

Abstract
In a screen for second site mutations capable of reducing the restrictive temperature of the fission yeast mutant cdc2-D217N, we have isolated a novel temperature-sensitive mutant, dim1-35. When shifted to restrictive temperature, dim1-35 mutant cells arrest before entry into mitosis or proceed through mitosis in the absence of nuclear division, demonstrating an uncoupling of proper DNA segregation from other cell cycle events. Deletion of dim1 from the Schizosaccharomyces pombe genome produces a lethal G2 arrest phenotype. Lethality is rescued by overexpression of the mouse dim1 homolog, mdim1. Likewise, deletion of the Saccharomyces cerevisiae dim1 homolog, CDH1, is lethal. Both mdim1 and dim1 + are capable of rescuing lethality in the cdh1::HIS3 mutant. Although dim1-35 displays no striking genetic interactions with various other G2/M or mitotic mutants, dim1-35 cells incubated at restrictive temperature arrest with low histone H1 kinase activity. Morevoer, dim1-35 displays sensitivity to the microtubule destabilizing drug, thiabendazole (TBZ). We conclude that Dim1p plays a fundamental, evolutionarily conserved role as a protein essential for entry into mitosis as well as for chromosome segregation during mitosis. Based on TBZ sensitivity and failed chromosome segregation in dim1-35, we further speculate that Dim1p may play a role in mitotic spindle formation and/or function.
The fission yeast Schizosaccharomyces pombe provides an excellent model system for analysis of mechanisms of cell cycle regulation. In particular, genes involved in the G2/M transition and in progression through mitosis itself have been identified and studied extensively. Entry into mitosis depends upon cdc2 gene function (Nurse et al., 1976). Temperature-sensitive (ts)1 cdc2 mutants arrest either exclusively at the G2/M boundary or at both the G2/M and G1/S boundaries (Nurse and Bissett, 1981). Likewise, temperature-sensitive cdc13 and cdc25 mutants arrest in G2 (Nurse et al., 1976; Russell and Nurse, 1986; Booher and Beach, 1987, 1988; Hagan et al., 1988). In contrast, temperature-sensitive mutations in wee1 result in premature entry into mitosis (Nurse, 1975; Thuriaux et al., 1978; Nurse and Thuriaux, 1980; Russell and Nurse, 1987). wee1 mik1 double mutants undergo lethal, premature entry into mitosis, termed mitotic catastrophe (Lundgren et al., 1991). Genetic and biochemical analyses have elucidated the molecular mechanisms that lie behind the observed phenotypes. cdc2 encodes a 34-kD protein serine/threonine kinase whose activity is required to drive progression into M phase (Hindley and Phear, 1984; Simanis and Nurse, 1986). Cdc2p activity depends both upon its association with Cdc13p, a B-type cyclin (Booher and Beach, 1987, 1988; Hagan et al., 1988), as well as upon the balance between positive and negative regulatory phosphorylation events. Activation of Cdc2p requires phosphorylation at T167, while phosphorylation at Y15, mediated by the wee1 and mik1 gene products, inhibits the Cdc2p kinase activity (Gould and Nurse, 1989; Gould et al., 1991; Featherstone and Russell, 1991; Lundgren et al., 1991; Lee et al., 1994). The cdc25 gene product, a protein tyrosine phosphatase, dephosphorylates Cdc2 on Y15, activating the Cdc2p/ Cdc13p complex and allowing entry into mitosis (for review see Millar and Russell, 1992; Coleman and Dunphy, 1994).
Progression through mitosis itself consists of a number of distinct processes. Early mitotic events in fission yeast include chromosome condensation, duplication of the spindle pole body (SPB; yeast equivalent of mammalian centrosome), and mitotic spindle formation. As cells progress through M phase, spindle elongation and chromosome segregation occur. Finally, septation and cytokinesis mark exit from mitosis. Just as genetic analyses first identified major players involved in coordinating entry into mitosis, analyses of mutant phenotypes have elucidated mechanisms of progression through mitosis. Temperaturesensitive cell untimely torn mutants (cut; Uemura and Yanagida, 1984; Hirano et al., 1986; Samejima et al., 1993) define a number of genes involved in mitotic processes, particularly the processes of chromosome condensation and segregation. For example, in cut1 (Uzawa et al., 1990), cut2 (Uzawa et al., 1990; Funabiki et al., 1996a ), cut3 (Saka et al., 1994), cut14 (Saka et al., 1994), and top2 (Uemura and Yanagida 1984, 1986; Uemura et al., 1987) mutants, spindle formation and elongation occur, but chromosomes fail to separate. Subsequently, a septum is layed down across the undivided nucleus to produce the characteristic cut phenotype. Cut1p and Cut2p interact physically; both gene products localize to the short mitotic spindle (Funabiki et al., 1996a ,b). Although Cut1p levels remain constant throughout the cell cycle (Funabiki et al., 1996b), proteolysis of Cut2p must occur at the metaphase to anaphase transition to allow chromosome segregation (Funabiki et al., 1996a ). cut3, cut14, and top2 are all required for proper chromosome condensation at mitosis. In cut3, cut14, and top2 mutants, mitotic spindles form and elongate normally, but uncondensed chromosomal material fails to segregate (Uemura and Yanagida 1984, 1986; Uemura et al., 1987; Saka et al., 1994).
The dis mutants, like the cut mutants, display defects in chromosome segregation (Okhura et al., 1988). In dis1 mutants, chromosome condensation as well as spindle formation and elongation appear to occur normally. Nevertheless, sister chromatids fail to separate at anaphase B, upon elongation of the mitotic spindle (Okhura et al., 1988; Nabeshima et al., 1995). The dis1 gene product, which displays no significant homology to previously characterized proteins, localizes to the mitotic spindle as well as to spindle pole bodies and cytoplasmic microtubules (Nabeshima et al., 1995).
In addition to mutants defective in chromosome segregation, mutants defective in spindle formation and/or elongation have been isolated and define a second class of genes involved in progression through mitosis. Products of such genes include Sad1p, which localizes to spindle pole bodies (Hagan and Yanagida, 1995); Nda2p and Nda3p, α1-tubulin, and β-tubulin, respectively (Toda et al., 1983; Umesono et al., 1983; Yanagida, 1989); Cut7p, a putative kinesin-like mitotic motor protein (Hagan and Yanagida 1990, 1992); Cut9p, Nuc2p, and Cut4p, S. pombe homologs of the Saccharomyces cerevisiae anaphase-promoting complex (APC) components CDC16, CDC23, and APC1, respectively, all of which are required for spindle elongation at the metaphase/anaphase transition (Hirano et al., 1988; Sikorski et al., 1990; Samejima and Yanagida, 1994; Yameshita et al., 1996; Zachariae et al., 1996); and Plo1p, the S. pombe homolog of the Drosophila polo kinase (Okhura et al., 1995).
Finally, genes involved in the events that mark exit from mitosis, namely septation and cytokinesis, include many initially identified by virtue of septation-defective mutant phenotypes (Nurse et al., 1976). In the septation-defective mutants (cdc7, cdc11, cdc14, and cdc15), septation and cytokinesis are blocked, but progression through the remainder of the cell cycle continues, such that cells elongate and accumulate multiple nuclei.
Interestingly, unlike cdc2 or cdc25 mutants (Nurse et al., 1976), many of the mitotic mutants described above do not block all of the events of mitosis or lead to mid-mitotic arrest. Rather, these mutants illustrate the separability of the events of mitosis, both from one another and from progression through the remainder of the cell cycle. Thus, in cut1, cut2, cut3, cut14, and top2 mutants, spindle elongation, septation, and, in the case of cut1 and top2, entry into a subsequent cell cycle occur in the absence of chromosome segregation (Uemura et al., 1987; Uzawa et al., 1990; Saka et al., 1994). Conversely, in cdc7, cdc11, cdc14, and cdc15, multiple rounds of mitosis occur without intervening rounds of septation and cytokinesis (Nurse et al., 1976).
Here we describe the isolation and characterization of a temperature-sensitive S. pombe mutant, dim1-35. Like mutations in cdc2 or cdc25, deletion of dim1 from the genome blocks entry into mitosis. Unlike cdc2 or cdc25 mutants, however, hypomorphic dim1-35 mutant cells also display the ability to uncouple mitotic events. That is to say, dim135 mutant cells incubated at restrictive temperature either arrest before entry into mitosis or proceed through mitosis and subsequent septation in the absence of proper chromosome segregation. Thus, dim1 + defines a previously undescribed gene essential both for G2/M progression as well as for chromosome segregation in mitosis.
