

Abstract
A cDNA encoding the Clostridium perfringens enterotoxin receptor gene (CPE-R) was cloned from an expression library of enterotoxin-sensitive Vero cells. The nucleotide sequence of CPE-R showed that the enterotoxin receptor consists of 209 amino acids with a calculated molecular mass of 22,029 D. This receptor is highly hydrophobic, contains four putative transmembrane segments, and has significant similarity to the rat androgen withdrawal apoptosis protein RVP1 and the mouse oligodendrocyte specific protein, the functions of which are unknown. The expression of CPE-R was detected in the enterotoxin-sensitive Vero, Hep3B, and Intestine 407 cell lines, but not in the enterotoxin-insensitive K562 and JY cell lines. The CPE-R gene product expressed in enterotoxin-resistant L929 cells bound to enterotoxin specifically and directly and with high affinity and rendered the cells sensitive to the toxin, indicating that the cloned receptor is functional. Results showed that enterotoxin could not assemble into a complex with a defined structure unless it interacted with the receptor. From these results, it is proposed that the enterotoxin receptor is required for both target cell recognition and poreformation in the cell membrane.
C lostridium perfringens enterotoxin (CPE)1, which consists of a single polypeptide chain and has a molecular weight of ∼35,000, is the causative agent of symptoms associated with C. perfringens food poisoning in man (McClane et al., 1988a ). CPE produced in the intestinal tract during sporulation injures intestinal epithelial cells and causes fluid accumulation in the intestinal cavity, resulting in diarrhea (Stark and Duncan, 1971). Morphological changes such as bleb balloon formation or complete destruction of intestinal epithelial cells induced by CPE has been observed in rat and rabbit models (McDonel and Duncan, 1975; McDonel et al., 1978). Similar cytotoxic effects have also been shown in cultured mammalian cells (Matsuda and Sugimoto, 1979; McClane and McDonel, 1979; McDonel, 1980; Tolleshaug et al., 1982). Several lines of evidence suggest that CPE increases membrane permeability by forming small pores and induces the release of intracellular molecules from sensitive cells (Matsuda and Sugimoto, 1979; McClane and McDonel, 1981; Sugimoto et al., 1985, 1988; Matsuda et al., 1986). The consequent loss of osmotic equilibrium is considered to lead to membrane destruction, resulting in morphological alterations and finally cell death.
The sensitivities of different cell types to CPE vary. Rabbit small intestinal epithelial cells (McDonel, 1980), rat liver cells (Tolleshaug et al., 1982), the monkey kidney cell line, Vero cells (McClane and McDonel, 1979), and HeLa cells (Matsuda and Sugimoto, 1979) have been shown to be sensitive to CPE. On the contrary, several types of cells such as the mouse fibroblast cell line L929 (Horiguchi et al., 1985) were found to show resistance to CPE. The sensitivity of cells to CPE is thought to depend on the presence of a specific receptor(s) for CPE on the cell surface (McDonel and McClane, 1979; Horiguchi et al., 1985). Pretreatment of CPE-sensitive cells with neuraminidase or addition of ganglioside, methyl β-galactoside, methyl α-mannose, or N-acetylglucosamine to the medium did not affect the binding ability of CPE, whereas extensive digestion of the sensitive cells with pronase did (McDonel, 1980; Wnek and McClane, 1986; Tolleshaug et al., 1982; McClane et al., 1988 b). These findings indicate that CPE binds to a specific protein receptor(s) on the surface of sensitive cells.
Cellular proteins of 50–60 kD derived from sensitive cells were reported to have affinity to CPE (Wnek and McClane, 1983; Sugii and Horiguchi, 1988). However, it is unknown whether these proteins act as a functional membrane receptor(s) for CPE. Wieckowski et al. (1994) reported that after its initial binding, CPE aggregates with several cellular proteins and forms a large hydrophobic complex with a molecular mass of 160 kD. By a simple calculation they concluded that the complex contained a 70-kD protein besides the 50-kD CPE binding substance and CPE (35 kD) and assumed that this complex contributed to pore formation in the membrane of target cells. But the composition and function of the CPE-induced complex can not be determined until the functional CPE receptor is identified.
To understand the molecular mechanism of the action of CPE, we isolated the CPE-receptor gene (CPE-R) and examined the function of the gene product. Here we report the identification and characterization of a functional CPE receptor.
Materials and Methods
Plasmid Construction
Genomic DNA was isolated from C. perfringens strain NCTC8239 (a gift from Dr. T. Asao, Osaka Prefectural Institute of Public Health, Osaka, Japan) by the method described by Marmur (1961). Approximately 10 ng of the genomic DNA was subjected to PCR using oligonucleotides 5′-CCGCTCGAGAGATGTGTTTTAACAGTTCCATCTAC-3′ (primer-S; the underline indicates XhoI site) and 5′-GGAAGATCTTAAAATTTTTGAAATAATATTGAATAAGGG-3′ (primer-A; the underline indicates BglII site) as sense and antisense primers to amplify the DNA fragment corresponding to amino acid residues 184–319 of CPE (Czeczulin et al., 1993). The amplified DNA fragment was digested with XhoI and BglII and then cloned into the XhoI–BamHI treated pET16b vector (Novagen Inc., Madison, WI) to fuse the CPE fragment to the down stream of the tag sequence with 10 histidine residues (Fig. 1 A). The resulting plasmid vector was designated as pETH10PER.
Figure 1.
