

Abstract
The MPS1 gene from Saccharomyces cerevisiae encodes an essential protein kinase required for spindle pole body (SPB) duplication and for the mitotic spindle assembly checkpoint. Cells with the mps1-1 mutation fail early in SPB duplication and proceed through monopolar mitosis with lethal consequences. We identified CDC37 as a multicopy suppressor of mps1-1 temperature-sensitive growth. Suppression is allele specific, and synthetic lethal interactions occur between mps1 and cdc37 alleles. We examined the cdc37-1 phenotype for defects related to the SPB cycle. The cdc37-1 temperature-sensitive allele causes unbudded, G1 arrest at Start (Reed, S.I. 1980. Genetics. 95: 561–577). Reciprocal shifts demonstrate that cdc37-1 arrest is interdependent with α-factor arrest but is not a normal Start arrest. Although the cells are responsive to α-factor at the arrest, SPB duplication is uncoupled from other aspects of G1 progression and proceeds past the satellite-bearing SPB stage normally seen at Start. Electron microscopy reveals side-by-side SPBs at cdc37-1 arrest. The outer plaque of one SPB is missing or reduced, while the other is normal. Using the mps2-1 mutation to distinguish between the SPBs, we find that the outer plaque defect is specific to the new SPB. This phenotype may arise in part from reduced Mps1p function: although Mps1p protein levels are unaffected by the cdc37-1 mutation, kinase activity is markedly reduced. These data demonstrate a requirement for CDC37 in SPB duplication and suggest a role for this gene in G1 control. CDC37 may provide a chaperone function that promotes the activity of protein kinases.
The centrosome serves as the microtubule-organizing center of the eukaryotic cell and must be precisely duplicated once during each cell cycle to provide the poles of the mitotic spindle (Kellogg et al., 1994). In the yeast Saccharomyces cerevisiae, centrosomal functions are provided by the spindle pole body (SPB)1 (for reviews see Kilmartin, 1994; Snyder, 1994). This trilaminar disk is embedded in the nuclear envelope throughout the cell cycle and nucleates microtubule arrays in the nucleus and cytoplasm. Electron microscopy reveals a darkly staining disk in the plane of the nuclear envelope, called the central plaque, which is flanked by less darkly staining disks on its nuclear and cytoplasmic surfaces. These structures, called the inner and outer plaques, respectively, are sites of microtubule association in vivo. The half-bridge, thought to be a modification of the nuclear envelope, lies next to the SPB on one side.
SPB duplication has been well characterized cytologically in wild-type and cell division cycle (cdc) mutant strains, leading to a proposed morphological assembly pathway (Byers and Goetsch, 1974; Winey and Byers, 1993). The first visible step in the pathway is formation of the satellite, a clump of electron-dense material found on the cytoplasmic surface of the half-bridge beside the existing SPB. The satellite is thought to be the precursor of the new SPB. This morphology is seen at Start, the point at which cells make a commitment to either mate or enter another round of division. After cells pass through Start, the SPB is duplicated in a conservative manner. Identical side-by-side SPBs are then seen, joined by a complete bridge structure. The cycle is complete when the SPBs later separate to form a short intranuclear spindle. These stages of duplication are also observed in various cdc mutants at the nonpermissive temperature, where the SPB cycle uniformly arrests at a stage appropriate to other indicators of cell cycle progression, such as budding and DNA synthesis. Mutation of CDC28 arrests cells at Start in G1 with a satellitebearing SPB, the same morphology that is seen when the mating pheromone α-factor is used to arrest cells at Start. In contrast, side-by-side SPBs are found at cdc4 or cdc34 arrest, which occurs late in G1 (Byers and Goetsch, 1974; Goebl et al., 1988).
A number of mutant strains have been identified that are specifically defective in the SPB duplication event, failing in duplication and assembling a monopolar spindle. The aberrant SPB structures formed in these strains have been examined by electron microscopy. These morphologies, along with order-of-function experiments, indicate where in the SPB duplication pathway each gene function is required (Winey et al., 1991). CDC31 and KAR1 are apparently required early in the pathway, possibly in deposition of the satellite, as mutations in these genes cause duplication to fail altogether (Byers, 1981b ; Rose and Fink, 1987; Winey et al., 1991). MPS1 is required for the transition from satellite-bearing to side-by-side SPBs; loss of function produces a single, large SPB with an enlarged halfbridge. MPS2 and NDC1 are required later in the pathway. In mps2 or ndc1 mutants, duplication proceeds, but the newly formed SPB is nonfunctional. It lacks an inner plaque structure and is not properly inserted into the nuclear envelope (Winey et al., 1991, 1993).
Generally, these mutants proceed through G1 and S phases at the nonpermissive temperature and transiently arrest in G2 as large-budded cells with monopolar spindles. However, cells carrying the mps1-1 mutation behave differently. Although SPB duplication has failed, these cells exhibit no cell cycle arrest and go on to commit an aberrant mitosis that results in rapid loss of viability. MPS1 has been implicated in a mitotic checkpoint that responds to spindle dysfunction by arresting the cell cycle (Hardwick et al., 1996; Weiss and Winey, 1996). MPS1 encodes an essential protein kinase (Poch et al., 1994; Lauzé et al., 1995). Significant sequence homology to the catalytic domains of the mammalian dual specificity kinases PYT/TTK (Mills et al., 1992; Lindberg et al., 1993) and esk (Douville et al., 1992) suggests that the MPS1 gene product, Mps1p, is itself a dual specificity kinase. In fact, Mps1p does autophosphorylate serine, threonine, and tyrosine residues in vitro (Lauzé et al., 1995).
We report here the identification of CDC37 as a multicopy suppressor of the mps1-1 temperature-sensitive (ts) phenotype. In addition to multicopy suppressor interactions, we find that alleles of CDC37 and MPS1 exhibit synthetic lethal interactions. CDC37 was originally isolated by Reed (1980a) (Ferguson et al., 1986) in a screen for mutations that cause cell cycle arrest at Start in G1 in a matingcompetent state, and a weak karyogamy defect is also seen in cdc37 mutants at the nonpermissive temperature (Dutcher and Hartwell, 1982, 1983). Functional homologues of this gene have recently been identified in Drosphilia melanogaster (Cutforth and Rubin, 1994) and mammals (Stepanova et al., 1996), and the mammalian gene encodes a subunit of the Hsp90 chaperone complex found in association with several protein kinases (Stepanova et al., 1996). Phenotypic analysis demonstrates that cdc37-1 arrest is a novel Start arrest, where the SPBs have begun duplication and formed an aberrant side-by-side structure, although the cell remains mating pheromone responsive. One SPB, apparently the newly formed one, displays an outer plaque defect. Mutation of CDC37 causes a loss of Mps1p kinase activity but does not affect protein stability. These findings demonstrate a requirement for CDC37 function in normal SPB duplication and may also suggest a more general role for this gene in G1 control.
Materials and Methods
Yeast Strains, Cell Culture, and Genetic Techniques
The yeast strains used in this study are listed in Table I. Yeast culture and genetic and molecular techniques were as described by Ausubel et al. (1994). Escherichia coli DH5α (Sikorski and Hieter, 1989) was cultured and transformed as described by Ausubel et al. (1994). Yeast 5-fluoroorotic acid (5-FOA) plates were prepared and used as described by Boeke et al. (1987) to select against yeast strains bearing a wild-type copy of the URA3 gene. The cdc37-1 strain 5962-1-1b was successively outcrossed to S288c-derived wild-type strains (Table I) until the G1 arrest phenotype of cells shifted to the nonpermissive temperature behaved consistently, as judged by budding index and flow cytometric determination of DNA content.
Table I.
Yeast Strain List
| Strain | Genotype | Source | ||
|---|---|---|---|---|
| 5962-1-1b | α cdc37-1 his4 ade2 lys2 trp1 tyr1 SUP4 | B. Byers* | ||
| AS126-2b | α mps1-1237 ura3-52 leu2-3, 112 his3Δ200 | This study‡ | ||
| AS138-11a | α cdc37-1 leu2-3, 112 ade2 | This study | ||
| AS146-1b | α cdc37-1 mps1-1 ura3-52 leu2-3, 112 hisΔ200 trp1Δ1; pC166NΔ (CDC37-URA3-2μ) | This study | ||
| AS146-26b | α cdc37-1 ura3-52 leu2-3, 112 lys2 | This study | ||
| AS151-4a | a cdc37-1 mps1-737 ura3-52 leu2-3, 112 his3Δ200 | This study‡ | ||
| AS153-12d | α cdc37-1 mps1-1237 ura3-52 leu2-3, 112 lys2 | This study‡ | ||
| AS156-3b | a cdc37-1 mps1-412 ura3-52 leu2-3, 112 trp1Δ1 lys2 | This study§ | ||
| AS157-10a | a cdc37-1 mps1-3796 ura3-52 leu2-3, 112 | This study‖ | ||
| AS158-2a | a cdc37-1 ura3-52 leu2-3, 112 his3Δ200 lys2 | This study | ||
| AS164-13a | α cdc37-2 mps1-737 ura3-52 leu2-3, 112 lys2 aro1 | This study‡ | ||
| AS165-3a | a cdc37-2 mps1-1237 ura3-52 leu2-3, 112 his3Δ200 aro1 | This study‡ | ||
| AS166-5b | a cdc37-2 mps1-412 ura3-52 leu2-3, 112 trp1 aro1 | This study§ | ||
| AS167-6b | a cdc37-2 mps1-3796 ura3-52 leu2-3, 112 his3Δ200 trp1 | This study‖ | ||
| AS171-3b | a cdc37-2 mps1-1 ura3-52; pC166NΔ (CDC37-URA3-2μ) | |||
| AS177-11b | a cdc37-1 mps2-1 ura3-52 leu2-3, 112 | This study | ||
| AS177-11b/p37 | a cdc37-1 mps2-1 ura3-52 leu2-3, 112; pC166NΔ (CDC37-URA3-2μ) | This study | ||
| AS217-5b | α cdc37-1 mps1-6 ura3-52 leu2-3, 112 | This study ¶ | ||
| AS218-9c | a cdc37-2 mps1-6 ura3-52 trp1Δ1 ; pC165 (CDC37-URA3-2μ) | This study ¶ | ||
| AS230-7a | a URA3-GAL1-NmycMPS1 leu2-3, 112 his3Δ200 trp1Δ1 ade2 | This study | ||
| AS230-7b | α cdc37-1 URA3-GAL1-NmycMPS1 leu2-3, 112 his3Δ 200 trp1Δ 1 ade2 | This study | ||
| AS231-2b | α cdc37-1 URA3-HIS3-ProA-MPS1 leu2-3, 112 | This study | ||
| AS231-2d | α URA3-HIS3-ProA-MPS1 leu2-3, 112 | This study | ||
| D8BX5CA | a/α ura3-52/ura3-52 leu2-3, 112/leu2-3, 112 his3Δ200/his3Δ200 trp1Δ1/trp1 Δ1 | This study | ||
| S288c | α gal2 | R.K. Mortimer** | ||
| SR674-2 | a cdc37-2 hom2 arol arg4 trp1 | S.I. Reed†† | ||
| Wx241-17a | α mps1-1 ura3-52 leu2-3, 112 his3Δ200 | This study | ||
| Wx257-2b | a ura3-52 leu2-3, 112 trp1Δ1 | This study | ||
| Wx257-2d | a ura3-52 leu2-3, 112 his3Δ200 | This study | ||
| Wx257-4c | α ura3-52 leu2-3, 112 his3Δ200 | This study |
University of Washington, Seattle, WA.