Materials and Methods
Yeast Methods, Strains, and Media
S. pombe strains used in this study are listed in Table I. Strains were grown in yeast extract medium or minimal medium with appropriate supplements (Moreno et al., 1991). Crosses were performed on malt extract medium (Moreno et al., 1991) or glutamate medium (minimal medium lacking ammonium chloride and containing 0.01M glutamate, pH 5.6). Random spore analysis and tetrad analysis were performed as described (Moreno et al., 1991). Double mutant strains were constructed and identified by tetrad analysis. Transformations were performed by electroporation (Prentice, 1991). Genomic DNA was isolated as described (Moreno et al., 1991; Hoffman, 1993).
Table I.
S. Pombe Strains Used in This Study
| Strain designation | Genotype | Source | ||
|---|---|---|---|---|
| KGY28 | h- 972 | P. Nurse* | ||
| KGY56 | h+ nda3-km311 leu1-32 | P. Nurse | ||
| KGY69 | h+ 975 | P. Nurse | ||
| KGY89 | h- nda2-km52 leu1-32 | P. Nurse | ||
| KGY90 | h- dis1-288 leu1-32 | P. Nurse | ||
| KGY107 | h- cdc11-119 | P. Nurse | ||
| KGY155 | h+ cdc25-22 | P. Nurse | ||
| KGY246 | h- ura4-D18 leu1-32 ade6-M210 | P. Nurse | ||
| KGY247 | h+ ura4-D18 leu1-32 ade6-M210 | P. Nurse | ||
| KGY248 | h- ura4-D18 leu1-32 ade6-M216 | P. Nurse | ||
| KGY249 | h+ ura4-D18 leu1-32 ade6-M216 | P. Nurse | ||
| KGY384 | h+ dim1-35 cdc25-22 | This study | ||
| KGY387 | h- dim1-35 dis1-288 | This study | ||
| KGY392 | h+ dim1-35 | This study | ||
| KGY394 | h+ dim1-35 ade6-M216 | This study | ||
| KGY396 | h+ dim1-35 leu1-32 | This study | ||
| KGY402 | h- dim1-35 | This study | ||
| KGY455 | h- cdc2-D217N ura4-D18 leu1-32 ade6-M210 | Berry and Gould, 1996 | ||
| KGY490 | h- dim1-35 ura4-D18 | This study | ||
| KGY517 | h+ rad1::ura4 + ura4-D18 leu1-32 | S. Subramani‡ | ||
| KGY533 | h- cdc2-22 leu1-32 ura4-D18 ade6-M216 | P. Nurse | ||
| KGY557 | h- dim1-35 ade6-M210 | This study | ||
| KGY621 | h- cdc13-A381V his3-237 ura4-D18 leu1-32 ade6-M210 | Berry and Gould, 1996 | ||
| KGY698 | h+ dim1-35 his3-237 ura4-D18 leu1-32 ade6-M210 | This study | ||
| KGY833 | h+/h- dim1-35/dim1::ura4 + his3-237/his3 + ura4-D18/ura4-D18 leu1-32/leu1-32 ade6-M210/ade6-M216 | This study | ||
| KGY838 | h- cdr1-76 leu1-32 ade6-M216 | Young and Fantes, 1987 | ||
| KGY841 | h+/h- dim1 +/dim1::ura4+ his3-237/his3 + ura4-D18/ura4-D18 leu1-32/leu1-32 ade6-M210/ade6-M216 | This study | ||
| KGY842 | h+ dim1::ura4 + carrying pKG709 ura4-D18 leu1-32 ade6-M216 | This study | ||
| KGY843 | h- cdc13-117 ura4-D18 leu1-32 ade6-M210 | P. Nurse | ||
| KGY858 | h+ dim1::ura4 + carrying pKG718 ura4-D18 leu1-32 ade6-M216 | This study | ||
| KGY872 | h- cdc25-22 dim1-35 his3-D1 ura4-D18 leu1-32 ade6-M210 | This study | ||
| KGY874 | h+ cdc13-117 dim1-35 ura4-D18 leu1-32 ade6-M210 | This study | ||
| KGY878 | h- cdc2-22 dim1-35 | This study | ||
| KGY879 | h- dim1-35 cdc13-A381V | This study | ||
| KGY895 | h- dim1-35 cdr1-76 | This study | ||
| KGY896 | h- cdc2-D217N dim1-35 | This study | ||
| KGY939 | h- cut1-RB5 ura4-D18 leu1-32 | I. Hagan§ | ||
| KGY942 | h+ cdc11-119 dim1-35 | This study | ||
| KGY943 | h- cut1-RB5 dim1-35 leu1-32 | This study | ||
| KGY957 | h+ cut3-477 | R. McIntosh‖ | ||
| KGY958 | h- cut1-205 leu1-32 | R. McIntosh | ||
| KGY982 | h+ nuc2-663 leu1-32 ura4-D18 ade6-M210 h+ | P. Nurse | ||
| KGY1019 | h- cut2-364 leu1-32 | R. McIntosh | ||
| KGY1020 | h- cut2-205 dim1-35 | This study | ||
| KGY1021 | h- cut2-364 dim1-35 | This study | ||
| KGY1022 | h- dim1-35 rad1::ura4 + ura4-D18 | This study | ||
| KGY1028 | h+/h- dim1-35/dim1 + his3-D1/his3-D1 ura4-D18/ura4-D18 leu1-32/leu1-32 ade6-M210/ade6-M216 | This study | ||
| KGY1087 | h+/h- dim1-35/dim1::his3 + his3-D1/his3-D1 ura4-D18/ura4-D18 leu1-32/leu1-32 ade6-M210/ade6-M216 | This study | ||
| KGY1088 | h+/h- dim1::his3 + /dim1 + his3-D1/his3-D1 ura4-D18/ura4-D18 leu1-32/leu1-32 ade6-M210/ade6-M216 | This study | ||
| KGY1089 | h- dim1::his3 + carrying pKG897 his3-D1 ura4-D18 leu1-32 ade6-M210 | This study | ||
| KGY1090 | h+ dim1::his3 + carrying pKG898 his3-D1 ura4-D18 leu1-32 ade6-M210 | This study | ||
| KGY1091 | h+ dim1::ura4 + carrying pKG815 ura4-D18 leu1-32 ade6-M216 | This study | ||
| KGY1092 | h+ dim1::ura4 + carrying pKG930 ura4-D18 leu1-32 ade6-M210 | This study | ||
| KGY1115 | h- leu1-32::pKG984 | This study | ||
| KGY1124 | h- dim1-35 leu1-32::pKG984 | This study | ||
| KGY1138 | h- dim1-35::pKG695 ura4-D18 leu1-32 ade6-M210 | This study | ||
| KGY1139 | h+ dim1-35::pKG695 ura4-D18 leu1-32 ade6-M210 | This study | ||
| KGY1155 | h- nda3-km311 dim1-35 leu1-32 | This study | ||
| KGY1156 | h- cut3-447 dim1-35 | This study | ||
| KGY1180 | h+ dim1::his3 + leu1-32::p980 his3-D1 ura4-D18 ade6-M210 | This study | ||
| KGY1216 | h+ leu1-32::0980 ura4-D18 ade6-M210 | This study |
Imperial Cancer Research Fund.
University of California, San Diego, CA.
University of Manchester, UK.
University of Colorado, Boulder, CO.
S. cerevisiae strains used in this study are listed in Table II. Strains were grown in YPD (1% yeast extract, 2% bactopeptone, 2% glucose), YPGR (1% yeast extract, 2% bactopeptone, 1% raffinose, 1% galactose), SD (0.67% yeast nitrogen base, 2% glucose), or SGR (0.67% yeast nitrogen base, 1% raffinose, 1% galactose) with appropriate supplements. For the MET25 promoter, inducing conditions refer to growth on media lacking methionine; repressing conditions refer to growth on media containing 2.5 mM methionine. For the GALS promoter, inducing conditions refer to growth on YPGR or SGR plus appropriate supplements; repressing conditions refer to growth on YPD or SD plus appropriate supplements. Diploids were induced to sporulate on sporulation medium (1% potassium acetate, 0.1% yeast extract, 0.05% glucose, 2% bacto-agar). Transformations were performed by the lithium acetate method (Becker and Lundblad, 1994). Genomic DNA was isolated as described (Hoffman, 1993).
