

Abstract
Utrophin is a large cytoskeletal protein that is homologous to dystrophin, the protein mutated in Duchenne and Becker muscular dystrophy. In skeletal muscle, dystrophin is broadly distributed along the sarcolemma whereas utrophin is concentrated at the neuromuscular junction. This differential localization, along with studies on cultured cells, led to the suggestion that utrophin is required for synaptic differentiation. In addition, utrophin is present in numerous nonmuscle cells, suggesting that it may have a more generalized role in the maintenance of cellular integrity. To test these hypotheses we generated and characterized utrophin-deficient mutant mice. These mutant mice were normal in appearance and behavior and showed no obvious defects in muscle or nonmuscle tissue. Detailed analysis, however, revealed that the density of acetylcholine receptors and the number of junctional folds were reduced at the neuromuscular junctions in utrophin-deficient skeletal muscle. Despite these subtle derangements, the overall structure of the mutant synapse was qualitatively normal, and the specialized characteristics of the dystrophin-associated protein complex were preserved at the mutant neuromuscular junction. These results point to a predominant role for other molecules in the differentiation and maintenance of the postsynaptic membrane.
Utrophin and dystrophin are large (>400 kD), homologous, membrane-associated cytoskeletal proteins (Blake et al., 1996a ; Ahn and Kunkel, 1993). In skeletal muscle, dystrophin binds to a large multimolecular complex (the dystrophin-associated protein complex or DPC)1 that spans the plasma membrane and links the cytoskeleton to the extracellular matrix (Ervasti and Campbell, 1993; Matsumura and Campbell, 1994). Disruption of the dystrophin gene leads to Duchenne or Becker muscular dystrophy (Hoffman et al., 1987). Genes encoding several components of the DPC are now known to be mutated in other congenital muscular dystrophies (Campbell, 1995). Together, these results suggest that dystrophin and the DPC provide crucial structural support to the contracting muscle fiber.
Utrophin was isolated by virtue of its similarity to dystrophin (Love et al., 1989; Khurana et al., 1990). Sequencing revealed the two proteins to be homologous along their entire length (Tinsley et al., 1992). Moreover, like dystrophin, utrophin is transcribed from multiple promoters (Pearce et al., 1993; Blake et al., 1995) and appears to associate with the DPC (Matsumura et al., 1992). Utrophin is expressed in a variety of muscle and nonmuscle tissues in both embryos and adults (Love et al., 1991; Clerk et al., 1993; Koga, 1993; Schofield et al., 1993; Mora et al., 1996; Ohlendieck et al., 1991; thiMan et al., 1991; Khurana et al., 1991). Despite a growing body of knowledge about the distribution and regulation of utrophin its functions remain unknown.
Interest in utrophin has largely centered on three issues. First, in adult skeletal muscle dystrophin is present throughout the sarcolemma whereas utrophin is exclusively associated with the neuromuscular junction (NMJ) (Ohlendieck et al., 1991; thiMan et al., 1991; Khurana et al., 1991; Byers et al., 1991; Sealock et al., 1991; Bewick et al., 1992). This selective localization, along with additional experiments on the expression and effects of utrophin in cultured cells (see Discussion), suggested the hypothesis that utrophin plays a role in synaptogenesis. Moreover, α-dystroglycan, a membrane component of the DPC, binds tightly to agrin, a neurally derived promoter of acetylcholine receptor (AChR) clustering (Campanelli et al., 1994; Gee et al., 1994; Sugiyama et al., 1994; Bowe et al., 1994). Together, these results led to the specific proposal that utrophin might be involved in the formation and/or maintenance of the high density aggregates of AChR with which it precisely codistributes in the postsynaptic membrane (Byers et al., 1991; Sealock et al., 1991; Bewick et al., 1992). Second, utrophin is present is several tissues, such as lung and kidney, in which dystrophin is undetectable (Love et al., 1991; Khurana et al., 1991; Schofield et al., 1993). This differential distribution together with the fact that some components of the DPC exist in these tissues (Ervasti and Campbell, 1993; Durbeej et al., 1995), suggests that utrophin could substitute for dystrophin to form a homologous membrane-spanning complex required for cellular integrity. Indeed, the absence of known spontaneous mutations in the utrophin gene, despite its enormous size (∼1.0 Mb; Pearce et al., 1993), has raised the possibility that its disruption might be embryonically lethal (Blake et al., 1996a ). Third, levels of utrophin are increased in muscles of dystrophindeficient humans and mice (Khurana et al., 1991; Matsumura et al., 1992; Pons et al., 1994b ). It has been suggested that this upregulation serves as a compensatory mechanism; and in fact, the muscles which have the greatest upregulation of utrophin show the least pathological changes in dystrophin-deficient (mdx) mice (Matsumura et al., 1992). If utrophin can compensate functionally for dystrophin, it might be possible to treat patients with Duchenne muscular dystrophy by increasing expression of their normal utrophin gene (Blake et al., 1996a ; Ahn and Kundel, 1993; Matsumura and Campbell, 1994).
As a first step toward testing these hypotheses, we have generated and characterized utrophin-deficient mice. Surprisingly, the mutants were viable and fertile, and displayed no severe abnormalities in muscle and nonmuscle tissues. Detailed analysis of the NMJ showed, however, that utrophin is required for complete differentiation of the postsynaptic membrane.
Materials and Methods
Generation of Mutant Mice
To construct a targeting vector we isolated a 14-kb genomic fragment from a 129 mouse genomic library (Stratagene, La Jolla, CA). The fragment contained a single 75-bp exon that encoded amino acids 2851-2875 of the mouse utrophin protein and corresponded to exon 64 of the dystrophin gene (Fig. 1 a). The long arm of homology in the targeting vector was a 9.5-kb SphI genomic fragment directly upstream of the isolated exon. The short arm was a 1.1-kb BstBI–BamHI fragment that included the distal 25-bp of the exon. These fragments were inserted into cloning sites flanking a PGKneomycin resistance cassette (Tybulewicz et al., 1991). The vector included a thymidine kinase cassette distal to the short arm. The mutant gene therefore lacked the ∼2.7-kb SphI–BstBI fragment which included the first 52 bp of the isolated exon. The vector was linearized with NotI and transferred into R1 type embryonic stem (ES) cells (Nagy et al., 1993) by electroporation. Homologous recombinants were isolated by double selection using G418 and FIAU. Chimeras from two independently derived ES cells gave rise to heterozygous and homozygous mutants. The phenotypes of the two lines were indistinguishable and the results obtained with both lines are presented together.
Figure 1.

