

Abstract
Actin-binding proteins of the actin depolymerizing factor (ADF)/cofilin family are thought to control actin-based motile processes. ADF1 from Arabidopsis thaliana appears to be a good model that is functionally similar to other members of the family. The function of ADF in actin dynamics has been examined using a combination of physical–chemical methods and actin-based motility assays, under physiological ionic conditions and at pH 7.8. ADF binds the ADPbound forms of G- or F-actin with an affinity two orders of magnitude higher than the ATP- or ADP-Pi– bound forms. A major property of ADF is its ability to enhance the in vitro turnover rate (treadmilling) of actin filaments to a value comparable to that observed in vivo in motile lamellipodia. ADF increases the rate of propulsion of Listeria monocytogenes in highly diluted, ADF-limited platelet extracts and shortens the actin tails. These effects are mediated by the participation of ADF in actin filament assembly, which results in a change in the kinetic parameters at the two ends of the actin filament. The kinetic effects of ADF are end specific and cannot be accounted for by filament severing. The main functionally relevant effect is a 25-fold increase in the rate of actin dissociation from the pointed ends, while the rate of dissociation from the barbed ends is unchanged. This large increase in the rate-limiting step of the monomer-polymer cycle at steady state is responsible for the increase in the rate of actin-based motile processes. In conclusion, the function of ADF is not to sequester G-actin. ADF uses ATP hydrolysis in actin assembly to enhance filament dynamics.
Actin filaments are a major cytoskeletal component of eukaryotic cells. Rapid changes in the levels of polymeric (F-) actin and monomeric (G-) actin are involved in the morphological changes of living cells that occur, in a spatially and temporally controlled fashion, in response to environmental signals. Observation of actin dynamics in motile cells indicates that not only are actin filaments actively polymerizing beneath the plasma membrane at the leading edge, but also the turnover of actin filaments is faster in the lamellipodia than in other regions of the cell (Condeelis, 1993; Fechheimer and Zigmond, 1993). While it is well appreciated that G-actin–binding proteins establish a pool of unassembled actin subunits (“sequestered actin”) that is used in site-directed actin assembly (Carlier and Pantaloni, 1994; Sun et al., 1995), the molecular mechanisms by which the local changes in critical concentration and the turnover rate of actin filaments are controlled in vivo remain largely unknown.
A family of related actin-binding proteins, actin depolymerizing factor (ADF)/cofilin1, appear as choice candidates for the control of actin-based motility processes in response to signaling (for review see Moon and Drubin, 1995). The recently solved tertiary structure of vertebrate ADF shows a folding similar to that of gelsolin segment-1, severin segment-2, and profilin (Hatanaka et al., 1996).
These conserved, essential proteins, initially characterized by their ability to depolymerize F-actin, possess the unique property, among other G-actin–binding proteins, to be regulated by reversible phosphorylation (Ohta et al., 1989; Agnew et al., 1995). In all cases investigated, the activity of ADF/cofilins was inhibited by phosphorylation of a serine in the NH2-terminal region. Rapid dephosphorylation occurs in response to stimuli. Consistently, ADF/cofilins are localized in ruffling membranes and at the leading edge of locomoting cells (Aizawa et al., 1995), and overexpression of ADF in Dictyostelium discoideum stimulates cell movement (Aizawa et al., 1996). In plants, ADF is encoded by a small multigene family, and at least one member is preferentially expressed in germinating pollen tubes (Lopez et al., 1996). Experiments with transgenic Arabidopsis thaliana indicate that ADF is involved in cell shape maintenance and cell elongation (Xia, G., Y. Ishimaru, L. Dong, Y. Hong, S. Ramachandran, A. Cleary, and N.-H. Chua, unpublished data).
In vitro studies of the interaction of ADF/cofilin with actin have revealed a broad complexity of functional properties. These proteins are identified as G-actin–binding, F-actin depolymerizing proteins; however, they have been shown to bind also to F-actin and proposed to sever filaments in a pH-sensitive fashion. The severing activity is thought to account for the ADF-induced rapid assembly and disassembly of filaments, as well as for the rapid drop in viscosity of F-actin solutions upon addition of ADF (Yonezawa et al., 1985; Cooper et al., 1986; Maciver et al., 1991; Hawkins et al., 1993; Hayden et al., 1993). The issue of the preferential binding of ADFs to ADP- or ATP-actin is still controversial (Hayden et al., 1993; Maciver and Weeds, 1994).
The present work addresses the function of ADF in actin dynamics using physical–chemical methods and in vitro motility assays, and Arabidopsis ADF1 is used as a model. We show that the main function of ADF is to increase 25fold the turnover rate of actin filaments by changing the kinetic parameters for actin assembly and disassembly in an end-directed fashion. As a result, ADF increases the rate of actin-based motility of Listeria monocytogenes in highly diluted platelet extracts. ADF does not act as a G-actin sequestering protein and does not appear to fragment filaments in vitro, but it changes their hydrodynamic parameters, which may account for its effects on the viscosity of F-actin solutions. Comparative assays carried out with other ADFs confirm that these proteins all act as “actin dynamizing factors.”
Materials and Methods
Proteins
Actin, purified from rabbit muscle acetone powder, was isolated as CaATP–G-actin by gel filtration over Sephadex G-200 in G-buffer (5 mM Tris Cl−, 0.2 mM ATP, 0.2 mM DTT, 0.1 mM CaCl2, 0.01% NaN3, pH 7.8) as described (Carlier et al., 1996). CaATP–G-actin was converted into MgATP–G-actin by the addition of 0.2 mM EGTA and 1 molar equivalent +10 μM excess of MgCl2 and used 3 min later. Actin was polymerized by the addition of 1 mM MgCl2 and 0.1 M KCl to Mg–G-actin. Gelsolincapped filaments were obtained by polymerizing Ca-actin without EGTA and 2 mM instead of 1 mM MgCl2. MgADP–G-actin was prepared by preincubation of MgATP–G-actin with 20 U/ml hexokinase and 5 mM glucose (Pollard et al., 1992). Actin was labeled by either pyrenyl iodoacetamide (Kouyama and Mihashi, 1981) or NBD-Cl (Detmers et al., 1981). Thymosin β4 (Tβ4) was purified from bovine spleen (Carlier et al., 1996). Spectrin-actin seeds were prepared from human erythrocytes and their molar concentration was determined as described (Casella et al., 1986) and by titration by gelsolin.
Recombinant ADF1 from Arabidopsis thaliana was prepared as follows. A cDNA clone 33D1T7 encoding a protein with homology to the ADF/cofilin family was obtained from the Arabidopsis Biological Resources Center at the Ohio State University. We determined the entire nucleotide sequence of both strands of the cDNA insert. The 0.69-kb cDNA was found to encode a full-length protein designated ADF1. The cDNA was expressed in Escherichia coli BL21DE3 strain by the use of pET 16b vector (Studier et al., 1990). Expression of 35S-labeled ADF1 (3.5 to 7 Ci/mol) was performed in M9 minimal medium containing 1.5 mCi [35S]methionine and [35S]cysteine in the presence of 0.2 mg/ml rifampicin, 30 min after the induction of T7 RNA polymerase expression by IPTG.
Sequence comparison of ADF1 with other members of the ADF/cofilin family (actophorin from Acanthamoeba castellanii, yeast, Dictyostelium discoideum, human cofilins) is displayed in Table I. ADF1 contains 139 amino acid residues, with a molecular mass of 16,113 D and an isoelectric point of 7.08.
Table I.
Sequence Comparison of ADF1 from Arabidopsis thaliana with Other Members of the ADF/Cofilin Family
| Organism | Amino acid sequence | |
|---|---|---|
| Yeast | MSRSGVAVADESLTAFNDLKLGK---------KYKFILFGLNDAKTEIVVK-----------ETSTDPSYDAFLEKLPENDCLYAIYDFEY | |
| Dict. D. | SSGIALAPNCVSTFNDLKLGR---------KYGGIIYRISDDSKEIIVD----------STLPAGCSFDEFTKCLPENECRYVVLDYQY | |
| Ac. Cast. | MSGIAVSDDCVQKFNELKLGHQH-------RYVTFKMNA--SNTEVVVEH--------VGG-PNAT-YEDFKSQLPERDCRYAIFDYEF | |
| A.T. | MANAASGMAVHDDCKLRFLELKAKRTH-------RFIVYKIEE--KQKQVVVE--K------VGQ-PIQT-YEEFAACLPADECRYAIYDFDF | |
| Human | MASGVQVADEVCRIFYDMKVRKCSTPEEIKKRKKAVIFCLSADKKCIIVEEGKEILVGDVGVTITDP-FKHFVGMLPEKDCRYALYDASF | |
| Yeast | EINGNEGKRSKIVFFTWSDDTAPVRSKMVYASSKDALRRALNGVSTDVQGTDFSEVSYDS-VLERVSRGAGSH | |
| Dict. D. | KEEG--AQKSKICFVAWCPDTANIKKKMMATSSKDSLRKACVGIQVEIQGTDASEVK-DSCFYEKCTK | |
| Ac. Cast. | QVDG--GQRNKITFILWAPDSAPIKSKMMYTSTKDSIKKKLVGIQVEVQATDAAEISEDA-VSERAKKDVK | |
| A.T. | VTAEN-CQKSKIFFIAWCPDIAKVRSKMIYASSKDRFKRELDGIQVELQATDPTEMDLDV-FRSRAN | |
| Human | ETKE--SRKEELMFFLWAPDLAPLKSKMIYASSKDAIKKKFQGIKHECQANGP-E-DLNRACIAEKLGGSLIVAFEGCPV |
Dashes are gaps introduced to optimize alignments. Identical or highly conserved residues (at least four out of the five) are shown in bold letters. Only K = R, D = E, V = I = L = M, Y = F are considered as highly conservative homologies. A.T., Arabidopsis Thaliana; Dict. D., Dictyostelium discoideum; Ac. Cost., Acanthamoeba castellanii.
A bacterial extract (80 ml) was dialyzed overnight against 2 liters of DEAE buffer (10 mM Tris Cl−, pH 7.8, 50 mM NaCl, 5 mM DTT, 0.2 mM EGTA, 0.1 mM PMSF, 0.01% NaN3) and chromatographed on DEAE cellulose equilibrated in the same buffer. ADF1 was recovered 85–90% pure in the flow-through. The solution was equilibrated in 10 mM Pipes, pH 6.5, 5 mM DTT, 0.2 mM EGTA, 0.01% NaN3, 25 mM NaCl and chromatographed on SP-trisacryl in the same buffer. ADF1 was recovered 99% pure in the flow-through. ADF1 was concentrated to 150–250 μM by ultrafiltration, dialyzed against 5 mM Tris Cl−, pH 7.5, 1 mM DTT, 0.01% NaN3, centrifuged at 400,000 g for 15 min, and stored on ice for up to 4 wk without any loss in activity. The concentration of ADF1 was determined spectrophotometrically, using the same extinction coefficient of 0.89 mg−1cm2 at 278 nm as for actophorin (Cooper et al., 1986).
Binding of ADF1 to G-actin
ADF1 binding to MgATP–G-actin or MgADP–G-actin was assayed by the quenching of fluorescence F of NBD-labeled actin. Experiments were carried out at 20°C, pH 7.8, in a Spex fluorolog 2 spectrofluorimeter (λexc 475 nm, λem 530 nm) using 0.3 ml samples of 0.8 μM G-actin in 4 × 4 mm square section cuvettes. The quenching of fluorescence,
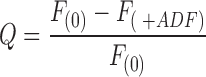 ,
,
was measured at each concentration of added ADF. Data were analyzed using Kaleidagraph software (Synergy Software, Reading, PA) and fitted using the following equation:
(1)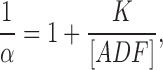
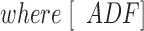
(2)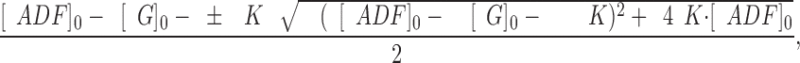
α = Q/Q max, Q max is the maximum quenching reached at saturation of G-actin by ADF, [ADF]0 and [G]0 are the total concentrations of ADF and G-actin, respectively, and K is the equilibrium dissociation constant for the G-actin–ADF complex.
Binding of ADF1 to F-actin
Sedimentation Assay.
