

Abstract
Calcium can activate mitochondrial metabolism, and the possibility that mitochondrial Ca2+ uptake and extrusion modulate free cytosolic [Ca2+] (Cac) now has renewed interest. We use whole-cell and perforated patch clamp methods together with rapid local perfusion to introduce probes and inhibitors to rat chromaffin cells, to evoke Ca2+ entry, and to monitor Ca2+-activated currents that report near-surface [Ca2+]. We show that rapid recovery from elevations of Cac requires both the mitochondrial Ca2+ uniporter and the mitochondrial energization that drives Ca2+ uptake through it. Applying imaging and single-cell photometric methods, we find that the probe rhod-2 selectively localizes to mitochondria and uses its responses to quantify mitochondrial free [Ca2+] (Cam). The indicated resting Cam of 100–200 nM is similar to the resting Cac reported by the probes indo-1 and Calcium Green, or its dextran conjugate in the cytoplasm. Simultaneous monitoring of Cam and Cac at high temporal resolution shows that, although Cam increases less than Cac, mitochondrial sequestration of Ca2+ is fast and has high capacity. We find that mitochondrial Ca2+ uptake limits the rise and underlies the rapid decay of Cac excursions produced by Ca2+ entry or by mobilization of reticular stores. We also find that subsequent export of Ca2+ from mitochondria, seen as declining Cam, prolongs complete Cac recovery and that suppressing export of Ca2+, by inhibition of the mitochondrial Na+/ Ca2+ exchanger, reversibly hastens final recovery of Cac. We conclude that mitochondria are active participants in cellular Ca2+ signaling, whose unique role is determined by their ability to rapidly accumulate and then release large quantities of Ca2+.
The Ca2+ content of mitochondria within resting cells is believed to be low, yet isolated mitochondria can accumulate large quantities of Ca2+ when it is provided to them (Lehninger et al., 1967; Carafoli, 1979; Gunter and Pfeiffer, 1990). During the 1970s and 1980s, however, uptake of Ca2+ into mitochondria was often considered to require pathological levels of cytoplasmic Ca2+. Because elevated [Ca2+] increases the activity of key metabolic enzymes of mitochondria, reversible uptake of Ca2+ into mitochondria is proposed to coordinate energy production to cellular needs (McCormack et al., 1990; Gunter et al., 1994; Hajnóczky et al., 1995). Recently, the availability of new optical indicators for Ca2+ ions has enabled studies of mitochondrial Ca2+ uptake in living cells and tests of possible roles for it. Signals from populations of cells expressing mitochondrially targeted aequorin reported rapid and large transient increases of free intramitochondrial [Ca2+] (Cam)1 and suggested that mitochondria sequester Ca2+ from locally high microdomains of cytosolic free [Ca2+] (Cac) associated with Ca2+ entry or with release from intracellular stores (Rizzuto et al., 1992, 1993, 1994; Rutter et al., 1993; Lawrie et al., 1996). In complementary studies, inhibitors that should block mitochondrial Ca2+ uptake severely compromised clearance of cytoplasmic Ca2+ after an imposed elevation, indicating that accumulation by mitochondria also is important for cellular Ca2+ homeostasis (Thayer and Miller, 1990; Friel and Tsien, 1994; Werth and Thayer, 1994; White and Reynolds, 1995; Herrington et al., 1996; Park et al., 1996). Comparing the rates of Cac decay after selective blockade of known mechanisms for cellular Ca2+ sequestration and extrusion showed that mitochondrial uptake (defined pharmacologically) accounts for as much as 70% of cytosolic Ca2+ removed during the initial rapid phase of recovery from large imposed Ca2+ loads in chromaffin cells (Herrington et al., 1996).
Here we introduce two fluorescent Ca2+ probes, one into the cytosol and the other into mitochondria of single rat chromaffin cells. Simultaneous optical monitoring demonstrates that even modest elevations of Cac produce rapid transient redistributions of Ca2+ from the cytosol into mitochondria. The mitochondrial Ca2+ uptake shapes and dampens the elevations of Cac during stimulation, and subsequent Ca2+ export from mitochondria delays complete recovery of Cac and may facilitate refilling of intracellular stores. Cellular Ca2+ signaling may best be considered as an interactive, multicompartmental network in which mitochondria are active participants.
Materials and Methods
All experiments were performed at a room temperature of 24–27°C. Results are reported as mean ± SEM.
Chemicals
CGP-37157 (7-chloro-3,5-dihydro-5-phenyl-1H-4,1-benzothiazepine-2-on) was the generous gift of Dr. Alain DePover, CIBA-GEIGY (Basel). Dyes were from Molecular Probes (Eugene, OR); ionomycin and BHQ (2,5 di(t-butyl)-1,4-hydroxyquinone) from Calbiochem (La Jolla, CA); CCCP (carbonyl cyanide m-chlorophenylhydrazone) and all other reagents from Sigma Chem. Co. (St. Louis, MO).
Cells and Media
Adrenal chromaffin cells were prepared as described previously (Herrington et al., 1996; Park et al., 1996) from ∼300 g male rats. Briefly, adrenal medullae were dissected and digested 50–70 min at 37°C with (mg/ml in modified Hanks' solution): 1.2 collagenase D; 0.4 trypsin; 0.1 DNase type I. Dispersed cells were plated on ∼5-mm square coverglass slips coated with poly-l-lysine and laminin, then maintained 1–4 d as primary cultures in a fortified DMEM medium.
Patch Clamp Procedures
Voltage clamp recordings used an Axopatch 1C amplifier (Axon Instruments, Foster City, CA) and the BASIC-FASTLAB analysis system (Indec Systems, Capitola, CA). The standard voltage protocol was a 0.05-1 s step to +10 mV from a holding potential of −80 mV. Tail currents in SKtype Ca2+-activated K–channels were recorded at a holding potential of −60 mV (Park et al., 1996).
For the whole-cell configuration of the patch clamp technique (Hamill et al., 1981), we usually used a standard pipette solution (mM): 75 Cs2SO4; 15 CsCl; 6.5 NaCl; 2.5 Na pyruvate; 2.5 malic acid; 1 NaH2PO4; 1 MgSO4; 50 Hepes; 5 MgATP; 0.3 Tris GTP; 0.5 Tris cAMP; 0.1 leupeptin; pH to 7.3 with CsOH. In some cases, 78 K2SO4 and 10 KCl replaced 75 Cs2SO4 and 15 CsCl. Perforated-patch recording (Horn and Marty, 1988) also used methods detailed previously (Park et al., 1996), and a simplified pipette solution: 111 Cs2SO4; 6 CsCl; 10 NaCl; 5 MgSO4; 20 Hepes; pH to 7.4 with CsOH (where noted, Cs+ salts were replaced with K+). Briefly, the tips of patch pipettes were filled by dipping for 1–3 s in simplified pipette solution, then backfilled with the same solution supplemented with 480 μg amphotericin/ml.
The standard external solution used during seal formation contained: 150 NaCl, 2.5 KCl, 2 mM CaCl2, 1 MgCl2, 10 Hepes, and 20 mM glucose, pH 7.4. For perforated-patch recordings external solutions also contained 2.5 mM Na pyruvate and 2.5 mM malic acid to optimize mitochondrial function (Villalba et al., 1994). For depolarization-evoked Ca2+ entry, the external CaCl2 was increased to 10 mM.
Activation of voltage-dependent Ca2+ channels by depolarization provides precisely timed, but imperfectly predictable control of Ca2+ entry, as judged by the peak Cac produced. The degree of buffering by introduced dyes and chelators, and the extent of time-dependent channel ‘rundown' (more severe in whole-cell- than in perforated-patch-recordings), and previous stimulus history all contribute to this variability. For these reasons, the durations of depolarizing stimuli often were adjusted upwards during the course of an experiment in an attempt to produce similar Cac challenges. In the interpretation of these experiments, differences in peak Cac reflect mainly imperfect compensation for this rundown.
