

Abstract
The events of myoblast fusion in Drosophila are dissected here by combining genetic analysis with light and electron microscopy. We describe a new and essential intermediate step in the process, the formation of a prefusion complex consisting of “paired vesicles.” These pairs of vesicles from different cells align with each other across apposed plasma membranes. This prefusion complex resolves into dense membrane plaques between apposed cells; these cells then establish cytoplasmic continuity by fusion of small areas of plasma membrane followed by vesiculation of apposed membranes. Different steps in this process are specifically blocked by mutations in four genes required for myoblast fusion. One of these genes, blown fuse, encodes a novel cytoplasmic protein expressed in unfused myoblasts that is essential for progression beyond the prefusion complex stage.
Although many examples are known of controlled cell fusion, from the development of the syncytial trophoblast in the placenta to the differentiation of osteoclasts, the molecular mechanisms controlling these events are not well understood. The fusion of myoblasts leading to the formation of skeletal muscle is of particular interest for three reasons. First, the fusion of myoblasts during development must be exquisitely controlled if the final muscles are to be patterned and sized correctly (Blau et al., 1993). Second, myoblasts fuse with mature muscle fibers during adult life as well, in response to either traumatic injury (Bischoff, 1979) or exercise (Schiaffino et al., 1979). The ability to influence this process would be of great therapeutic value. Finally, skeletal muscle is a prime target organ for gene therapy, as engineered myoblasts can be induced to fuse with mature muscle, forming a stable hybrid organ within the adult (Blau et al., 1993; Miller and Boyce, 1995).
The basic events surrounding muscle formation have been studied extensively (Abmayr et al., 1995; Ball and Goodman, 1985a ,b; Bate, 1993; Bischoff, 1978; Knudsen, 1992; Wakelam, 1985, 1988). In insects, myotubes form by fusion of myoblasts with specialized muscle precursor cells called muscle pioneers or founder cells, a subset of myoblasts that determine the final pattern of mature muscles (Ball et al., 1985; Bate, 1990; Ho et al., 1983; Rushton et al., 1995). New nuclei are added to existing muscles by subsequent fusion of additional myoblasts, since nuclei in myotubes are postmitotic (Ball and Goodman, 1985a ; Bate, 1993).
Myoblast fusion can be divided into a series of steps of differentiation, cell–cell recognition, adhesion, alignment, and membrane fusion (Knudsen and Horwitz, 1978; Wakelam, 1985). In the differentiation step, myoblasts begin to produce the proteins that make the cells competent to fuse. Myoblasts then locate and recognize an appropriate target for fusion, i.e., another myoblast, a previously extant myotube, or a pioneer cell. The cells adhere to each other through a specific calcium-dependent process (Knudsen and Horwitz, 1977). After adhesion, they assume a bipolar morphology and align along their long axes. The aligned plasma membranes come in close apposition and local membrane fusion events form small areas of cytoplasmic continuity between the cells. The excess plasma membrane in the fusion area then vesiculates (Przybylski and Blumberg, 1966; Rash and Fambrough, 1973) while the plasma membrane outside of the fusion area remains intact, resulting in the formation of a single multinucleated myotube. The remnants of the excess plasma membrane material are eliminated through an unknown process.
Although muscle development has been thoroughly described in vivo at the ultrastructural level in several species (reviewed by Knudsen, 1992), observation of specific fusion events are rare since each muscle in these organisms develops asynchronously over a period of weeks or months. This makes the observation of specific steps in the pathway of fusion between two cells difficult.
Ever since the observation that myoblast fusion occurs in primary tissue cultures (Holtzer et al., 1958; Firket, 1958; Cooper and Konigsberg, 1961), most ultrastructural and biochemical work has centered on in vitro systems in which cultures of myoblasts can be synchronized for fusion by lowering and subsequent readdition of extracellular calcium (Shainberg et al., 1971), or through the use of fusion-competent cell lines (Yaffe, 1968). Some progress has been made in identifying the proteins and signaling molecules involved in fusion (reviewed by Knudsen, 1992). These studies have also produced exciting observations on possible intermediate steps in the process of fusion (Engel et al., 1985; Gerson et al., 1976; Kalderon and Gilula, 1979; Lipton and Konigsberg, 1972; Rash and Fambrough, 1973), but the environment and morphology of cultured myoblasts differ greatly from the in vivo state. In addition, the established myogenic cell lines vary in fusion kinetics and morphology, from each other and from primary myoblast cultures (Wakelam, 1988).
Previous progress notwithstanding, the precise molecular mechanisms involved in myoblast fusion remain a mystery. Only a handful of the many proteins involved in the process have been unequivocally identified and many important questions remain unanswered. What is the nature of the signal identifying an appropriate target cell for fusion? What steps are required at the molecular level between alignment of pairs of myoblasts and the fusion and final breakdown of the plasma membranes? Which proteins and other molecules mediate those steps, and how do those molecules interact with each other?
The fruit fly Drosophila melanogaster is an excellent organism for the study of muscle development. The embryonic body wall musculature is simple, consisting of an array of only 30 individually identified muscle fibers repeated exactly in each abdominal hemisegment, each of which is formed by the fusion of between three and twenty myoblasts. The development of the Drosophila larval musculature has been well described at the light level (Bate, 1990). As in higher metazoans, myoblast fusion occurs asynchronously. Myoblasts in the ventral region of the embryo fuse earlier than those more dorsal, and myoblasts closer to the epithelium fuse before the more internal myoblasts. In flies, however, the entire process of muscle formation takes hours rather than days or weeks. Thus, many examples of fusion events in various stages of completion can be observed in single thin sections of developing muscle. This makes Drosophila a particularly attractive organism in which to define the ultrastructural steps of the myoblast fusion process.
Classical genetic mutant analysis is a powerful and specific tool for the identification of proteins involved in developmental and cell biological processes. Besides identifying novel proteins and demonstrating their role in specific processes, phenotypic analysis can “freeze” cells in intermediate steps of the process, helping to define the steps in a genetic and/or biochemical pathway. To date, two Drosophila mutants have been identified with specific defects in myoblast fusion: rolling stone (Paululat et al., 1995) and myoblast city (Rushton et al., 1995). We describe a third, blown fuse, in this paper. At least one more can be inferred from analysis of chromosomal deficiencies (Drysdale et al., 1993). In addition, expression of a dominant negative form of Drac1 in developing mesoderm blocks myoblast fusion (Luo et al., 1994). The phenotypes of these mutants at the light microscopic level have been well described, but no ultrastructural analysis has been published before this report.
By combining the advantages of classical and molecular genetic analysis with light and electron microscopy (EM) in Drosophila, we have identified new intermediate steps in the fusion process. We also describe the cloning and expression pattern of blown fuse, a gene essential for myoblast fusion. We propose a pathway for the steps of myoblast fusion and identify the step at which each mutant blocks this pathway.
Materials and Methods
Fly Stocks
The myoblast city stock mbcC1 (Rushton et al., 1995) was supplied by Susan Abmayr (Pennsylvania State University, State College, PA). rolling stone stocks (Paululat et al., 1995) were supplied by Renate RenkawitzPohl (Marburg, Germany). UAS:Drac1G12V flies (Luo et al., 1994) were supplied by Liqun Luo (Stanford University, Stanford, CA).
Histology
We visualized myoblasts and developing myotubes for light microscopy by immunochemical staining with a monoclonal antibody raised against Drosophila muscle myosin (FMM5, Kiehart and Feghali, 1986), and polyclonal antisera raised against a Blown Fuse fusion protein (see below). By adapting methods used for immunoelectron microscopic labeling, we were able to obtain strong staining of embryos dissected and then fixed by the periodate-lysine-paraformaldehyde (PLP)1 protocol of McLean and Nakane (1974).
Embryos (0–12 h) were dechorionated, rinsed with heptane, transferred to double-stick tape, placed inside a silicone rubber well on a polyl-lysine–coated slide, manually devitellinzed, and filleted. To retain antigenicity and morphology, the embryos were fixed 45 min at room temperature (RT) with PLP. After PLP fixation, the embryos were rinsed with 100 mM sodium cacodylate buffer (pH 7.4) and then fixed for 10 min at RT with 0.05% glutaraldehyde in sodium cacodylate buffer. After fixation, the embryos were rinsed with 100 mM sodium phosphate buffer (pH 7.4) containing 0.05% saponin (PO4/saponin) and treated to quench endogenous peroxidase activity by incubation for 10 min at RT in PO4/saponin buffer with 1 mM sodium azide and 0.01% H2O2. The embryos were then rinsed with PO4/saponin buffer and incubated in blocking solution (PO4/ saponin buffer containing 5% normal goat serum and 1% bovine serum albumin), with 50 mM glycine added to quench remaining aldehyde groups. Embryos were then incubated sequentially with rat antiserum to Blow (1: 500 or 1:1,000) or a 1:10 dilution of a mouse monoclonal supernatant raised against muscle myosin (Kiehart and Feghali, 1986) in blocking solution, followed by goat anti–rat or anti–mouse IgG conjugated to HRP (1: 200) in blocking solution. All antibody incubations were for 1 h at RT and were followed by extensive washes with PO4/saponin buffer.