Samples of 0.3 ml containing F-actin and 35S-labeled ADF at the indicated concentrations were incubated for 10 min to 2 h at most and sedimented at 20°C for 30 min at 400,000 g. When barbed ends were capped, a gelsolin/actin molar ratio of 1:300 to 1:500 was used. The amount of ADF bound to F-actin was derived from radioactivity measurements of the amounts of 35S-labeled ADF present in the samples before centrifugation and in the supernatant of sedimented samples. Supernatants and resuspended pellets were submitted to SDS-PAGE (15% acrylamide). Coomassie blue–stained gels were scanned (model Arcus II scanner; AGFA Corp., Orangeburg, NY), and the patterns were compared to those obtained for actin and ADF standards to estimate the amounts of actin and ADF in the pellets and supernatants, using the NIH Image program. The amount of G-actin in the supernatants was also derived from measurements of the inhibition of DNaseI activity (Blickstad et al., 1978).
Fluorescence Assay.
Samples of fully labeled pyrenyl– or NBD–F-actin at steady state in polymerization buffer were supplemented at time zero with ADF. Changes in light scattering at a 90° angle and fluorescence were recorded simultaneously using the “T” configuration of the Spex fluorimeter. The excitation monochromator was set at 366 nm for pyrenylactin and at 475 nm for NBD-actin. One of the two emission monochromators was set at the same wavelength as the excitation wavelength to monitor light scattering. The other monochromator was set at 387 or 530 nm to monitor pyrene or NBD fluorescence, respectively.
Polymerization and Depolymerization Assays
Kinetics of actin assembly and disassembly were monitored turbidimetrically at 310 nm in a spectrophotometer (model Uvikon; Kontron Instrs., Milan, Italy or model Cary 1; Varian Techtron, Victoria, Australia) using 1-cm path cuvettes thermostated at 20°C.
Measurements of initial rate of growth from barbed ends were carried out using spectrin-actin seeds. Assembly was started by adding ADF to preformed Mg–G-actin, followed by the seeds and salt. All buffer solutions were thoroughly filtered and degassed before the experiment.
The initial rate of growth is:
(3)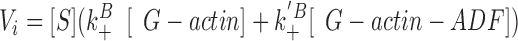 ,
,
where [S] represents the concentration of spectrin-actin seeds, and k B + and k′B + represent the association rate constants of G-actin and G-actin–ADF, respectively, to the barbed ends. The contribution of the off rate was neglected in Eq. 3 because the actin concentration (3.3 μM) is well above the critical concentration. The concentrations of G-actin and G-actin–ADF can be calculated knowing the values of [G]o, [ADF]o, and K:
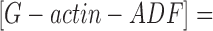
(4)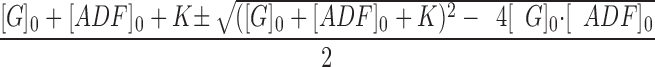 .
.
The value of k′B
+ can be derived from the plot of 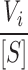 versus [G-actin– ADF]. Since initial rate measurements have been performed at a low percentage of ADF-actin, the impact of the difference in specific turbidities of F-actin and ADF–F-actin on the value of the rate parameters is minor.
versus [G-actin– ADF]. Since initial rate measurements have been performed at a low percentage of ADF-actin, the impact of the difference in specific turbidities of F-actin and ADF–F-actin on the value of the rate parameters is minor.
Elongation of filaments from the pointed ends was carried out using gelsolin-actin seeds (4 μM gelsolin + 8 μM Ca–G-actin in G buffer). A solution of 6.5 μM Mg–G-actin was supplemented at time zero with ADF, followed immediately by 0.2 mM CaCl2, 75 nM CapG (to cap the potential spontaneous barbed end nuclei), different amounts of gelsolin-actin seeds, and salt. The optimum amount of CapG was experimentally determined to efficiently cap spontaneous barbed end nuclei but avoid nucleation of new pointed ends, which we found to occur at concentrations of CapG above 150 nM.
Depolymerization of F-actin from the barbed ends was induced by adding 6 μM DNaseI and ADF as indicated to a solution of 6 μM F-actin. DNaseI binds tightly to monomeric actin (Blickstad et al., 1978) and to the pointed end subunits (Podolski and Steck, 1988; Weber et al., 1994); hence, the effect of ADF on the rate of depolymerization from the barbed ends specifically can be measured.
Depolymerization of F-actin from the pointed ends was induced by adding 26 μM Tβ4 and ADF as indicated to a solution of 3.5 μM F-actin polymerized in the presence of 7 nM gelsolin. The amount of Tβ4 added is low enough for this protein to act only as a G-actin sequestering agent.
Measurement of the Treadmilling Rate of Actin Filaments
The rate of filament turnover at steady state (treadmilling) was monitored by the decrease in fluorescence of εADP bound to F-actin after addition of a chase amount of ATP. Since ADP is nonexchangeable on F-actin (Pollard et al., 1992, and references therein) and since its fluorescence is sixfold higher in the actin-bound than in the free state, the rate of fluorescence decrease is a true measurement of filament turnover. εATP–G-actin (15 μM), prepared as described (Valentin-Ranc and Carlier, 1989), was polymerized in the presence of 30 μM free εATP. Samples of F-εADPactin were preincubated with the desired amounts of ADF for 20 min. The change in fluorescence of εADP (λexc = 350 nm; λem = 410 nm) was recorded versus time after addition of 1 mM ATP. Identical results, but with a lower time resolution, were obtained using a sedimentation assay (400,000 g for 15 min) of [3H]ADP–F-actin and measuring the increase in [3H]ADP in the supernatant at different time intervals after the onset of the ATP chase.
ATP Hydrolysis Measurements in F-actin Solutions at Steady State
Ca–G-actin (16 μM) was equilibrated in G buffer containing 0.2 mM γ 32P-labeled ATP, converted into Mg–G-actin, and polymerized by addition of 1 mM MgCl2 and 0.1 M KCl. ADF was added at 15 min, when steady state was reached. Acid labile [32Pi] resulting from ATP hydrolysis was monitored by extraction of the phosphomolybdate complex (Carlier et al., 1986) during assembly and at steady state over a period of 5 h.
Actin-based Motility Assay of Listeria monocytogenes in Platelet Extracts
Platelet extracts were prepared (Laurent and Carlier, 1997) by sonication of a suspension of washed unstimulated platelets in 10 mM Tris Cl−, pH 7.5, 2 mM MgCl2, 10 mM EGTA (8 × 109 ± 1 × 109 cells/ml) followed by centrifugation at 100,000 g for 35 min, 4°C. The amount of G-actin in the extracts was 35 μM, as derived from the DNaseI inhibition assay. The amount of Tβ4 was 61 μM as derived from HPLC (Carlier et al., 1996). The amount of unassembled actin and Tβ4 being 200 and 320–500 μM, respectively, in platelets (Weber et al., 1992), the cytoplasm was about sixfold diluted in the extracts. The extract was supplemented with extraction buffer, 5 mM ATP-Mg, 6 mM DTT, 3.25 μM rhodamine-labeled G-actin, oxygen scavengers (Isambert et al., 1995), methyl cellulose and ADF as indicated, and 108 bacteria/ml. The final dilution of the platelet cytoplasm could be varied by changing the volume of extraction buffer present in the motility assay. Sample preparation, fluorescence microscopy observation, and video recording of the formation of comet tails and bacteria propulsion were carried out as described (Marchand et al., 1995). 10–15 mobile bacteria were recorded per sample to derive the average rates and tail lengths. It was checked that ADF1 binds rhodamine-actin used in the motility assay. A 25% quenching of the fluorescence of rhodamine–F-actin occurred upon binding ADF1.
Sedimentation Velocity of Actin Filaments in the Analytical Ultracentrifuge—Effects of ADF1 and Gelsolin
Solutions of F-actin containing different amounts of ADF or gelsolin were centrifuged at 20°C at 20,000 rpm in an analytical ultracentrifuge (model Optima XLA; Beckman Instrs., Fullerton, CA). Scans were recorded at 295 nm at 4-min intervals. The average sedimentation coefficients of filaments present in the different samples were compared. Control samples for severed filaments were obtained using gelsolin at a 1:100, 1:500, or 1: 1,000 ratio to actin.
Results
ADF1 Binds ADP–G-actin with a 100-fold Higher Affinity than ATP–G-actin under Physiological Ionic Conditions
The fluorescence of NBD–G-actin was partially quenched upon binding ADF. Therefore, this parameter was used to determine the values of the equilibrium dissociation constant of the ADF–G-actin complex under a variety of conditions. The extent of quenching of NBD fluorescence versus ADF concentration displayed a saturation behavior, consistent with the formation of a tight 1:1 complex between G-actin and ADF. Under physiological ionic conditions (0.1 M KCl, 1 mM MgCl2, pH 7.8; Fig. 1), ADF bound to MgADP–G-actin with an affinity (K d = 0.1 μM) two orders of magnitude higher than MgATP–G-actin (K d = 8 μM). The quenching of NBD fluorescence was slightly higher in the ADP-bound complex (32 ± 2%) than in the ATPbound complex (27 ± 2%). At low ionic strength, the affinity of ADF for MgATP–G-actin was higher than at physiological ionic strength (K d = 0.08 μM) and only three- to fourfold lower than for MgADP–G-actin (K d = 0.025 μM). Binding constants at different ionic strength are displayed in Table II. Identical values of the binding constants (within 20%) were derived from kinetic measurements of the inhibition of nucleotide exchange on G-actin by ADF1 (data not shown).
Figure 1.

Interaction of ADF1 with G-actin. The quenching of fluorescence of 0.8 μM NBD-labeled MgATP–G-actin (○, abscissa bottom scale) or MgADP–G-actin (•, abscissa top scale) was measured at different concentrations of ADF, under physiological ionic conditions (0.1 M KCl, 1 mM MgCl2, pH 7.8). Symbols are data; lines are calculated binding curves using values of K d of 0.1 μM (•) and 8 μM (○).
Table II.
Equilibrium Parameters for the Interaction of ADF with G- and F-actin
| Parameter | Conditions | MgATP-actin | MgADP-actin | |||
|---|---|---|---|---|---|---|
| K DG (μM) | G buffer | 0.08 ± 0.02 | 0.025 ± 0.005 | |||
| F buffer (1 mM MgCl2) | 0.15 ± 0.03 | ND | ||||
| F buffer (0.1 M KCl, 1 mM MgCl2) | 8 ± 2 | 0.1 ± 0.02 | ||||
| F buffer (0.4 M KCl, 1 mM MgCl2) | 20 ± 5 | ND | ||||
| K DF (μM) | F buffer (0.1 M KCl, 1 mM MgCl2) | >10 (ND) | 0.3 (calculated) | |||
| C SS (μM) | Free barbed ends | 1.7 ± 0.2 | 4.5 ± 0.5 | |||
| Capped barbed ends | 3.5 ± 0.3 | 4.5 ± 0.5 |
K DG and K DF are the equilibrium dissociation constants for binding of ADF to G- and F-actin, respectively. C SS is the steady-state concentration of unassembled actin at saturation by ADF, measured at pH 7.8 under physiological ionic conditions.
The Different Binding of ADF1 to ADP–G-actin and ADP–F-actin Causes the Partial Depolymerization of Filaments
Sedimentation assays of the binding of 35S-labeled ADF to F-actin (Fig. 2 a) in the presence of 0.1 M KCl, 1 mM MgCl2, pH 7.8, show that ADF binds tightly to F-actin with a sigmoidal saturation curve. In agreement with others (Nishida et al., 1984; Hawkins et al., 1993; Hayden et al., 1993; Moon et al., 1993), one molar equivalent ADF per F-actin subunit was found in the pellet at saturation by ADF. The amount of F-actin decreased to a limited extent upon increasing the concentration of ADF. At saturating amounts of ADF, a constant amount of ADF–F-actin (1:1) coexisted at steady state with a constant amount of unassembled actin. Identical data were obtained when ADF was added to preassembled F-actin and when actin was assembled in the presence of ADF. The concentration of unassembled actin reached at saturation by ADF was independent of the total concentration of actin and was 1.7–2 μM at pH 7.8. The binding behavior of ADF was qualitatively identical to the above in a range of pH from 6.5 to 8.3 (Fig. 2, b and c). At pH 6.5, the amount of unassembled actin at steady state at saturating amounts of ADF was 1.2 ± 0.2 μM; it reached 3.5 ± 0.5 μM at pH 8.3.
