

Abstract
Listeria monocytogenes is a facultative intracellular bacterial pathogen that spreads cell to cell without exposure to the extracellular environment. Bacterial cell-to-cell spread is mediated in part by two secreted bacterial phospholipases C (PLC), a broad spectrum PLC (PC-PLC) and a phosphatidylinositolspecific PLC (PI-PLC). PI-PLC is secreted in an active state, whereas PC-PLC is secreted as an inactive proenzyme (proPC-PLC) whose activation is mediated in vitro by an L. monocytogenes metalloprotease (Mpl). Analysis of PI-PLC, PC-PLC, and Mpl single and double mutants revealed that Mpl also plays a role in the spread of an infection, but suggested that proPC-PLC has an Mpl-independent activation pathway. Using biochemical and microscopic approaches, we describe three intracellular proteolytic pathways regulating PCPLC activity. Initially, proPC-PLC secreted in the cytosol of infected cells was rapidly degraded in a proteasome-dependent manner. Later during infection, PCPLC colocalized with bacteria in lysosome-associated membrane protein 1–positive vacuoles. Activation of proPC-PLC in vacuoles was mediated by Mpl and an Mpl-independent pathway, the latter being sensitive to inhibitors of cysteine proteases. Lastly, proPC-PLC activation by either pathway was sensitive to bafilomycin A1, a specific inhibitor of vacuolar ATPase, suggesting that activation was dependent on acidification of the vacuolar compartment. These results are consistent with a model in which proPC-PLC activation is compartment specific and controlled by a combination of bacterial and host factors.
L isteria monocytogenes is a facultative intracellular bacterial pathogen that infects a variety of mammalian cells both in vivo and in vitro (Cossart and Mengaud, 1989; Gaillard et al., 1987; Havell, 1986; Kuhn et al., 1988; Portnoy et al., 1988). Microscopic studies of the intracellular growth cycle of L. monocytogenes have revealed a fascinating relationship between this microbe and its host (Mounier et al., 1990; Tilney and Portnoy, 1989). Subsequent to internalization, the bacterium mediates lysis of the surrounding vacuolar membrane and initiates rapid intracytosolic multiplication. Asymmetric polymerization of host actin at the bacterial surface results in actin-based motility and formation of filopodia-like structures that facilitate direct bacterial cell-to-cell spread (Dabiri et al., 1990; Mounier et al., 1990; Theriot et al., 1992; Tilney and Portnoy, 1989). During cell-to-cell spread, bacteria become transiently confined in secondary vacuoles, which are presumably formed upon phagocytosis of filopodia-like structures (Mounier et al., 1990; Tilney and Portnoy, 1989). Lastly, lysis of secondary vacuoles results in continuation of the intracellular cycle. This strategy of cell-to-cell spread facilitates propagation of the bacterial infection without exposure to the host's humoral immune response. A similar strategy has been adapted by a number of other microbial pathogens including Shigellae (Bernardini et al., 1989), Rickettsiae (Heinzen et al., 1993; Teysseire et al., 1992), and Vaccinia virus (Cudmore et al., 1995).
The primary virulence factors of L. monocytogenes have been identified (Portnoy et al., 1992). Lysis of the primary vacuole, formed upon initial phagocytosis, is largely mediated by a secreted pore-forming hemolysin called listeriolysin O (LLO)1 (Bielecki et al., 1990; Gaillard et al., 1987; Tilney and Portnoy, 1989). Nonhemolytic mutants of L. monocytogenes are unable to grow intracellularly in most cell types (Gaillard et al., 1987; Portnoy et al., 1988) and are completely avirulent in mice (Cossart et al., 1989; Gaillard et al., 1986; Kathariou et al., 1987; Michel et al., 1990; Portnoy et al., 1988). Escape from the primary vacuole is also enhanced by two secreted bacterial phospholipases C (PLC): a phosphatidylinositol-specific PLC (PI-PLC) (Camilli et al., 1993) and a broad-range PLC (PC-PLC) (Marquis et al., 1995; Smith et al., 1995). The ability to spread depends on the L. monocytogenes ActA protein that mediates actin polymerization (Brundage et al., 1993; Domann et al., 1992; Kocks et al., 1992). ActA mutants are able to escape from the primary vacuole, but they are unable to spread within cells or cell to cell. Bacterial factors mediating escape from the secondary vacuole formed during cell-to-cell spread are not as well defined as those for escape from the primary vacuole. The role of LLO in bacterial cell-to-cell spread has been difficult to address as nonhemolytic mutants fail to gain access to the cytosol (Gaillard et al., 1987; Tilney and Portnoy, 1989). However, the two PLCs are clearly important as mutants lacking either PC-PLC or both PLCs form small plaques in murine fibroblasts, presumably because they are less efficient in escaping from secondary vacuoles (Smith et al., 1995; Vazquez-Boland et al., 1992). The precise functions of the PLCs or their precise sites of action are not clear.
PC-PLC is secreted as an inactive precursor (proPCPLC), and proteolytic cleavage at its NH2 terminus generates the active form of the enzyme (Niebuhr et al., 1993; Raveneau et al., 1992). In vitro activation of proPC-PLC is mediated by a secreted bacterial metalloprotease (Mpl) with homology to members of a family of bacterial zinccontaining metalloproteases (Poyart et al., 1993; Domann et al., 1991; Häse and Finkelstein, 1993; Mengaud et al., 1991). The in vivo role of Mpl has not been determined.
In the present study we have defined the requirements for the intracellular activation of proPC-PLC. The intracellular activation of proPC-PLC was mediated by two different enzymes: a bacterial metalloprotease (Mpl), which is also active in vitro, and a cysteine protease, whose activity could only be detected during intracellular infection. The relative activity of PC-PLC generated by either protease on phosphatidylcholine (PC) or sphingomyelin was essentially the same. Furthermore, proPC-PLC activation by either pathway was dependent on (a) vacuolar bacterial localization and (b) vacuolar acidification. In the absence of bacterial cell-to-cell spread, proPC-PLC was synthesized but not activated. Instead, cytosolic proPC-PLC was rapidly degraded in a proteasome-dependent manner.
Materials and Methods
Bacterial Strains and Culture Conditions
Bacterial strains and relevant genotypes are listed in Table I. Two wildtype L. monocytogenes strains were used for this study: 10403S (Bishop and Hinrichs, 1987) and SLCC-5764 (Camilli et al., 1993). For all cell infection assays, strain 10403S and isogenic mutants were used. Strain SLCC-5764 and isogenic mutants were only used for detection of PC-PLC in broth culture. All strains were grown in brain heart infusion broth (BHI; Difco Laboratories, Detroit, MI) and maintained on BHI agar. Stock cultures were stored at −80°C.
Table I.
Bacterial Strains and Genotypes
| Strain | Genotype | Source of strain or reference | ||
|---|---|---|---|---|
| 10403S | Wild-type | Bishop and Hinrichs, 1987 | ||
| DP-L1552 | 10403S ΔplcA | Camilli et al., 1993 | ||
| DP-L1935 | 10403S ΔplcB | Smith et al., 1995 | ||
| DP-L1936 | 10403S ΔplcA, ΔplcB | Smith et al., 1995 | ||
| DP-L1942 | 10403S ΔactA | Brundage et al., 1993 | ||
| DP-L2296 | 10403S Δmpl | Marquis et al., 1995 | ||
| DP-L2483 | 10403S ΔplcA, Δmpl | This study | ||
| DP-L2484 | DP-L2296 complemented with the wild-type mpl allele | This study | ||
| DP-L2787 | 10403S ΔplcB, Δmpl | This study | ||
| DP-L2869 | 10403S ΔplcA, ΔplcB, Δmpl | This study | ||
| SLCC-5764 | Hypersecreting wild-type isolate | Camilli et al., 1993 | ||
| DP-L1545 | SLCC-5764 Δmpl | This study | ||
| DP-L1553 | SLCC-5764 ΔplcA | Camilli et al., 1993 | ||
| DP-L1938 | SLCC-5764 ΔplcA, ΔplcB | This study | ||
| DP-L1955 | SLCC-5764 ΔactA | This study |
Construction of L. monocytogenes Mutant Strains
Derivatives of pKSV7 (Smith and Youngman, 1992) were maintained in Escherichia coli DH5α (Life Technologies, Grand Island, NY) by growth in the presence of ampicillin (50 μg/ml). The internal in-frame deletion in mpl was generated as follows. Two fragments of mpl were amplified from 10403S chromosomal DNA by PCR and ligated sequentially into pKSV7. The 3′ end fragment encompassing bases 3,172–3,767 (Domann et al., 1991) was ligated into pKSV7 by use of BamHI- and SmaI-generated DNA ends creating pDP2263. Primers used were 5′-GGCGGGATCCGAATACGAA-3′ (primer 2239) and 5′-CCCCCGGGCCTTCTTTTTCACTAATT-3′ (primer 1701). The 5′ end fragment encompasses bases 1,857–2,487 (Domann et al., 1991), which was ligated into pDP2263 by use of SalI- and BamHI-generated DNA ends, creating pDP2264. Primers used were 5′-GAAGAATGTCGACACAGGCTTA-3′ (primer 1702) and 5′-GGACCGATGGATCCAAATGCTT-3′ (primer 2238). This resulted in a Δmpl allele with a 684-bp in-frame deletion (228 amino acids), encompassing 45% of the open reading frame including the putative propeptide cleavage site and the putative active site of the protease. The construct was confirmed by DNA sequence analysis.