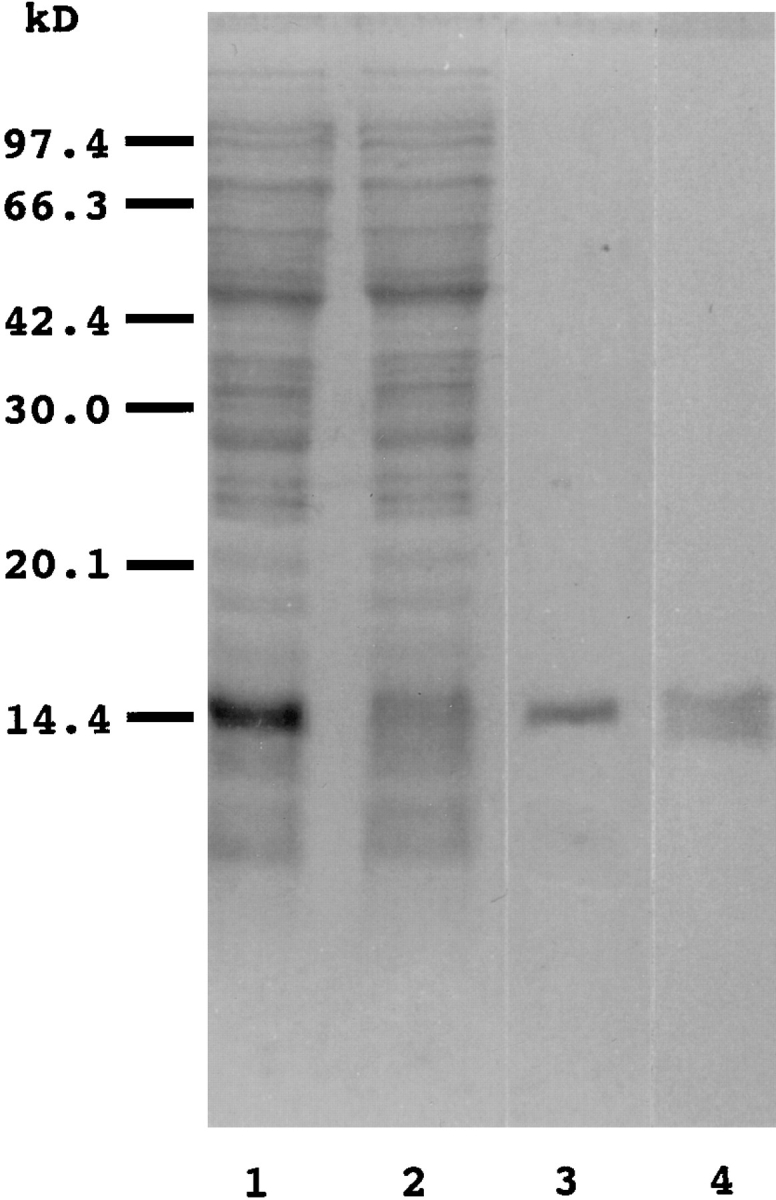
Expression and purification of a histidinetagged Clostridium perfringens enterotoxin COOH-terminal fragment (H10PER). H10PER was expressed in E. coli BL21 (DE3) and purified as described in Materials and Methods. A total cell lysate (lane 1), flow through fraction (lane 2), and purified H10PER (lane 3) were separated on SDS-15% polyacrylamide gel and stained with Coomassie brilliant blue. Purified H10PER was transferred to a PVDF membrane, and Western blot analysis was carried out using rabbit polyclonal anti-CPE antibody, followed by alkaline phosphatase–conjugated anti–rabbit IgG (lane 4). The positions of molecular weight standards are indicated on the left in kD.
For constructing pCMVβ-GAL, a HindIII-PstI fragment containing Escherichia coli β-galactosidase gene was isolated from pSGβ-galactosidase (Promega Biotech., Madison, WI) and was subcloned into the same site of pCDM8 (Invitrogen Corp., San Diego, CA).
For sequencing, the clone 706 encoding CPE-R was isolated by XhoI digestion followed by treatment with T4 DNA polymerase, and the fragment obtained was introduced into the EcoRV site of pBluescript SK(−) (Stratagene Cloning Systems, La Jolla, CA). Two clones containing the CPE-R gene in opposite orientations were obtained and named pBS70608 and pBS70614. Nested deletion mutants of these clones were prepared using a double-stranded Nested Deletion Kit (Pharmacia Biotech, Uppsala, Sweden) according to the manufacturer's manual.
The CPE receptor cDNA was introduced into pMEneo vector (Watanabe et al., 1996), and the resulting plasmid (pMEneo-CPE-R) was used to establish L929 cell lines stably expressing CPE-R.
For construction of a COOH-terminal FLAG peptide tagged CPE receptor expression plasmid, CPE-R in pBS70614 was amplified by PCR using the oligonucleotides 5′-GGGTCGACGCCTCCATGGGGCTACAGG3′ (the underline indicates the SalI site) and 5′-GGTCGCGACACGTAGTTGCTGGCAGCAG-3′ (the underline indicates the NruI site) as forward and back primers. The amplified fragment was treated with T4 DNA polymerase followed by T4 polynucleotide kinase and cloned into the EcoRV site of the pBluescript SK(−). The XhoI-FseI site of this plasmid was replaced with the fragment of the corresponding site (encoding NH2-terminal portion of native CPE receptor) of pBS70614 to generate p706NruI. The XhoI–NruI fragment of p706NruI was then isolated and recloned into the same site of pMEEB (Watanabe et al., 1996) into which NruI site, FLAG sequence, and stop codon (TCGCGAGACTACAAGGACGACGATGACAAGTAA; the underline indicates NruI site) was introduced. The resulting plasmid was named pMEEB-CPE-R-FLAG.
Plasmids pS7neo (Takahashi et al., 1996) was a gift from Dr. M. Takahashi (Department of Immunoregulation, Research Institute for Microbial Diseases, Osaka University). The construction of pMEPyoni18Sf(−) is described elsewhere (Ohishi et al., 1996). pMEPyoriLuc was constructed as described previously (Takahashi et al., 1996).
Expression of the CPE COOH-terminal Fragment in Escherichia coli
pETH10PER was introduced into the E. coli BL21 (DE3) strain, and expression of the CPE COOH-terminal fragment was induced by 1 mM isopropyl β-d-thiogalactopyranoside (Wako Pure Chemical Industry, Osaka, Japan). The E. coli cells were harvested, resuspended in buffer A (10 mM Tris-HCl, pH8.0, 400 mM NaCl, 5 mM MgCl2, 10% glycerol, 0.1 mM (p-amidinophenyl)methanesulfonyl fluoride hydrochloride, 1 mM β-mercaptoethanol), and then disrupted by sonication. After removal of cell debris by centrifugation, the cell lysate was applied to a Ni2+ column (HisBind Resin; Novagen Inc.), and the histidine-tagged COOH-terminal fragment of CPE (H10PER) was eluted in a 0–1 M imidazole gradient in buffer A. The purified 15-kD fragment (Fig. 1) was biotinylated with sulfo-NHS-LC biotin (Pierce, Rockford, IL) and used as a probe for flow cytometric analysis.
Flow Cytometric Analysis
Expression of the CPE receptor was examined by flow cytometric analysis after treatment of cells with biotinylated H10PER (0.01 mg/ml) followed by phycoerythrin (PE)-conjugated streptavidin (0.02 mg/ml; Biomeda Corp., Foster City, CA). The fluorescence intensity of the cells was examined in a FACS®can (Becton Dickinson, Mountain View, CA).