Generation of utrophin deficient mice. (a) Structure of the utrophin protein (line 1), a genomic fragment (line 2), the targeting vector (line 3), and the predicted product of homologous recombination (line 4). In line 1, A = actin-binding domain, R = rod domain, and C = cysteine-rich domain (hatched area). The solid box in line 2 represents the targeted exon that encodes the beginning of the cysteine-rich region. The probe used for Southern analysis is indicated in line 4. S, SphI; Bst, BstBI; B, BamHI; Nhe, NheI; Nco, NcoI. (b) Southern blot analysis of NcoI–NheI digested genomic DNA. The 4.2-kb fragment of the wild-type allele was altered to a 2.3-kb fragment in the mutant. (c) RT-PCR analysis of total RNA. Panel 1 shows the PCR results using primers a and a′. A 486-bp wild-type PCR fragment was altered to a 349-bp fragment in the mutant transcript. Sequencing of the PCR product revealed that the 137-bp deletion corresponded to bp 8624-8787 of the utrophin cDNA. Full-length utrophin cDNA was used as the template in lane 1 (wt). Panel 2 shows the location of the PCR primers in relation to utrophin's cysteine-rich domain (hatched box). The smaller bar indicates the region of utrophin protein encoded by the targeted exon, amino acids 2850-2875. The larger bar spans the region of utrophin deleted in the mutant RNA, corresponding to amino acids 2829-2875. This mutation results in a stop codon (*) at amino acid 2884. Both sets of flanking primers (a, a′ and b, b′) detected the same deletion. (d) Immunoblots of protein extracts from muscles and lungs of utrn+/− and utrn−/− animals using antibodies to the NH2 (N-UTR) and COOH-terminal (C-UTR) portion of utrophin and the COOH-terminal portion of dystrophin (DYST). Both utrophin and dystrophin migrate at ∼400 kD.
Molecular Analysis
For Southern analysis, DNA from adult liver was digested with NheI and NcoI and probed with a 32P-labeled BstBI–BamHI fragment of the utrophin genomic clone (Fig. 1 b).
For reverse transcription-polymerase chain reaction (RT-PCR) analysis, total RNA was isolated from adult tissues and cDNA was made using AMV-RT (Promega, Madison, WI) and random primers. Four oligonucleotides, which flanked the mutated exon by 597, 197, 214, and 554 bp (primers b, a, a′, and b′, respectively, in Fig. 1 c), were used pairwise to amplify reverse transcribed RNA from utrn+/− and utrn+/− muscle. The resulting fragments were then sequenced.
For immunoblots, tissue was homogenized and sonicated in extraction buffer (phosphate-buffered saline [PBS], 1% SDS, 5 mM EDTA, and protease inhibitors). Aliquots of a low speed supernatant were analyzed for total protein concentration using a BCA protein assay (Pierce, Rockford, IL); 30 μg of total protein was loaded per lane on 6% SDS polyacrylamide gels.
Histological Analysis
For immunohistochemical analysis, tissue was frozen in liquid nitrogencooled isopentane and sectioned at 7 μm. Sections were stained with antibodies diluted in PBS with 1% BSA for 2–12 h, then rinsed with PBS. Sections were then incubated with a mixture of contrasting flurophores: fluorescein-conjugated goat anti–mouse IgG1 or goat anti–rabbit IgG and rhodamine-conjugated α-bungarotoxin (BTX) or goat anti–rabbit IgG. For thick sections, the sternomastoid was fixed in 1% paraformaldehyde in PBS, cryoprotected in sucrose, frozen, and sectioned en face at 40 μm.
Sources of antibodies were as follows: mouse monoclonal antibodies to utrophin, dystrophin, β-dystroglycan, and α-sarcoglycan (adhalin) were purchased from Novocastra Laboratories Ltd. (Newcastle upon Tyne). A rabbit polyclonal anti-utrophin antibody was made in our laboratory to a peptide corresponding to the final 10 COOH-terminal amino acids from murine utrophin. A rabbit antiserum to dystrobrevin was made to a fusion protein containing a 292–amino-acid fragment from murine dystrobrevin. Sources for antibodies to agrin, laminin-β2, laminin-1, synaptophysin, and rapsyn are described in previous publications from our laboratory (Sanes et al., 1990; Gautam et al., 1995; Gautam et al., 1996). Antibodies to MuSK (DeChiara et al., 1996) and β2-syntrophin (Peters et al., 1994) were generous gifts of G. Yancopoulos (Regeneron) and S. Froehner (University of North Carolina), respectively.
For ultrastructural studies, sternomastoid and intercostal muscles were fixed in 4% glutaraldehyde/4% paraformaldehyde in PBS, washed, refixed in 1% OsO4, dehydrated, and embedded in resin. Thin sections were stained with lead citrate and uranyl acetate. Sections were systematically scanned in the electron microscope, and all NMJs encountered were photographed at 20,000×. Lengths of nerve-muscle apposition were measured from the micrographs, and junctional folds that opened into the primary cleft were counted and expressed as folds per micron of apposition. Results did not differ significantly between sternomastoid and intercostals, so data have been pooled.
Quantitation of AChR
AChR density was assessed by fluorescence imaging, a method devised and described by Turney et al. (1996). Briefly, live mice were anesthetized and the sternomastoid was incubated with a saturating dose of rhodamineBTX for 30 min. The neuromuscular junctions were visualized using a fluorescence microscope and a video camera. Quantitative images were obtained by storing the ratio of junctional fluorescence to that of a fluorescent standard. These images were stored in a digital image processor. For analysis, 2–4 intensity profiles were taken from separate parts of each neuromuscular junction. Dedicated computer software then gave each profile a single value representing its relative illumination intensity. Two utrn+/− (70 intensity profiles from 24 endplates) and two utrn−/− mice (67 intensity profiles from 21 endplates) were studied.
For AChR quantitation by 125I-BTX, the sternomastoid was dissected and placed in oxygenated buffer containing 50 nm 125I-BTX (Amersham). Muscles were incubated for 2 h, washed extensively, and fixed with 4% paraformaldehyde/5% sucrose in PBS. The muscles were then teased into small muscle bundles, endplates were visualized with an anticholinesterase stain, and counted. The small bundles were then divided into two equal sections, one with and one without endplates, and counted in a gamma counter. Specific counts were the difference between the two.
Results
Generation of utrn−/− Mice
Utrophin, like dystrophin, is comprised of four structural domains (Fig. 1 a; Tinsley et al., 1992; Blake et al., 1996): an amino-terminal segment that contains a putative actinbinding site, a long central rod, a highly conserved cysteine-rich region, and a COOH-terminal domain with homology to only two other known proteins, dystrobrevin (Wagner et al., 1993; Blake et al., 1996a ) and DRP2 (Roberts et al., 1996). In addition, both utrophin and dystrophin exist in shorter forms, reflecting both alternative splicing and transcription from multiple promoters (Pearce et al., 1993; Ahn and Kunkel, 1993; Blake et al., 1995, 1996a). These shorter forms lack some or all of the NH2-terminal, central rod, and COOH-terminal domains, but all known forms share the cysteine-rich region (Ahn and Kunkel, 1993; Blake et al., 1996a ). This region is required for binding of dystrophin and, by implication, of utrophin to the DPC (Suzuki et al., 1994; Rafael et al., 1996); furthermore, deletion of this part of dystrophin leads to a severe dystrophic phenotype (Bies et al., 1992). To maximize our chance of disrupting the function of all known utrophin isoforms, we targeted an exon that maps to the beginning of utrophin's cysteine-rich region (Fig. 1 a). The mutation was transferred by homologous recombination to embryonic stem (ES) cells, which were then used to generate germline chimeras. Heterozygous (utrn+/−) mice appeared normal and homozygous (utrn−/−) mice were produced in expected numbers. Southern analysis confirmed disruption of the utrophin gene (Fig. 1 b).