These mps1 alleles originally isolated by E.A. Siewert, University of Colorado, Boulder, CO.
This mps1 allele originally isolated by J.V. Kilmartin, MRC, Cambridge, England.
This mps1 allele originally isolated by T. Huffaker, Cornell University, Ithaca, NY.
This mps1 allele originally isolated by D. Koshland, Carnegie Institute of Washington, Baltimore, MD.
University of California, Berkeley, CA.
Scripps Research Institute, La Jolla, CA.
The mps1-1 strain Wx241-17a was transformed with genomic libraries constructed in the 2μ vectors YEp13 (Nasmyth and Tatchell, 1980) and YEp24 (Carlson and Botstein, 1982). Approximately 12,000 YEp13 library transformants and 96,000 YEp24 library transformants were obtained at permissive temperature. These colonies were replica plated to rich (YPD) medium at 30°C, the minimum nonpermissive temperature for this strain. Ts+ transformants were identified and retested on YPD at 30°C. YEp24 library plasmids were then selected against on 5-FOA medium to identify those isolates that depended upon the plasmid for growth at 30°C, and the plasmids were transferred to E. coli for further analysis. For YEp13 library constructs, plasmid dependence was assessed by loss of the plasmid during nonselective growth and subsequent testing for growth at 30°C.
In addition to 20 reisolations of MPS1, four suppressor constructs were isolated one or more times, for a total of 10 plasmids. Three contained overlapping inserts: pN7 (isolated once), pC165 (isolated three times), and pC166 (isolated five times; see Fig. 1), and they define the SMO1 gene. Creation of a variety of deletions in pC165 and pC166 narrowed the candidate region to the CDC37 open reading frame (ORF). A frameshift mutation at a unique BglII site early in the CDC37 ORF in the deletion construct pC166NΔ (see Fig. 1) abolished suppression. Ability of multicopy CDC37 to suppress other mps1 alleles was tested by transforming the strains with pC166 and testing for growth on YPD at various temperatures.
Figure 1.

SMO1 suppressor activity is provided by the CDC37 ORF. For the deletion (Δ) constructs, SphI and NheI sites from the vector were used to delete portions of the insert. The asterisk indicates the position of the frameshift mutation created in p166NΔBfs (Materials and Methods). Restriction enzyme sites: B, BglII; N, NheI; S, SphI. Suppression of mps1-1 was assessed by the ability of Wx241-17a (Table I) to grow at 30°C, and complementation of the cdc37-1 mutation was assessed in AS138-11a (Table I) at 38°C.
Cell Synchronization and Release from Arrest
Cells were arrested in G1 with α-factor (7–10 μM) obtained by a custom peptide synthesis using F-MOC chemistry on a peptide synthesizer (model 488; Applied Biosystems Inc., Foster City, CA). Efficiency of arrest was monitored by budding index determination. Arrest was deemed successful if >90% of a briefly sonicated sample of 100–200 cells was unbudded and was later confirmed by flow cytometric determination of DNA content. Cells were rinsed and released from the arrest into growth medium without α-factor that was equilibrated to the temperature being used for the release. Reentry into the cell cycle was monitored by budding index and flow cytometry.
Cell cycle arrest of cdc37-1, cdc37-1 mps1, and cdc37-1 mps2-1 strains was accomplished in two ways. For experiments where cells were not prepared for microscopy, asynchronous cultures were placed in fresh growth medium equilibrated to 38°C and incubated for 8 h. Cell cycle arrest was confirmed by budding index and flow cytometry as described above. In some cases, cells were then released into fresh medium at 25°C, with or without addition of α-factor. Because long incubations at the high temperature cause cdc37-1 cells to become enlarged and fragile, care was taken to minimize the time spent at 38°C when cells were to be prepared for electron microscopy or indirect immunofluorescence. For such experiments, cultures were prearrested in S phase at 25°C with 0.1 M hydroxyurea (Sigma Chemical Co., St. Louis. MO) until no unbudded cells remained in a sample of 100–200 cells. When rinsed and released into 38°C medium, this synchronous population traverses the cell cycle and reaches G1 arrest after 6 h rather than 8 h.
Cytological Techniques
Yeast cells were prepared for flow cytometry as described by Hutter and Eipel (1979) using the DNA stain propidium iodide (Sigma Chemical Co.). Stained cells were analyzed on a Becton-Dickinson (Mountain View, CA) FACScan® flow cytometer using the LYSYS software package to obtain and analyze data.
Yeast cells were prepared for thin sectioning as described by Byers and Goetsch (1974, 1975). After sectioning, some sections were stained a second time with uranyl acetate to increase contrast. Serial sections were viewed on an electron microscope (model CM10; Phillips Electronic Instrs. Co., Mahwah, NJ, or model 100 cx; JEOL, U.S.A. Inc., Peabody, MA). Immunofluorescent staining of microtubules was performed as described by Kilmartin and Adams (1984) as modified by Jacobs et al. (1988), using the rabbit polyclonal serum 206-1 (anti–β-tubulin, kindly provided by F. Solomon) (Bond et al., 1986).
Nucleic Acid Techniques
DNA was manipulated by standard techniques as described by Ausubel et al. (1994). Plasmid DNA was prepared from E. coli with Wizard prep kits (Promega Corp., Madison, WI) and from yeast as described by Hoffman and Winston (1987). DNA sequencing was performed with a Sequenase 2.0 kit (United States Biochemical Corp., Cleveland, OH) according to instructions provided by the supplier. To determine the identity of the SMO1 gene, limited sequencing was performed using pSMO1-LEU2 as template. A T7 sequencing primer was used to sequence from the pRS305 polylinker into the insert, beginning at the unique BglII site in the CDC37 gene and proceeding toward the 3′ end.
DNA hybridization was performed with a BglII fragment containing most of the CDC37 ORF and several kilobases of 3′ flanking sequence. DNA was labeled by random-primed incorporation of digoxigenin (DIG)– labeled deoxyuridine triphosphate and detected with the Genius System (Boehringer Mannheim Corp., Indianapolis, IN) as recommended by the supplier. The probe was hybridized to the Olson collection of overlapping genomic clones (Riles et al., 1993; American Type Culture Collection, Rockville, MD) in 5× SSC, 50% deionized formamide, 5% (wt/vol) Genius blocking reagent, 0.1% N-lauroylsarcosine (sodium salt), and 0.02% SDS at 42°C for 16 h. Anti-DIG antibody conjugated to alkaline phosphatase was bound to hybridized DIG-labeled DNA and detected by application of the chemiluminescent substrate Lumiphos 530.
To construct the integrant strain used for genetic mapping, the same BglII fragment was cloned into the integrative LEU2-marked vector pRS305 (Sikorski and Hieter, 1989) at the BamHI site to create pSMO1LEU2. This plasmid was linearized at a unique NheI site in the insert and used to transform the wild-type strain Wx257-2b (Table I). Proper integration was confirmed by genomic Southern blotting analysis. This strain was crossed to the cdc37-1 strain AS138-11a (Table I), and tetrads were dissected to examine the linkage of the LEU2 marker to cdc37-1.
Protein A Tagging of MPS1
The genomic copy of MPS1 was tagged by COOH-terminal in-frame integration of a fragment of the protein A gene encoding the IgG-binding domains, as described in Aitchison et al. (1995). Two primers were designed and used in PCR amplification from the protein A (ProA)/HU plasmid. The first primer (MPS1S) corresponds to the final 69 bp of the ORF of MPS1 (nucleotide numbers 2692–2762) and nucleotides 421–441 of the protein A ORF. Sequence of MPS1S: 5′-CCA CAT ATA TCA CAA GAT GAT CTC AAT GAT GTG GTA GAC ACT GTT TTA AGG AAA TTT GCA GAT TAC AAA ATT GGT GAA GCT CAA AAA CTT AAT-3′. The second primer (MPS1U) contains the reverse complement sequences of the 3′ untranslated region of MPS1, from nucleotide number 2868–2930, and the reverse complement sequences of the URA3 gene (nucleotide numbers 1027–1050). Sequence of MPS1U: 5′-GTA TTC AGT GTC TGT GAC GAA AAA TTG ATT GAT GAT TTA TTT AAA TGA ATA TAT ATA AAT TAG TTT ACT TAT AAT TCT GTT TTT TAG-3′.
The PCR product was gel purified and ethanol precipitated and then transformed directly into the haploid yeast strains Wx257-2d and Wx2574c, or the diploid strain D8BX5CA (Table I). Transformants were verified by Western blot against the protein A tag using rabbit IgG (Sigma Chemical Co.) as primary antibody, by PCR with primers to MPS1 and HIS3, and by linkage to mps1-1.
Protein Techniques and Kinase Assays
To examine ProA-Mps1p stability, cells carrying the tagged allele were grown at the appropriate temperatures and then lysed in 1× Laemmli sample buffer (Ausubel et al., 1994) by vortexing with 0.45–0.52-mm glass beads for 5 min at 4°C. Lysates were separated on 8% SDS-PAGE gels (Ausubel et al., 1994) and then electrophoretically transferred to Immobilon membranes (Millipore Corp., Bedford, MA). ProA-Mps1p was detected on Western blots with rabbit IgG as primary antibody and a horseradish peroxidase–conjugated secondary antibody using the ECL chemiluminescent system (Amersham Corp., Arlington Heights, IL). The half-life of myc-Mps1p was examined by inducing expression of the tagged gene with 4% galactose for 6 h at 25°C to create a pool of tagged protein (Hardwick et al., 1996). Glucose was then added to the culture at 2% to repress the promoter, and half the cells were transferred to 38°C medium. Cell lysates were made and protein stability monitored by Western blot as described for ProA-Mps1p, except that the anti-myc 9E10 antibody (Evan et al., 1985) was used. For both ProA-Mps1p and myc-Mps1p Western blots, equal protein loadings on SDS-PAGE gels were confirmed by staining of duplicate gels with Coomassie brilliant blue.