Figure 2.
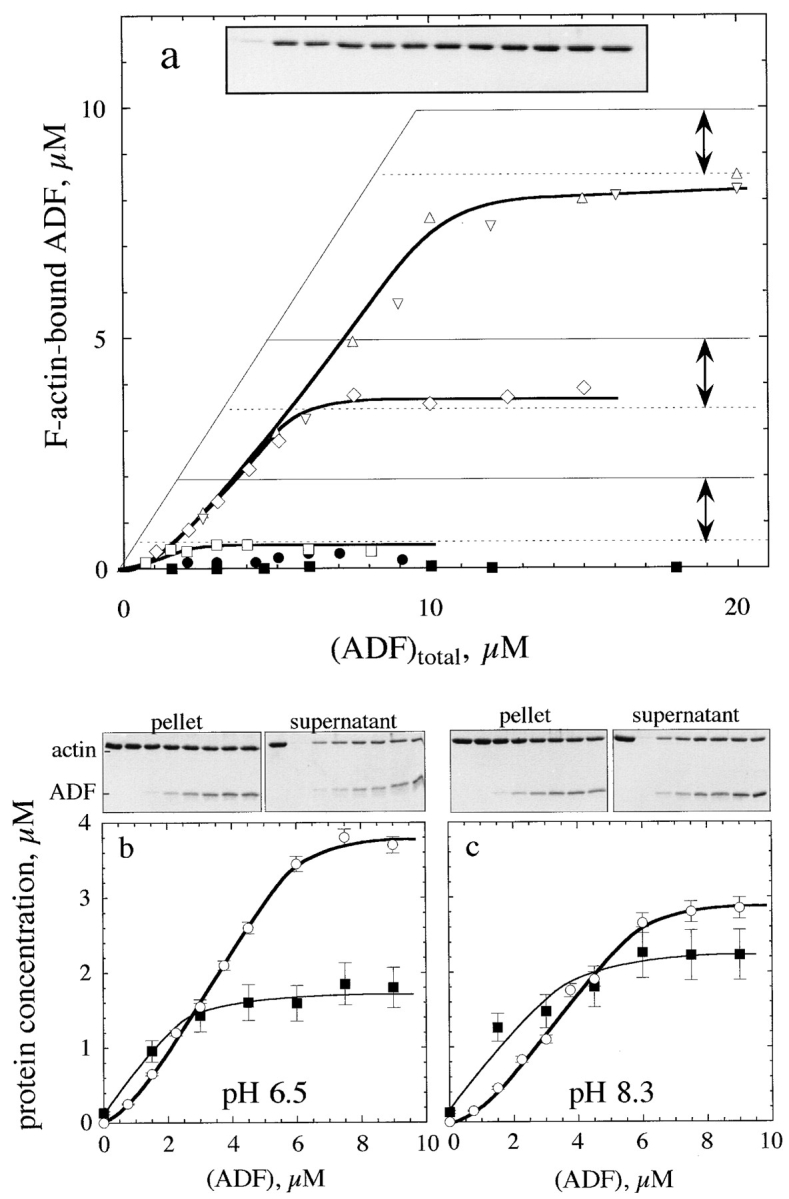
Interaction of ADF1 with F-actin. (a) Sedimentation assay for binding of ADF1 to F-actin. The binding of 35S-labeled ADF to F-actin was measured at the following concentrations of F-actin (μM): □, 2; ⋄, 5; ▵ and ▿, 10. •, 5 μM F-actin, 7.5 μM phalloidin. ▪, 7 μM F-actin-ADP-BeF3. The interval between the two arrows represents the amount of unassembled actin found at steady state in the supernatant of sedimented samples. Thin lines represent the high-affinity titration curves that would be obtained if ADF bound tightly to F-actin exclusively in a 1:1 molar ratio. (Inset) SDS-PAGE pattern of actin in the supernatant of sedimented samples containing 7 μM F-actin and ADF (in μM, left to right): 0, 1, 2, 3, 4, 5, 6, 8, 18, 12, 15, and 17. (b and c) pH dependence of ADF1 interaction with F-actin. SDS-PAGE of the pellets and supernatants of F-actin (5 μM) assembled at pH 6.5 (b) or pH 8.3 (c) in the presence of ADF. Left to right lanes: whole actin (5 μM); samples containing 0, 1.5, 3, 4.5, 6, 7.5, and 9 μM ADF. ▪, densitometered actin bands in the supernatants (in μM); ○, amount of ADF–F-actin, in μM (from 35S radioactivity measurements).
The partial depolymerization of actin by ADF is not in agreement with the behavior expected for a G-actin sequestering protein, which should eventually depolymerize F-actin totally in a concentration-dependent fashion (Carlier and Pantaloni, 1994). Rather, the results suggest that ADF-actin complex copolymerizes with actin and that a new steady-state concentration of unassembled actin is established in the presence of ADF. The sigmoidicity of the binding curves of ADF to F-actin is consistent with the observation of the gel patterns (Fig. 2) showing that addition of a small amount of ADF (∼1 μM) to an F-actin solution essentially causes depolymerization of F-actin (hence binding of ADF to G-actin), while very little ADF binds to the remaining filaments. At higher concentrations, ADF binds to F-actin. The sigmoidal curve therefore does not result from the cooperative binding of ADF to F-actin as previously thought (Hayden et al., 1993) but reflects the preferential interaction of ADF with G-ADP-actin over F-ADP-actin.
In the presence of BeF3−, a Pi analog that binds to F-ADPactin subunits and reconstitutes the F-ADP-P* transition state of ATP hydrolysis on F-actin (Carlier, 1991), ADF neither bound appreciably to F-actin nor depolymerized it, in agreement with other reports (Maciver et al., 1991; Maciver and Weeds, 1994). The binding of ADF to phalloidin– F-actin was very low. In conclusion, the affinity of ADF for F-actin, as well as for G-actin, is strongly dependent on the bound nucleotide and much higher for the ADP forms of both G- and F-actin.
The binding of ADF to gelsolin-capped filaments was examined next. A steady-state concentration of unassembled actin of 3.5 ± 0.3 μM was found at saturation by ADF. In the presence of ADP, a true critical concentration of 4.5 ± 0.5 μM for assembly of ADF-ADP-actin was found at saturation by ADF, both in the presence and absence of gelsolin. This value is threefold higher than the critical concentration for assembly of ADP-actin. Detailed balance therefore implies that the equilibrium dissociation constant for binding of ADF to ADP–F-actin subunits be threefold higher than to ADP–G-actin, i.e., 0.3 μM, as described by the following thermodynamic square scheme:
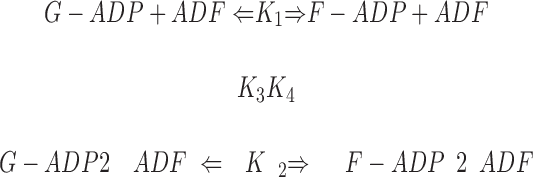
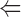
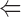
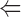
 ,
,
where K 1 and K 2 refer to the propagation constants (i.e., the critical concentrations for polymerization) of ADP-actin and ADF-ADP-actin, respectively, and K 3 and K 4 are the equilibrium dissociation constants for binding of ADF to G-ADP-actin and F-ADP-actin, respectively, with K 1·K 4 = K 2·K 3. All data are summarized in Table II.
Interaction of ADF with F-actin: Fluorescence and Light Scattering Measurements
The mechanism by which ADF causes partial depolymerization of F-actin was addressed in kinetic experiments. Since excess ADF causes depolymerization of a maximum of 1.7 μM actin, it was interesting to compare the effects of ADF addition (0.7 molar equivalent to actin) to either a low or a high amount of F-actin, at which depolymerization occurs to very different extents. Typical curves are shown in Fig. 3. The addition of ADF to 4.5 μM pyrenyl– F-actin (Fig. 3 a) caused a small instantaneous increase, followed by a time-dependent 38% decrease in light scattering, consistent with the depolymerization of 1.7 μM F-actin. Similarly, the addition of ADF to 15 μM F-actin (Fig. 3 b) caused a 10% decrease in light scattering, also consistent with 1.7 μM depolymerized actin.
Figure 3.

ADF1 binds to labeled F-actin with concomitant quenching of fluorescence followed by partial depolymerization. (a) Simultaneous recordings of light scattering (1) and pyrenyl fluorescence (2) upon addition of 3 μM ADF (arrow) to a 4.5 μM 100% pyrenyl-labeled F-actin solution. The curves are normalized by adjusting the light scattering and fluorescence intensities of F-actin recorded before addition of ADF to the same maximum level and subtracting the intensities corresponding to G-actin. (b) Same as in a, but 7.5 μM ADF was added to 13.5 μM fully labeled pyrenyl–F-actin. (c) Same as in a, with a 0.5% labeled pyrenyl– F-actin solution. (d) The extent of quenching of fluorescence of 100% labeled pyrenyl–F-actin (3.6 μM) upon binding ADF is plotted versus ADF concentration.
The changes in fluorescence did not quantitatively correlate with the changes in light scattering. A rapid large decrease was followed by a slower decrease. The slow phase was kinetically consistent with the decrease in light scattering. At all F-actin concentrations, the amplitude of the large rapid fluorescence decrease varied linearly with ADF and reached 95% quenching at a 1:1 molar ratio of ADF/F-actin (Fig. 3 d). This implies that the rapid binding of ADF to F-actin is linked to the quenching of pyrenyl– F-actin fluorescence to the level of G-actin and is followed by a slower partial depolymerization. The linear dependence of quenching on ADF concentration indicates that the ADF binds with high affinity (K a > 106 M−1) to F-ADPactin. The fast binding was noncooperative, which confirms our interpretation of the sigmoidal binding curves (Fig. 2) obtained when ADF binding is assayed by sedimentation after the slow relaxation to a new steady state. Identical results were obtained using NBD-labeled actin. The fluorescence of NBD–F-actin was quenched, upon binding ADF, to a level corresponding to 70% of the fluorescence of NBD–G-actin.
When ADF was added to partially labeled pyrenyl–Factin, the fluorescence change was more complex (Fig. 3 c). The rapid and slow decreases were then followed by a slow recovery to a higher stable limit, in agreement with others (Maciver et al., 1991; Aizawa et al., 1995). A straightforward interpretation is that ADF binds rapidly to both labeled and unlabeled F-actin subunits and then slowly redistributes from the labeled to the unlabeled F-actin subunits, with fluorescence recovery. This kinetic behavior is generated when the rate constants for association to unlabeled and labeled actins are similar, but the higher affinity for unlabeled actin is linked to a lower dissociation rate constant. A similar behavior is displayed by myosin subfragment-1 binding to partially labeled actin and was recently quantitatively analyzed with the same mechanism (Blanchoin et al., 1996). No changes in fluorescence nor light scattering were observed when ADF was added to phalloidin–F-actin or to F-ADP-BeF3-actin, which confirmed the observations made in Fig. 2.
Effect of ADF1 on the Kinetics of Actin Polymerization
The results presented above demonstrate that ADF modifies the steady state of actin assembly and point to the need to understand how the kinetics of assembly/disassembly at the barbed and pointed ends of the filaments are affected by ADF. Preliminary experiments (not shown) indicated that, in agreement with data in Fig. 3, the time courses of actin assembly followed simultaneously by light scattering and either NBD- or pyrenyl-actin fluorescence were no longer kinetically correlated in the presence of ADF because of the quenching of fluorescence that occurs upon binding of ADF to F-actin during polymerization.
Since fluorescence of labeled actin cannot be used reliably to monitor actin assembly in the presence of ADF, turbidimetry was used as an alternative tool. Fig. 4 shows the effects of ADF on the spontaneous assembly of MgATP-actin (a) and Mg-ADP-actin (b). In both cases, ADF increased the rate of assembly and the maximum extent of turbidity change. In the simple case of reversible polymerization of ADP-actin, monotonic time courses of assembly correspond to the copolymerization of ADP-actin and ADF– ADP-actin. In the presence of ATP, the overshoot kinetics suggest that partial depolymerization of ADF–F-actin occurs consecutive to some kinetic barrier. We know that: (a) Pi release after ATP hydrolysis associated with actin polymerization is a slow process (t 1/2 = 2 min) that takes place on the filaments after actin assembly (Carlier, 1991); and (b) ADF binds to ADP-actin with a 100-fold higher affinity than to ATP-actin. Hence, the most plausible explanation accounting for the different kinetics in ADP and ATP is that in the presence of ATP, ADF binds to F-actin and promotes its partial depolymerization only after Pi has been released. The final steady state concentration of unassembled actin in the presence of ADF (2 μM at this pH) is therefore established via overshoot kinetics. In the presence of inorganic phosphate, which maintains the filaments in the F-ADP-Pi state, the overshoot was abolished but the ADF-induced increase in initial rate of assembly was still observed.