The effects of the mutation on utrophin RNA were assessed by reverse transcription PCR (RT-PCR) (Fig. 1 c). We used primers that flanked the targeted 75-bp exon (corresponding to exon 64 of the dystrophin gene) by 197 and 214 bp. Reverse transcripted RNA from wild-type and mutant muscle was amplified by PCR using these primers. The expected 486-bp fragment was amplified from wild-type muscle, whereas a smaller (349 bp) fragment was amplified from mutant muscle. Sequencing of the PCR products revealed the mutant transcript to contain a 137-bp deletion encompassing not only the targeted exon but the preceding exon as well (corresponding to exon 63 of the dystrophin gene). These exons are located at the start of the cystein-rich region (Fig. 1 c). This deletion leads to a frameshift that introduces a stop codon 1.7-kb before the wildtype stop codon and theoretically results in a truncated utrophin protein missing both the cysteine-rich and the COOH-terminal domains. To seek transcripts with larger deletions, we repeated this analysis with primers that flanked the targeted exon by 597 and 554 bp. Only the 137bp-deleted product was recovered, suggesting that, at least in skeletal muscle, the mutated utrophin gene gives rise only to mRNAs that encode truncated proteins lacking the cysteine-rich and COOH-terminal domains.
Consistent with the RT-PCR analysis, immunoblotting with an antibody specific to the COOH terminus of utrophin failed to react with any protein in utrn−/− skeletal muscle (Fig. 1 d). However, an NH2-terminal specific antibody also failed to detect protein, indicating that truncated utrophin is either not produced or is unstable. Similar results were obtained in adult brain and lung and in neonatal muscle (Fig. 1 d and data not shown). Thus, the utrn−/− mutant we have generated is likely a severe hypomorph for all forms of utrophin.
Immunohistochemical Analysis of utrn+/−and utrn−/− Tissue
Despite the absence of utrophin, utrn−/− mice developed normally, were fertile and have now lived for 10 mo without any obvious pathology. To evaluate sublethal consequences of utrophin-deficiency, we examined several tissues that normally express utrophin: brain, heart, lung, kidney, and skeletal muscle (Love et al., 1989). We first assessed the cellular distribution of utrophin in normal adult tissue. In the brain, utrophin was present in the microvasculature (Fig. 2 a), the choroid plexus and the pia mater, as previously noted by others (Khurana et al., 1992; Uchino et al., 1994). This pattern suggests that utrophin may be a part of the blood-brain barrier. In cardiac muscle, we found utrophin associated with both intercalated discs and sarcolemma (Fig. 2 c); in contrast, a previous report found utrophin only at intercalated discs (Pons et al., 1994a ). In lung, utrophin localized to the membrane of most if not all the cells of the alveoli (Fig. 2 e) but was not detectable in bronchiolar or pulmonary arterial endothelium. In kidney, utrophin was localized to the cells adjacent to the glomerular basement membrane and to the basolateral membrane of a subset of tubules (Fig. 2 g). In skeletal muscle, utrophin was confined to the NMJ (Fig. 3, a and c), as detailed below.
Figure 2.

Immunohistochemical analysis of brain, heart, lung, and kidney from adult utrn+/− and utrn−/− mice. Tissues were doubly stained with a mixture of fluorescein-tagged antibodies to the NH2- and COOH-termini of utrophin (a–h) plus a rhodamine-tagged antibody to laminin-1 (a′–h′). Each pair of micrographs presents two views of a single field photographed with filters selective for each fluorophore. (a and b) A small cerebral vessel; (c and d) a longitudinal section through cardiac myocytes; (e and f) alveoli in lung; and (g and h) a renal glomerulus. In utrn+/− mice, utrophin-rich cells abut laminin-rich basal laminae (a′, c′, e′, and g′). In mutants, utrophin is undetectable, but laminin staining shows that normal tissue architecture is preserved (b′, d′, f′, and h′). Bar in h′ is 50 μm.
Figure 3.
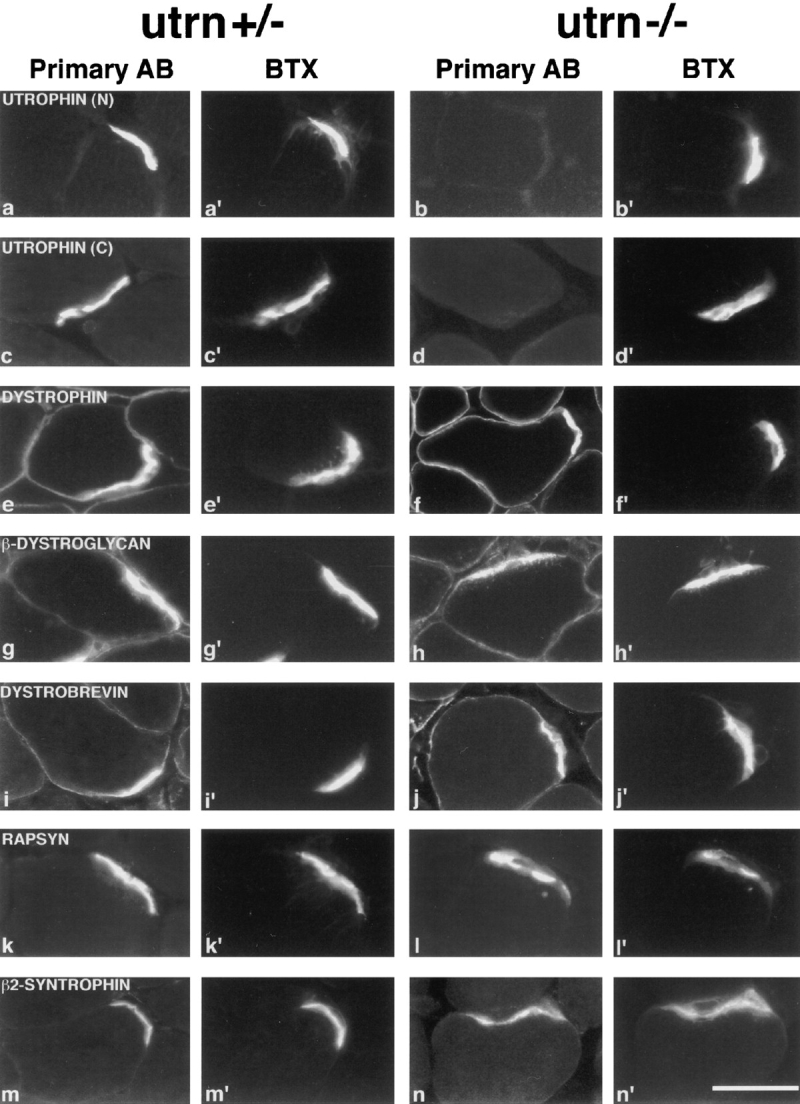
Immunohistochemical analysis of skeletal muscle from adult utrn+/− and utrn−/− mice. Sections of adult skeletal muscle were double stained with a fluorescein-tagged antibody to the protein listed in each row (a–n), plus with a rhodamine-BTX to label AChR (a′–n′). In a–d, N and C denote antibodies to the NH2- and COOH-termini of utrophin, respectively. Each pair of micrographs presents two views of a single field photographed with filters selective for each fluorophore. Utrophin staining was absent in utrn−/− muscle (b and d). The distribution of other proteins was similar in utrn+/− and utrn−/− muscle. Bar in n′ is 20 μm.