Allelic exchange of the 10403S chromosomal mpl allele with the Δmpl allele was performed as described previously (Camilli et al., 1993). Strain DP-L2296 (Δmpl) was derived from L. monocytogenes 10403S. Isogenic strains DP-L2787 (ΔplcB, Δmpl), DP-L2483 (ΔplcA, Δmpl), and DPL2869 (ΔplcA, ΔplcB, Δmpl) were derived from L. monocytogenes DPL1935 (ΔplcB) (Smith et al., 1995), DP-L1552 (ΔplcA) (Camilli et al., 1993), and DP-L1936 (ΔplcA, ΔplcB) (Smith et al., 1995), respectively.
An internal out-of-frame mpl deletion was generated in strain SLCC5764. The mpl gene was digested with BamHI and XbaI, treated with Klenow to produce blunt ends, and religated, generating pDP1434. Directly downstream of the 83-bp deletion were three new codons followed by a stop codon. The resulting open reading frame was 274 amino acids long, corresponding to ∼50% of the original protein and lacking the putative active site of the protease. Allelic exchange of the SLCC-5764 chromosomal mpl allele with the Δmpl allele was performed as described previously (Camilli et al., 1993), generating strain DP-L1545. Isogenic strain DP-L1938 (ΔplcA, ΔplcB) was constructed by transforming DP-L1553 (ΔplcA) (Camilli et al., 1993) with pDP1888 (ΔplcB) (Smith et al., 1995), generating the equivalent of DP-L1936 (Smith et al., 1995), but in an SLCC-5764 background. Isogenic strain DP-L1955 is the equivalent of DP-L1942 (Brundage et al., 1993), but in an SLCC-5764 background.
The correct gene deletions on the L. monocytogenes chromosome were confirmed by determining the size of the corresponding chromosomal restriction fragment by Southern blotting, and by amplifying the deleted allele using PCR.
Complementation of L. monocytogenes Δmpl Mutant Strains
The L. monocytogenes mpl gene was amplified from 10403S chromosomal DNA by PCR and ligated into pKSV7 by use of SalI- and SmaI-generated DNA ends to generate pDP2437. The primers used were 1701 and 1702 (see above). Allelic exchange was used to replace the Δmpl allele of strain DP-L2296 with 10403S mpl allele as described (Camilli et al., 1993). Strain DP-L2484, a wild-type revertant, was identified by PCR amplification of the mpl allele and by Southern blotting. DP-L2484 formed wild-type size plaques, whereas DP-L2296 formed smaller plaques as reported in the Results section.
SDS-PAGE and Western Immunoblotting
Secreted L. monocytogenes polypeptides were prepared from strain SLCC-5764 and isogenic mutants as previously described with minor modifications (Camilli et al., 1993). After an overnight incubation in BHI broth, the cultures were diluted 1:10 and incubated at 37°C with shaking for an additional 5 h. Secreted bacterial polypeptides were precipitated on ice in 10% TCA and suspended in 2× SDS-PAGE sample buffer/0.2 N NaOH, to 1% of the original volume. The secreted polypeptide preparations were fractionated by electrophoresis on a 10% SDS-polyacrylamide gel. For Coomassie blue staining, 25 μl of the preparation was loaded per well (equivalent to 2.5 ml of bacterial culture supernatant), whereas for the Western immunoblot and egg yolk overlay assay, 10 μl was loaded per well (equivalent to 1.0 ml of bacterial culture supernatant).
For the Western immunoblot, proteins were electrotransferred to polyvinylidene difluoride membrane (Millipore Corp., Bedford, MA) and reacted with affinity-purified anti–PC-PLC rabbit antibodies. Alkaline phosphatase–labeled affinity-purified goat antibody to rabbit IgG (Kirkegaard & Perry Laboratories, Inc., Gaithersburg, MD) was used as secondary antibody, and the enzymatic reaction was induced with nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate (Sigma Chemical Co., St. Louis, MO).
Plaque Formation and Virulence Assay
The plaquing assay was performed as previously described (Sun et al., 1990). Plaques formed in monolayers of mouse fibroblast L2 cells were visualized by staining the cells with neutral red at 4 d after infection. The mean plaque diameter formed by each strain was compared with the mean plaque diameter of strain 10403S. The relative plaque size is reported as a percentage of wild-type plaque size. 50% lethal doses (LD50) were determined in BALB/c mice by intravenous injection of bacteria as described previously (Portnoy et al., 1988).
Infection of J774 Cells and Metabolic Labeling of PC-PLC
J774 mouse macrophage-like cells were propagated in a spinner flask as described (Brundage et al., 1993). The evening before use, cells were seeded in appropriate tissue-culture dishes. For detection of PC-PLC activity by the egg yolk gel overlay assay, 3.5–4 × 106 J774 cells in 100-mm tissue-culture dishes were infected with 4–8 × 107 bacteria grown overnight at 30°C, achieving an initial infection of approximately two to four bacteria per cell. The cells were washed with PBS at 30 min after infection, and gentamicin (50 μg/ml) was added at 1 h after infection. At 5.5 h after infection, the cells were washed with PBS, lysed in 2× SDS-PAGE sample buffer, immediately frozen on dry ice, and then boiled for 5 min. The samples were resolved on a 10% SDS-polyacrylamide gel.
For [35S]methionine protein labeling, 106 J774 cells in 35-mm tissue-culture dishes were infected with 5–10 × 106 bacteria grown overnight at 30°C, achieving an initial infection of approximately one to two bacteria per cell. At 30 min after infection, the cells were washed three times with PBS, and methionine-free DME with 10% dialyzed FBS (Hyclone Laboratories, Logan, UT) was added. Gentamicin (50 μg/ml) was added at 1 h after infection. Cells were pulse labeled for 30 min at 4 h after infection with 200 μl of the same medium containing gentamicin and 90 μCi [35S]methionine (Express 35S protein labeling mix; NEN® Research Products, Boston, MA). In pulse-chase experiments, cells were pulse labeled for 10 min, and then chased with unlabeled methionine (5 mM) and chloramphenicol (20 μg/ml) to prevent further incorporation of labeled methionine. Host cell protein synthesis was not blocked at any time. Bacterial counts were determined in triplicate for each strain in each experiment as described previously (Portnoy et al., 1988). Samples were resolved on 11% SDS-polyacrylamide gels. The amount of sample loaded per lane was normalized for the number of bacteria. After electrophoresis, the gel was processed for fluorography (EN3HANCE; NEN® Research Products).
Inhibitor Studies
Inhibitors were present for 1 h before and during the 30-min pulse labeling. N-acetyl-leucine-leucine-norleucine (LLnL), also called calpain inhibitor I, and N-acetyl-leucine-leucine-methionine (LLM), also called calpain inhibitor II, were obtained from Boehringer Mannheim Biochemicals (Indianapolis, IN). Benzyloxycarbonyl-phenylalanine-alanine-diazomethane (Z-FA-CHN2) was obtained from Bachem Biochemicals (King of Prussia, PA). A 25-mM solution of each inhibitor was prepared in methanol immediately before use. Bafilomycin A1 was obtained from Sigma Chemical Co. A 100-μM solution was prepared in DMSO and stored in a desiccator at −20°C. These inhibitors were used at concentrations indicated in the text. An equal volume of methanol and/or DMSO was added to cells not treated with inhibitors.
Affinity Purification of Antibody
The active form of PC-PLC was purified as described (Geoffroy et al., 1991; Goldfine et al., 1993), and antiserum was raised in rabbits (Cocalico Biologicals, Inc., Reamstown, PA). Specific PC-PLC antibodies were affinity purified (Olmsted, 1981), using the rabbit polyclonal antiserum and purified PC-PLC protein. The purified protein (25–50 μg) was spotted directly on a small strip of polyvinylidene difluoride membrane and blocked with 5% nonfat dry milk in TBS. The membrane was incubated with 1 ml of the rabbit polyclonal immune serum for 2 h at 4°C, and then washed four times in TBS-TX (TBS, 0.1% Triton X-100) and twice in TBS-EDTA (TBS, 1 mM EDTA). Antibodies specific to PC-PLC were eluted twice in 1 ml of 0.2 M glycine-HCl, pH 3.0, at 4°C. After 2-min incubation times, each aliquot was rapidly neutralized with 2 N NaOH. Aliquots were pooled and diluted in 2 vol of PBS, 0.25% BSA, and 0.02% sodium azide. The antibody solution was transferred into a Centriprep 10 concentrator (Amicon, Inc., Beverly, MA) and centrifuged at 3,000 g for 30 min at 4°C. The final antibody solution was in a volume of ∼1.2 ml. Each preparation of affinity-purified antibodies was titrated to determine the optimal volume required for immunoprecipitation of PC-PLC from a lysate of 106 J774 cells infected with wild-type L. monocytogenes.