Cell Culture and Establishment of Stable Cell Lines
Cells were cultured in DMEM supplemented with 10% FCS at 37°C under 5% CO2 in air. Clonal L929 cell lines stably expressing the polyoma large T antigen were established by electroporation of linearized plasmid pS7neo, followed by G418 (Geneticin; GIBCO BRL, Gaithersburg, MD) selection. Of 20 cell lines tested, the L929 cell line, which exhibited the highest β-galactosidase and firefly luciferase activity of the transfected pCMVβGAL and pMEPyoriLuc, respectively, was selected. This cell line was named L929pyT18 and used as the host cell line for cDNA library screening. L929 cell lines expressing CPE-R and its FLAG peptide-tagged version (CPE-R-FLAG) were established in the same manner, except that pMEneo-CPE-R and pMEEB-CPE-R-FLAG were introduced by electroporation followed by G418 or hygromycin (Wako Pure Chemical Industry) selection. The clonal cell lines expressing CPE receptor and FLAG-tagged CPE receptor were identified by flow cytometric analysis and were designated as 706Neo and 706FLAG, respectively.
cDNA Libraries
A cDNA library was constructed by the method of Gubler and Hoffmann (1983). An exponentially growing Vero cell culture was used as an RNA source. Total RNA was isolated by cesium trifluoroacetic acid isopycnic centrifugation. PolyA+ RNA was purified by the spinning of two successive oligo-dT cellulose columns (Pharmacia Biotech). PolyA+ RNA was reverse transcribed by Superscript reverse transcriptase II (GIBCO BRL) at 45°C and then converted to double-stranded cDNA with the aid of RNase H, E. coli DNA polymerase, and E. coli DNA ligase (Takara Shuzo Co., Shiga, Japan). After treatment with T4 DNA polymerase (Toyobo Inc., Osaka, Japan) followed by BstXI linker (Invitrogen Corp.) ligation, cDNAs were size selected and inserted into BstXI treated pMEPyori18Sf(−) vector to construct a cDNA library for eukaryotic expression.
Expression Cloning
The cDNA library was introduced into the L929pyT18 cell line by electroporation and cultured for 2 d before subsequent analysis. Expression of the CPE receptor was examined by flow cytometric analysis as described. Positive cells (the 0.1% most fluorescent of the transfected cells) were initially sorted by FACS®Vantage (Becton Dickinson) and pooled, and the transfected plasmids were recovered by the method of Hirt (1967). The rescued plasmids were reintroduced into E. coli strain MC1061, amplified, and then used for subsequent screening. This screening was repeated three times. After the third screening, the rescued plasmid clones were divided into 24 pools of 24 plasmids each, and the positive pools were identified by flow cytometric analysis. Finally, positive clones were identified by separating positive pools into single clones.
DNA Sequencing and Sequence Analysis
The nucleotide sequences of both strands of the cloned gene were determined by the dideoxy chain termination method using Thermo Sequenase (Amersham International, Amersham, UK) and a DNA sequencer (model 373A; Applied Biosystems, Foster City, CA). Sequence comparisons were made using the BLAST search program (Altschul et al., 1990) on an nr–nt data base. Multiple alignment was done by the CLUSTAL method (Higgins and Sharp, 1989).
Northern Blot Analysis
Total RNA was extracted from various cell lines by guanidinium thiocyanate-CsTFA isopycnic centrifugation using a QuickPrep Total RNA Extraction Kit (Pharmacia Biotech). Samples of 15 μg of total RNAs were subjected to agarose/formaldehyde gel electrophoresis. Northern blot analysis was performed by the method of Sambrook et al. (1989). The 1.7-kb XhoI fragment of clone 706, containing the entire coding sequence and 5′- and 3′-untranslated regions, was radiolabeled by the random priming method (Megaprime DNA labeling system; Amersham Intl.) and used as a probe. The blot was rehybridized with an elongation factor-1 α probe (Uetsuki et al., 1989) to determine the amount of RNA applied. The blots were exposed to an imaging plate for 18 h and analyzed with a Bioimaging Analyzer (BAS1500; Fuji Film Co., Tokyo, Japan).
Antibodies
Mouse monoclonal anti-FLAG peptide antibody M2 and M2-conjugated Sepharose were purchased from Eastman Kodak Co. (New Haven, CT). Alkaline phosphatase–conjugated goat anti–mouse IgG and streptavidin were from Organon Teknika (Turnhout, Belgium) and Oncogene Science Inc. (Uniondale, NY), respectively. Rabbit polyclonal antibody was raised against CPE toxoid and purified by protein A–Sepharose chromatography (Senda et al., 1995). Biotinylation of anti-CPE antibody was done as described.
Purification of CPE, Cytotoxicity Assay, and Binding Assay
CPE was purified by the method of Sakaguchi et al. (1973). The cytotoxic effect of CPE on cells was determined by examination of morphological alterations. CPE was radio-iodinated as described previously (Horiguchi et al., 1985). The ability of CPE binding to Neo706 cells (0.4 × 106 cells/assay) expressing CPE-R was measured by the method of Horiguchi et al. (1985), except that the binding reaction was done in PBS(−). Scatchard analysis was performed by the SP123 program (Ikeda et al., 1991).
Ligand Overlay Assay
Binding of CPE to the CPE-R gene product in vitro was examined by ligand overlay assay (Manser et al., 1992). Cell lysates (200 μg protein/ lane) of L929 and 706FLAG cells were subjected to SDS-15% polyacrylamide gel electrophoresis and then transferred to nitrocellulose membranes. After a denaturation and renaturation cycle (Manser et al., 1994), the blots were soaked in PBS-1% BSA containing 125I-labeled CPE (5 nM) with or without 100-fold molar excess of cold CPE. The membranes were then washed and exposed to an imaging plate, and the radioactive band was located with a Bioimaging Analyzer.
Immunoprecipitation
706FLAG cells (1 × 106) were labeled for 8 h with 50 μCi/ml of [35S]methionine. Cells were harvested after brief trypsinization and suspended in 1 ml of DME-10% FCS. 150 pM of purified CPE or H10PER was then added to the cell suspension. After incubating at 4° or 37°C for 30 min, the cells were pelleted and lysed with 400 μl of PBS containing 0.5% NP-40 (PBS-N), and the mixture was centrifuged at 12,000 g for 15 min to remove cell debris. The lysate was mixed with 5 μl of anti-FLAG antibody– conjugated Sepharose which had been blocked with PBS-N containing 5% skim milk. The mixtures were incubated at 4°C for 3 h with mild agitation, and the beads were washed three times with PBS-N at 4°C. The precipitated proteins were boiled in SDS-PAGE sample buffer containing 5% β-mercaptoethanol for 3 min and loaded onto SDS-12% polyacrylamide gel. Radioactive bands were visualized with a Bioimaging analyzer.
In immunological studies, L929 and 706FLAG cells were harvested and treated with CPE or H10PER at 37°C. Cell lysates were prepared and immunoprecipitated as described. The precipitated proteins were separated by electrophoresis on SDS-15% polyacrylamide gel or SDS-2–15% polyacrylamide gradient gel (Multi Gel 2/15; Daiichi Pure Chemicals Co., Tokyo, Japan) and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were probed with biotinylated anti-FLAG or anti-CPE antibodies and then treated with alkaline phosphatase–conjugated streptavidin; and color was developed with NBT/BCIP.