In all tissues studied, utrophin-rich membranes abutted basal laminae, as revealed by counterstaining with antibodies to the ubiquitous basal lamina component, laminin (Fig. 2, a′, c′, e′, and g′ and data not shown). Based on this association, we used laminin counterstaining to seek residual utrophin expression in utrn−/− mice and to look for any histological abnormalities in cells that normally express utrophin. No utrophin immunoreactivity was detectable in any of the utrn−/− tissues studied (Fig. 2, b, d, f, and h, and 3, b and d). Moreover, the arrangement of basal laminae and, by implication, of the cells that abut them, were normal in all utrn−/− tissue (Fig. 2, b′, d′, f′, and h′ and data not shown). In support of this conclusion, no structural abnormalities were detected in hematoxylineosin stained sections (data not shown). Similarly, utrophin was undetectable and the tissues histologically normal in heart, brain, lung, and skeletal muscles from neonatal (P1) utrn−/− mice (data not shown).
Limited investigations have failed so far to reveal any significant functional consequences of utrophin-deficiency: (1) Serial observations detected no behavioral, motor or cardiorespiratory defects. (2) Urinalysis showed no significant proteinuria in the mutant animals. (3) Mutant mice had no evidence of a disrupted blood-brain barrier as shown by the exclusion of Evans Blue from the brain parenchyma (Chiueh et al., 1978). (4) Utrophin-deficient mothers bore normal-sized litters and reared their offspring appropriately. (5) Mutant neonates remained viable and active even when separated from their mothers for ⩾4 h.
Decreased AChR Density and Junctional Folds at the utrn−/− Synapse
Given the selective association of utrophin with the NMJ (Fig 3, a and c) and its putative role in synaptogenesis (see Discussion), we suspected that subtle defects might be present and therefore analyzed the NMJ in detail. Labeling the AChR with rhodamine-BTX (Fig. 4, a and b) and the nerve terminals with antibodies to synaptic vesicle proteins (not shown) revealed the utrn−/− synapse to be of normal size and geometry (Fig. 4, a and b). However, the density of AChR was ∼30% lower in utrn−/− than in utrn+/− muscle as assessed by fluorescence-imaging following incubation with rhodamine-BTX (Turney et al., 1996) (utrn−/−: 39.5 ± 0.9, n = 67; utrn+/−: 55.5 ± 1.6, n = 70; illumination intensity ± standard error of the mean, n = number of synaptic images analyzed; P < .0001 by Mann-Whitney). This difference appeared to reflect a moderate decrease in AChR density at most synapses rather than a drastic decrease in density at a specific subset of synapses (Fig. 4 c). Preliminary measurements of a total number of AChR using 125I-BTX gave results suggesting a similar reduction (data not shown). The outwardly normal motor behavior of the utrn−/− mice despite the significant decrease in AChR is consistent with the known safety margin in the number of AChR required for normal muscle function (Patton and Waud, 1967).
Figure 4.

Structure and AChR density of the NMJ in utrn+/− and utrn−/− mice. (a and b) Longitudinal sections of adult muscle stained with rhodamine-BTX. Size and geometry of utrn−/− endplates were similar to those in utrn+/− muscle. (c) Histogram depicting the illumination intensity for each observation made from synapses of utrn+/− and utrn−/− mice. Arrows depict average illumination intensity ± SEM for each genotype. Utrn−/− synapses have ∼30% fewer AChR than utrn+/− synapses. Bar in b is 10 μm for a and b.
Further abnormalities were discovered when muscles were analyzed by electron microscopy. In normal muscle, junctional folds invaginate the postsynaptic membrane; AChR and utrophin are concentrated at the tops of these folds (Sealock et al., 1991; Bewick et al., 1992). In utrn−/− sternomastoid and intercostal muscles, the number of folds was reduced by ∼50% compared to utrn+/− and utrn+/+ synapses (Fig. 5). This difference was apparent by P11 and persisted into adulthood. Despite the decrease in junctional folds, the mutant synapse displayed normal apposition of Schwann cell to nerve and of nerve to muscle as well as normally differentiated nerve terminals (Fig. 5, a–d). Thus, although utrophin is clearly not essential for neuromuscular synaptogenesis, the utrn−/− mice show morphological as well as molecular alterations in their postsynaptic apparatus.
Figure 5.

Ultrastructure of the NMJ in utrn+/− and utrn+/− mice. (a–d) Electron micrographs of NMJs from P11 (a and b) and P50 mice (c and d). Nerve terminals and Schwann cells appeared normal in the mutant, but there were fewer junctional folds in mutant than in control postsynaptic membranes. (e) Average number of folds per micron of primary synaptic cleft in utrn+/+, utrn+/−, and utrn−/− mice at P11 and P50. Each bar shows mean ± SEM of measurements from 37–136 synapses. Bar in d is 1 μm for a–d.
The Dystrophin-associated Protein Complex at utrn−/− Synapses
In skeletal muscle, dystrophin and the dystrophin-associated protein (DPC) join the actin cytoskeleton to the extracellular matrix protein laminin-2 providing structural support to the sarcolemma (Ervasti and Campbell, 1993; Matsumura and Campbell, 1994). The DPC is found at the synapse as well, but in an altered form. Intracellularly, dystrophin and α1-syntrophin are present throughout the sarcolemma, whereas utrophin and β2-syntrophin are exclusively synaptic (Ohlendieck et al., 1991; thiMan et al., 1991; Khurana et al., 1991; Peters et al., 1994). Extracellularly, the β1 chain of the laminin heterotrimer is replaced by laminin-β2 in synaptic basal lamina (Sanes et al., 1990). In addition, agrin, a protein responsible for AChR clustering (Gautam et al., 1996), is concentrated in the synaptic cleft and binds to the extracellular portion of the DPC (Campanelli et al., 1994; Gee et al., 1994; Sugiyama et al., 1994; Bowe et al., 1994). Finally, studies with recombinant proteins have suggested that the synaptic DPC associates with the AChR-associated protein, rapsyn (Apel et al., 1995), which in turn associates with MuSK (Gillespie et al., 1996), a tyrosine kinase receptor thought to mediate the effects of agrin (DeChiara et al., 1996).
The utrophin-deficient mice allowed us to ask whether utrophin is needed for the DPC to acquire its specialized synaptic characteristics. We immunostained for the synapse-specific proteins rapsyn, β2-syntrophin, laminin β2, agrin, and MuSK. All of these antigens were concentrated at synaptic sites in both utrn+/− and utrn−/− mice (Fig. 3, k–n, and data not shown). In addition, we assayed the more broadly distributed DPC proteins β-dystroglycan, dystrobrevin, α-sarcoglycan, and dystrophin. These proteins were also normally distributed in utrn−/− skeletal muscle (Fig. 3, e–j, and data not shown). Thus, the synaptic DPC can exist and maintain its specialized character in the absence of utrophin.