Table II.
S. cerevisiae Strains Used in This Study
| Strain designation | Genotype | Source | ||
|---|---|---|---|---|
| KGY311 | MATα cdh1::HIS3 leu2-3, 112::pKG914 ura3-52 his3-d200 trp1-d901, suc2-D9, lys2-801 | This study | ||
| KGY572 | MATα cdh1::HIS3 leu2-3, 112::pKG913 ura3-52 his3-D200 trp1-D901, suc2-D9, lys2-801 | This study | ||
| KGY820 | MATα his3-d200 leu2-3, 112 lys2-801 ura3-52 trp1-d901 suc2-d9 | T. Graham | ||
| KGY821 | MATα his3-d200 leu2-3, 112 ade2-101 ura3-52 trp1-d901 suc2-d9 | T. Graham | ||
| KGY823 | MATα/MATa his3-d200/his3-d200 leu2-3, 112/leu2-3, 112 leu2-801/+ade2-101/+ ura3-52/ | T. Graham | ||
| ura3-52 trp1-d901/trp1-d901 suc2-d9/suc2-d9 | ||||
| KGY937 | MATα/MATa CDH1/cdh1::HIS3 his3-d200/ his3-d200 leu2-3, 112/ leu2-3, 112 lys2-801/+ | This study | ||
| ade2-101/1 ura3-52/ura3-52 trp1-d901/trp1-d901 suc2-d9/suc2-d9 | ||||
| KGY965 | MATα cdh1::HIS3 leu2-3,112::pKG912 ura3-52 his3-D200 trp1-D901, suc2-D9, lys2-801 | This study | ||
| KGY966 | MATα cdh1::HIS3 leu2-3,112::pKG911 ura3-52 his3-D200 trp1-D901, suc2-D9, lys2-801 | This study | ||
| KGY1023 | MATα cdh1::HIS3 leu2-3,112::pKG903 ura3-52 his3-D200 trp1-D901, suc2-D9, lys2-801 | This study | ||
| KGY1026 | MATα cdh1::HIS3 leu2-3,112::pKG882 ura3-52 his3-D200 trp1-D901, suc2-D9, lys2-801 | This study | ||
| KGY1093 | MATα cdh1::HIS3 carrying pKG900 leu2-3,112 ura3-52 his3-D200 trp1-D901, suc2-D9, lys2-801 | This study |
Plasmids and Molecular Biological Techniques
Plasmids used in this study are listed in Table III. All plasmid manipulations and bacterial transformations were by standard techniques (Sambrook et al., 1989). Essential features of plasmid construction are described below; details of plasmid construction are available from the authors upon request. All sequencing was performed using Sequenase 2.0 (USB, Cleveland, OH) according to manufacturer's instructions. All PCR reactions were performed using Taq DNA polymerase and the GeneAmp PCR reagent kit (Perkin Elmer, Norwalk, CT) in a PTC-100 programmable thermal controller (PTC-100; MJ Research, Watertown, MA) programmed as follows: 94°C, 1 min; 50°C, 2 min; 72°C, 2 min (40 cycles); 72°C, 10 min.
Table III.
Plasmids Constructed in This Study
| Plasmid | Parent vector | Insert | ||
|---|---|---|---|---|
| pKG402 | pREP41 | dim1 + cDNA | ||
| pKG681 | pUR19 | dim1 3.7 kb genomic fragment | ||
| pKG683 | pUR19 | dim1 2 kb genomic fragment | ||
| pKG695 | pJK210 | dim1 3.7 kb genomic fragment | ||
| pKG703 | pBS-SK+ | dim1::ura4 + (1 kb dim1 5′ flank::ura4 +::1.2 kb dim1 3′ flank) | ||
| pKG709 | pMNS21L | dim1 + cDNA | ||
| pKG718 | pRHA41 | dim1 + cDNA | ||
| pKG745 | pMNS21L | dim1-35 genomic PCR product | ||
| pKG815 | pREP81 | dim1 + cDNA | ||
| pKG836 | pMNS21L | CDH1 cDNA | ||
| pKG837 | pRS415GALS | CDH1 cDNA | ||
| pKG843 | pRS415GALS | dim1 + cDNA | ||
| pKG849 | pRS415MET25 | CDH1 cDNA | ||
| pKG850 | pRS415MET25 | dim1 + cDNA | ||
| pKG872 | pBS-SK+ | N-degron/Ubiquitin tag | ||
| pKG875 | pRS415GALS | UBCDH1 cDNA | ||
| pKG876 | pRS415GALS | UBdim1 + cDNA | ||
| pKG882 | pRS405 | GALS::UBCDH1 cDNA | ||
| pKG889 | pREP81 | CDH1 cDNA | ||
| pKG894 | pREP41 | CDH1 cDNA | ||
| pKG897 | pREP42 | dim1 + cDNA | ||
| pKG898 | pREP82 | dim1 + cDNA | ||
| pKG900 | pRS415GALS | mdim1 cDNA | ||
| pKG901 | pREP81 | mdim1 cDNA | ||
| pKG903 | pRS405 | GALS::UBdim1 + cDNA | ||
| pKG911 | pRS405 | MET25::dim1 + cDNA | ||
| pKG912 | pRS405 | GALS::CDH1 cDNA | ||
| pKG913 | pRS405 | MET25::CDH1 cDNA | ||
| pKG914 | pRS405 | GALS::dim1 + cDNA | ||
| pKG916 | pBS-SK+ | dim1::HindIII (1 kb dim1 5′ flank::HindIIIsite::1.2 kb 3′ flank) | ||
| pKG917 | pBS-SK+ | dim1::his3 + (1 kb dim1 5′ flank::his3 +::1.2 kb 3′ flank) | ||
| pKG930 | pREP1 | mdim1 cDNA | ||
| pKG931 | pREP81 | mdim1 cDNA | ||
| pKG980 | pJK148 | nmtl-T81::dim1 + cDNA | ||
| pKG984 | pJK148 | nmtl::CDH1 cDNA |
Isolation of the dim1-35 Mutant
The temperature-sensitive strain cdc2-D217N (restrictive temperature, 36°C) was treated with the chemical mutagen nitrosoguanidine (NTG) as decribed (Moreno et al., 1991). Cells mutagenized to 50% viability were plated on YE at a density of 300 colonies/plate and incubated at 27°C. Colonies were replica plated to 32°C. Colonies unable to grow at 32°C were picked and outcrossed extensively. Upon outcrossing of colony number 35, an extragenic ts mutation (restrictive temperature, 36.5°C) was observed to segregate away from cdc2-D217N. At 32°C, this mutant (later termed dim1-35) proved semiviable in a cdc2 + background and lethal in a cdc2-D217N background. Hence, it was chosen for further characterization.
Physiological Experiments
For analysis of synchronous cell populations, 4 liters of cells were grown to midlog phase (8 × 106 cells/ml) at permissive temperature (25°C) in YE medium. Cells were separated on the basis of size by centrifugal elutriation in an elutriator rotor (JE 5.0; Beckman Instr., Fullerton, CA). Cells synchronized in early G2 (i.e., the smallest cells in the population) were collected and inoculated into YE medium at 25 or 36°C. Synchrony was monitored at 20- to 25-min intervals by scoring 100 cells for the presence of a septum.
For nitrogen starvation and release experiments, cells were grown to midlog phase in rich medium at permissive temperature. Cells were collected by centrifugation, washed three times in minimal medium lacking nitrogen, and then inoculated into minimal medium lacking nitrogen. Cells were starved for nitrogen at permissive temperature for 19 h and then shifted to restrictive temperature. After 30 min at restrictive temperature, cells were collected by centrifugation and inoculated into rich medium (prewarmed to 36°C). Cultures were incubated at 36.5°C; samples were collected at 30-min intervals to monitor septation index, total cell number, viable cell number, cell morphology, and DNA content (see below).
For dim1-35 spore germination experiments, dim1-35 mutant spores were generated by mating KGY394 and KGY557 and then selecting for diploids on media lacking adenine. Diploid cells were allowed to sporulate. Sporulated diploids were collected and then treated with glusulase. Free spores were purified by centrifugation through 40% glucose. Purified spores were inoculated into rich medium and incubated at 36.5°C. Samples were collected at 1-h intervals to monitor septation index, total cell number, viable cell number, cell morphology, and DNA content of cells.