The embryos were developed in PO4/saponin buffer containing 0.3 mg/ ml diaminobenzidine and 0.01% H2O2 and allowed to react for 10 min at RT. Embryos were mounted after staining and photographed on a Zeiss Axiophot microscope.
Conventional Electron Microscopy
Mutant embryos collected from blow2/CyOβgal, mbcc 1/TM3βgal, and rost15/CyO7.1 stocks were screened and processed for electron microscopy as described by Lin et al. (1994). Embryos expressing Drac1G12V were obtained from a UAS-Drac1G12V × 24B-GAL4 cross and did not have to be screened.
Cloning and Sequencing of blown fuse
We mapped the blow gene to 43E by deficiency analysis, and determined that blow is identical to l(2)43Eb (Heitzler et al., 1993) by its failure to complement the original BB034 allele. We obtained a P element lethal stock, P3427, from the Berkeley Drosophila Genome Center. P3427 is a zygotic lethal allele of scraps (Field, C.M., B.M. Alberts, and S.K. Doberstein, manuscript in preparation), which maps close to blow (Heitzler et al., 1993). We isolated genomic DNA flanking the scraps3427 P element by the inverse polymerase chain reaction (Dalby et al., 1995) and used that DNA to screen a genomic DNA library in λDASH at high stringency. cDNAs were isolated from a 9–12-h embryo λgt11 library (Zinn et al., 1988) using the genomic phage inserts as probes. Genomic Southern and RNA blots were performed as described by Sambrook et al. (1989).
The phage inserts were subcloned into pBluescriptII KS+ (Stratagene, La Jolla, CA). We sequenced the cDNAs and some genomic subclones using the dideoxynucleotide chain termination method (Sanger et al., 1977) using the AutoRead kit (Pharmacia LKB Biotechnology, Piscataway, NJ) following the protocol of the manufacturer. Reactions were analyzed on a Pharmacia/LKB automated laser fluorescent DNA sequencer. Two separate full-length cDNAs were sequenced completely on both strands. Sequences were compiled using Intelligenetics LaserGene software. Database searches were performed using the BLAST program (Altschul et al., 1990) as implemented on the National Center for Biotechnology Information World Wide Web page.
RNA localization in embryos was performed exactly as described by Tear et al. (1996). Mutant lines were counterstained with mouse anti–βgalactosidase after RNA localization to reveal and eliminate embryos containing β-galactosidase marked balancer chromosomes.
Antibody Production
We raised polyclonal antisera against a fusion protein consisting of Blow amino acids 136-605 fused to glutathione-S-transferase (Smith and Johnson, 1988). The resultant fusion protein was insoluble, and we purified the inclusion bodies (Harlow and Lane, 1989) and immunized rats and mice. Animals were boosted with antigen and bled on alternating weeks in a two week cycle.
Results
Normal Myoblast Fusion
We examined myoblasts and developing myotubes as early as the middle of stage 12, when muscle myosin becomes detectable by immunochemical staining. At this stage, staining appears first in a subset of ventral myoblasts, which appear in each segment as large clusters of teardrop-shaped cells just dorsal to the central nervous system. Muscle myosin is expressed in some Drosophila myoblasts before fusion begins, in distinction to vertebrates, where muscle myosin is expressed only in myoblasts that have already begun the fusion process. Myotubes become visible in the ventral region by early stage 13, with most myotubes concentrated adjacent to the epidermis (Fig. 1 A). Large numbers of unfused myoblasts are present attached on the interior surface of the myotubes. By stage 14, the ventral myotubes have apparently attached to their epidermal insertion sites, and some myoblasts remain unfused in the region (Fig. 1 B). The ventral muscles are essentially complete by stage 15 (Fig. 1 C), with very few unfused myoblasts present. Fusion in more dorsal regions begins later and continues into stage 16 (Fig. 1 D).
Figure 1.
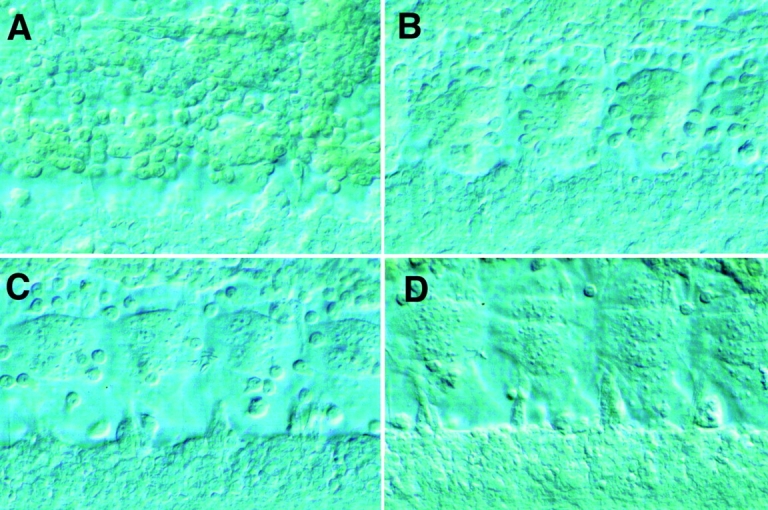
Myoblast fusion in the developing Drosophila embryo. Light level micrographs of myoblast fusion in the ventral muscle region of wild-type Drosophila embryos. Developing muscles are imaged by Nomarski optics, and the plane of focus is close to the epidermis. (A) Wild-type early stage 13 embryo. Small early myotubes are present, with many unfused myoblasts attached to the surface of the myotubes. (B) Wild-type stage 14 embryo. (C) Wild-type stage 15 embryo. Myotubes are substantially larger, with few unfused myoblasts remaining. (D) Wild-type stage 16 embryo.
Since the fusion process begins asynchronously in ventral, lateral, and dorsal muscle regions, and the individual myoblasts fuse asynchronously within those regions, it is possible to see various stages of cell fusion within a single cross section through an abdominal segment (Fig. 2). Unfused myoblasts are teardrop-shaped, with a single pseudopod, and are morphologically very similar to vertebrate myoblasts.
Figure 2.
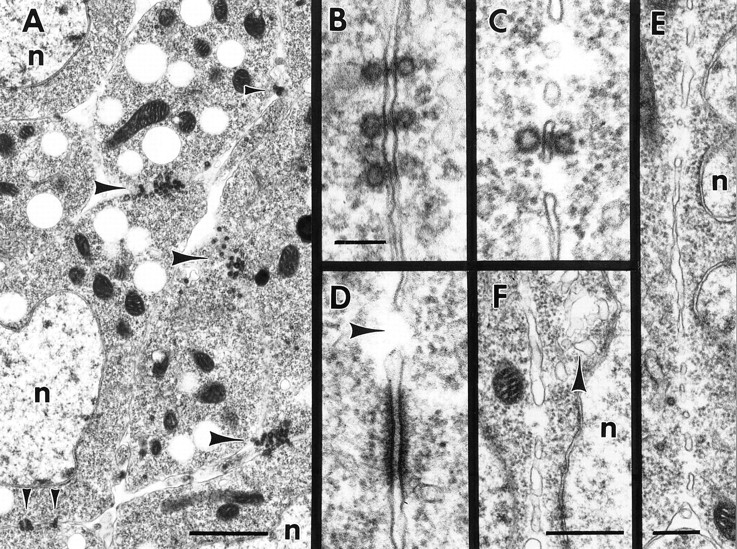
Ultrastructure of intermediate steps in myoblast fusion. Electron micrographs of wild-type myoblast fusion in early stage 13 embryos. All stages of the fusion process occur simultaneously in various parts of the developing musculature. (A) Myoblasts in early stage of fusion. Note prefusion complexes at points of cell–cell contact (arrowheads); n indicates myoblast nuclei. (B) Three sets of paired vesicles. Note electron-dense material in the extracellular space between pairs of vesicles. (C) Paired vesicles oriented across a vesiculating pair of plasma membranes. (D) An electron-dense plaque near a region of actively fusing membrane; note fusion pore (arrow). (E) Fusion pores in a vesiculating plasma membrane. The cytoplasm within and beneath the pore is free of staining material such as ribosomes. (F) Later stage vesiculating plasma membrane. The membrane sacs have increased in width and a group of irregular clear vesicles is present (arrowhead). Bars: (A) 1 μm; (B–D) 100 nm; (E) 250 μm; (F) 500 μm.