One possible explanation for the maintenance of postsynaptic specialization in the utrn−/− mouse is that a synaptic concentration of dystrophin substitutes for utrophin. Indeed, utrophin and dystrophin are both concentrated at the adult synapse (Fig. 3, a, c, and e), although their precise distribution differs: utrophin colocalizes with AChRs at the crests of the junctional folds whereas dystrophin is primarily at the base of the folds (Byers et al., 1991; Sealock et al., 1991; Bewick et al., 1992). In newborn (P1) animals, utrophin was already enriched at the endplate, although some extrasynaptic staining was evident as well (Fig. 6 a). Dystrophin, on the other hand, was not yet enriched at the endplate at birth (Fig. 6 c), presumably reflecting the postnatal formation of junctional folds which continues for several weeks after birth. If dystrophin compensated for utrophin, then one might expect dystrophin to become enriched at the neonatal mutant synapse. In utrn−/− neonates, however, dystrophin remained evenly distributed throughout the sarcolemma with no synaptic enrichment (Fig. 6 d). Nonetheless, rapsyn and AChR remained concentrated at the neonatal utrn−/− synapse (Fig. 6, e, e′, f, and f′ ). Thus, the localization of the AChRrapsyn complex to the postsynaptic membrane can occur when neither utrophin nor dystrophin is synaptically concentrated.
Figure 6.

Immunohistochemcial analysis of neonatal skeletal muscle from utrn+/− and utrn−/− mice. (a and b) Muscle stained with an NH2-terminal utrophin antibody. Utrophin is concentrated at synaptic sites in utrn+/− muscle by P1, but low levels are also present extrasynaptically. (c and d) Muscle stained for dystrophin shows no synaptic enrichment in either utrn+/− or utrn−/− muscle. (e and f) Rapsyn maintains its synaptic localization in utrn−/− muscle. (a′–f′) AChR labeled with rhodamine-BTX. Bar in d is 20 μm.
Dystrophin and the DPC in Tissues Other than Skeletal Muscle
Functional compensation by dystrophin may help explain the normal structure of tissues other than skeletal muscle in utrn−/− mice. We examined this hypothesis by assessing the distribution of dystrophin in several tissues that normally express utrophin. In normal heart and brain, dystrophin and utrophin had overlapping distributions; both were localized to the sarcolemma and intercalated discs in cardiac myocytes and to endothelial cells in the cerebral vasculature (Fig. 2 c, 7, a and c and data not shown). The distribution of dystrophin in heart and brain of utrn−/− mutants did not differ detectably from that in controls (Fig. 7 d, and data not shown). Moreover, the DPC component β-dystroglycan was distributed along the sarcolemma at similar levels in utrn+/− and utrn−/− cardiac myocytes (Fig. 7, e and f). Thus, in these tissues, dystrophin is appropriately placed to compensate for the loss of utrophin.
Figure 7.

Immunohistochemical analysis of neonatal heart (a–f) and lung (g–l) from utrn+/− and utrn−/−mice. Utrophin is widely distributed in control heart and lung (a and g), but is undetectable in utrn−/− heart and lung (b and h). Dystrophin was associated with the sarcolemma in both utrn+/− and utrn−/− hearts (c and d), but was absent from both utrn+/− and utrn−/− lung (i and j). β-Dystroglycan was associated with the membrane of cardiac myocytes (e and f) and aveoli (k and l) in both utrn+/− and utrn−/− mice.
In contrast, we detected no dystrophin in lung or kidney of either utrn+/− or utrn−/− mice (Fig. 7, i and j, and data not shown). The antibody we used was specific for the COOH terminus of dystrophin, and therefore should have detected short forms as well as full-length dystrophin. Low levels of dystrophin have been detected in these tissues by immunoblot analysis (e.g., Hoffman et al., 1988); these levels may have been too low to be detected by our immunohistochemical assay. Nonetheless, compensation by upregulation of dystrophin is unlikely in these tissues. In light of this result, we used the lung to determine whether components of the DPC persist in the apparent absence of both utrophin and dystrophin. In dystrophic muscle, loss of dystrophin leads to a marked decrease in the levels of DPC components in the sarcolemma (Matsumura et al., 1992). In lung, however, one component of the DPC, β-dystroglycan, was present in similar levels in utrn+/− and utrn−/− mice (Fig. 7, k and l). This result implies the existence of a utrophin- and dystrophin-independent mechanism for retention of the DPC.
Discussion
Mice lacking a functional utrophin gene are viable and fertile, but have subtle defects in the postsynaptic apparatus of their skeletal neuromuscular junctions. In an accompanying paper, Deconinck et al. (1997) reported similar results. The allele described here removes the COOH-terminal cysteine-rich region that is shared by both forms of utrophin (Blake et al., 1995), and is likely, by analogy to dystrophin, to be important for its function (Bies et al., 1992; Suzuki et al., 1994; Rafael et al., 1996). In fact, no utrophin at all was detectable in our mutant, either because insertion of the neomycin cassette led to reduced levels of mRNA or because the mutant protein was unstable. We cannot rule out, however, the possibility that truncated utrophin is present in some tissues or at some stages of development. Deconinck et al. (1997) deleted an NH2terminal exon, leading to complete loss of full-length utrophin. They, however, cannot exclude the possibility that shorter forms of utrophin, transcribed from a promoter COOH-terminal to their deletion (Blake et al., 1995), are present in the mutant. Thus, the similarity of the phenotype reported here to that reported by Deconinck et al. (1977) provides a strong argument that both alleles are effectively nulls.
Utrophin and Synaptogenesis
Four sets of studies had suggested that utrophin might be critical for neuromuscular synaptogenesis and particularly for the differentiation of the postsynaptic membrane. First, agrin, a critical organizer of postsynaptic differentiation (Gautam et al., 1996), binds to dystroglycan, a component of the DPC (Campanelli et al., 1994; Gee et al., 1994; Sugiyama et al., 1994; Bowe et al., 1994). Dystroglycan, in turn, appears to associate with utrophin at the NMJ and with dystrophin extrasynaptically (Matsumura et al., 1992; Ervasti and Campbell, 1993). It seemed possible, therefore, that utrophin converted synaptic dystroglycan into a functional agrin receptor. Second, in cultured muscle cells, large but not small AChR clusters are associated with utrophin, suggesting that utrophin is important for the growth or stabilization of high density AChR aggregates (Phillips et al., 1993; Campanelli et al., 1994). Third, mice incapable of forming postsynaptic AChR clusters through targeted mutagenesis of rapsyn (Gautam et al., 1995), MuSK (DeChiara et al., 1996), or agrin (Gautam et al., 1996), lack synaptic accumulations of utrophin. Finally, forced expression of the putative dystroglycan binding domain of utrophin in cultured myotubes leads to fewer AChR clusters in response to agrin (Namba and Scheller, 1997). This presumptive dominant negative effect suggested that interfering with the utrophin-dystroglycan association attenuates the agrin-mediated AChR cluster transduction pathway.