For analysis of the dim1::his3 + terminal phenotype, KGY1180 and KGY1216 cells were grown in minimal medium (lacking thiamine) at 32°C. Cells in log phase growth were inoculated into YE medium (containing thiamine) and incubated at 32°C. Samples were collected at 1-h intervals to monitor total cell number, cell morphology, DNA content, and histone H1 kinase activity.
For determination of total cell number in the above experiments, cells were collected and fixed in 0.12 M NaCl, 3% formaldehyde, diluted appropriately, sonicated briefly, and then counted in triplicate using the Coulter® Multisizer II (Coulter Electronics Limited, UK). Total cell number was taken as the average of each triplicate. For determination of viable cell number, cells collected at specified intervals were diluted appropriately, sonicated briefly, and then plated in triplicate onto appropriate media. For S. pombe cultures, cells were plated onto YE and incubated at 25°C. For S. cerevisiae GALS promoter shut off experiments, cells were plated onto YPGR and incubated at 32°C. After incubation, colonies were counted and viable cell number taken as the average of each triplicate. Total cell number and viable cell number were plotted on a linear scale; percent viability was taken as viable cells/ml divided by total cells/ml at each time point.
For analysis of thiabendazole (TBZ) sensitivity, dim1-35 or wild-type strains were streaked to YE agar containing 9 μg/ml TBZ dissolved in DMSO or an equivalent amount of DMSO alone. Plates were incubated at 29°C for 3 d. For analysis of TBZ sensitivity in liquid culture, cells were grown to mid-log phase in YE at 29°C. TBZ was added to cultures to a final concentration of 10 μg/ml. After 6 h, cells were collected, fixed with ethanol, and stained with DAPI (see below).
Flow Cytometry and Microscopy
For flow cytometric analysis, cells were fixed in ice-cold 70% ethanol, washed in 50 mM sodium citrate, incubated with 0.1 μg/ml RNaseA in 50 mM sodium citrate for 2 h at 37°C, and then stained with 2 μg/ml propidium iodide in 50 mM sodium citrate at 4°C in the dark overnight. Cells were sonicated and analyzed by flow cytometry as described (Sazer and Sherwood, 1990). All fluoresence microscopy was performed on a Zeiss microscope (Axioscope; Zeiss, Inc., Thornwood, NY) using appropriate filters. To visualize DNA, ethanol-fixed cells were washed in PBS and then stained with the fluorescent DNA-binding dye DAPI at 1 μg/ml. For immunofluorescence, cells were fixed in 70% ethanol at 4°C or in 100% methanol at −20°C for 8 min and then washed with PBS and processed as described (Moreno et al., 1991). For staining of microtubules, fixed cells were incubated in a 1:10 dilution of the monoclonal TAT-1 primary antibody (Woods et al., 1989; a generous gift of Dr. K. Gull) followed by a 1: 100 dilution of Texas red–conjugated goat anti–mouse secondary antibody (Molecular Probes, Eugene, OR). For staining of spindle pole bodies, fixed cells were incubated in a 1:25 dilution of the polyclonal anti-Sad1p primary antibody (Hagan and Yanagida, 1995; a generous gift of Dr. I. Hagan) followed by a 1:100 dilution of FITC-conjugated goat anti–rabbit secondary antibody (Molecular Probes).
Cloning and DNA Sequence of dim1
dim1-35 mutant cells transformed with a pUR19-based S. pombe genomic library (Barbet et al., 1992) were selected at 25°C on media lacking uracil and then replica plated to 36°C. Plasmids were recovered from 4 Ura+ Dim1+ colonies and analyzed by restriction digest and Southern blot analysis. Deletion constructs generated from rescuing plasmids were retransformed into dim1-35 to identify a minimal rescuing fragment. The minimal rescuing fragment was sequenced; sequence was analyzed for coding potential by comparison to sequences available in nucleic acid sequence databases accessible through the Baylor College of Medicine (Houston, TX) and the National Center for Biotechnology Information (Bethesda, MD).
For integration mapping, 3.7 kb of dim1-proximal genomic sequence encompassing the 2-kb minimal rescuing fragment was subcloned into the S. pombe ura4-based integrating vector pJK210 (Keeney and Boeke, 1994) to generate pKG695. pKG695 was linearized at the unique Sac1 site within the dim1 genomic fragment and the linearized plasmid transformed into the dim1-35 ura4-D18 mutant strain KGY698. Transformants were selected on media lacking uracil. Dim1+Ura+ transformants were picked and outcrossed to either dim1 + or dim1-35 strains to verify cosegregation of the dim1 + phenotype with the ura4 + marker.
A putative full length dim1 cDNA was isolated by PCR-mediated amplification from a pDB20-based S. pombe cDNA library (Fikes et al., 1990) using the oligos 35START (5′-TCAGTTGATCATATGAGTTATTTTTTACC-3′) and 35STOP (5′-ATAAGCAAATGGATCCTTCTAGTAGCG-3′). PCR products were digested with Nde1 and BamH1 and then subcloned into the Nde1 and BamH1 sites of the S. pombe expression vector pMNS21L under control of the thiamine-repressible nmt1 promoter (Maundrell, 1991, 1993) to generate pKG709. Two independent subclones were sequenced to verify accurate PCR amplification of the appropriate cDNA. In the construction of all subsequent dim1 cDNA-containing plasmids, pKG709 was utilized as the source of dim1 cDNA, excised as an Nde1/BamH1 fragment.
To determine the sequence of the dim1-35 allele, genomic DNA was isolated from the dim1-35 mutant strain KGY698. The dim1-35 gene was PCR amplified using the oligonucleotide primers 35START and 35STOP and subcloned into pMNS21L. Two independent subclones were sequenced.
Deletion of dim1 from the Genome and Analysis of Rescue by dim1+, CDH1, and mdim1 cDNAs
To generate a deletion construct containing dim1 genomic 5′ and 3′ flanking sequence but lacking dim1 coding sequence, a 3-kb genomic fragment consisting of the dim1 coding region plus ∼1-kb 5′ flank and 1.2-kb 3′ flank was subcloned into the plasmid pBS-SK+ (Stratagene, La Jolla, CA) to create pKG748. pKG748 was subjected to PCR amplification using the oligonucleotide primers 35Δ5′flank (5′-GCAATTCAGCTTCAGTTGATATAA) and 35Δ3′flank (5′-AGCCACCATTTGCTTATGCAAGAA). The PCR product was digested with HindIII and religated to create pKG916, in which the entire dim1 coding region was replaced with a HindIII site. A 1.8-kb fragment encoding the ura4 + gene or a 2.0-kb fragment encoding the his3 + gene (Ohi et al., 1996) was subcloned into pKG916 at the HindIII site to create pKG703 or pKG917, respectively. Sac1/HincII fragments consisting of dim1 5′ flank::ura4 + (or his3 +)::dim1 3′ flank were isolated from pKG703 (or pKG917) and transformed into the diploid strains dim1-35/dim1 + his3-237/his3 + ura4-D18/ura4-D18 leu1-32/leu1-32 ade6-M210/ade6-M216 (or dim1-35/dim1 + his3-D1/his3-D1 ura4-D18/ ura4-D18 leu1-32/leu1-32 ade6-M210/ade6-M216). Ura+ (or His+) transformants were selected on media lacking uracil (or lacking histidine). Uracil prototrophs KGY833 and KGY841 and histidine prototrophs KGY1087 and 1088 were isolated, and genomic DNA was prepared and digested with HincII. HincII digests were separated by agarose gel electrophoresis, blotted to membranes, and hybridized with an α-[32P]dCTPlabeled, 700-bp Sac1/Xba1 fragment of dim1 5′ flank (see Fig. 3) to verify replacement of one allele of dim1 with either the ura4 + or his3 + cassette in each diploid strain.
Figure 3.