Immunoprecipitation of PC-PLC from Infected J774 Cells
Immunoprecipitation was performed as previously described (Brundage et al., 1993) with some modifications. Cell lysates and slurries of protein A–Sepharose CL-4B beads (Sigma Chemical Co.) were prepared in RIPA buffer (150 mM NaCl, 50 mM Tris-HCl, pH 8.0, 1% NP-40, 0.5% deoxycholate, 0.1% SDS) supplemented with a cocktail of protease inhibitors (Sigma Chemical Co.): 10 mM EDTA, 1 mM PMSF, 0.3 μM aprotinin, 1 μM leupeptin, and 1 μM pepstatin. The affinity-purified antibody was preadsorbed with a lysate of uninfected unlabeled cells (2 × 106) for 1 h at 4°C. The volume of affinity-purified antibody used per reaction was determined as described above. The lysate of infected labeled cells (106) was mixed with the adsorbed antibody for 1–1.5 h at 4°C, and then with a slurry of protein A–Sepharose beads (10 mg per lysate) for 1 h at 4°C. Protein A beads were washed four times in the same buffer, and the immunoprecipitates were recovered in 2× SDS sample buffer. The samples were boiled for 5 min and stored at −80°C. Bacterial counts were determined in parallel dishes, and equivalent numbers of bacteria were loaded per lane of an 11% SDS-polyacrylamide gel.
Immunofluorescence
J774 cells (106) grown on 12-mm square coverslips in 35-mm dishes were infected with 0.5–1 × 106 bacteria grown overnight at 30°C, achieving an initial infection of approximately one bacterium per 5 to 10 cells. At 5 h after infection, infected cells were fixed with 3.3% formaldehyde EMGrade (Electron Microscopy Sciences, Ft. Washington, PA) in PBS and permeabilized in TBS-TX. The samples were blocked with 1% BSA in TBS-TX and all the antibodies were diluted in the same buffer. PC-PLC was detected by reacting the samples successively with affinity-purified rabbit anti–PC-PLC antibodies, donkey anti–rabbit IgG conjugated with biotin (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA), and rhodamine streptavidin (Molecular Probes, Eugene, OR). The lysosome-associated membrane protein 1 (Lamp1) was detected using a rat mAb 1D4B (developed by J. Thomas August and obtained from the Developmental Studies Hybridoma Bank, Iowa City, IA) and donkey anti– rat conjugated with FITC (Jackson ImmunoResearch Laboratories, Inc.). L. monocytogenes staining was performed last with a rabbit anti–L. monocytogenes antibody conjugated with fluorescein (Difco Laboratories). Between each reaction, the coverslips were extensively washed with TBSTX. The coverslips were mounted with Testog FITC-Guard (Testog, Inc., Chicago, IL) and sealed on glass slides. Confocal microscopy was performed using a TCS 4D apparatus (Leica Inc., Deerfield, IL) equipped with an Image Graphics image recorder. (Focus Graphics Inc., Foster City, CA). Selected fields were scanned with 0.07 μM pixel size and 0.5 μm step size. The intensity settings were constant for all samples tested within each experiment.
Enzyme Assays
Two types of assays were performed to detect PC-PLC activity: an egg yolk gel overlay assay (Kathariou et al., 1990) and an isotope-based assay (Goldfine et al., 1993). For the egg yolk overlay assay, samples were boiled and fractionated by SDS-PAGE. Immediately after electrophoresis, the gel was washed once in 25% isopropanol and twice in PBS (Ca2+ and Mg2+ free) for 30 min each time. The gel was then overlaid with egg yolk soft agar (0.7% agarose, 2.5% egg yolk, 50 μM ZnCl2, 0.05 μM CaCl2, 1 mM DTT, and 25 μg/ml gentamicin, in 1× PBS) and incubated at 37°C. PC-PLC activity was detected by the formation of zones of opacity. Periods of incubation varied from 2 h for the in vitro grown bacteria to 2 d for the intracellularly grown bacteria.
The isotope-based enzyme assay was performed as described elsewhere (Goldfine et al., 1993) with the exception that sodium deoxycholate (0.15% final concentration) was used instead of Triton X-100 for the mixed micelle suspension, and the concentration of PC and sphingomyelin was 0.15 mM. PC-PLC was immunoprecipitated from infected J774 cells as described above with some modifications. J774 cells were not deprived of methionine during infection and crude anti–PC-PLC rabbit hyperimmune serum was used. The optimal concentration of crude antiserum was determined to be 1 μl. After immunoprecipitation, the pellets of protein A beads were washed three times in RIPA buffer and once in 140 mM NaCl/10 mM Tris-HCl, pH 8.0. Enzyme assays were done directly in the microfuge tubes (1.5 ml) containing the pellets of protein A beads to which PC-PLC– antibody complexes were bound. Hydrolysis of [choline-methyl-3H]-phosphatidylcholine (3H-PC) and [choline-methyl-14C]-sphingomyelin (14Csphingomyelin) (NEN® Research Products) was measured as described (Goldfine et al., 1993) after a 10-min incubation period at 37°C. Under these conditions, hydrolysis of PC was linear for at least 10 min and the hydrolysis of sphingomyelin showed a lag of 2 to 5 min.
Results
Role of Mpl and the PLCs in Mouse Virulence and Plaque Formation
A series of deletions was generated into the PLC-, Mpl-, and ActA-encoding genes to evaluate the requirements for proPC-PLC proteolytic activation. The mutations were introduced into the chromosome of L. monocytogenes strains SLCC-5764 and 10403S. Proteolytic activation of proPC-PLC was first evaluated in vitro, using SLCC-5764 and isogenic mutant strains. SLCC-5764 is a hypersecreting strain of L. monocytogenes that facilitates in vitro analysis of virulence factors, such as ActA and PC-PLC, not normally expressed in vitro (Camilli et al., 1993). ProPCPLC (M r 33 kD) and the processed form of the enzyme (M r 28 kD) were identified by Coomassie blue staining and Western immunoblotting. ProPC-PLC was secreted in comparable amounts from each plcB positive strain (Fig. 1, a and b), indicating that the internal deletions in mpl and actA genes, which are located within the same operon and upstream of the plcB gene (Portnoy et al., 1992), did not affect plcB gene expression. Similarly, the internal deletion in plcA, which is located within the same operon and upstream of prfA, the transcriptional regulator encoding gene (Portnoy et al., 1992), did not affect plcB gene expression (Fig. 1, a and b, lane 3). The processed form of PC-PLC was secreted in comparable amounts from SLCC5764 and isogenic plcA and actA mutant strains (Fig. 1, a and b, lanes 2, 3, and 5). Enzymatic activity, as determined by the egg yolk gel overlay assay, comigrated with the processed form of PC-PLC (compare Fig. 1 b to 1 c, lanes 2, 3, and 5), confirming that PI-PLC and ActA are not required for the proteolytic activation of proPC-PLC in vitro and that the processed form is the active form of the enzyme. A processed form of PC-PLC was detected in minute amounts from the mpl mutant strain (Fig. 1, a and b, lane 6). However, there was no detectable PC-PLC activity associated with this mutant (Fig. 1 c, lane 6), even after increasing the amount of protein loaded on the gel fourfold (data not shown). This result is consistent with Mpl being required for activation of proPC-PLC in broth culture as previously reported (Poyart et al., 1993).
Figure 1.

Detection of PC-PLC in the supernatant of L. monocytogenes broth cultures. Strain SLCC-5764 and isogenic mutants were used for this experiment. For each assay, an equal volume of concentrated bacterial secreted proteins was loaded per lane. (a) Coomassie blue staining of secreted proteins resolved by SDS-PAGE. (Lane 1) 31-kD protein marker; (lane 2) SLCC5764; (lane 3) DP-L1553 (PI-PLC−); (lane 4) DP-L1938 (PIPLC−, PC-PLC−); (lane 5) DP-L1955 (ActA−); (lane 6) DPL1545 (Mpl−). (b) Alkaline phosphatase Western immunoblot. (Lane 1) 32-kD prestained molecular mass marker; (lanes 2–6) same as in a. (c) Egg yolk gel overlay assay. (Lane 1) 32- and 42-kD prestained molecular mass markers; (lanes 2–6) same as in a.
The virulence of wild-type strain 10403S and isogenic mutants was evaluated in vivo using the mouse infection model. The ability of these mutants to spread cell to cell was evaluated in a mouse fibroblast cell line by a plaquing assay. The results are reported in Fig. 2 and Table II. The single plcA and plcB mutants show respective reductions of 12% and 33% in plaque sizes, and the double plcA, plcB mutant shows a reduction of 66% in plaque size. In the mouse infection model, the plcA mutant shows a minor increase in LD50, the plcB mutant is 1 log less virulent, whereas the double plcA, plcB mutant is 2.5 logs less virulent than the wild-type strain. These results are consistent with previously reported data, suggesting that the PLCs have overlapping functions (Smith et al., 1995). The double mpl, plcB mutant was phenotypically identical to the single plcB mutant, consistent with Mpl function being related to the activation of proPC-PLC. Similarly, the triple plcA, plcB, mpl mutant was phenotypically identical to the double plcA, plcB mutant. A single mutation in the mpl gene resulted in a 29% decrease in plaque size, also consistent with Mpl being required for proPC-PLC activation. However, the mpl mutant was as virulent as the wild-type strain in the mouse infection model, which is inconsistent with Mpl function being required for the activation of proPC-PLC. In addition, a double plcA, mpl mutant had a 47% decrease in plaque size, which differs significantly (P < 0.0001) from the 66% decrease observed with the double plcA, plcB mutant. The double mpl, plcA mutant was ∼2 logs less virulent than wild type but not as attenuated as the double plcA, plcB mutant in the mouse infection model. Taken together, these data clearly show a role for Mpl in bacterial cell-to-cell spread and reveal a role for Mpl in virulence as seen in the double mpl, plcA mutant. However, unlike the plcB mutant, the virulence of the single mpl mutant was not attenuated in mice, and that of the double mpl, plcA mutant was not as attenuated as the double plcA, plcB mutant. These results raised the possibility that intracellular activation of proPC-PLC may proceed in the absence of Mpl.
