

Abstract
The key gluconeogenic enzyme, fructose1,6-bisphosphatase (FBPase), is induced when Saccharomyces cerevisiae are starved of glucose. FBPase is targeted from the cytosol to the yeast vacuole for degradation when glucose-starved cells are replenished with fresh glucose. Several vid mutants defective in the glucose-induced degradation of FBPase in the vacuole have been isolated. In some vid mutants, FBPase is found in punctate structures in the cytoplasm. When extracts from these cells are fractionated, a substantial amount of FBPase is sedimentable in the high speed pellet, suggesting that FBPase is associated with intracellular structures in these vid mutants. In this paper we investigated whether FBPase association with intracellular structures also existed in wild-type cells. We report the purification of novel FBPase-associated vesicles from wild-type cells to near homogeneity. Kinetic studies indicate that FBPase association with these vesicles is stimulated by glucose and occurs only transiently, suggesting that these vesicles are intermediate in the FBPase degradation pathway. Fractionation analysis demonstrates that these vesicles are distinct from known organelles such as the vacuole, ER, Golgi, mitochondria, peroxisomes, endosomes, COPI, or COPII vesicles. Under EM, these vesicles are 30–40 nm in diam. Proteinase K experiments indicate that the majority of FBPase is sequestered inside the vesicles. We propose that FBPase is imported into these vesicles before entering the vacuole.
The key regulatory enzyme in the gluconeogenesis pathway, fructose-1,6-bisphosphatase (FBPase)1, is induced when Saccharomyces cerevisiae are grown in medium containing poor carbon sources (Gancedo, 1971). When cells are transferred to medium containing fresh glucose, FBPase is rapidly inactivated and degraded (Gancedo, 1971; Chiang and Schekman, 1991). Regulated degradation of FBPase is mediated by a selective targeting of FBPase from the cytosol to the lysosome (vacuole) for degradation (Chiang and Schekman, 1991, 1994; Chiang et al., 1996). The glucose-induced FBPase redistribution into the vacuole has been demonstrated by cell fractionation, immunofluorescence microscopy, and immunoelectron microscopy (Chiang and Schekman, 1991, 1994; Chiang et al., 1996).
Glucose also inactivates peroxisomal enzymes in Hasenula polymorpha, Pichia pastoris, Candida boidinii, or S. cerevisiae (Bormann and Sahm, 1978; Veenhuis et al., 1983; Tuttle and Dunn, 1995; Chiang et al., 1996). Peroxisomes are taken up by the vacuole by autophagy. In P. pastoris, two modes of autophagy operate to deliver peroxisomes to the vacuole for degradation (Tuttle and Dunn, 1995). Peroxisomes can be taken up by the vacuole by microautophagy when cells are transferred from methanol to glucose. When cells are shifted from methanol to ethanol, peroxisomes are surrounded by a layer of membrane, and then internalized by the vacuole by macroautophagy (Tuttle and Dunn, 1995). In addition to FBPase and peroxisomes, the maltose transporter and the galactose transporter are also degraded in the vacuole in response to glucose (Riballo et al., 1995; Chiang et al., 1996). These transporters are delivered to the vacuole by the endocytic pathway, as mutants defective in the endocytic process impair the glucoseinduced degradation of these sugar transporters (Riballo et al., 1995; Chiang et al., 1996). Accumulation of autophagic bodies inside the vacuole has been observed when S. cerevisiae are starved of nitrogen (Takeshige et al., 1992).
Proteins can be sorted to the vacuole through the secretory pathway. The vacuolar protein carboxypeptidase Y (CPY) is synthesized, processed, and transported from the ER to the Golgi (Hasilik and Tanner, 1978; Hemmings et al., 1981; Rothman and Stevens, 1986; Banta et al., 1988; Jones, 1991). Sorting occurs in the late Golgi by the Pep1p/ Vps10p (CPY receptor protein). CPY is delivered to the vacuole through the prevacuolar compartments, and the CPY receptor recycles back to the Golgi compartment (Marcusson et al., 1994; Cooper and Stevens, 1996). Other vacuolar proteins such as aminopeptidase I (API) and α-mannosidase are transported directly from the cytosol to the vacuole, independent of the secretory pathway (Yoshihisa and Anraku, 1990; Klionsky et al., 1992). Mutants defective in API targeting to the vacuole have been isolated. They process and sort CPY normally, suggesting that the API targeting pathway is different from the CPY sorting pathway (Harding et al., 1995).
In mammalian cells, serum starvation induces protein degradation by lysosomes (Dice, 1990; Haynes and Dice, 1996). This protein degradation pathway requires a pentapeptide sequence (Chiang and Dice, 1988; Chiang et al., 1989; Terlecky et al., 1992). Proteins are translocated to the lysosomal lumen by a heat shock protein–mediated process (Terlecky and Dice, 1993; Cuervo et al., 1994). The receptor protein for the selective uptake of RNase A and glyceraldehyde-3-phosphate dehydrogenase by lysosomes has been identified to be the lysosomal glycoprotein LGP96 (Cuervo and Dice, 1996). Overexpression of LGP96 increases the activity of the selective lysosomal degradation pathway both in vivo and in vitro (Cuervo and Dice, 1996).
To study the pathway of FBPase degradation in S. cerevisiae, mutants defective in the glucose-regulated degradation of FBPase were isolated using a colony blotting procedure (Hoffman and Chiang, 1996). The vid (vacuolar import and degradation) mutations are all recessive. They process and sort CPY normally, suggesting that they are distinct from the mutants affecting vacuolar proteolysis (pep), protein secretion (sec), and vacuolar protein sorting (vps). FBPase is inactivated by the cAMP-dependent signal transduction pathway (Lamponi et al., 1987; Toyoda et al., 1987). Since vid mutants all inactivate FBPase at rates indistinguishable from wild-type cells, the vid mutants appear to transduce signal properly. Immunolocalization experiments demonstrated that FBPase is found in the cytosol in most vid mutants. In some vid mutants, FBPase is found in punctate structures in the cytoplasm (Hoffman and Chiang, 1996). When extracts from these cells are further fractionated, a substantial amount of FBPase is sedimentable in the high speed pellet, suggesting that FBPase is associated with intracellular structures in these mutants (Hoffman and Chiang, 1996).