Dye Loading and Photometry
Rhod-2-AM (0.2 mM) and Calcium GreenTM-AM (10 mM) were dispensed from DMSO stocks, dispersed in 10% Pluronic 147, and diluted to 1 and 12 μM in 0.25 ml standard external solution. A single coverslip of cells was immediately added, incubated 35–50 min at 22–25°C in the dark, then transferred to an examination chamber containing 2–4 ml fresh buffer. Indo-1 (100 μM), or either rhod-2 or Calcium Green dextran (3,000 M r) at 10 μM together with 90 μM BAPTA, were introduced by inclusion in the whole-cell pipette solution. All photometric measurements were performed on an inverted microscope using a 1.3 NA 40× oil objective (Nikon), an attenuated (1.0 NDF) 100 W Hg source, and paired photon-counting detectors (Hamamatsu). An electronic shutter (Uniblitz) restricted excitation to the sampling interval (50–100 ms; duty cycles of 0.5% in normal and 50% in fast mode). Pinhole diaphragms limited the excitation and emission fields to ∼25 μm diameter regions. Indo-1 was excited at 365 ± 10 nm and detected at 405 ± 35 and 500 ± 40 nm. A filter cube with dual band excitation (490 ± 15 and 560 ± 15 nm) and dual band emission (530 ± 15 and 650 ± 75 nm) (XF-52; Omega Optical) allowed simultaneous excitation of Calcium Green and rhod-2. Detection at 525 ± 10 and 580 ± 30 nm was accomplished with additional interference filters separated by a 540nm dichroic mirror.
Calibration of Indo-1, Rhod-2, and Calcium Green Signals
The ratiometric probe indo-1 was calibrated as detailed previously (Herrington et al., 1996; Park et al., 1996). Briefly, the ratio R (F405/F500) of background-corrected signals was applied to the standard calibration equation (Grynkiewicz et al., 1985):
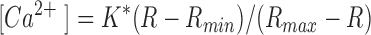
where Rmin (0.47), Rmax (7.15), and K* (2540 nM) were determined empirically from cells dialyzed with pipette solutions containing 50 mM EGTA, 15 mM CaCl2, or 20 mM EGTA and 15 mM CaCl2 (calculated free [Ca2+] of 251 nM), respectively.
Fluorescence signals from the single-wavelength indicators rhod-2 and Calcium Green were corrected first for autofluorescence observed with nonloaded cells (<5% of total signal), then for the spillover of fluorescence from Calcium Green (22–48%) or its dextran conjugate (10–15%) into the long-wavelength (rhod-2) detector channel, as determined from cells loaded only with these probes. The extent of spillover increased with age of the dual pass excitation filter set, presumably as a consequence of deterioration. Spillover of fluorescence from rhod-2 into the short wavelength (Calcium Green) channel was negligible. Compensation also was applied for time-dependent loss of signal (primarily photobleaching) as determined from exponential fits of resting-state fluorescence before and after recovery from stimulus (mean rate constants: rhod-2, 4.89 ± 0.10 × 10−4 s−1; Calcium Green, 5.80 ± 0.17 × 10−4 s−1). The corrected signals (F) were converted to [Ca2+] using the simplified calibration equation:
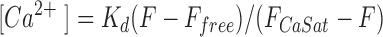
where the K d determined in vitro for rhod-2 (500 nM) or Calcium Green (240 nM; Eberhard and Erne, 1991) was assumed to apply, and Ffree and FCaSat were calculated from postexperimental responses of each cell as follows. Cells were sequentially perfused with a Mg2+-free saline containing 10 mM CaCl2 and 10 μM ionomycin, then with the same solution in which 2 mM MnCl2 replaced CaCl2.
These treatments elicited fluorescence in an average ratio (FMnSat/FCaSat) of 0.75 ± 0.05 (n = 11) and 0.78 ± 0.04 (n = 9) for Calcium Green and its dextran conjugate, indistinguishable from FMnSat/FCaSat ratios determined in vitro in a 150 mM KCl, 1 mM MgCl2, pH 7.4 medium, containing 1 μM dye and 10 mM CaCl2 or MnCl2. Compared with dye in the same medium containing 10 mM EGTA, Ca2+-free:Ca2+-saturated fluorescence (Ffree/FCaSat) was 0.091. Assuming this ratio also applies in vivo, cellular Ffree was calculated from the observed FCaSat elicited by ionophore.
For the K+ salt of rhod-2, Ffree/FCaSat determined in vitro was 0.068. When the rhod-2 salt was applied to the cytosol by whole cell dialysis, the FMnSat/FCaSat ratio (0.29 ± 0.01; n = 3) agreed well with that determined fluorimetrically for rhod-2 in vitro (0.24). FMnSat/FCaSat also measured in vitro was unchanged from pH 7-8.2 and altered little or not at all by variation of ionic strength (0.1–0.2 M KCl), viscosity (0–2 M sucrose), or dielectric constant (0–10% ethanol) that might be encountered intramitochondrially. However, for rhod-2 introduced into mitochondria by AM-loading, FMnSat/FCaSat was much larger either before (0.50 ± 0.13; n = 14) or after (0.68 ± 0.11; n = 4) whole cell dialysis, presumably indicating that the much higher affinity (>100-fold) of rhod-2 for Mn2+ than for Ca2+ and the greater efficacy of the ionophore in Mn2+ transport (Erdahl et al., 1996) allowed full saturation of the compartmentalized probe with Mn2+ but not with Ca2+. Therefore, cellular Ffree and FCaSat were both calculated from the FMnSat observed for rhod-2AM loaded, ionophore-treated cells, using the FMnSat/FCaSat and Ffree/FCaSat ratios observed in vitro for rhod-2. These determined ratios (and the K d for Ca2+ used here) agree with the specifications now provided by the manufacturer, but differ from those reported in the original description of rhod-2 (Minta et al., 1989), presumably due to removal of a Ca2+-insensitive contaminant present in the initial preparations of this probe.
Imaging
Images were collected on a Deltavision SA3.1 wide-field deconvolution microscope system (Applied Precision, Inc., Issaquah, WA) using a Nikon PlanApo 60× 1.4 NA objective on an Olympus IMT-2 inverted microscope with a Photometrics PXL-2 cooled CCD camera containing a Kodak KAF1400 chip. Images were collected with an intermediate magnification of 1.5×. Binning was used to increase signal-to-noise. For every 2-s time point, four optical sections were collected at 0.2 μm spacing each with 0.3-s exposure. Final voxel resolution was 0.157 μm (planar) × 0.200 μm (axial). Images of each z-axis series were corrected for photobleaching and variations in lamp intensity; only intensity corrections were applied between time points. The deconvolution reconstruction algorithms of Agard and Sedat (Agard et al., 1989; Hiraoka et al., 1990, 1991; Chen et al., 1995) were applied to each z-series using an optical transform function determined empirically from a 0.1-μm bead.
Results
We begin with the Ca2+-sensitive dye rhod-2 as a probe of Cam. Extrusion of protons during electron transport creates a large inside-negative potential difference (Δψm) across the inner mitochondrial membrane that is subsequently harnessed by the ATP synthase in the production of ATP. This negative Δψm has been exploited in the past to label energized mitochondria with membrane-permeant cations like Rhodamine 123 (Johnson et al., 1981). Similarly, the permeant cation rhod-2 acetoxymethyl ester (rhod-2AM) should also accumulate preferentially in mitochondria where it should become trapped when subsequent esterolysis generates zwitterionic rhod-2 (Minta et al., 1989). Unlike the membrane permeant Rhodamine 123, the trapped rhod-2 should not be sensitive to subsequent changes in mitochondrial membrane potential. The following experiments confirm these predictions for rat chromaffin cells.
Rhod-2 Localizes to Mitochondria and Responds to Ca2+ Entry
To verify compartmentalization of dye, cells loaded with rhod-2AM were visualized by fluorescence deconvolution imaging. In Fig. 1, rhod-2 fluorescence was found primarily in punctate and filamentous extranuclear regions, consistent with a mitochondrial localization, and in 1–3 small (∼1 μm) intranuclear bodies, tentatively identified as nucleoli. With long exposure images, the characteristic vermiform mitochondrial morphology (Loew et al., 1995) was more apparent (Fig. 1 E). Pharmacological validation of the mitochondrial localization of rhod-2 is found in the following sections below. When the neutral ester of Calcium Green was coloaded in the same cell together with cationic rhod-2AM, the Calcium Green fluorescence showed a more pancellular distribution (Fig. 1 F), consistent with a localization in the cytosol and nucleus as well as some additional association with mitochondria.
Figure 1.