Results
Detection of the CPE-R Expressed on the Surface of CPE-sensitive Cell Lines by Flow Cytometric Analysis with a Recombinant CPE COOH-terminal Fragment
We attempted to identify a putative CPE-R by expression cloning. For this, we used flow cytometric analysis with a biotinylated CPE COOH-terminal fragment peptide as a probe, since intact CPE might kill cells expressing the CPE receptor, making it difficult to isolate cells with the CPE-R. On the basis of reports that the most COOH-terminal part of CPE functions as the receptor binding domain (Horiguchi et al., 1986, 1987; Hanna et al., 1991, 1992), we used amino acid residues 184–319 of CPE as a probe and constructed a CPE COOH-terminal peptide (H10PER) expression system in which the COOH-terminal fragment was fused to 10 consecutive histidine residues followed by linker peptides to facilitate its purification. H10PER was expressed in E. coli and was purified by Ni2+ chelating column chromatography. As expected, a single polypeptide of ∼15 kD was purified to almost homogeneity (Fig. 1, lane 3). The purified protein was recognized by rabbit anti-CPE serum (Fig. 1, lane 4) and protected Vero cells from CPE-induced cell lysis (data not shown).
The purified H10PER was biotinylated, and its cell surface binding ability was examined by flow cytometric analysis. When Vero cells were treated with biotinylated H10PER followed by phycoerythrin-conjugated streptavidin, the intensity of fluorescence increased markedly, whereas no increase was observed when biotinylated H10PER was omitted from the reaction (Fig. 2 A). The intensity of fluorescence decreased after pretreatment of the cells with unlabeled H10PER (Fig. 2 A, thin line). When the human hepatoma cell line Hep3B (Fig. 2 B) or the human intestinal epithelial cell line Henle Intestine 407 (Fig. 2 C), which are sensitive to CPE (data not shown), was used as target cells in this analysis, the intensity of fluorescence increased, although the levels of intensity were somewhat lower than those of Vero cells. The binding of biotinylated H10PER to L929 cells, which are known to be CPE-insensitive (Horiguchi et al., 1985), did not increase the fluorescence intensity (Fig. 2 D). H10PER did not bind to several cell lines that are resistant to CPE-induced cell lysis (data not shown), including K562 cells (Fig. 2 E) and JY cells (Fig. 2 F). These results show that H10PER specifically bound to the cell surface receptors on Vero, Hep3B, and Intestine 407 cells.
Figure 2.

Detection of the CPE receptor by a flow cytometric analysis. Vero (A), Hep3B (B), Henle Intestine 407 (C), L929 (D), K562 (E), and JY (F) cells were treated with biotinylated H10PER (thick lines) or buffer alone (dotted lines) followed by PE-conjugated streptavidin. For (A), cells were preincubated with unlabeled H10PER as a competitor (thin line). The plots show cell numbers on the ordinate and relative fluorescences on a log scale on the abscissa.
CPE-R Encodes a Highly Hydrophobic Membrane Protein
To identify CPE-R, we performed expression cloning. A cDNA library from the CPE-sensitive Vero cell line was introduced into 107 L929pyT18 cells that stably express polyoma virus large T antigen (data not shown; see Materials and Methods) and do not bind CPE (Fig. 3 A, thick line). 48 h after transfection, expression of CPE receptor was monitored by flow cytometric analysis, as described, and bright cells were sorted. By this screening, the brightest 1,000 cells among 107 transfected cells were initially selected. After three rounds of screening (Fig. 3 A, thin line), several positive clones were obtained. Clone 706 contained a fragment of ∼1.7-kb DNA. Other positive clones were screened by Southern blot analysis using the 1.7-kb XhoI fragment from clone 706 as a probe and found to encode the same gene (data not shown).
Figure 3.




Cloning and sequence analysis of the CPEreceptor cDNA. (A) Cloning of the CPE receptor. L929pyT18 cells were transfected with 10 μg of pME18Sf(−)pyori vector (thick line) or 10 μg of the Vero cDNA library rescued after the third round of screening (thin line). Flow cytometric analysis was done as described in Materials and Methods. The plot displays cell number on the ordinate and relative fluorescence on a log scale on the abscissa. (B) Nucleotide and deduced amino acid sequence of CPE-receptor cDNA. The deduced amino acid sequence (single letter code) is shown under the nucleotide sequence. The underlines indicate possible phosphorylation sites. A putative polyadenylation signal is indicated by bold letters. These sequence data are available from GenBank/EMBL/DDBJ under accession number D88492. (C) Hydrophobicity profile of CPE receptor. The hydrophobicity index was determined by the Kyte and Doolittle algorithm (Kyte and Doolittle, 1982). The four putative transmembrane segments are indicated by numbers above the line. (D) Alignment of the amino acid sequence of CPE receptor with those of the rat androgen withdrawal apoptosis protein RVP1 (Genbank/EMBL/DDBJ accession number M74067) and the mouse oligodendrocyte specific protein (GenBank/EMBL/DDBJ accession number U19582). Identical amino acids are shown by white letters in black boxes, and conservative replacements are shadowed. Triple alignment was done by the CLUSTAL method (Higgins and Sharp, 1989).
Sequence analysis revealed that the longest open reading frame of this clone was 630 bp and that it contained a polyadenylation signal followed by a polyA tail at its 3′ end (Fig. 3 B), suggesting that the gene is functionally expressed in Vero cells. Two potential AUG initiation codons are present at nucleotide positions 137–139 and 146–148 in the same reading frame. Since the neighboring sequence of the first AUG is consistent with the consensus sequence proposed by Kozak (1987), this was assigned as the initiation codon of CPE-R. Thus, CPE-R encodes a protein of 209–amino acid residues with a calculated molecular mass of 22,029 D. The protein consists mainly of hydrophobic amino acids and contains four putative transmembrane domains (Fig. 3 C) and several potential phosphorylation sites in the stretches of hydrophilic residues (Fig. 3 B). A search of data bases indicated that the amino acid sequence of the CPE-R product showed homology to proteins designated as the androgen withdrawal apoptosis protein RVP1 (67.8% homology, 95.6% similarity) and the oligodendrocyte-specific protein (28.7% homology, 63.8% similarity; Fig. 3 D). The latter half of the nucleotide sequence of clone 706, however, showed considerable difference from the RVP1 gene and contained a termination codon at nucleotide position 764–766. Accordingly, clone 706 lacked about one fifth of the COOH-terminal amino acid sequence of RVP1.
We examined the expression of CPE-R in various cell lines by Northern blot analysis with the clone 706 probe. Of five primate cell lines tested, Vero cells expressed the highest level of a transcript of about 1.8 kb. Its expression was also observed in CPE-sensitive Hep3B and Intestine 407 cell lines but at considerably lower levels than in Vero cells. No expression of CPE-R was detectable in the CPEinsensitive K562 and JY cell lines (Fig. 4, upper panel). The levels of expression of human elongation factor 1α gene in these cell lines were almost the same (Fig. 4, lower panel). The levels of expression of CPE-R correlated well with the CPE-binding abilities of these cell lines (Fig. 2), indicating that clone 706 encodes a functional CPE receptor protein.