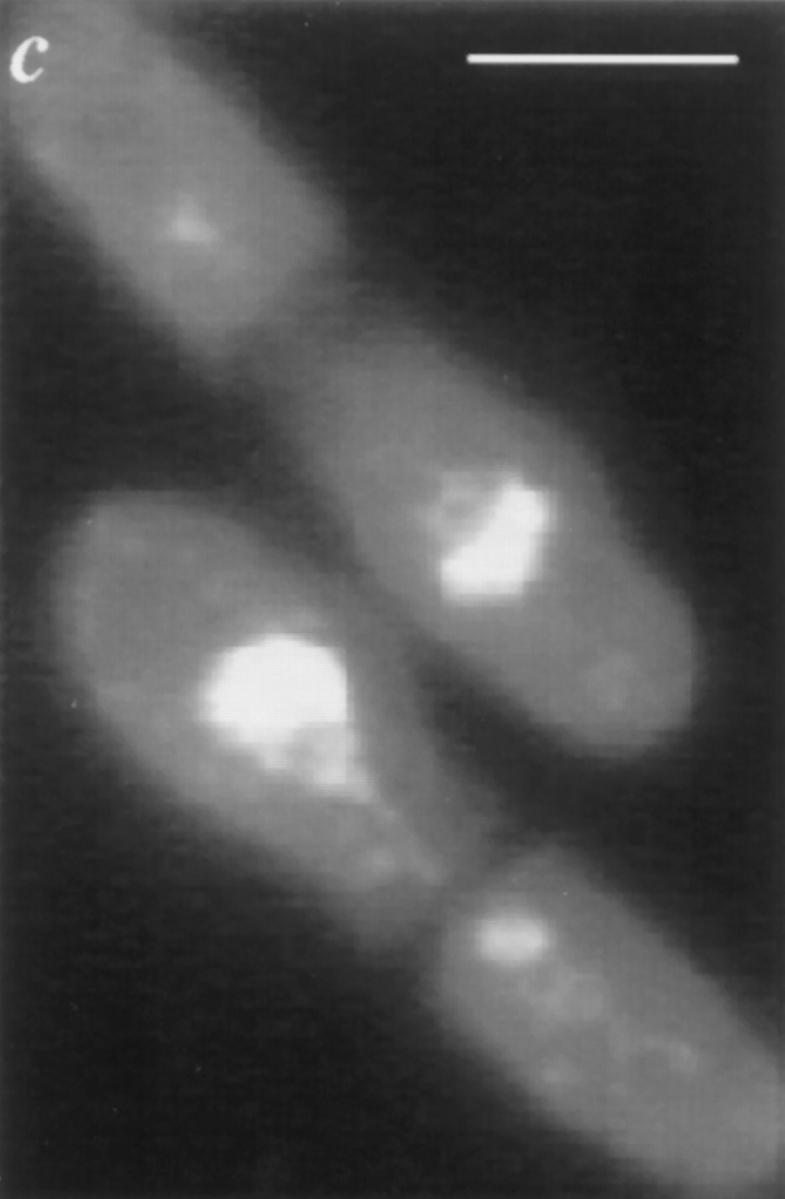
Upon release from nitrogen starvation, dim1-35 cells incubated at restrictive temperature accumulate 2C DNA but display first cell cycle arrest at or just before entry into mitosis. dim1-35 cells were starved for nitrogen at permissive temperature and then shifted to rich medium at restrictive temperature. Samples were collected for analysis at 30-min intervals. (a) DNA content of dim1-35 cells after shift. (b) Viability (○), 2C DNA content (□), and septation index (•) of dim1-35 cells after shift. Note that viability remains high until the majority of cells have completed S phase (i.e., accumulated 2C DNA) and then begins to decline ∼4 h into the time course, just before septated cells are first observed (at t = 4.5 h). (c) Cut phenotype of septated cells at t = 5 h. Bar, 5 μm.
To test for rescue of putative dim1::ura4 + or dim1::his3 + mutants by the dim1 cDNA, by a mouse dim1 (mdim1) cDNA obtained from Genome Systems (these sequence data available from EMBL/GenBank/DDBJ under accession number W85636; clone ID number, 40729; Genome Systems ID number, CD4549; Genome Systems Inc., St. Louis, MO), or by the CDH1 cDNA PCR amplified from S. cerevisiae genomic DNA (see below), diploids carrying the dim1::ura4 + or dim1::his3 + deletions were transformed with LEU2- or ura4-based plasmids containing the cDNA of interest under control of the full strength nmt1 promoter, the partially attenuated nmt1-T4 promoter, or the fully attentuated nmt1-T81 promoter in the pREP series of S. pombe expression vectors (Maundrell, 1991, 1993; Basi et al., 1993; plasmids: pKG402, pKG709, pKG718, pKG815, pKG836, pKG897, pKG898, pKG901, pKG930, pKG931; see Table III). Tranformants identified on selective media were allowed to sporulate. Spores were harvested by glusulase treatment of sporulated diploids (Moreno et al., 1991) and plated on appropriate selective media to isolate the dim1:: ura4 + or dim1::his3 + haploids of interest. (dim1::ura4 + haploids transformed with LEU2-based plasmids were selected on media lacking uracil and leucine; dim1::his3 + haploids transformed with LEU2-based plasmids were selected on media lacking histidine and leucine; dim1::his3 + haploids transformed with ura4-based plasmids were selected on media lacking histidine and uracil.) Lack of endogenous dim1 sequence in putative deletion haploids rescued by plasmid-borne cDNAs was verified by Southern blot analysis. In brief, genomic DNA was isolated, digested with HincII, separated by agarose gel electrophoresis, blotted to membranes, and then hybridized with α-[32P]dCTP–labeled dim1 cDNA or the 700-bp Sac1/Xba1 fragment described above.
To generate a dim1::his3 + strain carrying a single integrated copy of nmt1-T81-dim1 +, the dim1 cDNA was first subcloned as an Nde1/BamH1 fragment into pREP81 to generate pKG815. The nmt1-T81-dim1 + cassette was excised from pKG815 as a Pst1/BamH1 fragment and subcloned into the leu1-based S. pombe integrating vector pJK148 (Keeney and Boeke, 1994) to generate pKG980. pKG980 was linearized at the unique Nru1 site within leu1 and tranformed into KGY1090 (dim1::his3 + carrying pKG898; see Table II). Leu+ transformants were selected on media lacking leucine and then streaked to media containing 1 mg/ml 5-fluoroorotic acid to evict the ura4-based pKG898 plasmid. A Leu+ Ura− colony was isolated. Genomic DNA was prepared and analyzed by Southern hybridization as described above to verify lack of endogenous dim1 sequence and integration of pKG980 in single copy at the leu1 locus.
Isolation of a Putative Full Length CDH1 cDNA
A full length CDH1 cDNA was PCR amplified from S. cerevisiae genomic DNA using the primers CDH1Start (5′-GAGTAAGAAGACTAGTTCATATGGCTAGTG-3′) and CDH1Stop (5′-CATAGTTATGTCGACTTATGAAAC-3′). PCR products were digested with the enzymes Nde1 and Sal1 or Spe1 and Sal1. Nde1/Sal1 fragments were subcloned into the S. pombe expression vector pMNS21L to generate pKG836; Spe1/Sal1 fragments were subcloned into the S. cerevisiae expression vector pRS415 GALS (Mumberg et al., 1994) to generate pKG837. For pKG836, one subclone, and for pKG837, two independent subclones were sequenced. All additional CDH1 cDNA constructs were generated using the CDH1 cDNA excised from pKG836 as an Nde1/Sal1 fragment or from pKG837 as a Spe1/Sal1 fragment.
Deletion of CDH1 and Analysis of Rescue of CDH1 by CDH1, dim1, and mdim1 cDNAs
A deletion construct consisting of the S. cerevisiae HIS3 gene flanked by 36 bp of genomic sequence 5′ of the CDH1 initiating ATG and 36 bp of sequence 3′ of the CDH1 stop codon was generated by PCR amplification of the HIS3 gene from the HIS3-based S. cerevisiae expression vector pRS313 (Sikorski and Hieter, 1989) using the oligonucleotide primers CDH1KOStart (5′-ATAGTACGTAGAGAAAGAGTAAGAAGTCAAGTTTTGGCAGATTGTACTGAC-3′) and CDH1KOStop (5′-AGACATCGTGCGCCTAGCCTACATAGTTATTATGAACTCCTTACGCAT- CTG-3′). The PCR product was gel-purified and transformed into the diploid strain KGY823. Transformants were selected on media lacking histidine. The His+ transformant KGY937 was isolated, and genomic DNA was prepared. Genomic DNA was digested with HindIII and analyzed by Southern hybridization using the CDH1 cDNA and the cdh1::HIS3 deletion construct as probes, to verify replacement of one allele of the CDH1 coding region with the HIS3 gene.
To test for rescue of the cdh1::HIS3 mutant by CDH1, dim1 +, or mdim1, cDNAs under control of the heterologous MET25 or GALS promoter in the LEU2-based S. cerevisiae expression vector pRS415MET25 or pRS415GALS (Mumberg et al., 1994) were transformed into KGY937, and transformants were selected on media lacking leucine. (Plasmids tested for rescue: pKG837, pKG843, pKG849, pKG850, and pKG930; see Table III) Transformants were induced to sporulate, and tetrads were dissected. His+ Leu+ colonies were identified by replica plating to media lacking histidine or leucine.