Rhod-2 images of chromaffin cell responses to depolarization and coimages of rhod-2 with Calcium Green. (A–D) Fluorescence deconvolution images of a single cell after loading with AM ester of rhod-2. Images were collected before, during, ∼4 s after, and ∼1 min after a 10-s, fast local perfusion with a depolarizing (70 mM KCl) medium. (E–F) After coloading with AM esters of rhod-2 and Calcium Green, images were collected from a central plane of a single cell not subjected to stimulus, using excitation and emission filters for rhod-2 in E and then for Calcium Green in F. Scale bar represents 4 μm.
Calcium entry induced by KCl depolarization increased the rhod-2 fluorescence within the presumptive mitochondria (Fig. 1 B). The fluorescence returned to baseline levels within a minute after the depolarization (Fig. 1, C and D). No fluorescence increase occurred during application of KCl in the nominal absence of external Ca2+ (not shown). Hence, rhod-2 reports a transient rise of mitochondrial calcium following depolarization-induced Ca2+ entry. This is the phenomenon we wish to study.
Rapid Clearance of Cytosolic Ca2+ Requires Energized Mitochondria and the Ca2+ Uniporter
Uptake of Ca2+ by isolated mitochondria involves electrophoretic transport through a Ca2+ uniporter driven by the mitochondrial membrane potential Δψm (Gunter and Pfeiffer, 1990; Gunter et al., 1994; but also see Sparagna et al., 1995). We now show that Ca2+ uptake by mitochondria within the cell also depends on the negative Δψm and the uniporter. We perturb mitochondrial function with four agents, oligomycin, cyanide, carbonyl cyanide m-chlorophenylhydrazone (CCCP), and ruthenium red as illustrated in Fig. 2. Oligomycin blocks mitochondrial ATP production by direct inhibition of the ATP synthase but should not decrease Δψm by itself. Cyanide blocks electron transport but should allow maintenance of Δψm by an alternate route, the reversal of the ATP synthase fueled by cytoplasmic ATP. However, oligomycin and cyanide in combination block both routes for maintaining Δψm and should depolarize mitochondria. Likewise, the ionophore CCCP collapses Δψm by making membranes permeable to protons. Finally, ruthenium red blocks the Ca2+ uniporter directly.
Figure 2.

Pathways of Ca2+ and H+ transport in the inner mitochondrial membrane and diagnostic inhibitors. Abbreviations: CU, Ca2+ uniporter inhibited by ruthenium red, RR; F1F0, ATP synthase inhibited by oligomycin; NCE, Na+-Ca2+ exchanger inhibited by CGP-37157, CGP; e.t.c., electron transport chain inhibited by cyanide; CCCP, a protonophore that collapses the mitochondrial membrane potential, Δψm.
The first of these experiments (Fig. 3) showed that CCCP and ruthenium red block rapid Ca2+ clearance. Ca2+ entry into the cell was induced by a step depolarization of the plasma membrane, and removal of cytosolic Ca2+ was monitored with the Ca2+ probe indo-1 introduced directly into the cytoplasm from a whole-cell pipette. In Fig. 3 A, spatially averaged Cac rose to >4 μM during a brief depolarizing voltage step (first arrow) and then decayed within a few seconds to submicromolar levels. As reported previously (Herrington et al., 1996), addition of CCCP produced a small transient Cac increase of variable duration and size apparently dependent on mitochondrial Ca2+ content at the time of addition. CCCP also greatly slowed the recovery of Cac after a second depolarization. When this experiment was repeated with other cells internally dialyzed with ruthenium red to block the uniporter, the initial decay was already slowed even without the addition of CCCP (Fig. 3 B). Subsequent treatment with CCCP had no apparent further effect on recovery from Ca2+ entry (n = 4). These experiments show that mitochondria cannot clear Ca2+ rapidly from the cytoplasm when the Δψm is collapsed or the Ca2+ uniporter is blocked.
Figure 3.

Prolongation of Ca2+ clearance by blockade of mitochondrial energization or of the mitochondrial uniporter. (A–B) Time course of cytoplasmic [Ca2+] monitored with indo-1 (100 μM) introduced into the cytosol by inclusion in the pipette solution alone (A) or, in a different cell, together with 2 μM RR (B). Beginning 3–5 min after the wholecell patch-clamp configuration was established, dual emission fluorescence signals were recorded. One or two 500–600-ms depolarizing pulses were applied (arrows) while each cell was perfused with control medium alone. Another one or two pulses were applied after 2 μM CCCP was included in the perfusion medium. (C– D) Effect of mitochondrial inhibitors on clearance of submembrane Cac as measured by the decay (tail) of a Ca2+-activated K+ current. Tail currents were recorded in perforated-patch configuration in response to 1-s depolarizations to 0 mV applied every 2 min (Park, 1996). After 9.5 min, the perfusion medium was supplemented with 3 μM oligomycin or 5 mM NaCN and at 11.5 min, with both. (D) Peak amplitude and half-decay times of currents observed after treatment were normalized to those obtained in the preceding control stimulus applied to the same cell. The numbers of cells examined are shown in parentheses.
The second set of experiments (Fig. 3, C and D) tested the ability of oligomycin and cyanide to block Ca2+ clearance. Ca2+ entry was again induced by a brief depolarization, but here clearance of submembrane cytosolic Ca2+ was monitored electrophysiologically by the decay (tail) of a Ca2+-activated K+ current that is prominent in rat chromaffin cells (SK current: Neely and Lingle, 1992a ; Park, 1994). This method requires neither cell dialysis nor introduction of dyes into the cytoplasm and thus provides a useful, non-invasive indication of [Ca2+] at the plasma membrane. As in previous work (Park et al., 1996), the K+ current that was activated by Ca2+ entry during a 1-s depolarization then declined with a half-time of 2–7 s as the [Ca2+] fell and the SK channels closed. The decay was not slowed by oligomycin or cyanide alone but was prolonged greatly by the combination of these agents. Blockade of both available routes for mitochondrial energization increased the half-time of current decay 5.4 ± 1.8-fold (n = 4), without appreciably affecting the peak amplitude. These experiments without cell dialysis show again that rapid clearance of cytoplasmic Ca2+ depends on the existence of a negative Δψm and not, in the short term, on the ongoing oxidative production of ATP.
Monitoring of the CCCP-sensitive Mitochondrial Compartment
Using compartmentalized dyes, we can now verify the assumption that CCCP slows Cac clearance by preventing uptake of Ca2+ into mitochondria. We have seen that coloading of chromaffin cells by coincubation with AM esters compartmentalizes rhod-2 into mitochondria and disperses Calcium Green throughout the cell. Signals from the two dyes were followed at high time resolution using photometry and a filter cube designed for simultaneous dual-wavelength excitation and dual-wavelength detection. After sequential corrections for autofluorescence, signal cross-talk, and photobleaching, Cam and global free [Ca2+] were calculated from rhod-2 and Calcium Green fluorescence by application of calibration parameters determined for each cell examined. Voltage-clamp depolarizations controlled Ca2+ entry, and inhibitors were applied and removed rapidly by fast, local perfusion. We applied CCCP in the presence of oligomycin to prevent any accelerated consumption of cellular ATP by reverse-mode operation of the ATP synthase in proton-permeant mitochondria (Budd and Nichols, 1996; Park et al., 1996). As expected, oligomycin alone did not affect Cam or Cac of resting cells (Fig. 4).
Figure 4.

Reciprocal actions of CCCP on mitochondrial and cytosolic Ca2+. Fluorescence signals from rhod-2 and Calcium Green, coloaded as AM esters in a preliminary incubation, were recorded simultaneously from a single cell, beginning 3–5 min after the perforated patch clamp configuration was established. The perfusion medium then was supplemented with 10 mM Ca2+ and 3 μM oligomycin and then with 2 μM CCCP as indicated. At the arrows 500-ms depolarizations were applied. A postexperimental calibration procedure (see Materials and Methods) provided parameters for conversion of rhod-2 and Calcium Green fluorescence to the indicated mitochondrial and cytosolic free Ca2+ concentrations (Cam and Cac, respectively). The results shown are representative of four similar experiments. (Insets) The initial portions of Cam and Cac responses are aligned in time to the depolarizing stimulus.
