

Abstract
While much is known about the role of nuclear pore complexes (NPCs) in nucleocytoplasmic transport, the mechanism of NPC assembly into pores formed through the double lipid bilayer of the nuclear envelope is not well defined. To investigate the dynamics of NPCs, we developed a live-cell assay in the yeast Saccharomyces cerevisiae. The nucleoporin Nup49p was fused to the green fluorescent protein (GFP) of Aequorea victoria and expressed in nup49 null haploid yeast cells. When the GFP–Nup49p donor cell was mated with a recipient cell harboring only unlabeled Nup49p, the nuclei fused as a consequence of the normal mating process. By monitoring the distribution of the GFP–Nup49p, we could assess whether NPCs were able to move from the donor section of the nuclear envelope to that of the recipient nucleus. We observed that fluorescent NPCs moved and encircled the entire nucleus within 25 min after fusion. When assays were done in mutant kar1-1 strains, where nuclear fusion does not occur, GFP–Nup49p appearance in the recipient nucleus occurred at a very slow rate, presumably due to new NPC biogenesis or to exchange of GFP– Nup49p into existing recipient NPCs. Interestingly, in a number of existing mutant strains, NPCs are clustered together at permissive growth temperatures. This has been explained with two different hypotheses: by movement of NPCs through the double nuclear membranes with subsequent clustering at a central location; or, alternatively, by assembly of all NPCs at a central location (such as the spindle pole body) with NPCs in mutant cells unable to move away from this point. Using the GFP–Nup49p system with a mutant in the NPCassociated factor Gle2p that exhibits formation of NPC clusters only at 37°C, it was possible to distinguish between these two models for NPC dynamics. GFP– Nup49p-labeled NPCs, assembled at 23°C, moved into clusters when the cells were shifted to growth at 37°C. These results indicate that NPCs can move through the double nuclear membranes and, moreover, can do so to form NPC clusters in mutant strains. Such clusters may result by releasing NPCs from a nuclear tether, or by disappearance of a protein that normally prevents pore aggregation. This system represents a novel approach for identifying regulators of NPC assembly and movement in the future.
Nuclear pore complexes (NPCs)1 regulate the exchange of macromolecules across the nuclear envelope (Forbes, 1992; Melchior and Gerace, 1995; Gorlich and Mattaj, 1996). These large proteinaceous assemblies are presumably anchored via integral membrane proteins in ∼90-nm-diam pores that join the inner and outer nuclear membranes. High resolution EM studies of amphibian oocyte NPCs have yielded a three-dimensional reconstruction of NPC ultrastructure. The NPC is a cylindrical entity with a superficial octagonal symmetry that is characterized by distinct substructures referred to as spokes, rings, a central plug, cytoplasmic fibrils, and a nuclear basket (Ris, 1991; Stewart, 1992; Akey, 1995; Goldberg and Allen, 1995; Pante and Aebi, 1996). To account for this organizational complexity, NPC biogenesis is likely a highly regulated process that coordinates the formation of the nuclear pore with the assembly of soluble NPC proteins (nucleoporins). In addition, formation of NPCs is finely controlled to account for the maintenance of NPC density over the nuclear surface during cell growth and for reversible NPC disassembly during mitosis in higher eukaryotic cells (Maul, 1977; Forbes, 1992; Wiese and Wilson, 1993; Gerace and Foisner, 1994).
Considerable progress has been achieved using fractionated Xenopus egg extracts to characterize the assembly of nuclear envelopes in vitro (Lohka and Masui, 1983). Both vesicular and soluble components are necessary for the in vitro assembly of transport-competent NPCs (Sheehan et al., 1988; Dabauvalle et al., 1990; Finlay and Forbes, 1990; Finlay et al., 1991; Vigers and Lohka, 1991). Moreover, nuclear vesicle fusion requires energy, an N-ethylmaleimide–sensitive factor, and possibly localized Ca+2 gradients (Newport, 1987; Newmeyer and Forbes, 1990; Pfaller et al., 1991; Boman et al., 1992a,b; Newport and Dunphy, 1992; Vigers and Lohka, 1992; Sullivan et al., 1993). To order the steps of NPC assembly, Macaulay and Forbes (1996) developed an anchored nuclear assembly assay using Xenopus egg extracts. Their work demonstrated that the formation of the double nuclear membrane precedes the Ca+2-dependent, GTPγS-sensitive assembly of NPCs (Macaulay and Forbes, 1996). However, in all cell types, the components required for NPC assembly into intact nuclear envelopes remain largely undefined.
NPC and/or nuclear envelope structural perturbations have been observed in many different yeast mutants (Wente et al., 1996). In some cases, these phenotypes may reflect effects on NPC biogenesis such that NPC assembly is inhibited or an accumulation of NPC assembly intermediates results. Depletion of the nucleoporin Nsp1p or of mutant alleles of the gene encoding the nucleoporin Nic96p results in a reduction of NPC density over the nuclear surface (Mutvei et al., 1992; Zabel et al., 1996). In nup116 null mutants or when NUP170 is overexpressed in a pom152 null mutant, stacks of intranuclear double membranes with NPC-like structures are observed (Wente and Blobel, 1993; Aitchison et al., 1995b ). These stacks resemble the intranuclear annulate lamellae found in vertebrate cells (Kessel, 1983), which possibly serve as a storage form for NPCs. Mutations in several nucleoporin genes can also alter the distribution of NPCs over the nuclear surface. In wild-type yeast cells, NPCs are distributed over the nuclear surface at a density of ∼15 NPC per μm2, and they are excluded from areas where the vacuolar and nuclear membranes abut (Severs et al., 1976). This distribution is altered in cells expressing mutant alleles of nup145, nup133, nup159, nup120, nup84, nup85, and gle2 (Doye et al., 1994; Wente and Blobel, 1994; Aitchison et al., 1995a ; Gorsch et al., 1995; Heath et al., 1995; Li et al., 1995; Pemberton et al., 1995; Goldstein et al., 1996; Murphy et al., 1996; Siniossoglou et al., 1996), where large clusters of NPCs in localized patches of nuclear envelope are observed. Vertebrate NPCs are also found in localized patches in certain specialized cell types, such as spermatocytes (Fawcett, 1981). Further analysis will be required to demonstrate whether any of these yeast nucleoporins are bona fide NPC assembly factors.
Several fundamental aspects of NPC physiology also have not been adequately examined. Integral membrane proteins can diffuse within the lipid bilayer of the yeast outer nuclear membrane (Latterich and Schekman, 1994). Do the NPCs anchored in the nuclear pore via integral membrane proteins move over the nuclear surface? Alternatively, is NPC position fixed after assembly (are they locked in place)? Moreover, studies have not directly monitored the rate of NPC assembly. This information is important for a molecular understanding of the cellular control of NPC biogenesis. To address these issues, we developed assays to monitor NPC dynamics in live cells of the yeast Saccharomyces cerevisiae. NPCs in wild-type and mutant yeast cells were fluorescently labeled by a functional fusion of the green fluorescent protein (GFP) of Aequorea victoria to the nucleoporin Nup49p. Rates of NPC movement were assessed by monitoring GFP–Nup49p location during mating and nuclear membrane fusion with an unlabeled yeast strain. We found that wild-type NPCs moved freely within the nuclear envelope at a rate distinct from that for incorporating GFP–Nup49p into NPCs. Finally, cluster formation in gle2 cells was due to the movement of NPCs into a localized area of the nuclear envelope. The development of this technology has allowed the first analysis of NPC dynamics in live cells.
Materials and Methods
Strains and Plasmids
General yeast manipulations were conducted by standard methods (Sherman et al., 1986), with transformations by the lithium acetate method (Ito et al., 1983). DH5α was used as the bacterial host for all plasmids. The S. cerevisiae strains used in this study are described in Table I. The kar1-1 allele was identified by quantitative mating assays (Meluh and Rose, 1990). The gle2-1 allele was identified by monitoring the temperature sensitivity of the mutants. All plasmids were made by standard methods (Sambrook et al., 1989). Chromosomal integration of the ADE2 gene used pSW286 (an ∼3,600-bp BamHI ADE2 fragment from pLSD257 in the integrating vector pRS306; Sikorski and Hieter, 1989). pSW240 and pSW241 were constructed by ligating a 2,415-bp BamHI/SalI fragment containing the entire NUP49 locus from pSW40 (Wente et al., 1992) into the 2 μ plasmids pRS423 and pRS424 (Christianson et al., 1992), respectively. pSW441 was made by insertion of the 735-bp GFP–S65T BamHI fragment from pBJ646 (Waddle et al., 1996) into pNLS-E1 (Underwood and Fried, 1990). A BamHI/PstI-digested 715-bp PCR product (containing the NUP49 promoter and initiation methionine) and a BamHI/KpnI PCR product (containing the last 764 bp of the NUP49 coding sequence [corresponding to amino acids 473–726] and 211 bp of 3′ sequence) were ligated into PstI/KpnI-digested pRS304 (Sikorski and Hieter, 1989) to yield pSW420. pSW442 was made by inserting the BamHI GFP–S65T fragment into BamHI–digested pSW420.
Table I.
