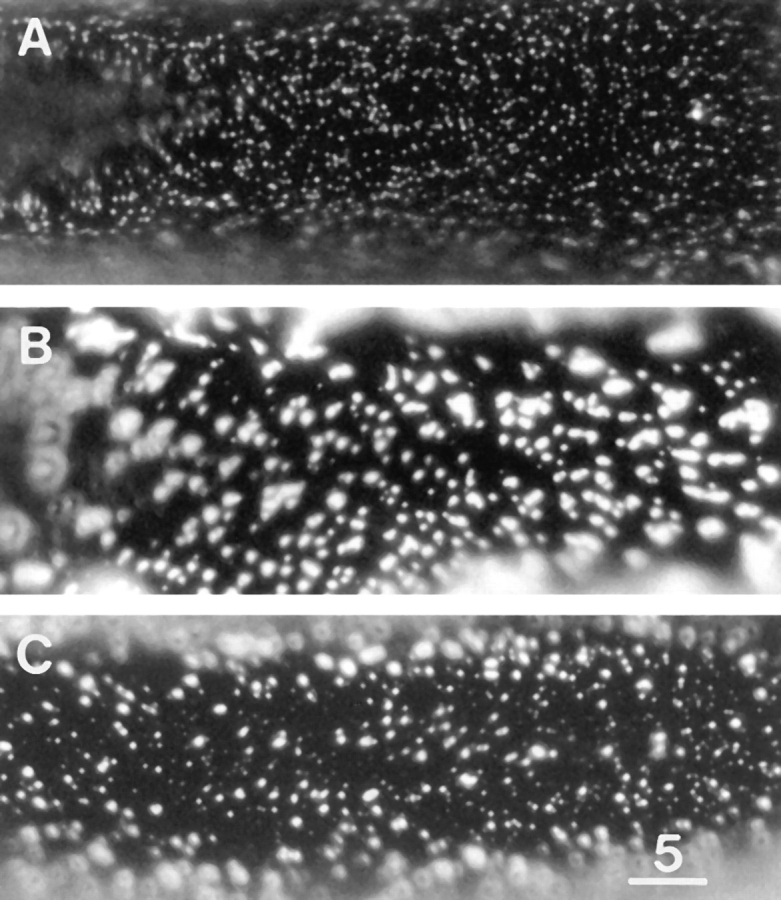
E3 lacks clustering activity and inhibits laminin-induced clustering. (A) E3 binding sites after treatment with 3.3 μM E3 for 2 d. Note the relatively high density of microclusters and the paucity of macroclusters. (B) LBS after treatment with 0.6 nM laminin for 1 d. (C) Laminin and E3 binding sites after treatment with 0.6 nM laminin and 3.3 μM E3 for 1 d. E3 inhibited the laminininduced clustering. Bar, 5 μm.
In view of its lack of clustering activity and its known competition with laminin for binding to αDG, E3 would be expected to inhibit laminin-induced clustering. This was found to be the case. On muscle cells treated with 0.6 nM laminin for 1 d (Fig. 8 B), macroclusters occupied 4.9 ± 0.7% (n = 36) of the surface area, whereas in comparison, cultures treated with a combination of 0.6 nM laminin and 3.3 μM E3 for 1 d (Fig. 8 C), macroclusters occupied only 0.9 ± 0.3 (n = 28) of the surface area. Thus E3 inhibited formation of macroclusters by >80%. Lower concentrations of E3 also inhibited the laminin-induced clustering, but to a lesser extent.
Overall, the results indicate that laminin-induced clustering depends not only on that portion of the laminin molecule which binds to αDG but also on other portions which are not contained in the E3 fragment. The simplest proposal is that these other portions are the self-binding sites on the laminin molecule (see Discussion).