We investigated whether FBPase association with intracellular structures also existed in wild-type cells. Since this association might occur rapidly at 30°C, we monitored FBPase distribution after transfer of wild-type cells to glucose at timed intervals at 22°C, based on our earlier observation that FBPase targeting to the vacuole is delayed at this temperature. We report the purification of FBPase-associated vesicles to near homogeneity from wild-type cells. Proteinase K experiments demonstrate that a substantial amount of FBPase is sequestered inside the vesicles. We propose that FBPase is imported into the vesicles before entering the vacuole.
Materials and Methods
Strains, Media, and Antibodies
The strain used in this study was W303 (MATα leu2-3,112 ade2 his3-200 trp1-1 ura3-52). YPD contained 1% yeast extract (Difco Laboratories, Inc., Detroit, MI), 2% peptone (Difco Laboratories, Inc.), and 2% glucose. YPKG contained 1% yeast extract, 2% peptone, 1% potassium acetate, and 0.5% glucose. Anti-FBPase antibodies were produced by injecting rabbits with affinity-purified FBPase. Anti-Sec21p and Sec22p antibodies were kindly provided by Dr. C. Barlowe (Dartmouth Medical School, Hanover, NH), anti-Sec62p by Dr. T. Rapoport (Harvard Medical School, Boston, MA), anti-Mnn1p by Dr. T. Graham (Vanderbilt University, Nashville, TN), and anti-Pep12p by Dr. S. Emr (University of California at San Diego). Antibodies were used at 2,000 dilution for Sec21p, Sec22p, Mnn1p, and Pep12p, and 10,000 dilution for Sec62p, Pma1p, 3-oxoacyl CoA thiolase, cytochrome C, and FBPase. Antibodies directed against plasma membrane ATPase, 3-oxoacyl CoA thiolase were produced as described (Chiang et al., 1996). Anti-CPY and cytochrome C were produced by injecting rabbits with CPY (Sigma Chemical Co., St. Louis, MO) and cytochrome C (Sigma Chemical Co.).
Cell Fractionation
Yeast cells were grown in 1 liter of YPKG for 2 d at 30°C in an environmental shaker (300 rpm). Cells (OD600 = 10,000) were harvested by centrifugation at 500 g for 5 min and divided. Cells were shifted to YPD for 0, 30, 60, or 90 min at 22°C. At the end of incubation, 10 mM NaN3 was added to the culture medium and yeast cells were collected by centrifugation. Spheroplasts were prepared as described (Hoffman and Chiang, 1996). Spheroplasts were homogenized using a Dounce homogenizer with 20 strokes on ice. Cell lysates were centrifuged first at 15,000 g for 20 min. The supernatant was further centrifuged at 100,000 g for 2 h to obtain the high speed supernatant and pellet. The pellet was resuspended in 1 ml of TEA buffer (10 mM triethanolamine, pH 7.5, 100 mg/ml PMSF, 10 mM NaF, 10 mM NaN3, 1 mM EDTA, and 0.8 M sorbitol), loaded on a 90 cm × 1.5 cm Sephacryl S-1000 (Pharmacia Fine Chemicals, Piscataway, NJ) column, and eluted with 200 ml of TEA buffer with a flow rate of 8 ml/h. Fractions (4 ml) were collected, and 1.5 ml of samples was precipitated with TCA at the final concentration of 10% at 4°C for 1 h on ice. Samples were centrifuged at 13,000 g for 10 min at 4°C, and pellets were washed once with 1 ml ice-cold acetone and resuspended in 50 μl of SDS sample buffer. Proteins (15 μl) were separated on SDS-PAGE gels and immunoblotted with antibodies directed against FBPase and organelles, followed by 1:10,000 dilution of HRP-conjugated anti–rabbit antibodies and exposure on x-ray films after enhanced chemiluminescence (ECL) reactions. For the detection of CPY, Sec21p, plasma membrane ATPase, and Mnn1p, 7.5% SDS-PAGE gels were used; 12.5% SDS-PAGE gels were used for Sec22p and Sec62p, and 15% SDS-PAGE gels were used for cytochrome C. For the detection of FBPase, 3-oxoacyl CoA thiolase, and Pep12p, 10% SDS-PAGE gels were used. Fractions are shown from 16–30.
Sucrose Equilibrium Gradients
Wild-type cells were shifted to glucose for 0, 30, 60, or 90 min at 22°C. Fractions 25–30 were pooled from the S-1000 columns and centrifuged at 100,000 g for 4 h. The pellets were resuspended in 0.5 ml of TEA buffer and loaded onto the top of 20–50% sucrose gradients. The gradients were prepared by adding 3 ml of 50% sucrose, 3 ml of 40% sucrose, 3 ml of 30% sucrose, and 2 ml of 20% sucrose sequentially. Samples were centrifuged at 100,000 g for 20 h at 4°C using a Sorvall TH 641 rotor (Sorvall Instruments Division, DuPont Co., Newton, CT). Samples were collected from the top (0.8 ml), precipitated with TCA, and processed as described above. Proteins were resolved on SDS-PAGE gels and detected by immunoblotting with antibodies specific to organelle markers and FBPase.
Electron Microscopy
Fractions 25–30 were pooled from the S-1000 columns and separated on two sequential 20–50% sucrose gradients. Vesicles were isolated from the second sucrose gradient (fractions 10 and 11) and examined directly by EM. Samples (5 μl) were adsorbed to a carbon-coated grid that was made hydrophilic by a 30-s exposure to a glow discharge in an Auto 306 vacuum evaporator (Edwards High Vacuum, Inc., Grand Island, NY). After a brief rinse with water to wash away excess sucrose, the samples were stained with 1% uranyl acetate for 1 min. The grids were examined using a 1200 EX transmission electron microscope (JEOL USA, Peabody, MA). Micrographs were recorded at magnifications of 40,000 or 60,000.