For ProA-Mps1p kinase assays, cultures were grown at 25° or 38°C, and 6 × 108 cells were collected for each assay. Cells were disrupted by vortexing for 5 min at 4°C in 0.5 ml buffer B (Lauzé et al., 1995) with 300 μl glass beads. The lysate was clarified by centrifuging for 10 min at 13,000 g at 4°C, and the supernatant was incubated with 5% IgG-Sepharose (Pharmacia LKB Biotechnology, Piscataway, NJ) for 2 h at 4°C. The resin was then washed three times in buffer B, five times in TST (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.05% Tween-20), and three times in nondetergent buffer (Lauzé et al., 1995). Immunoprecipitated material was used for kinase assays as described in Lauzé et al. (1995), except that reactions were carried out for 20 min. To quantitate ProA-Mps1p, a small amount of lysate (described above) was separated on SDS-PAGE gels. Proteins were transferred to Immobilon and detected with rabbit IgG as primary antibody, followed by alkaline phosphatase secondary antibody and the quantitative chemiluminescent substrate Attaphos (JBL Scientific, San Luis Obispo, CA). Autoradiograms and Western blots were scanned using a phosphorimager (model Storm 860; Molecular Dynamics, Sunnyvale, CA) and quantitated with the ImageQuaNT analysis package.
Results
Isolation of Multicopy Suppressor Plasmids
Yeast genomic libraries constructed in the multicopy 2μ vectors YEp13 and YEp24 (see Materials and Methods) were transformed into an mps1-1 mutant strain that is ts for growth (Wx241-17a; Table I). Plasmids that conferred growth at 30°C, the lower limit of the nonpermissive temperature for mps1-1, were identified. Only constructs containing wild-type MPS1 enabled growth at temperatures above 30°C. In addition to MPS1 itself, four constructs were identified that could suppress the mutation. Three of these (pN7, from the YEp13 library; and pC165 and pC166, from the YEp24 library) were found by restriction digest to contain overlapping inserts. Restriction maps for pC165 and pC166 are shown in Fig. 1. The gene common to these was designated suppressor of mps one (SMO1). The fourth was unique and was designated SMO2. Because pN7, pC165, and pC166 were stronger suppressors, conferring better growth at 30°C than pN8 (SMO2), this group was chosen for further study.
CDC37 Is a Multicopy Suppressor of mps1-1
Deletion of regions from the pC165 and pC166 genomic inserts identified a 3.5-kb region required for suppression (Fig. 1). A probe from this region was physically mapped by hybridization to the overlapping set of λ genomic clones (Riles et al., 1993). This mapped SMO1 to the Olson clones 5879 (American Type Culture Collection clone 70620) and 5219 (American Type Culture Collection clone 70543), on the right arm of chromosome IV. A frameshift mutation created by filling in and religation of a unique BglII site in pC166NΔ destroyed suppressor activity (Fig. 1). DNA sequence analysis of the region adjacent to this BglII site revealed that the suppressor activity is encoded by the CDC37 gene.
The physical map position on chromosome IV is consistent with the genetic map position of CDC37. Further confirmation of the identity of the gene was achieved in two ways. First, p166NΔ (Fig. 1) was transformed into a cdc37-1 mutant strain (5962-1-1b; Table I) and fully complemented the ts phenotype. Second, the BglII fragment from pC165 (Fig. 1) was placed in the integrative LEU2 vector pRS305 (Sikorski and Hieter, 1989) to create pSMO1-LEU2. This construct was linearized and transformed into the wildtype strain Wx257-2b (Table I) to be integrated at the SMO1 locus. The resulting strain was crossed to cdc37-1 (AS138-11a; Table I), and the integrated LEU2 marker behaved tightly linked to cdc37-1 as expected (data not shown).
In addition to the mps1-1 allele used for the suppressor screen, several other alleles of this gene are available. A group of ts isolates was identified based upon their failure to complement the mps1-1 mutation. An x-ray–induced mitotic recombination assay (Mannis and Mortimer, 1964) was used to divide these isolates into recombination groups, defining a total of at least six distinct alleles: mps1-1, mps1-6, mps1-737, mps1- 1237, mps1-412, and mps1-3796. Multicopy CDC37 was able to suppress only one other allele, mps1-1237 (Table I), and that suppression was weak; these cells grew very poorly at 34°C, which is the minimum nonpermissive temperature for mps1-1237 strains. The ability of CDC37 to act as a bypass suppressor of a mps1 disruption allele was also tested. A 2μ CDC37 plasmid could not rescue the inviability of strains carrying a HIS3marked disruption allele (Lauzé et al., 1995), indicating that CDC37 does not bypass the requirement for MPS1 in mitotic growth.
Two ts alleles of CDC37 have previously been identified, each causing cdc arrest at Start in the G1 phase of the cell cycle (Reed, 1980a , b). We used these alleles to look for additional genetic evidence that MPS1 and CDC37 interact. We found that multicopy MPS1 was unable to suppress the ts phenotype of cdc37-1 or cdc37-2. Double mutant strains were constructed between the two alleles of CDC37 and the six alleles of MPS1 to look for synthetic phenotypes (Table I). These double mutant strains exhibited synthetic phenotypes of varying severity (Table II). Combination of the mps1-1 allele with either cdc37-1 or cdc37-2 was lethal, as was combination of mps1-6 with cdc37-2. When haploid strains carrying these mps1 mutations were mated to cdc37-1 or cdc37-2 haploids and the resulting diploids were sporulated, no viable double mutant segregants were obtained. If the same mps1/MPS1 cdc37/CDC37 diploids were transformed with a 2μ URA3 plasmid bearing CDC37 before sporulation, double mutant segregants could be generated, but these strains were dependent on the plasmid for survival (assayed by inability to grow on 5-FOA medium). Double mutant combinations between mps1-6 and cdc37-1, or between mps1-737 and either cdc37 allele, showed enhanced temperature sensitivity; the nonpermissive temperature for these strains was lowered by several degrees compared to that for each allele alone. These strains also grew very slowly at room temperature. Combination of mps1-1237 or mps1-3796 with cdc37-2 also caused enhanced temperature sensitivity, but these strains grew normally at room temperature. The remaining combinations of alleles caused no synthetic phenotypes.
Table II.
Synthetic Growth Phenotypes of cdc37 mps1 Double Mutants
| mps1 allele | cdc37 allele | |||
|---|---|---|---|---|
| cdc37-1 | cdc37-2 | |||
| mps1-1 | lethality* | lethality* | ||
| mps1-6 | enhanced ts‡ | lethality* | ||
| mps1-412 | none | none | ||
| mps1-737 | enhanced ts‡ | enhanced ts‡ | ||
| mps1-1237 | none | enhanced ts‡ | ||
| mps1-3796 | none | enhanced ts‡ | ||
Strain names and genotypes are listed in Table I.
These strains are inviable and can only be cultured when one wild-type gene is present on a plasmid. ‡ The nonpermissive temperature of these strains is lowered by several degrees relative to that of either allele alone.
Side-by-side SPBs Are Found at cdc37-1 Arrest
Like MPS1, the CDC37 gene might be required for proper SPB duplication. To address this, we examined the state of the SPB at cdc37-1 arrest by electron microscopy of serial thin sections. We initially expected to see a single SPB with a satellite in each cell (Fig. 2 a). The cdc37-1 mutation was originally identified by its ability to arrest the cell cycle at Start in G1, with the characteristic “shmoo” morphology seen in cells responding to mating pheromone (Reed, 1980a ), and we have confirmed this G1 arrest in our strains by cell morphology and flow cytometric analysis of DNA content (data not shown). Such a Start arrest is ultrastructurally defined by the presence of a satellitebearing SPB (Byers and Goetsch, 1974, 1975). These cells are also reportedly somewhat competent to mate at the arrest (Reed, 1980a ), which implies the presence of the single satellite-bearing SPB structure normally required to participate in karyogamy (Byers and Goetsch, 1975).
Figure 2.

cdc37-1 causes an aberrant SPB phenotype. (a) Schematic representation of the SPB duplication pathway (after Byers, 1981a ). The outer and central plaques are shown, and microtubules are depicted only on the nuclear surface of the SPB to show orientation. (b) Normal side-by-side SPB structure seen in a cdc37-1 mutant strain (AS158-2a; Table I) grown at 25°C, the permissive temperature. One complete outer plaque (arrow) and part of the second have been captured in this section. OP, outer plaque; CP, central plaque; B, bridge. The inner plaque is difficult to see under these fixation and staining conditions. (c–e) Side-by-side SPB structures seen in a cdc37-1 mutant strain (AS158-2a; Table I) incubated at 38°C for 6 h. One prominent outer plaque is present in each cell (arrows). In e, a small amount of outer plaque material can be seen on the second SPB (arrowhead). In each case (c–e), examination of serial sections adjacent to the sections shown here revealed no additional outer plaque material. Bars, 0.2 μm.
When cdc37-1 cells (AS158-2a; Table I) were incubated at the nonpermissive temperature and the nuclei examined by electron microscopy, we found duplicated side-byside SPBs joined by a full bridge structure, distinct from the expected satellite-bearing SPBs (Fig. 2, c–e). Of 15 nuclei examined, 14 contained clear side-by-side SPBs and one contained a spindle, consistent with the degree of G1 arrest seen in this experiment by flow cytometry and budding index (data not shown). Although the cells were presumably arrested at Start, their SPBs had proceeded past that point.