Comparison with Agrin-induced Clustering
Previous studies on other culture systems have indicated that agrin-induced clusters contain DG and several other molecules including tyrosine phosphorylated AChRs, other tyrosine phosphorylated proteins, and utrophin (Fallon and Hall, 1994; Carbonetto and Lindenbaum, 1995; Glass et al., 1996). As shown in Fig. 9, we confirmed that agrininduced clusters on embryonic Xenopus muscle cells contain DG, AChRs, utrophin, and phosphotyrosine (PY), as well as LBS. These clusters spanned the macrocluster range in size and also varied considerably in shape and distribution. Unlike the laminin-induced clusters, they were not restricted to the dorsal surface and actually occurred more frequently on the ventral surface. In regions of the dorsal surface lacking agrin-induced clusters, the density of the widespread LBS microclusters was almost unchanged (1.43 ± 0.07/μm2 for agrin-treated cultures compared with 1.58 ± 0.04/μm2 for control cultures). However, decreases in the density of microclusters were apparent in regions of the dorsal surface where the incidence of agrin-induced clusters was relatively high (Fig. 9 A2).
Figure 9.

Agrin-induced clusters on the dorsal surface. (A1 and A2) βDG and LBS immunofluorescence, revealing several patterned clusters, excellent colocalization, and some decline in the density of microclusters in surrounding regions. (B1 and B2) AChR stain and utrophin immunofluorescence, revealing excellent colocalization. (C1 and C2) AChR stain and PY immunofluorescence, revealing excellent colocalization. The agrin treatment was 3 d in A and B and 2 d in C. Similar results were obtained when the agrin treatment was 1 d. Bar, 5 μm.
In contrast to agrin-induced clusters, the vast majority of laminin-induced clusters did not contain detectable concentrations of AChRs, utrophin, or PY (Fig. 10, B–D). However, the laminin-induced clusters did contain dystrophin (Fig. 10, A and B). The effect of the tyrphostin RG50864, a tyrosine kinase inhibitor (Lyall et al., 1989), was also different in the two cases. As shown in Table I, 100 μM RG50864 markedly inhibited the agrin-induced clustering (also Daggett et al., 1996). On the other hand, 100 μM RG50864 did not significantly inhibit the laminin- induced clustering (Fig. 6, striped columns). These findings indicate that the clustering of DG by agrin and by laminin involves two different sets of intermolecular linkages.
Table I.
Inhibition of Agrin–induced Clustering by a Tyrphostin
| Treatment | Cells with more than five AChR-containing clusters | Inhibition | ||||
|---|---|---|---|---|---|---|
| No inhibitor | 100 μM RG50864 | |||||
| None | 0.6 ± 0.2% | — | — | |||
| (five cultures) | ||||||
| 6 nM Laminin | 1.9 ± 1.1% | — | — | |||
| (four cultures) | ||||||
| 1/200 Agrin | 96% | 57% | 40.6% | |||
| 1/200 Agrin | 94% | 21% | 77.7% | |||
| 1/1000 Agrin | 55% | 8% | 85.5% | |||
The treatments with laminin and agrin were for 1 d. At least 100 muscle cells were assessed in each culture. All AChR-containing clusters were counted, independent of their size.
Agrin and laminin also differed in their action on spontaneously formed megaclusters. In agreement with previous studies on this culture system, AChR staining revealed that most of the muscle cells in control cultures typically have one or two megaclusters, usually on their ventral surface (Fig. 11 A). We also confirmed that agrin (Daggett et al., 1996), like innervation (Anderson and Cohen, 1977; Moody-Corbett and Cohen, 1982), inhibits the formation and/or survival of these megaclusters (Fig. 11 B). By contrast, the incidence of megaclusters was not reduced by chronic treatment with laminin (Fig. 11 C). Based on examination of 100 cells/culture, ventral surface megaclusters were seen on 79.5 ± 1.8% (mean ± SEM; n = 17) of the cells in control cultures compared to only 15.0 ± 7.3% (n = 7) for cultures treated with agrin for 1 d. The corresponding values for cultures treated with laminin (2.4–6 nM) for 1 d (78.3 ± 4.3%; n = 7) or for 2–3 d (81.3 ± 3.5%; n = 3) were similar to the control value. Thus, unlike agrin-induced clustering, laminin-induced clustering does not interfere with the occurrence of spontaneously formed megaclusters.