To generate strains carrying integrated copies of CDH1 or dim1 under control of the MET25 or GALS promoter, promoter-cDNA cassettes were isolated from pRS415GALS or pRS415MET25 plasmid derivatives (pKG837, pKG843, pKG849, and pKG850; see Table III) as Sac1/Sal1 fragments and subcloned into the S. cerevisiae integrating vector pRS405 (Sikorski and Hieter, 1989). The resultant constructs (pKG912, pKG914, pKG913, and pKG911, respectively) were linearized at the unique EcoRV or BstX1 site within the LEU2 gene, and linearized plasmids were transformed into KGY937. Putative integrants in the cdh1::HIS3 background were isolated as described above for isolation of cdh1::HIS3 haploids carrying a rescuing plasmid. Lack of endogenous CDH1 sequence and integration of rescuing constructs in single copy at the leu2-3,112 locus were confirmed by preparing genomic DNA from putative integrants, digesting DNA with Xho1 and HindIII, and subjecting digested DNA to Southern blot analysis using the CDH1 cDNA as a probe.
To generate Ubiquitin/N-degron–tagged versions of CDH1 and dim1 + cDNAs under control of the GALS promoter, a Ubiquitin/N-degron tag was generated by PCR-mediated amplification of Ubiquitin encoding sequence from S. cerevisiae genomic DNA using the oligonucleotide primers UBIQ1 (5′-GCTCTAGAATGCAGATCTTTGTCAAGACTTTGACTGG-3′) and UBIQ2 (5′-GGACTAGTCATATGACCACTACCACCTCTCAATCTCAAGAC-3′). PCR products were subcloned into pBSSK+ (Stratagene, La Jolla, CA) to create pKG872. The Ubiquitin tag was removed from pKG872 as an Xba1/Nde1 fragment and cloned in frame upstream of either the CDH1 cDNA or the dim1 + cDNA in pRS415 GALS, to generate pKG875 or pKG876, respectively. Integrating versions of GALS::UBCDH1 or GALS::UBdim1 + (pKG882 or pKG903, respectively) were generated as described above, and cdh1::HIS3 haploids carrying integrated copies of GALS::UBCDH1 or GALS::UBdim1 + were generated and verified as described above.
Histone H1 Kinase Assays
For analysis of KGY1180 and KGY1216, lysates were prepared in HB buffer, and total cell lysates were assayed for histone H1 kinase activity as described (Moreno et al., 1989). For temperature shift experiments, cells were grown in YE at permissive temperature (25°C) to mid-log phase, diluted in YE to 2 × 106 cells/ml, and then shifted to restrictive temperature (36.5°C) for 4.5 h. Cells were pelleted, lysed with glass beads, and extracted with NP-40 buffer as described (Gould et al., 1991). Active Cdc2p/ Cdc13p complexes were immunoprecipitated from cell lysates by incubation with the anti-Cdc13p antibody GJG56 (DenHaese, G., and K.L. Gould, unpublished results), followed by incubation with protein A–Sepharose. Immunoprecipitates were washed extensively with NP-40 buffer and then split in two. One half of each immunoprecipitate was used as the source of histone H1-directed kinase activity in histone H1 kinase assays, as described (Gould et al., 1991). The second half was retained for use in Western blotting analysis to verify equal recovery of Cdc2 protein. Both kinase assays and untreated immunoprecipitates were resolved on 6 to 20% SDSPAGE gradient gels and then transferred to Immobilon-P (Millipore Corp., Bedford, MA). For detection of Cdc2p in the untreated immunoprecipitates, the blot was incubated consecutively with anti-Cdc2p antibody 4711 (1:10,000), alkaline phosphatase-conjugated goat anti–rabbit IgG (1:10,000; Molecular Dynamics, Sunnyvale, CA, and Amersham Life Science, Pittsburgh, PA), and enhanced chemifluorescence (ECF) detection reagents (Molecular Dynamics and Amersham Life Science), followed by chemifluorescence detection and quantitation using the Molecular Dynamics StormTM scanner. For quantitation of kinase activity, blots were exposed to phosphorimager screens (Molecular Dynamics) overnight and then phosphorylated histone H1 bands, visualized, and quantitated on a phosphorimager using MD ImageQuant Software Version 3.3 (Molecular Dynamics). For graphic presentation, histone H1 kinase activity was taken as phosphorylated histone H1 band intensity normalized to corresponding Cdc2p level, and kinase activity in the dim1-35 strain was assigned a value of one arbitrary unit, as the standard of comparison for all other strains.
Results
dim1-35 Mutant Cells Fail to Segregate DNA Properly during Mitosis
In a screen for second site mutations that would reduce the restrictive temperature of the ts strain cdc2-D217N (see Materials and Methods), we isolated a recessive ts mutant, which we have named dim1-35 (for defective entry into mitosis). At the restrictive temperature of 36.5°C, dim1-35 mutant cells arrested with one of the following phenotypes: unequal division of DNA to daughter cells as judged by DAPI staining; DNA strung out along the length of a mitotic spindle, in either a cut1/cut2-like “archery bow” conformation (Uzawa et al., 1990) or a top2/cut3/cut14-like “φ” conformation (Saka et al., 1994); DNA “cut” by a medial septum laid down across the undivided nucleus; or a single undivided interphase nucleus (data not shown).
To characterize the arrest phenotype of dim1-35 more precisely, synchronized G2 populations of dim1-35 mutant cells or wild-type cells were inoculated into rich medium at 36.5°C. After inoculation, samples were collected at 20- to 25-min intervals to monitor septation index, total cell number, viable cell number, DNA content of cells, and cell and nuclear morphology. The results of these analyses are summarized in Fig. 1. In the dim1-35 mutant, the first peak of septation occurred at time 155 min. In contrast, in wildtype cells the first peak of septation occured 95 min after inoculation (Fig. 1 c). At the first peak of septation, little loss of viability was observed in dim1-35 cells, and most cells maintained a 2C DNA content (Fig. 1, a and d). (The transient decrease in percentage of cells with 2C DNA content at the peak of septation presumably reflects the brief accumulation of daughter cells that have completed S phase of the next cell cycle while still adjoined at the septum, resulting in cells of apparent 4C DNA content.) DAPI staining revealed that cells passing through a first round of septation did not cut (Fig. 1 b, top). At the end of the first peak of septation, cell number had approximately doubled, from 9 × 105 to 16.3 × 105 total cells/ml, suggesting that ∼80% of cells divided in the first cell cycle after shift to restrictive temperature.
Figure 1.