The modest reduction in AChR density that we and Deconinck et al. (1977) find in the utrn−/− mice provides limited support for the involvement of utrophin and the DPC in postsynaptic differentiation. The nature of this involvement, however, remains unclear. One possibility is that utrophin-DPC dependent processes are required for complete AChR clustering but that other pathways play a dominant role in transmitting agrin's signals. For example, there is now evidence that a synaptically localized tyrosine kinase, MuSK, is part of or associated with a functional agrin receptor (DeChiara et al., 1996; Glass et al., 1996). Moreover, agrin fragments that are incapable of binding dystroglycan retain their AChR clustering activity in vitro (Sugiyama et al., 1994; Gesemann et al., 1995, 1996; Campanelli et al., 1996).
A second possibility is that utrophin and the synaptic DPC might actually be crucial for postsynaptic differentiation, but that dystrophin substitutes, albeit imperfectly, for utrophin in the utrn−/− mice. In this context, it is noteworthy that mutation of the dystroglycan gene leads to a far more severe phenotype than mutation of either the utrophin or the dystrophin gene (cited in Henry and Campbell, 1996). Moreover, a reduction in the density of junctional folds is also seen at NMJs of dystrophin-deficient (mdx) mice. In this case, however, the synaptic alterations are thought to result from muscle fiber necrosis and regeneration, rather than from the absence of dystrophin per se (Torres and Duchen, 1987; Lyons and Slater, 1991).
A third possibility is that the decreased density of AChRs in utrn−/− mice may result from the decreased number of junctional folds. In normal skeletal muscle, AChRs are concentrated not only at the tops of folds but also partway down their sides (Fertuck and Salpeter, 1976). A loss of folds would, therefore, lead to a decrease in the total AChR-rich area within the NMJ. In this scenario, the decreased AChR density in utrn−/− muscle would not result from any functionally important interaction of utrophin or the DPC with AChRs. Instead, the direct effect of utrophin in muscle might be to promote the initial invagination of the postsynaptic membrane that leads to the generation of folds.
Utrophin in Nonskeletal Tissues
In contrast to the neuromuscular phenotype, we detected no abnormalities in other tissues that express utrophin. The early and widespread expression of utrophin led to the speculation that utrophin-deficiency might lead to embryonic lethality (Blake et al., 1996a ). This hypothesis, which we have now disproven, provided an attractive explanation for the fact that no mutations of the human utrophin gene have yet been reported despite its extremely large size (Pearce et al., 1993). It is premature, however, to conclude that utrophin is unimportant for the function of nonskeletal tissues. In fact, the histological analysis so far applied to nonskeletal tissue also failed to detect defects in muscle. A more detailed analysis will be required to determine whether utrophin plays a role in such structures as the glomerular filter or the blood-brain barrier.
Functional Redundancy
Utrophin levels are increased in dystrophin-deficient animals, suggesting that it may functionally compensate, in part, for the missing dystrophin (Ahn and Kunkel, 1993; Matsumura and Campbell, 1994; Blake et al., 1996a ). We asked whether the reverse was true in utrn−/− mice. We obtained, however, no evidence that dystrophin was upregulated in utrophin-deficient tissue. Indeed, lung and kidney appeared histologically and functionally normal in the absence of detectable utrophin or dystrophin. Moreover, the NMJ can maintain its specialized character even when neither utrophin nor dystrophin is concentrated at synaptic sites. We still cannot exclude the possibility that an undetected isoform of dystrophin, or a dystrophin homologue such as dystrobrevin (Wagner et al., 1993; Blake et al., 1996b ) or DRP2 (Roberts et al., 1996), might be compensating for the loss of utrophin. Fortunately, it will be possible to test this hypothesis since dystrophin-deficient (mdx) mice are available, and we have now generated dystrobrevin-deficient mice which are viable and fertile (Grady, R.M., Merlie, J.P., and Sanes, J.R., manuscript in preparation). We are now breeding these animals to generate doubly mutant mice deficient in utrophin and either dystrophin or dystrobrevin.
Acknowledgments
We thank M. Nichol and J. Mudd for a generation of chimeras; M. Elam, B. Klocke, and other members of the Merlie and Sanes laboratories for assistance; A. Nage for R1-Es cells; and A. Guo, S. Froehner, and G. Yancopoulos for gifts of antisera. We are especially grateful to M. Nichol for her management of the animal facility; to J. Cunningham for the electron microscopy; and to S. Culican for the fluorescence-imaging of AChR. We thank A. Vincent, K. Davies, and C. Slater for communicating results before publication, including their initial discovery that the density of AChR is decreased at utrn−/− endplates.
This work was supported by a National Research Service Award grant (R.M. Grady), and grants from the MDA of America and the National Institutes of Health (J.R. Sanes and J.P. Merlie).
Abbreviations used in this paper
- AChR
acetylcholine receptors
- DPC
dystrophin-associated protein complex
- ES
embryonic stem
- NMJ
neuromuscular junction
- RT-PCR
reverse transcription-polymerase chain reaction
Footnotes
†J.P. Merlie died on 27 May 1995.
Please address all correspondence to J.R. Sanes, Department of Anatomy and Neurobiology, Washington University School of Medicine, 660 South Euclid Avenue, Box 8108, St. Louis, MO 63110. Fax: (314) 7471150.