Upon examination of a number of side-by-side SPBs from cells at cdc37 arrest, it became apparent that the structure was not completely normal. Although one SPB in each cell appeared intact and all of its expected structures were seen, this was not true of the other. The second SPB had a defective outer plaque structure, which was absent in some nuclei and reduced to varying degrees in others. Examples of the range of this phenotype are shown in Fig. 2, c–e. For comparison, an example of normal side-byside SPBs is shown in Fig. 2 b. The single outer plaque observed on one SPB at cdc37-1 arrest was much more prominent than that seen in wild-type or cdc37-1 cells grown at 25°C, suggesting that accumulation of excess outer plaque material is an aspect of this mutant phenotype. Continued incorporation of material into the SPB and half-bridge during incubation at the nonpermissive temperature has previously been observed in monopolar spindle mutants (Byers, 1981a ; Winey et al., 1991); some of these display very prominent outer plaques. Viability of cdc37-1 cells remained high after 8 h at the nonpermissive temperature. When cells were then plated at the permissive temperature, they recovered from arrest with a haploid DNA content, suggesting that the aberrant SPB structure could somehow be completed or circumvented and a functional spindle could be assembled to carry out nuclear division.
cdc37-1 Arrest Is Interdependent with Start
Since the presence of duplicated side-by-side SPBs in these cells seemed at odds with their previously reported arrest at Start, we reexamined the cdc37-1 arrest phenotype to determine if it was consistent with a Start arrest. Reed (1980a) used a reciprocal shift experiment to examine where the cdc37 arrest point lies in the cell cycle, relative to Start. However, the Coulter counter measurements of cell number used to track cell cycle progression did not demonstrate clear cell cycle arrest, possibly because cdc37 cells become elongated and may fragment during long incubations at the nonpermissive temperature. We repeated this experiment using flow cytometric determination of DNA content as a potentially more accurate way to determine if cells proceeded through the cycle into S phase or remained arrested.
This reciprocal shift experiment (see Hereford and Hartwell, 1974) made use of the mating pheromone α-factor, which causes MAT a cells to arrest in G1 at Start, the point where cells make a commitment to mate or enter the next round of mitotic division. Mating pheromone reversibly blocks SPB duplication at the satellite-bearing SPB stage (Fig. 2 a). The reciprocal shift used two distinct methods to block the cell cycle, α-factor and cdc37 arrest, first in one order and then in the reciprocal order. Behavior of the cells in response to these manipulations allows us to compare the timing of α-factor arrest relative to cdc37 arrest. For the first shift, a population of log phase cdc37-1 cells (AS158-2a; Table I) was arrested in G1 by incubation at the nonpermissive temperature. The cells were then returned to the permissive temperature in the presence of an excess of α-factor. Cell cycle progression was monitored by flow cytometric analysis of DNA content and by cell morphology. In this experiment, the cells remained arrested in G1 (Fig. 3 a), indicating that the cdc arrest point must be before or equivalent to α-factor arrest.
Figure 3.

Reciprocal shift data indicate that the cdc37-1 arrest point is interdependent with Start. (a) A cdc37-1 strain (AS158-2a; Table I) was arrested in G1 by incubation at 38°C for 8 h and then released at 25°C in the presence of α-factor. Samples were taken for flow cytometry at the arrest (0 h) and at 1-h intervals (1 h–4 h). These cells remain G1-arrested. In the control (C; filled curve), arrested cells were released at 25°C with no α-factor and sampled at 4 h. These cells have a 2 N DNA content; they have recovered from cdc37 arrest and moved into G2/M. After 9 h at the permissive temperature, the population has completely recovered and displays a normal cell cycle distribution (not shown). (b) The same strain was arrested at Start with α-factor at 25°C and then released from this arrest at 38°C. Samples were taken at the arrest (0 h) and at 1-h intervals (1 h–4 h). The cells again remain G1 arrested. Drifting of the G1 peak to the right is eliminated when the experiment is performed with rho° derivatives of AS158-2a, indicating that the drift results from accumulation of mitochondrial DNA as the cells become enlarged, not from progression into S phase (not shown). In the control (C; filled curve), α-factor–arrested cells were released at 25°C and sampled at 4 h. The cells have recovered from α-factor arrest and show a typical G1 and G2/M distribution of DNA content. In these histograms, the x-axis is the relative DNA content determined by propidium iodide fluorescence, and the y-axis is the relative number of cells (Materials and Methods). Each sample represents 10,000 cells.
In the reciprocal experiment, log phase cultures of the same strain were exposed to α-factor at room temperature to arrest them at Start. Cells were then removed from mating pheromone and placed at the nonpermissive temperature, and cell cycle progression was monitored as described above. The cells remained arrested in G1 (Fig. 3 b), indicating that the arrest must be subsequent or equivalent to α-mating pheromone arrest. Integration of data from both halves of the reciprocal shift confirms the previous observation that the cdc37-1 arrest point is equivalent to, or interdependent with, Start. However, because of the duplicated or partially duplicated nature of the SPBs, this represents a novel Start arrest.
Epistatic Relationship between CDC37 and MPS1
Although CDC37 and MPS1 have been found to interact genetically, mutations in these two genes have distinct cell cycle and SPB phenotypes. The epistatic relationship between cdc37 and mps1 was examined with respect to both phenotypes. All eight viable combinations between cdc37-1 and the six alleles of mps1 (Table II) were shifted to the nonpermissive temperature to examine their cell cycle phenotype. An mps1 single mutant strain shows no delay in cell cycle progression at high temperature (Winey et al., 1991). In contrast, all cdc37-1 mps1 double mutant strains arrested in G1 as a cdc37-1 single mutant would (Fig. 4 a). Thus, cdc37-1 is epistatic to mps1 for cell cycle phenotype. SPB morphology was examined in one of these strains, a cdc37-1 mps1-1237 double mutant (AS153-12d; Table I). The double mutant strain was incubated at the nonpermissive temperature, and nuclei were examined by electron microscopy. These cells exhibited the mps1 phenotype (Fig. 4, b and c); only one SPB was present, and it lay adjacent to an enlarged and clearly visible half-bridge (Fig. 4 c, arrow; n = 9). Thus, the epistasis for SPB morphology is reversed compared to cell cycle behavior. Both of the epistatic relationships described are consistent with the execution points for these two genes.
Figure 4.
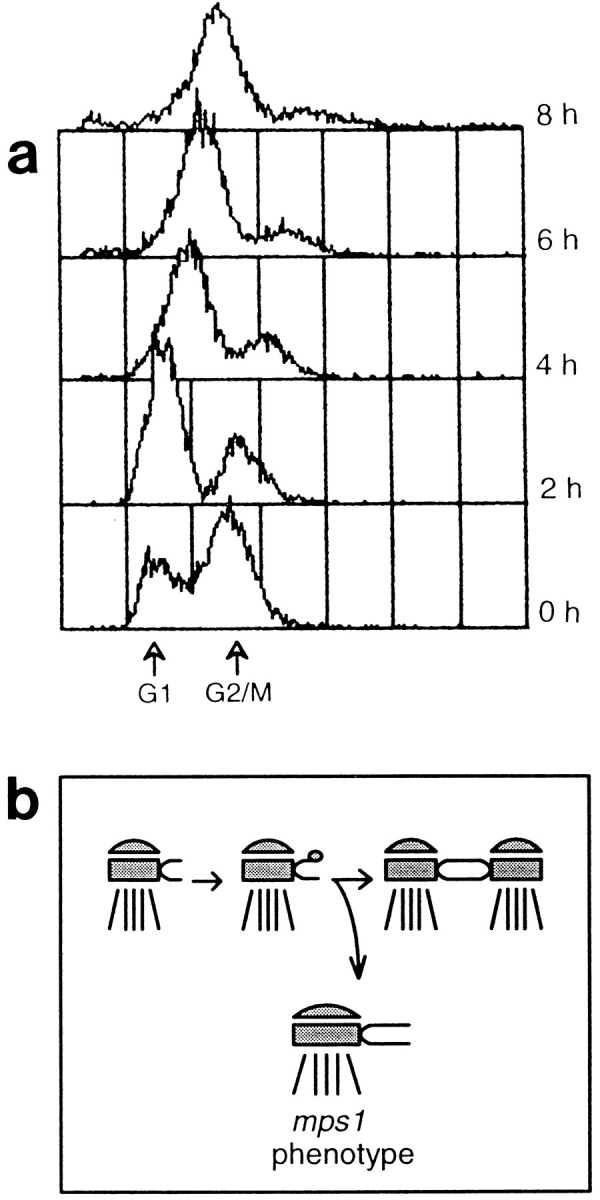

The epistatic relationship between mps1-1237 and cdc37-1 is different for different phenotypes. (a) An asynchronous culture of the mps1-1237 cdc37-1 double mutant strain AS153-12d (Table I) was shifted to 38°C, and cell cycle distribution was monitored by flow cytometry. Samples were taken before the shift (0 h) and at 2-h intervals (2 h–8 h). These cells exhibit the cdc37-1 phenotype; they arrest in G1. A similar G1 arrest is seen in double mutant strains that carry other alleles of mps1. The drift of G1 and G2/M peaks to the right at later time points is attributed to accumulation of mitochondrial DNA as the cells become enlarged. The x-axis is the relative DNA content determined by propidium iodide fluorescence, and the y-axis is the relative number of cells (Materials and Methods). Each sample represents 5,000 cells. (b) Schematic representation of the mps1 SPB phenotype. (c) The same mps1-1237 cdc37-1 strain was incubated at 38°C for 6 h and examined by EM. These cells display the typical mps1 phenotype: a single SPB, with an enlarged halfbridge (arrow). Bar, 0.2 μm.
The Outer Plaque of the Newly Formed SPB Is Defective
Since asymmetry was observed between the two SPBs at cdc37-1 arrest, with one being normal and the other defective, we reasoned that the defect might be specific to either the old or new SPB. It seemed likely that the newly formed SPB would be the defective one, given that the existing SPB had been assembled before CDC37 function was lost and presumably remained intact. To test this idea, we made use of the mps2-1 mutation (Winey et al., 1991) to distinguish between the existing and newly formed SPBs. In mps2-1 strains at the nonpermissive temperature, one SPB is completely normal, but the other—the new SPB—is morphologically distinct. This SPB lacks an inner plaque structure, is not properly inserted into the nuclear envelope (Fig. 5 a), and nucleates only cytoplasmic microtubules. At early time points after shift to the nonpermissive temperature, the mps2 defective SPB can be seen perched on the end of the half-bridge, on its cytoplasmic face. Fig. 5 b shows an example of this morphology in an mps2-1 cdc37-1 double mutant strain with the cdc37 mutation complemented by a wild-type copy of this gene on a plasmid (AS177-11b/p37; Table I). In response to this spindle defect, mps2-1 cells arrest in G2.
Figure 5.