Contact sites between unfused myoblasts frequently have dramatic concentrations of vesicles of a ∼40-nm (38.1 ± 2.6 nm, n = 27) diameter near the cytoplasmic face of each of the juxtaposed plasma membranes (Figs. 2, A–C and 3). These vesicles have a distinctive and thick electron dense margin, and although similar in general size to synaptic vesicles, are clearly distinguishable from the latter based on their characteristic electron density. The vesicles are exclusively present in myoblasts and their fusion partners (pioneer cells and myotubes). Based on serial reconstructions of prefusion complexes, the groups range in a number of up to 50 vesicles per cell at the contact point (Fig. 3). Since as many as six single myoblasts can form pseudopodial cell contacts at the same site, it is not uncommon for the same prefusion complex to range across three or four cells. Since muscle pioneer cells are apparently morphologically indistinguishable from other myoblasts, it is not possible for us to determine how many of the myoblasts within a group are pioneers.
Figure 3.
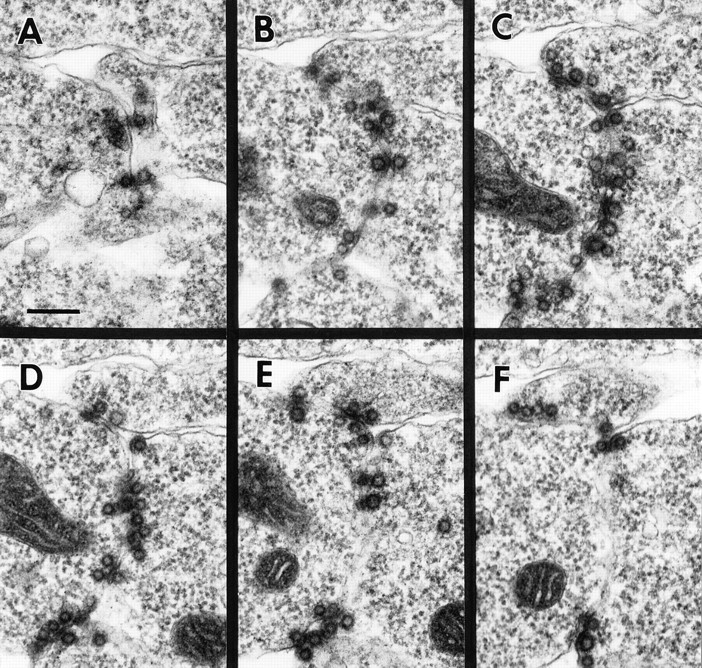
The prefusion complex contains paired vesicles. Serial section electron micrographs through a prefusion complex in a wild-type stage 13 embryo. This complex contains about 45 pairs of vesicles distributed among three cells. Bar: (A) 200 nm.
Pairs of the 40-nm vesicles, one in each cell, line up with each other across the apposed plasma membranes (Fig. 2, B and C). The paired vesicles appear to contact the internal leaflet of their respective plasma membranes, and electron-dense material is present associated with the plasma membranes and in the extracellular space between paired vesicles. Occasionally, a single unpaired vesicle comes in close contact with the plasma membrane without a partner in the opposite cell. The plasma membrane beneath these single vesicles has no electron-dense extracellular material associated with it, suggesting a role for the extracellular material in aligning the pairs of vesicles. Groups of paired vesicles can spread across as much as 1 μm2 of the cell surface. We hereafter refer to groups of paired vesicles and the associated electron dense material as “prefusion complexes.”
We observed electron-dense stretches of 10-nm-thick material along apposed plasma membranes, extending for ∼500 nm along the cytoplasmic face of the membranes (Fig. 2 D). These regions are similar to membrane plaques described previously in vertebrate myoblasts (Rash and Fambrough, 1973). There is also substantial electrondense material in the extracellular space between cells in the plaques. Plaques are rare relative to prefusion complexes, and we have observed them in areas of plasma membrane breakdown (Fig. 2 D). The electron-dense material in the plaques appears similar to the material making up the paired vesicles, and we suspect that the plaques result from fusion of the paired vesicles with the plasma membranes.
After the initial cell–cell contact the cells elongate and align with each other. The cells establish cytoplasmic continuity through multiple small zones (pores) of local fusion between the apposed plasma membranes (Fig. 2, C–F). The plasma membranes vesiculate along the zone of contact, forming sacs of membrane enclosing the previously extracellular space (Fig. 2 E). Paired vesicles are sometimes seen associated with the membrane sacs (Fig. 2 C). The pore regions and cytoplasm immediately beneath the fusing plasma membranes remain free of staining cytoplasmic components such as ribosomes.
The plasma membrane sacs become progressively rounder in profile as the membranes break up. Groups of irregular clear vesicles are occasionally present in the region beneath late stage vesiculating plasma membranes (Fig. 2 F). These vesicle groups may be the recycling system for excess plasma membrane components after fusion.
Mutant Phenotypes
myoblast city.
Myoblasts in embryos homozygous for mutations in the myoblast city (mbc) gene fail to fuse, and form loose clusters of myosin-positive cells in locations roughly corresponding to the ventral, lateral, and dorsal muscle groups (Fig. 4 C and 5 B). Many single myoblasts are removed during dissection of the embryo unless great care is taken. The myoblasts express myosin robustly, and usually have the typical single pseudopodium seen in electron micrographs of normal myoblasts before alignment.
Figure 4.

Mutations in genes that are essential for myoblast fusion. Light level micrographs of myoblast fusion in the ventral muscle region of wild-type (A–B) and mutant (C–F) Drosophila embryos. Myoblasts are stained with anti-myosin monoclonal antibody FMM5 (Kiehart and Feghali, 1986). The plane of focus is more superficial (closer to the gut) than in Fig. 1 to discern individual unfused myoblasts. (A) Wild-type stage 13 embryo. Fusion has begun and the early ventral myotubes are beginning to extend towards their attachment sites. (B) Wild-type stage 14 embryo. Myotubes have attached to the epidermis and unfused myoblasts are present on the superficial surface of the myotubes. (C) mbcC1 stage 14 embryo. Compare to B; little or no fusion has occurred. (D) blow2 stage 14 embryo. The myoblasts are more tightly clustered than in mbc mutants. (E) rost15 stage 14 embryo. The morphology of the unfused myoblast clusters is different from other mutants due to the alignment of myoblasts close to the epidermis. (F) Drac1G12V:GAL4-24B stage 14 embryo. Most of the unfused myoblasts have been removed by macrophages. Bar, 25 μm.
Typical of mbc myoblasts is the nearly complete absence of prefusion complexes, consistent with the prefusion complex forming after recognition and/or adhesion of myoblasts to target cells (Fig. 6 A). Although an occasional complex can be seen at apparently random locations, the number of prefusion complexes is reduced by at least 90%. The few prefusion complexes that do exist contain roughly wild-type numbers of paired vesicles, suggesting that the defect in mbc myoblasts lies upstream of the actual assembly of the prefusion complex. We observed no electrondense plaques between myoblasts in mbc embryos.
Figure 6.
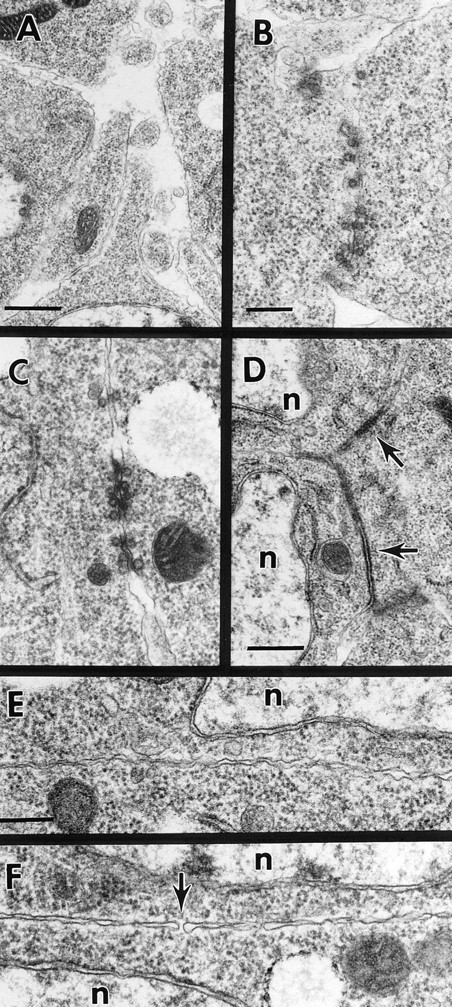
Different mutants block specific steps in myoblast fusion. (A) Representative cell–cell contacts between myoblasts in an early stage 13 mbcC1 mutant embryo. Prefusion complexes are absent. (B) Prefusion complex in a stage 13 blow2 mutant embryo. The complexes in this mutant are indistinguishable from those in wild-type embryos. (C) Prefusion complex in a stage 13 rost4 mutant embryo. The prefusion complexes in this mutant are also indistinguishable from wild type. (D) Membrane plaques (arrows) between three myoblasts in a rost15 embryo. n indicates myoblast nuclei. (E) Close apposition of plasma membranes in a stage 14 rost15 embryo. (F) Abortive plasma membrane fusion in a Drac1G12V/24B embryo. A single fusion pore is visible (arrow). At certain places the apposed plasma membranes are so close, they are indistinguishable from a single membrane. Bars: (A) 500 nm; (B and C) 250 nm; (D) 500 nm; (E and F) 250 nm.