Discussion
This study has indicated that treatment with laminin leads to a clustering of DG on the surface of embryonic muscle cells. DG consists of the extracellular peripheral membrane protein αDG tightly bound to its transmembrane partner βDG, both of which are derived from the same precursor protein (Ibraghimov-Beskrovnaya et al., 1992, 1993; Bowe et al., 1994; Yoshida et al., 1994). Laminin, which in known to bind to αDG (Ibraghimov-Beskrovnaya et al., 1992; Ervasti and Campbell, 1993; Gee et al.,1993), is present at these clusters, as is dystrophin, which is known to bind to βDG (Suzuki et al., 1994; Jung et al., 1995). In contrast to agrin-induced clustering, laminin-induced clustering did not involve AChRs, utrophin, or phosphotyrosine and was not inhibited by the tyrphostin RG50864, a tyrosine kinase inhibitor. Clustering was apparent within 150 min of treatment with 6 nM laminin, was inhibited by low temperature, and occurred at concentrations as low as 30 pM, the lowest tested. The laminin-induced increase in the formation of macroclusters was accompanied by a decrease in the density of microclusters. This inverse relationship implies that the macroclusters were derived from the microclusters.
On the other hand, it is highly unlikely that the macroclusters were derived from megaclusters. The laminin- induced macroclusters were distributed over the entire dorsal surface of the muscle cells, whereas megaclusters occupied a very limited region of the cell and were situated most often on the ventral surface. Moreover, in contrast to the case for agrin treatment and for innervation, laminin treatment did not stimulate the disappearance of megaclusters. An alternative possibility, that laminin-induced macroclusters resulted simply from an increased incorporation of DG at the cell surface, also seems unlikely in view of the fact that the formation of macroclusters was accompanied by a marked decline in the density of microclusters.
The simplest explanation of these findings is that DG microclusters on the surface of embryonic muscle cells are mobile or are unstable and give rise to single, mobile DG complexes (Fig. 12). In fact, some of the smallest microclusters may actually have been single molecules. In the absence of laminin, collisions between the mobile DG fail to result in the formation of stable, long-lived aggregates. However, as indicated in the model of Fig. 12, when laminin is bound to the mobile DG, stable and hence larger aggregates do form, necessarily resulting in a decline in the density of the microclusters. Also consistent with this interpretation is the paucity of endogenous laminin (Fig. 3 C; Vogel et al., 1983) and extracellular matrix (Kullberg et al., 1977; Weldon and Cohen, 1979; Hall and Sanes, 1993) on the surface of the embryonic muscle cells. The extensive clustering at the cell periphery, where the dorsal and ventral surfaces meet, can also be readily explained by this diffusion trap mechanism. Fluorescent staining for laminin, after briefly exposing culture chambers to laminin at higher concentrations than used in the present study, reveals that laminin binds tightly to the culture substrate (Cohen, M.W., unpublished observations). Accordingly, when mobile laminin–DG complexes on the dorsal surface happen to reach the cell periphery and contact the culture substrate, they become at least partially immobilized. The presence of this additional trapping factor at the cell periphery necessarily leads to an extensive clustering at this site.
Figure 12.

Schematic representation of some of the surface proteins clustered by laminin and agrin, both of which bind to αDG. Before clustering, DG complexes of αDG and βDG are considered to be mobile, like AChRs. Laminin-induced clusters of DG and dystrophin are considered to be linked by laminin–laminin binding. They do not contain utrophin, AChRs, or tyrosine phosphorylated molecules. Not shown is that the end of the vertical short arm of laminin also participates in self binding; that laminin may bind to other sarcolemma-associated molecules; that dystrophin binds to cytoskeletal actin; and that sarcoglycans and syntrophins may also be present at laminin-induced clusters. Agrin-induced clusters contain DG, sarcoglycans (unlabeled), utrophin, β2 syntrophin (syn), tyrosine phosphorylated AChRs, rapsyn (rn), and tyrosine phosphorylated muscle specific kinase (MuSK). Their formation unlike the formation of laminin-induced clusters, involves stimulation of tyrosine kinase activity.
Our observations with the laminin fragment E3 provide additional insight into the mechanism of the laminin- induced clustering. E3 consists of the last two G repeats at the end of laminin's long arm (Yurchenco and Cheng, 1993) and competes with laminin for binding to αDG (Gee et al., 1993). Since E3 inhibited the laminin-induced clustering, it follows that laminin's αDG binding domain is required for clustering DG. Furthermore, E3 itself did not exhibit any clustering activity, thereby indicating that other portions of the laminin molecule are also required.