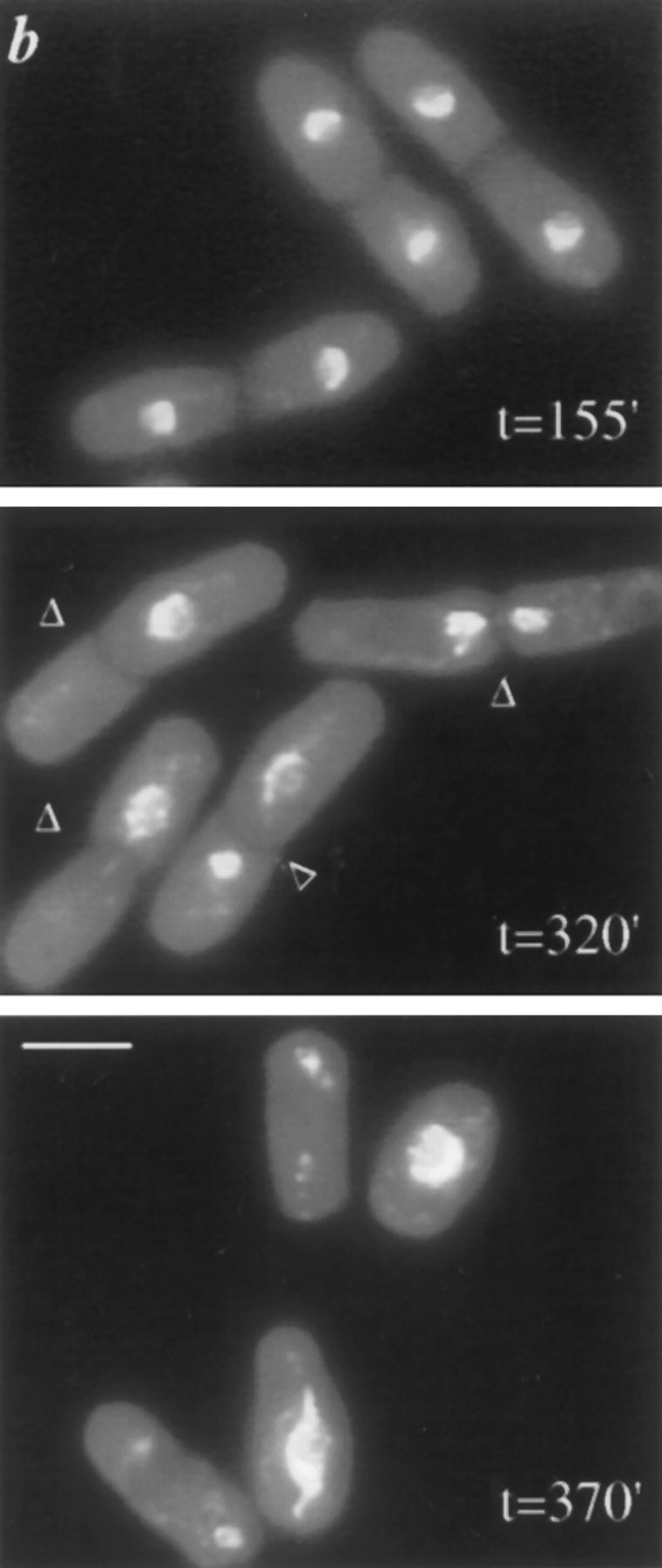


A synchronized population of dim1-35 mutant cells shifted to restrictive temperature displays a second cell cycle arrest. dim1-35 mutant (KGY392) or wildtype (KGY28) cells were grown to midlog phase in rich medium at permissive temperature (25°C). Cells were synchronized in early G2 by centrifugal elutriation and then inoculated into rich medium at 36.5°C. Samples were collected at 20- to 25min intervals and subjected to various analyses. (a) DNA content of dim1-35 mutant cells after shift to restrictive temperature. Cells were fixed with ethanol, stained with propidium iodide, and subjected to flow cytometric analysis. DNA content, expressed in arbitrary units, is shown on the horizontal axis. Cell number is shown on the vertical axis. (b) Cell and nuclear morphology of dim1-35 cells at the first peak of septation (top), at the second peak of septation (middle), and after the second round of septation (bottom). Cells were fixed with ethanol and stained with DAPI. Arrowheads indicate septa. (c) Septation index of dim1-35 (○) or wild type (•) cells after shift. (d) Percent viability (○), percentage of cells containing 2C DNA (▵), and septation index (□) of dim1-35 mutant cells after shift. Note that viability begins to decline sharply at t = 230 min, just before septated cells are first observed at t = 250 min. (e) Tubulin and SPB staining of dim1-35 mutant cells at second peak of septation after shift to restrictive temperature. (Left) DNA stained with DAPI. (Middle) Tubulin stained with TAT1 antibody. (Right) SPBs stained with α-Sad1p antibody. Note that septation has occurred, bisecting the elongated spindle and producing two daughter cells, one containing the undivided nucleus (*) and one containing no chromatin (**). Arrowheads indicate septa; arrows indicate SPBs. Bars: (b) 5 μm; (e) 3 μm.
In dim1-35, the second peak of septation occurred 145 min after the first peak of septation, in reasonable accord with wild-type cells in which the second peak of septation occurred 135 min after the first (Fig. 1 c). Thus, dim1-35 cells were delayed relative to wild type in the first peak of septation but not in the second. In dim1-35, the second round of septation coincided with a significant loss of viability; viability began to decline rapidly just before septated cells were first observed in the population (i.e., late G2 and/or early M phases of the cell cycle) and continued to decline throughout the second round of septation (Fig. 1 c). Flow cytometric analysis revealed a decrease in percentage of cells containing 2C DNA, with an accumulation of cells of greater or less than 2C DNA content (Fig. 1 d). Consistent with flow cytometric analysis, DAPI staining revealed a cut phenotype or missegregation of DNA in ∼70% of septated cells at the second peak of septation (Fig. 1 b, middle). At the end of the second round of septation, cell number had increased from 16.5 × 105 to 25.4 × 105 cells/ml, suggesting that ∼50% of cells divided in the second cell cycle. Thus, 50% of cells appeared not to enter a second round of mitosis and subsequent cell division. Consistent with the conclusion that 50% of cells failed to enter mitosis, 50% of cells maintained a 2C DNA content (Fig. 1, a and d), and ∼50% of cells appeared as single cells with an intact interphase nucleus, rather than as V-doublets in which gross missegregation or cutting of DNA had occurred (Fig. 1 b, bottom; and data not shown).
In a separate experiment, dim1-35 cells synchronized by centrifugal elutriation were fixed and processed for immunostaining to examine both microtubules as well as the number of spindle pole bodies. No gross defects in SPB duplication, spindle formation, or spindle elongation were observed at either the first or the second mitosis, despite the failure of chromosomal material to segregate properly on the mitotic spindle at the second mitosis (Fig. 1 e).
In summary, in a synchronous population of dim1-35 cells shifted to restrictive temperature, 80% of cells completed one round of mitosis; 20% of cells failed to divide in the first cell cycle after shift. Cells appeared to lose viability at or just before entry into a second round of mitosis (i.e., at the G2/M transition). Viability began to decline just before cells undergoing a second round of septation were observed and continued to decline throughout the second round of septation. These cells were unable to complete mitosis successfully. 50% of cells failed to enter mitosis, while in the 50% of cells that underwent a second round of mitosis, 70% “cut” or missegregated their DNA, resulting in accumulation of cells of non-2C DNA content (Fig. 1).
In the Absence of Septation and Cutting, the dim1-35 Mutation Blocks Multiple Rounds of Nuclear Division
In the synchronous block described above, the dim1-35 mutant shifted to restrictive temperature lost viability just before development of a “cut” phenotype in septated cells. We wished to determine what effect the dim1-35 mutation might have in the absence of septation. To answer this question, we made use of the cdc11-119 mutant. cdc11-119 cells shifted to the restrictive temperature of 36°C fail to make septa but rather undergo repeated rounds of nuclear division without septation, accumulating multiple nuclei in each cell (Nurse et al., 1976). A dim1-35 cdc11-119 double mutant was constructed, and the double mutant strain examined after shift to restrictive temperature, in comparison to cdc11-119 alone. As expected, cdc11-119 cells elongated and accumulated multiple nuclei. dim1-35 cdc11-119 cells elongated to a comparable extent but failed to accumulate more than two nuclei per cell (Fig. 2, a and b). Binucleate cells did accumulate in both cdc11-119 single and dim1-35 cdc11-119 double mutant cells, although accumulation of binucleate cells was somewhat delayed in cdc11119 dim1-35 as compared to cdc11-119 (Fig. 2 b). During the remaining 4 h at restrictive temperature, cdc11-119 single mutants went on to accumulate four and eight nuclei/ cell, while the majority of dim1-35 cdc11-119 double mutants remained binucleate (Fig. 2 b). (Note that in the cdc11-119 dim1-35 double mutant, ∼20% of cells remained uninucleate throughout the time course of the experiment; at later time points, up to 20% of cells leaked through the apparent dim1-35 block to accumulate three or four nuclei/cell.) Thus, consistent with the behavior of the dim1-35 single mutant, the dim1-35 mutation in the absence of septation allowed a single, delayed round of nuclear division in ∼80% of cells at restrictive temperature but blocked subsequent nuclear division in the majority of cells.
Figure 2.