At both the light microscopic and EM level, mbc myoblasts do not appear to align and become elongate as wildtype myoblasts do. In electron micrographs of early stage 13 embryos, mbc myoblasts have characteristic teardrop morphology, with a single pseudopod per cell. However, there appears to be slightly more extracellular space between myoblast cell bodies in mbc embryos. By stage 14, there is no sign of specific attachment sites for unfused myoblasts, as in blown fuse embryos (see below) although clusters are present in the locations corresponding to the main muscle groups (Fig. 5 B). We hypothesize that the random orientation of myoblasts relative to pioneer cells is due to a failure of one of two processes, either target recognition or cell adhesion. By the end of stage 16, most unfused myoblasts have been cleared by macrophages, revealing a rough scaffolding of muscle pioneer cells, some binucleate, which are apparently unaffected by the mutation (Rushton et al., 1995).
Figure 5.
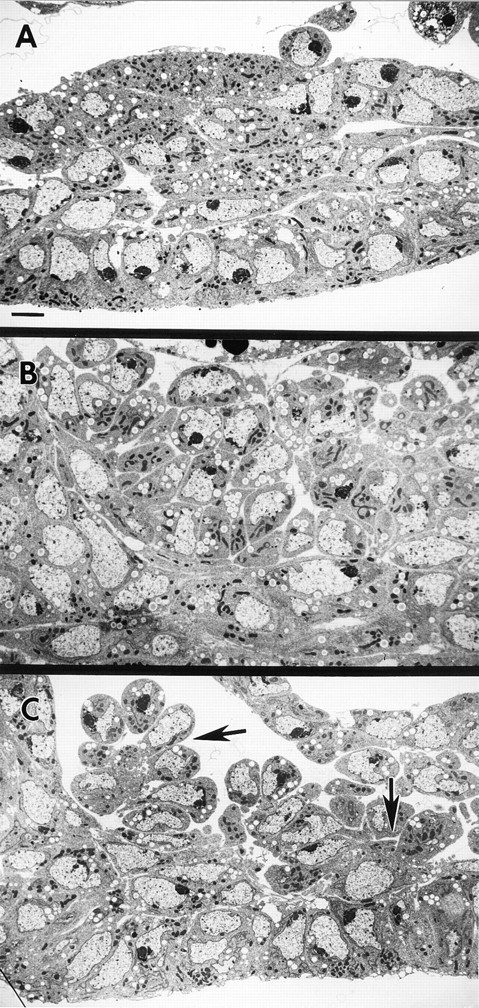
mbc is required for recognition and/or attachment of pioneer cells by myoblasts. Electron micrographs of ventral muscle region in stage 14 homozygous embryos. (A) Wild-type embryo. The ventral nerve cord is to the left side of the frame. (B) mbcC1 embryo. The unfused myoblasts are oriented in an apparently random manner, indicating that recognition and/or attachment of myoblasts to pioneer cells is disrupted in this mutant. (C) blow2 embryo. Note groups of myoblasts attached to single pioneer cells (arrows). Bar, 2 μm.
blown fuse.
In blown fuse (blow) embryos, myoblasts fail to fuse, forming clusters of teardrop-shaped cells in roughly the same locations as in mbc embryos (Figs. 4 D and 5 C). The myoblasts are less prone to removal during dissection compared to mbc myoblasts. In early stage 13 blow embryos, normal numbers of prefusion complexes are present, and we observe no change in morphology or number of paired vesicles in the complexes (Fig. 6 B). We observed no electron-dense plaques in blow embryos. We hypothesize that blown fuse blocks the formation of normal electron-dense plaques from prefusion complexes, and that the complexes disperse after inactivity. However, the relative scarcity of plaques in wild-type embryos raises the alternative possibility that plaques may exist in the blow mutants but are either more scarce or shorter-lived than in wild-type embryos.
By stage 14, the myoblasts have sorted out into groups of teardrop-shaped cells that share attachment sites on a single pioneer cell (Fig. 5 C), with a morphology reminiscent of bunches of grapes. The “bunch of grapes” morphology suggests that these myoblasts are competent to recognize and adhere to pioneer cells much as wild-type myoblasts do. No prefusion complexes are present by stage 14. It is not clear whether the prefusion complexes complete their function before their disappearance or whether they are blocked at some stage before functioning and then disperse after some time of inactivity. As in mbc mutants, most unfused cells are cleared by macrophages by the end of stage 16, and the scaffold of muscle precursors is apparently unaffected.
rolling stone.
In rolling stone (rost) embryos, paired vesicles are present at wild-type levels during early stage 13 and disappear by stage 14 as they do in wild type (Fig. 6 C). However, the plasma membrane never vesiculates (Figs. 4 E and 6 E). Instead, extensive electron-dense plaques are present along apposed plasma membranes between pairs of myoblasts during stage 13 (Fig. 6 D). These are much more common and are often significantly larger than the electron-dense plaques in wild-type embryos, becoming nearly ubiquitous in later stage myoblasts. Except for their larger size and higher frequency, the plaques in rost embryos are indistinguishable from plaques in wild-type embryos.
By stage 14 the electron-dense plaques have disappeared, and groups of myoblasts in rost embryos are aligned in the same positions as mature myotubes in wildtype embryos. The cells are no longer teardrop-shaped, and at low magnification the cell clusters close to the epidermis can be mistaken for normal myotubes. However, little fusion has occurred, and although plasma membranes between aligned myoblasts are much more closely apposed than in wild-type unfused aligned myoblasts, the membranes are intact (Fig. 6 E). It seems likely that substantial removal of membrane glycoproteins is necessary before close apposition of plasma membranes whether by proteolysis or by physical movement of those proteins. Protein removal is probably completed in rost mutants, explaining the abnormally close plasma membrane apposition after dispersal of the plaques.
Drac1G12V.
Substitution of valine for glycine at position 12 in rac family proteins creates a dominantly active gainof-function form of the molecule (Ridley et al., 1992), and expression of Drac1G12V in developing myoblasts blocks myoblast fusion (Luo et al., 1994). We used the GAL424B line as described by Luo et al. (1994) to drive expression of Drac1G12V exclusively in myoblasts.
In embryos which contain myoblasts expressing Drac1G12V, myoblasts distribute themselves in groups as in the other mutants described here and generally fail to fuse, but the clusters of unfused cells have a markedly different morphology from other fusion mutants (Fig. 4 F). We estimate that roughly 10% of myoblasts in these embryos do fuse, so the rudimentary myotubes are substantially more robust in this mutant than in the loss-of-function mutants we studied. The remaining unfused cells have a more elongate morphology, and tend to be more spread out along each other in groups of cells relative to the other mutants we observed.
Many prefusion complexes are present in Drac1G12V embryos, in wild-type abundance and locations. Electrondense membrane plaques are present, and the cells appear to elongate and align themselves normally. However, apposed plasma membranes between fusion partners have aberrant morphology, with few or no pores (Fig. 6 F). The membranes come very close or into direct contact with each other, with many small sections of paired membranes in such close contact as to be indistinguishable from single bilayers. A few examples of single fusion pores can be found, so in some cases the cytoplasm of pairs of myoblasts is technically continuous. However, in only a few cases does the plasma membrane vesiculate in any serious way. We suggest that Drac1G12V blocks a very late step in plasma membrane pore formation.
The stage 14 membranes in rost mutants are very similar in appearance to stage 13 plasma membranes in Drac1G12V embryos (Fig. 6, E and F). By stage 14, many if not most myoblasts in Drac1G12V embryos are either dead, dying, or have already been cleared by macrophages (Fig. 4 E). This might be due to premature cell death caused by membrane instability. Rac1 has been shown in vertebrate cells to mediate cytoskeletal/membrane dynamic interactions such as membrane ruffling (Ridley et al., 1992). Perhaps the combination of fusing plasma membranes and interfering membrane ruffling produces an inherently unstable set of membranes, leading to premature lysis of the abortively fused cells.
Interpretation of a possible role for wild-type Drac1 in myoblast fusion is problematic. Loss of function alleles of Drac1 have not yet been reported. Although dominant negative forms of the protein appear to cause aberrations in myoblast fusion when expressed in myoblasts, the effects are subtle and variable (Luo et al., 1994). It seems possible that the presence of a constitutively active form of Rac might confuse membrane dynamics in myoblasts sufficiently to block the process without having a specific role in fusion. However, the constitutively active form of Dcdc42, a closely related protein, does not block fusion, suggesting some specific role for Drac1 (Luo et al., 1994). Further analysis of the role of Drac1 in fusion awaits better genetic tools.