The rapid rise and decline of [Ca2+] calculated from the Calcium Green signal during and following Ca2+ entry (Fig. 4, lower traces) resemble the Cac responses reported from indo-1 applied directly in the cytosol (Fig. 3 A). Other common features include a delayed final return to the resting level (Figs. 4, 6, and 8) and a small [Ca2+] increase when CCCP was applied late in the recovery from brief Ca2+ entry (Fig. 4). Calcium Green and indo-1 signals also both report that CCCP slows the initial clearance of Cac yet hastens the final recovery to resting levels. These similarities in the signals from the two probes confirm the usefulness of AM-loaded Calcium Green to measure Cac. We note, however, that in the experiments of Fig. 3, A and B and in previous studies using pipetteloaded indo-1 (Herrington et al., 1996), resting Cac was lower (by 100–200 nM) and peak Cac evoked by Ca2+ entry usually was higher than the values reported here by Calcium Green loaded as its AM ester and examined in perforated patch protocols. It is not yet clear whether these differences reflect alterations in the extent of Ca2+ entry, in cytosolic buffering or clearance mechanisms (all of which may be affected by the dye loading and patch clamp procedures employed), or by differences in dye localization and inaccuracies in calibration.
Figure 6.

Reversible elimination of the sustained phase of Cac recovery by blockade of mitochondrial Ca2+ extrusion. Cells were coloaded with rhod-2 and Calcium Green esters, examined in the perforated patch clamp configuration, and the data analyzed as in Fig. 4. Responses are representative of five similar experiments. (A) Arrows mark the application of sequential depolarizations of 1.5, 2.0, and 2.5 s duration, applied to a single cell in the presence or absence of perfusion with 10 μM CGP-37157 as indicated. (B) Another cell received 0.5, 1.0, and 1.5 s stimuli before, during, and after application of inhibitor.
Figure 8.

Calcium-induced release of Ca2+ and its mitochondrial sequestration. Cells were coloaded with rhod-2 and Calcium Green and examined in perforated patch as in Fig. 4. (A) Calibrated (left) and aligned (right) Cam and Cac records are from a single cell subjected to sequential depolarizations of 50 (diamond), 100 (box), 250 (circle), and 500 (triangle) ms depolarizations, applied at the arrows. Results are representative of four similar experiments. (B) Averaged responses from four cells treated exactly as above. The enlarged, closed symbols mark the observed or interpolated Cam and Cac values at the termination of stimulus.
In comparison to the quick changes of Cac during and after brief plasma membrane depolarizations, rhod-2 reported a more limited but far more sustained elevation of Cam (Fig. 4, insets). Subsequent addition of CCCP caused a simultaneous fall in Cam and rise in Cac (Fig. 4; n = 4), presumably reflecting release of Ca2+ from depolarized mitochondria. CCCP also strongly attenuated the rise of Cam in response to elevation of Cac. The small residual signal may result from incomplete inhibition by CCCP, from incomplete compensation for crossover of the Calcium Green signal, or from rhod-2 reporting from a nonmitochondrial compartment.
Redistribution of Ca2+ during and after Ca2+ Entry
We further refined the compartmentation of the two dyes by using the patch pipette in whole-cell rather than perforated patch mode to allow introduction of the Calcium Green as a dextran conjugate in the pipette solution. This strategy should restrict Calcium Green to the cytosol and also remove any cytosolic rhod-2 and remaining ester by dialysis. Little or no loss of rhod-2 fluorescence occurred during entry of Calcium Green (not shown), confirming the extensive compartmentation of this dye indicated by imaging in Fig. 1. Subsequent responses of the two dye signals to Ca2+ entry evoked by depolarizations in the presence and absence (Fig. 5) of CCCP were qualitatively similar to those in Fig. 4. Because removal of CCCP restored Cam responses to Ca2+ entry (not shown), mitochondrial depolarization apparently does not result in artifactual release and diffusional loss of rhod-2.
Figure 5.

Cam increases during and following Ca2+ entry and decreases during late clearance. Rhod-2 was loaded as its AM ester and Calcium Green Dextran by whole cell dialysis. Calcium Green fluorescence observed in the on-cell patch clamp configuration was subtracted before calibration of signals simultaneously recorded from a single cell 5 min after the whole-cell configuration was established. (A) Cam and Cac responses to a 500-ms depolarization applied at the arrow. The final portion of Cac recovery is shown (dots) on a 20× expanded scale. (B) The initial portions of Cam (open circle) and Cac (open triangle) traces from A are aligned in time to the depolarizing stimulus. (C) The relationship of Cam to Cac during this response to stimulus and subsequent recovery; a closed diamond marks the Cam and Cac values at the termination of stimulus. The curved arrow indicates the approximate nonlinear time course; the lines divide elapsed time into segments of ∼0.5, 5, and 50 s duration.
Under these more stringent conditions of dye compartmentation, simultaneous recording of Cac and Cam at high temporal resolution suggests that three phases of Ca2+ traffic are initiated by Ca2+ entry. Fig. 5 A shows typical (n = 4) time courses of rhod-2 and Calcium Green dextran responses to a 500-ms depolarization, and Fig. 5 B shows overlaid initial segments of these records (sampled every 100 ms) on an expanded time scale. The three phases are best seen by plotting each Cam value against Cac (Fig. 5 C). During the 500-ms depolarization-induced Ca2+ entry, Cam rose without delay and in direct proportion to the rise in Cac. In the following 5 s, Cam continued to rise as Ca2+ was cleared from the cytosol, and Cac fell proportionately. In the final >50 s, Cam fell slowly during the final return of Cac to its resting value.
Our previous work suggested that mitochondria sequester the majority of the Ca2+ that enters the cell in response to depolarization (Herrington et al., 1996; Park et al., 1996). As was expected, we now see directly that Cam increases as Cac decreases. It now also is clear that half of mitochondrial uptake is accomplished while entry is still in progress and that uptake thus must attenuate the peak Cac reached.
Ca2+ Is Extruded from Mitochondria by Na+-Ca2+ Exchange
In the simplest interpretation, the fall in Cam that occurs when Cac returns to near resting values would represent export of mitochondrial Ca2+. Alternatively, Cam might fall as a consequence of conversion of Ca2+ to an inaccessible form within the mitochondrial matrix (Lehninger, 1970; Friel and Tsien, 1994). The following experiments distinguish between these possibilities.
The benzothiazepine CGP-37157 selectively and potently inhibits the Na+-Ca2+ exchanger that exports Ca2+ from isolated mitochondria (Cox and Matlib, 1993). Fig. 6, A and B show that this agent has no effect on the rise of Cam evoked by Ca2+ entry into chromaffin cells but reversibly slows the Cam fall (n = 5), providing good evidence that extrusion of Ca2+ by the mitochondrial Na+-Ca2+ exchanger mediates the normal fall in Cam. While Cam is elevated and Ca2+ is being extruded from mitochondria, Cac often remains slightly above its resting value (Figs. 3–5). This small elevation has been attributed to a steady flux of Ca2+ into the cytoplasm from mitochondria (Freil and Tsien, 1994; Werth and Thayer, 1994; Herrington et al., 1996). The reversible reduction of this persistent Cac elevation by the exchange inhibitor CGP-37157 (Fig. 6) confirms this hypothesis. Unexpectedly, the fall in Cam that followed removal of CGP-37157 was accompanied by no elevation (Fig. 6 A) or only minor elevation (Fig. 6 B) of Cac. It would be useful to repeat these experiments in cells where the other mechanisms of Ca2+ clearance are inhibited.
Cam Responds to Release of Ca2+ from Intracellular Stores
Having established that mitochondria take up Ca2+ that enters through voltage-gated Ca2+ channels of the plasma membrane, we now consider if they also take up Ca2+ released from intracellular stores. Chromaffin cells have IP3sensitive Ca2+ stores that can be mobilized by agonists acting at G protein–coupled receptors (O'Sullivan et al., 1989; Malgaroli et al., 1990; Neely and Lingle, 1992b ). We tested the selectivity of mitochondrial Ca2+ uptake for this source of Ca2+ by comparing responses of Cam and Cac to depolarizing steps and to agonists. For these experiments we used the perforated-patch configuration for recording since intracellular stores tend to run down quickly in wholecell recording so responses to agonists vanish (Herrington, J., Y.B. Park, D.F. Babcock, and B. Hille, unpublished observations).