The possible identity of these other portions is suggested by studies on the aggregation of laminin molecules in solution. It has been found that this aggregation involves self binding domains at the ends of the short arms of the laminin molecule (Yurchenco and Cheng, 1993; Colognato-Pyke et al., 1995). These self binding domains are obvious candidates for participation in the clustering of mobile laminin–DG complexes. At first glance, one apparent inconsistency is that aggregation of laminin in solution occurs at a critical concentration of ∼60 nM (Yurchenco and Schittny, 1990), whereas the critical concentration for laminin-induced clustering on embryonic muscle cells was some 1,000 fold lower. However, at least part of this difference in critical concentrations can be explained by the alignment of laminin's self binding domains when it is bound to αDG (Fig. 12) and by the restriction of movement to two dimensions rather than three. Under such conditions a much greater proportion of the laminin–laminin collisions would involve the self binding domains than would be the case for unbound laminin molecules in solution. In fact, laminin aggregates into macroclusters when it binds to synthetic lipid bilayers, and the critical concentration for this aggregation is about an order of magnitude lower than self aggregation in solution (Kalb and Engel, 1991).
The probability of laminin–laminin binding might be increased even further by the interaction of mobile laminin– DG complexes with other molecules. One such molecule is dystrophin, which was found to be present at the laminininduced clusters presumably bound to βDG. Since dystrophin can bind to cytoskeletal actin (Hemmings et al., 1992) it would be expected to facilitate the aggregation of colliding laminin–DG complexes. In the absence of laminin, however, this interaction by itself is apparently too weak to promote the formation of large, stable aggregates between colliding DG molecules. This would account for the relatively low incidence of macroclusters and the high density of microclusters in control cultures.
That cluster formation on the surface of embryonic muscle cells involves an aggregation of preexisting mobile membrane proteins has been established for nerve- and agrin-induced clustering of AChRs (Anderson and Cohen, 1977; Godfrey et al., 1984; Ziskind-Conhaim et al., 1984; Kidokoro et al., 1986). The present study supports the proposal (Cohen et al., 1995a ) that the DG, which accumulates together with AChRs at these clusters, is also derived from a preexisting pool of mobile DG. While recruitment of mobile DG may be common to both agrin- and laminininduced clustering, the molecular interactions responsible for trapping DG are clearly different. Agrin's action depends on activation of the transmembrane receptor tyrosine kinase MuSK (DeChiara et al., 1996; Glass et al., 1996), although the identity of the tyrosine phosphorylated intermediates which promote the trapping of DG and AChRs at the same site is unclear. By contrast, laminin-induced clustering does not appear to involve tyrosine kinase activation or the participation of tyrosine phosphorylated proteins. Instead, as discussed above, laminin–laminin binding may be the key molecular interaction involved in the trapping of mobile laminin–DG complexes.
Based on this diffusion trap scheme, other surface molecules which bind laminin, such as integrins and perlecan (Timpl and Brown, 1994), might also be expected to participate in laminin-induced clustering. Where such molecules are already anchored they could act as foci for trapping mobile laminin–DG complexes. Where these other lamininbinding molecules are themselves mobile, laminin could cluster them together with DG. It might be expected as well that different isoforms of laminin (Timpl and Brown, 1994) will have different clustering activities from the laminin-1 isoform studied here.
Another difference between the action of agrin and laminin is that utrophin, like AChRs and PY, accumulates only at agrin-induced clusters. Both utrophin and dystrophin have homologous domains for binding to βDG (Ozawa et al., 1995), yet only dystrophin accumulated at the laminin- induced clusters. If there are preexisting populations of DG–dystrophin and DG–utrophin complexes on the muscle cell surface, then laminin may selectively recruit the former, whereas agrin may selectively recruit the latter. Alternatively, laminin and agrin may differentially promote the formation of these two different complexes. Other molecules which are preferentially associated with agrin-induced clusters (e.g., tyrosine phosphorylated proteins) may also contribute to the accumulation of utrophin by binding it directly or by facilitating its binding to βDG. In this regard it is interesting to note that utrophin is concentrated together with AChRs, DG, and phosphotyrosine at spontaneously formed megaclusters (Cohen, M.W., unpublished observations) even though agrin is not detected at these sites (Cohen and Godfrey, 1992). This lends support to the suggestion that molecules in addition to agrin play a role in promoting utrophin accumulation at AChR-containing clusters.