The mps2 mutation causes failure of SPB duplication at a later stage. This phenotype is depicted schematically in a. The old and new SPBs can be distinguished by their location relative to the nuclear envelope and by the absence of nuclear microtubules on the new (defective) SPB. (b) An mps2-1 cdc37-1 strain carrying the wild-type CDC37 gene on a plasmid (AS177-11b/p37; Table I) was incubated at the nonpermissive temperature for 4 h and examined by EM. In this micrograph, the new, defective SPB (arrow) has not yet migrated away from its sibling. Bar, 0.2 μm.
When an mps2-1 single mutant strain is shifted to the nonpermissive temperature, the defective SPB eventually migrates away from its sibling to a distal location on the nuclear envelope periphery. The defective SPB is easily detected in these cells because its cytoplasmic microtubules are seen as a second focus of staining when microtubule organization is observed by immunofluorescent staining of tubulin (Winey et al., 1991). We examined tubulin staining patterns in an mps2-1 cdc37-1 double mutant strain (AS177-11b; Table I) shifted to the nonpermissive temperature to see if the second focus of staining was affected by the cdc37 mutation. If formation of the outer plaque on the defective SPB was disrupted, the structure would no longer be competent to nucleate microtubules. At all time points, only one focus of staining was observed, with the long cytoplasmic microtubule arrays characteristic of a Start arrest (data not shown). This observation suggests that the formation or function of the mps2 defective SPB has been impaired. When the same strain was transformed with CDC37 on a plasmid and then shifted to the nonpermissive temperature, a typical mps2 mutant tubulin staining pattern with two foci was restored (data not shown), and the mps2 SPB phenotype was seen by electron microscopy (as in Fig. 5 b), indicating that background effects in the double mutant strain are not contributing to the phenotype.
To further explore the SPB phenotype, the double mutant strain was shifted to the nonpermissive temperature, and SPB morphology was examined by electron microscopy. The cells arrest in G1; cdc37-1 is epistatic to mps2-1 for cell cycle phenotype (data not shown). Proper position in the nuclear envelope was used to differentiate the old SPB from the new (Fig. 5). By this method we were able to assign the outer plaque defect specifically to the newly formed SPB. In each cell (n = 14), one normal SPB was seen residing within the nuclear envelope, bearing an obvious outer plaque (Fig. 6), leading us to conclude that the existing SPB is unaffected by the cdc37-1 mutation. No other clear SPB structure was present. Instead, a clump of electron-dense material, similar to a satellite but larger, was often seen near the open end of the half-bridge (Fig. 6, arrowheads). This clump varied in size, and in some cases appeared more diffuse than others. We suggest that this clump may represent the remnants of the typical mps2 defective SPB. Migration of this clump away from its sibling SPB was not detected by electron microscopy. Movement of the defective SPB to a distal location in an mps2 single mutant is attributed to cytoplasmic microtubules, and these are absent in the double mutant cells. These observations suggest that CDC37 gene function is required for successful formation of the new SPB.
Figure 6.
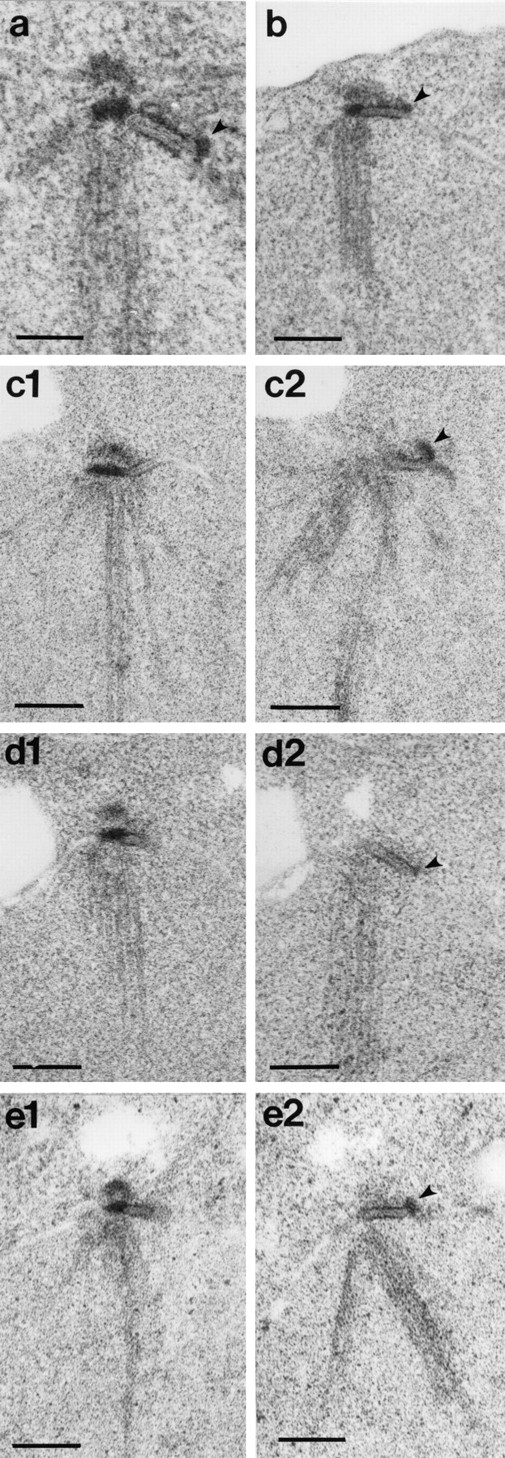
mps2-1 cdc37-1 cells display a synthetic SPB phenotype. The mps2-1 cdc37-1 strain AS177-11b (Table I) was incubated at 38°C for 6 h and examined by EM. In each cell, one SPB appears normal and intact. Adjacent to each half-bridge is a clump of material that may constitute the remnants of the new SPB (arrowheads). This clump varies somewhat in size and shape. In a and b, the entire structure has been captured in one thin section. Each of the remaining pairs of images (c1 and c2; d1 and d2; and e1 and e2) is a set of adjacent serial sections through a single nucleus, where part of the SPB structure was captured in the first section and the remainder can be seen in the next section. Bars: (a) 0.1 μm; (c1–e2), 0.2 μm.
The cdc37-1 Mutation Affects Mps1 Kinase Activity
Loss of CDC37 function has previously been shown to affect the stability of two protein kinases in budding yeast, Cdc28p (Gerber et al., 1995) and the heterologously expressed oncogenic tyrosine kinase pp60v -src (Boschelli et al., 1993; Dey et al., 1996 b). For Cdc28p, reduced steady-state levels of the protein have been observed in a cdc37-1 background (Gerber et al., 1995). We examined the effect of this mutation on Mps1p stability. To do this, Mps1p was tagged at its COOH terminus with the IgG-binding domains of Staphylococcus aureus protein A. This was achieved by integration of the protein A coding sequence into the chromosomal copy of MPS1 just before the stop codon, creating a single-copy tagged allele expressed from the endogenous promoter (Aitchison et al., 1995; see Materials and Methods). The resulting ProA-Mps1p fusion protein was functional; haploid strains that carried only this allele of MPS1 grew normally (data not shown). Endogenous levels of ProA-Mps1p could be detected by Western blotting of whole cell lysates using rabbit IgG as the primary antibody (Materials and Methods). Steady-state levels of ProAMps1p were not affected by the cdc37-1 mutation, even after extended incubation at the nonpermissive temperature (Fig. 7 a).
Figure 7.

Steady-state levels and half-life of Mps1 fusion proteins are unaffected by mutation of CDC37. (a) Steady-state levels of ProA-Mps1p were assessed in wild-type and cdc37-1 strains. AS231-2d and AS231-2b (Table I), which carry the integrated protein A–tagged allele of MPS1, were grown at 25°C and then shifted to 38°C for 4 and 8 h. ProA-Mps1p was detected in whole cell lysates using rabbit IgG as primary antibody. Protein levels were unaffected even after an 8-h incubation at the nonpermissive temperature. (b) The half-life of a pool of galactose-induced myc-Mps1p was monitored in wild-type (AS230-7a) and cdc37-1 (AS230-7b) strains upon shift to the nonpermissive temperature. Expression of the fusion protein was induced with galactose at 25°C for 6 h; glucose was then added to the culture to repress the promoter, and the culture was divided. Cells were incubated at 25 or 38°C and sampled every 15 min for 1 h. Lysates were made and myc-Mps1p detected with 9E10 antibody. No reduction in halflife is seen at the nonpermissive temperature.
It has recently been shown that inactivation of the mammalian homologue of Cdc37p markedly reduces the halflife of the Cdk4 kinase (Stepanova et al., 1996). Because of this result, we examined specifically the contribution of half-life to steady-state levels of Mps1p in wild-type and cdc37-1 cells. This experiment made use of a functional, galactose-inducible myc-tagged MPS1 allele (Hardwick et al., 1996). Wild-type and cdc37-1 cells (AS230-7a and AS230-7b, respectively; Table I) were given a 6-h “pulse” of galactose to induce myc-Mps1p, then “chased” by transfer to medium containing glucose to repress the promoter, and incubated at permissive and nonpermissive temperatures. Stability of myc-Mps1p after the “chase” was monitored by Western blotting with anti-myc antibody. Mutation of CDC37 did not significantly alter the half-life of myc-Mps1p at either temperature (Fig. 7 b). Intriguingly, we found that incubation of the wild-type strain at high temperature caused a modest but reproducible increase in the amount of myc-Mps1p that remained after 6 h of chase (Fig. 7 b), suggestive of some type of a heat shock response contribution to protein stability. We also examined galactose-induced accumulation of myc-Mps1p in cdc37-1 strains induced at the nonpermissive temperature, but we discovered that this mutation causes a general defect in galactose-induced expression similar to that reported for mutations in YDJ1 (data not shown; Dey et al., 1996a ).
Beyond its effect on kinase stability, a requirement for CDC37 to promote the enzymatic activity of protein kinases has been observed; for example, Cdc28p kinase activity is reduced two- to threefold in a cdc37-1 strain (Gerber et al., 1995). With this observation in mind, we examined the effect of the cdc37-1 mutation on Mps1p kinase activity, using the protein A–tagged allele described above. In this experiment, we assayed ProA-Mps1p kinase activity from wild-type and cdc37-1 cells growing at the permissive temperature, and after 1 h of incubation at the nonpermissive temperature. This high-temperature incubation was kept brief to avoid caveats arising from the different cell cycle distributions of actively growing wild-type cultures and cdc37-1 cultures that are becoming arrested in G1. Kinase assays were performed with material immunoprecipitated from wild-type and mutant cells (AS231-2d and AS231-2b, respectively; Table I). Activity was then quantitated with a phosphorimager and normalized for the amount of ProA-Mps1p present in the lysates as determined by Western blotting with rabbit IgG (Materials and Methods).