Figure 2.

Formation of plaques in infected L2 mouse fibroblasts. L2 cells were infected with strain 10403S and isogenic mutants. Cell monolayers were stained with neutral red at 4 d after infection, highlighting clear areas of dead cells resulting from L. monocytogenes intracellular growth and cell-to-cell spread. The mean plaque diameters were calculated from several individual experiments (see Table II) and are indicated below each well.
Table II.
LD50s and Plaquing Results
| Strain | LD50 in mice | Plaque size | Source or reference§ | |||
|---|---|---|---|---|---|---|
| (CFU) | %*± SD (n)‡ | |||||
| 10403S | 1–3 × 104 | 100 | Portnoy et al., 1988 | |||
| DP-L1552 | 3.2 × 104 | 88.3 ± 6.0‖ (10) | Camilli et al., 1993 | |||
| DP-L1935 | 2 × 105 | 66.7 ± 5.8‖ (18) | Smith et al., 1995 | |||
| DP-L2296 | <5 × 104 | 71.0 ± 6.4 (17) | This study | |||
| DP-L2787 | 3.5 × 105 | 66.4 ± 4.3 (10) | This study | |||
| DP-L1936 | 5 × 106 | 34.0 ± 5.2‖ (15) | Smith et al., 1995 | |||
| DP-L2483 | 1.6 × 106 | 53.1 ± 5.8 (16) | This study | |||
| DP-L2869 | ND | 36.7 ± 3.3 (5) | This study |
Relative percentage of diameter of plaques formed in L2 cells by mutant strains in comparison with the wild-type strain.
(n) = number of plaquing experiments.
For LD50s' data.
Plaquing results from this study were combined with previously reported results from this laboratory (Smith et al., 1995).
Intracellular Activation of ProPC-PLC
To directly evaluate the role of Mpl in the intracellular activation of proPC-PLC, we examined the production and processing of proPC-PLC in a tissue-culture model of infection. At 4 h after infection, J774 cells were pulse labeled for 30 min with [35S]methionine and lysed, and both forms of PC-PLC were immunoprecipitated using affinity-purified antibodies. The precursor and processed forms of PCPLC were identified based on their relative migration on SDS-PAGE, and by comparison with cells infected with the plcB mutant strain. Both forms of the enzyme were present in cells infected with either the wild-type strain or the mpl mutant (Fig. 3, lanes 1 and 3), indicating that Mpl was not essential for intracellular processing of proPCPLC. Furthermore, results from an egg yolk gel overlay assay from infected cells indicated that the processed form of PC-PLC generated in cells infected with either the wildtype strain or the mpl mutant had enzymatic activity (Fig. 4, lanes 2 and 5). Phospholipase activity was not detected in uninfected cells or cells infected with the plcB mutant (Fig. 4, lanes 1 and 3).
Figure 3.

Detection of PC-PLC from lysates of infected cells. J774 cells were infected with strain 10403S and isogenic mutants. At 4 h after infection, cells were pulse labeled for 30 min with [35S]methionine. Immediately after the pulse, the cells were lysed, PC-PLC was immunoprecipitated using affinity-purified antibodies, and proteins were fractionated by SDS-PAGE. PC-PLC was detected by fluorography. (Lane 1) 10403S; (lane 2) DP-L1935 (PC-PLC−); (lane 3) DP-L2296 (Mpl−). Number of colony forming units (CFU) per sample: 2.8 × 107 (lanes 1 and 3), and 2.6 × 107 (lane 2).
Figure 4.

PC-PLC activity in lysates of infected cells. J774 cells were infected with strain 10403S and isogenic mutants. Cells were harvested in 2× SDS sample buffer at 5.5 h after infection. After electrophoresis, the gel was successively soaked in isopropanol and PBS and overlaid with egg yolk agar. Zones of opacity are indicative of phospholipase activity. (Lane 1) Uninfected cells; (lane 2) 10403S; (lane 3) DP-L1935 (PC-PLC−); (lane 4) DPL1942 (ActA−); (lane 5) DP-L2296 (Mpl−). Number of CFU per sample: 1.5 × 108 (lane 2), 2.0 × 108 (lane 3), 2.7 × 108 (lane 4), and 1.9 × 108 (lane 5).
Subcellular Localization of PC-PLC
Double immunofluorescence staining and confocal microscopy was performed at 5 h after infection to determine the site of PC-PLC and L. monocytogenes localization. Distinctively, only a small proportion of intracellular bacteria stained positively for PC-PLC, and PC-PLC staining colocalized with L. monocytogenes (Fig. 5 A). The pattern of PC-PLC staining suggested a vacuolar localization. Double immunofluorescence staining of PC-PLC and Lamp1 was performed to define more precisely the site of PC-PLC localization. Lamp1 is an endosomal/lysosomal marker (Chen et al., 1985; Lewis et al., 1985) that increases in concentration with endosomal maturation (Berón et al., 1995; Pitt et al., 1992). Colocalization of PC-PLC and Lamp1 was observed (Fig. 5, D and E), but the amount of Lamp1 colocalizing with PC-PLC varied considerably. This staining pattern suggested that PC-PLC was concentrated in vacuoles that had fused with endosomes and lysosomes. PC-PLC staining was not observed in filopodia-like structures, but we cannot eliminate the possibility that this could be due to a lack of sensitivity of the technique used.
Figure 5.

Immunolocalization of PC-PLC in infected cells. J774 cells were infected with strain 10403S (A, B, and D), or isogenic mutants DP-L1935 (PC-PLC−) (C) and DP-L2296 (Mpl−) (E). Cells were fixed in formalin at either 5 (A and C–E) or 6 h after infection (B), stained for immunofluorescence analysis as described in Materials and Methods, and analyzed by confocal microscopy (0.5-μm sections). (A–E) PC-PLC (rhodamine); (A–C) L. monocytogenes (fluorescein); (D and E) Lamp1 (fluorescein). Overlapping of both fluors (rhodamine and fluorescein) generates yellow. (A, arrows) Examples of bacteria/PC-PLC colocalized staining suggesting vacuolar localization; (arrowheads) PC-PLC staining at the septum of dividing bacteria. (B) In this experiment, cells were infected with 10-fold less bacteria to facilitate identification of primary (highly infected central cell) and secondary infected cells (peripheral cells). The background of this particular field was increased by computer to facilitate visualization of the cells. (Arrows) Examples of bacteria/PC-PLC colocalizing in three different secondary infected cells. (D and E) Large arrows show examples of PC-PLC–containing vacuoles with no Lamp1 staining, whereas small arrows show examples suggesting advanced fusion events between PC-PLC–containing vacuoles and Lamp1-positive endosomes. Arrowhead at the bottom of D shows a PC-PLC–containing vacuole surrounded by Lamp1. Bars, 10 μm.
To directly address whether PC-PLC localized to secondary vacuoles formed upon bacterial cell-to-cell spread, J774 cells were infected with a 10-fold lower multiplicity of infection to facilitate identification of primary and secondary infected cells at 5 and 6 h after infection. Double immunofluorescence staining of PC-PLC and L. monocytogenes indicated that the majority of PC-PLC localized to cells at the periphery of infected foci, presumably in secondary vacuoles formed during bacterial cell-to-cell spread. An example of an infection focus at 6 h after infection is shown in Fig. 5 B.
The Mpl-dependent and -independent Pathways of ProPC-PLC Proteolytic Activation
The above results suggested that secondary vacuoles formed during bacterial cell-to-cell spread fused with vesicles of the endocytic pathway (Berón et al., 1995; Gruenberg and Maxfield, 1995; Pitt et al., 1992). To assess the role of vacuolar acidification and lysosomal enzymes in proPC-PLC processing, we used two enzyme inhibitors. The first, bafilomycin A1, is a specific inhibitor of the vacuolar proton pump ATPase (Yoshimori et al., 1991), which serves to acidify a vacuolar compartment. The second is a peptidyldiazomethane, Z-FA-CHN2, which specifically inactivates cysteine proteases by alkylation of the reactive site cysteine residue (Leary et al., 1977). Z-FA-CHN2 has strong affinity for cathepsins B and L (Crawford et al., 1988; Kirschke and Shaw, 1981; Wilcox and Mason, 1992), which are lysosomal acid cysteine proteases, but does not react with the calpains (Crawford et al., 1988), which are cytosolic calcium-activated neutral cysteine proteases. The results showed that both bafilomycin A1 and Z-FA-CHN2 blocked processing of proPC-PLC in cells infected with the mpl mutant (Fig. 6, lanes 5 and 6). In wild-type infected cells, processing of proPC-PLC was not affected by Z-FACHN2, but no processing was detected in cells treated with bafilomycin A1 (Fig. 6, lanes 2 and 3), although the protein was detected by immunofluorescence in secondary vacuoles (data not shown). Neither bafilomycin A1 nor Z-FACHN2 inhibited activation of proPC-PLC in broth culture (data not shown). These results indicate that there are two intracellular pathways of activation of proPC-PLC: an Mpl-mediated pathway and a cysteine protease-mediated pathway. Both pathways were blocked by a specific inhibitor of the vacuolar proton pump ATPase, suggesting that vacuolar acidification is a prerequisite to the intracellular processing of proPC-PLC.