Cells were first briefly perfused with bradykinin (n = 5) or muscarine (n = 3) and 3–4 min later given a brief depolarizing step (Fig. 7). The Ca2+-mobilizing agonists and voltage steps raised Cac to similar, but not identical, levels (see Materials and Methods), and the amplitudes of the corresponding Cam signals also were similar. Hence, mitochondria appear to sequester cytoplasmic Ca2+ ions, whether entering from outside or released from intracellular stores. The initial recovery of Cac after stimulation by agonist was consistently slower than after depolarizationinduced Ca2+ entry, very likely because IP3-dependent mobilization continues for some seconds after the agonist is washed away.
Figure 7.

Similar mitochondrial responses to calcium entry and to calcium mobilization by agonists. Cells were coloaded with rhod-2 and Calcium Green esters, examined in perforated patch, and the data analyzed, all as in Fig. 4. Each record is representative of 3–5 similar experiments. (A) At the indicated times a single cell was perfused with 100 nM bradykinin (BK) for 5 s. After recovery, a 250-ms depolarization (ΔV) was applied. (B) Cam and Cac responses of another cell to a 5-s perfusion with 50 μM muscarine (musc) and subsequent depolarization.
Chromaffin cells also contain a Ca2+ store that can be mobilized by caffeine or by Ca2+ entry during perforatedpatch recording (Park et al., 1996). This Ca2+-induced Ca2+ release (CICR) is especially apparent in cells with impaired mitochondrial Ca2+ uptake (as in Figs. 3 C and 4) and can be recognized in experiments like that in Fig. 8 A where depolarizations of increasing duration were applied to a cell in perforated-patch mode. The plots of averaged Cam vs Cac from four such experiments (Fig. 8 B) show that Cac continued to rise for several hundred milliseconds after the Ca2+ entry evoked by depolarization had ceased (large filled symbols). These plots can be compared with the similar plot for whole-cell recording (Fig. 5 C) where Cac began to fall as soon as the membrane depolarization ended. In the perforated patch experiments (Fig. 8 B), Cam also rose more steeply during the period of CICR, as if mitochondria were taking up the additional Ca2+ released from stores. While spatially averaged Cac was still highly elevated, Cam then began to fall, as if reuptake into the depleted stores was creating a local sink for Ca2+ fed from mitochondria.
To substantiate the interpretation that mitochondria sequester Ca2+ released from CICR stores, we coapplied caffeine and the reticular Ca2+-ATPase inhibitor BHQ to deplete CICR stores and prevent their refilling (Fig. 9). When caffeine and BHQ were added, Cam and Cac increased transiently (Fig. 9 A; n = 4), showing that when caffeine induces release of Ca2+ from stores, mitochondria will take it up. For the preparation of cells used in these experiments, channel rundown (see Materials and Methods) was more severe than for those of Fig. 8. A control stimulus of 500 ms was needed to produce a Cac challenge similar to that found with a 250-ms pulse in the previous experiments (compare the trajectories marked with circles in Figs. 8 B and 9 B). Lengthening of the depolarization to 1 and 1.5 s then was required for subsequent stimuli applied in the presence of inhibitors.
Figure 9.
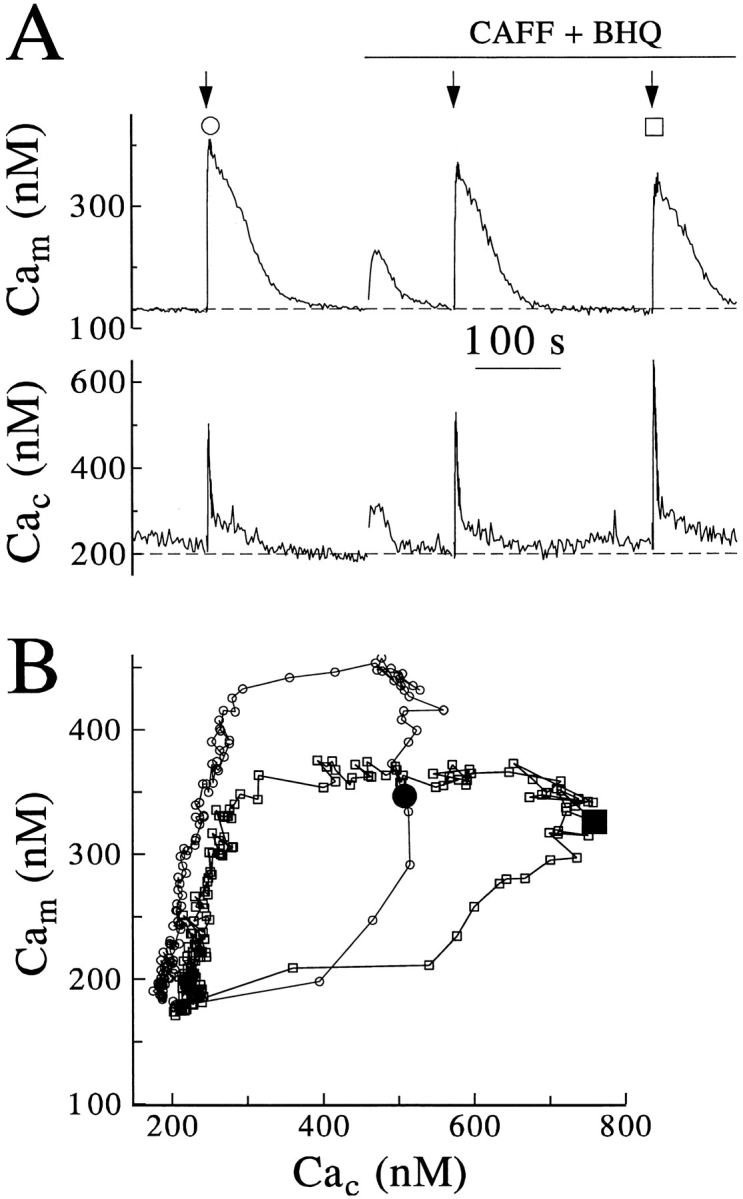
Mitochondrial sequestration of Ca2+ released from caffeine-sensitive stores. Cells were coloaded with rhod-2 and Calcium Green and examined in perforated patch as in Fig. 4. (A) Cam and Cac responses from another cell subjected to sequential depolarizations of 0.5 s applied before (circle), and of 1.0 or 1.5 s (box) applied after perfusion with 10 mM caffeine and 10 μM BHQ. (B) Averaged responses from three cells treated exactly as above. The enlarged, closed symbols mark the observed Cam and Cac values at the termination of stimulus. The intermediate response to the first depolarization applied after treatment is not shown, to avoid excessive overlap of records.
Consider the trajectories of Cac and Cam that follow the depolarizing pulses (Fig. 9 B). Under the control conditions, there was a full second during which Cac was relatively constant while Cam continued to rise. In the presence of caffeine and BHQ, subsequent depolarizations still gave large averaged peak Cac responses but the rise of Cam after the end of the depolarization was greatly reduced (Fig. 9 B), confirming that much of the late rise of Cam represents uptake of Ca2+ released by CICR.
The initial rate of rise of Cam during Ca2+ entry is faster in these perforated-patch experiments than it was with the whole-cell recordings of Fig. 5. Using perforated-patch it was 674 ± 150 nM s−1 (n = 9), and in whole-cell mode it was 79 ± 30 nM s−1 (n = 6). We suspect that the faster rise reflects a combination of CICR and greater proximity of mitochondria to locally high microdomains of Cac around Ca2+ channels (Neher and Augustine, 1992; Robinson et al., 1995) in the undialyzed cells. Dialysis may disrupt cytoskeletal elements that establish a mitochondrial localization near the plasma membrane and other organelles. In the first second after entry ceased, when such microdomains should be dispersed, Cam continued to rise at ∼40 nM s−1 in the experiment of Fig. 5 B.
Discussion
The original description of the synthesis and characterization of rhod-2 noted that its AM-ester might be selectively accumulated, hydrolyzed, and trapped in mitochondria (Minta et al., 1989), and recent confocal imaging has shown compartmentation of this probe (Burnier et al., 1994; Tsien and Bacskai, 1995). It has also been shown qualitatively that Ca2+ entry (Tsien and Bacskai, 1995) and Ca2+ mobilization (Hajnóczky et al., 1995, Rutter et al., 1996) increase the signal from compartmentalized rhod-2. Our imaging of rhod-2 by fluorescence deconvolution methods and our quantitative photometric observations during application of pharmacological agents further validate and extend the use of rhod-2 as a probe of mitochondrial [Ca2+] in living cells.