Cloning and Expression of blown fuse
blown fuse is the first gene cloned that is essential for myoblast fusion as identified by classical genetic analysis. blown fuse was originally isolated as BB034 in a screen for homozygous lethal mutations defective in motoneuron axon guidance (Van Vactor et al., 1993). Staining of muscle precursors with anti-myosin antibodies (see above) revealed that the motoneuron guidance defects are a secondary consequence of a myoblast fusion defect, since intact muscles act both as substrates and targets for growing axons.
We determined by deficiency mapping that BB034 is located in 43E and is allelic to l(2)43Eb (Heitzler et al., 1993). The 43A-E region has been saturation mapped for lethal mutations (Heitzler et al., 1993), and none of the other mutations in the region cause defects in myoblast fusion. We renamed the locus blown fuse, and designated l(2)43Eb1 as blow1 and BB034 as blow2. Complementation analysis of lethal P elements in the 43 region uncovered a P element zygotic lethal allele of scraps, which is adjacent to blow by deficiency analysis (Heitzler et al., 1993). We recovered ∼25 kb of genomic DNA flanking this P element (Fig. 7 A).
Figure 7.
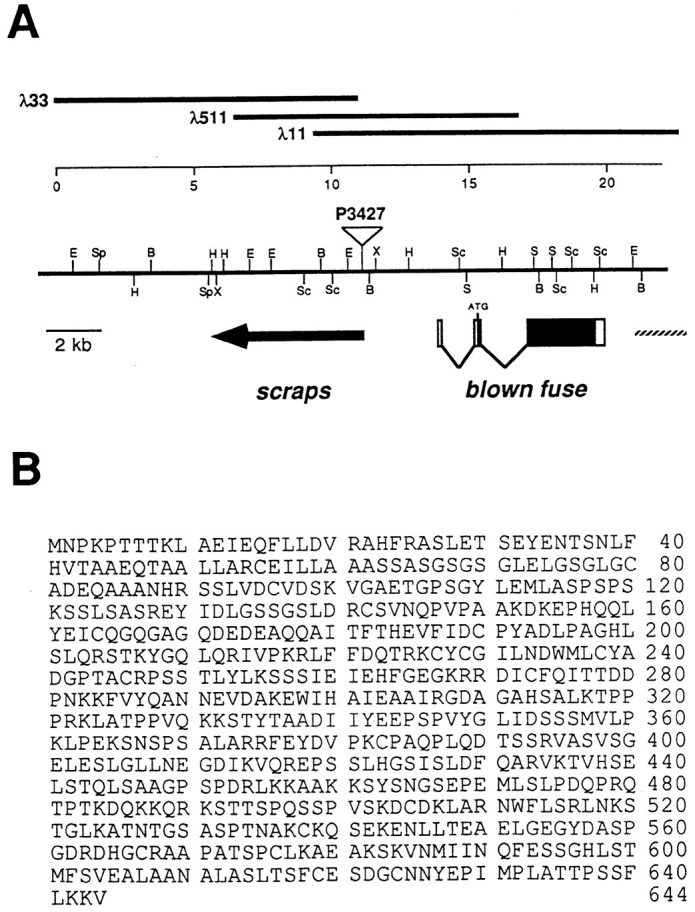
Genomic organization and sequence of the blown fuse gene. (A) Genomic map of the region in 43E containing scraps and blown fuse. At top, numbers represent scale in kilobases. Thick lines indicate representative λ phage clones isolated during the chromosomal walk. Single letter abbreviations indicate restriction sites: B, BamHI; E, EcoRI; H, HindIII; S, SalI; Sc, SacI; Sp, SpeI; X, XbaI. The insertion site of scrapsP3427 is indicated by an open triangle. Beneath the chromosomal map, the location of the scraps gene is indicated by a solid arrow. The blown fuse gene is represented by boxes, with open boxes indicating noncoding regions and solid boxes indicating the coding region. Part of an unidentified third gene is indicated by a hatched line. (B) Amino acid sequence of the Blown Fuse protein.
We isolated three distinct groups of cDNAs from this region by using the genomic DNA to probe an embryonic cDNA library (Zinn et al., 1988; Fig. 7 A). The P element is inserted into the 5′ end of the scraps gene, which encodes a single 4-kb transcript which is expressed ubiquitously in embryos. There exist both maternal effect and zygotic lethal alleles of scraps, and both alleles of blow complement all existing alleles of scraps. blown fuse is the nearest neighboring gene and is expressed solely in myoblasts just before and during myoblast fusion (see below). The third transcript has at least two splice forms, of 2.8 kb and 3.4 kb, and gives no signal in in situ hybridization to whole embryos.
The blown fuse gene contains two introns and produces a single 2.6-kb transcript that encodes a 69.5-kD protein (Fig. 7 B). The Blow protein has no significant sequence similarity to any known proteins. There is no signal sequence and no significant hydrophobic stretches, consistent with an intracellular localization for the protein (see below).
The blow transcript begins to be expressed in late stage 10 in 13 distinct clusters of mesodermal cells, presumably myoblast and pioneer cell precursors (Fig. 8 A). During stage 11 and 12 these clusters resolve into segmental stripes of fusing myoblasts (Fig. 8 B). Expression is strongest during early stage 13 (Fig. 8, C and D), is reduced by late stage 13 (Fig. 8 E), and is absent by the end of stage 14.
Figure 8.

Expression of blow mRNA. Expression pattern of blow mRNA. (A) In situ hybridization of the blow cDNA to a whole mount stage 10 embryo. The mRNA is expressed in 12 cell clusters in the developing mesoderm. (B) In situ hybridization to a stage 12 embryo. (C) In situ hybridization to a stage 13 embryo. The mRNA is expressed at high levels in myoblasts, and is not expressed in other cells. (D) In situ hybridization to a stage 13 embryo, ventral view. (E) In situ hybridization to a stage 14 embryo. Expression level is lower than in previous stages. (F) In situ hybridization to a homozygous blow2 stage 13 embryo. No mRNA is detectable.
We generated polyclonal antibodies to a fragment of the Blow protein which detect a single band of ∼70 kD on protein immunoblots (data not shown). These antibodies detect the Blow protein in unfused myoblasts (including pioneer cells) during stages 11-14, with small amounts of residual protein in early myotubes (Fig. 9, A and B). The protein is located in the cytoplasm, as predicted by the amino acid sequence, and is excluded from the nucleus (Fig. 9 C).
Figure 9.
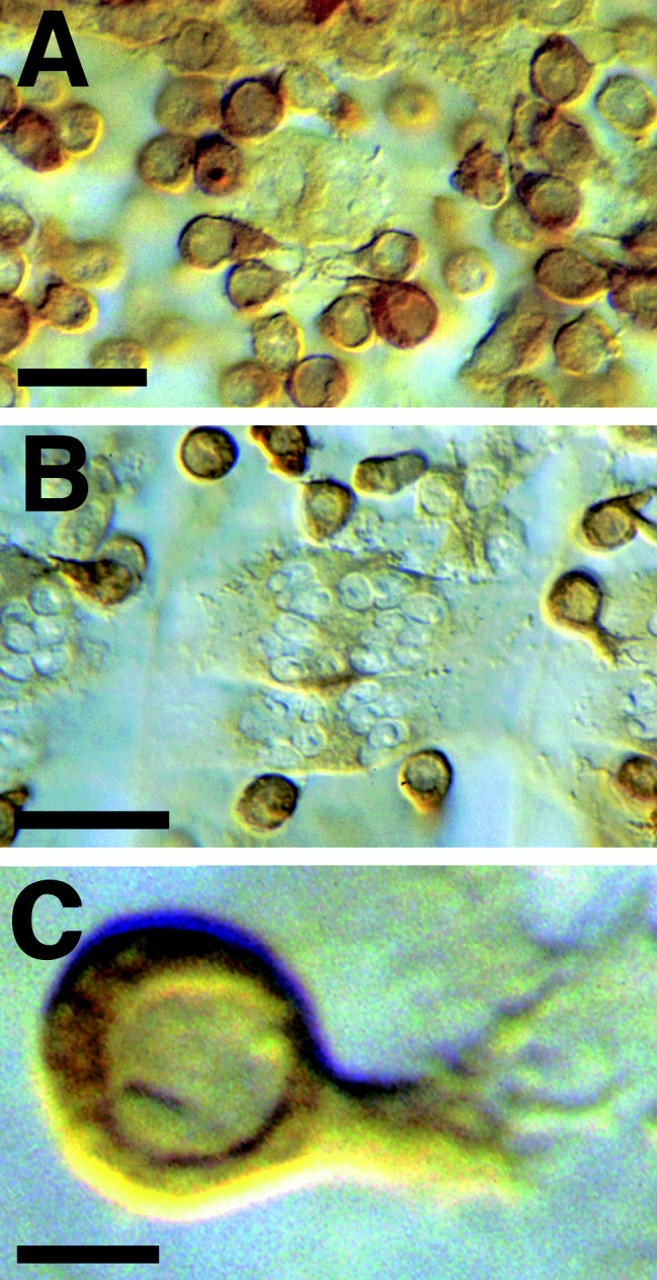
Localization of Blow protein. Subcellular localization of Blow protein. (A and B) Anti-Blow staining in a dissected stage 13 wild-type embryo. Little protein is present in myotubes relative to the level in unfused myoblasts. (C) Anti-Blow staining in a wild-type myoblast. The protein is distributed evenly throughout the cytoplasm of the myoblasts and is excluded from the nucleus. Note the large single pseudopodium. Bars: (A and B) 10 μm; (C) 2 μm.