Whereas treatment with agrin resulted in a marked reduction in the incidence of spontaneously formed megaclusters, treatment with laminin did not. Previous studies have indicated that the formation and survival of spontaneously formed, AChR-containing megaclusters is inhibited when there is extensive induction of new AChR-containing clusters elsewhere on the muscle cell surface. The latter relationship has been noted for AChR clustering induced by innervation (Anderson and Cohen, 1977; Moody-Corbett and Cohen, 1982), by application of appropriately coated beads (Peng et al., 1981; Peng, 1986), by pathways of substrate-bound, endogenous neural agrin (Cohen et al., 1995b ), and by diffusely applied agrin (Daggett et al., 1996; present study). The mechanism of this competition between newly induced and spontaneously formed AChR-containing clusters remains to be elucidated but presumably involves some of the intracellular signalling events associated with AChR clustering. The present results suggest that laminin's AChR-clustering activity is insufficient to initiate this competition in cultures of Xenopus muscle cells. Only a small and variable increase in AChR-containing clusters was observed (Table I), consistent with the small (twofold) laminin-induced increase previously observed on rat myotubes in culture (Vogel et al., 1983). The results also indicate that although spontaneously formed megaclusters contain DG, extensive clustering of DG elsewhere on the cell surface does not interfere with their formation and/or survival.
Laminin-induced clustering occurred at a laminin concentration of 30 pM and probably occurs at even lower concentrations. Such concentrations may be generated in vivo when developing cells secrete laminin into a confined extracellular space. The resulting changes in the distribution of surface molecules to which laminin binds may be important in the assembly of extracellular matrix and could have developmental consequences. In this regard it is interesting to note that the molecular basis of muscular dystrophy in the dy2J mouse has been ascribed to partial deletion of one of the self binding domains of laminin-2 (Xu et al., 1994).
Acknowledgments
This work was supported by grants to M.W. Cohen from the Medical Research Council of Canada, to P.D. Yurchenco from National Institutes of Health (DK36425), to G.E. Morris from the Muscular Dystrophy Group of Great Britain and Northern Ireland, and to S. Carbonetto from the Muscular Dystrophy Association (USA) and the Canadian Centres of Excellence.
Abbreviations used in this paper
- AChR
acetylcholine receptor
- DAP
dystrophin associated protein
- DG
dystroglycan
- LBS
laminin binding sites
- PY
phosphotyrosine
- ROI
region of interest
Footnotes
D. McDonald, M. Ignatova, and T. Inoue provided excellent technical assistance. We thank K.P. Campbell for the generous gift of anti-αDG antibody.
Please address all correspondence to M.W. Cohen, Department of Physiology, McGill University, 3655 Drummond Street, Montreal, Quebec, Canada H3G1Y6. Tel.: (514) 398-4342; Fax: (514) 398-7452.