Yeast Strain Genotypes
| Strain | Genotype | Derivation | ||
|---|---|---|---|---|
| W303a | Mata ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 | |||
| W303α | Matα ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 | |||
| W303a/α | Mata/Matα ade2-1/ade2-1 ura3-1/ura3-1 his3-11,15/his3-11,15 trp1-1/trp1-1 | |||
| leu2-3,112/leu2-3,112 can1-100/can1-100 | ||||
| MS739 | Matα ade2-101 ura3-52 leu2-3,112 kar1-1 | Vallen et al., 1992 | ||
| SWY1 | Mata/Matα ade2-1/ade2-1 ura3-1/ura3-1 his3-11,15/his3-11,15 trp1-1/trp1-1 | Wente et al., 1992 | ||
| leu2-3,112/leu2-3,112 can1-100/can1-100 nup49-1::URA3/+ | ||||
| SWY458 | Mata ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 ade2-1 ADE2-URA3 | W303a transformed with the integrating plasmid | ||
| pSW286 digested with BglII | ||||
| SWY459 | Mata ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 ade2-1 ADE2-URA3 | W303α transformed with the integrating plasmid | ||
| pSW286 digested with BglII | ||||
| SWY518 | Mata ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 | 5-FOAr clone of SWY458 which forms white colonies | ||
| SWY519 | Matα ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 | 5-FOAr clone of SWY458 which forms white colonies | ||
| SWY595 | Mata/Matα ura3-1/ura3-1 his3-11,15/his3-11,15 trp1-1/trp1-1 leu2-3,112/leu2-3,112 | SWY518 crossed with SWY519 | ||
| can1-100/can1-100 | ||||
| SWY731 | Mata/Matα ade2-1/ade2-1 ura3-1/ura3-1 his3-11,15/his3-11,15 trp1-1/trp1-1 | SWY1 transformed with the integrating plasmid | ||
| leu2-3,112/leu2-3,112 can1-100/can1-100 nup49-1::URA3 | pSW442 digested with AvrII (see Materials and | |||
| nup49ΔGLFG::GFP-S65T-TRP1/+ | Methods and Fig. 2) | |||
| SWY734 | Mata ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 nup49-1::URA3 | segregant of SWY731 | ||
| nup49ΔGLFG::GFP-S65T-TRP1 | ||||
| SWY737 | Matα ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 nup49-1::URA3 | segregant of SWY731 | ||
| nup49ΔGLFG::GFP-S65T-TRP1 | ||||
| SWY757 | Mata ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 ADE2-URA3 pSW241 (TRP1) | pSW241 transformed into SWY458 | ||
| SWY759 | Matα ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 ADE2-URA3 pSW241 (TRP1) | pSW241 transformed into SWY459 | ||
| SWY809 | Mata ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 nup49-1::URA3 | segregant of diploid created by | ||
| nup49ΔGLFG::GFP-S65T-TRP1 | crossing SWY737 with SWY518 | |||
| SWY865 | Mata/Mata ura3-1/ura3-1 his3-11,15/his3-11,15 trp1-1/trp1-1 | SWY595 transformed with the PCR product created | ||
| leu2-3,112/leu2-3,112 can1-100/can1-100 GAL10-GFP-S65T-nup49-URA3/+ | with oligos GFB and 49U using pSW441 as template | |||
| (see Materials and Methods and Fig. 7) | ||||
| SWY869 | Mata ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 GAL10-GFP-S65T-nup49-URA3 | segregant of SWY865 | ||
| SWY1136 | Matα ade2-1 ura3-1 his3-11,15 leu2-3,112 gle2-1 | Murphy et al., 1996 | ||
| SWY1308 | Matα ura3-1 trp1-1 leu2-3,112 kar1-1 nup49-1::URA3 nup49ΔGLFG::GFP-S65T-TRP1 | segregant of diploid created by crossing MS739 with | ||
| SWY809 | ||||
| SWY1310 | Matα ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 GAL10-GFP-S65T-nup49-URA3 | segregant of diploid created by crossing | ||
| gle2-1 | SWY869 with SWY1136 | |||
| SWY1324 | Matα ura3-1 his3-11,15 trp1-1 leu2-3,112 gle2-1 GAL10-GFP-S65T-nup49-URA3 | pSW240 transformed into SWY1310 | ||
| pSW240 (HIS3) |
GFP–Nup49p under control of the galactose-inducible promoter (GAL10) was integrated at the NUP49 locus by the gene deletion method of Baudin et al. (1993). Wild-type SWY595 cells were transformed with a PCR product generated with oligos GFB (5′ TCCTGCAGGTTTAGAGCCAAATAGTCCAGCGCTTGCATTATTCATCCCTTT GT A T AGTTCATCC 3′) and 49U (5′ CGATAAAAACAGTAATTGAGAGGGTTTTCACGGGTAGTGGGCACACAATTCAATTCATCATTTT 3′) using pSW441 as template. This template contained the 1,173-bp URA3 gene and GFP–S65T in frame with the initiation ATG supplied by a portion of CYC1 under control of GAL10. Sequences flanking the PCR product are complimentary to nucleotides of the NUP49 locus and placed GAL10 GFP–S65T in frame with the NUP49 open reading frame. Integration at the NUP49 locus was confirmed by genomic colony PCR.
Characterization of GFP–Nup49p and 2 μ NUP49
To determine doubling times, cells were grown in YEP (1% yeast extract, 2% bactopeptone) containing 2% glucose, diluted to 106 cells per ml, and incubated at 23°, 30°, or 37°C. Aliquots were taken from cultures every hour for 12 h, and finally after 23 h. Cell number was determined by measuring the optical density at 600 nm and by counting. Colocalization of GFP–Nup49p with Nup116p was performed by indirect immunofluorescence in wild-type (SWY809) cells as described previously (Wente et al., 1992), using a 1:10 dilution of affinity-purified rabbit polyclonal antibody against the carboxy terminus of Nup116p (Iovine et al., 1995). Texas red– conjugated goat anti–rabbit IgG at a 1:200 dilution was used to visualize bound antibody. Photographs were taken using a microscope (Olympus Corp., Lake Success, NY) with a ×100 oil-immersion objective, a CCD camera, and the public domain NIH Image 1.59 program (developed at the United States National Institutes of Health and available from the Internet by anonymous FTP from zippy.nimh.gov) on a Power Macintosh 9500/132 with a framegrabber card (LG-3; Scion Corp., Frederick, MD).
To compare expression levels of Nup49p, total yeast cell extracts and immunoblotting were conducted as described (Iovine et al., 1995). Bands were visualized by developing with nitro blue tetrazolium and 5-bromo-4chloro-3-indolyl-1-phosphate (Promega, Madison, WI).
Movement and Assembly Assays
For live-time experiments with mating cells, ADE2 cells were grown in synthetic complete (SC) media lacking tryptophan and containing 2% glucose and 0.1 mg/ml 4′,6-diamidino-2-phenylindole (DAPI). Matings were performed by mixing ∼107 cells of each mating type for 5 h in a 25-ml culture. Mating mixtures were concentrated by centrifugation and briefly sonicated. Slide preparation was conducted as described (Waddle et al., 1996). Briefly, cells were dispensed onto a pad containing 2% agarose and 2% glucose in SC media (Bio101, Vista, CA) that was mounted onto a double-well Teflon-coated slide (Cell-Line Associates, Newfield, NJ). Coverslips with a thin line of Vaseline at the edges were used to disperse the small drop of culture and to prevent drying. Slides at room temperature were scanned for zygotes or schmooing cells. Cells of opposite mating types were distinguishable because one strain expressed the directly visible GFP–Nup49p, whereas the other did not. Uptake of DAPI into cells of both mating types was used to determine if nuclear fusion had occurred in the zygotes. Videos of GFP fluorescence were taken as described (Waddle et al., 1996) by scanning z-axis focal planes, 0.5 μm apart (typically ten total), at each 2-min time point using the Olympus microscope and NIH Image 1.59. Image Math was used to project all the z-axis focal planes onto one two-dimensional image. In this final image, each pixel represents the brightest value for that position in all of the focal planes (“min” operator). The microscope was equipped with optivars allowing magnifications of up to 6,000 with the ×100 objective and an ISIT-68 camera (Dage-MTI Inc., Wabash, MI). The stage and shutters were controlled by a Ludl box (MAC2000; Ludl Electronic Products Ltd., Hawthorne, NY). Photobleaching was minimized using neutral density filters. Videos ranged from 1–4 h and selected frames are shown in the figures. Complete time-lapse videos can be viewed on our World Wide Web page at http://www.wentelab. wustl.edu. NIH Image 1.60 was used to quantify fluorescence values of areas encircling half-nuclei in the case of wild-type zygotes or entire nuclei in the case of kar1-1 zygotes, resulting in average pixel brightness values. For quantifying, pixel values were first inverted such that grayscale values ranged from 0–256, with 0 being no fluorescence signal and 256 being the maximum fluorescence value. There were no saturated pixels in the images used for quantification (theoretically above 256 or below 0). The fluorescence ratio is defined as the ratio of the average fluorescence value of pixels contained within the recipient nucleus divided by the average fluorescence value of pixels contained within the donor nucleus.
Visualization of NPC Clusters in Live Cells
To monitor formation of NPC clusters using indirect immunofluorescence, gle2-1 cells expressing GFP–Nup49p under the control of the GAL10 promoter and NUP49 on a 2 μ plasmid (SWY1324) were grown at 23°C overnight in SC media lacking histidine (− his) and containing 0.5% galactose and 1.5% raffinose. Cells were then shifted for 1 h to SC − his containing 2% glucose. Samples were processed for indirect immunofluorescence before and after a shift to 37°C. GFP fluorescence was visualized directly, and Nup116p was visualized using the polyclonal antibody derived against the carboxy terminus as described above. Photographs were taken on an Olympus microscope with T-Max 400 film (Eastman Kodak Co., Rochester, NY). To monitor formation of NPC clusters in live cells, gle2-1 cells expressing GFP–Nup49p from the GAL10 promoter were grown at 23°C in media containing 0.5% galactose and 1.5% raffinose. After a 5-h shift into media containing 2% glucose, cells were mounted on an agarose pad as described for the movement assay. The slide was shifted to 37°C using a temperature-controlled stage (MicroIncubator 5000; 20/20 Technology, Inc., Wilmington, NC) on the Olympus microscope described above. Videos were taken as described for the movement assay with frames every 20 min. To monitor the disappearance of gle2-1 clusters in live cells, gle2-1 GAL–GFP-nup49 cells were grown in SC − his with 0.5% galactose and 1.5% raffinose at 23°C. Cells were then washed into SC − his with 2% glucose and simultaneously shifted to 37°C for 5 h. Slides were prepared as for the movement assay. The temperature-controlled stage maintained the slide at 37°C until the videos were started and the stage was returned to 23°C. The videos monitored GFP fluorescence in the cells at 23°C as described above.