Previous work using blockers of mitochondrial function implicated mitochondria in the recovery of neurons and chromaffin cells from imposed Ca2+ entry (Thayer and Miller, 1990; Friel and Tsien, 1994; Werth and Thayer, 1994; White and Reynolds, 1995; Park et al., 1996; Herrington et al., 1996). For chromaffin cells we had found that blockers of mitochondrial Ca2+ uptake but not blockers of other cellular Ca2+ transporters greatly slow the initial clearance of Cac yet shorten the total time to return to the resting state (Park et al., 1996; Herrington et al., 1996). Now our simultaneous monitoring of Cam and Cac clearly establishes that Ca2+ uptake through the mitochondrial uniporter underlies rapid removal of cytosolic Ca2+. It shows further that subsequent Ca2+ export from mitochondria requires the mitochondrial Na+-Ca2+ exchanger and accounts for the slowness of the final decrease in Cac. With the less-invasive perforated-patch method we find that mitochondria accumulate Ca2+ mobilized from either IP3- or caffeine-sensitive internal stores, and speculate that stores may be refilled with Ca2+ exported from mitochondria. A picture emerges of a highly interactive network of cellular Ca2+ signaling that involves mitochondria and at least two mobilizable pools. The rapid rise of Cam to even modest physiological Cac loads presumably also means that mitochondrial energy metabolism adjusts itself to cellular activities from moment to moment (McCormack et al., 1990; Gunter et al., 1994; Hajnóczky et al., 1995; Rutter et al., 1996).
What Is the Free Calcium Level in Mitochondria?
In the next sections we show that the Ca2+-handling properties of mitochondria within cells are similar to those found in detailed studies of isolated mitochondria. Some previous work used permeant precursors to load the fluorescent Ca2+ probes indo-1 and fura-2 into isolated mitochondria (Lukács et al., 1987; Gunter et al., 1988; Reers et al., 1989; McCormack et al., 1989; Leisey et al., 1993). Mitochondria within cells have also been loaded nonselectively with these dyes and studied after quench of cytosolic dye with Mn2+ (Miyata et al., 1991) or by imaging of mitochondria-rich regions (Sheu and Jou, 1994) to estimate a compartmentalized signal. For both isolated and cellular mitochondria, these studies with ratiometric dyes obtained low resting values of Cam in the range of 80–200 nM, much as for nonratiometric rhod-2 in our work. Thus it is agreed that there is hardly any gradient of free [Ca2+] across the resting mitochondrial membrane, despite a large, insidenegative Δψm. Presumably the uptake via the uniporter is in balance with extrusion via exchange and other mechanisms, and perhaps the uniporter is largely shut down at rest (Gunter and Pfeiffer, 1990; Gunter et al., 1994).
When cytoplasmic or extramitochondrial [Ca2+] is raised to 1–2 μM, all studies with dyes report elevations in Cam to 250–1,000 nM, again as we have found. The general agreement supports the calibration protocols we have used for the nonratiometric rhod-2 dye. On the other hand, work with cells expressing mitochondrially targeted aequorin reports that Cam increases (for only a few seconds) to >5 μM upon Ca2+ entry into excitable cells or mobilization of Ca2+ stores in nonexcitable cells (Rizzuto et al., 1992, 1993, 1994; Rutter et al., 1993). This higher level could be reconciled with the dye results if heterogeneity among dye-containing compartments allowed some to rise briefly to very high Ca2+ levels, which would saturate dyes and give high aequorin emission, while others had only moderate and longer lasting [Ca2+] elevations, keeping the spatially averaged dye signal below saturation. Alternatively, there may be subtle difficulties in calibrating aequorin signals. For single CHO cells responding to a Ca2+ mobilizing agonist, Rutter et al. (1996) found that rhod-2 fluorescence transiently increased 2–3-fold. Assuming a resting Cam of 100 nM and using the spectral properties and affinities of rhod-2 that we observe in vitro, we estimate that this corresponds to a peak Cam of 400– 800 nM, much like the responses found in the present study, but much smaller than the >10 μM peak Cam estimated from the responses of the same CHO cells transfected with mitochondrial aequorin.
What is the State of Mitochondrial Calcium?
Mitochondria can accumulate enormous quantities of Ca2+. Classical experiments showed that isolated mitochondria exposed to modest Ca2+ concentrations, socalled “limited loading,” accumulate up to 100 nmol Ca2+ (mg protein)−1 in half a min without damage (Lehninger et al., 1967; Gunter and Pfeiffer, 1990). Exposure to millimolar Ca2+ (“massive loading”) can lead to accumulations up to 2,600 nmol Ca2+ (mg protein)−1, accompanied by irreversible damage, morphological changes, and precipitation of hydroxyapatite crystals. These values would be equivalent to Ca2+ contents of 36 and >900 mM within the mitochondria, respectively!
We now calculate how much the mitochondrial Ca2+ content should rise in our experiments if, for example, 70% (Herrington et al., 1996) of the Ca2+ in a single 1.5-μM cytoplasmic challenge were taken up into the mitochondria. The mitochondria would actually remove much more than the 1.5-μM free Ca2+ because almost all the cytoplasmic Ca2+ is reversibly bound to proteins and other Ca2+ buffers. In rat chromaffin cells (see Figs. 2 and 3 of Park et al., 1996) and in other excitable cells (Neher, 1995), typical values for cytoplasmic Ca2+-binding ratios are near 100. Thus, mitochondria would have to remove 0.7 × 100 × 1.5 μM = 105 μM total Ca2+. Since the mitochondrial volume is only 6% of the combined cytosolic and nuclear volumes in rat chromaffin cells (Tomlinson et al., 1987), this uptake should raise the mitochondrial Ca2+ content by 1.75 mM. These numbers emphasize how remarkable it is that compartmentalized dyes report Cam rises of only 250–450 nM during such a challenge.
Why is the rise of Cam several thousand times less than expected? Like the cytoplasm, the mitochondrial matrix must contain Ca2+ buffers. These could include membrane phospholipids, Ca2+-binding proteins, and phosphate compounds. The inner leaflet of the inner mitochondrial membrane contributes 6–20 mM of phospholipids (Carafoli, 1979; Schwerzmann et al., 1986), and the matrix contains tens of millimolar of phosphate compounds. The kind and concentration of Ca2+-binding proteins is not well known.
We would suggest that the Ca2+ taken up in our experiments binds quickly and reversibly to sites in the mitochondrion and does not precipitate as insoluble compounds. If this were not true, the full Ca2+ load would not be able to re-enter the cytoplasm within seconds after application of CCCP (e.g., Figs. 1 D and 5 B of Herrington et al., 1996). We conclude therefore that the mitochondrial matrix buffers Ca2+ reversibly with an effective Ca2+ binding ratio of ∼4,000 (see also Coll et al., 1982; Lukács and Kapus, 1987). This is 40 times the Ca2+-binding ratio of cytoplasm.
How Fast Can Mitochondria Transport Ca2+?
Rates of Ca2+ transport for isolated mitochondria have been extensively studied. Taking typical reported values, the Ca2+ uniporter is half maximally activated around 10– 20 μM Ca2+ and has a maximum velocity at room temperature ∼10–30 nmol (mg protein)−1 s−1 in energized cardiac and liver mitochondria (Carafoli, 1979; Gunter and Pfeiffer, 1990). The maximum velocity of the mitochondrial Na+-Ca2+ exchanger is 30–400 times slower. Although the maximum velocity may be significantly affected by the bathing phosphate, Mg2+, pH, and other conditions, we will use it to compare with the uptake rates we measure in cells.
What would be the effect of a uniporter operating near its maximum velocity, at 20 nmol Ca2+ (mg protein)−1 s−1? Since 1 nmol (mg protein)−1 corresponds to 385 μmol/liter of mitochondria (Schwerzmann et al., 1986), we would expect that a liter of mitochondria could take up 7,700 μmol Ca2+ s−1. Once again we apply the mitochondrial volume (6%) and the cytoplasmic Ca2+-binding ratio (100) and predict that Cac (the free Ca2+) should fall at 4.6 μM s−1 when the uniporter is working at full velocity.