Figure 6.

The effect of bafilomycin A1 and Z-FA-CHN2 on proPC-PLC processing in infected cells. J774 cells were infected with strain 10403S and isogenic mutants. At 4 h after infection, cells were processed as described in legend of Fig. 3. Bafilomycin A1 (Baf) (lanes 2 and 5) or Z-FA-CHN2 (Z-FA) (lanes 3 and 6) was added at 3 h after infection at final concentrations of 1 μM and 10 μM, respectively. The inhibitors were also present during pulse labeling. (Lanes 1–3) 10403S; (lanes 4–6) DP-L2296 (Mpl−); (lane 7) DP-L1935 (PC-PLC−). Number of CFU per sample was 3.9 × 107.
Intracellular Activity of PC-PLC on Phosphatidylcholine and Sphingomyelin
Intracellular activation of proPC-PLC was mediated by either Mpl or a cysteine protease, raising the possibility that PC-PLC activity might vary depending on the activating protease. To address that point, we investigated the activities of PC-PLC generated in cells infected with either the wild-type strain or the mpl mutant. PC-PLC was immunoprecipitated from infected cells, and the enzymatic assay was performed directly on PC-PLC bound to antibodies on protein A–Sepharose beads. Hydrolysis of 3H-PC and 14C-sphingomyelin by immunoprecipitated PC-PLC was measured as described in Materials and Methods. In either case, PC-PLC was capable of mediating PC and sphingomyelin hydrolysis (Table III). When corrected for the number of bacteria per dish, the ability of PC-PLC generated by the wild-type strain and the mpl mutant to hydrolyze PC and sphingomyelin was essentially the same, although a small shift in substrate preference was observed. Phospholipase activity was not detected on immunoprecipitates from uninfected cells or cells infected with the plcB mutant (data not shown).
Table III.
Phospholipase Activities of Immunoprecipitated PC-PLC from Cells Infected with Either Wild-Type Strain or the mpl Mutant
| Strain | PC-PLC activity on PC: nmol/ 10 min/108 CFU (n)* | PC-PLC activity on sphingomyelin: nmol/10 min/108 CFU (n)* | Relative activity sphingomyelin/PC (n)* | |||
|---|---|---|---|---|---|---|
| 10403S | 1.00 ± 0.58 (4) | 0.091 ± 0.052 (5) | 0.095 ± 0.018 (4) | |||
| DP-L2296 | 1.32 ± 0.82‡ (5) | 0.098 ± 0.075‡ (5) | 0.068 ± 0.011§ (5) |
(n) = number of experiments.
P > 0.05 by unpaired t test (NS).
P < 0.05 by unpaired t test (significant).
Proteolytic Activation of ProPC-PLC in the Absence of Bacterial Cell-to-Cell Spread
The above results indicated that PC-PLC localized to Lamp1-positive vacuoles (Fig. 5) and that vacuolar acidification was a prerequisite to proPC-PLC activation (Fig. 6, lanes 2 and 5). These results suggested that active PC-PLC would not be generated in the cytosol of infected cells. To further investigate the intracellular requirements for proPCPLC activation, we monitored the presence of PC-PLC in cells infected with an actA mutant of L. monocytogenes that is defective in actin-based motility, and consequently fails to spread cell to cell. ProPC-PLC was immunoprecipitated in similar amounts in cells infected with either the wild-type strain or the actA mutant, indicating that proPCPLC was synthesized by cytosolic bacteria, but processing of proPC-PLC, although observable, was very inefficient in the cytosol of cells infected with the actA mutant (Fig. 7, compare lanes 1 and 5).
Figure 7.

Detection of PC-PLC from lysates of cells infected with nonspreading bacteria. J774 cells were infected with strain 10403S and isogenic mutants. At 4 h after infection, cells were processed as described in legend of Fig. 3. Cytochalasin D was added at times relative to the beginning of labeling (as indicated on the figure) and during labeling at a final concentration of 1 μM (lanes 2–4). (Lanes 1–4) 10403S; (lane 5) DP-L1942 (ActA−); (lane 6) DP-L1935 (PC-PLC−). Number of CFU per sample was 1.9 × 107 for lanes 1–4, 1.3 × 107 for lane 5, and 2.2 × 107 for lane 6.
To eliminate the possibility that ActA was directly involved in proPC-PLC processing, we infected cells with the wild-type strain, and then blocked bacterial cell-to-cell spread by adding cytochalasin D (Dabiri et al., 1990; Tilney and Portnoy, 1989), an inhibitor of actin polymerization, 30 min before labeling. Again, the precursor form of PC-PLC was present in these infected cells but the processed form was not detectable, indicating that ActA was not directly responsible for proPC-PLC processing (Fig. 7, lane 4). Moreover, the amount of processed PC-PLC was largely reduced when actin polymerization was blocked as late as 10 min into the pulse, and it was barely detectable when actin polymerization was blocked 10 min before labeling (Fig. 7, lanes 2 and 3). Results from the cytochalasin D time course experiment indicated that continuous bacterial spreading was required for proPC-PLC processing. Not surprisingly, PC-PLC activity, as measured by the egg yolk gel overlay assay, was detected neither in cells infected with the actA mutant strain (Fig. 4, lane 4) nor in wild-type infected cells treated with cytochalasin D (data not shown). Therefore, proPC-PLC proteolytic activation, but not synthesis, occurred predominantly in secondary vacuoles.
Cytosolic Degradation of ProPC-PLC
Our observations indicated that proPC-PLC proteolytic activation was inefficient in the absence of bacterial cellto-cell spread (Fig. 7). However, in the absence of proteolytic activation, proPC-PLC did not appear to accumulate intracellularly, suggesting that proPC-PLC was rapidly degraded when secreted in the cytosol of the host cell. Proteolytic degradation of cytosolic proteins is mostly proteasome dependent (Rock et al., 1994). We investigated the role of the proteasome in the intracellular degradation of proPC-PLC using two aldehyde tripeptide inhibitors: LLnL and LLM. LLnL blocks the proteasome activity, while LLM does not (Rock et al., 1994; Vinitsky et al., 1992).
The intracellular stability of proPC-PLC was evaluated by a pulse-chase experiment. Cells infected with ActA− bacteria were treated with either LLnL or LLM at 3 h after infection and the inhibitors were present during the entire pulse-chase experiment. At 3 h and 50 min after infection, cells were pulse labeled with [35S]methionine for 10 min, and then chased for specific periods of time. A role for the proteasome in proPC-PLC degradation was demonstrated by the observation that proPC-PLC was stabilized in infected cells treated with LLnL (Fig. 8, compare lanes 5–8 to lanes 1–4), but not in those treated with LLM (Fig. 8, lanes 9–12). Indeed, the half-life of proPC-PLC was <15 min in either untreated or LLM-treated cells, and >60 min in LLnL-treated cells. The intracellular stability of proPC-PLC was comparable in cells infected with either the wild-type strain, the mpl mutant, or the actA mutant (data not shown). Yet, in wild-type and Mpl− infected cells, proPC-PLC chased into active PC-PLC (data not shown). These results suggested an additional level of PC-PLC regulation by proteolytic degradation of the precursor. ProPC-PLC secreted into the cytosol was degraded by host proteases and consequently had a short half-life.
Figure 8.

Determination of proPC-PLC intracellular stability in cells infected with nonspreading bacteria. J774 cells were infected with 10403S isogenic mutants. Cells were pulse labeled for 10 min with [35S]methionine at 3 h and 50 min after infection, and then chased with methionine and chloramphenicol to prevent further incorporation of labeled methionine. Host protease inhibitors were added 1 h before labeling and during labeling at a final concentration of 50 μM. Samples were harvested for PC-PLC immunoprecipitation at specific times during the chase. PC-PLC was detected by fluorography. (Lanes 1–12) DP-L1942 (ActA−); (lane 13) DP-L1935 (PC-PLC−). Number of CFU per sample was 1.4 × 107.