Results
Experimental Rationale for an NPC Movement Assay
To characterize NPC dynamics, an approach was designed based on GFP technology (Stearns, 1995) and the observation that nucleoporins (NUP) can be functionally fused to a heterologous polypeptide sequence (Nehrbass et al., 1990; Wente et al., 1992; Grandi et al., 1993). If an in-frame fusion of an NUP to the coding sequence for GFP was expressed in yeast cells, we predicted that NPCs could be monitored by direct fluorescence microscopy of live cells. To detect NPC movement, we exploited an aspect of the budding yeast life cycle. In yeast, the nuclear envelope remains intact throughout all stages of the life cycle. Upon mating to form a diploid, the nuclear envelopes of the two haploid nuclei fuse. For the NPC movement assay (Fig. 1, left), one haploid strain expresses a GFP-labeled Nup (donor nucleus), whereas the strain of the opposite mating type (recipient nucleus) expresses only unlabeled Nup. Movement of GFP-labeled NPCs would be detected by the spreading of fluorescent signal over the entire surface of the newly formed diploid nucleus. If the fluorescent signal remained confined to the portion of the nuclear envelope from the donor nucleus, this would reflect a lack of NPC movement. This approach is similar to that used by Latterich and Schekman (1994) to monitor the fate of two proteins of the ER (which is contiguous with the outer nuclear membrane). Using differentially epitope-tagged haploid strains and indirect immunofluorescence microscopy, Sec61p (an integral membrane protein of the outer nuclear membrane) and Eug1p (a soluble protein in the lumen of the nuclear envelope) both diffuse over the entire surface of a newly formed diploid nucleus.
Figure 1.
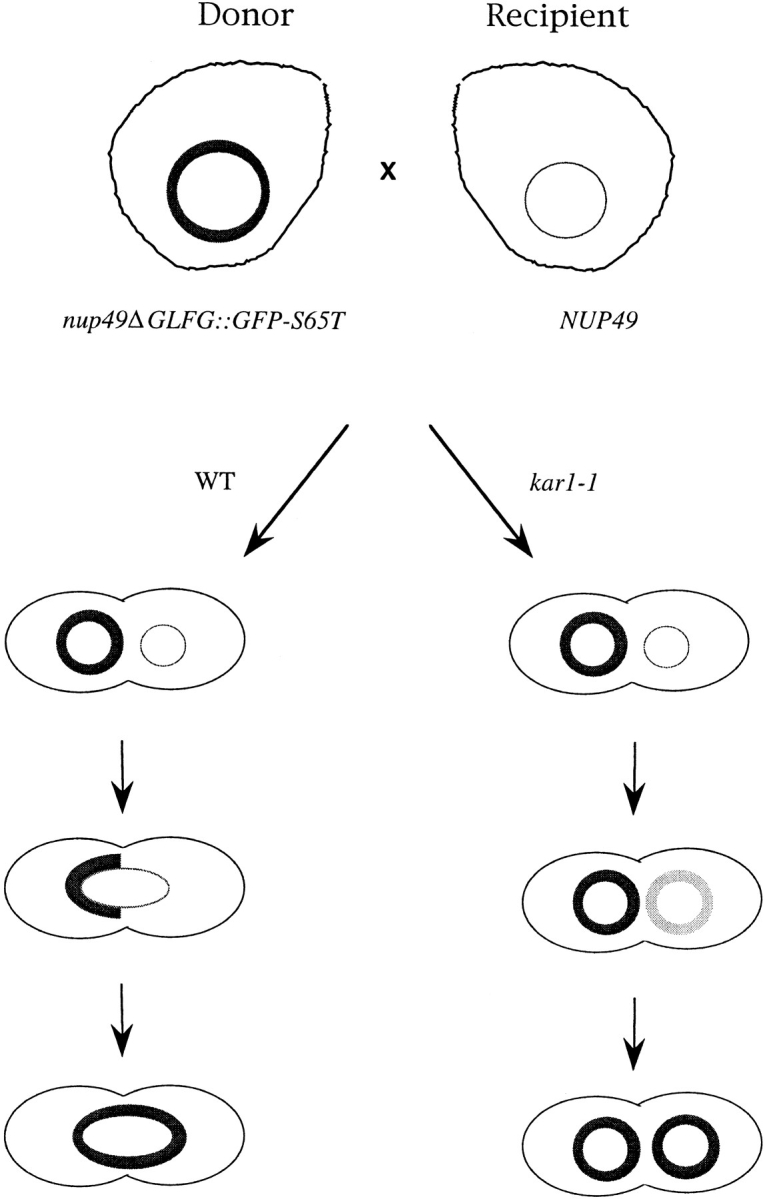
NPC movement and assembly assay. A haploid yeast strain expressing only GFP–Nup49p (Donor) is mated with a haploid strain expressing unlabeled Nup49p (Recipient). In the case where both strains are otherwise wild type (WT), the labeled nucleus (thick circle) will fuse with the recipient nucleus (thin circle) upon mating. Movement of NPCs can then be monitored by watching GFP–Nup49p redistribution in live cells. In a kar1-1 background (kar1-1), the nuclei are unable to fuse and GFP- labeled NPCs are obtained by the recipient nucleus only by incorporation into preexisting/new NPCs. The rate of acquisition of GFP–NPCs in the recipient nuclei reflects either the movement of NPCs or the assembly of NPCs, respectively.
Besides the movement of preexisting NPCs, an increase in GFP–Nup signal in the recipient nuclear envelope may also be due to assembly of new NPCs from individual components including GFP–Nup or to the exchange of individual GFP–Nup for unlabeled Nup components between existing NPC structures. To measure the rate of such assembly/ exchange, a modification of the movement assay was designed (Fig. 1, right). The unilateral karyogamy mutant kar1-1 was incorporated into the genotype of the donor strain. Kar1p is a well-characterized component of yeast spindle pole bodies, and it is required for early nuclear migration steps in karyogamy (Conde and Fink, 1976; Rose and Fink, 1987; Vallen et al., 1992; Kurihara et al., 1994). Since the nuclear envelopes do not fuse in zygotes formed from the mating of a wild-type and a kar1-1 cell, redistribution of the GFP signal would only be observed from direct assembly/exchange of the GFP–Nup into NPCs. Thus, in kar1-1 cells, the rate of GFP signal appearance in the NPCs of the unlabeled recipient nucleus will reflect the assembly/exchange of GFP–Nup into NPCs. An assumption in this strategy is that the kar1-1 mutant does not perturb NPC biogenesis. In all cases, the kar1-1 mutant was incorporated into the genotype of the donor strain. Both schemes in Fig. 1 could also be conducted in the presence of excess unlabeled Nup expressed in the recipient cells on a multicopy (2 μ) plasmid. The excess unlabeled Nup would effectively act as a “chase” during and after cell fusion, and lower the apparent contribution of GFP–Nup assembly/exchange. Thus, any change in GFP–Nup distribution in the movement assay would likely not be due to incorporation of GFP–Nup into NPCs of the recipient nucleus.
GFP–Nup49p Is Functional and Localizes to NPCs
Previous studies have shown that the carboxy-terminal domain of Nup49p is sufficient for NPC function (Iovine et al., 1995). Therefore, the amino-terminal glycine-leucine-phenylalanine-glycine (GLFG) region of Nup49p was replaced with the S65T variant of GFP (Heim and Tsien, 1996) as diagrammed in Fig. 2 A. The nup49ΔGLFG:: GFP-S65T allele was integrated into the nup49 null chromosome by transformation and homologous recombination. Three criteria were used to test whether the GFP- labeled protein had the ability to function in place of the wild-type protein. First, the nup49ΔGLFG::GFP-S65T allele rescued the lethal phenotype of the null mutant. Second, the nup49ΔGLFG::GFP-S65T haploid strains grew at rates comparable to wild-type strains at 23°, 30°, or 37°C (Fig. 2 B). Finally, GFP–Nup49p localization was determined by fluorescence microscopy. The GFP signal showed a nuclear rim staining pattern typical of nucleoporin localization. Moreover, indirect immunofluorescence with a polyclonal antibody against Nup116p showed that the GFP–Nup49p colocalized with the nucleoporin Nup116p (Fig. 2 C). These data suggested that GFP–Nup49p maintained the full functionality of wild-type Nup49p and was an appropriate marker for analyzing NPC dynamics.
Figure 2.