Figure 4.

Northern blot analysis of CPE-R. Total RNA samples (15 μg/lane) from Vero (lane 1), K562 (lane 2), Hep3B (lane 3), JY (lane 4), and Intestine 407 (lane 5) cell lines were subjected to Northern blot analysis. The blots were hybridized with 32Plabeled DNA probes from the entire sequence of clone 706 containing CPE-R cDNA (top). The blots were rehybridized with 32P-labeled human EF-1α probe (bottom) to confirm the amounts and integrities of the samples. The positions of 28 and 18S ribosomal RNA are indicated on the right.
Both CPE Binding Ability and CPE Sensitivity of an L929 Cell Line Stably Expressing CPE-R
To determine whether the CPE-R product was functional, we established an L929 cell line stably expressing CPE-R (designated as the Neo706 cell line). Flow cytometric analysis revealed that the H10PER probe specifically bound to the cell surface of Neo706 cells (Fig. 5 A, thick line). Then we examined the CPE sensitivity of the Neo706 cell line. Purified CPE at 100 ng/ml caused Neo706 cells to form bleb balloons (compare Fig. 5 B, upper left with right) which were indistinguishable from those observed in CPEsensitive cell lines (Matsuda and Sugimoto, 1979; McClane and McDonel, 1979). As reported previously (Horiguchi et al., 1985), the parental L929 cell line did not exhibit any detectable morphological changes in the presence of even 5 μg/ ml of purified CPE (compare Fig. 5 B, lower left with right).
Figure 5.



Functional analysis of stable cell lines expressing the CPE receptor. (A) Clonal L929 cell lines expressing the CPE receptor were isolated as described in Materials and Methods. Expression of the CPE-R gene product was examined by flow cytometric analysis in a typical cell line expressing CPE-R (thick line, indicated as Neo706) and parental L929 cell line (thin line, indicated as control), as shown in Fig. 2. Cell numbers are shown on the ordinate and relative fluorescence on a log scale on the abscissa. (B) Comparison of sensitivities of typical cell line expressing the CPE receptor (top) and the parental L929 cell line (bottom) to CPE. The cells were not treated with CPE (top and bottom, denoted as none) or with CPE at 100 ng/ml (top, denoted as CPE), or 5 μg/ml (lower right, denoted as CPE) in DME supplemented with 10% FCS for 30 min at 37°C. Bar, 30 μm. (C) Scatchard plot analysis of the binding of 125I-CPE to the L929 cell line expressing CPE-R. Scatchard plot analysis giving a Ka value of 1.49 × 108 M−1 for 125I-CPE binding. (Inset) Various concentrations of 125I-CPE were incubated with the L929 cell line expressing CPE-R (0.4 × 106 cells each concentration), and specific binding was determined as described before (Horiguchi et al., 1985). Symbols represent means ± SD (n = 6).
To analyze the biochemical properties of CPE and CPE receptor interaction, we prepared radioiodinated CPE and performed binding studies as described by Horiguchi et al. (1985). Equilibrium binding data (Fig. 5 C, inset) analyzed by Scatchard plots (Fig. 5 C) showed the presence of a single order of binding sites with high affinity (Ka = 1.49 × 108 M−1). The Ka value is similar to those reported previously (Horiguchi et al., 1985). From these results, we concluded that the CPE-R gene product was actually functional.
Direct Interaction of the CPE-R Gene Product with CPE
We performed a ligand overlay assay to prove that the CPE-R gene product interacts directly with CPE. For this, an L929 cell line stably expressing COOH-terminal FLAG peptide tagged CPE receptor (706FLAG cell line) was established. The 706FLAG cells formed bleb balloons to the same extent as Neo706 cells (data not shown) on treatment with 100 ng/ml of native CPE. This indicates that introduction of the FLAG peptide sequence into its COOHterminal end did not affect the function of CPE receptor.
The parental cell line L929 and 706FLAG cells were solubilized with 0.5% NP-40, and the cell lysates were separated by SDS-PAGE and immobilized on a nitrocellulose membrane. Anti-FLAG peptide antibody specifically recognized a 22-kD band that was expressed only in the 706FLAG cell line (Fig. 6, compare lanes 1 and 2). The membranes were blotted simultaneously and subjected to ligand overlay assay with 125I-labeled CPE as a probe. The labeled CPE specifically recognized the 22-kD protein from 706FLAG cells (Fig. 6, compare lanes 3 and 4), and addition of 100-fold molar excess of cold CPE completely blocked the interaction (Fig. 6, compare lanes 4 and 6), indicating that CPE interacts directly and specifically with the CPE-R gene product.
Figure 6.

Specific and direct binding of CPE to the CPEreceptor. L929 cell lines expressing the CPE receptor with FLAG tag peptide at the COOH-terminal end were established as described in Materials and Methods, and a typical cell line (706FLAG) was selected. Cell lysates were prepared from the parental L929 (lanes 1, 3, and 5) and 706FLAG (lanes 2, 4, and 6) cell lines and subjected to Western blot analysis with anti-FLAG antibody (lanes 1 and 2) and ligand overlay assay (lanes 3 to 6). Ligand overlay assay was carried out using 125I-CPE with (lanes 5 and 6) or without (lanes 3 and 4) cold CPE. The positions of molecular weight standards are indicated on the right in kD.
Inclusion of the CPE-R Gene Product in the CPE-induced Large Complex
After binding to the cell surface, CPE is reported to form a large hydrophobic complex, which may possibly constitute the pore in the cell membrane (Wieckowski et al., 1994). Therefore we examined whether CPE formed a complex containing the CPE receptor. For this the 706FLAG cells were labeled with [35S]methionine, treated with CPE or H10PER at 4° or 37°C for 30 min, and then lysed. The cell lysates were treated with anti-FLAG antibody, and the precipitates were separated by SDS-PAGE. The CPE receptor migrated as a band of ∼22 kD (Fig. 7, A and B, lanes 5–7). In addition to this band, a higher molecular weight protein band was observed when CPE was incubated with the 706FLAG cells (Fig. 7 A, lanes 5 and 6), suggesting that cellular proteins form a complex with CPE. This band was intense when the 706FLAG cells were treated with CPE at 37°C, but was not detectable when the 706FLAG cells were treated with H10PER (Fig. 7 A, lanes 3 and 4). An additional band of ∼42 kD was precipitated from all the samples. This was probably a protein that reacted nonspecifically with anti-FLAG resin, because it was also detected in a precipitate prepared from the parental L929 cell line (data not shown). Decrease in intensity of this band was probably due to leakage of cell contents on CPE-induced cell lysis during the incubation (Fig. 7 A, lane 6). The components of this complex were examined by Western blot analysis of the immunoprecipitates. AntiFLAG antibody recognized the CPE receptor (Fig. 7 B, lanes 5–7, CPE-R) as well as the large complex (Fig. 7 B, lane 7). The complex also reacted with anti-CPE antibody (Fig. 7 B, lane 3). These results indicate that the complex contained at least CPE and the CPE receptor. As expected, neither the CPE receptor nor the large complex was detected in CPE-insensitive L929 cells treated with CPE (Fig. 7 B, lanes 4 and 8). H10PER was also coprecipitated with the CPE receptor (Fig. 7 B, lane 2, H10PER), but the large complex was not formed (Fig. 7 B, lanes 2 and 6, and A, lanes 3 and 4). When CPE alone was subjected to SDS-PAGE, the large protein band was not detected, although it aggregated and migrated as broad bands (Fig. 7 B, lower panel, lane 9) as reported previously (Enders and Duncan, 1976). From these results we concluded that the complex was formed by specific interaction between CPE and the CPE receptor. The NH2-terminal half of CPE, which is known to be necessary for cytotoxic activity, was essential for formation of the complex.