Figure 4.

Effects of ADF1 on the polymerization of ATP- and ADP-actin. (a) Turbidimetric recording of the spontaneous polymerization of 9.6 μM MgATP-actin in the presence of ADF1. (b) Spontaneous polymerization of 20 μM MgADP-actin in the presence of ADF1. The concentrations of ADF1 (in μM) are indicated on the curves. (c) Critical concentration plots for actin assembly derived from turbidimetric measurements. The extent of turbidity change (A t= − A t=0) over the time course of polymerization was plotted for actin alone (○) and actin polymerized in the presence of a saturating (1.5 molar equivalent) amount of ADF (•).
The polymerization time courses are not consistent with fragmentation of filaments by ADF because they do not exhibit the acceleration and symmetric shape around the half-polymerization time point characteristic of such a process, which has been observed and mathematically analyzed (Carlier et al., 1985).
The validity of the turbidity change as a measure of the mass amount of assembled actin in the absence and presence of ADF is illustrated by the linearity of the critical concentration plots shown in Fig. 4 c. The extent of turbidity change per unit mass of assembled actin was 1.65-fold greater for ADF–F-actin than for F-actin. This figure is quantitatively consistent with the increase in mass per unit length of filaments decorated by ADF. Indeed, the turbidity is expected to be proportional to the square of the molecular mass per unit length of the polymer (Carlier et al., 1994). Assuming that the structure factor of F-actin filaments remains unchanged, in a first approximation, when ADF is bound to F-actin, the ratio of the specific turbidity of ADF–F-actin and F-actin is expected to be equal to the ratio of the square of the molecular mass of the polymerizing unit, which in this case is equal to (58/42)2 = 1.9, in reasonable agreement with the experimental value of 1.65. The steady-state concentrations of monomeric actin, as derived from the plot shown in Fig. 4 c, were 0.15 and 2.5 μM in the absence and presence of a saturating amount of ADF, respectively, consistent with the sedimentation data.
Note that the turbidity reached at steady state (Fig. 4 a) in the presence of different concentrations of ADF is fully consistent with the sedimentation data (Fig. 2 a) and the increase in specific turbidity of F-actin upon binding ADF (Fig. 4 c), as follows. Low concentrations of ADF essentially promote depolymerization of F-actin; hence, filaments are eventually poorly decorated by ADF at steady state, and the final turbidity is low. At higher concentrations of ADF, the final turbidity is higher because of the increased binding of ADF to F-actin.
ADF1 Increases the Association Rate Constant of Actin to Barbed Ends, Not to Pointed Ends of Actin Filaments
To determine the association rate constant of actin-ADF to barbed ends, the effect of ADF on the initial rate of growth from spectrin-actin seeds was examined. Data (Fig. 5 a) were analyzed using Eqs. 3 and 4 (Materials and Methods). The association rate constant of G-actin–ADF complex to barbed ends appeared to be 12 ± 3–fold higher than that of G-actin. This result may seem puzzling since the association rate constant of actin to barbed ends (107 M−1·s−1) has been shown to be diffusion-limited (Drenkhahn and Pollard, 1986). It is plausible that, upon binding to G-actin, ADF induces a dipolar moment in the actin monomer, which modifies the charge distribution at the interface of G-actin with the barbed end, thus enhancing long-range electrostatic interactions and steering the association reaction. As an example, the electrostatically assisted association of barnase to barstar has recently been described (Schreiber and Fersht, 1996) and modeled (Janin, 1997). This interpretation was challenged by testing the shielding effect of ionic strength. In a low ionic strength F buffer (1 mM MgCl2), ADF–G-actin associated 20-fold faster to barbed ends than G-actin. At high ionic strength (0.4 M KCl, 1 mM MgCl2), the steering effect of ADF was abolished. Specifically, no change was observed in the rate of filament elongation when the concentration of ADFATP-G-actin, calculated using Eq. 4 with the experimentally determined value of K d at 20 μM (Table I), represented up to 40% of the total amount of G-actin in the elongation assay. The strong ionic strength dependence of the kinetic facilitation of actin association to barbed ends by ADF therefore supports the view that electrostatic forces are involved in its action.
Figure 5.
ADF1 increases the rate of filament growth at the barbed end, not at the pointed end of actin filaments. (a) Barbed end growth. The initial rate of elongation of G-actin (3.3 μM) was measured turbidimetrically at the indicated concentrations of spectrin-actin seeds, and the following concentrations of ADF (μM): ▪, 0; ▵, 0.5; •, 1; ○, 1.5. (Bottom inset) Typical raw data at 0, 1, and 1.5 μM ADF. (Top inset) The increase in slope of the data shown in the main panel was plotted versus the concentration of MgATP-G-actin-ADF complex calculated using Eq. 3, with K = 8 μM. (b) Pointed end growth. The assembly of 6.5 μM MgATP–G-actin was initiated by the addition of 20 nM (1 and 3) or 4 nM (2 and 4) gelsolin-actin seeds in the absence (1 and 2) and in the presence (3 and 4) of 3 μM ADF. The reaction was started by addition of salt. The turbidity was measured before the addition of salt and subtracted from the polymerization time course. The dead time was 5 s.
The effect of ADF on the rate of actin association to the pointed ends (Fig. 5 b) was evaluated using gelsolin-actin seeds to nucleate pointed end growth. The effect of ADF was much less pronounced than at the barbed ends. The presence of the overshoot indicated that partial depolymerization induced by ADF occurs at least partly from the pointed ends.
If the increase in the rate of elongation from spectrinactin seeds had been due to a severing effect of ADF rather than an effect on the association rate, then the same severing action would have occurred when filaments were induced to grow from their pointed ends. Clearly the data eliminate this possibility.
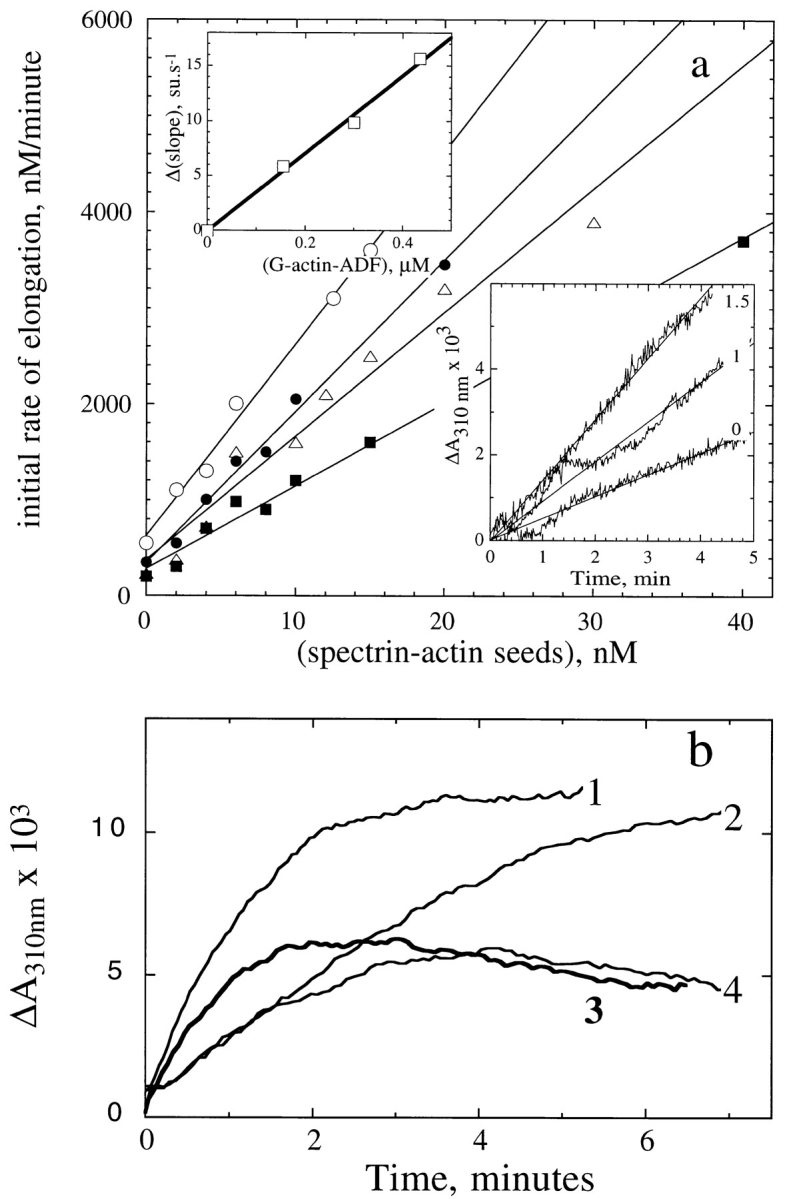
ADF1 Increases the Rate of Filament Depolymerization from the Pointed Ends, Not from the Barbed Ends
Depolymerization of gelsolin-capped actin filaments (3.5 μM F-actin) was induced by addition of ADF at different concentrations and monitored turbidimetrically (Fig. 6 a). The critical concentration at the pointed ends being increased up to 3.5 μM by ADF, addition of ADF to capped F-actin promotes, in these conditions, total depolymerization. The initial rate of depolymerization then truly represents the off rate at the pointed ends. Identical time courses were obtained in the additional presence of Tβ4 used as a sequestering agent (not shown). The rate of depolymerization, in absorbance U/min, was increased up to 36 ± 5–fold by ADF, compared with a control in which total depolymerization was promoted by Tβ4. Once corrected for the 65% higher specific turbidity of ADF–F-actin as compared to F-actin (Fig. 4 c), the rate of depolymerization is actually increased 22 ± 3–fold by ADF. The ADF concentration dependence of the increase in rate reflected the high-affinity 1:1 binding of ADF to ADP–F-actin.
Figure 6.
ADF1 increases the rate of depolymerization from the pointed ends, not from the barbed ends of actin filaments. (a) Depolymerization from the pointed ends. ADF1 was added at the indicated concentrations to a solution of 3.5 μM F-actin and 7 nM gelsolin. The initial rate of depolymerization was measured turbidimetrically. Typical curves are shown in the inset. 0, no ADF; 1, 0.3 μM; 2, 2.4 μM; 3, 6.5 μM; 4, control curve without ADF in which depolymerization was induced by adding 26 μM Tβ4. (The time course shown was recorded over 200 min, i.e., with a 10-fold contracted scale.) (b) Depolymerization from the barbed ends. Depolymerization of F-actin (6 μM) was induced by addition of 6 μM DNaseI in the absence (thin line) and in the presence (thick line) of 10 μM ADF.
The effect of ADF on the rate of depolymerization from the barbed ends was examined next using DNaseI (Fig. 6 b). The rate of DNaseI-induced depolymerization was unaffected by ADF in saturating amounts. From the turbidity data, the initial rate of disassembly was about 100 nM subunits/s, consistent with a population of filaments of 2.5-μm average length depolymerizing at a rate of 12 subunits/s (Pollard and Cooper, 1986). After capping the barbed ends of the same F-actin solution by 120 nM CapG in the presence of 0.2 mM Ca2+ ions (Carlier et al., 1996), depolymerization from the pointed ends was induced by Tβ4 in the presence and absence of ADF. The same 22-fold increase in rate of depolymerization as in Fig. 6 a was observed.
If the increase in the rate of depolymerization from the pointed ends (Fig. 6 a) had been due to a severing action of ADF, creating a large number of uncapped, rapidly depolymerizing filaments, then the same severing action would have caused a large increase in the rate of DNaseIinduced depolymerization from the barbed ends (Fig. 6 b). Clearly the data again eliminate this possibility. Also note that if ADF had a weak severing efficiency of 0.1% (Hawkins et al., 1993), a stoichiometric effect (Fig. 6 a) would not be observed. In conclusion, the effects of ADF on actin assembly and disassembly are end specific.