Discussion
L. monocytogenes has the remarkable ability to spread from one cell to another without exposure to the extracellular environment. The currently accepted model proposes that L. monocytogenes exploits a host system of actinbased motility to move into cellular projections that are internalized by neighboring cells, resulting in bacteria transiently confined within double membrane vacuoles (Tilney and Portnoy, 1989). The molecular and cellular mechanisms that govern bacterial cell-to-cell spread are not yet understood. However, in this and other studies, three L. monocytogenes secreted polypeptides have been identified that mediate bacterial cell-to-cell spread. Two of these polypeptides are phospholipases of the C class: PI-PLC and PC-PLC (Smith et al., 1995; Vazquez-Boland et al., 1992). PC-PLC is secreted as an inactive precursor (proPCPLC), which is proteolytically activated by a third L. monocytogenes secreted product, a metalloprotease (Mpl) (Poyart et al., 1993). In this study we show that there are three potential fates for proPC-PLC subsequent to secretion by intracellular L. monocytogenes. In the absence of bacterial cell-to-cell spread, proPC-PLC secreted in the host cytosol was rapidly degraded in a proteasome-dependent manner. During bacterial cell-to-cell spread, proPCPLC activation was mediated by either Mpl or a cysteine protease presumably of host origin. The majority of PCPLC staining was found in Lamp1-positive vacuoles. Lastly, proPC-PLC activation by either pathway was blocked by bafilomycin A1, a specific inhibitor of the vacuolar proton pump ATPase.
It was previously shown that Mpl is responsible for mediating proPC-PLC activation during growth in liquid medium (Poyart et al., 1993). Based on this observation, we hypothesized that Mpl was responsible for mediating intracellular activation of proPC-PLC. The results of this study clearly show that Mpl is not essential for the proteolytic activation of proPC-PLC during infection, as the active form of PC-PLC was detected in cells infected with the mpl mutant. The Mpl-independent pathway of proPCPLC activation was blocked with an inhibitor of cysteine proteases. However, this inhibitor did not block proPCPLC activation in cells infected with the wild-type strain. These results are consistent with the existence of two intracellular pathways of proPC-PLC activation: an Mpl- mediated pathway and a cysteine protease-mediated pathway. The presence of an Mpl-independent pathway is also consistent with our genetic data in which an mpl mutant had a different phenotype than a plcB mutant. Lastly, these results do not exclude the possibility that Mpl has another role than proPC-PLC activation.
Two different classes of proteases, a metalloprotease and a cysteine protease, were capable of mediating proPCPLC activation. We considered the possibility that the two activating proteases might have different cleavage recognition sites, resulting in altered PC-PLC activity. Our results revealed retention of PC and sphingomyelin hydrolytic capacity by the host-processed enzyme, with a small shift in substrate preference. Therefore, if there was a difference in cleavage recognition sites, the consequences for PC-PLC enzymatic activity were minor. We also need to consider the possibility that Mpl-mediated activation of proPC-PLC occurs earlier than Mpl-independent activation of proPC-PLC, with the bacterial protease acting upon secretion, and the cysteine protease acting after secretion. The consequence would be Mpl-mediated activation of proPC-PLC in a different cellular compartment than the cysteine protease-mediated activation. In mice, L. monocytogenes probably spreads from macrophages to hepatocytes and from hepatocyte to hepatocyte (North, 1970; Rosen et al., 1989). The importance of the cysteine protease pathway in the intracellular activation of proPCPLC may vary among cell types.
Immunofluorescence studies revealed that PC-PLC was concentrated in host cell vacuoles which contained L. monocytogenes. Based on our current model (Tilney and Portnoy, 1989), we propose that these vacuoles are formed during bacterial cell-to-cell spread. The observation that these vacuoles stained positive for Lamp1 indicates that they fused with vesicles of the endocytic pathway (Berón et al., 1995; Gruenberg and Maxfield, 1995; Pitt et al., 1992). This is consistent with proPC-PLC activation being mediated by the lysosomal proteases, cathepsins B and L, in cells infected with the mpl mutant. This is also consistent with proPC-PLC activation being dependent on vacuolar acidification as the cathepsins are acid hydrolases (Bohley and Seglen, 1992). However, it was surprising that vacuolar acidification was a prerequisite for the Mpl-mediated activation of proPC-PLC. Presumably, Mpl is secreted as a proenzyme that becomes activated by autocatalytic processing as demonstrated for homologous bacterial metalloproteases (Mengaud et al., 1991; Häse and Finkelstein, 1993). Perhaps, Mpl activation and/or catalytic activity are pH dependent. Alternatively, proPC-PLC may be more susceptible to proteolytic activation when secreted in the acidic vacuolar environment. Another possibility is that bafilomycin A1 blocks the action of the pore-forming protein, LLO, which is known to have an acidic pH optimum (Geoffroy et al., 1987). Perhaps, LLO-induced pore formation is a prerequisite for Mpl and/or proPC-PLC expression. Unfortunately, we are unable to directly assess the role of LLO because LLO− mutants fail to escape from the primary vacuole, and thus fail to grow (Gaillard et al., 1987; Tilney and Portnoy, 1989). Interestingly, bafilomycin A1 blocks L. monocytogenes escape from the primary vacuole in Caco-2 cells, a human carcinoma cell line (Conte et al., 1996). Lastly, bafilomycin A1 blocks acidification of the vacuole (Yoshimori et al., 1991) and endosomal maturation (Clague et al., 1994; van Weert et al., 1995). Perhaps additional host factors are required for proPC-PLC activation. Based on these observations, we propose that conventional vacuolar maturation (Berón et al., 1995; Desjardins et al., 1994; Gruenberg and Maxfield, 1995) occurs before bacterial escape from the secondary vacuole, and we speculate that the activation of proPCPLC by either Mpl or the cysteine protease depends on the stage of maturation of the phagosome.
In the absence of bacterial cell-to-cell spread, proPCPLC was still synthesized but was rapidly degraded with a half-life of <15 min. Degradation of proPC-PLC was blocked by an inhibitor of the host proteasome, which is the major cytosolic proteolytic system of eukaryotic cells (Rock et al., 1994). There is precedent for this finding in that the L. monocytogenes hemolysin is also released into the cytosol and degraded in a proteasome-dependent manner (Villanueva et al., 1995). Interestingly, even when the degradation of proPC-PLC was prevented, no mature PC-PLC appeared. Thus, there are two mechanisms to prevent PC-PLC production in the cytosol: degradation and lack of activation. Hence, active PC-PLC is restricted to a vacuolar compartment. These observations are consistent with previous results showing that sustained elevation of ceramide, a product of sphingomyelin hydrolysis, was observed in cells infected with the wild-type strain but not in cells infected with the actA mutant (Smith et al., 1995).
Intracellular parasites by necessity use host processes for the regulation of pathogenesis. The results of this study define two potential levels of regulation that are dependent on bacterial subcellular localization (Fig. 9). The first level of regulation is proteolytic activation. ProPC-PLC secreted in vacuoles is activated by either a bacterial or a host protease subsequent to acidification of the vacuole. The second level of regulation is proteolytic degradation. ProPC-PLC secreted into the cytosol is degraded by host proteases, and consequently has a short half-life and is essentially inactive in the cytosol. The results presented in this study are the first to demonstrate proteolytic regulation of a product secreted intracellularly by a bacterial pathogen.
Figure 9.
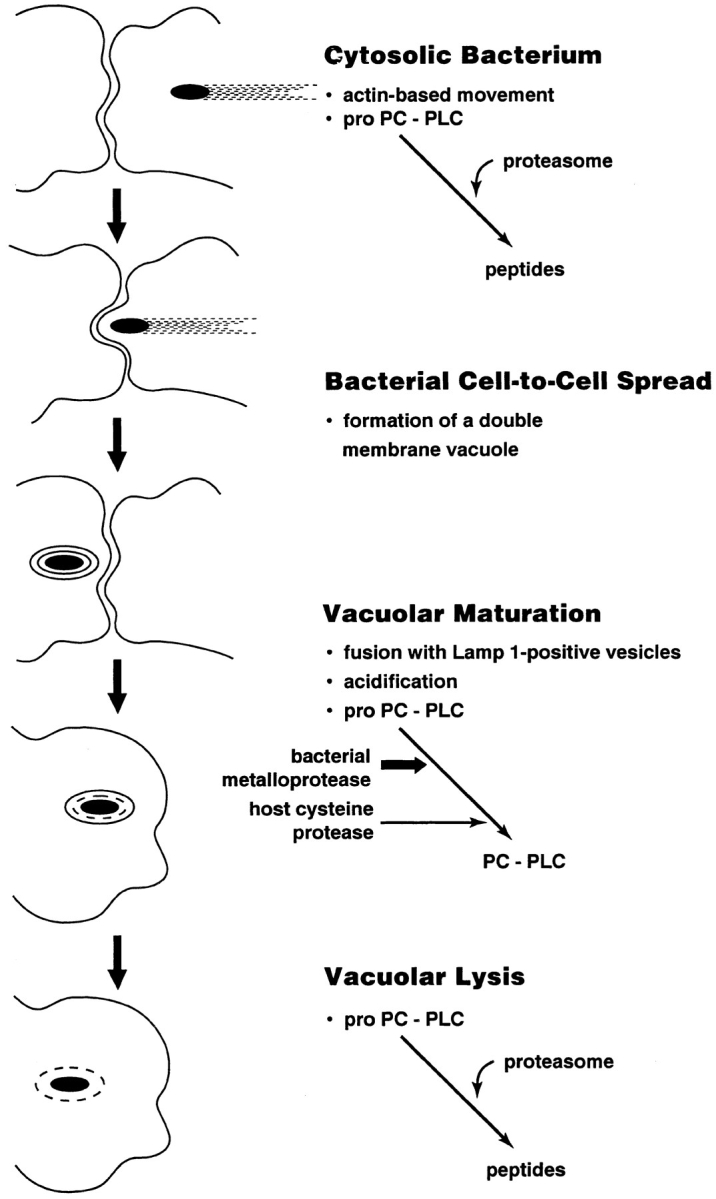
Schematic representation of the intracellular regulation of proPC-PLC activity. Asymmetric polymerization of host actin at the bacterial surface results in actin-based motility, which facilitates direct bacterial cell-to-cell spread. During cell-to-cell spread, the bacterium becomes transiently trapped in a double membrane vacuole, which fuses with Lamp1-positive vesicles. ProPC-PLC secreted in the vacuole is activated by either a bacterial or a host protease subsequent to acidification of the vacuole. Upon lysis of the vacuole, which is mediated in part by the phospholipases, the bacterium gains access to the cytosol where secreted proPC-PLC is degraded by the proteasome.