The blow transcript is absent in blow2 embryos as determined by in situ hybridization with a blow cDNA probe (Fig. 8 F), while transcript levels in blow1 appear to be normal (data not shown). Antibody staining of mutant embryos reveals a reduced level of protein in embryos homozygous for blow1, and no staining in embryos homozygous for either blow2 or Df(2L)88/07×4 (a small deficiency removing both blow and scraps, data not shown), consistent with blow2 being a transcript and protein null allele. The histological phenotypes of blow1 and blow2 are indistinguishable. There are no large chromosomal rearrangements in either allele as determined by genomic Southern blots.
Discussion
Drosophila larval musculature, like skeletal muscle in vertebrate embryos, forms in the embryo by the fusion of mononuclear myoblasts to produce syncytial myotubes (for review see Bate, 1990, 1993). After recognizing a partner for fusion, pairs of myoblasts establish a unique organelle we term the prefusion complex, which consists of groups of paired vesicles (one vesicle in each cell, aligned across closely apposed plasma membranes) and associated electron dense material, both inside and outside the cells. The complex resolves into electron-dense plaques along the plasma membranes of the apposed cells, most likely by fusion of the paired vesicles with their respective plasma membranes in response to a signal. The fusing cells align along their long axes, and pores form between the apposed plasma membranes. The plasma membranes vesiculate along their shared lengths, and the plasma membrane remnants are disposed of and presumably recycled.
We analyzed embryos mutant in four different genes essential for fusion: mbc, blow, rost, and Drac1G12V. Each of these mutations blocks a different step in the fusion process. We cloned one of these genes, blow, which encodes a novel cytoplasmic protein expressed in unfused myoblasts whose phenotype suggests an important role in normal functioning of the prefusion complex.
The myoblast fusion process in the Drosophila embryo shares many characteristics with myoblast fusion in vertebrates. The major steps of differentiation, recognition, adhesion, alignment, and plasma membrane breakdown previously described for vertebrates all occur in the fly embryo. Since embryonic fusion occurs over such a short time span in Drosophila, many fusions occur simultaneously in each segment. This organism therefore allows us the additional benefit of being able to document multiple fusion events simultaneously.
Although only a few proteins have been unequivocally implicated in myoblast fusion in vivo (such as Blow), several classes of macromolecules have been suggested to be essential for fusion based on in vitro studies of vertebrate myoblasts. These include cell adhesion molecules (Knudsen et al., 1990a ,b; Mege et al., 1992; Rosen et al., 1992), metalloproteases (Couch and Strittmatter, 1983, 1984; Knudsen, 1985; Yagami-Hiromasa et al., 1995), phosopholipases (Wakelam, 1983; Wakelam and Pette, 1982, 1984), and calmodulin (Bar-Sagi and Prives, 1983; Knudsen, 1985). It remains to be seen whether mutations in genes encoding the Drosophila homologues of these proteins influence myoblast fusion in vivo.
Intracellular fusion of vesicles during vesicle sorting and both endo- and exocytosis has been extensively studied (reviewed in Rothman and Warren, 1994), and many proteins essential for the process have been identified. We expect that few or none of the proteins required for intracellular membrane fusion will also be involved in intercellular fusion, since in the intracellular case, membranes fuse with their cytoplasmic faces interacting first, while in intercellular fusion the extracellular face (which is topologically identical to the lumen of intracellular vesicles) fuses first.
Paired Vesicles and the Prefusion Complex
In this paper we describe the discovery of distinctive groups of paired vesicles at sites of myoblast–myoblast contact. The behavior of these vesicles is unprecedented, with pairs of vesicles from different cells aligning with each other across a pair of plasma membranes. We believe that the paired vesicles are of prime importance to later steps in the myoblast fusion process since mbc myoblasts (which have no prefusion complexes) also lack electron-dense plaques, and do not align or fuse. Vesicles with electrondense material along their cytoplasmic surfaces have been reported in primary cultures of quail myoblasts (Lipton and Konigsberg, 1972) and in the muscle cell line L6 (Engel et al., 1985). The pairing behavior and the electrondense material between cells were not described in either case. The quail vesicles were shown to fuse with the plasma membrane, and in at least one case, a pair of those vesicles in apposed cells were shown in the act of fusing simultaneously with their respective plasma membranes (Lipton and Konigsberg, 1972). It is unclear whether the vesicles described by these previous workers are analogues of the paired vesicles we describe. Prefusion complexes are present in blow embryos, and absent in mbc embryos (which are defective in recognition and/or adhesion). It therefore seems clear that the prefusion complex forms only after the recognition of (and perhaps adhesion to) an appropriate fusion target cell.
What is the function of the paired vesicles? First, the paired vesicles may contain the essential components of the fusion apparatus destined for the plasma membrane, particularly the electron-dense material making up the plaques that sometimes appear in later steps of the fusion process. Alternatively, the paired vesicles might have a specific mechanistic role in the fusion process in excess of simple delivery of components to the apposed plasma membranes. A third possibility is that the vesicles might have a role in the recognition and/or attachment phase of the process. If the recognition phase were aborted by lack of vesicles, we would expect to see no further progression to the attachment phase.
The 1:1 pairing of vesicles in different cells across their apposed plasma membranes suggests some hypotheses for the function of these organelles. If the vesicles have a mechanistic role in later fusion events, the exact geometry of paired vesicles in the prefusion complex relative to the plasma membranes and each other might be of prime importance. If the paired vesicles have a simple role of delivering fusion components to the plasma membranes, the pairing might serve two functions. First, docking the vesicles to a prefusion complex would serve to restrict the plasma membrane distribution of potentially fusogenic macromolecules to the small area where fusion is necessary and not to regions where fusion would be inappropriate. Second, pairing of vesicles might enable a strict 1:1 ratio of molecules essential for fusion in the fusing region of each cell.
In either case, the presence of paired vesicles and the apparent symmetry of the prefusion complex strongly argues for a bidirectional function of the fusion event, that is, that there is not a “donor/receiver” relationship between the fusing cells once the prefusion complex is formed. We therefore hypothesize that the protein and lipid composition of the two plasma membranes in the fusing areas are nearly identical, and that the mechanics of the fusion process take place in a symmetrical fashion. This theoretical homotypic fusion is quite different from heterotypic fusion, for example, infection of cells by enveloped viruses, in which the viral membrane contains different components of the fusion process than the membrane of the target cell. The apparent bidirectional nature of the fusion process also implies that the fusing myoblasts are able to identify appropriate targets for fusion (i.e., myotubes or muscle pioneer cells) before the formation of the prefusion complex. This concept is supported by the absence of prefusion complexes in mbc mutants, which appear to be defective in recognition and/or adhesion to fusion targets.
Electron-dense Plaques
In some cases, we observed accumulations of electrondense material lining the cytoplasmic side of apposed plasma membranes, with diffuse electron-dense material present in the extracellular space as well. These electrondense plaques, while relatively rare in wild-type Drosophila embryos, were previously described in developing rat intercostal muscle (Kelly and Zacks, 1969), primary cultures of rat myoblasts (Rash and Fambrough, 1973), and in a rat myogenic cell line while absent from a nonfusing variant of that line (Engel et al., 1985). In addition, electron-dense staining was reported along the length of membrane sacs (presumably vesiculated plasma membranes) in fusing primary myoblasts (Rash and Fambrough, 1973), indicating that plasma membrane breakdown occurs in plaque regions.
These plaques may normally be an intermediate between fusion of the paired vesicles with the plasma membranes and the formation of pores between the apposed plasma membranes. Since embryos homozygous for the rolling stone mutation accumulate extensive electrondense plaques, greatly in excess of those seen in wild-type embryos, we believe that the rost mutation blocks the immediate next step in the fusion process. The relative scarcity of the plaques relative to prefusion complexes on one hand and fusion pores on the other suggests that the plaque intermediate is short-lived compared to either the prefusion complex or the fusion pores. Alternatively, the few plaques in wild-type embryos may be the result of a small number of abortive fusion events, and the rost mutation might increase the number of those aborted fusions. Since rost myoblasts form aberrantly extensive and long-lived plaques, but also complete cellular alignment, the normal function of the plaques is not required for alignment.