Proteinase K Treatment
The titration of proteinase K was performed using isolated vesicles (50 μg) incubated with increasing amounts of proteinase K in the absence or presence of 2% Triton X-100 for 30 min on ice. Proteinase K was added at the concentration of 0, 0.5, 1, 1.5, 2, or 2.5 mg/ml in 100-μl reaction mixtures. Reactions were terminated by adding 10% TCA and processed as described. To determine the time course of proteinase K sensitivity, vesicles (50 μg) were isolated from the sucrose gradient and treated with or without proteinase K (2 mg/ml) in the absence or presence of 2% Triton X-100 for 0, 10, 20, 30, or 40 min on ice. Reactions (100 μl) were terminated by adding 10% TCA, and samples were processed as described above. FBPase was detected by immunoblotting. The 75-kD protein was detected by staining with Coomassie blue.
Results
FBPase Is Associated with Intracellular Structures after Glucose Shift
We investigated whether FBPase was associated with intracellular structures in wild-type cells. Since the association may occur rapidly at 30°C, cells were shifted to glucose at 22°C, based on our earlier observation that FBPase targeting to the vacuole is delayed at this temperature. Wild-type cells were shifted to glucose for 0, 30, 60, or 90 min at 22°C. Cells were homogenized and centrifuged to obtain high speed pellets. The pellets were further fractionated on Sephacryl S-1000 columns to separate intracellular organelles based on size. The distribution of FBPase on the S-1000 columns was followed by immunoblotting with anti-FBPase antibodies. We showed previously that FBPase was found in two distinct peaks on the S-1000 columns in vid mutants (Hoffman and Chiang, 1996). A similar distribution of FBPase was also found in wild-type cells. It remains to be determined whether two types of vesicles are involved in the FBPase degradation pathway; we focused on the second FBPase peak and attempted to purify the vesicles from the second peak. Kinetic studies show that, at t = 0 min, very little FBPase was detected on the S-1000 columns in wild-type cells (Fig. 1 a). However, 30 min after glucose shift, FBPase appeared in fractions 26–30 (Fig. 1 b). The amount of FBPase was further increased at t = 60 min (Fig. 1 c), and then decreased at t = 90 min (Fig. 1 d). Under the same conditions, the amount of the vacuolar CPY was constant before and after glucose shift (Fig. 1, e–h).
Figure 1.

Glucose induces FBPase association with intracellular structures. Wildtype cells were shifted to 2% glucose for 0, 30, 60, and 90 min at 22°C. Cells were homogenized and centrifuged, and the high speed pellets were separated on S-1000 columns as described (Hoffman and Chiang, 1996). The second FBPase peak was analyzed (fractions 16–30). Aliquots from each fraction were TCA precipitated, washed, and electrophoresed on SDS-PAGE gels. Proteins were blotted onto nitrocellulose membranes. The positions of FBPase and CPY at t = 0, 30, 60, or 90 min were followed by ECL reactions with anti-FBPase (a–d) and anti-CPY antibodies (e–h).
Organelle Distribution on the S-1000 Columns
We examined the distribution of FBPase and organelles by immunoblotting with antibodies directed against FBPase and organelle markers. We used CPY for the vacuole (Hasilik and Tanner, 1978; Hemmings et al., 1981; Rothman and Stevens, 1986; Banta et al., 1988; Jones, 1991) and Sec62p, an integral membrane on the ER membrane, for the ER (Deshaies and Schekman, 1989, 1990). To detect the Golgi complex, we used antibodies produced against Mnn1p (α-1,3-mannosyltransferase), a membrane protein that marks the medial Golgi compartment (Graham et al., 1994). We determined the positions of peroxisomes and mitochondria by using antibodies directed against 3-oxoacyl CoA thiolase for peroxisomes, and cytochrome C for mitochondria. The plasma membrane was detected by the distribution of the plasma membrane ATPase (Pma1p). We also examined the positions of transport vesicles by immunoblotting techniques. The COPI vesicles are involved in the retrograde and anterograde transport between the ER and the Golgi in yeast (Barlowe et al., 1994; Letourneur et al., 1994; Bednarek et al., 1995). These vesicles were detected by antibodies directed against Sec21p (Bednarek et al., 1995). The ER-derived COPII vesicles were identified using antibodies produced against Sec22p (Barlowe et al., 1994; Bednarek et al., 1995). Endosomes were followed by immunoblotting of fractions with antibodies raised against Pep12p, a syntaxin homologue required for sorting of luminal hydrolases to the vacuole (Becherer et al., 1996). Total protein profile was visualized by Coomassie blue staining.
Fig. 2 shows that, on the S-1000 columns, the fractions containing FBPase also contained most organelles such as the vacuole (CPY), ER (Sec62p), Golgi (Mnn1p), peroxisomes (3-oxoacyl CoA thiolase), and mitochondria (cytochrome C) (Fig. 2, a–f). Fractions 26–30 also contained the COPI vesicles (Sec21p) and the COPII vesicles (Sec22p) (Fig. 2, h and i). The FBPase-containing fractions showed some overlap with the endosomal marker Pep12p (Fig. 2 j). The plasma membrane ATPase (Pma1p) was partitioned primarily in early fractions (Fig. 2 g). Protein staining demonstrated that most proteins were distributed in fractions 25–30 (Fig. 2 k). Therefore, FBPase overlaps significantly with many organelles on the S-1000 columns, with the exception of the plasma membrane marker Pma1p.
Figure 2.