In conclusion, it appears that much of the regulation of L. monocytogenes pathogenesis occurs at levels subsequent to secretion. Regulatory proteolysis and pH requirements of L. monocytogenes virulence factors facilitate bacterial cell-to-cell spread with minimal damage to the host cell. We recently proposed that cytosolic half-life and pH optimum are factors preventing LLO cytotoxicity (Jones et al., 1996). The present study emphasizes the importance of host-mediated posttranslational control in the pathogenesis of intracellular parasites.
Acknowledgments
We thank Archie Bouwer and David Hinrichs for the animal studies, Nancy Freitag for strain DP-L1545, Gregory Smith for strain DP-L1938, and Andrew Camilli for strain DP-L1955. We also thank Peter Bannerman and Tracey Oliver of the Confocal Core of the Children's Hospital of Philadelphia and the Cancer Center of the University of Pennsylvania for help with the confocal microscopy.
This work was supported by U.S. Public Health Services grants AI26919 (to D.A. Portnoy), AI-27655 (to D.A. Portnoy), and GM-52797 (to H. Goldfine) from The National Institutes of Health. H. Marquis was a postdoctoral fellow of the Medical Research Council of Canada.
Footnotes
1. Abbreviations used in this paper: BHI, brain heart infusion broth; CFU, colony forming unit; Lamp1, lysosome-associated membrane protein 1; LD50, 50% lethal dose; LLM, N-acetyl-leucine-leucine-methionine; LLnL, N-acetyl-leucine-leucine-norleucine; LLO, listeriolysin O; Mpl, Listeria monocytogenes metalloprotease; PC, phosphatidylcholine; PLC, phospholipase C; PC-PLC, broad-range PLC; PI-PLC, phosphatidylinositol-specific PLC; proPC-PLC, secreted proenzyme form of PC-PLC.
Please address all correspondence to Daniel A. Portnoy (at his present address), Department of Molecular and Cell Biology and School of Public Health, Room 401, Barker Hall, University of California, Berkeley, CA 94720-3202.
H. Marquis' present address is Department of Microbiology, School of Medicine, Campus Box B175, University of Colorado Health Sciences Center, Denver, CO 80262.
References
- Bernardini ML, Mounier J, d'Hauteville H, Coquis-Rondon M, Sansonetti PJ. Identification of icsA, a plasmid locus of Shigella flexnerithat governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci USA. 1989;86:3867–3871. doi: 10.1073/pnas.86.10.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berón W, Alvarez-Dominguez C, Mayorga L, Stahl PD. Membrane trafficking along the phagocytic pathway. Trends Cell Biol. 1995;5:100–104. doi: 10.1016/s0962-8924(00)88958-8. [DOI] [PubMed] [Google Scholar]
- Bielecki J, Youngman P, Connelly P, Portnoy DA. Bacillus subtilis expressing a haemolysin gene from Listeria monocytogenescan grow in mammalian cells. Nature (Lond) 1990;345:175–176. doi: 10.1038/345175a0. [DOI] [PubMed] [Google Scholar]
- Bishop DK, Hinrichs DJ. Adoptive transfer of immunity to Listeria monocytogenes: the influence of in vitrostimulation on lymphocyte subset requirements. J Immunol. 1987;139:2005–2009. [PubMed] [Google Scholar]
- Bohley P, Seglen PO. Proteases and proteolysis in the lysosome. Experientia (Basel) 1992;48:151–157. doi: 10.1007/BF01923508. [DOI] [PubMed] [Google Scholar]
- Brundage RA, Smith GA, Camilli A, Theriot JA, Portnoy DA. Expression and phosphorylation of the Listeria monocytogenesActA protein in mammalian cells. Proc Natl Acad Sci USA. 1993;90:11890–11894. doi: 10.1073/pnas.90.24.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilli A, Tilney LG, Portnoy DA. Dual roles of plcA in Listeria monocytogenespathogenesis. Mol Microbiol. 1993;8:143–157. doi: 10.1111/j.1365-2958.1993.tb01211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JW, Murphy TL, Willingham MC, Pastan I, August JT. Identification of two lysosomal membrane glycoproteins. J Cell Biol. 1985;101:85–95. doi: 10.1083/jcb.101.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clague MJ, Urbé S, Aniento F, Gruenberg J. Vacuolar ATPase activity is required for endosomal carrier vesicle formation. J Biol Chem. 1994;269:21–24. [PubMed] [Google Scholar]
- Conte MP, Petrone G, Longhi C, Valenti P, Morelli R, Superti F, Seganti L. The effects of inhibitors of vacuolar acidification on the release of Listeria monocytogenesfrom phagosomes of Caco-2 cells. J Med Microbiol. 1996;44:418–424. doi: 10.1099/00222615-44-6-418. [DOI] [PubMed] [Google Scholar]
- Cossart P, Mengaud J. Listeria monocytogenes.A model system for the molecular study of intracellular parasitism. Mol Biol Med. 1989;6:463–474. [PubMed] [Google Scholar]
- Cossart P, Vicente MF, Mengaud J, Baquero F, Perez-Diaz JC, Berche P. Listeriolysin O is essential for virulence of Listeria monocytogenes: direct evidence obtained by gene complementation. Infect Immun. 1989;57:3629–3636. doi: 10.1128/iai.57.11.3629-3636.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford C, Mason RW, Wikstrom P, Shaw E. The design of peptidyldiazomethane inhibitors to distinguish between the cysteine proteinases calpain II, cathepsin L and cathepsin B. Biochem J. 1988;253:751–758. doi: 10.1042/bj2530751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudmore S, Cossart P, Griffiths G, Way M. Actin-based motility of vaccinia virus. Nature (Lond) 1995;378:636–638. doi: 10.1038/378636a0. [DOI] [PubMed] [Google Scholar]
- Dabiri GA, Sanger JM, Portnoy DA, Southwick FS. Listeria monocytogenesmoves rapidly through the host-cell cytoplasm by inducing directional actin assembly. Proc Natl Acad Sci USA. 1990;87:6068–6072. doi: 10.1073/pnas.87.16.6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins M, Huber LA, Parton RG, Griffiths G. Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J Cell Biol. 1994;124:677–688. doi: 10.1083/jcb.124.5.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domann E, Leimeister-Wächter M, Goebel W, Chakraborty T. Molecular cloning, sequencing, and identification of a metalloprotease gene from Listeria monocytogenesthat is species specific and physically linked to the listeriolysin gene. Infect Immun. 1991;59:65–72. doi: 10.1128/iai.59.1.65-72.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domann E, Wehland J, Rohde M, Pistor S, Hartl M, Goebel W, Leimeister-Wächter M, Wuenscher M, Chakraborty T. A novel bacterial virulence gene in Listeria monocytogenesrequired for host cell microfilament interaction with homology to the proline-rich region of vinculin. EMBO (Eur Mol Biol Organ) J. 1992;11:1981–1990. doi: 10.1002/j.1460-2075.1992.tb05252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard J-L, Berche P, Sansonetti P. Transposon mutagenesis as a tool to study the role of hemolysin in the virulence of Listeria monocytogenes. . Infect Immun. 1986;52:50–55. doi: 10.1128/iai.52.1.50-55.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard J-L, Berche P, Mounier J, Richard S, Sansonetti P. In vitro model of penetration and intracellular growth of Listeria monocytogenesin the human enterocyte-like cell line Caco-2. Infect Immun. 1987;55:2822–2829. doi: 10.1128/iai.55.11.2822-2829.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy C, Gaillard J-L, Alouf JE, Berche P. Purification, characterization, and toxicity of the sulfhydryl-activated hemolysin listeriolysin O from Listeria monocytogenes. . Infect Immun. 1987;55:1641–1646. doi: 10.1128/iai.55.7.1641-1646.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy C, Raveneau J, Beretti J-L, Lecroisey A, Vazquez-Boland J-A, Alouf JE, Berche P. Purification and characterization of an extracellular 29-kilodalton phospholipase C from Listeria monocytogenes. . Infect Immun. 1991;59:2382–2388. doi: 10.1128/iai.59.7.2382-2388.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfine H, Johnston NC, Knob C. Nonspecific phospholipase C of Listeria monocytogenes: activity on phospholipids in Triton X-100-mixed micelles and in biological membranes. J Bacteriol. 1993;175:4298–4306. doi: 10.1128/jb.175.14.4298-4306.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenberg J, Maxfield FR. Membrane transport in the endocytic pathway. Curr Opin Cell Biol. 1995;7:552–563. doi: 10.1016/0955-0674(95)80013-1. [DOI] [PubMed] [Google Scholar]