ADF1 Increases the Turnover of Actin Filaments and the Steady-State ATPase of F-actin
The rate of treadmilling at steady state was derived from the rate at which the nonexchangeable, F-actin–bound εADP was replaced, after an ATP chase, by nonfluorescent ADP as a result of subunit flux through the filaments (Wegner, 1976). The decrease in fluorescence of εADP proceeded linearly with time, at all concentrations of ADF, for over 60% of the total renewal of F-εADP-actin, suggesting that under physiological conditions monomer–polymer exchange is essentially due to treadmilling (Brenner and Korn, 1983). A very large increase in the rate of treadmilling was induced by ADF. The increase was concentration dependent (Fig. 7) and reached a maximum of 25-fold, which corresponded to an average flux of 2 subunits/s. At higher concentrations of ADF, an apparent decrease in the treadmilling rate was recorded, most likely as a result of the inhibition by ADF of εADP dissociation from G-actin after its dissociation from the pointed ends. The exchange of nucleotide on G-actin then becomes rate limiting in the monomer– polymer exchange process. The 25-fold increase in treadmilling rate is consistent with the 22-fold increase in the dissociation rate at the pointed ends.
Figure 7.

ADF1 increases the treadmilling and steady state ATPase rates of F-actin. The rate of treadmilling (□) was derived from the decrease in fluorescence of εADP bound to F-actin (13 μM) after a chase of ATP. The rate of ATP hydrolysis (•) was measured under the same conditions (13.5 μM F-actin) in polymerization buffer containing 0.17 mM γ[32P]ATP. A rate of 0.014 μM/min was measured in the absence of ADF.
The steady-state ATPase of F-actin provides an alternative measure of filament turnover. The ATPase of F-actin was greatly increased by ADF. ATP hydrolysis was linear with time for over 5 h at all ADF concentrations. The data (Fig. 7) superimpose onto the turnover measurements. At the maximum, ∼0.8 ATP was hydrolyzed per second per average filament of 8–10-μm length. After 18 h of incubation at 20°C, at least 90% of the ATP was found hydrolyzed in samples containing 5–10 μM ADF.
ADF1 Increases the Rate of Listeria Propulsion in Platelet Extracts
The actin-based movement of Listeria monocytogenes can be reconstituted in human platelet extracts (Laurent and Carlier, 1997). Assays were carried out increasing the dilution of the extract in extraction buffer supplemented with rhodamine-actin. As documented in detail elsewhere (Laurent and Carlier, 1997; Table I), the rate at which actin “clouds” are formed around the bacteria as well as the rate of propulsion of the bacteria increase upon increasing the dilution of the extracts, reach a maximum, and then decrease at high dilution, most likely because of the limiting amounts of one or several of the cellular components necessary for efficient movement. At a 48-fold dilution of the platelet cytoplasm, Listeria moved at an average steady rate of 4 μm/min, i.e., 2.5-fold lower than the maximum rate observed at a lower dilution, and displayed actin tails of 16-μm length. The fact that both the length of the actin tails and the rate of movement are constant over several hours indicates that movement of Listeria results from a steady state of actin assembly, in which the measured rate of actin polymerization, which drives the movement (Theriot et al., 1992), is equal to the rate of the kinetically limiting step in the steady-state cycle. The ADF/actin molar ratio is 0.1 in platelets (Davidson and Haslam, 1994); hence, the concentration of endogenous ADF might have been at most 200·(0.1)/48 μM = 0.4 μM in the assay. When added to the diluted platelet extract, ADF1 increased the rate of actin-based motility in a concentration-dependent fashion and caused a shortening of the length of the actin tail (Table III). In the presence of 0.75 μM ADF1, the bacteria moved twice as fast and the actin tails were fourfold shorter. Typical pictures are shown in Fig. 8. No effect of ADF on the rate of movement was observed when it was added to less diluted extracts in which Listeria moved at 10 μm/min. These results indicate that in the highly diluted extracts the rate of movement is low because of the limited amounts of endogenous ADF. Platelet extracts appeared to be more convenient than Xenopus egg extracts for monitoring the movement of Listeria at high dilution, presumably because platelets are specialized cells for actin-based motility and contain high amounts of actin-binding proteins.
Table III.
Effect of ADF on the Rate of Propulsion of L. monocytogenes in Platelet Extracts
| Additions | ADF | Rate of Movement | Actin tail length | |||
|---|---|---|---|---|---|---|
| μM | μM/min | μm | ||||
| methyl cellulose 0.13% | 0 | 2.7 ± 0.4 (10) | 28.5 ± 14.8 (10) | |||
| 0.25 | 3.3 ± 0.5 (13) | 8.6 ± 2.9 (15) | ||||
| 0.75 | 5.0 ± 0.5 (8) | 5.3 ± 1.2 (15) | ||||
| No methyl cellulose | 0 | 4 ± 0.7 (10) | 16.3 ± 6.0 (10) | |||
| 0.5 | 5.5 ± 0.9 (9) | 5.9 ± 1 (7) | ||||
| 1 | 10.0 ± 3.5 (5) | 5.0 ± 0.9 (12) | ||||
| 1.5 | cannot be estimated | 3.7 ± 0.6 (10) | ||||
| 3 | cannot be estimated | <1 |
All measurements were made over a period of 30–40 min, starting 30 min after mixing the components of the assay and sealing the samples. Total actin concentration was 6.5 μM. The number of measurements made to derive the average values of the rate of propulsion or the actin tail length is given in parentheses. The same 48-fold dilution of platelet cytoplasm was used in all samples.
Figure 8.

ADF1 increases the rate of propulsion of Listeria monocytogenes in platelet extracts and shortens the length of the actin tails. (a and b) Images of Listeria moving in diluted platelet extracts (no ADF added). (c–e) Images of Listeria in diluted platelet extracts supplemented with 0.75 μM ADF. Times are indicated in min. The trajectories of bacteria are visualized as white lines. Bar, 5 μm.
The effects of ADF on the motility of Listeria are consistent with our biochemical data showing that ADF increases the rate of depolymerization at the pointed ends, which is the kinetically limiting step in the turnover rate of actin filaments and therefore limits the rate of assembly at the barbed ends at the surface of the bacteria. The shortening of the tail is consistent with results (Marchand et al., 1995) indicating that filaments are capped in the tail body and depolymerize from their pointed ends. We checked that capping proteins are functional in platelet extracts by measuring the shift in critical concentration of a pyrenyllabeled F-actin solution upon addition of increasing amounts of platelet extracts. The critical concentration of the pointed ends was established as soon as 5% in volume of the platelet extract (corresponding to a 120-fold dilution of the platelet cytoplasm) was added to F-actin.
As the tails grew shorter and the bacteria moved faster upon addition of ADF, the trajectories of the bacteria became less straight, and the frequent changes in direction made it more difficult to measure the rate of propulsion with accuracy. When high concentrations of ADF were added, the actin tails never reached a size (i.e., acquired a friction coefficient) sufficient to support unidirectional movement. The lower limit size of the actin tail required to start movement was of the same magnitude as the length of the bacterium, i.e., 1 μm. Data are summarized in Table III.
Does ADF1 Fragment Filaments In Vitro?
In agreement with reports on other ADFs (Cooper et al., 1986; Hawkins et al., 1993; Moon et al., 1993; Aizawa et al., 1995), the addition of Arabidopsis ADF to F-actin resulted in a rapid drop in viscosity (data not shown), which temporally correlated with binding of ADF to F-actin and preceded depolymerization. No recovery of viscosity with time was observed. Even after overnight incubation, the viscosity of ADF–F-actin solutions (20 μM, pH 6.5–8.0) remained low, while the F-actin controls were solid gels.
The rapid decrease in viscosity has generally been attributed to the severing activity of ADF. However, the fact that ADF affects the rates of assembly/disassembly differently at both ends (Figs. 5 and 6) cannot be simply accounted for by a severing activity. Consistently, electron microscopy observation of negatively stained samples of ADF-decorated filaments (Fig. 9) failed to display a 20fold decrease in filament length, which should have been observed to account for the kinetic data in Figs. 5 a, 6 a, and 7. The electron micrographs of ADF1-decorated actin filaments show the same features as those obtained by Ohta et al. (1984), i.e., a thickened appearance and contorted shape, but no appreciable change in average length.
Figure 9.
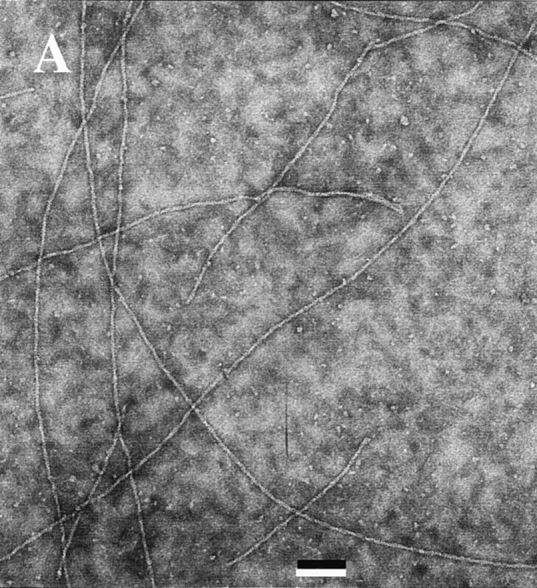
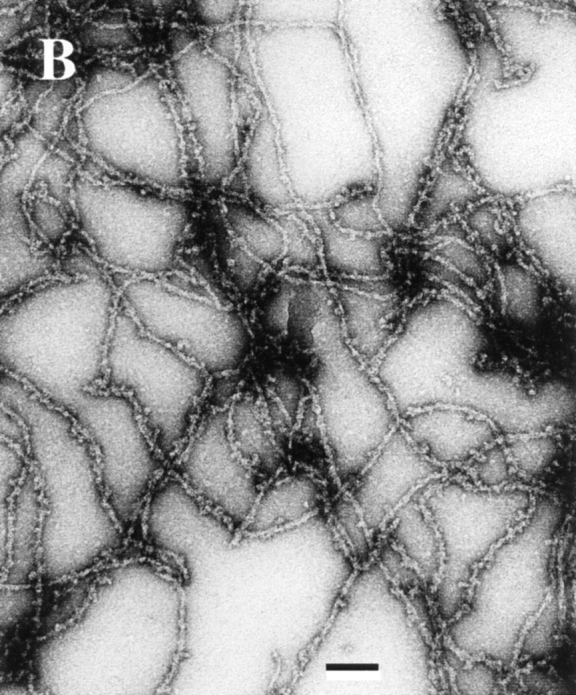
Electron micrographs of F-actin and ADF–F-actin filaments. A solution of 4 μM F-actin polymerized in the absence (A) and in the presence (B) of 5 μM ADF1 in physiological ionic strength buffer, pH 7.8, was deposited on the grid using a largely truncated pipet tip and processed for negative staining as described (Carlier et al., 1994). Bar, 0.1 μm.
To get more insight into the putative severing activity of ADF1, the sedimentation velocity of samples of F-actin (15 μM) containing different concentrations of ADF or gelsolin was examined in the analytical ultracentrifuge at pH 7.8 in physiological ionic strength buffer. The apparent sedimentation coefficient of F-actin at 15 μM was 60 ± 5 S, and increased to 93 ± 7 S in the presence of 20 μM ADF. In contrast, it decreased to 53 ± 5 and 32 ± 2 S in the presence of gelsolin at molar ratios to actin of 1:500 and 1:100, respectively. These data show that actin filaments severed by gelsolin sediment more slowly, while they sediment faster in the presence of ADF. In an additional experiment, the sedimentation velocity of F-actin (7.7 μM) containing gelsolin at a 1:1,000 molar ratio to actin and increasing amounts of ADF was examined. Average sedimentation coefficients of 76 ± 4, 96 ± 5, and 121 ± 6 S were measured for F-actin samples containing 0, 4, and 8 μM ADF, respectively. The concentration dependence of the sedimentation coefficients of F-actin and ADF–F-actin was derived from these data, considering the 3 μM difference in concentration of assembled actin in the presence and absence of ADF. Extrapolation to zero actin concentration yielded the values of S0,20 of 96 ± 6 and 135 ± 10 S for F-actin and ADF–F-actin in the presence of gelsolin at a 1: 1,000 molar ratio to actin. Actin filaments have a persistence length of 7–8 μm (Isambert et al., 1995); hence, they can be considered as rods in this experiment where their average length is 3 μm. The sedimentation coefficient of rods is given by the following equation (Garcia de la Torre, 1992):
(5)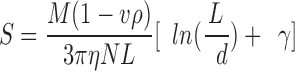 ,
,
where M is the molecular mass of the polymer, L its length, and d its diameter. v is the partial specific volume, ρ the density, η the solvent viscosity, and N the Avogadro's number. For very long rods (L ≫10d), γ takes the value 0.386. The ratio of the molecular masses of ADF–Factin and F-actin subunits being 1.38, the ratio of the average diameters of the corresponding filaments is 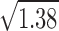 = 1.17 (assuming that the binding of ADF to F-actin thickens the filament but does not increase its length).