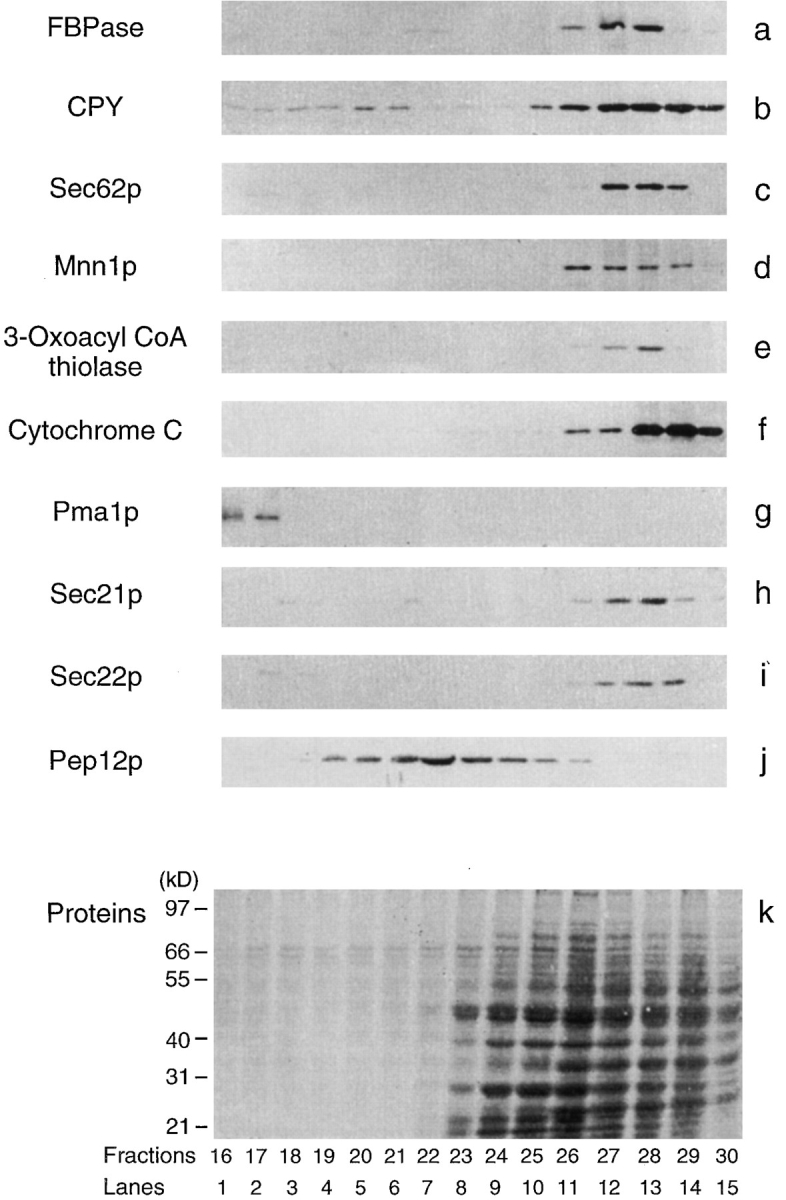
Organelle distribution on the S-1000 columns. The distributions of FBPase and organelles were followed by immunoblotting with antibodies specific to the organelles. CPY for the vacuole (61 kD, b), Sec62p for the ER (30 kD, c), Mnn1p for the Golgi (106 kD, d), 3-oxoacyl CoA thiolase for peroxisomes (45 kD, e), cytochrome C for mitochondria (13 kD, f), Pma1p for the plasma membrane (106 kD, g), Sec21p for COPI vesicles (105 kD, h), Sec22p for COPII vesicles (24 kD, i), and Pep12p for endosomes (35 kD, j). Total protein was monitored by Coomassie blue R-250 staining (k). The cells had been shifted to glucose for 30 min.
Transient Association of FBPase with Dense Structures
To purify the FBPase vesicles, fractions 25–30 from the S-1000 columns were pooled and recentrifuged. High speed pellets were centrifuged to equilibrium on 20–50% sucrose density gradients. The positions of FBPase and CPY after glucose shift were detected by immunoblotting with antibodies directed against FBPase and CPY. The kinetics of FBPase distribution in response to glucose is shown in Fig. 3, a–d. FBPase was undetectable on the sucrose gradients at t = 0 min (Fig. 3 a); the amount was increased at t = 30 min (Fig. 3 b). On the sucrose gradients, the FBPase antigen was in fractions 10–12, which corresponds to a density of ∼1.22 g/cm3. At t = 60 min, two peaks of FBPase were found; one was in fractions 9–12, the other in fractions 1–5 (Fig. 3 c). At t = 90 min, FBPase disappeared from fractions 9–12; a smaller amount of FBPase was found in fractions 2–5 (Fig. 3 d). In contrast with FBPase, the distribution of CPY was not affected by glucose (Fig. 3, e–h), nor the amount of CPY altered by the addition of glucose for up to 90 min. Thus, FBPase distribution to high density fractions 9–12 is induced by glucose, occurs only transiently, and precedes the association with the vacuole.
Figure 3.

Transient association of FBPase with dense structures on the sucrose gradients. Wild-type cells were shifted to glucose for 0, 30, 60, or 90 min at 22°C. Fractions 25–30 were pooled from the S-1000 columns and centrifuged. The pellets were resuspended, loaded onto the top of 20–50% sucrose gradients, and centrifuged at 100,000 g for 20 h. Samples were collected from the top and treated as described in Materials and Methods. The positions of FBPase and CPY at t = 0, 30, 60, or 90 min were followed by immunoblotting with anti-FBPase (a–d) and CPY antibodies (e–h) using ECL reactions.
Purification of the FBPase-associated Vesicles
FBPase was associated transiently with high density structures on the sucrose density gradients. To determine which organelles FBPase was associated with, we examined the distribution of organelle markers and FBPase in a sucrose gradient using immunoblotting with antibodies directed against organelle markers and FBPase. Since FBPase was found in the denser part of the sucrose gradients, the sucrose gradients were designed to separate the FBPase peak away from most organelles. Fig. 4 shows that most organelles were distributed in earlier fractions on the sucrose gradients. The vacuole (CPY), ER (Sec62p), Golgi (Mnn1p), peroxisomes (3-oxoacyl CoA thiolase), mitochondria (cytochrome C), COPI (Sec21p) and COPII (Sec22p) vesicles, and endosomes (Pep12p) were all found in early fractions (Fig. 4, b–i). Coomassie-stained gels showed that most proteins were distributed in the first eight fractions; smaller amounts of proteins were found in the FBPase-containing fractions 9–12 (Fig. 4 j).
Figure 4.