References
- Anderson MJ, Cohen MW. Nerve-induced and spontaneous redistribution of acetylcholine receptors on cultured muscle cells. J Physiol. 1977;268:757–773. doi: 10.1113/jphysiol.1977.sp011880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowe MA, Deyst KA, Leszyk JD, Fallon JR. Identification and purification of an agrin receptor from Torpedopostsynaptic membranes: a heteromeric complex related to dystroglycans. Neuron. 1994;12:1173–1180. doi: 10.1016/0896-6273(94)90324-7. [DOI] [PubMed] [Google Scholar]
- Campanelli JT, Roberds SL, Campbell KP, Scheller RH. A role for dystrophin-associated glycoproteins and utrophin in agrin-induced AChR clustering. Cell. 1994;77:663–674. doi: 10.1016/0092-8674(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Campbell KP. Three muscular dystrophies: loss of cytoskeleton–extracellular matrix linkage. Cell. 1995;80:675–679. doi: 10.1016/0092-8674(95)90344-5. [DOI] [PubMed] [Google Scholar]
- Carbonetto S, Lindenbaum M. The basement membrane at the neuromuscular junction: a synaptic mediatrix. Curr Opin Neurobiol. 1995;5:596–605. doi: 10.1016/0959-4388(95)80064-6. [DOI] [PubMed] [Google Scholar]
- Cohen MW, Godfrey EW. Early appearance of and neuronal contribution to agrin-like molecules at embryonic frog nerve–muscle synapses formed in culture. J Neurosci. 1992;12:2982–2992. doi: 10.1523/JNEUROSCI.12-08-02982.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MW, Moody-Corbett F, Godfrey EW. Neuritic deposition of agrin on culture substrate: implications for nerve-muscle synaptogenesis. J Neurosci. 1994;14:3293–3303. doi: 10.1523/JNEUROSCI.14-05-03293.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MW, Jacobson C, Godfrey EW, Campbell KP, Carbonetto S. Distribution of α-dystroglycan during embryonic nerve–muscle synaptogenesis. J Cell Biol. 1995a;129:1093–1101. doi: 10.1083/jcb.129.4.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MW, Moody-Corbett F, Godfrey EW. Former neuritic pathways containing endogenous neural agrin have high synaptogenic activity. Dev Biol. 1995b;167:458–468. doi: 10.1006/dbio.1995.1041. [DOI] [PubMed] [Google Scholar]
- Colognato-Pyke H, O'Rears JJ, Yamada Y, Carbonetto S, Cheng Y-S, Yurchenco PD. Mapping of network-forming, heparin-binding, and α1β1 integrin-recognition sites within the α-chain short arm of laminin-1. J Biol Chem. 1995;270:9398–9406. doi: 10.1074/jbc.270.16.9398. [DOI] [PubMed] [Google Scholar]
- Daggett DF, Stone D, Peng HB, Nikolics K. Full length agrin isoform activities and binding site distributions on cultured Xenopusmuscle cells. Mol Cell Neurosci. 1996;7:75–88. doi: 10.1006/mcne.1996.0006. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon JR, Hall ZW. Building synapses: agrin and dystroglycan stick together. TINS (Trends Neurosci) 1994;17:469–473. doi: 10.1016/0166-2236(94)90135-x. [DOI] [PubMed] [Google Scholar]
- Ferns M, Deiner M, Hall Z. Agrin-induced acetylcholine receptor clustering in mammalian muscle requires tyrosine phosphyrylation. J Cell Biol. 1996;132:937–944. doi: 10.1083/jcb.132.5.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- Gee SH, Blacher RW, Douville PJ, Provost PR, Yurchenco PD, Carbonetto S. Laminin-binding protein 120 from brain is closely related to the dystrophin-associated glycoprotein, dystroglycan, and binds with high affinity to the major heparin binding domain of laminin. J Biol Chem. 1993;268:14972–14980. [PubMed] [Google Scholar]
- Gee SH, Montanaro F, Lindenbaum MH, Carbonetto S. Dystroglycan-α, a dystrophin-associated glycoprotein, is a functional agrin receptor. 1994. Cell. 1994;77:675–686. doi: 10.1016/0092-8674(94)90052-3. [DOI] [PubMed] [Google Scholar]
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattsson K, Burden SJ, et al. Agrin acts via a MuSK receptor complex. Cell. 1996;85:513–523. doi: 10.1016/s0092-8674(00)81252-0. [DOI] [PubMed] [Google Scholar]