This analysis demonstrated that autophosphorylation by ProA-Mps1p was compromised in cdc37-1 cells, even when these cells were growing at the permissive temperature (Fig. 8 a). Kinase activity in lysates of cdc37-1 cells was two- to threefold lower than in lysates of wild-type cells. After 1 h at the nonpermissive temperature, cell cycle distribution in the mutant culture was not yet appreciably affected (Fig. 8 b), but kinase activity was reduced even further, to a level four- to fivefold less than that seen in wild-type lysates, a more dramatic effect than reported for Cdc28p (Gerber et al., 1995). This difference in kinase activity was emphasized by the appearance of other, slower-migrating species in the wild-type assays (Fig. 8 a, bracket). Substantial autophosphorylation by Mps1p fusion proteins produces higher molecular weight forms of the protein (Lauzé et al., 1995). In the cdc37-1 assays, where kinase activity was significantly reduced, few hyperphosphorylated species were produced and ProA-Mps1p migrated primarily as a single band (Fig. 8 a, arrow). Although these experiments were performed with epitope-tagged Mps1p, the tagged proteins are functional and for kinase activity experiments were expressed at endogenous levels, so we believe that these data reflect the behavior of wild-type Mps1p. Our observations indicate that loss of Mps1p protein kinase activity contributes to the cdc37-1 mutant phenotype described here and suggest that this effect occurs not through destabilization of Mps1p but by some other type of posttranslational regulation.
Figure 8.

The cdc37-1 mutation reduces ProA-Mps1p kinase activity. (a) Wild-type (AS231-2d, wt) and mutant (AS231-2b, cdc37-1; Table I) strains carrying the protein A–tagged allele of MPS1 were cultured at 25°C, and some cells were shifted to 38°C for 1 h. Cells were lysed, ProA-Mps1p was immunoprecipitated and assayed for autophosphorylation activity, and kinase activity was normalized for the amount of ProA-Mps1p present in the lysates (Materials and Methods). Autophosphorylation signal is reduced in the cdc37-1 strain even at 25°C (two- to threefold lower activity than wild-type), and drops further after incubation at the nonpermissive temperature (four- to fivefold less than wild-type; 1 h 38°C). Normalized activity values from four independent assays were averaged for each strain and temperature. (b) Cell cycle distribution in each culture was monitored by flow cytometry. The reduction in kinase activity seen after 1 h at 38°C occurs before any G1 bias has appeared in the cdc37-1 culture. The x-axis is the relative DNA content determined by propidium iodide fluorescence, and the y-axis is the relative number of cells (Materials and Methods). Each sample represents 10,000 cells.
Discussion
We describe here the identification of CDC37 as an allelespecific multicopy suppressor of a temperature-sensitive mutation in MPS1. These two genes also show allele-specific synthetic lethal interactions. CDC37 was originally identified by Reed (1980a) as a gene required for the execution of Start. Examination of the SPBs in cdc37-1 cells incubated at the nonpermissive temperature reveals an unusual phenotype: SPB duplication has begun in these arrested cells, but they are still mating pheromone responsive. Thus, the cdc37-1 arrest point represents a novel Start arrest, where events at Start have become uncoordinated and the side-by-side SPB structure that is produced is not completely normal. The outer plaque on one SPB is reduced or absent, and the mps2-1 cdc37-1 synthetic phenotype suggests that this defect belongs to the newly formed SPB. Mutation of CDC37 reduces the kinase activity of a ProA-Mps1p fusion protein, indicating that low Mps1p activity may cause this SPB duplication defect.
Cell Cycle and SPB Phenotypes of cdc37-1 Cells
At the nonpermissive temperature, the cdc37-1 mutation confers a unique SPB phenotype. At cdc37-1 arrest, the duplication event has already begun. Two central plaques joined by a full bridge are seen in these cells, and microtubules emanate from the nuclear surfaces of both SPBs. (This phenotype has also been observed by Johnson [1991].) However, the cells harboring this side-by-side SPB structure behave as if they are blocked at Start in that they are responsive to mating pheromone. Thus, this phenotype defines a novel Start arrest. The point of Start has been previously defined by the presence of a single, satellitebearing SPB (Byers and Goetsch, 1974, 1975). In this case, that morphological marker is not present. However, the reciprocal shift experiment examining cdc and α-factor arrest demonstrates that the cells are functionally arrested at Start. One aspect of the cell cycle, SPB duplication, has been uncoupled from other indicators of cell cycle progression through G1.
Although SPB duplication has begun in these cells, it has not been successfully completed. The central plaques appear normal, and both SPBs nucleate microtubules on their nuclear surfaces; however, one structural aspect is obviously abnormal. The outer plaque on one SPB is missing or reduced. Several explanations could account for this defect. The cdc37-1 mutation might cause a faulty duplication event to take place, where an aberrant structure is formed when outer plaque assembly is disrupted and proceeds only partially or not at all. Alternatively, the outer plaque may have been assembled properly but is unstable in some way, and part or all of it is lost during sustained incubation at the nonpermissive temperature. Another possibility is that the cdc37-1 mutation blocks the SPB duplication cycle at an intermediate step in assembly—a step that occurs normally, but has not yet been captured in asynchronous wild-type populations. Satellite-bearing SPBs are seen in cycling populations of cells, but the transition to duplicated side-by-side SPBs is thought to occur rapidly because no structure corresponding to a transition state has been reported (Byers and Goetsch, 1975). In other cdc mutants such as cdc4 and cdc34, which arrest in G1 with two side-by-side SPBs, both SPBs bear outer plaques on their cytoplasmic faces (Byers and Goetsch, 1974; Goebl et al., 1988). It is unclear how the SPBs seen at cdc37-1 arrest could mediate the low level of successful karyogamy reported by Reed (1980a), since that event begins with fusion of unduplicated SPBs beginning at their satellites (Byers and Goetsch, 1975). One possibility is that this incomplete structure can behave as an unduplicated SPB, although not efficiently. Alternatively, our observation that cdc37-1 cells require a long incubation at the nonpermissive temperature to reach uniform arrest might explain the mating competence. The reported mating assays involved a relatively short preincubation at high temperature, and observed mating events might have been contributed by a small number of cells that had not yet reached the arrest.
The fact that only one SPB is affected by the cdc37-1 mutation could result from the presumed conservative nature of the SPB duplication event (Winey et al., 1991; Vallen et al., 1992). Phenotypic analysis of an mps2-1 cdc37-1 double mutant strain allowed us to address this question. The mps2-1 mutation makes the SPBs nonequivalent, with the apparently preexisting SPB marked by its proper location in the nuclear envelope (Winey et al., 1991). Using this marker, we conclude that the existing SPB is unaffected by the cdc37-1 mutation, because the SPB found in the nuclear envelope appears completely normal. The nascent SPB, however, is affected. The defective SPB morphology seen in an mps2-1 single mutant is not observed. Instead, a clump of electron-dense material on the cytoplasmic face of the half-bridge is seen in many cells. We suggest that this material may be the remnants of the new SPB. Given the mps2 phenotype, we would expect the new SPB to lie outside the plane of the nuclear envelope and to bear no microtubules on its “nuclear” face. The structure might also lack an outer plaque because of the cdc37-1 mutation. We might therefore expect to see a clump of electron-dense central plaque material that is discontinuous with the nuclear envelope, consistent with the observed morphology.
Although MPS1 and CDC37 interact genetically, their mutant phenotypes differ with respect to cell cycle behavior and SPB morphology. Epistasis analysis shows that mps1 cdc37 double mutant strains exhibit G1 arrest but display a typical mps1 SPB structure, with a single enlarged SPB and half-bridge. Presence of the mps1 phenotype in the double mutant strain indicates that the mps1 mutation blocks SPB duplication earlier in the pathway than the cdc37-1 mutation (Fig. 9). This precludes expression of the cdc37 phenotype, which requires some later step or steps in duplication to develop. Since mps1 cdc37 strains arrest efficiently in G1, the cdc37-terminal SPB phenotype need not be reached for G1 arrest to occur.
Figure 9.

Proposed SPB duplication pathway, showing the requirement for CDC37 function in timing and execution of this event (adapted from Winey and Byers, 1993). CDC28 and MPS1 act at nearly the same time in the early stages of duplication. The cdc37-1 mutant phenotype may result from effects on both kinases, and CDC37 might also affect CDC28 function later in the cell cycle. The “+” signs indicate both genetic and biochemical links but do not necessarily imply physical interaction between the gene products. Note that the requirement for CDC37 function in SPB duplication occurs later than the requirement for CDC37 in the execution of Start. The aberrant SPB structures in mps2 and cdc37 strains are shown branching off from the pathway at the same point. Because mps2 cdc37 double mutant strains display a synthetic phenotype rather than a clear epistatic relationship, the execution points of these two genes cannot be ordered relative to one another.
The Molecular Role of CDC37 in the Cell Cycle
CDC37 shows interactions not only with MPS1, but with several other protein kinase genes. CDC37 has been linked with genes encoding the cyclin-dependent kinase Cdc28p (Gerber et al., 1995; Reed et al., 1985), Kin28p (Valay et al., 1995), casein kinase II (McCann, R., and C. Glover, personal communication), and mammalian pp60v -src heterologously expressed in yeast (Boschelli et al., 1993; Dey et al., 1996 b). The complicity of CDC37 with this collection of protein kinases has led to the idea that the gene may play a somewhat general role in kinase function, perhaps in protein folding or complex assembly. Circumstantial evidence supports this idea; CDC37 has been implicated in several situations that also involve the action of molecular chaperones. One example derives from the expression of pp60v-src in yeast. Activity of this kinase requires two yeast chaperone genes: HSP90 (Xu and Lindquist, 1993; Nathan and Lindquist, 1995) and YDJ1, a DnaJ homologue (Bohen et al., 1995; Caplan et al., 1995; Kimura et al., 1995; Dey et al., 1996a ); it also requires CDC37 function (Boschelli et al., 1993; Dey et al., 1996 b). Interestingly, we too have found a connection with YDJ1. An allele of this gene was recovered as a synthetic lethal with mps1-1 (Bachant, J., and M. Winey, unpublished observations). A similar link to chaperones is found in Drosophila, where mutation of the fly Cdc37 homologue enhances the sevenless receptor tyrosine kinase mutant phenotype, and mutation of Hsp83 (a member of the Hsp90 family) has the same effect (Cutforth and Rubin, 1994).