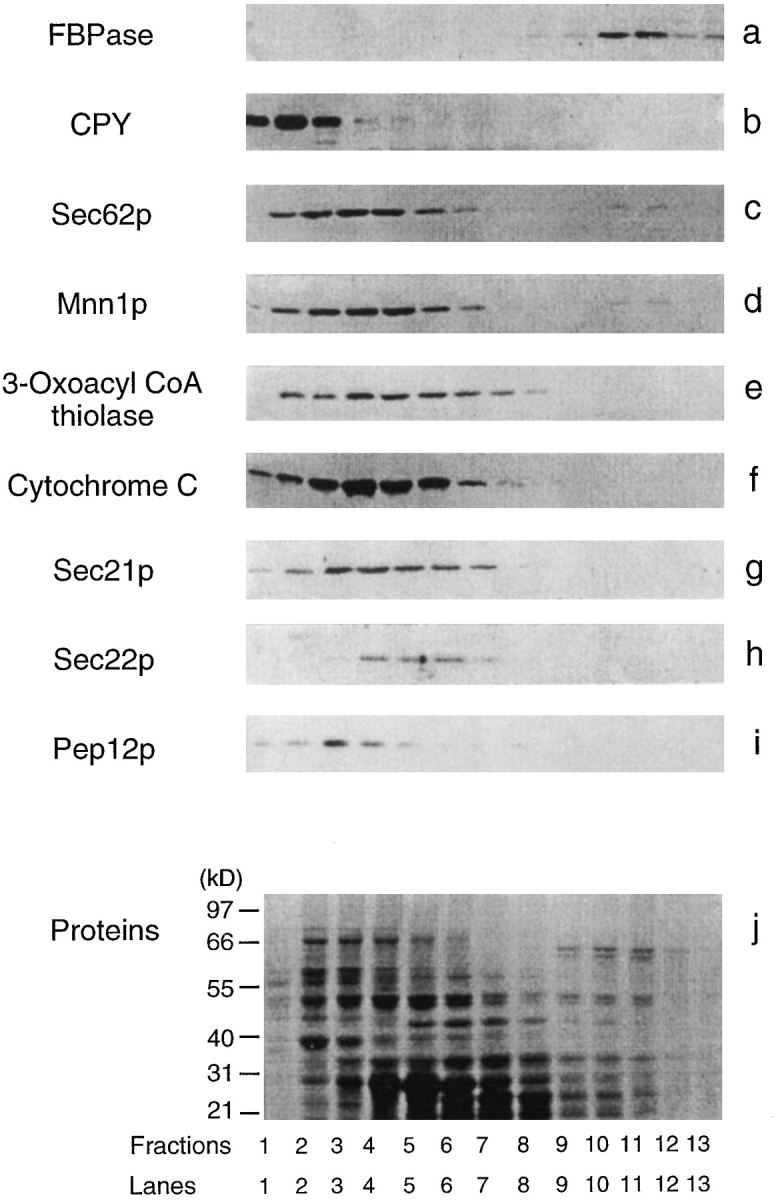
Organelle distribution on the sucrose gradients. Wildtype cells were shifted to glucose for 30 min. Fractions 25–30 from t = 30 min were pooled from the S-1000 columns and resolved on 20–50% sucrose equilibrium gradients. The distributions of various organelles were detected by immunoblotting with antibodies directed against organelle markers described in Fig. 2. Total protein was visualized by staining with Coomassie blue R-250 (j).
Although the sucrose gradient was not optimal to separate most intracellular organelles apart, it did separate the FBPase peak from most organelles (Fig. 4). To minimize the contamination of minor proteins that might be trailed off from other fractions, the FBPase-containing fractions (fractions 10 and 11) from the first sucrose gradient were pooled and refractionated on a second 20–50% equilibrium sucrose gradient. Proteins were separated by SDSPAGE gel electrophoresis and stained with Coomassie blue. Fig. 5 shows that several protein bands were enriched in the FBPase-containing fractions 10 and 11 on the second sucrose gradient, as compared with the protein patterns obtained from the S-1000 column (Fig. 2 k), or the first sucrose gradient (Fig. 4 j). When organelle markers were assayed, FBPase was the only marker that appeared in fractions 10 and 11; the other organelle markers were undetectable on the second sucrose gradient (not shown).
Figure 5.

Protein staining on the second sucrose gradient. Fractions 10 and 11 from the first sucrose gradient were pooled, pelleted at high speed, and recentrifuged on a second equilibrium sucrose gradient. Fractions were collected from the top and processed as described. Proteins were separated by electrophoresis on 10% SDS-PAGE gels and stained by Coomassie blue R-250.
The purity of the FBPase-associated structures was examined by EM. Fractions 10 and 11 from the second sucrose gradient were isolated, stained with uranyl acetate, and visualized by transmission electron microscopy. A homogeneous population of vesicles of 30–40 nm in diam was found by EM (Fig. 6, a and b). At higher magnification, these vesicles appear to be surrounded by a layer of membrane (Fig. 6 b). Since very little contamination of other structures was found under EM, these vesicles have been purified to near homogeneity. Table I summarizes the purification procedure for the isolation of the 30–40-nm vesicles from 10 liters of wild-type yeast culture. After differential centrifugation at 100,000 g and subsequent separation on the S-1000 columns, 4.2% of the proteins were recovered. Upon further purification on two sequential sucrose gradients, 0.1% of the proteins were recovered. Vesicles of the same size and protein composition have also been purified from a vid mutant (not shown).
Figure 6.