GFP–Nup49p is functional and localizes to NPCs. (A) Plasmid DNA encoding for GFP–Nup49p fusion protein was cut with AvrII for chromosomal integration into a diploid strain heterozygous for the NUP49 null allele, nup49Δ::URA3. Haploids expressing GFP– Nup49p were obtained by sporulation and dissection of Trp+ Ura+ diploids in which TRP1 and URA3 markers cosegregated. (B) Cells expressing GFP–Nup49p grow at rates comparable to wildtype cells at all temperatures. Growth curves are plotted as the log10 of the cell concentration over time. (Circles) SWY809, nup49ΔGLFG:: GFP–S65T cells; (squares) SWY518, wild-type cells. (C) GFP–Nup49p colocalizes with the nucleoporin Nup116p. Immunofluorescence was performed on cells expressing GFP–Nup49p using an affinity-purified polyclonal antibody against Nup116p. GFP fluorescence was visualized directly, and the antiNup116p antibody was visualized using a Texas red– conjugated goat anti–rabbit antibody. Nuclear DNA was visualized by DAPI staining. Bar, 10 μm.
Wild-Type NPCs Move within the Nuclear Envelope
To monitor changes in NPC distribution after nuclear fusion, cells expressing GFP–Nup49p were mated at 30°C to wild-type cells expressing both chromosomal unlabeled Nup49p and excess unlabeled Nup49p from a 2 μ plasmid. Matings were conducted in synthetic minimal media containing 0.1 mg/ml DAPI and lacking tryptophan (to ensure maintenance of the 2 μ NUP49 plasmid). Cells were concentrated onto an agarose-coated slide and scanned at room temperature using Nomarski optics and DAPI filters for the presence of zygotes in which the two nuclei had not fused. Time-lapse videos of GFP–Nup49p fluorescence were acquired by projecting 10 focal planes 0.5 μm apart onto a single two-dimensional image every 2 min until the new diploid nucleus budded. The objective was to standardize each zygote video by the following criteria: the zero time point was at or before the point of nuclear fusion, and the last time point was at or after the point of nuclear division. Fig. 3 shows an example of two zygotes that were found in the same field with representative frames over a 98-min video shown. The bottom zygote had already undergone nuclear fusion at time 0. The nuclei in the top zygote fused at 8 min (between the 6- and 12-min time points shown). By the end of the video, the diploid nucleus had undergone mitosis, and the new daughter nucleus was entering the bud (98-min frame). At this point, the top nucleus was also starting to divide.
Figure 3.

Wild-type NPCs move within the nuclear envelope. Cells expressing GFP–Nup49p (SWY809) were mixed with cells of the opposite mating type expressing wild-type Nup49p from the chromosome and a high copy number plasmid (SWY759). After 5 h in culture, cells were prepared for video microscopy by incubating mating mixtures on an agarose-covered slide. Both Nomarski and fluorescence images for the 0- and 98-min time points are shown. Two newly formed zygotes are shown. GFP–Nup49p distribution was recorded every 2 min for 98 min, and selected frames are shown here. Each fluorescent image is a two-dimensional projection of all z-axis planes. By the end of the sequence (98 min panel), the two zygotes are at various stages of nuclear division, allowing nuclear segregation into the newly formed daughter buds (Nomarski image, 98 min). Numbers indicate time in min. Bar, 5 μm.
Since key nuclear fusion events take place in a time scale of minutes, we monitored NPC dynamics over the same time scale. At the 0-min time point of the video in Fig. 3, the GFP–Nup49p signal was just beginning to distribute around the entire circumference of the diploid nucleus in the bottom zygote. At later times (24 min), it is clear that the diploid nucleus of the bottom zygote was entirely encircled by GFP-labeled NPCs. The top zygote appeared to follow similar kinetics for acquiring GFPlabeled NPCs around the entire new diploid nucleus: fusion occurred at ∼8 min and complete redistribution was achieved at ∼34 min. Data from observing 19 other independent zygotes were identical in terms of the time frame for redistribution of the GFP–Nup49p signal. In general, the entire nuclear surface area of the newly formed diploid was encircled in GFP fluorescence within 25 min after nuclear fusion. The distinct shape of the fused diploid nucleus served as a marker for nuclear movements, and focusing through the z-axis confirmed an even distribution of GFP–Nup49p signal over the entire nuclear surface. Because the signal did not stay confined to the area of the nuclear envelope from the donor nucleus, this suggested that NPCs move over the surface of the nucleus in the nuclear envelope.
NPC Movement Is Distinguishable from NPC Assembly
Inherent in the NPC movement hypothesis is the assumption that the redistribution of GFP–Nup49p was not simply due to assembly of GFP–Nup49p into NPCs in the nuclear surface of the recipient nucleus. Several experiments were conducted to test this possibility. First, if the redistribution was due to movement, the GFP fluorescent signal should spread across the surface of the recipient nucleus with the point in the recipient nucleus farthest from the fusion junction acquiring GFP fluorescent signal last. In contrast, incorporation of either newly synthesized GFP–Nup49p or GFP–Nup49p disassembled from donor NPCs would likely be random and appear uniformly over the entire surface of the recipient nucleus. To test this hypothesis, the redistribution of signal for the top zygote in the Fig. 3 video was quantified. At time points during the redistribution, measurements of the fluorescence signal intensity were taken at ∼0.3-μm increments across a line spanning the fused diploid nucleus. An image of the zygote at time 0 is shown in Fig. 4 with a representative line for data collection designated. The data from multiple time points during the video are displayed in a compiled histogram (Fig. 4). Each colored ribbon represents a time point. Over time (y-axis), the total donor signal decreased while the recipient signal increased. However, it is clear that the signal over the entire surface area of the recipient did not increase uniformly. The region closest to the nuclear membrane fusion point was the first region to begin increasing, and the far edge of the recipient nuclear surface (Fig. 4, right) was the last to gain signal. Moreover, the far edge of the donor nucleus (left) was the last region to decrease in signal. These redistribution results suggest that preexisting donor GFPlabeled NPCs are moving over the surface of the recipient.
Figure 4.
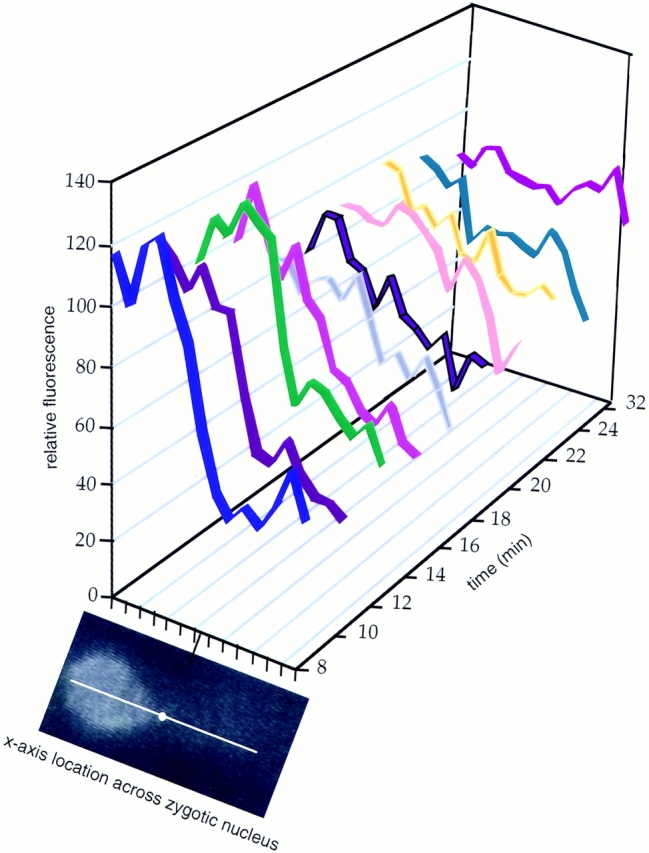
Time course of GFP–Nup49p redistribution in the zygotic nucleus. A representative image is shown from early in the video of the top wild-type zygote in Fig. 3. At each time point during the video (y-axis), a line spanning the donor and recipient nuclear surfaces was designated (shown in white with the dot marking the fusion junction). Distance along the line is graphed on the x-axis. For each line in the respective zygotic nucleus, NIH Image 1.60 was used to quantify the fluorescence intensity at 28 points ∼0.3 μm apart. The fluorescence values of each two sequential measurements along the x-axis were averaged, and the 14 resulting relative fluorescence data points were graphed on the z-axis. The first time point at 8 min in the video (blue ribbon) represents the approximate time of nuclear fusion. Later time points are shown in different colors along the y-axis. Microsoft Excel 4.0 (Seattle, WA) was used to display the graph.
Second, the rate of GFP–Nup49p incorporation into NPCs was directly determined in experiments with a kar1-1 donor strain expressing GFP–Nup49p. When kar1-1 nup49ΔGLFG::GFP-S65T cells were mated with wild-type cells expressing both chromosomal unlabeled Nup49p and excess unlabeled Nup49p on a 2 μ plasmid, the nuclei did not fuse. Fig. 5 shows frames from a representative 98-min video, with the 0 time point representing the point at which the zygote was found. The recipient and donor nuclei were both dividing by the last time point (98 min). With continued incubation, the zygotes in the kar1-1 crosses typically acquired multiple haploid nuclei as a result of continued nuclear division in the absence of nuclear fusion (data not shown). Thus, after division, assembly into a single nucleus could not be assessed and the video was terminated. GFP fluorescence signal was monitored over this time frame. At time 0, the recipient (unlabeled) nucleus was barely visible by GFP staining (arrow in the zygote). With time, the recipient nucleus slowly gained GFP fluorescence signal. Data was collected from seven independent trials. In all cases with kar1-1 cells (Fig. 1, right), the time course for fluorescence accumulation in the recipient nucleus was notably longer than when wild-type cells were mated (Fig. 1, left). By the 30-min time point shown in Fig. 5, the recipient nucleus was only one-fourth as bright as the kar1-1 donor nucleus (a fluorescence ratio of 0.26 recipient to donor; see below). In contrast, at the 30-min time point in the wild-type video (Fig. 3), the fluorescence signal over the surface area of the recipient nucleus was equivalent to that of the donor nucleus. The recipient nuclei in the kar1-1 experiments actually divided before they reached the GFP fluorescence level of the donor nuclei. Overall, the fluorescence signal increase in the nuclear surface area of the recipient nucleus was significantly more rapid when fused to a donor nucleus harboring GFP-Nup49p–NPCs. Therefore, the fluorescence signal redistribution observed in the wild-type zygotes of Fig. 3 was probably due to the movement of NPCs with minimal contribution from the assembly of GFP–Nup49p into NPCs.