Of the genes we studied, rolling stone encodes the protein most likely to be directly involved in the actual membrane dynamics of the plasma membrane fusion . The extensive electron-dense plaques that accumulate in rost embryos argue for a direct role for Rolling Stone protein in the function of the plaques, perhaps in mediating lipid dynamics during formation of fusion pores, the defining step in the fusion process.
The Fusion Pore
Fusion pores appear frequently in stages 13 and 14 between adherent and aligned myoblasts. The exclusion of ribosomes and other stained particles from the pore regions makes vesiculating regions of plasma membrane obvious even at low magnifications. These pores are identical to those seen in vertebrate myoblasts (Shimada, 1971; Kalderon and Gilula, 1979).
It is not clear from 3D reconstruction of serial-sections whether the pores are noncontiguous and circular in cross section, or whether the openings in each pore are contiguous with each other, forming an irregular cross-section. If the pores are in fact noncontiguous, a second membrane fusion event is required to resolve each pore into receding front of plasma membranes. If the pores are contiguous, no additional fusion event is needed.
Occasionally, groups of irregular, non-electron dense vesicles appear in the cytoplasm beneath the plasma membranes where fusion pores are present. We do not know the function of these vesicles, although it is tempting to speculate that they represent a mechanism for disposal of the excess plasma membranes.
A Model for Myoblast Fusions
In Fig. 10 we propose a model for the sequence of events at the ultrastructural level leading to myotube formation. First, myoblasts identify and adhere to fusion targets, either muscle pioneer cells or existing myotubes. This step may very well involve multiple separate stages, including chemoattraction of myoblasts to fusion targets, cell–cell communication for identification of target cells, and cell adhesion. The stage 14 EM phenotype of mbc is consistent with a block somewhere in the process before cell adhesion. Pairs of cells that have correctly identified appropriate fusion targets then set up prefusion complexes at contact points where fusion will eventually begin. These complexes include paired vesicles and their associated electron dense material. The myoblasts become elongated, and align themselves along their long axes. Defects in the blown fuse gene stop the process before alignment takes place.
Figure 10.

Model of intermediate steps in myoblast fusion. Proposed schematic of the steps of myoblast fusion at the ultrastructural level, indicating action points of each mutant.
What might the function of the Blown Fuse protein be in normal myoblasts? We hypothesize that Blown Fuse is required for the normal function of the prefusion complex, while not an integral component of that complex. Blown Fuse might have an enzymatic activity necessary for prefusion complex function. The structure of the prefusion complex taken along with the relative scarcity of plaques suggests that paired vesicles and other complex components are accumulated at contact sites and remain quiescent for a relatively long period of time before dispersing by forming a plaque. Perhaps a signal transduction cascade must be activated before the complex can complete its normal function, with Blown Fuse being an essential part of that cascade. A third possibility is that the Blown Fuse protein is part of a checkpoint system that allows progress through the fusion process only after proper function of the prefusion complex, and that later steps are inhibited due to improper functioning of the checkpoint system.
After an unknown signal, the prefusion complex resolves into a short-lived electron-dense plaque. It is not clear from this work whether alignment must take place before the plaque stage or whether the two events happen independently of each other. The rolling stone mutation causes aberrant accumulation of plaques in stage 13 embryos, although the plasma membranes are able to become closely apposed as seen when the accumulated plaques disperse by stage 14. Next, fusion pores form, making the cytoplasm of the fusing cells continuous. Drac1G12V blocks the formation of the pores. The pores expand and the plasma membrane breaks down into smooth sacs of membrane. These sacs become rounder in profile through time and eventually are accumulated in groups of clear, irregularly shaped vesicles before recycling or disposal.
Acknowledgments
We thank Beth Blankemeier for technical assistance, and Meg Winberg, Doug Fambrough, Kathleen Ryan, and Jennifer Doyle for comments on the manuscript. We thank David Van Vactor, Helen Sink, Pascal Heitzler, Susan Abmayr, Renate Renkawitz-Pohl, and the Berkeley Drosophila Genome Project for providing Drosophila stocks.
This work was supported by the Muscular Dystrophy Association Carl M. Pearson Neuromuscular Disease Research Fellowship to S.K. Doberstein. R.D. Fetter is a Senior Research Associate, and C.S. Goodman is an Investigator with the Howard Hughes Medical Institute.
Abbreviations used in this paper
- blow
blown fuse
- mbc
myoblast city
- PLP
periodate-lysine-paraformaldehyde
- rost
rollingstone
- RT
room temperature
Footnotes
Please address all correspondence to C. Goodman, Howard Hughes Medical Institute, Department of Molecular and Cell Biology, University of California, Berkeley, CA 94720.
References
- Abmayr SM, Erickson MS, Bour BA. Embryonic development of the larval body wall musculature of Drosophila melanogaster. . Trends Genet. 1995;11:153–159. doi: 10.1016/s0168-9525(00)89030-7. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Ball EE, Goodman CS. Muscle development in the grasshopper embryo: II. Syncytial origin of the extensor tibiae muscle pioneers. Dev Biol. 1985a;111:399–416. doi: 10.1016/0012-1606(85)90493-2. [DOI] [PubMed] [Google Scholar]
- Ball EE, Goodman CS. Muscle development in the grasshopper embryo: III. Sequential origin of the flexor tibiae muscle pioneers. Dev Biol. 1985b;111:417–424. doi: 10.1016/0012-1606(85)90494-4. [DOI] [PubMed] [Google Scholar]
- Ball EE, Ho RK, Goodman CS. Muscle development in the grasshopper embryo. I. Muscles, nerves and apodemes in the metathoracic leg. Dev Biol. 1985c;111:383–398. doi: 10.1016/0012-1606(85)90492-0. [DOI] [PubMed] [Google Scholar]
- Bar-Sagi D, Prives J. Trifluoperazine, a calmodulin antagonist, inhibits muscle cell fusion. J Cell Biol. 1983;97:1375–1380. doi: 10.1083/jcb.97.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bate M. The embryonic development of larval muscles in Drosophila. . Development. 1990;110:791–804. doi: 10.1242/dev.110.3.791. [DOI] [PubMed] [Google Scholar]
- Bate, M. 1993. The mesoderm and its derivatives. In The Development of Drosophila melanogaster. M. Bate and A. Martinez Arias, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 1013–1090.
- Bischoff, R. 1978. Myoblast fusion. In Membrane Fusion. G. Poste and G.L. Nicholson, editors. Elsevier/North-Holland Biomedical Press. 127–179.
- Bischoff, R. 1979. Tissue culture studies on the origin of myogenic cells during muscle regeneration in the rat. In Muscle Regeneration. A. Mauro, editor. Raven Press, New York. 13.
- Blau HM, Dhawan J, Pavlath GK. Myoblasts in pattern formation and gene therapy. Trends Genet. 1993;9:269–274. doi: 10.1016/0168-9525(93)90012-7. [DOI] [PubMed] [Google Scholar]
- Cooper WG, Konigsberg IR. Dynamics of myogenesis in vitro. . Anat Rec. 1961;140:195–205. doi: 10.1002/ar.1091400305. [DOI] [PubMed] [Google Scholar]
- Couch CB, Strittmatter WJ. Rat myoblast fusion requires metalloendoprotease activity. Cell. 1983;32:257–265. doi: 10.1016/0092-8674(83)90516-0. [DOI] [PubMed] [Google Scholar]
- Couch CB, Strittmatter WJ. Specific blockers of myoblast fusion inhibit a soluble and not the membrane-associated metalloendoprotease in myoblasts. J Biol Chem. 1984;259:5396–5399. [PubMed] [Google Scholar]
- Dalby B, Pereira AJ, Goldstein LSB. An inverse PCR screen for the detection of P element insertions in cloned genomic intervals in Drosophila melanogaster. . Genetics. 1995;139:757–766. doi: 10.1093/genetics/139.2.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drysdale R, Rushton E, Bate M. Genes required for embryonic muscle development in Drosophila melanogaster: a survey of the X chromosome. Roux's Arch Dev Biol. 1993;202:276–295. doi: 10.1007/BF00363217. [DOI] [PubMed] [Google Scholar]
- Engel LC, Egar MW, Przybylski RJ. Morphological characterization of actively fusing L6 myoblasts. Eur J Cell Biol. 1985;39:360–365. [PubMed] [Google Scholar]
- Firket H. Recherches sur la synthèse des acides désoxyribonucléiques et la préparation à la mitose dans des cellules cultiveés in vitro. . Arch Biol. 1958;69:1–8. [PubMed] [Google Scholar]
- Gerson I, Seecof RL, Teplitz RL. Ultrastructural differentiation during embryonic Drosophilamyogenesis in vitro. In Vitro. 1976;12:615–622. doi: 10.1007/BF02797459. [DOI] [PubMed] [Google Scholar]
- Harlow, E., and D. Lane. 1989. Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. pp. 90–91.