Figure 7.

Presence of both the CPE and CPE receptor in a CPE-induced high molecular weight complex. (A) 706 FLAG cells were labeled with [35S]methionine and treated with buffer alone (lanes 1 and 2) or buffer containing either 140 nmol of H10PER (lanes 3 and 4) or CPE (lanes 5 and 6) for 30 min at 4°C (odd-numbered lanes) or 37°C (even-numbered lanes). The cells were solubilized with PBS-0.5% NP-40, and the lysates were immunoprecipitated with anti-FLAG antibody. Precipitated proteins were separated by SDS-15% PAGE, and protein bands were located with Bioimaging Analyzer. The positions of molecular weight standards are indicated on the left in kD. (B) 706FLAG cells (lanes 1–3 and 5–7), and parental L929 cells (lanes 4 and 8) were treated with buffer alone (lanes 1 and 5) or buffer containing either 140 nmol of H10PER (lanes 2 and 6) or CPE (lanes 3, 4, 7, and 8). The cells were solubilized with PBS-0.5% NP-40 and immunoprecipitated with anti-FLAG antibodies. The precipitates were separated by SDS-15% PAGE (top) or SDS-2–15% gradient PAGE (bottom; note that only the relevant area is shown), transferred to a PVDF membrane, and then treated with biotinylated anti-CPE (lanes 1–4) or anti-FLAG (lanes 5–8) antibodies followed by alkaline phosphatase–conjugated streptavidin. As a control, 7 nmol of purified CPE was applied to SDS-2–15% polyacrylamide gradient gel, and then subjected to Western blotting with biotinylated anti-CPE antibody (bottom, lane 9). The positions of molecular weight standards are indicated on the left in kD.
Discussion
The present study describes a novel CPE receptor gene product, the expression of which confers CPE-resistant L929 cells with both ability to bind CPE and CPE sensitivity. This 22-kD receptor was essential for the cytotoxic action of CPE, as demonstrated by induction of its stable expression in CPE-insensitive L929 cells and showed all the characteristics of the CPE receptor so far reported. The present results also suggest that CPE acts through a unique mechanism.
Functionality of the CPE-R Gene Product
Several groups have reported that CPE uses a high affinity binding site on the target cell membrane as a specific receptor (McDonel and McClane, 1979; McDonel, 1980; Horiguchi et al., 1985). The present results are consistent with these previous reports, because the 22-kD CPE receptor could act as a high affinity binding site (Ka = 1.49 × 108 M−1) for CPE. In addition, we demonstrated the in vitro binding of CPE to the CPE receptor by ligand overlay assay. Thus CPE binds directly and specifically to the CPE receptor as the initial step in CPE-induced intoxication.
On analysis, the primary structure of this small 22-kD CPE receptor was found to contain 4 putative transmembrane segments. No possible glycosylation site was identified in its primary amino acid sequence of the receptor, indicating that it is not modified by carbohydrate. In fact, treatment with tunicamycin did not significantly alter its electrophoretic mobility on SDS-PAGE (Katahira, J., N. Inoue, Y. Horiguchi, M. Matsuda, and N. Sugimoto, unpublished results). These characteristics of our cloned CPE-R gene product are consistent with reports that the sensitivities and the binding abilities of CPE-sensitive cells were not affected by neuraminidase treatment or the additions of various sugars to the media for binding reactions (Tolleshaug et al., 1982; Wnek and McClane, 1986).
Most importantly, we showed here that the CPE-insensitive L929 cell line became CPE sensitive on expression of CPE-R. Thus we conclude that the cloned gene product is the functional CPE receptor.
Induction by CPE of a Large Protein Complex Containing Both CPE and the CPE-Receptor
Wieckowski et al. (1994) detected a large hydrophobic complex after treatment of CPE-sensitive cells with CPE. By coimmunoprecipitation assay, we demonstrated here that a large complex contains both CPE and the 22-kD CPE receptor. This complex was fairly stable, not being dissociated completely by SDS treatment. The COOH-terminal half of CPE, which can bind directly to the CPE receptor but has no cytotoxic activity, did not form a complex. Moreover, formation of the complex was promoted by incubation of the cells with CPE at 37°C, a temperature at which the cytotoxic action is much greater than at 4°C (Horiguchi et al., 1985; McClane and Wnek, 1990). Thus this complex must be essential for CPE intoxication and form a CPE-induced pore in the cell membrane. Purified intact CPE shows an anomalous electrophoretic migration pattern, but the COOH-terminal fragment does not (Fig. 7 B). Thus we consider that the NH2-terminal half of CPE may have a role in self assembly but that it is not sufficient alone for formation of a defined structure. It is not yet clear whether the CPE receptor is the sole requisite for CPE intoxication. The interaction of CPE with its receptor might be necessary not only for the recognition of target cells but also for the regulated assembly of the CPE-induced complex in the plasma membrane. Thus we propose that CPE acts through a novel mechanism differing from those of other pore-forming toxins (for review see Bhakdi et al., 1996). Analyses of the stoichiometric features of CPE and its receptor in the complex, and investigations on its further interaction with other components are now in progress in our laboratory.