Figure 5.

NPC assembly follows a slower time course than NPC movement. kar1-1 mutant cells expressing GFP–Nup49p (SWY1308) were mixed with cells of the opposite mating type expressing both genomic unlabeled Nup49p and unlabeled Nup49p from a high copy 2 μ plasmid (SWY757). GFP–NPC distribution was recorded in two-dimensional projection videos as described in Fig. 3. Selected frames from a 98-min video are shown. The first and last frames are Nomarski images taken at the beginning and end of the video: note that the two nuclei in the zygote have not fused. The recipient nucleus (arrows) slowly acquired GFP fluorescence but divided before it reached the intensity of the GFP–Nup49p donor nucleus. Numbers indicate time in min. Bar, 5 μm.
Rate of NPC Movement
To compare apparent NPC movement rates in wild-type zygotes to apparent NPC assembly rates in kar1-1 zygotes, videos were quantified for the total relative fluorescence values of the recipient and donor nuclei (or the relevant nuclear surface from each). The fluorescence ratio (recipient to donor) was plotted vs time, and the data from one representative video for each zygote type are shown in Fig. 6. Data collection began at a similar relative time point (when the donor nucleus was approximately four times as bright as the recipient nucleus). In zygotes with a wild-type background, the recipient nucleus acquired GFP fluorescence to equal the GFP fluorescence of the donor nucleus (ratio of 1) before nuclear division occurred (top arrow). In the kar1-1 zygotes, however, the two haploid nuclei divided (lower arrow) before the recipient nucleus acquired GFP fluorescence comparable to that associated with the donor nucleus. The change in the fluorescence ratio (F.R.) per min was calculated from the slopes of multiple data sets. The rate of apparent movement (in wild-type cell experiments with the Nup49p 2 μ plasmid, n = 19) was 0.017 ± 0.009 F.R./min. In comparison, the relative rate of GFP– Nup49p incorporation (in kar1-1 cell experiments with the Nup49p 2 μ plasmid, n = 5) was only 0.0025 ± 0.0006 F.R./ min. Therefore, the rate of apparent NPC movement was at least sixfold greater than the rate of GFP–Nup49p incorporation into NPCs.
Figure 6.

NPC movement rates are distinct from GFP–Nup49p incorporation rates. Quantification of NPC movement and assembly was conducted using NIH Image to calculate the average pixel brightness value of an area encircling the donor or recipient nucleus. The fluorescence ratio was determined by dividing the average brightness value of the recipient nucleus by the average brightness value of the donor nucleus. The data for one zygote for each type of mating are shown. (Circles) Wild-type zygote with 2 μ NUP49 plasmid; (diamonds) kar1-1 zygote with 2 μ NUP49 plasmid; (squares) kar1-1 zygote with no plasmid. The Cricket Graph program was used to fit a line through the linear portion of the wild-type data set, and through all the points for the kar1-1 experiments. (Arrows) Approximate time of nuclear division for wild-type (top arrow) and kar1-1 zygotes (bottom arrow).
Similar experiments were also performed using a recipient strain with only chromosomally expressed levels of unlabeled Nup49p. In such zygotes formed by crossing kar1-1 nup49ΔGLFG::GFP-S65T cells to wild-type cells, the recipient nucleus never acquired GFP–Nup49p to the same level as was associated with the donor nucleus (Fig. 6). The relative rate of GFP–Nup49p incorporation in the absence of excess unlabeled Nup49p was 0.005 ± 0.002 F.R./ min (n = 6). This rate is approximately twofold greater than in the presence of excess unlabeled Nup49p. Immunoblot analysis was conducted to measure the relative levels of unlabeled Nup49p (Fig. 7). The strain with the 2 μ plasmid (+, lane 2) expressed approximately threefold more unlabeled Nup49p compared with a similar strain expressing only chromosomal Nup49p (−, lane 1). In both strains, the levels of Nup116p were the same. Therefore, the decreased assembly/exchange rate correlated with increased unlabeled Nup49p expression levels. This suggested that the excess unlabeled Nup49p served as an inhibitor of GFP–Nup49p incorporation into NPCs of the recipient nucleus.
Figure 7.

Cells harboring the 2 μ NUP49 plasmid have elevated levels of Nup49p. Yeast strains SWY459 (wild type, lane 1) and SWY759 (SWY459 cells harboring the 2 μ NUP49 plasmid, lane 2) were grown in synthetic media lacking uracil or tryptophan, respectively. Total protein extracts were prepared from equivalent numbers of cells, separated on a 9% SDS polyacrylamide gel, and transferred to nitrocellulose. Blots were probed with an affinitypurified polyclonal antibody raised against the GLFG region of Nup116p that also recognizes Nup49p (gift of K. Iovine and J. Watkins, Washington University, St. Louis, MO).
NPCs Move to Form Clusters in Temperature-arrested gle2-1 Cells
Mutations in the gene encoding the NPC-associated factor Gle2p have recently been characterized (Murphy et al., 1996). Interestingly, gle2-1 cells show temperature-dependent formation of NPC clusters. This clustering phenotype may result from perturbations of NPC dynamics. If NPCs are assembled at one location on the nuclear surface, the clusters may arise from the lack of movement by the mutant NPCs. In contrast, if NPCs are inserted randomly over the surface, clusters may form via aggregation of mutant NPCs moving in the nuclear envelope. To determine if the gle2-1 phenotype was due to the movement of randomly distributed NPCs into clusters, we monitored NPC distribution with GFP–Nup49p in gle2-1 cells. In contrast with the experiments conducted with mating cells, these experiments monitored redistribution of GFP–Nup49p in single cells.
First, we tested if clusters in gle2-1 cells were formed from preexisting NPCs. GFP–Nup49p was placed under control of the GAL10 promoter in gle2-1 cells, allowing GFP– Nup49p synthesis to be strictly controlled by specific galactose induction and glucose repression of the GAL10 promoter (Fig. 8 B). With GAL-GFP-nup49 as the only source of Nup49p, wild-type growth was obtained in media containing 0.5% galactose and 1.5% raffinose (data not shown). A 2 μ plasmid harboring NUP49 was transformed into the GAL-GFP-nup49 gle2-1 cells to ensure that any phenotypes detected after addition of glucose were not simply due to the depletion of Nup49p (an essential nucleoporin). The NUP49 2 μ plasmid supported growth of the GAL-GFP-nup49 gle2-1 cells in media containing glucose as the sole carbon source (data not shown). The GALGFP-nup49 gle2-1 cells were grown at 23°C in media containing galactose, and then maintained in galactose media or washed into media containing 2% glucose for 1 h before a 5-h temperature shift to 37°C. Cells were processed for direct visualization and indirect immunofluorescence using the anti-Nup116p antibody as described for Fig. 2 C. As shown in Fig. 8 A, cells grown in either glucose- or galactose-containing media at 23°C did not exhibit the clustering phenotype (first and third columns). In contrast, cells shifted to 37°C for 5 h exhibit the NPC clustering phenotype whether they were grown in glucose- or galactosecontaining media (second and fourth columns). Similar results were seen for cells grown at 23°C in glucose or galactose media for 5 h before the 37°C shift (data not shown). These results suggested that NPC clusters in gle2-1 cells formed by movement of preexisting NPCs into aggregates rather than by the assembly of new NPCs into a distinct area of the nuclear envelope.
Figure 8.


NPCs move into clusters in the gle2-1 temperature-sensitive mutant. (A) gle2-1 cells expressing GFP– Nup49p under control of the galactose-inducible promoter and Nup49p from a 2 μ plasmid (SWY1324) were grown in synthetic minimal medium lacking histidine and containing 0.5% galactose and 1.5% raffinose. Half of the cells were washed out of galactose and into glucosecontaining media for 1 h before a shift to the nonpermissive temperature of 37°C. Cells were maintained at 37°C in glucose or galactose for 5 h before processing for direct visualization and immunofluorescence as described for Fig. 2 C. Clusters of NPCs appear in cells shifted to glucose as well as those maintained in galactose. (B) PCR was performed on pSW441 to create a fragment flanked by sequences complementary to NUP49. The fragment was transformed into wild-type SWY595 cells for integration onto the chromosome. Bar, 10 μm.
To directly monitor NPC movement into clusters, cluster formation in live gle2-1 cells was observed. The gle2-1 cells expressing GFP–Nup49p under control of the GAL10 promoter were grown in galactose media as for the fixed cell experiment in Fig. 8 A. Cells were washed into media containing 2% glucose for 5 h before being concentrated onto an agarose-coated slide. Cells were shifted to 37°C using a temperature-controlled microscope stage. Videos of GFP fluorescence were acquired as in the movement and assembly assays, with time points taken every 20 min for 5 h. Selected frames are shown in Fig. 9. At time points up to 100 min after the 37°C shift, no GFP-labeled clusters were observed. However, by 140 min, NPC clusters were detectable (Fig. 9, arrowheads) and appeared to become larger at later time points (300 min). To determine if NPC cluster formation was reversible, gle2-1 cells grown at 37°C were shifted back to growth at 23°C for extended time periods. The GAL-GFP-nup49 gle2-1 cells were grown in media containing galactose, and then shifted for 5 h to 37°C in media containing 2% glucose. Cells were prepared for live microscopy, and the temperature-controlled microscope stage was used to shift to growth at 23°C. A field of cells is shown in Fig. 10. Four cells each with distinct NPC clusters were present at the 0 time point (after the 5-h shift to 37°C). After 8 h at 23°C, the cells had clearly divided (compare the Nomarski images in the first and last panels). However, the NPC clusters were still present. Therefore, the clusters did not disassemble over the same time frame that they assembled.