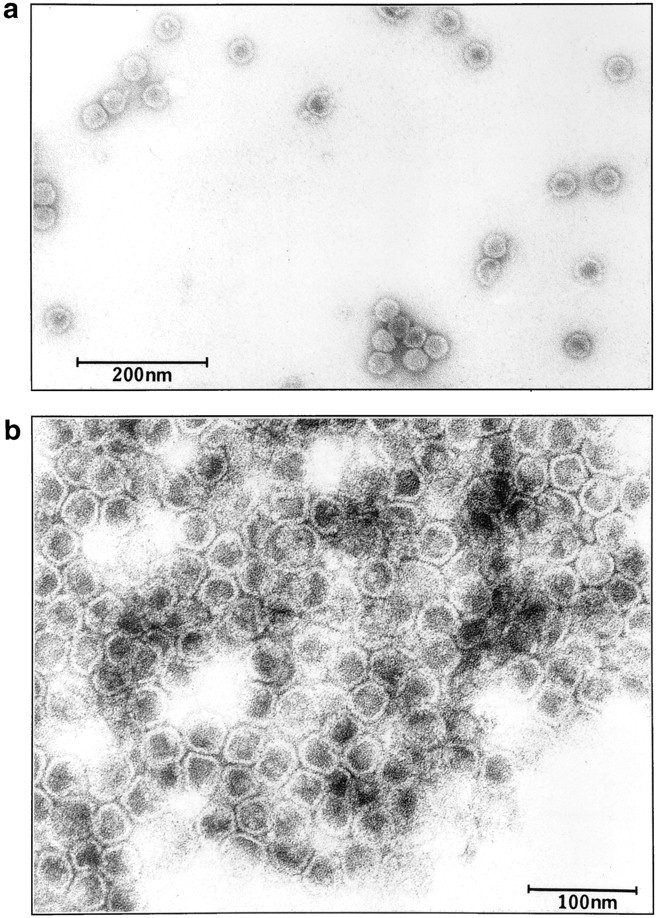
Electron micrograph of purified vesicles. Vesicles were isolated from the second sucrose gradient (fractions 10 and 11). Samples (5 μl) were adsorbed to a carbon-coated grid and examined using a JEOL 1200 EX transmission electron microscope. Bars: (a) 200 nm; (b) 100 nm.
Table I.
Scheme for the Purification of the 30–40-nm Vesicles
| Purification steps | Protein concentrations | Volumes | Protein amounts | Recovery | ||||
|---|---|---|---|---|---|---|---|---|
| mg/ml | ml | mg | % | |||||
| Postnuclear supernatant | 7.9 | 380 | 3,000 | 100 | ||||
| 15,000 g supernatant | 6.8 | 350 | 2,380 | 79.3 | ||||
| 100,000 g pellet | 15.8 | 14 | 221 | 7.4 | ||||
| Fractions 25–30 from | ||||||||
| S-1000 column | 4.2 | 30 | 126 | 4.2 | ||||
| Fractions 10 and 11 from | ||||||||
| first sucrose gradient | 3.6 | 1.6 | 5.8 | 0.2 | ||||
| Fractions 10 and 11 from | ||||||||
| second sucrose gradient | 1.8 | 1.6 | 2.8 | 0.1 |
Wild-type cells were grown in 10 liters of YPKG for 2 d in a fermentor (New Brunswick Scientific Co., Edison, NJ). Cells (OD600 = 100,000) were harvested and shifted to YPD for 30 min at 22°C. Purification procedures were performed as described in Materials and Methods. Protein concentrations were determined by protein assay kits (Bio Rad Laboratories, Hercules, CA).
FBPase Is Sequestered inside the Vesicles
FBPase is localized in the cytosol before transfer of cells to glucose (Chiang and Schekman, 1991; Chiang et al., 1996). In the cytosol, FBPase is sensitive to proteinase K digestion (Chiang and Schekman, 1991). If these vesicles function as carriers to deliver FBPase to the vacuole for degradation, FBPase might enter the lumen of the vesicles. To determine whether this was the case, we performed proteinase K experiments. If FBPase was sequestered inside the vesicles, FBPase would be resistant to proteinase K digestion. By contrast, if FBPase was associated with the vesicles peripherally, FBPase would be sensitive to proteinase K digestion. Since FBPase was the only marker to detect the vesicles, we followed total protein bands by staining gels with Coomassie blue, before and after proteinase K treatment. We found that the 75-kD protein that copurified with FBPase served as a useful control, since the 75-kD protein was readily digested by proteinase K. Fig. 7 a shows that the 75-kD protein was digested by proteinase K at concentrations ranging from 0.5–2.5 mg/ml (Fig. 7 a, lanes 1–6); the sensitivity was not affected whether or not Triton X-100 was present (Fig. 7 a, lanes 7–12). By contrast, FBPase was stable when incubated with 0.5–2.5 mg/ml proteinase K in the absence of Triton X-100 (Fig. 7 a, lanes 1–6). However, when 2% Triton X-100 was added to solubilize the membranes, FBPase became sensitive to proteinase K; it was digested completely by proteinase K at a concentration as low as 0.5 μg/ml (Fig. 7 a, lanes 7–12).
Figure 7.


FBPase is sequestered inside the vesicles. (a) Isolated vesicles (50 μg) were incubated with increasing amounts of proteinase K in the absence (lanes 1–6) or presence of 2% Triton X-100 (lanes 7–12) for 30 min on ice. Proteinase K was added at the concentration of 0, 0.5, 1, 1.5, 2, or 2.5 mg/ml in 100-μl reaction mixtures. Reactions were terminated by adding 10% TCA and processed as described. FBPase was detected by immunoblotting. The 75-kD protein was detected by staining with Coomassie blue R-250. (b) Vesicles (50 μg) isolated from the sucrose gradient were incubated with or without proteinase K (2 mg/ml) in the absence or presence of 2% Triton X-100 on ice for 0, 10, 20, 30, or 40 min. Reactions (100 μl) were terminated by adding 10% TCA, and samples were processed as described above. (Lanes 1–5) Without proteinase K, without Triton X-100. (Lanes 6–10) Without proteinase K, with Triton X-100. (Lanes 11–15) With proteinase K, without Triton X-100. (Lanes 16–20) With proteinase K, with Triton X-100.
We determined the time course of proteinase K sensitivity in the presence or absence of Triton X-100. Isolated vesicles were incubated with or without 2 mg/ml proteinase K for 0–40 min on ice in the absence or presence of 2% Triton X-100. We found that FBPase was stable in the absence of proteinase K for up to 40 min (Fig. 7 b, lanes 1–10). FBPase remained resistant to proteinase K in the absence of Triton X-100 (Fig. 7 b, lanes 11–15). It became sensitive to proteinase K when Triton X-100 was added to solubilize the membranes (Fig. 7 b, lanes 16–20); most FBPase was digested within 10 min of treatment of proteinase K. The 75-kD protein was digested within 10 min by proteinase K, either in the absence or the presence of Triton X-100 (Fig. 7 b, lanes 11–20). These results suggest that FBPase was sequestered inside the vesicles, and the 75-kD protein was not.