- Heitzler P, Coulson D, Saenz-Robles M-T, Ashburner M, Roote J, Simpson P, Gubb D. Genetic and cytogenetic analysis of the 43A-E region containing the segment polarity gene costa and the cellular polarity genes prickle and spiny-legs in Drosophila melanogaster. . Genetics. 1993;135:105–115. doi: 10.1093/genetics/135.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho RK, Ball EE, Goodman CS. Muscle pioneers: large mesodermal cells that erect a scaffold for developing muscles and motoneurones in grasshopper embryos. Nature (Lond) 1983;301:66–69. doi: 10.1038/301066a0. [DOI] [PubMed] [Google Scholar]
- Holtzer H, Abbott J, Lash J. On the formation of multinucleated myotubes. Anat Rec. 1958;131:567. [Google Scholar]
- Kalderon N, Gilula NB. Membrane events involved in myoblast fusion. J Cell Biol. 1979;81:411–425. doi: 10.1083/jcb.81.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly AM, Zacks SI. The histogenesis of rat intercostal muscle. J Cell Biol. 1969;41:135–153. doi: 10.1083/jcb.42.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiehart DP, Feghali R. Cytoplasmic myosin from Drosophila melanogaster. . J Cell Biol. 1986;103:1517–1525. doi: 10.1083/jcb.103.4.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen KA. The calcium-dependent myoblast adhesion that precedes fusion is mediated by glycoproteins. J Cell Biol. 1985;101:891–897. doi: 10.1083/jcb.101.3.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen, K.A. 1992. Fusion of myoblasts. In Membrane Fusion. J. Wilschut and D. Hoekstra, editors. Marcel Dekker, Inc., New York. 601–626.
- Knudsen KA, Horwitz AF. Tandem events in myoblast fusion. Dev Biol. 1977;58:328–338. doi: 10.1016/0012-1606(77)90095-1. [DOI] [PubMed] [Google Scholar]
- Knudsen KA, Horwitz AF. Towards a mechanism of myoblast fusion. J Supramol Struct. 1978;8:563–568. [PubMed] [Google Scholar]
- Knudsen KA, McElwee SA, Smith L. A role for the neural cell adhesion molecule, NCAM, in myoblast interaction during myogenesis. Dev Biol. 1990a;138:159–168. doi: 10.1016/0012-1606(90)90185-l. [DOI] [PubMed] [Google Scholar]
- Knudsen KA, Myers L, McElwee SA. A role for the Ca2+-dependent adhesion molecule, N-cadherin, in myoblast interaction during myogenesis. Exp Cell Res. 1990b;188:175–184. doi: 10.1016/0014-4827(90)90157-6. [DOI] [PubMed] [Google Scholar]
- Lin DM, Fetter RD, Kopczynski C, Grenningloh G, Goodman CS. Genetic analysis of fasciclin II in Drosophila: defasciculation, refasciculation, and altered fasciculation. Neuron. 1994;13:1055–1069. doi: 10.1016/0896-6273(94)90045-0. [DOI] [PubMed] [Google Scholar]
- Lipton BH, Konigsberg IR. A fine-structural analysis of the fusion of myogenic cells. J Cell Biol. 1972;53:348–364. doi: 10.1083/jcb.53.2.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Liao YJ, Jan LY, Jan YN. Distinct morphogenetic functions of similar small GTPases: DrosophilaDrac1 is involved in axonal outgrowth and myoblast fusion. Genes & Dev. 1994;8:1787–1802. doi: 10.1101/gad.8.15.1787. [DOI] [PubMed] [Google Scholar]
- McLean IW, Nakane PK. Periodate-lysine-paraformaldehyde fixative: a new fixative for immunoelectron microscopy. J Histochem Cytochem. 1974;22:1077–1083. doi: 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- Mege RM, Goudou D, Diaz C, Nicolet M, Garcia L, Geraud G, Rieger F. N-cadherin and N-CAM in myoblast fusion: compared localisation and effect of blockade by peptides and antibodies. J Cell Sci. 1992;103:897–906. doi: 10.1242/jcs.103.4.897. [DOI] [PubMed] [Google Scholar]
- Miller JB, Boyce FM. Gene therapy by and for muscle cells. Trends Genet. 1995;11:163–165. doi: 10.1016/s0168-9525(00)89032-0. [DOI] [PubMed] [Google Scholar]
- Paululat A, Burchard S, Renkawitz-Pohl R. Fusion from myoblasts to myotubes is dependent on the rolling stone gene (rost) of Drosophila. . Development. 1995;121:2611–2620. doi: 10.1242/dev.121.8.2611. [DOI] [PubMed] [Google Scholar]
- Przybylski RJ, Blumberg JM. Ultrastructural aspects of myogenesis in the chick. Lab Invest. 1966;15:836–863. [PubMed] [Google Scholar]
- Rash JE, Fambrough D. Ultrastructural and electrophysiological correlates of cell coupling and cytoplasmic fusion during myogenesis in vitro. . Dev Biol. 1973;30:166–186. doi: 10.1016/0012-1606(73)90055-9. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Patterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Rosen GD, Sanes JR, LaChance R, Cunningham JM, Roman J, Dean DC. Roles for the integrin VLA-4 and its counter receptor VCAM-1 in myogenesis. Cell. 1992;69:1107–1119. doi: 10.1016/0092-8674(92)90633-n. [DOI] [PubMed] [Google Scholar]
- Rothman JE, Warren G. Implications of the SNARE hypothesis for intracellular membrane topology and dynamics. Curr Biol. 1994;4:220–233. doi: 10.1016/s0960-9822(00)00051-8. [DOI] [PubMed] [Google Scholar]
- Rushton E, Drysdale R, Abmayr SM, Michaelson AM, Bate M. Mutations in a novel gene, myoblast city, provide evidence in support of the founder cell hypothesis for Drosophilamuscle development. Development. 1995;121:1979–1988. doi: 10.1242/dev.121.7.1979. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chainterminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffino, S., S. Pierobon Bormioli, and M. Aloisi. 1979. Fibre branching and the formation of new fibres during compensatory muscle hypertrophy. In Muscle Regeneration. A. Mauro, editor. Raven Press, New York. pp. 177.
- Shainberg A, Yagil G, Yaffe D. Control of myogenesis in vitro by Ca2+concentrations in nutritional medium. Exp Cell Res. 1971;58:163–167. doi: 10.1016/0014-4827(69)90127-x. [DOI] [PubMed] [Google Scholar]
- Shimada Y. Electron microscope observations on the fusion of chick myoblasts in vitro. J Cell Biol. 1971;48:128–142. doi: 10.1083/jcb.48.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene (Amst) 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- Tear G, Harris R, Sutaria S, Kilomanski K, Goodman CS, Seeger MA. commisureless controls growth cone guidance across the CNS midline in Drosophilaand encodes a novel membrane protein. Neuron. 1996;16:501–514. doi: 10.1016/s0896-6273(00)80070-7. [DOI] [PubMed] [Google Scholar]
- Van Vactor D, Sink H, Fambrough D, Tsoo R, Goodman CS. Genes that control neuromuscular specificity in Drosophila. . Cell. 1993;73:1137–1153. doi: 10.1016/0092-8674(93)90643-5. [DOI] [PubMed] [Google Scholar]
- Wakelam MJO. Inositol phospholipid metabolism and myoblast fusion. Biochem J. 1983;214:77–82. doi: 10.1042/bj2140077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakelam MJO. The fusion of myoblasts. Biochem J. 1985;228:1–12. doi: 10.1042/bj2280001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakelam MJO. Myoblast fusion—a mechanistic analysis. Curr Top Memb Transp. 1988;32:87–112. [Google Scholar]
- Wakelam MJO, Pette D. The breakdown of phosphatidylinositol in myoblasts stimulated to fuse by the addition of Ca2+ . Biochem J. 1982;202:723–729. doi: 10.1042/bj2020723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakelam, M.J.O., and D. Pette. 1984. Myoblast fusion and inositol phospholipid breakdown: causal relationship of coincidence? In Symposium on Cell Fusion. D. Evered and J. Whelen, editors. Pitman Books, London. 100–118. [DOI] [PubMed]
- Yaffe D. Retention of differentiation potentialities during prolonged cultivation of myogenic cells. Proc Natl Acad Sci USA. 1968;61:477–483. doi: 10.1073/pnas.61.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagami-Hiromasa T, Sato T, Kurisaki T, Kamijo K, Nabeshima Y, Fujisawa-Sehara A. A metalloprotease-disintegrin participating in myoblast fusion. Nature (Lond) 1995;377:652–656. doi: 10.1038/377652a0. [DOI] [PubMed] [Google Scholar]
- Zinn K, McAllister L, Goodman CS. Sequence analysis and neuronal expression of fasciclin I in grasshopper and Drosophila. . Cell. 1988;53:577–587. doi: 10.1016/0092-8674(88)90574-0. [DOI] [PubMed] [Google Scholar]