Figure 9.

Monitoring movement of NPCs into clusters. SWY1324 cells were grown as in Fig. 8 A to induce expression of GFP–Nup49p. Cells were then washed into media containing 2% glucose for 5 h. Cells were shifted to 37°C on a slide set on a temperature-controlled microscope stage. Videos were taken with time points every 20 min for 5 h as described for Fig. 3. Selected frames 40 min apart are shown. The first and last frames are Nomarski images before and after the video. Numbers indicate time after the 37°C shift in min. (Arrowheads) NPC clusters formed by NPC movement. Bar, 5 μm.
Figure 10.

Cluster formation in gle2-1 cells is not rapidly reversed. SWY1324 cells were induced at 23°C to express GFP–Nup49p by growing in SC − his containing 0.5% galactose/1.5% raffinose. Cells were then shifted to 37°C in SC − his with 2% glucose for 5 h before a return to 23°C. Videos were taken as described for the movement assay in Fig. 3. Frames were taken every 30 min for 8 h after returning the cells to 23°C. Both Nomarski and fluorescence images were taken at the 0- and 8-h time points. Selected frames are shown here. Bar, 5 μm.
Discussion
We have developed a new assay to measure NPC dynamics in live yeast cells. The assay involves mating a cell expressing a GFP–Nup with a cell expressing an unlabeled Nup and monitoring GFP-labeled NPC distribution in the zygotic nucleus. Studies using this assay have yielded two important conclusions concerning NPC physiology: first, that NPCs have a remarkable degree of mobility within the double nuclear membrane; and, second, that NPC clustering in at least one mutant results from the migration of existing NPCs into groups rather than NPC biogenesis at a fixed site on the envelope. The fact that NPCs can move over the nuclear surface presents a significant change in thinking about NPC dynamics.
Previous studies of yeast clustering mutants have suggested two different mechanisms for NPC cluster formation. If NPCs are assembled at a single site on the nuclear surface (such as near the spindle pole body), the clusters may arise from the inability of these newly assembled NPCs to migrate away. Alternatively, clusters may form via aggregation of mutant NPCs moving in the nuclear envelope. Using the GFP–Nup49p system and the temperature-dependent gle2-1 clustering mutant, we were able to distinguish between these two models for pore dynamics. Clusters in gle2-1 cells form by the movement of preexisting NPCs. The movement of NPCs into such clusters occurs after release from a nuclear tether. Alternatively, the clusters may result from the disappearance of a protein that normally prevents pore aggregation. Interestingly, the NPC clusters formed in gle2-1 cells at 37°C did not rapidly disappear after shifting back to growth at 23°C. This suggests that once clusters were formed in gle2-1 cells, the NPCs in those clusters lost the ability to move. Incorporation of newly synthesized Gle2p at 23°C may be required for movement of the NPCs out of clusters. If this occurs at a rate similar to that for GFP–Nup49p incorporation into NPCs, the observed slow reversibility would be expected.
Understanding the mechanism of NPC cluster formation is especially important because clustering has thus far proven a fairly prevalent phenotype for mutations in NPCassociated factors. At this point, roughly one-third of the identified genes encoding NPC-associated proteins have mutant alleles that result in NPC cluster formation. It is possible that the mechanism for cluster formation may be different among the known yeast mutants and will reflect distinct functional roles for the individual components. Three general types of clustering phenotypes have been reported: nup145, nup133, nup120, nup84, and nup85 mutants form clusters constitutively at all growth temperatures (Doye et al., 1994; Wente and Blobel, 1994; Aitchison et al., 1995a ; Heath et al., 1995; Li et al., 1995; Pemberton et al., 1995; Goldstein et al., 1996; Siniossoglou et al., 1996); the rat7-1/nup159 mutant has clusters at 23°C that disappear after ∼1 h at 37°C (Gorsch et al., 1995); and gle2 mutants exhibit cluster formation at 37°C (Murphy et al., 1996). The GFP–Nup assay system could be used to test whether rat7-1/nup159 cluster disappearance at 37°C is due to disassembly of NPCs within the cluster or to movement of NPCs out of clusters.
Rapid movement (up to 1.48 μm/min) has been previously characterized for the spindle pole body, a large organelle embedded in the yeast nuclear envelope (Kahana et al., 1995; Yeh et al., 1995). With respect to the complexity of NPC ultrastructure, it is surprising that NPCs also move in the nuclear envelope across the surface of the nucleus. Vertebrate NPCs have extensive connections to intranuclear filamentous networks (including the lamins and the lattice) as well as proposed connections to the cytoskeleton (for review see Goldberg and Allen, 1995). Nuclear basket-like structures and cytoplasmic filaments have also been observed on yeast NPCs (Rout and Blobel, 1993), but searches of the complete yeast DNA sequence database have not revealed any predicted polypeptides with significant similarity to the vertebrate nuclear lamins. Thus, if the presence of lamins inhibits movement, vertebrate NPCs may not move over the nuclear surface. Alternatively, vertebrate NPCs may move, but the rate of movement could be cell type specific. Interestingly, the vertebrate lamins are not required for the assembly of NPCs in Xenopus annulate lamellae (Dabauvalle et al., 1991; Meier et al., 1995). Measurement of vertebrate NPC movement will require the development of analogous assays in vertebrate cells.
There are several interesting functional consequences to consider, knowing that NPCs have the capacity to move within the nuclear membrane. If de novo assembly of NPCs occurs at one or a few sites of the nuclear envelope, the ability of NPCs to move within the nuclear envelope may serve to distribute NPCs around the nuclear periphery. It is also possible that the cell may respond to environmental or developmental stimuli by allowing NPCs to move, e.g., to active sites of transcription. The localized patches of NPCs in spermatocytes (Fawcett, 1981) may reflect constraints on NPC movement in this specialized cell type.
Several independent criteria were used to distinguish between NPC movement and incorporation of GFP– Nup49p into NPCs. These include experiments with kar1-1 cells, the addition of excess unlabeled Nup49p, and quantitative analysis of the signal spreading over the surface of the fused nuclei. In the kar1-1 assay, the incorporation of GFP–Nup49p into NPCs was directly monitored. Measurements of the rate of redistribution in wild-type vs kar1-1 cell types were clearly distinct, and thus support the hypothesis that NPCs move. In fact, the GFP-labeled NPCs moved over the surface of the nuclear envelope at a rate approximately sixfold faster than the rate for incorporating GFP–Nup49p into NPCs. However, the kar1-1 experimental strategy could not distinguish between incorporation due to exchange into preexisting NPCs vs that due to new NPC assembly. Therefore, the analysis yields a combined assembly rate from both sources. In wild-type yeast cells, no cytoplasmic pool of nucleoporins from either the GLFG or the FXFG family has been detected (Aris and Blobel, 1989; Wente et al., 1992), and it is presumed that, after synthesis of the polypeptides, the nucleoporins are rapidly assembled into NPCs. In this report the incorporation rate for GFP–Nup49p into NPCs has been inferred from measuring incorporation over the entire nuclear surface. To test whether other nucleoporins exhibit the same relative incorporation rate as GFP–Nup49p, additional nucleoporins must be tagged and similarly assayed. If all GFP–Nups display similar incorporation rates, this behavior may reflect that of the entire NPC. It should also be noted that apparent assembly rates may vary at different points in the cell cycle, given that measurements of total pore number per cell have suggested yeast NPC formation peaks during and after mitosis (Jordan et al., 1977). Since vertebrate nuclear pore and NPC assembly requires the prior formation of a double nuclear membrane (Macaulay and Forbes, 1996) and NPCs are architecturally and functionally similar in all eukaryotic organisms (Maul, 1977; Forbes, 1992; Osborne and Silver, 1993; Davis, 1995), NPC assembly into the intact nuclear envelope of budding yeast could occur by a similar mechanism. We predict that studying NPC dynamics in yeast will lend insight into NPC biogenesis in all organisms.
What is the mechanism of NPC movement? Because limitations on in vivo microscopy do not allow resolution of individual NPCs, the rate of NPC movement in this report has been inferred from the redistribution of a population of NPCs. Using this assay, we will be able to directly test whether NPC movement requires energy or any known motor proteins. Constraints on freedom of movement can also be examined, such as in areas where the nucleus abuts the vacuole (Severs et al., 1976), in yeast cells that overexpress HMG-CoA reductase and produce extensive nuclear envelope–associated karmellae (Wright et al., 1988; Hampton et al., 1996), or in yeast cells that overexpress vertebrate lamins (Smith and Blobel, 1994). Moreover, existing mutants can be analyzed for perturbations of NPC movement or assembly rates, and genetic screens can be conducted to isolate novel NPC assembly mutants on the basis of their distinct fluorescent properties as compared with wild-type cells. Our long range goal is to define the network of structural interactions that mediate NPC biogenesis by testing the involvement of particular nucleoporins in NPC assembly and movement.