= 1.17 (assuming that the binding of ADF to F-actin thickens the filament but does not increase its length).
The ratio of the sedimentation coefficients of ADF–Factin and F-actin can be calculated using Eq. 5, in two cases, using an average filament length of 3,000 nm (fixed by gelsolin) and values of 8 and 9.4 nm for the diameters of F-actin and ADF–F-actin: (a) If ADF does not fragment filaments, the ratio of the sedimentation coefficients is 1.38 [ln(3,000/9.4) + 0.386]/[ln(3,000/8) + 0.386] = 1.35, which is in good agreement with the experimental value of 135/ 96 = 1.4. (b) If ADF had a weak severing activity of 0.1% (Hawkins et al., 1993), the addition of 20 μM ADF to a 15 μM F-actin solution would generate 20 nM fragments, thus decreasing the average length by about threefold. It can then be calculated that the sedimentation coefficient would be decreased by 18% if ADF only severed filaments without binding F-actin, as was proposed to occur at pH above 7.0. It would be increased by only 10% if ADF both severed and bound tightly to F-actin.
In conclusion, although the drop in viscosity of F-actin upon binding ADF1 is suggestive of severing of filaments, electron microscopy and sedimentation velocity fail to confirm the fragmentation hypothesis.
Other Members of the ADF/Cofilin Family Are Biochemically Similar to ADF1 from A. thaliana and Increase the Turnover Rate of Actin Filaments by the Same Mechanism
Yeast cofilin, Acanthamoeba actophorin, and human ADF (destrin) were tested using the assays described in this paper. The following results were obtained: All three proteins bound to F-ADP-actin, causing a quenching of fluorescence of NBD- and pyrenyl-labeled F-actin. In physiological ionic strength buffer, pH 7.8, all three proteins partially depolymerized F-actin like ADF1. The concentration of unassembled actin at steady state was 0.7 and 1.2 μM for the yeast and ameba proteins and 3 μM for human ADF, values close to the one found for ADF1 (1.8 μM). The pH dependence was similar too. The steady-state ATPase of F-actin was increased up to 12-fold by all three proteins.
Turbidimetric kinetic measurements showed that these three proteins increased the rate of disassembly of F-ADPactin from the pointed end. Therefore, the enhancement of actin dynamics is a common feature to all ADF/cofilins, and it has the same mechanistic origin, which indicates that all ADFs/cofilins might have the same function in actin-based motility in different species. This conclusion of biochemical work is in agreement with recent genetic studies (Iida et al., 1993).
Ameba, yeast, and vertebrate ADFs also increased the rate of assembly at the barbed end to different extents, which reflected their different affinities for ATP–G-actin. (Actophorin did not increase the rate of actin assembly at the barbed ends, while yeast and human ADFs did.) All three proteins behaved very similarly to ADF1 turbidimetrically and showed overshoot polymerization time courses.
In conclusion, interesting quantitative differences in the biochemistry of these different ADF/cofilins are worth investigating further, using the quantitative assays that we set up here, to understand how their primary function in the enhancement of actin dynamics may be modulated in different species.
Discussion
The recombinant ADF1 protein from A. thaliana studied here exhibits phenomenological properties similar to other members of the ADF/cofilin protein family. Since it has been largely documented that the native and recombinant ADF/cofilins from other species have identical properties, we can be reasonably confident that the native plant protein is functionally identical to the recombinant protein. The new data reported here lead us to propose a comprehensive interpretation of the present and previous results obtained on a variety of ADF/cofilin variants. A novel view of the mechanism of action of ADF is proposed, according to which the main functional property of ADF in actinbased motility is to increase the turnover rate of actin filaments at steady state. This function is mediated by the large increase in the rate of depolymerization from the pointed ends, which is the rate-limiting step in the steady-state monomer–polymer cycle of actin in the presence of ATP.
The detailed biochemical analysis of ADF1 function has been made possible by the development of new tools (fluorescence quenching, turbidimetry) and by the use of kinetics to understand the mechanism of action of ADF in a quantitative fashion. This kind of study required the use of muscle actin, which is easily available and whose kinetics have been extensively studied. While the physiological relevance of our approach may be questioned, our results compare well with previous works in which muscle actin has been used to study ADF/cofilins from amebas (Maciver et al., 1991), yeast (Moon et al., 1993), starfish oocytes (Sutoh and Mabuchi, 1989), plant (Lopez et al., 1996), and vertebrate nonmuscle tissues (Nishida et al., 1985; Yonezawa et al., 1985; Moriyama et al., 1990; Hayden et al., 1993). To temper the concern one might have about the relevance of such in vitro studies, one should note that the biochemical properties of Acanthamoeba actophorin (Cooper et al., 1986) and D. discoideum cofilin (Aizawa et al., 1995) remained the same when these proteins were assayed with homologous actin. The lethality of yeast cofilin-minus mutants was rescued by mammalian cofilin or ADF (Iida et al., 1993). However, actophorin reacts differently with muscle and ameba actins when Ca2+ is bound to actin (Mossakowska and Korn, 1996).
ADF Does Not Act as a G-actin Sequestering Protein
In a broad pH range (6.5–8.3), the ADF/cofilins assayed here all cause only partial depolymerization of F-actin. Our understanding of ADF function therefore is conceptually different from the one derived from previous in vitro work (for review see Sun et al., 1995). Thus far, ADF from various sources has been described as a protein which completely depolymerized actin in a 1:1 molar ratio at pH 8.0 (Nishida et al., 1985; Yonezawa et al., 1985), which is the exact definition of a high-affinity G-actin sequestering protein. This conclusion was derived from experiments carried out at actin concentrations of 3 μM at most. Our data are in perfect agreement with those; however, our interpretation differs because we observe that at higher actin concentrations, ADF fails to further depolymerize actin. ADF binds to both F- and G-actin, hence the ADF-actin complex should be considered as another polymerizable rather than a sequestered form of actin. This conceptual difference is important in the analysis of the data, as follows.
The action of a G-actin sequestering protein is usually visualized by a shift in critical concentration plots, from which the value of its affinity for G-actin can be derived. The exclusive binding of the protein to ATP–G-actin is implicit in this calculation (Carlier and Pantaloni, 1994). This analysis cannot apply to ADF, which binds to both G- and F-actin, preferentially in their ADP-bound forms. Therefore, the ADF-induced shift in critical concentration plots that have been routinely observed (Hawkins et al., 1993; Hayden et al., 1993) led to an incorrect estimate of the equilibrium dissociation constant for binding of ADF to ATP– G-actin because they were interpreted within a G-actin sequestering activity of ADF. In addition, a strong bias would be introduced in the interpretation by assuming that the changes in fluorescence of labeled actin reflect changes in the amount of F-actin, which we have seen is not the case.
The pH dependence of the F-actin/G-actin ratio in the presence of ADF1, like for other ADFs, is simply quantitative and does not reflect a switch in function from an F- to a G-actin–binding activity of ADF upon increasing pH.
Although ADF tightly binds ADP–G-actin, it cannot be considered as an ADP–G-actin sequestering protein either. Indeed, in the presence of ADP, ADF also binds to F-actin and a true polymerization equilibrium of ADFADP-G-actin into ADF-ADP-F-actin is established, with a critical concentration of 4 μM (see the thermodynamic square scheme presented in Results section).
The Enhancement of Actin Dynamics by ADF Cannot Be Accounted for by Severing of Filaments
The severing of filaments by ADF/cofilins was originally proposed for the following reasons: It was first noticed that ADF accelerated actin polymerization and promoted a rapid drop in fluorescence of NBD- or pyrenyl-labeled F-actin (Cooper et al., 1986; Maciver et al., 1991; Moon et al., 1993; Quirk et al., 1993; Maciver and Weeds, 1994), but both properties were interpreted in terms of an increase in filament number due to fragmentation. The fragmentation hypothesis was enticing because it also provided a satisfactory explanation for the rapid drop in viscosity of F-actin solutions after addition of substoichiometric amounts of ADF, similar to the effect of the severing protein gelsolin. Further effort was made to visualize the fragmentation in electron or optical microscopy. In some instances, filaments were observed to be shorter, which was thought to be because of depolymerization (Abe and Obinata, 1989) or severing (Cooper et al., 1986). In other instances, filaments did not appear shorter (Ohta et al., 1984). In fluorescence optical microscopy, filaments immobilized on myosin-coated glass surfaces and partly stabilized by rhodamine-phalloidin appeared fragmented by flushing ADF in the flow-cell (Maciver et al., 1991).
The fragmentation hypothesis, however, did not provide a description of ADF effects fully consistent with all data. No increase in the number of ends could be detected using cytochalasin B. The severing was then thought to be transient and followed by reannealing (Hawkins et al., 1993; Hayden et al., 1993). Other works rejected the reannealing hypothesis (Nishida et al., 1985; Maciver et al., 1991).
The present work shows that some of the effects of ADF that were attributed to severing should be reinterpreted. First, the rapid decrease in fluorescence of pyrenyl- or NBD-labeled F-actin appears to be due to a quenching of fluorescence linked to ADF binding to F-actin. The depolymerization process is partial at all pHs and occurs on a slower time scale. The possibility of quenching has been evoked in other reports (Ohta et al., 1984; Cooper et al., 1986; Moon et al., 1993) but was not considered quantitatively.
Second, the apparent acceleration previously noted in the time courses of spontaneous polymerization in the presence of ADF/cofilins is explained, in view of the present work, by two independent reasons. First, in previous works, polymerization was started by addition of KCl and MgCl2 to mixtures of ADF and Ca–G-actin. ADF, which slows down metal/nucleotide exchange on G-actin, thereby slowed down the production of rapidly nucleating Mg-actin (Tobacman and Korn, 1983). Second, the tight binding of ADF/cofilin to F-ADP-actin, produced late in the polymerization process (Carlier, 1991), causes a delayed increase in the specific light scattering of the filament, which was not appreciated in earlier works.
Third, the present kinetic data showing that the increases in rates are different at the two ends rule out the fragmentation hypothesis as an interpretation of the effects of ADF on actin assembly/disassembly. Fourth, both electron microscope observations and sedimentation velocity data fail to show evidence for appreciable fragmentation of filaments by ADF1.
The absence of evident severing activity of ADF argues against the structural model recently proposed on the basis of a severing activity of ADF (Hatanaka et al., 1996), according to which ADF would bind actin like gelsolin segment-1, at the pointed end of the actin monomer.
Proposals can be made to reconcile the above discrepancies about the severing activity of ADFs. The possibility cannot be discounted that some F-actin–binding proteins enhance photobleaching-induced fragmentation. It is also possible that ADF-decorated filaments are more fragile than native filaments and break more easily when submitted to the mechanical stresses involved in the preparation of samples for electron microscopy, to shearing forces in Ostwald-type viscometers (Ohta et al., 1984), or to Brownian movement. Such side effects, however, are different from a gelsolin-like activity. More experiments are needed to explain these discrepancies.
If the drop in viscosity cannot be interpreted by severing of filaments, an alternative explanation should be sought for the change in viscosity of the filaments linked to ADF binding to F-actin. It is possible that ADF binding induces a large change in the flexibility of the filaments. As noted by Ohta et al. (1984), the binding of ADF to F-actin may change the surface properties of the filaments, affecting their electroviscosity, thus preventing the filament–filament interactions that lead to the formation of a gel. It is also possible that the ADF-induced increase in treadmilling rate changes the rheological properties of actin (Isambert and Maggs, 1996). Experiments are in progress to address these questions. Interestingly, the structural/mechanical change in actin filaments linked to the binding of an accessory protein may provide an alternative mechanistic description of the gel–sol transition, thus far understood generally in terms of filament severing (Bray, 1992).