We have previously measured the rates of Cac decay in chromaffin cells attributable to mitochondrial uptake after depolarization-induced Ca2+ entry (Herrington et al., 1996). At 1.5 μM Cac the mean rate was 0.8 μM s−1. In Park et al. (1996), we gave evidence for appreciable uptake during a 200-ms depolarization, and in the present paper, we find that Cam rises more rapidly during the Ca2+ entry than afterward; indeed more than half of the rise in Cam occurs during a 500-ms depolarizing pulse (Figs. 5, 8, and 9). Thus, we presume that early uptake proceeds at rates equivalent to a decay of Cac > 0.8 μM s−1. These velocities for Ca2+ import by mitochondria in living cells seem quite compatible with the 4.6 μM s−1 maximum velocity predicted from uptake by isolated mitochondria; however, they are higher than would be expected at 1.5 μM Ca2+ if we take into account the reported Ca2+ affinity and cooperativity of uptake. A model in which a subset of mitochondria lies so close to the plasma membrane that they transiently see elevated Cac values well above those reported by the cytoplasmic dye (Lawrie et al., 1996) could explain these results. Perhaps also Ca2+ transport is more effective for mitochondria bathed in cytoplasm than for isolated mitochondria.
Exploring the Cellular Ca2+ Network
We propose that mitochondria have a unique role in the cytoplasmic Ca2+ network. The plasma membrane and reticular stores deliver Ca2+ rapidly to the cytoplasm, by opening ion channels, and then remove it slowly by primary and secondary active transport. Conversely, and consistent with their origins as a cellular symbiont within eukaryotic cytoplasm, mitochondria rapidly remove Ca2+ by a uniporter that acts like a channel, then return it more slowly by secondary active transport. Hence, the plasma membrane and internal stores create pulses of Cac with microdomains of high concentration suitable for initiating exocytosis or rapid muscle contraction. Mitochondria, on the other hand, act in an apparently constitutive manner, capturing Ca2+ pulses and then returning the Ca2+ slowly to the cytoplasm where it can prolong activation of highaffinity, Ca2+-dependent processes and perhaps feed refilling of stores. In some cells this transient sequestration into mitochondria is important in generation of Ca 2+ waves and oscillations (Jouaville et al., 1995; Hehl et al., 1996). In addition, the transiently elevated Cam probably stimulates mitochondrial ATP production in anticipation of cellular needs. Such a major role for mitochondria in Ca2+ clearance holds for chromaffin cells and neurons but not for skeletal and cardiac muscles with a highly elaborated sarcoplasmic reticulum. We need now to examine the importance of mitochondria in other cell types and to determine if mitochondrial Ca2+ uptake is a regulated process.
Acknowledgments
We thank Applied Precision Inc. (Issaquah, WA) for generous use of their Deltavision image analysis system. The authors also thank Drs. P.B. Detwiler, J. Howard, J.S. Isaacson, and W.W. Parson for critical evaluation of this manuscript and Lea Miller, Don Andersen, and Paulette Brunner for technical assistance.
This work was supported by National Institutes of Health grants AR17803 and HD07878, the W.M. Keck Foundation, and the Royalty Research Fund of the University of Washington.
Abbreviations used in this paper
- Cac
free cytosolic [Ca2+]
- Cam
free intramitochondrial [Ca2+]
- CGP-37157
7-chloro-3,5-dihydro-5-phenyl-1H4,1-benzothiazepine-2-on
- BHQ
2,5 di(t-butyl)-1,4-hydroxyquinone
- CCCP
carbonyl cyanide m-chlorophenylhydrazone
Footnotes
Please address all correspondence to B. Hille, Department of Physiology & Biophysics, G424 Health Sciences Building, University of Washington, Box 357290, Seattle, WA 98195-7290. Tel.: (206) 543-8639. Fax: (206) 6850619. E-Mail: hille@u.washington.edu
References
- Agard DA, Hiraoka Y, Shaw P, Sedat JW. Fluorescence microscopy in three dimensions. Methods Cell Biol. 1989;30:353–377. doi: 10.1016/s0091-679x(08)60986-3. [DOI] [PubMed] [Google Scholar]
- Budd SL, Nichols DG. A reevaluation of the role of mitochondria in neuronal Ca2+homeostasis. J Neurochem. 1996;66:403–411. doi: 10.1046/j.1471-4159.1996.66010403.x. [DOI] [PubMed] [Google Scholar]
- Burnier M, Centeno G, Burki E, Brunner HR. Confocal microscopy to analyze cytosolic and nuclear calcium in cultured vascular muscle cells. Am J Physiol. 1994;266:C1118–C1127. doi: 10.1152/ajpcell.1994.266.4.C1118. [DOI] [PubMed] [Google Scholar]
- Carafoli E. The calcium cycle of mitochondria. FEBS Lett. 1979;104:1–5. doi: 10.1016/0014-5793(79)81073-x. [DOI] [PubMed] [Google Scholar]
- Chen, H., J.R. Swedlow, M. Gote, J.W. Sedat, and D.A. Agard. 1995. The collection, processing, and display of digital three-dimensional images of biological specimens. In Handbook of Biological Confocal Microscopy. J.B.Pawley, editor. Plenum Press, NY. 197–210.
- Coll KE, Joseph SK, Corkey BE, Williamson JR. Determination of the matrix free Ca2+ concentration and kinetics of Ca2+efflux in liver and heart mitochondria. J Biol Chem. 1982;257:8696–8704. [PubMed] [Google Scholar]
- Cox DA, Matlib MA. A role for the mitochondrial Na+-Ca2+exchanger in the regulation of oxidative phosphorylation in isolated heart mitochondria. J Biol Chem. 1993;268:938–947. [PubMed] [Google Scholar]
- Eberhard M, Erne P. Calcium binding to fluorescent calcium indicators: calcium green, calcium orange and calcium crimson. Biochem Biophys Res Commun. 1991;180:209–215. doi: 10.1016/s0006-291x(05)81278-1. [DOI] [PubMed] [Google Scholar]
- Erdahl WL, Chapman CJ, Wang E, Taylor RW, Pfeiffer DR. Ionophore 4-BrA23187 transports Zn2+ and Mn2+ with high selectivity over Ca2+ . Biochemistry. 1996;35:13817–13825. doi: 10.1021/bi961391q. [DOI] [PubMed] [Google Scholar]
- Friel DD, Tsien RW. An FCCP-sensitive Ca2+ store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [Ca2+]i . J Neurosci. 1994;14:4007–4024. doi: 10.1523/JNEUROSCI.14-07-04007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Peonie M, Tsien RY. A new generation of Ca2+indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Restrepo D, Gunter KK. Conversion of esterified fura-2 and indo-1 to Ca2+forms by mitochondria. Am J Physiol. 1988;255:C304–C310. doi: 10.1152/ajpcell.1988.255.3.C304. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Gunter KK, Sheu S-S, Gavin CE. Mitochondrial calcium transport: physiological and pathological relevance. Am J Physiol. 1994;267:C313–C339. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- Hajnóczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hehl S, Golard A, Hille B. Involvement of mitochondria in intracellular calcium sequestration by rat gonadotropes. Cell Calcium. 1996;20:515–524. doi: 10.1016/s0143-4160(96)90094-9. [DOI] [PubMed] [Google Scholar]
- Herrington J, Park YB, Babcock DF, Hille B. Dominant role of mitochondria in clearance of large Ca2+loads from rat adrenal chromaffin cells. Neuron. 1996;16:219–228. doi: 10.1016/s0896-6273(00)80038-0. [DOI] [PubMed] [Google Scholar]
- Hiraoka Y, Sedat JW, Agard DA. Determination of three-dimensional imaging properties of a light microscope system. Partial confocal behavior in epifluorescence microscopy. Biophys J. 1990;57:325–333. doi: 10.1016/S0006-3495(90)82534-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka Y, Swedlow JR, Paddy MR, Agard DA, Sedat JW. Three-dimensional multiple-wavelength fluorescence microscopy for the structural analysis of biological phenomena. Semin Cell Biol. 1991;2:153–165. [PubMed] [Google Scholar]
- Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording technique. J Gen Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LV, Walsh ML, Bockus BJ, Chen LB. Monitoring of relative mitochondrial membrane potential in living cells by fluorescence microscopy. J Cell Biol. 1981;88:526–535. doi: 10.1083/jcb.88.3.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouaville LS, Ichas F, Holmuhamedov EL, Camacho P, Lechleiter JF. Synchronization of calcium waves by mitochondrial substrates in Xenopus laevisoocytes. Nature. 1995;377:438–441. doi: 10.1038/377438a0. [DOI] [PubMed] [Google Scholar]
- Lawrie AM, Rizzuto R, Pozzan T, Simpson AWM. A role for calcium influx in the regulation of mitochondrial calcium in endothelial cells. J Biol Chem. 1996;271:10753–10759. doi: 10.1074/jbc.271.18.10753. [DOI] [PubMed] [Google Scholar]
- Lehninger AL. Mitochondria and calcium ion transport. Biochem J. 1970;119:129–138. doi: 10.1042/bj1190129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehninger AL, Carafoli E, Rossi CS. Energy linked ion movements in mitochondrial systems. Adv Enzymol. 1967;29:259–320. doi: 10.1002/9780470122747.ch6. [DOI] [PubMed] [Google Scholar]
- Leisey JR, Grotyohann LW, Scott DA, Scaduto RC., Jr Regulation of cardiac mitochondrial calcium by average extramitochondrial calcium. Am J Physiol. 1993;265:H1203–H1208. doi: 10.1152/ajpheart.1993.265.4.H1203. [DOI] [PubMed] [Google Scholar]
- Loew LM, Carrington W, Tuft RA, Fay FS. Physiological cytosolic Ca2+transients evoke concurrent mitochondrial depolarizations. Proc Natl Acad Sci USA. 1995;91:12579–12583. doi: 10.1073/pnas.91.26.12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukács GL, Kapus A. Measurement of the matrix free Ca2+concentration in heart mitochondria by entrapped fura-2 and quin2. Biochem J. 1987;248:609–613. doi: 10.1042/bj2480609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malgaroli A, Fesce R, Meldolesi J. Spontaneous [Ca2+]i fluctuations in rat chromaffin cells do not require inositol 1,4,5 trisphosphate elevations but are generated by a caffeine- and ryanodine-sensitive intracellular Ca2+store. J Biol Chem. 1990;265:3005–3008. [PubMed] [Google Scholar]
- McCormack JG, Browne H, Dawes NJ. Studies on mitochondrial Ca2+-transport and matrix Ca2+using fura-2-loaded rat heart mitochondria. Biochim Biophys Acta. 1989;973:420–427. doi: 10.1016/s0005-2728(89)80384-6. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–420. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- Minta A, Kao JP, Tsien RY. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J Biol Chem. 1989;264:8171–8178. [PubMed] [Google Scholar]
- Miyata H, Silverman HS, Sollot SJ, Lakatta EG, Stern MD, Hansford RG. Measurement of mitochondrial free Ca2+concentration in living single rat cardiac myocytes. Am J Physiol. 1991;261:H1123–H1134. doi: 10.1152/ajpheart.1991.261.4.H1123. [DOI] [PubMed] [Google Scholar]
- Neely A, Lingle CJ. Two components of calcium-activated potassium current in rat adrenal chromaffin cells. J Physiol. 1992a;453:97–131. doi: 10.1113/jphysiol.1992.sp019220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely A, Lingle CJ. Effects of muscarine on single rat adrenal chromaffin cells. J Physiol. 1992b;453:133–166. doi: 10.1113/jphysiol.1992.sp019221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. The use of fura-2 for estimating Ca buffers and Ca fluxes. Neuropharmacology. 1995;34:1423–1442. doi: 10.1016/0028-3908(95)00144-u. [DOI] [PubMed] [Google Scholar]
- Neher E, Augustine GJ. Calcium gradients and buffers in bovine chromaffin cells. J Physiol. 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan AJ, Cheek TR, Moreton RB, Berridge MJ, Burgoyne RD. Localization and heterogeneity of agonist-induced changes in cytosolic calcium concentration in single bovine adrenal chromaffin cells from video imaging of fura-2. EMBO (Eur Mol Biol Organ) J. 1989;8:401–411. doi: 10.1002/j.1460-2075.1989.tb03391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YB. Ion selectivity and gating of small conductance Ca2+-activated K+channels in cultured rat adrenal chromaffin cells. J Physiol. 1994;481:555–570. doi: 10.1113/jphysiol.1994.sp020463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YB, Herrington J, Babcock DF, Hille B. Ca2+clearance mechanisms in isolated rat adrenal chromaffin cells. J Physiol. 1996;492:329–346. doi: 10.1113/jphysiol.1996.sp021312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reers M, Kelly RA, Smith TW. Calcium and proton activities in rat cardiac mitochondria. Effect of matrix environment on behavior of fluorescent probes. Biochem J. 1989;257:131–142. doi: 10.1042/bj2570131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson IM, Finnegan JM, Monck JR, Wightman RM, Fernandez JM. Colocalization of calcium entry and exocytotic release sites in adrenal chromaffin cells. Proc Natl Acad Sci USA. 1995;92:2474–2478. doi: 10.1073/pnas.92.7.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Simpson AVM, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+revealed by specifically targeted recombinant aequorin. Nature (Lond) 1992;358:325–327. doi: 10.1038/358325a0. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science (Wash DC) 1993;262:744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Bastianutto C, Brini M, Murgia M, Pozzan T. Mitochondrial Ca2+homeostasis in intact cells. J Cell Biol. 1994;126:1183–1194. doi: 10.1083/jcb.126.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter GA, Theler J-M, Murgia M, Wolheim CB, Pozzan T, Rizzuto R. Increased Ca2+ influx raises mitochondrial free Ca2+to micromolar levels in a pancreatic beta-cell line. J Biol Chem. 1993;268:22385–22390. [PubMed] [Google Scholar]
- Rutter GA, Burnett P, Rizzuto R, Brini M, Murgia M, Pozzan T, Tavaré JM, Denton RM. Subcellular imaging of intramitochondrial Ca2+with recombinant targeted aequorin: significance for the regulation of pyruvate dehydrogenase activity. Proc Natl Acad Sci USA. 1996;93:5489–5494. doi: 10.1073/pnas.93.11.5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu S-S, Jou M-J. Mitochondrial free Ca2+concentration in living cells. J Bioenerg Biomembr. 1994;26:487–493. doi: 10.1007/BF00762733. [DOI] [PubMed] [Google Scholar]
- Sparagna GC, Gunter KK, Sheu S-S, Gunter TE. Mitochondrial calcium uptake from physiological-type pulses of calcium. A description of the rapid uptake mode. J Biol Chem. 1995;270:27510–27515. doi: 10.1074/jbc.270.46.27510. [DOI] [PubMed] [Google Scholar]
- Schwerzmann K, Cruz-Orive LM, Eggman R, Sänger A, Weibel ER. Molecular architecture of the inner membrane of mitochondria from rat liver: a combined biochemical and stereological study. J Cell Biol. 1985;102:97–103. doi: 10.1083/jcb.102.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer SA, Miller RJ. Regulation of the intracellular free calcium concentration in single rat dorsal root ganglion neurones in vitro. . J Physiol. 1990;425:85–116. doi: 10.1113/jphysiol.1990.sp018094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson A, Durbin J, Coupland RE. A quantitative analysis of rat adrenal chromaffin tissue: morphometric analysis at tissue and cellular level correlated with catecholamine content. Neuroscience. 1987;20:895–907. doi: 10.1016/0306-4522(87)90250-8. [DOI] [PubMed] [Google Scholar]
- Tsien, R.Y., and B.J. Bacskai. 1995. Video-rate confocal microscopy. In Handbook of Biological Confocal Microscopy, J.B. Pawley, editor. Plenum Press, New York. 459–478.
- Villalba M, Martinez-Serrano A, Gómez-Puertas P, Blanco P, Börner C, Villa A, Casado M, Giménez C, Pereira R, Bogonez E, et al. The role of pyruvate in neuronal calcium homeostasis. Effect on intracellular calcium pools. J Biol Chem. 1994;269:2468–2476. [PubMed] [Google Scholar]
- Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured dorsal root ganglion neurons. J Neurosci. 1994;14:348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondria and Na+/Ca2+exchange buffer glutamate-induced calcium loads in cultured cortical neurons. J Neurosci. 1995;15:1318–1328. doi: 10.1523/JNEUROSCI.15-02-01318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]