Discussion
We provide evidence for the importance of novel vesicles in the degradation of the regulatory enzyme, FBPase, after transfer of wild-type cells from poor carbon sources to glucose. We have purified one type of FBPase-associated vesicles of 30–40 nm in diam to near homogeneity. Kinetic studies indicate that FBPase association with the vesicles is induced by glucose and disappears as FBPase is delivered to the vacuole for degradation. These vesicles are distinct from the vacuole, peroxisomes, mitochondria, ER, Golgi, endosomes, COPI, or COPII vesicles as shown by fractionation experiments. Morphological analysis also demonstrates that the FBPase-associated vesicles are smaller than the early or late endosomes that are 100–400 nm in diam (Singer-Kruger et al., 1993), the COPI or COPII vesicles that are 50 nm in diam (Kaiser and Schekman, 1990; Bednarek et al., 1995), or the post-Golgi vesicles that are 100 nm in diam (Walworth and Novick, 1987).
A substantial amount of FBPase is sequestered inside the vesicles 30 min after transfer of wild-type cells to glucose. If FBPase binds to the surface of the 30–40-nm vesicles first and then enters the lumen of the vesicles, it might be possible to enrich for the vesicles containing FBPase on the surface using mutants defective in FBPase degradation. The vesicles might fuse with the vacuole. After fusion, the sequestered FBPase could be released directly into the lumen of the vacuole for degradation. If a small amount of FBPase is present on the surface of the vesicles, after fusion, FBPase would be distributed on the vacuolar membrane. The membrane-bound FBPase could then be internalized by autophagy. Cloning of VID genes and studies of vid mutants may shed light onto the mechanisms of the FBPase degradation pathway.
Acknowledgments
We thank Maria Ericsson for performing EM work, Kathleen J. Barrett for editing this manuscript, and members in the Chiang laboratory for helpful discussions.
This work was supported by American Cancer Society grant BE-212A to H.-L. Chiang.
Abbreviations used in this paper
- API
aminopeptidase I
- CPY
carboxypeptidase Y
- ECL
enhanced chemiluminescence
- FBPase
fructose-1,6bisphosphatase
Footnotes
Address all correspondence to Hui-Ling Chiang, Department of Cell Biology, Harvard Medical School, 240 Longwood Avenue, Boston, MA 02115. Tel. and Fax: (617) 432-0140. e-mail: chiangne@warren.med.harvard.edu
References
- Banta LM, Robinson JS, Klinosky DJ, Emr S. Organelle assembly in yeast: characterization of yeast mutants defective in vacuolar biogenesis and protein sorting. J Cell Biol. 1988;107:1369–1383. doi: 10.1083/jcb.107.4.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach M, Ravazzola M, Amherdt M, Schekman R. COP II: a membrane coat formed by Sec proteins that drive vesicle budding of the endoplasmic reticulum. Cell. 1994;77:895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- Becherer KA, Rieder SE, Emr SD, Jones EW. Novel syntaxin homologue, Pep12p, required for the sorting of lumenal hydrolases to the lysosome-like vacuole in yeast. Mol Biol Cell. 1996;7:579–594. doi: 10.1091/mbc.7.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarek SY, Ravazzola M, Hosobuchi M, Amherdt A, Schekman R, Orci L. COPI and COPII-coated vesicles bud directly from the endoplasmic reticulum in yeast. Cell. 1995;83:1183–1196. doi: 10.1016/0092-8674(95)90144-2. [DOI] [PubMed] [Google Scholar]
- Bormann C, Sahm H. Degradation of microbodies in relation to activities of alcohol oxidase and catalase in Candida boidinii. . Arch Microbiol. 1978;117:67–72. doi: 10.1007/BF00689353. [DOI] [PubMed] [Google Scholar]
- Chiang H-L, Dice JF. Peptide sequences that target proteins for enhanced degradation during serum withdrawal. J Biol Chem. 1988;263:6797–6805. [PubMed] [Google Scholar]
- Chiang H-L, Schekman R. Regulated import and degradation of a cytosolic protein in the yeast vacuole. Nature (Lond) 1991;350:313–318. doi: 10.1038/350313a0. [DOI] [PubMed] [Google Scholar]
- Chiang H-L, Schekman R. Site of catabolite inactivation. Nature (Lond) 1994;369:284. doi: 10.1038/369283a0. [DOI] [PubMed] [Google Scholar]
- Chiang H-L, Terlecky SR, Plant CP, Dice JF. A role for a 70 kD heat shock protein in lysosomal degradation of intracellular proteins. Science (Wash DC) 1989;246:382–385. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- Chiang H-L, Schekman R, Hamamoto S. Selective uptake of cytosolic, peroxisomal and plasma membrane proteins by the yeast vacuole. J Biol Chem. 1996;271:9934–9941. doi: 10.1074/jbc.271.17.9934. [DOI] [PubMed] [Google Scholar]
- Cooper AA, Stevens TH. Vps10p cycles between the late-Golgi and prevacuolar compartments in its function as the sorting receptor for multiple yeast vacuolar hydrolases. J Cell Biol. 1996;133:529–541. doi: 10.1083/jcb.133.3.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science (Wash DC) 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Terlecky SR, Dice JF, Knecht E. Selective binding and uptake of RNase A and glyceraldehyde-3-phosphate dehydrogenase by isolated rat liver lysosome. J Biol Chem. 1994;269:26374–26380. [PubMed] [Google Scholar]
- Deshaies R, Schekman R. SEC62 encodes a putative membrane protein required for protein translocation into the yeast endoplasmic reticulum. J Cell Biol. 1989;109:2653–2664. doi: 10.1083/jcb.109.6.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies R, Schekman R. Structural and functional dissection of Sec62p, a membrane-bound component of the yeast endoplasmic reticulum protein import machinery. Mol Cell Biol. 1990;10:6024–6035. doi: 10.1128/mcb.10.11.6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. 1990;15:305–309. doi: 10.1016/0968-0004(90)90019-8. [DOI] [PubMed] [Google Scholar]