Similarities of the CPE-R Gene Product to Several Other Gene Products with No Known Function
The CPE-R gene product shows sequence similarities to the rat androgen withdrawal apoptosis protein RVP1 (Briehl and Miesfeld, 1991) and mouse oligodendrocyte specific protein (these sequence data are available from GenBank/EMBL/DDBJ under accession number U19582). These have been reported to be expressed in a highly restricted manner: RVP1 gene is expressed in ventral prostate cells after withdrawal of androgen from the culture medium in vitro or castration in vivo (Briehl and Miesfeld, 1991); oligodendrocyte specific protein is expressed specifically in oligodendrocyte. CPE-R was constitutively expressed in various cell lines tested in this study and in tissues including kidney, liver, and intestine (Katahira, J., N. Inoue, Y. Horiguchi, M. Matsuda, and N. Sugimoto, unpublished results). On searching the expressed sequence tag data base, we also obtained CPE-R and putative RVP1 homologues of human and mouse origin. We found that the human CPE receptor homologue showed higher homology to monkey CPE receptor (99.0% identity, 99.5% similarity) than to human RVP1 (70.9% identity, 96.6% similarity) and that the mouse CPE receptor showed higher homology to the monkey CPE receptor (83.8% identity, 98.6% similarity) than to rat RVP1 (66.8% identity, 94.6% similarity; Katahira, J., N. Inoue, Y. Horiguchi, M. Matsuda, and N. Sugimoto, manuscript in preparation). Thus we consider that RVP1 may be a different gene product, although CPE receptor and RVP1 share a structural similarity and might be members of a functionally identical gene family. Further functional analysis of the CPE-R product may provide a clue to not only the physiological function of the CPE receptor but also the functions of RVP1 and the oligodendrocyte specific protein.
Abbreviations used in this paper
- CPE
Clostridium perfringens enterotoxin
- CPE-R
CPE-receptor gene
- H10PER
histidine-tagged CPE COOHteminal fragment
- PE
phycoerythrin
Footnotes
We are indebted to Dr. T. Kinoshita and members of the Department of Immunoregulation for their helpful discussions and providing plasmids. We are grateful to the following persons of Research Institute for Microbial Diseases, Osaka University; Dr. H. Nojima (Department of Molecular Genetics) for helpful suggestions on library construction and Drs. N. Wakamiya (Department of Viral Infections) and K. Nagayama (Department of Bacterial Infections) for providing cell lines. We are also grateful to Dr. T. Asao (Osaka Prefectural Institute of Public Health) for providing the C. perfringens strain. We also thank K. Nakamura, Central Laboratory, Research Institute for Microbial Diseases, Osaka University, for cell sorting. We are grateful to Dr. S. Kozaki (University of Osaka Prefecture) for critical reading of this manuscript.
Please address all correspondence to Jun Katahira, Department of Bacterial Toxicology, Osaka University, 3-1 Yamadaoka, Suita, Osaka 565, Japan. Tel.: 81-6-879-8285; Fax.: 81-6-879-8283; E-mail: katahira@biken.osaka-u.ac.jp
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Bhakdi S, Bayley B, Valeva A, Walev I, Walker B, Weller U, Kehoe M, Palmer M. Staphylococcal alpha-toxin, streptolysin-O, and Escherichia colihemolysin: prototypes of pore-forming bacterial cytolysins. Arch Mikrobiol. 1996;165:73–79. doi: 10.1007/s002030050300. [DOI] [PubMed] [Google Scholar]
- Briehl MM, Miesfeld RL. Isolation and characterization of transcripts induced by androgen withdrawal and apoptotic cell death in the rat ventral prostate. Mol Endocrinol. 1991;5:1381–1388. doi: 10.1210/mend-5-10-1381. [DOI] [PubMed] [Google Scholar]
- Czeczulin JR, Hanna PC, McClane BA. Cloning nucleotide sequencing and expression of the Clostridium perfringens enterotoxin gene in Escherichia coli. . Infect Immun. 1993;61:3429–3439. doi: 10.1128/iai.61.8.3429-3439.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enders GL, Duncan CL. Anomalous aggregation of Clostridium perfringensenterotoxin under dissociation conditions. Can J Microbiol. 1976;22:1410–1414. doi: 10.1139/m76-209. [DOI] [PubMed] [Google Scholar]
- Gubler U, Hoffmann BJ. A simple and very efficient method for generating cDNA libraries. Gene (Amst) 1983;25:263–269. doi: 10.1016/0378-1119(83)90230-5. [DOI] [PubMed] [Google Scholar]
- Hanna PC, Mietzner TA, Schoolnik GK, McClane BA. Localization of the receptor-binding region of Clostridium perfringensenterotoxin utilizing cloned toxin fragments and synthetic peptides. The 30 C-terminal amino acids define a functional binding region. J Biol Chem. 1991;266:11037–11043. [PubMed] [Google Scholar]
- Hanna PC, Wieckowski EH, Mietzner TA, McClane BA. Mapping of functional regions of Clostridium perfringenstype A enterotoxin. Infect Immun. 1992;60:2110–2114. doi: 10.1128/iai.60.5.2110-2114.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins DG, Sharp PM. Fast and sensitive multiple sequence alignments on a microcomputer. Comput Appl Biosci. 1989;5:151–153. doi: 10.1093/bioinformatics/5.2.151. [DOI] [PubMed] [Google Scholar]
- Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Horiguchi Y, Uemura T, Kozaki S, Sakaguchi G. The relationship between cytotoxic effect and binding to mammalian cultured cells of Clostridium perfringensenterotoxin. FEMS Microbiol Lett. 1985;28:131–135. [Google Scholar]
- Horiguchi Y, Uemura T, Kamata Y, Kozaki S, Sakaguchi G. Production and characterization of monoclonal antibodies to Clostridium perfringensenterotoxin. Infect Immun. 1986;52:31–35. doi: 10.1128/iai.52.1.31-35.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiguchi Y, Akai T, Sakaguchi G. Isolation and function of a Clostridium perfringensenterotoxin fragment. Infect Immun. 1987;55:2912–2915. doi: 10.1128/iai.55.12.2912-2915.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda S, Oka J, Nagao T. Effects of four diltiazem stereoisomers on binding of d-cis-[3H]diltiazem and (+)-[3H]PN200-110 to rabbit T-tuble calcium channels. Eur J Pharmacol. 1991;208:199–205. doi: 10.1016/0922-4106(91)90096-z. [DOI] [PubMed] [Google Scholar]
- Kozak M. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987;15:8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydrophobic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Manser E, Leung T, Monfries C, Teo M, Hall C, Lim L. Diversity and versatility of GTPase activating proteins for the p21 rho subfamily of rasG proteins detected by novel overlay assay. J Biol Chem. 1992;267:16025–16028. [PubMed] [Google Scholar]