Acknowledgments
This work was supported by a Beckman Young Investigator grant to S.R. Wente from the Arnold and Mabel Beckman Foundation, and an American Cancer Society Junior Faculty Research Award to S.R. Wente.
Abbreviations used in this paper
- DAPI
4′,6-diamidino-2-phenylindole
- F.R.
fluorescence ratio
- GLFG
glycine-leucine-phenylalanine-glycine
- NPC
nuclear pore complex
- NUP
nucleoporin
- SC
synthetic complete medium
Footnotes
We are indebted to J. Waddle and J. Cooper for advice in monitoring GFP in live yeast cells, and to J. Waddle and G. Baxter for writing the computer macros for collecting and analyzing the data. We thank M. Rose for the kar1-1 strain; R. Murphy for the gle2-1 strain; L. Riles, J. Waddle, R. Heim, and R. Tsien for plasmids; and J. Cooper, J. Waddle, K. Wilson, M. Winey, and colleagues in the Wente laboratory for insightful discussion and comments on the manuscript.
Address all correspondence to Susan R. Wente, Department of Cell Biology and Physiology, Box 8228, Washington University School of Medicine, 660 S. Euclid Ave., St. Louis, MO 63110. Tel.: (314) 362-2713. Fax: (314) 362-7463. e-mail: swente@cellbio.wustl.edu
References
- Aitchison JD, Blobel G, Rout MP. Nup120p: a yeast nucleoporin required for NPC distribution and mRNA transport. J Cell Biol. 1995a;131:1659–1675. doi: 10.1083/jcb.131.6.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitchison JD, Rout MP, Marelli M, Blobel G, Wozniak RW. Two novel related yeast nucleoporins Nup170p and Nup157p : complementation with the vertebrate homologue Nup155p and functional interactions with the yeast nuclear pore–membrane protein Pom152p. J Cell Biol. 1995b;131:1133–1148. doi: 10.1083/jcb.131.5.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey CW. Structural plasticity of the nuclear pore complex. J Mol Biol. 1995;248:273–293. doi: 10.1016/s0022-2836(95)80050-6. [DOI] [PubMed] [Google Scholar]
- Aris JP, Blobel G. Yeast nuclear envelope proteins cross-react with an antibody against mammalian pore complex proteins. J Cell Biol. 1989;108:2059–2067. doi: 10.1083/jcb.108.6.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin A, Ozier KO, Denouel A, Lacroute F, Cullin C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. . Nucleic Acids Res. 1993;21:3329–3330. doi: 10.1093/nar/21.14.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boman AL, Delannoy MR, Wilson KL. GTP hydrolysis is required for vesicle fusion during nuclear envelope assembly in vitro. J Cell Biol. 1992a;116:281–294. doi: 10.1083/jcb.116.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boman AL, Taylor TC, Melancon P, Wilson KL. A role for ADP-ribosylation factor in nuclear vesicle dynamics. Nature (Lond) 1992b;358:512–514. doi: 10.1038/358512a0. [DOI] [PubMed] [Google Scholar]
- Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P. Multifunctional yeast high-copy-number shuttle vectors. Gene (Amst) 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- Conde J, Fink GR. A mutant of Saccharomyces cerevisiaedefective for nuclear fusion. Proc Natl Acad Sci USA. 1976;73:3651–3655. doi: 10.1073/pnas.73.10.3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabauvalle MC, Loos K, Merkert H, Scheer U. Spontaneous assembly of pore complex-containing membranes (“annulate lamellae”) in Xenopusegg extract in the absence of chromatin. J Cell Biol. 1991;112:1073–1082. doi: 10.1083/jcb.112.6.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabauvalle MC, Loos K, Scheer U. Identification of a soluble precursor complex essential for nuclear pore assembly in vitro. Chromosoma (Berl) 1990;100:56–66. doi: 10.1007/BF00337603. [DOI] [PubMed] [Google Scholar]
- Davis LI. The nuclear pore complex. Annu Rev Biochem. 1995;64:865–896. doi: 10.1146/annurev.bi.64.070195.004245. [DOI] [PubMed] [Google Scholar]
- Doye V, Wepf R, Hurt EC. A novel nuclear pore protein Nup133p with distinct roles in poly(A)+RNA transport and nuclear pore distribution. EMBO (Eur Mol Biol Organ) J. 1994;13:6062–6075. doi: 10.1002/j.1460-2075.1994.tb06953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett, D.W. 1981. The Cell. Second edition. W.B. Saunders Company, Philadelphia.
- Finlay DR, Forbes DJ. Reconstitution of biochemically altered nuclear pores: transport can be eliminated and restored. Cell. 1990;60:17–29. doi: 10.1016/0092-8674(90)90712-n. [DOI] [PubMed] [Google Scholar]
- Finlay DR, Meier E, Bradley P, Horecka J, Forbes DJ. A complex of nuclear pore proteins required for pore function. J Cell Biol. 1991;114:169–183. doi: 10.1083/jcb.114.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes DJ. Structure and function of the nuclear pore complex. Annu Rev Cell Biol. 1992;8:495–527. doi: 10.1146/annurev.cb.08.110192.002431. [DOI] [PubMed] [Google Scholar]
- Gerace L, Foisner R. Integral membrane proteins and dynamic organization of the nuclear envelope. Trends Cell Biol. 1994;4:127–131. doi: 10.1016/0962-8924(94)90067-1. [DOI] [PubMed] [Google Scholar]
- Goldberg MW, Allen TD. Structural and functional organization of the nuclear envelope. Curr Opin Cell Biol. 1995;7:301–309. doi: 10.1016/0955-0674(95)80083-2. [DOI] [PubMed] [Google Scholar]
- Goldstein AL, Snay CA, Heath CV, Cole CN. Pleiotropic nuclear defects associated with a conditional allele of the novel nucleoporin Rat9p/Nup85p. Mol Biol Cell. 1996;7:917–934. doi: 10.1091/mbc.7.6.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich D, Mattaj IW. Nucleocytoplasmic transport. Science (Wash DC) 1996;271:1513–1518. doi: 10.1126/science.271.5255.1513. [DOI] [PubMed] [Google Scholar]
- Gorsch LC, Dockendorff TC, Cole CN. A conditional allele of the novel repeat-containing yeast nucleoporin RAT7/NUP159causes both rapid cessation of mRNA export and reversible clustering of nuclear pore complexes. J Cell Biol. 1995;129:939–955. doi: 10.1083/jcb.129.4.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandi P, Doye V, Hurt EC. Purification of NSP1 reveals complex formation with ‘GLFG' nucleoporins and a novel nuclear pore protein NIC96. EMBO (Eur Mol Biol Organ) J. 1993;12:3061–3071. doi: 10.1002/j.1460-2075.1993.tb05975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Koning A, Wright R, Rine J. In vivo examination of membrane protein localization and degradation with green fluorescent protein. Proc Natl Acad Sci USA. 1996;93:828–833. doi: 10.1073/pnas.93.2.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath CV, Copeland CS, Amberg DC, Delpriore V, Snyder M, Cole CN. Nuclear pore complex clustering and nuclear accumulation of poly(A)+ RNA associated with mutation of the Saccharomyces cerevisiae rat2/nup120gene. J Cell Biol. 1995;131:1677–1697. doi: 10.1083/jcb.131.6.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim R, Tsien RY. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol. 1996;6:178–182. doi: 10.1016/s0960-9822(02)00450-5. [DOI] [PubMed] [Google Scholar]
- Iovine MK, Watkins JL, Wente SR. The GLFG repetitive region of the nucleoporin Nup116p interacts with Kap95p, an essential yeast nuclear import factor. J Cell Biol. 1995;131:1699–1713. doi: 10.1083/jcb.131.6.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan EG, Severs NJ, Williamson DH. Nuclear pore formation and the cell cycle in Saccharomyces cerevisiae. . Exp Cell Res. 1977;104:446–449. doi: 10.1016/0014-4827(77)90114-8. [DOI] [PubMed] [Google Scholar]
- Kahana JA, Schnapp BJ, Silver PA. Kinetics of spindle pole body separation in budding yeast. Proc Natl Acad Sci USA. 1995;92:9707–9711. doi: 10.1073/pnas.92.21.9707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessel RG. The structure and function of annulate lamellae: porous cytoplasmic and intranuclear membranes. Int Rev Cytol. 1983;82:181–303. doi: 10.1016/s0074-7696(08)60826-8. [DOI] [PubMed] [Google Scholar]
- Kurihara LJ, Beh CT, Latterich M, Schekman R, Rose MD. Nuclear congression and membrane fusion: two distinct events in the yeast karyogamy pathway. J Cell Biol. 1994;126:911–923. doi: 10.1083/jcb.126.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latterich M, Schekman R. The karyogamy gene KAR2and novel proteins are required for ER-membrane fusion. Cell. 1994;78:87–98. doi: 10.1016/0092-8674(94)90575-4. [DOI] [PubMed] [Google Scholar]
- Li O, Heath CV, Amberg DC, Dockendorff TC, Copeland SC, Snyder M, Cole CN. Mutation or deletion of the Saccharomyces cerevisiae RAT3/NUP133 gene causes temperature-dependent nuclear accumulation of poly(A)+RNA and constitutive clustering of nuclear pore complexes. Mol Biol Cell. 1995;6:401–417. doi: 10.1091/mbc.6.4.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohka MJ, Masui Y. Formation in vitro of sperm pronuclei and mitotic chromosomes induced by amphibian ooplasmic components. Science (Wash DC) 1983;220:719–721. doi: 10.1126/science.6601299. [DOI] [PubMed] [Google Scholar]