References
- Ahn AH, Kunkel LM. The structural and functional diversity of dystrophin. Nature Genet. 1993;3:283–291. doi: 10.1038/ng0493-283. [DOI] [PubMed] [Google Scholar]
- Apel ED, Roberds SL, Campbell KP, Merlie JP. Rapsyn may function as a link between the acetylcholine receptor and agrin-binding dystrophin-associated glycoprotein complex. Neuron. 1995;15:115–126. doi: 10.1016/0896-6273(95)90069-1. [DOI] [PubMed] [Google Scholar]
- Bewick GS, Nicholson LVB, Young C, O'Donnell E, Slater CR. Different distributions of dystrophin and related proteins at nervemuscle junctions. NeuroReport. 1992;3:857–860. doi: 10.1097/00001756-199210000-00009. [DOI] [PubMed] [Google Scholar]
- Bies RD, Caskey CT, Fenwick R. An intact cysteine-rich domain is required for dystrophin function. J Clin Invest. 1992;90:666–672. doi: 10.1172/JCI115909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake DJ, Schofield JN, Zuellig RA, Gorecki DC, Phelps SR, Barnard EA, Edwards YH, Davies KE. G-utrophin, the autosomal homologue of dystrophin Dp 116, is expressed in sensory ganglia and brain. Proc Natl Acad Sci USA. 1995;92:3697–3701. doi: 10.1073/pnas.92.9.3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake DJ, Tinsley JM, Davies KE. Utrophin: a structural and functional comparison to dystrophin. Brain Pathology. 1996a;6:37–47. doi: 10.1111/j.1750-3639.1996.tb00781.x. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Nawrotzki R, Peters MF, Froehner SC, Davies KE. Isoform diversity of dystrobrevin, the murine 87-kDa postsynaptic protein. J Biol Chem. 1996b;271:7802–7810. doi: 10.1074/jbc.271.13.7802. [DOI] [PubMed] [Google Scholar]
- Bowe MA, Deyst KA, Leszyk JD, Fallon JR. Identification and purification of an agrin receptor from Torpedopostsynaptic membranes: a heteromeric complex related to the dystroglycans. Neuron. 1994;12:1173–1180. doi: 10.1016/0896-6273(94)90324-7. [DOI] [PubMed] [Google Scholar]
- Byers TJ, Kunkel LM, Watkins SC. The subcellular distribution of dystrophin in mouse skeletal, cardiac, and smooth muscle. J Cell Biol. 1991;115:411–421. doi: 10.1083/jcb.115.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanelli JT, Gayer GG, Scheller RH. Alternative RNA splicing that determines agrin activity regulates binding to heparin and α-dystroglycan. Development. 1996;122:1663–1672. doi: 10.1242/dev.122.5.1663. [DOI] [PubMed] [Google Scholar]
- Campanelli JT, Roberds SL, Campbell KP, Scheller RH. A role for dystrophin-associated glycoproteins and utrophin in agrin-induced AChR clustering. Cell. 1994;77:663–674. doi: 10.1016/0092-8674(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Campbell KP. Molecular basis of three muscular dystrophies: disruption of cytoskeleton-extracellular matrix linkage. Cell. 1995;80:675–679. doi: 10.1016/0092-8674(95)90344-5. [DOI] [PubMed] [Google Scholar]
- Chiueh CC, Sun CL, Kopin IJ, Fredericks WR, Rapoport SI. Entry of [3H]norepinephrine, [125]albumin and evans blue from blood into brain following unilateral osmotic opening of the blood-brain barrier. Brain Res. 1978;145:291–301. doi: 10.1016/0006-8993(78)90863-6. [DOI] [PubMed] [Google Scholar]
- Clerk A, Morris GE, Dubowitz V, Davies KE, Sewry CA. Dystrophin-related protein, utrophin, in normal and dystrophic human fetal skeletal muscle. Histochem J. 1993;25:554–561. [PubMed] [Google Scholar]
- DiChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. . Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- Deconinck, A.E., A.C. Potter, J.M. Tinsley, S.J. Wood, R. Vater, C. Young, L. Metzinger, A. Vincent, C.R. Slater, and K.E. Davies. Postsynaptic abnormalities at the neuromuscular junctions of utrophin-deficient mice. J. Cell Biol. In press. [DOI] [PMC free article] [PubMed]
- Durbeej M, Larsson E, Ibraghimov-Beskrovanaya O, Roberds SL, Campbell KP, Ekblom P. Non-muscle α-dystroglycan is involved in epithelial development. J Cell Biol. 1995;130:79–91. doi: 10.1083/jcb.130.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fertuck HC, Salpeter MM. Quantitation of junctional and extrajunctional acetylcholine receptors by electron microscope autoradiography after 125Iα-bungarotoxin binding at mouse neuromuscular junctions. J Cell Biol. 1976;69:144–158. doi: 10.1083/jcb.69.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Mudd J, Nichol M, Chu GC, Sanes JR, Merlie JP. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature (Lond) 1995;377:232–236. doi: 10.1038/377232a0. [DOI] [PubMed] [Google Scholar]
- Gee SH, Montanaro F, Lindenbaum MH, Carbonetto S. Dystroglycan-α, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell. 1994;77:675–686. doi: 10.1016/0092-8674(94)90052-3. [DOI] [PubMed] [Google Scholar]
- Gesemann M, Denzer AJ, Ruegg MA. Acetylcholine receptor- aggregating activity of agrin isoforms and mapping of the active site. J Cell Biol. 1995;128:625–636. doi: 10.1083/jcb.128.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesemann M, Cavalli V, Denzer AJ, Brancaccio A, Schumacher B, Ruegg MA. Alternative splicing of agrin alters its binding to heparin, dystroglycan, and the putative agrin receptor. Neuron. 1996;16:755–767. doi: 10.1016/s0896-6273(00)80096-3. [DOI] [PubMed] [Google Scholar]
- Gillespie SK, Balasubramanian S, Fung ET, Huganir RL. Rapsyn clusters and activates the synapse-specific receptor tyrosine kinase MuSK. Neuron. 1996;16:953–962. doi: 10.1016/s0896-6273(00)80118-x. [DOI] [PubMed] [Google Scholar]
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattsson K, Burden SJ, et al. Agrin acts via a MuSK receptor complex. Cell. 1996;85:513–523. doi: 10.1016/s0092-8674(00)81252-0. [DOI] [PubMed] [Google Scholar]
- Henry MD, Campbell KP. Dystroglycan: an extracellular matrix receptor linked to the cytoskeleton. Curr Opin Cell Biol. 1996;8:625–631. doi: 10.1016/s0955-0674(96)80103-7. [DOI] [PubMed] [Google Scholar]
- Hoffman EP, Hudecki MS, Rosenberg PA, Pollina CM, Kunkel LM. Cell and fiber-type distribution of dystrophin. Neuron. 1988;1:411–420. doi: 10.1016/0896-6273(88)90191-2. [DOI] [PubMed] [Google Scholar]
- Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- Khurana TS, Hoffman EP, Kunkel LM. Identification of a chromosome 6-encoded dystrophin-related protein. J Biol Chem. 1990;265:16717–16720. [PubMed] [Google Scholar]
- Khurana TS, Watkins SC, Chafey P, Chelly J, Tomé F, Fardeau M, Kaplan J, Kunkel LM. Immunolocalization and development expression of dystrophin related protein in skeletal muscle. Neuromus Dis. 1991;1:185–194. doi: 10.1016/0960-8966(91)90023-l. [DOI] [PubMed] [Google Scholar]
- Khurana TS, Watkins SC, Kunkel LM. The subcellular distribution of chromosome 6-encoded dystrophin-related protein in the brain. J Cell Biol. 1992;119:357–366. doi: 10.1083/jcb.119.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga R, Ishiura S, Takemitsu M, Kamakura K, Matsuzaki T, Arahata K, Nonaka I, Sugita H. Immunoblot analysis of dystrophin-related protein (DRP) Biochim Biophys Acta. 1993;1180:257–261. doi: 10.1016/0925-4439(93)90047-5. [DOI] [PubMed] [Google Scholar]