- Häse CC, Finkelstein RA. Bacterial extracellular zinc-containing metalloproteases. Microbiol Rev. 1993;57:823–837. doi: 10.1128/mr.57.4.823-837.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havell EA. Synthesis and secretion of interferon by murine fibroblasts in response to intracellular Listeria monocytogenes. . Infect Immun. 1986;54:787–792. doi: 10.1128/iai.54.3.787-792.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzen RA, Hayes SF, Peacock MG, Hackstadt T. Directional actin polymerization associated with spotted fever group Rickettsiainfection of Vero cells. Infect Immun. 1993;61:1926–1935. doi: 10.1128/iai.61.5.1926-1935.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Preiter K, Portnoy DA. Conversion of an extracellular cytolysin into a phagosome-specific lysin which supports the growth of an intracellular pathogen. Mol Microbiol. 1996;21:1219–1225. doi: 10.1046/j.1365-2958.1996.00074.x. [DOI] [PubMed] [Google Scholar]
- Kathariou S, Metz P, Hof H, Goebel W. Tn916-induced mutations in the hemolysin determinant affecting virulence of Listeria monocytogenes. . J Bacteriol. 1987;169:1291–1297. doi: 10.1128/jb.169.3.1291-1297.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathariou S, Pine L, George V, Carlone GM, Holloway BP. Nonhemolytic Listeria monocytogenesmutants that are also noninvasive for mammalian cells in culture: evidence for coordinate regulation of virulence. Infect Immun. 1990;58:3988–3995. doi: 10.1128/iai.58.12.3988-3995.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschke H, Shaw E. Rapid inactivation of cathepsin L by Z-PhePheCHN2 and Z-Phe-AlaCHN2 . Biochem Biophys Res Commun. 1981;101:454–458. doi: 10.1016/0006-291x(81)91281-x. [DOI] [PubMed] [Google Scholar]
- Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. L. monocytogenes-induced actin assembly requires the actAgene product, a surface protein. Cell. 1992;68:521–531. doi: 10.1016/0092-8674(92)90188-i. [DOI] [PubMed] [Google Scholar]
- Kuhn M, Kathariou S, Goebel W. Hemolysin supports survival but not entry of the intracellular bacterium Listeria monocytogenes. . Infect Immun. 1988;56:79–82. doi: 10.1128/iai.56.1.79-82.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leary R, Larsen D, Watanabe H, Shaw E. Diazomethyl ketone substrate derivatives as active-site directed inhibitors of thiol proteases. Papain. Biochemistry. 1977;16:5857–5861. doi: 10.1021/bi00645a033. [DOI] [PubMed] [Google Scholar]
- Lewis V, Green SA, Marsh M, Vihko P, Helenius A, Mellman I. Glycoproteins of the lysosomal membrane. J Cell Biol. 1985;100:1839–1847. doi: 10.1083/jcb.100.6.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquis H, Doshi V, Portnoy DA. The broad-range phospholipase C and a metalloprotease mediate listeriolysin O-independent escape of Listeria monocytogenesfrom a primary vacuole in human epithelial cells. Infect Immun. 1995;63:4531–4534. doi: 10.1128/iai.63.11.4531-4534.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengaud J, Geoffroy C, Cossart P. Identification of a new operon involved in Listeria monocytogenesvirulence: its first gene encodes a protein homologous to bacterial metalloproteases. Infect Immun. 1991;59:1043–1049. doi: 10.1128/iai.59.3.1043-1049.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel E, Reich KA, Favier R, Berche P, Cossart P. Attenuated mutants of the intracellular bacterium Listeria monocytogenesobtained by single amino acid substitutions in listeriolysin O. Mol Microbiol. 1990;4:2167–2178. doi: 10.1111/j.1365-2958.1990.tb00578.x. [DOI] [PubMed] [Google Scholar]
- Mounier J, Ryter A, Coquis-Rondon M, Sansonetti PJ. Intracellular and cell-to-cell spread of Listeria monocytogenesinvolves interaction with F-actin in the enterocytelike cell line Caco-2. Infect Immun. 1990;58:1048–1058. doi: 10.1128/iai.58.4.1048-1058.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niebuhr K, Chakraborty T, Köllner P, Wehland J. Production of monoclonal antibodies to the phosphatidylcholine-specific phospholipase C of Listeria monocytogenes, a virulence factor for this species. Med Microbiol Lett. 1993;2:9–16. [Google Scholar]
- North RJ. Suppression of cell-mediated immunity to infection by an antimitotic drug. J Exp Med. 1970;132:535–545. doi: 10.1084/jem.132.3.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmsted JB. Affinity purification of antibodies from diazotized paper blots of heterogeneous protein samples. J Biol Chem. 1981;256:11955–11957. [PubMed] [Google Scholar]
- Pitt A, Mayorga LS, Stahl PD, Schwartz AL. Alterations in the protein composition of maturing phagosomes. J Clin Invest. 1992;90:1978–1983. doi: 10.1172/JCI116077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy DA, Jacks PS, Hinrichs DJ. Role of hemolysin for the intracellular growth of Listeria monocytogenes. . J Exp Med. 1988;167:1459–1471. doi: 10.1084/jem.167.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy DA, Chakraborty T, Goebel W, Cossart P. Molecular determinants of Listeria monocytogenespathogenesis. Infect Immun. 1992;60:1263–1267. doi: 10.1128/iai.60.4.1263-1267.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyart C, Abachin E, Razafimanantsoa I, Berche P. The zinc metalloprotease of Listeria monocytogenesis required for maturation of phosphatidylcholine phospholipase C: direct evidence obtained by gene complementation. Infect Immun. 1993;61:1576–1580. doi: 10.1128/iai.61.4.1576-1580.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveneau J, Geoffroy C, Beretti J-L, Gaillard J-L, Alouf JE, Berche P. Reduced virulence of a Listeria monocytogenesphospholipase-deficient mutant obtained by transposon insertion into the zinc metalloprotease gene. Infect Immun. 1992;60:916–921. doi: 10.1128/iai.60.3.916-921.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- Rosen H, Gordon S, North RJ. Exacerbation of murine listeriosis by a monoclonal antibody specific for the type 3 complement receptor of myelomonocytic cells. J Exp Med. 1989;170:27–37. doi: 10.1084/jem.170.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GA, Marquis H, Jones S, Johnston NC, Portnoy DA, Goldfine H. The two distinct phospholipases C of Listeria monocytogeneshave overlapping roles in escape from a vacuole and cell-to-cell spread. Infect Immun. 1995;63:4231–4237. doi: 10.1128/iai.63.11.4231-4237.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K, Youngman P. Use of a new integrational vector to investigate compartment-specific expression of the Bacillus subtilis spoIIMgene. Biochimie (Paris) 1992;74:705–711. doi: 10.1016/0300-9084(92)90143-3. [DOI] [PubMed] [Google Scholar]
- Sun AN, Camilli A, Portnoy DA. Isolation of Listeria monocytogenessmall-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect Immun. 1990;58:3770–3778. doi: 10.1128/iai.58.11.3770-3778.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teysseire N, Chiche-Portiche C, Raoult D. Intracellular movements of Rickettsia conorii and R. typhibased on actin polymerization. Res Microbiol. 1992;143:821–829. doi: 10.1016/0923-2508(92)90069-z. [DOI] [PubMed] [Google Scholar]
- Theriot JA, Mitchison TJ, Tilney LG, Portnoy DA. The rate of actin-based motility of intracellular Listeria monocytogenesequals the rate of actin polymerization. Nature (Lond) 1992;357:257–260. doi: 10.1038/357257a0. [DOI] [PubMed] [Google Scholar]
- Tilney LG, Portnoy DA. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. . J Cell Biol. 1989;109:1597–1608. doi: 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Weert AWM, Dunn KW, Geuze HJ, Maxfield FR, Stoorvogel W. Transport from late endosomes to lysosomes, but not sorting of integral membrane proteins in endosomes, depends on the vacuolar proton pump. J Cell Biol. 1995;130:821–834. doi: 10.1083/jcb.130.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Boland J-A, Kocks C, Dramsi S, Ohayon H, Geoffroy C, Mengaud J, Cossart P. Nucleotide sequence of the lecithinase operon of Listeria monocytogenesand possible role of lecithinase in cell-to-cell spread. Infect Immun. 1992;60:219–230. doi: 10.1128/iai.60.1.219-230.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva MS, Sijts AJAM, Pamer EG. Listeriolysin is processed efficiently into an MHC class I-associated epitope in Listeria monocytogenes-infected cells. J Immunol. 1995;155:5227–5233. [PubMed] [Google Scholar]
- Vinitsky A, Michaud C, Powers JC, Orlowski M. Inhibition of the chymotrypsin-like activity of the pituitary multicatalytic proteinase complex. Biochemistry. 1992;31:9421–9428. doi: 10.1021/bi00154a014. [DOI] [PubMed] [Google Scholar]
- Wilcox D, Mason RW. Inhibition of cysteine proteinases in lysosomes and whole cells. Biochem J. 1992;285:495–502. doi: 10.1042/bj2850495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H+-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem. 1991;266:17707–17712. [PubMed] [Google Scholar]