A chaperone function for mammalian Cdc37 has recently been demonstrated directly. Stepanova et al. (1996) have found that Cdc37 is the long-known but previously unidentified p50 subunit of the Hsp90 molecular chaperone complex, and they suggest that this protein targets Hsp90 to the Cdk4 kinase. Cdc37/Hsp90 binds the fraction of Cdk4 that is not associated with cyclins, and pharmacological disruption of Cdc37/Hsp90 function in cultured cells decreases Cdk4 half-life. This effect on kinase stability appears to extend to the oncoprotein kinases Raf-1 and pp60v -src as well. Both associate with the p50Cdc37/ Hsp90 complex and are destabilized when that complex is inactivated (Whitesel et al., 1994; Nathan and Lindquist, 1995; Schulte et al., 1995).
Does this stability mechanism hold true in yeast? The relationship between chaperone function and kinase activity has been previously examined for both Cdc28p and pp60v -src. Although kinase stability is affected in both instances, destabilization may not be the primary reason for loss of kinase activity in the cell. Cdc28p levels do drop somewhat in cdc37-1 cells, but while reduction in kinase activity is temperature dependent, the reduced protein level is not. At the nonpermissive temperature, Cdc28p is still present in excess over the Cln2p cyclin but does not bind normally to it, suggesting a role for Cdc37p in assembly of active kinase complexes (Gerber et al., 1995). In the case of pp60v -src, cdc37 and hsp90 mutations can influence both accumulation and activity of the kinase, but these effects are distinct and separable (Nathan and Lindquist, 1995; Dey et al., 1996 b). We now report that Mps1p kinase activity, but not stability, depends upon CDC37 function. Although the epitope tags we used to monitor Mps1p could potentially affect stability, these proteins were functional and behaved similarly with two different tags, suggesting that their properties resemble those of the wildtype protein. The difference observed in CDC37 and/or HSP90 function between yeast and mammals could be explained in several ways. It may be that the mechanisms of action are different in the two organisms, but it is important to bear in mind that unlike Cdk4, direct physical interaction between Cdc28p and Cdc37p has not been reported, and we have not detected binding of Cdc37p to Mps1p (Schutz, A., and M. Winey, unpublished observation). Cdc37p might actually do its work one or more steps away from Mps1p in the SPB duplication pathway, perhaps by stabilizing an upstream kinase, and thus we would not observe a requirement for Mps1p stability. This relationship could be complex; even if Cdc37p does act directly on Mps1p, it may also have indirect effects on Mps1p activity through modulation of other G1 kinases, possibly including Cdc28p. Experimental evaluation of the relative contributions of direct and indirect effects may be difficult.
Multiple requirements for CDC37 function in the cell may be responsible for the unusual phenotype observed in cdc37-1 mutant strains, which includes defects both in coordination of events at Start and in SPB duplication. We and others have shown that the cdc37-1 mutation reduces the in vitro kinase activity of both Cdc28p and Mps1p, enzymes that are required for these events and act very close together in G1 (Fig. 9). A reduced level of phosphorylation by Cdc28p at cdc37-1 arrest, possibly combined with effects on other G1 kinases such as casein kinase II, may cause confusion about the G1 status of a cdc37-1 cell. In this situation, some aspects of the cell cycle would be blocked at Start, but residual Cdc28p activity might be sufficient to trigger the SPB duplication pathway. Why then would SPB duplication not proceed to completion? The structural and genetic evidence presented here suggests that CDC37 functions downstream of MPS1, since a cdc37-1 mutant proceeds considerably farther in SPB duplication than an mps1-1 mutant does before the process stalls, but the requirement of CDC37 for Mps1p kinase activity contradicts this interpretation. We propose that the cdc37-1 SPB phenotype arises from significant but not complete loss of Mps1p kinase activity. Phenotypic analysis of mps1 alleles indicates that this kinase is required not only to begin SPB duplication, but also to carry it to completion (Schutz, A., and M. Winey, unpublished observation). At the nonpermissive temperature, a cdc37-1 mutant strain may retain sufficient kinase activity to initiate duplication, but too little to actually finish the process.
This study addresses G1 functions of CDC37, but this gene may also be important in other stages of the cell cycle. The alleles discussed here were specifically selected for their behavior at Start (Reed 1980a , b) and may not address all the requirements for CDC37. Several of the protein kinases intimate with CDC37 perform functions later in the cell cycle, raising the possibility that CDC37 could also be important after G1. Mps1p participates in a spindle assembly checkpoint in G2/M (Weiss and Winey, 1996), and casein kinase II is also required around this time (Hanna et al., 1995). Similarly, Cdc28p is required to promote cell cycle progression not only at Start, but also in S phase and in G2/M (for review see Nasmyth, 1993; Fig. 9). Gerber et al. (1995) report biochemical evidence for a CDC37 requirement in G2/M: cdc37-1 mutant strains also exhibit a defect in binding and activation of Cdc28p by the mitotic cyclin Clb2p. The recent isolation of CDC37 alleles that affect pp60v -src activity and cause a G2/M cell cycle arrest provides genetic evidence to bolster this idea (Dey et al., 1996 b). A variety of alleles of this gene may be needed to explore and understand the functions of CDC37 in G1 and throughout the cell cycle.
Acknowledgments
We thank Tim Huffaker, John Kilmartin, Doug Koshland, and Elizabeth Siewert for mps1 strains; Steve Reed for the cdc37-2 strain; Frank Solomon for the gift of anti-tubulin antibody; and John Aitchison for the ProA tagging construct. Communication of unpublished data by Jeff Bachant, Rick McCann, and Claiborne Glover is greatly appreciated. We are grateful to Susan Dutcher, Lorraine Pillus, and Rob West for critical reading of the manuscript, to Xueqing Wang and Tom Bier for technical assistance, and to the members of the Winey lab for useful discussions.
Abbreviations used in this paper
- 5-FOA
5-fluoro-orotic acid
- cdc
cell division cycle
- DIG
digoxigenin
- ORF
open reading frame
- ProA
protein A
- SMO1
suppressor of mps one
- SPB
spindle pole body
- ts
temperature sensitive for growth
Footnotes
This work was initiated under an American Cancer Society grant (MV63940) and completed with support from the National Institutes of Health (NIH GM51312). Additional support was from an American Cancer Society JFRA (A70760) and the Pew Scholars Program in the Biomedical Sciences award (P0020SC) to M. Winey. An NIH Training Grant (GM07135) supported A.R. Schutz and E. Steiner. Further support for A.R. Schutz was provided by a National Science Foundation Predoctoral Fellowship and Truman Scholarship, and for E. Steiner by the Achievement Rewards for College Scientists Foundation.
Address all correspondence to Mark Winey, Department of Molecular, Cellular, and Developmental Biology, University of Colorado–Boulder, Boulder, CO 80309-0347. Tel.: (303) 492-3409. Fax: (303) 492-7744. E-mail: Mark.Winey@Colorado.edu
References
- Aitchison JD, Blobel G, Rout MP. Nup120p: a yeast nucleoporin required for NPC distribution and mRNA transport. J Cell Biol. 1995;131:1659–1675. doi: 10.1083/jcb.131.6.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel, F.M., R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl. 1994. Current Protocols in Molecular Biology. John Wiley & Sons, New York.
- Boeke JD, Trueheart J, Natsoulis G, Fink GR. 5-fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 1987;154:64–175. doi: 10.1016/0076-6879(87)54076-9. [DOI] [PubMed] [Google Scholar]
- Bohen SP, Kralli A, Yamamoto KR. Hold'em and fold'em: chaperones and signal transduction. Science (Wash DC) 1995;268:1303–1304. doi: 10.1126/science.7761850. [DOI] [PubMed] [Google Scholar]
- Bond JF, Fridovich-Keil JL, Pillus L, Mulligan RC, Solomon F. A chicken-yeast chimeric β-tubulin protein is incorporated into mouse microtubules in vivo. Cell. 1986;44:461–468. doi: 10.1016/0092-8674(86)90467-8. [DOI] [PubMed] [Google Scholar]
- Boschelli F, Uptain SM, Lightbody JJ. The lethality of p60v -src in Saccharomyces cerevisiae and the activation of p34CDC28kinase are dependent on the integrity of the SH2 domain. J Cell Sci. 1993;105:519–528. doi: 10.1242/jcs.105.2.519. [DOI] [PubMed] [Google Scholar]
- Byers, B. 1981a. Cytology of the yeast life cycle. In The Molecular Biology of the Yeast Saccharomyes: Life Cycle and Inheritance. J.N. Strathern, E.W. Jones, and J.R. Broach, editors. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York. 59–96.
- Byers, B. 1981b. Multiple roles of the spindle pole bodies in the life cycle of Saccharomyces cerevisiae. In Molecular Genetics in Yeast. Alfred Benzon Symposia 16. D. von Wettstein, A. Stenderup, M. Kielland-Brandt, and J. Friis, editors. Munksgaard, Copenhagen. 119–133.