- Gancedo C. Inactivation of fructose-1,6-bisphosphatase by glucose in yeast. J Bacteriol. 1971;107:401–405. doi: 10.1128/jb.107.2.401-405.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TR, Seeger M, Payne G, MacKay V, Emr S. Clathrin- dependent localization of α-1,3-mannosyltransferase to the Golgi complex of Saccharomyces cerevisiae. . J Cell Biol. 1994;127:667–678. doi: 10.1083/jcb.127.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding TM, Morano KA, Scott SV, Klionsky DJ. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasilik A, Tanner W. Biosynthesis of the vacuolar yeast glycoprotein. Carboxypeptidase Y conversion of precursor into the enzyme. Eur J Biochem. 1978;85:599–608. doi: 10.1111/j.1432-1033.1978.tb12275.x. [DOI] [PubMed] [Google Scholar]
- Haynes S, Dice JF. Roles of molecular chaperones in protein degradation. J Cell Biol. 1996;132:255–258. doi: 10.1083/jcb.132.3.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmings B, Zubenko G, Hasilik A, Jones EW. Mutants defective in processing of an enzyme located in the lysosome-like vacuole of Saccharomyces cerevisiae. . Proc Natl Acad Sci USA. 1981;78:435–439. doi: 10.1073/pnas.78.1.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman M, Chiang H-L. Isolation of degradation-deficient mutants defective in the targeting of fructose-1,6-bisphosphatase into the vacuole for degradation in Saccharomyces cerevisiae. . Genetics. 1996;143:1555–1566. doi: 10.1093/genetics/143.4.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EW. Three proteolytic systems in the yeast Saccharomyces cerevisiae. . J Biol Chem. 1991;266:7963–7966. [PubMed] [Google Scholar]
- Kaiser C, Schekman R. Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell. 1990;61:723–733. doi: 10.1016/0092-8674(90)90483-u. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Cueva R, Yaver DS. Aminopeptidase I of Saccharomyces cerevisiaeis localized to the vacuole independent of the secretory pathway. J Cell Biol. 1992;119:287–299. doi: 10.1083/jcb.119.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamponi S, Galassi P, Tortora P, Guerritore A. Glucose induced degradation of yeast fructose-1,6-bisphosphatase requires additional triggering events besides protein phosphorylation. FEBS Lett. 1987;216:265–269. doi: 10.1016/0014-5793(87)80703-2. [DOI] [PubMed] [Google Scholar]
- Letourneur F, Gayner E, Hennecke S, Demolliere C, Duden R, Emr S, Riezman H, Cosson P. Coatmer is essential for retrieval of dilysinetagged proteins to the endoplasmic reticulum. Cell. 1994;79:1199–1207. doi: 10.1016/0092-8674(94)90011-6. [DOI] [PubMed] [Google Scholar]
- Marcusson EG, Horadovsky BF, Cereghino JL, Gharakhanian E, Emr S. The sorting receptor for yeast vacuolar carboxypeptidase Y is encoded by the VPS10 gene. Cell. 1994;77:579–586. doi: 10.1016/0092-8674(94)90219-4. [DOI] [PubMed] [Google Scholar]
- Riballo E, Herweijer M, Wolf D, Lagunas R. Catabolite inactivation of the yeast maltose transporter occurs in the vacuole after internalization by endocytosis. J Bacteriol. 1995;177:5622–5627. doi: 10.1128/jb.177.19.5622-5627.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman J, Stevens T. Protein sorting in yeast: mutants defective in vacuolar biogenesis mislocalize vacuolar proteins into the late secretory pathway. Cell. 1986;47:1041–1051. doi: 10.1016/0092-8674(86)90819-6. [DOI] [PubMed] [Google Scholar]
- Singer-Kruger B, Frank R, Crausaz F, Riezman H. Partial purification and characterization of early and late endosomes from yeast. J Biol Chem. 1993;268:14376–14386. [PubMed] [Google Scholar]
- Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol. 1992;119:301–311. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terlecky SR, Dice JF. Polypeptide import and degradation by isolated lysosomes. J Biol Chem. 1993;268:23490–23495. [PubMed] [Google Scholar]
- Terlecky SR, Chaing H-L, Oslon TS, Dice JF. Protein and peptide binding and stimulation of in vitro lysosomal proteolysis by 73 kD heat shock cognate protein. J Biol Chem. 1992;267:9202–9209. [PubMed] [Google Scholar]
- Toyoda Y, Fuji H, Miwa I, Okuda J, Sy J. Anomeric specificity of glucose effect on cAMP, fructose-1,6-bisphosphatase and trehalase in yeast. Biochem Biophys Res Commun. 1987;143:212–217. doi: 10.1016/0006-291x(87)90652-8. [DOI] [PubMed] [Google Scholar]
- Tuttle DL, Dunn WA. Divergent modes of autophagy in the methylotrophic yeast Pichia pastoris. . J Cell Sci. 1995;108:25–35. doi: 10.1242/jcs.108.1.25. [DOI] [PubMed] [Google Scholar]
- Veenhuis M, Douma AC, Harder W, Osumi M. Degradation and turnover of peroxisomes in the yeast Hasenula polymorphainduced by selective inactivation of peroxisomal enzymes. Arch Microbiol. 1983;134:193–203. doi: 10.1007/BF00407757. [DOI] [PubMed] [Google Scholar]
- Walworth N, Novick P. Purification and characterization of constitutive secretory vesicles from yeast. J Cell Biol. 1987;105:163–174. doi: 10.1083/jcb.105.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihisa T, Anraku Y. A novel pathway of import of α-mannosidase, a marker enzyme for vacuolar membrane, in Saccharomyces cerevisiae. . J Biol Chem. 1990;265:22418–22425. [PubMed] [Google Scholar]