- Macaulay C, Forbes DJ. Assembly of the nuclear pore: biochemically distinct steps revealed with NEM, GTPγS, and BAPTA. J Cell Biol. 1996;132:5–20. doi: 10.1083/jcb.132.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul GG. The nuclear and the cytoplasmic pore complex: structure, dynamics, distribution, and evolution. Int Rev Cytol (Suppl) 1977;5:75–186. [PubMed] [Google Scholar]
- Meier E, Miller BR, Forbes DJ. Nuclear pore complex assembly studied with a biochemical assay for annulate lamellae formation. J Cell Biol. 1995;129:1459–1472. doi: 10.1083/jcb.129.6.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchior F, Gerace L. Mechanisms of nuclear protein import. Curr Opin Cell Biol. 1995;7:310–318. doi: 10.1016/0955-0674(95)80084-0. [DOI] [PubMed] [Google Scholar]
- Meluh PB, Rose MD. KAR3, a kinesin-related gene required for yeast nuclear fusion. Cell. 1990;60:1029–1041. doi: 10.1016/0092-8674(90)90351-e. [DOI] [PubMed] [Google Scholar]
- Murphy R, Watkins JL, Wente SR. GLE2, a S. cerevisiae homologue of the S. pombe export factor RAE1, is required for nuclear pore complex structure and function. Mol Biol Cell. 1996;7:1921–1937. doi: 10.1091/mbc.7.12.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutvei A, Dihlmann S, Herth W, Hurt EC. NSP1 depletion in yeast affects nuclear pore formation and nuclear accumulation. Eur J Cell Biol. 1992;59:280–295. [PubMed] [Google Scholar]
- Nehrbass U, Kern H, Mutvei A, Horstmann H, Marshallsay B, Hurt EC. NSP1: a yeast nuclear envelope protein localized at the nuclear pores exerts its essential function by its carboxy-terminal domain. Cell. 1990;61:979–989. doi: 10.1016/0092-8674(90)90063-k. [DOI] [PubMed] [Google Scholar]
- Newmeyer DD, Forbes DJ. A N-ethylmaleimide-sensitive cytosolic factor necessary for nuclear protein import: requirement in signal- mediated binding to the nuclear pore. J Cell Biol. 1990;110:547–557. doi: 10.1083/jcb.110.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newport J. Nuclear reconstitution in vitro: stages of assembly around protein-free DNA. Cell. 1987;48:205–217. doi: 10.1016/0092-8674(87)90424-7. [DOI] [PubMed] [Google Scholar]
- Newport JW, Dunphy W. Characterization of the membrane binding and fusion events during nuclear envelope assembly using purified components. J Cell Biol. 1992;116:295–306. doi: 10.1083/jcb.116.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne MA, Silver PA. Nucleocytoplasmic transport in the yeast Saccharomyces cerevisiae. . Annu Rev Biochem. 1993;62:219–254. doi: 10.1146/annurev.bi.62.070193.001251. [DOI] [PubMed] [Google Scholar]
- Pante N, Aebi U. Molecular dissection of the nuclear pore complex. Crit Rev Biochem Mol Biol. 1996;31:153–199. doi: 10.3109/10409239609106583. [DOI] [PubMed] [Google Scholar]
- Pemberton LF, Rout MP, Blobel G. Disruption of the nucleoporin gene NUP133results in clustering of nuclear pore complexes. Proc Natl Acad Sci USA. 1995;92:1187–1191. doi: 10.1073/pnas.92.4.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller R, Smythe C, Newport JW. Assembly/disassembly of the nuclear envelope membrane: cell cycle-dependent binding of nuclear membrane vesicles to chromatin in vitro. Cell. 1991;65:209–217. doi: 10.1016/0092-8674(91)90155-r. [DOI] [PubMed] [Google Scholar]
- Ris H. The three dimensional structure of the nuclear pore complex as seen by high voltage electron microscopy and high resolution low voltage scanning electron microscopy. EMSA Bull. 1991;21:54–56. [Google Scholar]
- Rose MD, Fink GR. KAR1, a gene required for function of both intranuclear and extranuclear microtubules in yeast. Cell. 1987;48:1047–1060. doi: 10.1016/0092-8674(87)90712-4. [DOI] [PubMed] [Google Scholar]
- Rout MP, Blobel G. Isolation of the yeast nuclear pore complex. J Cell Biol. 1993;123:771–783. doi: 10.1083/jcb.123.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Second edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 545 pp.
- Severs NJ, Jordan EG, Williamson DH. Nuclear pore absence from areas of close association between nucleus and vacuole in synchronous yeast cultures. J Ultrastruct Res. 1976;54:374–387. doi: 10.1016/s0022-5320(76)80023-8. [DOI] [PubMed] [Google Scholar]
- Sheehan MA, Mills AD, Sleeman AM, Laskey RA, Blow JJ. Steps in the assembly of replication-competent nuclei in a cell-free system from Xenopuseggs. J Cell Biol. 1988;106:1–12. doi: 10.1083/jcb.106.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, F., G.R. Fink, and J.B. Hicks. 1986. Methods in Yeast Genetics. Vol. 35. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siniossoglou S, Wimmer C, Rieger M, Doye V, Tekotte H, Weise C, Emig S, Segref A, Hurt EC. A novel complex of nucleoporins, which includes Sec13p and a Sec13p homolog, is essential for normal nuclear pores. Cell. 1996;84:265–275. doi: 10.1016/s0092-8674(00)80981-2. [DOI] [PubMed] [Google Scholar]
- Smith S, Blobel G. Colocalization of vertebrate lamin B and lamin B receptor (LBR) in nuclear envelopes and in LBR-induced membrane stacks of the yeast Saccharomyces cerevisiae. . Proc Natl Acad Sci USA. 1994;91:10124–10128. doi: 10.1073/pnas.91.21.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns T. Green fluorescent protein. The green revolution. Curr Biol. 1995;5:262–264. doi: 10.1016/s0960-9822(95)00056-x. [DOI] [PubMed] [Google Scholar]
- Stewart M. Nuclear pore structure and function. Semin Cell Biol. 1992;3:267–277. doi: 10.1016/1043-4682(92)90028-t. [DOI] [PubMed] [Google Scholar]
- Sullivan KM, Busa WB, Wilson KL. Calcium mobilization is required for nuclear vesicle fusion in vitro: implications for membrane traffic and IP3 receptor function. Cell. 1993;73:1411–1422. doi: 10.1016/0092-8674(93)90366-x. [DOI] [PubMed] [Google Scholar]
- Underwood MR, Fried HM. Characterization of nuclear localizing sequences derived from yeast ribosomal protein L29. EMBO (Eur Mol Biol Organ) J. 1990;9:91–99. doi: 10.1002/j.1460-2075.1990.tb08084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallen EA, Hiller MA, Scherson TY, Rose MD. Separate domains of KAR1 mediate distinct functions in mitosis and nuclear fusion. J Cell Biol. 1992;117:1277–1287. doi: 10.1083/jcb.117.6.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigers GP, Lohka MJ. A distinct vesicle population targets membranes and pore complexes to the nuclear envelope in Xenopuseggs. J Cell Biol. 1991;112:545–556. doi: 10.1083/jcb.112.4.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigers G P, Lohka MJ. Regulation of nuclear envelope precursor functions during cell division. J Cell Sci. 1992;102:273–284. doi: 10.1242/jcs.102.2.273. [DOI] [PubMed] [Google Scholar]
- Waddle JA, Karpova TS, Waterston RH, Cooper JA. Movement of cortical actin patches in yeast. J Cell Biol. 1996;132:861–870. doi: 10.1083/jcb.132.5.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wente SR, Blobel G. A temperature–sensitive NUP116null mutant forms a nuclear envelope seal over the yeast nuclear pore complex thereby blocking nucleocytoplasmic traffic. J Cell Biol. 1993;123:275–284. doi: 10.1083/jcb.123.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wente SR, Blobel G. NUP145encodes a novel yeast glycine-leucine-phenylalanine-glycine (GLFG) nucleoporin required for nuclear envelope structure. J Cell Biol. 1994;125:955–969. doi: 10.1083/jcb.125.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wente, S.R., S.M. Gasser, and A.J. Caplan. 1996. The nucleus and nucleocytoplasmic transport in Saccharomyces cerevisiae. In The Molecular and Cellular Biology of the Yeast Saccharomyces. Vol. 3. J.R. Broach, E. Jones, and J. Pringle, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. In press.
- Wente SR, Rout MP, Blobel G. A new family of yeast nuclear pore complex proteins. J Cell Biol. 1992;119:705–723. doi: 10.1083/jcb.119.4.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese C, Wilson KL. Nuclear membrane dynamics. Curr Opin Cell Biol. 1993;5:387–394. doi: 10.1016/0955-0674(93)90002-8. [DOI] [PubMed] [Google Scholar]
- Wright R, Basson M, D'Ari L, Rine J. Increased amounts of HMG–CoA reductase induce “karmellae”: a proliferation of stacked membrane pairs surrounding the yeast nucleus. J Cell Biol. 1988;107:101–114. doi: 10.1083/jcb.107.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E, Skibbens RV, Cheng JW, Salmon ED, Bloom K. Spindle dynamics and cell cycle regulation of dynein in the budding yeast, Saccharomyces cerevisiae. . J Cell Biol. 1995;130:687–700. doi: 10.1083/jcb.130.3.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel U, Doye V, Tekotte H, Wepf R, Grandi P, Hurt EC. Nic96p is required for nuclear pore formation and functionally interacts with a novel nucleoporin, Nup188p. J Cell Biol. 1996;133:1141–1152. doi: 10.1083/jcb.133.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]