- Godfrey EW, Nitkin RM, Wallace BG, Rubin LL, McMahan UJ. Components of Torpedoelectric organ and muscle that cause aggregation of acetylcholine receptors on cultured muscle cells. J Cell Biol. 1984;99:615–627. doi: 10.1083/jcb.99.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, Z.W., and J.R. Sanes. 1993. Synaptic structure and development: the neuromuscular junction. Cell 72/Neuron 10 (Supl.). 99–121. [DOI] [PubMed]
- Hemmings L, Kuhlman PA, Critchley DR. Analysis of the actinbinding domain of α-actinin by mutagenesis and demonstration that dystrophin contains a functionally homologous domain. J Cell Biol. 1992;116:1369–1380. doi: 10.1083/jcb.116.6.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature (Lond) 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, Milatovitch A, Ozielik T, Yang B, Koepnick K, Francke U, Campbell KP. Human dystroglycan: skeletal muscle cDNA, genomic structure, origin of tissue isoforms and chromosomal localization. Hum Mol Genet. 1993;2:1651–1657. doi: 10.1093/hmg/2.10.1651. [DOI] [PubMed] [Google Scholar]
- James M, Nguyen thi Man, Wise CJ, Jones GE, Morris GE. A utrophin-glycoprotein complex in membranes of adherent cultured cells. Cell Motil Cytoskeleton. 1996;33:163–174. doi: 10.1002/(SICI)1097-0169(1996)33:3<163::AID-CM1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Jung D, Yang B, Meyer J, Chamberlain JS, Campbell KP. Identification and characterization of the dystrophin anchoring site on β-dystroglycan. J Biol Chem. 1995;270:27305–27310. doi: 10.1074/jbc.270.45.27305. [DOI] [PubMed] [Google Scholar]
- Kalb E, Engel J. Binding and calcium-induced aggregation of laminin onto lipid bilayers. J Biol Chem. 1991;266:19047–19052. [PubMed] [Google Scholar]
- Kidokoro Y, Brass B, Kuromi H. Concanavalin A prevents acetylcholine receptor redistribution in Xenopusnerve–muscle cultures. J Neurosci. 1986;6:1941–1951. doi: 10.1523/JNEUROSCI.06-07-01941.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramarcy NR, Vidal A, Froehner SC, Sealock R. Association of utrophin and multiple dystrophin short forms with mammalian Mr58,000 dystrophin-associated protein (syntrophin) J Biol Chem. 1994;269:2870–2876. [PubMed] [Google Scholar]
- Kullberg RW, Lentz TL, Cohen MW. Development of the myotomal neuromuscular junction in Xenopus laevis: an electrophysiological and fine-structural study. Dev Biol. 1977;60:101–129. doi: 10.1016/0012-1606(77)90113-0. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (Lond) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lyall RM, Zilberstein A, Gazit A, Gilon C, Levitski A, Schlessinger J. Tyrphostins inhibit epidermal growth factor (EGF)-receptor tyrosine kinase activity in living cells and EGF-stimulated cell proliferation. J Biol Chem. 1989;264:14503–14509. [PubMed] [Google Scholar]
- Matsumura K, Yamada H, Shimizu T, Campbell KP. Differential expression of dystrophin, utrophin, and dystrophin-associated proteins in peripheral nerve. FEBS (Fed Eur Biochem Soc) Lett. 1993;334:281–285. doi: 10.1016/0014-5793(93)80695-q. [DOI] [PubMed] [Google Scholar]
- Moody-Corbett F, Cohen MW. Influence of nerve on the formation and survival of acetylcholine receptor and cholinesterase patches on Xenopusmuscle cells in culture. J Neurosci. 1982;2:633–646. doi: 10.1523/JNEUROSCI.02-05-00633.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GE, Simmons C, Nguyen thi Man Apo-dystrophins (Dp140 and Dp71) and dystrophin splicing isoforms in developing brain. Biochem Biophys Res Commun. 1995;215:361–367. doi: 10.1006/bbrc.1995.2474. [DOI] [PubMed] [Google Scholar]
- Nguyen thi Man AJ, Cartwright, Morris GE, Love D, Bloomfield JF, Davies KE. Monoclonal antibodies against defined regions of the muscular dystrophy protein, dystrophin. FEBS (Fed Eur Biochem Soc) Lett. 1990;262:237–240. doi: 10.1016/0014-5793(90)80199-s. [DOI] [PubMed] [Google Scholar]