- Love DR, Hill DF, Dickson G, Spurr NK, Byth BC, Marsden RF, Walsh FS, Edwards YH, Davies KE. An autosomal transcript in skeletal muscle with homology to dystrophin. Nature (Lond) 1989;339:55–57. doi: 10.1038/339055a0. [DOI] [PubMed] [Google Scholar]
- Love DR, Morris GE, Ellis JM, Fairbrother U, Marsden RF, Bloomfield JF, Edwards YH, Slater CP, Parry DJ, Davies KE. Tissue distribution of the dystrophin-related gene product and expression in the mdx and dy mouse. Proc Natl Acad Sci USA. 1991;88:3243–3247. doi: 10.1073/pnas.88.8.3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons PR, Slater CR. Structure and function of the neuromuscular junction in young adult mdxmice. J Neurocytol. 1991;20:969–981. doi: 10.1007/BF01187915. [DOI] [PubMed] [Google Scholar]
- Matsumura K, Campbell KP. Dystrophin-glycoprotein complex: its role in the molecular pathogenesis of muscular dystrophies. Muscle and Nerve. 1994;17:2–15. doi: 10.1002/mus.880170103. [DOI] [PubMed] [Google Scholar]
- Matsumura K, Ervasti JM, Ohlendieck K, Kahl SD, Campbell KP. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature (Lond) 1992;360:588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- Mora M, DiBlasi C, Barresi R, Morandi L, Brombati B, Jarre L, Ferdinando C. Developmental expression of dystrophin, dystrophin-associated glycoproteins and other cytoskeletal proteins in human skeletal and heart muscle. Dev Brain Res. 1996;91:70–82. doi: 10.1016/0165-3806(95)00169-7. [DOI] [PubMed] [Google Scholar]
- Nagy A, Rossant J, Nagy T, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namba, T., and R.H. Scheller. 1997. Inhibition of agrin-mediated acetylcholine receptor clustering by utrophin C-terminal peptides. Genes to Cells. In press. [DOI] [PubMed]
- Ohlendieck K, Ervasti JM, Matsumura K, Kahl SO, Leveille CJ, Campbell KP. Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron. 1991;7:499–508. doi: 10.1016/0896-6273(91)90301-f. [DOI] [PubMed] [Google Scholar]
- Patton WDM, Waud DR. The margin of safety of neuromuscular transmission. J Physiol. 1967;191:59–90. doi: 10.1113/jphysiol.1967.sp008237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce M, Blake DJ, Tinsley JM, Byth BC, Campbell L, Monaco AP, Davies KE. The utrophin and dystrophin genes share similarities in genomic structure. Human Molecular Gene. 1993;2:1765–1772. doi: 10.1093/hmg/2.11.1765. [DOI] [PubMed] [Google Scholar]
- Peters MF, Kramarcy NR, Sealock R, Froehner SC. β2-syntrophin: localization at the nueromuscular junction in skeletal muscle. NeuroReport. 1994;5:1577–1580. [PubMed] [Google Scholar]
- Phillips WD, Noakes PG, Roberds SL, Campbell KP, Merlie JP. Clustering and immobilization of acetylcholine receptors by the 43-kD protein: a possible role for dystrophin-related protein. J Cell Biol. 1993;123:729–740. doi: 10.1083/jcb.123.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons F, Robert A, Fabbrizio E, Hugon G, Califano J, Fehrentz JA, Martinez J, Mornet D. Utrophin localization in normal and dystrophin-deficient heart. Circulation. 1994a;90:369–374. doi: 10.1161/01.cir.90.1.369. [DOI] [PubMed] [Google Scholar]
- Pons F, Robert A, Marini JF, Léger JJ. Does utrophin expression in muscles of mdx mice during postnatal development functionally compensate for dystrophin deficiency? . J Neurolog Sciences. 1994b;122:162–170. doi: 10.1016/0022-510x(94)90295-x. [DOI] [PubMed] [Google Scholar]
- Rafael JA, Cox GA, Corrado K, Jung D, Campbell KP, Chamberlain JS. Forced expression of dystrophin deletion constructs reveals structure-function correlations. J Cell Biol. 1996;134:93–102. doi: 10.1083/jcb.134.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RG, Freeman TC, Kendall E, Vetrie DLP, Dixon AK, ShawSmith C, Bone Q, Bobrow M. Characterization of DRP2, a novel dystrophin homologue. Nature Genet. 1996;13:223–226. doi: 10.1038/ng0696-223. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Engvall E, Butkowski R, Hunter DD. Molecular heterogeneity of basal laminae: isoforms of laminin and collagen IV at the neuromuscular junction and elsewhere. J Cell Biol. 1990;111:1685–1699. doi: 10.1083/jcb.111.4.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield J, Houzelstein D, Davies KE, Buckingham M, Edwards YH. Expression of the dystrophin-related protein (utrophin) gene during mouse embryogenesis. Dev Dyn. 1993;198:254–264. doi: 10.1002/aja.1001980403. [DOI] [PubMed] [Google Scholar]
- Sealock R, Butler MH, Kramarcy NR, Gao KX, Murnave AA, Douville K, Froehner SC. Localization of dystrophin relative to acetylcholine receptor domains in electric tissue and adult cultured skeletal muscle. J Cell Biol. 1991;113:1133–1144. doi: 10.1083/jcb.113.5.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama J, Bowen DC, Hall ZW. Dystroglycan binds nerve and muscle agrin. Neuron. 1994;13:103–115. doi: 10.1016/0896-6273(94)90462-6. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Yoshida M, Hayashi K, Mizuno Y, Hagiwara Y, Ozawa E. Molecular organization at the glycoprotein-complex-binding site of dystrophin. Eur J Biochem. 1994;220:283–292. doi: 10.1111/j.1432-1033.1994.tb18624.x. [DOI] [PubMed] [Google Scholar]
- thiMan N, Ellis JM, Love DR, Davies KE, Gatter KC, Dickson G, Morris GE. Localization of the DMDL gene-encoded dystrophin- related protein using a panel of nineteen monoclonal antibodies: presence at neuromuscular junctions, in the sarcolemma of dystrophic skeletal muscle, in vascular and other smooth muscles, and in proliferating brain cell lines. J Cell Biol. 1991;115:1695–1700. doi: 10.1083/jcb.115.6.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinsley JM, Blake DJ, Roche A, Fairbrother U, Riss J, Byth BC, Knight AE, Kendrick-Jones J, Suthers GK, Love DR, et al. Primary structure of dystrophin-related protein. Nature (Lond) 1992;360:591–593. doi: 10.1038/360591a0. [DOI] [PubMed] [Google Scholar]
- Torres LF, Duchen LW. The mutant mdx: inherited myopathy in the mosue. Morphological studies of nerves, muscles and end-plates. Brain. 1987;110:269–299. doi: 10.1093/brain/110.2.269. [DOI] [PubMed] [Google Scholar]
- Turney SG, Culican SM, Lichtman JW. A quantitative fluorescence-imaging technique for studying acetylcholine receptor turnover at neuromuscular junctions in living animals. J Neurosci Methods. 1996;64:199–208. doi: 10.1016/0165-0270(95)00135-2. [DOI] [PubMed] [Google Scholar]
- Tybulewicz VLJ, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- Uchino M, Yoshioka K, Miike T, Tokunaga M, Uyama E, Teramoto H, Naoe H, Ando M. Dystrophin and dystrophin-related protein in the brains of normal and mdx mice. Muscle and Nerve. 1994;17:533–538. doi: 10.1002/mus.880170511. [DOI] [PubMed] [Google Scholar]
- Wagner KR, Cohen JB, Huganir RL. The 87 k postsynaptic membrane protein from Torpedois a protein-tyrosine kinase substrate homologous to dystrophin. Neuron. 1993;10:511–522. doi: 10.1016/0896-6273(93)90338-r. [DOI] [PubMed] [Google Scholar]