ADF Functions as an Actin Dynamizing Factor
We show that ADF can increase up to 25-fold the rate of treadmilling of actin filaments at steady state. This result implies that the rate-limiting step in the steady-state monomer–polymer exchange process is increased by ADF. The rate-limiting step is the dissociation of ADP-actin subunits from the pointed ends, which we show to be consistently 22-fold higher when ADF is bound to F-actin. It is remarkable that the effect of ADF is end specific, the rate of dissociation of ADP-actin from the barbed ends being unaffected by ADF.
The effect of ADF on the steady-state turnover of F-actin is displayed in Fig. 10. In the absence of ADF, it is known that at steady state, barbed ends contain predominantly ADP-Pi subunits, while ADP subunits are present at the pointed ends. Net slow association of ATP–G-actin at the barbed ends is compensated by net slow depolymerization of ADP-actin at the pointed ends, which is the rate-limiting step in the cycle. To be specific, at the measured steady-state concentration of ATP-actin (C SS), the on rate at the barbed end is k B + ·(C SS − C B C), where C B C is the critical concentration at the barbed end. The off rate at the pointed end is very close to k P −, and these two fluxes are equal and of opposite sign. Using established values of 10 μM−1 for k B + , 0.1 μM for C SS, and 0.2 s−1 for k P − (Pollard and Cooper, 1986), we conclude that the value of C B C must be 0.08 μM to support a steady-state flux of 0.2 s−1.
Figure 10.

ADF increases the treadmilling of actin filaments. T, D-Pi, and D represent the ATP, ADP-Pi, and ADP, respectively, bound to actin. The different sizes of the different species drawn are meant to give an idea of their relative steady-state concentrations.
In the presence of ADF, the steady-state cycle is faster as a result of the faster depolymerization of ADF-F-ADPactin from the pointed ends. Subsequent dissociation of ADF from its complex with ADP–G-actin, followed by exchange of ATP for bound ADP, results in a larger association flux of ATP–G-actin (at a new steady-state concentration C′SS) and ADF-ATP-G-actin to barbed ends. The new barbed end association flux is:
(6)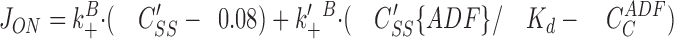
where {ADF} is the concentration of free ADF, C c ADFis the critical concentration for polymerization of ADF-ATP-Gactin, and k B + is the association rate constant of ADFATP-G-actin to barbed ends. Assuming {ADF} = 1 μM as an example, since K d = 10 μM, the concentration of ADFATP-G-actin is 10-fold lower than the concentration of ATP–G-actin. Since its critical concentration for polymerization ( C c ADF) is probably much larger than zero, the contribution of ADF-ATP-G-actin to barbed end growth at steady state (second term in Eq. 6) is much lower than that of ATP–G-actin. Within this simplification, a 25-fold faster steady-state flux of subunits through the filament (i.e., 5 s−1) will be established at a steady-state concentration of ATP–G-actin C SS′ given by the following equation.
(7)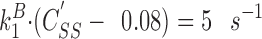 ,
,
which leads to C SS′ = 0.58 μM, to be compared to the value of 0.1 μM found in the absence of ADF. This calculation emphasizes that a 25-fold faster treadmilling rate can be obtained at a steady-state concentration of ATP– G-actin less than sixfold higher than in the absence of ADF. In summary, an off rate of 5 s−1 at the pointed end is generated by the depolymerization of ADF-ADP-F-actin, and the concentration of ATP–G-actin self-adjusts to a higher value that allows the on-flux at the barbed end to exactly compensate the off-flux from the pointed ends.
In the presence of ADF, the steady-state concentration of monomeric actin is a mixture of ATP–G-actin, ADP–Gactin, and their complexes with ADF. While the total concentration of all these components (1.7 μM) is obtained in sedimentation assays, the exact partial concentration of each of these species is the steady-state solution of a set of differential equations describing the kinetics of association and dissociation of each species to the two ends of the filament (in preparation). According to the above calculation, in the presence of 1 μM free ADF, if ATP–G-actin is equal to 0.58 μM at steady state, and if the amounts of ADF-ATP-G-actin and ADP–G-actin represent only 10% of the amounts of ATP–G-actin and ADF-ADP-G-actin, respectively (see the values of the equilibrium dissociation constants, Table II), the measured amount of 1.7 μM unassembled actin would imply that the concentration of ADFADP-G-actin is close to 1.12 μM. These values should be considered as plausible approximates that reasonably account for the data.
We should note that because ADF has a much higher affinity for ADP-actin than for ATP-actin, the nature of the major actin species that associates to the filament (mostly ATP-actin) is not the same as the one that dissociates from the filament (mostly ADF-ADP-actin), both in terms of bound nucleotide and in terms of bound ligand that potentially allows ADF to modulate the nonlinearity in the J(c) plots (Carlier, 1991).
ADF Activates Actin-based Motility
Do the above in vitro biochemical properties of ADF provide a clue to understanding its cellular function in actinbased motility? It has long been recognized (Wang, 1985; Fechheimer and Zigmond, 1993) that net polymerization of actin occurs at the front of locomoting cells, while net depolymerization occurs throughout the lamella, but puzzlingly the rate of actin flux in this treadmilling-like process was one order of magnitude faster than expected from the in vitro measurements of actin filament treadmilling. Similarly, the rate of Listeria movement in Xenopus egg extracts appeared 10-fold faster than the calculated rate of actin assembly (Marchand et al., 1995) in the medium. The present data showing that ADF increases the rate of treadmilling in vitro up to values of the same magnitude as the one observed in vivo in the lamellipodium (Small, 1995; Small et al., 1995) lead us to conclude that endogenous ADF is responsible for this fast rate. The actin-based propulsion of Listeria monocytogenes in acellular extracts offers an opportunity to test the effects of ADF in an integrated system closer to the in vivo situation. ADF increases the rate of propulsion because it increases the rate of depolymerization from the pointed ends of the bulk population of filaments, which kinetically limits the rate of barbed end assembly at the bacterium surface. This in turn causes the shortening of the capped filaments present in the medium, in particular those in the body of the actin tail attached to the bacteria, hence a shortening of the tail itself. Assuming that the motility assay of Listeria in acellular extracts is a good model for actin-based motile processes in response to signaling, these results indicate that ADF might, by the same mechanism, control the dynamics and the length of actin filaments in vivo. They also account for the enhanced motility of cells overexpressing cofilin (Aizawa et al., 1996), and for the high levels of ADF in developmental stages (Bamburg and Bray, 1987; Abe et al., 1989, 1996), in which extensive actin dynamics are involved.
In conclusion, it is emphasized that different actin-binding proteins amazingly use the conformational switch of ATP hydrolysis on actin in a variety of ways to modulate actin dynamics. Profilin, in binding preferentially to ATPactin, promotes assembly at the barbed ends (Pantaloni and Carlier, 1993; Perelroizen et al., 1996). ADF, in binding preferentially to ADP-actin, enhances the directional shuttling of subunits through the filaments.
Acknowledgments
We thank Drs. Yukio Doi, David Drubin, Thomas D. Pollard, and Helen Yin for generous gifts of gelsolin, yeast cofilin, actophorin, and CapG, respectively, and Dr. Sally Zigmond for a detailed protocol of the preparation of spectrin-actin seeds. We are grateful to Jean Lepault for the electron microscopy work. We thank Matt Welch and Tim Mitchison for communication of the use of platelets to monitor actin-based motility of Listeria. We acknowledge helpful discussions with Drs. François Amblard, Joël Janin, Anthony Maggs, and Annemarie Weber, and the constructive comments of Dr. E.D. Korn on an earlier version of the manuscript.
Abbreviations used in this paper
- ADF
actin depolymerizing factor
- NBD
7-chloro-4-nitrobenzeno-2-oxa-1,3-diazole
- Tβ4
thymosin β4
Footnotes
This work was funded in part by the Association pour la Recherche contre le Cancer (ARC), the Association Française contre les Myopathies (AFM), the EC (grant No. CHRX-CT94-0652), the Ligue Nationale Française contre le Cancer, and a grant from the National Science and Technology Board, Singapore.
Address all correspondence to M.-F. Carlier, LEBS, CNRS, Gif-surYvette 91198, France. Tel.: 33-1-69 82 34 65. Fax: 33-1-69 82 31 29. E-mail: carlier@lebs.cnrs-gif.fr
References
- Abe H, Obinata T. An actin-depolymerizing protein in embryonic chicken skeletal muscle: purification and characterization. J Biochem. 1989;106:172–180. doi: 10.1093/oxfordjournals.jbchem.a122810. [DOI] [PubMed] [Google Scholar]
- Abe H, Oshima S, Obinata T. A cofilin-like protein is involved in the regulation of actin assembly in developing skeletal muscle. J Biochem. 1989;106:696–702. doi: 10.1093/oxfordjournals.jbchem.a122919. [DOI] [PubMed] [Google Scholar]
- Abe H, Obinata T, Minamide LS, Bamburg JR. Xenopus laevisactin-depolymerizing factor/cofilin: a phosphorylation-regulated protein essential for development. J Cell Biol. 1996;132:871–885. doi: 10.1083/jcb.132.5.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnew BJ, Minamide LS, Bamburg JR. Reactivation of phosphorylated actin depolymerizing factor and identification of the regulatory site. J Biol Chem. 1995;270:17582–17587. doi: 10.1074/jbc.270.29.17582. [DOI] [PubMed] [Google Scholar]
- Aizawa H, Sutoh K, Tsubuki S, Kawashima S, Ishii A, Yahara I. Identification, characterization and intracellular distribution of cofilin in Dictyostelium discoideum. . J Biol Chem. 1995;270:10923–10932. doi: 10.1074/jbc.270.18.10923. [DOI] [PubMed] [Google Scholar]
- Aizawa H, Sutoh K, Yahara I. Overexpression of cofilin stimulates bundling of actin filaments, membrane ruffling and cell movement in Dictyostelium. . J Cell Biol. 1996;132:335–344. doi: 10.1083/jcb.132.3.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamburg JR, Bray D. Distribution and cellular localization of actin depolymerizing factor. J Cell Biol. 1987;105:2817–2825. doi: 10.1083/jcb.105.6.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchoin L, Didry D, Carlier M-F, Pantaloni D. Kinetics association of myosin subfragment-1 to unlabeled and pyrenyl-labeled actin. J Biol Chem. 1996;271:12380–12386. doi: 10.1074/jbc.271.21.12380. [DOI] [PubMed] [Google Scholar]
- Blickstad I, Markey F, Carlsson L, Persson T, Lindberg U. Selective assay of monomeric and filamentous actin in cell extracts, using inhibition of deoxyribonuclease I. Cell. 1978;15:935–943. doi: 10.1016/0092-8674(78)90277-5. [DOI] [PubMed] [Google Scholar]
- Bray, D. 1992. Cell Movements. Garland Publishing, Inc., New York. 143–155.