- Byers B, Goetsch L. Duplication of spindle plaques and integration of the yeast cell cycle. Cold Spring Harbor Symp Quant Biol. 1974;38:123–131. doi: 10.1101/sqb.1974.038.01.016. [DOI] [PubMed] [Google Scholar]
- Byers B, Goetsch L. Behavior of the spindles and spindle plaques in the cell cycle and conjugation of Saccharomyces cerevisiae. . J Bacteriol. 1975;124:511–523. doi: 10.1128/jb.124.1.511-523.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan AJ, Langley E, Wilson EM, Vidal J. Hormone-dependent transactivation by the human androgen receptor is regulated by a dnaJ protein. J Biol Chem. 1995;270:5251–5257. doi: 10.1074/jbc.270.10.5251. [DOI] [PubMed] [Google Scholar]
- Carlson M, Botstein D. Two differentially regulated mRNAs with different 5′ ends encode secreted and intracellular forms of yeast invertase. Cell. 1982;28:145–154. doi: 10.1016/0092-8674(82)90384-1. [DOI] [PubMed] [Google Scholar]
- Cutforth T, Rubin GM. Mutations in Hsp83 and cdc37 impair signaling by the sevenless receptor tyrosine kinase in Drosophila. . Cell. 1994;77:1027–1036. doi: 10.1016/0092-8674(94)90442-1. [DOI] [PubMed] [Google Scholar]
- Dey B, Caplan AJ, Boschelli F. The Ydj1 molecular chaperone facilitates formation of active p60v -srcin yeast. Mol Biol Cell. 1996a;7:91–100. doi: 10.1091/mbc.7.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey B, Lightbody JJ, Boschelli F. b. CDC37 is required for p60v-srcactivity in yeast. Mol Biol Cell. 1996;7:1405–1417. doi: 10.1091/mbc.7.9.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douville EMJ, Afar DEH, Howell BW, Letwin K, Tannock L, Bendavid Y, Pawson T, Bell JC. Multiple cDNAs encoding the eskkinase predict transmembrane and intracellular enzyme isoforms. Mol Cell Biol. 1992;12:2681–2689. doi: 10.1128/mcb.12.6.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutcher SK, Hartwell LH. The role of S. cerevisiaecell division cycle genes in nuclear fusion. Genetics. 1982;100:175–184. doi: 10.1093/genetics/100.2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutcher SK, Hartwell LH. Genes that act before conjugation to prepare the Saccharomyces cerevisiaenucleus for caryogamy. Cell. 1983;33:203–210. doi: 10.1016/0092-8674(83)90349-5. [DOI] [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsey G, Bishop JM. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson J, Ho J-Y, Peterson TA, Reed SI. Nucleotide sequence of the yeast cell division cycle start genes CDC28, CDC36, CDC37, and CDC39, and a structural analysis of the predicted proteins. Nucleic Acids Res. 1986;14:6681–6697. doi: 10.1093/nar/14.16.6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber MR, Farrell A, Deshaies RJ, Herskowitz I, Morgan DO. Cdc37 is required for association of the protein kinase Cdc28 with G1 and mitotic cyclins. Proc Natl Acad Sci USA. 1995;92:4651–4655. doi: 10.1073/pnas.92.10.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebl MG, Yochem J, Jentsch S, McGrath JP, Varshavsky A, Byers B. The yeast cell cycle gene CDC34encodes a ubiquitin-conjugating enzyme. Science (Wash DC) 1988;241:1331–1335. doi: 10.1126/science.2842867. [DOI] [PubMed] [Google Scholar]
- Hanna DE, Rethinaswamy A, Glover CVC. Casein kinase II is required for cell cycle progression during G1 and G2/M in Saccharomyces cerevisiae. . J Biol Chem. 1995;270:25905–25914. doi: 10.1074/jbc.270.43.25905. [DOI] [PubMed] [Google Scholar]
- Hardwick KG, Weiss E, Luca FC, Winey M, Murray AW. Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption. Science (Wash DC) 1996;273:953–956. doi: 10.1126/science.273.5277.953. [DOI] [PubMed] [Google Scholar]
- Hereford LM, Hartwell LH. Sequential gene function in the initiation of S. cerevisiaeDNA synthesis. J Mol Biol. 1974;84:445–461. doi: 10.1016/0022-2836(74)90451-3. [DOI] [PubMed] [Google Scholar]
- Hoffman CS, Winston F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. . Gene. 1987;57:267–272. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]
- Hutter KJ, Eipel HE. Microbial determination by flow cytometry. J Gen Microbiol. 1979;113:369–375. doi: 10.1099/00221287-113-2-369. [DOI] [PubMed] [Google Scholar]
- Jacobs CW, Adams AEM, Szaniszlo PJ, Pringle JR. Functions of microtubules in the Saccharomyces cerevisiaecell cycle. J Cell Biol. 1988;107:1409–1426. doi: 10.1083/jcb.107.4.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, S.L. 1991. Ph.D. thesis. University of Washington, Seattle, WA. 130 pp.
- Kellogg DR, Moritz M, Alberts BM. The centrosome and cellular organization. Annu Rev Biochem. 1994;63:639–674. doi: 10.1146/annurev.bi.63.070194.003231. [DOI] [PubMed] [Google Scholar]
- Kilmartin JV. Genetic and biochemical approaches to spindle function and chromosome segregation in eukaryotic microorganisms. Curr Opin Cell Biol. 1994;6:50–54. doi: 10.1016/0955-0674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- Kilmartin JV, Adams AEM. Structural rearrangements of tubulin and actin during the cell cycle of the yeast Saccharomyces. . J Cell Biol. 1984;98:922–933. doi: 10.1083/jcb.98.3.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Yahara I, Lindquist S. Role of the protein chaperone YDJ1in establishing Hsp90-mediated signal transduction pathways. Science (Wash DC) 1995;268:1362–1365. doi: 10.1126/science.7761857. [DOI] [PubMed] [Google Scholar]
- Lauzé E, Stoelcker BS, Luca FC, Weiss E, Schutz AR, Winey M. Yeast spindle pole body duplication gene MPS1encodes an essential dual specificity protein kinase. EMBO (Eur Mol Biol Organ) J. 1995;14:1655–1663. doi: 10.1002/j.1460-2075.1995.tb07154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg RA, Fischer WH, Hunter T. Characterization of a human protein threonine kinase isolated by screening an expression library with antibodies to phosphotyrosine. Oncogene. 1993;8:351–359. [PubMed] [Google Scholar]
- Mannis TR, Mortimer RK. Allelic mapping in yeast by x-ray induced mitotic reversion. Science (Wash DC) 1964;143:581–582. doi: 10.1126/science.143.3606.581. [DOI] [PubMed] [Google Scholar]
- Mills GB, Schmandt R, McGill M, Amendola A, Hill M, Jacobs K, May C, Rodricks A, Campbell S, Hogg D. Expression of TTK, a novel human protein kinase, is associated with cell proliferation. J Biol Chem. 1992;267:16000–16006. [PubMed] [Google Scholar]
- Nasmyth K. Control of the yeast cell cycle by the Cdc28 protein kinase. Curr Opin Cell Biol. 1993;5:166–179. doi: 10.1016/0955-0674(93)90099-c. [DOI] [PubMed] [Google Scholar]
- Nasmyth KA, Tatchell K. The structure of transposable yeast mating type loci. Cell. 1980;19:753–764. doi: 10.1016/s0092-8674(80)80051-1. [DOI] [PubMed] [Google Scholar]
- Nathan DF, Lindquist S. Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol Cell Biol. 1995;15:3917–3925. doi: 10.1128/mcb.15.7.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poch O, Schwob E, de Fraipont F, Camasses A, Bordonné R, Martin RP. RPK1, an essential yeast protein kinase involved in the regulation of the onset of mitosis, shows homology to mammalian dual-specificity kinases. Mol Gen Genet. 1994;243:641–653. doi: 10.1007/BF00279573. [DOI] [PubMed] [Google Scholar]
- Reed SI. The selection of S. cerevisiaemutants defective in the Start event of cell division. Genetics. 1980a;95:561–577. doi: 10.1093/genetics/95.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SI. The selection of amber mutations in genes required for completion of Start, the controlling event of the cell division cycle of S. cerevisiae. . Genetics. 1980b;95:579–588. doi: 10.1093/genetics/95.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SI, de Barro MA, Lopes, Ferguson J, Hadwiger JA, Ho J-Y, Horwitz R, Jones CA, Lorincz AT, Mendenhall MD, Peterson TA, et al. Genetic and molecular analysis of division control in yeast. Cold Spring Harbor Symp Quant Biol. 1985;50:627–634. doi: 10.1101/sqb.1985.050.01.076. [DOI] [PubMed] [Google Scholar]
- Riles L, Dutchik JE, Baktha A, McCauley BK, Thayer EC, Leckie MP, Braden VV, Depke JE, Olson MV. Physical maps of the six smallest chromosomes of Saccharomyces cerevisiaeat a resolution of 2.6 kilobase pairs. Genetics. 1993;134:81–150. doi: 10.1093/genetics/134.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose MD, Fink GR. KAR1, a gene required for function of both intranuclear and extranuclear microtubules in yeast. Cell. 1987;48:1047–1060. doi: 10.1016/0092-8674(87)90712-4. [DOI] [PubMed] [Google Scholar]
- Schulte TW, Blagosklonny MV, Ingui C, Neckers L. Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J Biol Chem. 1995;270:24585–24588. doi: 10.1074/jbc.270.41.24585. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder M. The spindle pole body of yeast. Chromosoma (Berl) 1994;103:369–380. doi: 10.1007/BF00362281. [DOI] [PubMed] [Google Scholar]
- Stepanova L, Leng X, Parker SB, Harper JW. Mammalian p50Cdc37is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev. 1996;10:1491–1502. doi: 10.1101/gad.10.12.1491. [DOI] [PubMed] [Google Scholar]
- Valay J-G, Simon M, Dubois M-F, Bensaude O, Facca C, Faye G. The KIN28gene is required for both RNA polymerase II mediated transcription and phosphorylation of the Rpb1p CTD. J Mol Biol. 1995;249:535–544. doi: 10.1006/jmbi.1995.0316. [DOI] [PubMed] [Google Scholar]
- Vallen EA, Scherson TY, Roberts T, van Zee K, Rose MD. Asymmetric mitotic segregation of the yeast spindle pole body. Cell. 1992;69:505–515. doi: 10.1016/0092-8674(92)90451-h. [DOI] [PubMed] [Google Scholar]
- Weiss E, Winey M. The Saccharomyces cerevisiae spindle pole body duplication gene MPS1is part of a mitotic checkpoint. J Cell Biol. 1996;132:111–123. doi: 10.1083/jcb.132.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesel L, Mimnaugh EG, DeCosta B, Myers CE, Neckers LN. Inhibition of heat shock protein Hsp90-pp60v -srcheteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci USA. 1994;91:8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winey M, Byers B. Assembly and functions of the spindle pole body in budding yeast. Trends Genet. 1993;9:300–304. doi: 10.1016/0168-9525(93)90247-f. [DOI] [PubMed] [Google Scholar]
- Winey M, Goetsch L, Baum P, Byers B. MPS1 and MPS2: novel yeast genes defining distinct steps of spindle pole body duplication. J Cell Biol. 1991;114:745–754. doi: 10.1083/jcb.114.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winey M, Hoyt MA, Chan C, Goetsch L, Botstein D, Byers B. NDC1: a nuclear envelope component required for yeast spindle pole body duplication. J Cell Biol. 1993;122:743–751. doi: 10.1083/jcb.122.4.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Lindquist S. Heat-shock protein hsp90 governs the activity of pp60v -srckinase. Proc Natl Acad Sci USA. 1993;90:7074–7078. doi: 10.1073/pnas.90.15.7074. [DOI] [PMC free article] [PubMed] [Google Scholar]