- Manser E, Leung T, Salihuddin H, Zhao Z, Lim L. A brain serine/ threonine protein kinase that inihibits the GTPase activity of p21cdc42 . Nature (Lond) 1994;367:40–46. [Google Scholar]
- Marmur J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J Mol Biol. 1961;3:208–218. [Google Scholar]
- Matsuda M, Sugimoto N. Calcium-independent and dependent steps in action of Clostridium perfringensenterotoxin on Hela and Vero cells. Biochem Biophys Res Commun. 1979;91:629–636. doi: 10.1016/0006-291x(79)91568-7. [DOI] [PubMed] [Google Scholar]
- Matsuda M, Ozutsumi K, Iwahashi H, Sugimoto N. Primary action of Clostridium perfringenstype A enterotoxin on Hela and Vero cells in the absence of extracellular calcium: rapid and characteristic changes in membrane permeability. Biochem Biophys Res Commun. 1986;141:704–710. doi: 10.1016/s0006-291x(86)80229-7. [DOI] [PubMed] [Google Scholar]
- McClane BA, McDonel JL. The effects of Clostridium perfringensenterotoxin on morphology, viability, and macromolecular synthesis in Vero cells. J Cell Physiol. 1979;99:191–199. doi: 10.1002/jcp.1040990205. [DOI] [PubMed] [Google Scholar]
- McClane BA, McDonel JL. Protective effects of osmotic stabilizers on morphological and permeability alterations induced in Vero cells by Clostridium perfringensenterotoxin. Biochim Biophys Acta. 1981;641:401–409. doi: 10.1016/0005-2736(81)90496-x. [DOI] [PubMed] [Google Scholar]
- McClane BA, Wnek AP. Studies of Clostridium perfringensenterotoxin action at different temperatures demonstrate a correlation between complex formation and cytotoxicity. Infect Immun. 1990;58:3109–3115. doi: 10.1128/iai.58.9.3109-3115.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClane BA, Wnek AP, Hulkower KI, Hanna PC. Divalent cation involvement in the action of Clostridium perfringenstype A enterotoxin. J Biol Chem. 1988a;263:2423–2435. [PubMed] [Google Scholar]
- McClane BA, Hanna PC, Wnek AP. b. Clostridium perfringenstype A enterotoxin. Microb Pathog. 1988;4:317–323. doi: 10.1016/0882-4010(88)90059-9. [DOI] [PubMed] [Google Scholar]
- McDonel JL. Binding of Clostridium perfringens 125I-enterotoxin to rabbit intestinal cells. Biochemistry. 1980;19:4801–4807. doi: 10.1021/bi00562a014. [DOI] [PubMed] [Google Scholar]
- McDonel JL, Duncan CL. Histopathological effect of Clostridium perfringensenterotoxin in the rabbit ileum. Infect Immun. 1975;12:1214–1218. doi: 10.1128/iai.12.5.1214-1218.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonel JL, McClane BA. Binding versus biological activity of Clostridium perfringensenterotoxin in Vero cells. Biochem Biophys Res Commun. 1979;87:497–504. doi: 10.1016/0006-291x(79)91823-0. [DOI] [PubMed] [Google Scholar]
- McDonel JL, Chang LW, Pounds JG, Duncan CL. The effect of Clostridium perfringensenterotoxin in rat and rabbit ileum: an electron microscopic study. Lab Invest. 1978;39:210–217. [PubMed] [Google Scholar]
- Ohishi K, Kurimoto Y, Inoue N, Endo Y, Takeda J, Kinoshita T. Cloning and characterization of the murine GPI anchor synthesis gene Pigf, a homologue of the human PIGFgene. Genomics. 1996;34:340–346. doi: 10.1006/geno.1996.0296. [DOI] [PubMed] [Google Scholar]
- Sakaguchi G, Uemura T, Riemann H. Simplified method for purification of Clostridium perfringenstype A enterotoxin. Appl Microbiol. 1973;27:762–767. doi: 10.1128/am.26.5.762-767.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. 2nd Ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- Senda T, Sugimoto N, Horiguchi Y, Matsuda M. The enterotoxin of Clostridium perfringenstype A binds to the presynaptic nerve endings in neuromuscular junctions of mouse phrenic nerve-diaphragm. Toxicon. 1995;33:499–506. doi: 10.1016/0041-0101(94)00165-5. [DOI] [PubMed] [Google Scholar]
- Stark RL, Duncan CL. Purification and biochemical properties of Clostridium perfringenstype A enterotoxin. Infect Immun. 1971;4:89–96. doi: 10.1128/iai.6.5.662-673.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugii S, Horiguchi Y. Identification and isolation of the binding substance for Clostridium perfringensenterotoxin on Vero cell. FEMS Microbiol Lett. 1988;52:85–90. [Google Scholar]
- Sugimoto N, Ozutsumi K, Matsuda M. Morphological alterations and changes in cellular cations induced by Clostridium perfringenstype A enterotoxin in tissue culture cells. Eur J Epidemiol. 1985;1:264–273. doi: 10.1007/BF00237101. [DOI] [PubMed] [Google Scholar]
- Sugimoto N, Takagi M, Ozutsumi K, Harada S, Matsuda M. Enterotoxin of Clostridium perfringenstype A forms ion-permeable channels in a lipid bilayer membrane. Biochem Biophys Res Commun. 1988;156:551–556. doi: 10.1016/s0006-291x(88)80877-5. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Inoue N, Ohishi K, Maeda Y, Nakamura N, Endo Y, Fujita T, Takeda J, Kinoshita T. PIG-B, a membrane protein of the endoplasmic reticulum with a large lumenal domain, is involved in transferring the third mannose of the GPI anchor. EMBO (Eur Mol Biol Organ) J. 1996;15:4254–4261. [PMC free article] [PubMed] [Google Scholar]
- Tolleshaug H, Skjelkvale R, Berg T. Quantitation of binding and subcellular distribution of Clostridium perfringensenterotoxin in rat liver cells. Infect Immun. 1982;37:486–491. doi: 10.1128/iai.37.2.486-491.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetsuki T, Naito A, Nagata S, Kaziro Y. Isolation and characterization of the human chromosomal gene for polypeptide chain elongation factor-1 alpha. J Biol Chem. 1989;264:5791–5798. [PubMed] [Google Scholar]
- Watanabe R, Kinoshita T, Masaki R, Yamamoto A, Takeda J, Inoue N. PIG-A and PIG-H, which participate in glycosylphosphatidylinositol anchor biosynthesis, form a protein complex in the endoplasmic reticulum. J Biol Chem. 1996;271:26868–26875. doi: 10.1074/jbc.271.43.26868. [DOI] [PubMed] [Google Scholar]
- Wieckowski EU, Wnek AP, McClane BA. Evidence that an ∼50 kDa mammalian plasma membrane protien with receptor-like properties mediates the amphiphilicity of specifically-bound Clostridium perfringensenterotoxin. J Biol Chem. 1994;269:10838–10848. [PubMed] [Google Scholar]
- Wnek AP, McClane BA. Identification of a 50,000 Mr protein from rabbit brush border membranes that binds Clostridium perfringensenterotoxin. Biochem Biophys Res Commun. 1983;112:1099–1105. doi: 10.1016/0006-291x(83)91731-x. [DOI] [PubMed] [Google Scholar]
- Wnek AP, McClane BA. Comparison of receptors for Clostridium perfringenstype A and cholera enterotoxins in rabbit intestinal brush border membranes. Microb Pathog. 1986;1:89–100. doi: 10.1016/0882-4010(86)90035-5. [DOI] [PubMed] [Google Scholar]