- Brenner SL, Korn ED. On the mechanism of actin monomer-polymer subunit exchange at steady state. J Biol Chem. 1983;258:5013–5020. [PubMed] [Google Scholar]
- Carlier M-F. Actin: protein structure and filament dynamics. J Biol Chem. 1991;266:1–4. [PubMed] [Google Scholar]
- Carlier M-F, Pantaloni D. Actin assembly in response to extracellular signals: role of capping proteins, thymosin β4and profilin. Semin Cell Biol. 1994;5:183–191. doi: 10.1006/scel.1994.1023. [DOI] [PubMed] [Google Scholar]
- Carlier M-F, Pantaloni D, Korn ED. Polymerization of ADPactin and ATP-actin under sonication and characteristics of the ATP-actin polymer. J Biol Chem. 1985;260:6565–6571. [PubMed] [Google Scholar]
- Carlier M-F, Pantaloni D, Korn ED. The effects of Mg2+at the high-affinity and low-affinity sites on the polymerization of actin and associated ATP hydrolysis. J Biol Chem. 1986;261:10785–10792. [PubMed] [Google Scholar]
- Carlier M-F, Didry D, Erk I, Lepault J, Pantaloni D. Myosin subfragment-1-induced polymerization of G-actin. Formation of partially decorated filaments at high actin-S1 ratios. J Biol Chem. 1994;269:3829–3837. [PubMed] [Google Scholar]
- Carlier M-F, Didry D, Erk I, Lepault J, Van Troys ML, Vandekerckhove J, Perelroizen I, Yin H, Doi Y, Pantaloni D. Tβ4is not a simple G-actin sequestering protein and interacts with F-actin at high concentration. J Biol Chem. 1996;271:9231–9239. doi: 10.1074/jbc.271.16.9231. [DOI] [PubMed] [Google Scholar]
- Casella JF, Maack DJ, Lin S. Purification and initial characterization of a protein from skeletal muscle that caps the barbed ends of actin filaments. J Biol Chem. 1986;261:10915–10921. [PubMed] [Google Scholar]
- Condeelis J. Life at the leading edge: the formation of cell protrusions. Annu Rev Cell Biol. 1993;9:411–444. doi: 10.1146/annurev.cb.09.110193.002211. [DOI] [PubMed] [Google Scholar]
- Cooper JA, Blum JD, Williams RC, Pollard TD. Purification and characterization of Actophorin, a new 15,000 dalton actin-binding protein from Acanthamoeba castellanii. . J Biol Chem. 1986;261:477–485. [PubMed] [Google Scholar]
- Davidson MM, Haslam TJ. Dephosphorylation of cofilin in stimulated platelets: role for a GTP-binding protein and Ca2+ . Biochem J. 1994;301:41–47. doi: 10.1042/bj3010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmers P, Weber A, Elzinga M, Stephens RE. 7-chloro-4-nitrobenzeno-2-oxa-1,3-diazole actin as a probe for actin polymerization. J Biol Chem. 1981;256:99–105. [PubMed] [Google Scholar]
- Drenkhahn D, Pollard TD. Elongation of actin filaments is a diffusion-limited reaction at the barbed end and is accelerated by inert macromolecules. J Biol Chem. 1986;261:12754–12758. [PubMed] [Google Scholar]
- Fechheimer M, Zigmond SH. Focusing on unpolymerized actin. J Cell Biol. 1993;123:1–5. doi: 10.1083/jcb.123.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia de la Torre, J. 1992. Sedimentation coefficients of complex biological particles. In Analytical Centrifugation in Biochemistry and Polymer Science. S.E. Harding, A.J. Rowe, and J.C. Horton, editors. Royal Society of Chemistry, Cambridge. 333–345.
- Hatanaka H, Ogura K, Moriyama K, Ichikawa S, Yahara I, Inagaki F. Tertiary structure of destrin and structural similarity between two actin-regulating protein families. Cell. 1996;85:1047–1055. doi: 10.1016/s0092-8674(00)81305-7. [DOI] [PubMed] [Google Scholar]
- Hawkins M, Pope B, Maciver SK, Weeds AG. Human actin depolymerizing factor mediates a pH-sensitive destruction of actin filaments. Biochemistry. 1993;32:9985–9993. doi: 10.1021/bi00089a014. [DOI] [PubMed] [Google Scholar]
- Hayden SDM, Miller PS, Brauweiler A, Bamburg JR. Analysis of the interactions of actin depolymerizing factor with G- and F-actin. Biochemistry. 1993;32:9994–10004. doi: 10.1021/bi00089a015. [DOI] [PubMed] [Google Scholar]
- Iida K, Moriyama K, Matsumoto S, Kawasaki H, Nishida E, Yahara I. Isolation of a yeast essential gene, COF1, that encodes a homolog of mammalian cofilin, a low molecular weight actin-binding and depolymerizing protein. Gene. 1993;124:115–120. doi: 10.1016/0378-1119(93)90770-4. [DOI] [PubMed] [Google Scholar]
- Isambert H, Maggs AC. Dynamics and rheology of actin solutions. Macromolecules. 1996;29:1036–1040. [Google Scholar]
- Isambert H, Venier P, Maggs AC, Fattoum A, Kassab R, Pantaloni D, Carlier M-F. Flexibility of actin filaments derived from thermal fluctuations. J Biol Chem. 1995;270:11437–11444. doi: 10.1074/jbc.270.19.11437. [DOI] [PubMed] [Google Scholar]
- Janin, J. 1997. The kinetics of protein-protein recognition. Proteins. Struct. Funct. Genet. In press. [DOI] [PubMed]
- Kouyama T, Mihashi K. Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labeled F-actin. Eur J Biochem. 1981;114:33–38. [PubMed] [Google Scholar]
- Laurent, V., and M.-F. Carlier. 1997. Use of platelet extracts for actin-based motility of Listeria monocytogenes. In Cell Biology: A Laboratory Handbook. J. Celis, editor. Academic Press, New York. In press.
- Lopez I, Anthony RG, Maciver SK, Jiang C-J, Khan S, Weeds AG, Hussey PJ. Pollen specific expression of maize genes encoding actin depolymering factor-like proteins. Proc Natl Acad Sci USA. 1996;93:7415–7420. doi: 10.1073/pnas.93.14.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciver SK, Weeds AG. Actophorin preferentially binds monomeric ADP-actin over ATP-bound actin: consequences for cell locomotion. FEBS (Fed Exp Biol Soc) Lett. 1994;347:251–256. doi: 10.1016/0014-5793(94)00552-4. [DOI] [PubMed] [Google Scholar]
- Maciver SK, Zot HG, Pollard TD. Characterization of actin filament severing by actophorin from Acanthamoeba castellanii. . J Cell Biol. 1991;115:1611–1620. doi: 10.1083/jcb.115.6.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand J-B, Moreau P, Paoletti A, Cossart P, Carlier M-F, Pantaloni D. Actin-based movement of Listeria monocytogenes: actin assembly results from the local maintenance of uncapped barbed ends at the bacterium surface. J Cell Biol. 1995;130:331–343. doi: 10.1083/jcb.130.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon A, Drubin DG. The ADF/cofilin proteins: stimulus-responsive modulations of actin dynamics. Mol Biol Cell. 1995;6:1423–1431. doi: 10.1091/mbc.6.11.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon AL, Janmey PA, Louie KA, Dubin DG. Cofilin is an essential component of the yeast cortical cytoskeleton. J Cell Biol. 1993;120:421–435. doi: 10.1083/jcb.120.2.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama K, Nishida E, Yonezawa N, Sakai H, Matsumoto S, Iida K, Yahara I. Destrin, a mammalian actin-depolymerizing protein, is closely related to cofilin: cloning and expression of porcine brain destrin cDNA. J Biol Chem. 1990;265:5768–5773. [PubMed] [Google Scholar]
- Mossakowska M, Korn ED. Ca++ bound to the high-affinity divalent cation binding site of actin enhances actophorin-induced depolymerization of muscle F-actin but inhibits actophorin-induced depolymerization of AcanthamoebaF-actin. J Mus Res Cell Motil. 1996;17:383–389. doi: 10.1007/BF00123355. [DOI] [PubMed] [Google Scholar]
- Nishida E, Maekawa S, Sakai H. Cofilin, a protein in porcine brain that binds to actin filaments and inhibits their interactions with myosin and tropomyosin. Biochemistry. 1984;23:5307–5313. doi: 10.1021/bi00317a032. [DOI] [PubMed] [Google Scholar]
- Nishida E, Mineyuki E, Mekawa S, Ohta Y, Sakai H. An actindepolymerizing protein (destrin) from porcine kidney. Its action on F-actin containing or lacking tropomyosin. Biochemistry. 1985;24:6624–6630. doi: 10.1021/bi00344a049. [DOI] [PubMed] [Google Scholar]
- Ohta Y, Endo S, Nishida E, Murofushi H, Sakai H. An 18K protein from ascites hepatoma cell depolymerizes actin filaments rapidly. J Biochem. 1984;96:1547–1588. doi: 10.1093/oxfordjournals.jbchem.a134984. [DOI] [PubMed] [Google Scholar]
- Ohta Y, Nishida E, Sakai H, Miyamoto E. Dephosphorylation of cofilin accompanies heat shock-induced nuclear accumulation of cofilin. J Biol Chem. 1989;264:16143–16148. [PubMed] [Google Scholar]
- Pantaloni D, Carlier M-F. How profilin promotes actin filament assembly in the presence of thymosin β4 . Cell. 1993;75:1007–1014. doi: 10.1016/0092-8674(93)90544-z. [DOI] [PubMed] [Google Scholar]
- Perelroizen I, Didry D, Christensen HE, Chua NH, Carlier M-F. Role of nucleotide exchange and hydrolysis in the function of profilin in actin assembly. J Biol Chem. 1996;271:12302–12309. doi: 10.1074/jbc.271.21.12302. [DOI] [PubMed] [Google Scholar]
- Podolski JL, Steck TL. Association of DNaseI with the pointed ends of actin filaments in human red blood cell membrane skeletons. J Biol Chem. 1988;263:638–645. [PubMed] [Google Scholar]
- Pollard TD, Cooper JA. Actin and actin binding proteins: a critical evaluation of mechanisms and functions. Annu Rev Biochem. 1986;55:987–1035. doi: 10.1146/annurev.bi.55.070186.005011. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Goldberg I, Schwarz WH. Nucleotide exchange, structure and mechanical properties of filaments assembled from ATP-actin and ADP-actin. J Biol Chem. 1992;267:20339–20345. [PubMed] [Google Scholar]
- Quirk S, Maciver SK, Ampe C, Doberstein SK, Kaiser DA, Van Damme J, Vandekerckhove JS, Pollard TD. Primary structure of and studies on Acanthamoebaactophorin. Biochemistry. 1993;32:8525–8533. doi: 10.1021/bi00084a019. [DOI] [PubMed] [Google Scholar]
- Schreiber G, Fersht AR. Rapid, electrostatically assisted association of proteins. Nat Struct Biol. 1996;3:427–431. doi: 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- Small JV. Getting the actin filaments straight: nucleation-release or treadmilling. Trends Cell Biol. 1995;5:52–54. doi: 10.1016/s0962-8924(00)88939-4. [DOI] [PubMed] [Google Scholar]
- Small JV, Herzog M, Anderson K. Actin filament organization in the fish keratocyte lamellipodium. J Cell Biol. 1995;129:1275–1286. doi: 10.1083/jcb.129.5.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- Sun H-Q, Kwiatkowska K, Yin HL. Actin monomer binding proteins. Curr Opin Cell Biol. 1995;7:102–110. doi: 10.1016/0955-0674(95)80051-4. [DOI] [PubMed] [Google Scholar]
- Sutoh K, Mabuchi I. End-label fingerprintings show that an N-terminal segment of depactin participates in interaction with actin. Biochemistry. 1989;28:102–106. doi: 10.1021/bi00427a015. [DOI] [PubMed] [Google Scholar]
- Theriot JA, Mitchison TJ, Tilney LG, Portnoy DA. The rate of actin-based motility of intracellular Listeria monocytogenesequals the rate of actin polymerization. Nature (Lond) 1992;357:257–260. doi: 10.1038/357257a0. [DOI] [PubMed] [Google Scholar]
- Tobacman LS, Korn ED. The kinetics of actin nucleation and polymerization. J Biol Chem. 1983;258:3207–3214. [PubMed] [Google Scholar]
- Valentin-Ranc C, Carlier M-F. Evidence for the direct interaction between tightly bound divalent metal ion and ATP on actin. J Biol Chem. 1989;264:20871–20880. [PubMed] [Google Scholar]
- Wang Y-L. Exchange of actin subunits at the leading edge of living fibroblasts: possible role of treadmilling. J Cell Biol. 1985;101:597–602. doi: 10.1083/jcb.101.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A, Nachmias VT, Pennise CR, Pring M, Safer D. Interaction of thymosin β4with muscle and platelet actin: implications for actin sequestration in resting platelets. Biochemistry. 1992;31:6179–6185. doi: 10.1021/bi00142a002. [DOI] [PubMed] [Google Scholar]
- Weber A, Pennise CR, Pring M. DNaseI increases the rate constant of depolymerization at the pointed ends of actin filaments. Biochemistry. 1994;33:4780–4786. doi: 10.1021/bi00182a005. [DOI] [PubMed] [Google Scholar]
- Wegner A. Head to tail polymerization of actin. J Mol Biol. 1976;108:139–150. doi: 10.1016/s0022-2836(76)80100-3. [DOI] [PubMed] [Google Scholar]
- Yonezawa N, Nishida E, Sakai H. pH control of actin polymerization by cofilin. J Biol Chem. 1985;260:14410–14412. [PubMed] [Google Scholar]