- Nguyen thi Man JM, Ellis, Love DR, Davies KE, Gatter KC, Dickson G, Morris GE. Localization of the DMDL gene-encoded dystrophin-related protein using a panel of 19 monoclonal antibodies. Presence at neuromuscular junctions, in the sarcolemma of dystrophic skeletal muscle, in vascular and other smooth muscles, and in proliferating brain cell lines. J Cell Biol. 1991;115:1695–1700. doi: 10.1083/jcb.115.6.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa E, Yoshida M, Suzuki A, Mizuno Y, Hagiwara Y, Noguchi S. Dystrophin-associated proteins in muscular dystrophy. Hum Mol Genet. 1995;4:1711–1716. doi: 10.1093/hmg/4.suppl_1.1711. [DOI] [PubMed] [Google Scholar]
- Peng HB. Elimination of preexistent acetylcholine receptor clusters induced by the formation of new clusters in the absence of nerve. J Neurosci. 1986;6:581–589. doi: 10.1523/JNEUROSCI.06-02-00581.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng HB, Cheng P-C, Luther PW. Formation of ACh receptor clusters induced by positively charged latex beads. Nature (Lond) 1981;292:831–834. doi: 10.1038/292831a0. [DOI] [PubMed] [Google Scholar]
- Reist NE, Werle MJ, McMahan UJ. Agrin released by motor neurons induces the aggregation of acetylcholine receptors at neuromuscular junctions. Neuron. 1992;8:865–868. doi: 10.1016/0896-6273(92)90200-w. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Yoshida M, Hayashi K, Misuno Y, Hagiwara Y, Ozawa E. Molecular organization of the glycoprotein-complex-binding site of dystrophin. Three dystrophin-associated proteins bind directly to the carboxy-terminal portion of dystrophin. Eur J Biochem. 1994;220:283–292. doi: 10.1111/j.1432-1033.1994.tb18624.x. [DOI] [PubMed] [Google Scholar]
- Timpl R, Brown JC. The laminins. Matrix Biol. 1994;14:275–281. doi: 10.1016/0945-053x(94)90192-9. [DOI] [PubMed] [Google Scholar]
- Tinsely JM, Blake DJ, Zuellig RA, Davies KE. Increasing complexity of the dystrophin-associated protein complex. Proc Natl Acad Sci USA. 1994;91:8307–8313. doi: 10.1073/pnas.91.18.8307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel Z, Christian CN, Vigny M, Bauer HC, Sonderegger P, Daniels MP. Laminin induces acetylcholine receptor aggregation on cultured myotubes and enhances the receptor aggregation activity of a neuronal factor. J Neurosci. 1983;3:1058–1068. doi: 10.1523/JNEUROSCI.03-05-01058.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace BG. Staurosporine inhibits agrin-induced acetylcholine receptor phosphorylation and aggregation. J Cell Biol. 1994;125:661–668. doi: 10.1083/jcb.125.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace BG. Regulation of the interaction of nicotinic acetylcholine receptors with the cytoskeleton by agrin-activated protein tyrosine kinase. J Cell Biol. 1995;128:1121–1129. doi: 10.1083/jcb.128.6.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weldon PR, Cohen MW. Development of synaptic ultrastructure at neuromuscular contacts in an amphibian cell culture system. J Neurocytol. 1979;8:239–259. doi: 10.1007/BF01175564. [DOI] [PubMed] [Google Scholar]
- Worton R. Muscular dystrophies: diseases of the dystrophin-glycoprotein complex. Science (Wash DC) 1995;270:755–756. doi: 10.1126/science.270.5237.755. [DOI] [PubMed] [Google Scholar]
- Xu H, Wu X-R, Wewer UM, Engevall E. Murine muscular dystrophy caused by a mutation in the laminin α2 (Lama 2) gene. Nat Genet. 1994;8:297–301. doi: 10.1038/ng1194-297. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Suzuki A, Yamamoto H, Naguchi S, Mizuno Y, Ozawa E. Dissociation of the complex of dystrophin and its associated proteins into several unique groups by n-octyl β-D-glucoside. Eur J Biochem. 1994;222:1055–1061. doi: 10.1111/j.1432-1033.1994.tb18958.x. [DOI] [PubMed] [Google Scholar]
- Yurchenco PD, Schittny JC. Molecular architecture of laminin binding. FASEB (Fed Am Soc Exp Biol) J. 1990;4:1577–1590. doi: 10.1096/fasebj.4.6.2180767. [DOI] [PubMed] [Google Scholar]
- Yurchenco PD, Cheng Y-S. Self-assembly and calcium-binding sites in laminin. J Biol Chem. 1993;268:17286–17299. [PubMed] [Google Scholar]
- Ziskind-Conhaim L, Geffen I, Hall ZW. Redistribution of acetylcholine receptors on developing rat myotubes. J Neurosci. 1984;4:2346–2349. doi: 10.1523/JNEUROSCI.04-09-02346.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]