cdc11-119 dim1-35 double mutants shifted to restrictive temperature arrest primarily as binucleates. cdc11-119 (KGY107) or cdc11-119 dim1-35 (KGY942) mutant cells were grown to midlog phase in rich medium at 25°C and then shifted to 37°C. Samples were taken at hourly intervals to monitor progression through the cell cycle after shift. (a) Cell and nuclear morphology of cdc11-119 (left) or cdc11-119 dim1-35 (right) 6 h after shift. Cells were fixed with ethanol and stained with DAPI. (b) Nuclear content of cdc11119 (left) or cdc11-119 dim135 (right) after shift. Horizontal axis shows time after shift. Vertical axis shows percent of cells containing one nucleus (□), two nuclei (▵), three or four nuclei (for cdc11-119 dim1-35; ○), four nuclei (for cdc11-119, ○), or 8 nuclei (▪). (c) DNA content of cdc11-119 (left) or cdc11-119 dim1-35 (right) after shift. Horizontal axis shows time after shift. Vertical axis shows percent of cells containing 2C DNA (□), 4C DNA (▵), or 8C DNA (○). (d) Cell and nuclear morphology of cdc11-119 (left) or cdc11-119 dim1-35 (right) 5 h after shift. (Top) DNA stained with DAPI. (Middle) Tubulin stained with the monoclonal antibody TAT1. (Bottom) SPBs stained with polyclonal α-Sad1p antibodies. (e) Flow cytometric analysis of cdc11-119 (left) or cdc11-119 dim1-35 (right) after shift. Note that DNA content data for cdc11-119 at the 7 h time point were not collected, as most cells had accumulated >8C DNA. (f) cdc11-119 dim1-35, 6 h after shift, showing trinucleate phenotype. Note that this phenotype occurred in only 10% of cells at the 6-h time point. Bars: (a) 10 μm; (d) 5 μm; (f) 5 μm.
Given the arrest phenotype of cdc11-119 dim1-35 mutant cells as binucleates, we wished to ask whether the dim1-35 mutation resulted in failure to enter a second round of mitosis, in an arrest during the second round of mitosis itself, or in failed nuclear division despite entry into mitosis. To address the possibility of a mid-mitotic arrest, cdc11-119 and cdc11-119 dim1-35 mutants were incubated at restrictive temperature for 5 h, at which point cdc11-119 dim1-35 cells had reached the terminal arrest phenotype, with ∼20% of cells arrested as uninucleates and ∼70% arrested as binucleates (Fig. 2 b). At hourly intervals after shift to restrictive temperature, cells were collected, fixed, and processed for immunostaining. Throughout the time course, the majority of cells in both cdc11-119 and cdc11-119 dim1-35 mutants contained interphase nuclei with uncondensed chromatin, an interphase array of microtubules, and a single SPB per nucleus (Fig. 2 d and data not shown). In both populations, however, there was evidence of continuing passage through M phase; in either population, 5 to 7% of cells at each time point contained a mitotic spindle and/or duplicated SPBs (data not shown). By these criteria, the dim1-35 mutation did not lead to midmitotic arrest, although some cells continued to pass through M phase, with or without segregation of DNA (see below and Discussion).
In the synchronous shift experiment described above, accumulation of cells of greater than 2C DNA content was observed (Fig. 1, a and d). Accumulation of polyploid DNA content suggested that cells that failed to segregate DNA properly during mitosis nevertheless entered S phase of a subsequent cell cycle. We wished to determine whether cdc11-119 dim1-35 cells likewise entered subsequent cell cycles after proceeding through mitosis without segregation of DNA. cdc11-119 dim1-35 cells shifted to restrictive temperature were collected and fixed for flow cytometric analysis. In cdc11-119, peaks of DNA content rose and fell coincident with peaks of nuclear content; peaks of 1, 2, and 4 nuclei/cell reached a maximum just before peaks of 2C, 4C, and 8C DNA, respectively (Fig. 2, b, c, and e, left graphs). In contrast, cdc11-119 dim1-35 double mutants accumulated a significant population of cells of 8C DNA content by 7 h after shift to restrictive temperature, despite the fact that cdc11-119 dim1-35 double mutant cells remained predominantly binucleate from the 4 h time point on (Fig. 2, b, c,and e, right graphs).
Consistent with the hypothesis that at least a subpopulation of cdc11-119 dim1-35 mutant cells entered and completed mitosis without DNA segregation, trinucleate cells were observed at some frequency in the cdc11-119 dim135 population. Of the 20% of cdc11-119 dim1-35 cells that leaked through the dim1-35 block to accumulate greater than 2 nuclei/cell, 50% contained three rather than four nuclei, suggesting a failure of nuclear division in one of two nuclei despite entry into M phase (Fig. 2 f, see Discussion).
In Nitrogen Starvation and Release Experiments, dim1-35 Cells Display a First Cell Cycle Arrest
As described above, in synchronous shift experiments as well as in the dim1-35 cdc11-119 double mutant, the dim135 mutation produced a second cell cycle arrest phenotype. Therefore, we considered the possibility that the dim1 gene product may play a role in S phase. According to this hypothesis, cells in G2 shifted to restrictive temperature would complete one round of mitosis successfully but would fail at the second mitosis due to defects that occur upon passage through S phase at restrictive temperature. To test this hypothesis, we performed both nitrogen starvation/release as well as spore germination experiments. In nitrogen starvation/release experiments, dim1-35 cells were starved for nitrogen at permissive temperature to accumulate cells in G1, incubated at restrictive temperature for 30 min to initiate inactivation of the temperaturesensitive Dim1-35p, and then released into rich medium at restrictive temperature. Samples were collected at 30-min intervals to monitor septation index, viability, cell morphology, and DNA content. As shown in Fig. 3, cells reentered the cell cycle and accumulated 2C DNA with little loss in viability. Viability then began to decline just before septated cells were first observed in the population (i.e., coincident with late G2 and/or early M phases of the cell cycle; Fig. 3 b). As in the synchronous shift experiments described above, loss of viability was followed by a loss of 2C DNA content, as cells cut or missegregated their DNA (Fig. 3).
In spore germination experiments, dim1-35 spores were inoculated into rich medium at restrictive temperature and samples collected for analysis at hourly intervals. Similar to the results of the nitrogen starvation/release experiment, spores were able to germinate and pass through G1/S, as evidenced by the accumulation of cells with 2C DNA content (data not shown). Again, loss of viability occurred coincident with late G2/early M; viability began to decline just before the appearance of septated (cut) cells (data not shown). Thus, in both nitrogen starvation/release and spore germination experiments, a first cell cycle arrest occurred, suggesting the possibility that the G2/M phenotype observed in dim1-35 may be the result of S phase defects. More likely, however, the first cell cycle arrest in these experiments reflected the length of time spent at restrictive temperature before entry into the first mitosis: ∼85 min in the synchronous shift, ∼3.5 h in the nitrogen starvation/release experiment, and ∼7 h in the spore germination experiment (Figs. 1 and 3; see Discussion).
dim1 Encodes a Highly Conserved Predicted Polypeptide of 142 Amino Acids
To explore the molecular basis of the dim1-35 phenotype in greater detail, as well as to determine the phenotype of a dim1 null allele, we cloned the dim1 gene by complementation of the dim1-35 mutant phenotype. A pUR19-based S. pombe genomic library (Barbet et al., 1992) was transformed into the dim1-35 mutant strain. Four genomic clones that allowed growth of dim1-35 at 36°C were recovered and shown by restriction digest and Southern blot analysis to contain overlapping fragments. A minimal rescuing fragment of 2 kb was identified (Fig. 4 a) and sequenced to reveal a single coding region consisting of five exons separated by four introns, each containing perfect 5′ splice, 3′ splice, and branch site consensus sequences (Zhang and Marr, 1994; Fig. 4 b).
Figure 4.


Cloning of dim1. (a) Restriction map of dim1 genomic locus and identification of minimal rescuing fragment. □, dim1 exon; H, HincII; S, Sac1; X, Xba1; E, EcoRV; C, ClaI. (b) Sequence of minimal rescuing fragment, showing putative coding region, intron/exon structure, and coding potential. Consensus 5′ splice, 3′ splice, and branch site sequences are underlined. The nucleotide and amino acid altered in dim1-35 are indicated in bold. These sequence data are available from GenBank/EMBL/ DDBJ under accession number AF001214. (c) Alignment of S. pombe Dim1p and putative Dim1 homologs. Mus musculus, S. cerevisiase, and partial Homo sapiens cDNAs were obtained and sequenced. The remaining cDNAs represent ESTs available in the databases. For any given Dim1p homolog, only those residues not identical to the corresponding residue in S. pombe Dim1p are indicated by a one letter amino acid code. Conserved residues are indicated by a dash (–). Note that the Arabidopsis thaliana, Orzia sativa, and H. sapiens sequences are incomplete. Blank spaces indicate unknown sequence or stretches of sequence in which a frame shift, likely due to error in the available sequence, results in loss of homology or introduction of a nonsense mutation. Of the full length clones sequenced in this lab, S. pombe and M. musculus dim1 share 79% identity at the amino acid level; S. pombe and S. cerevisiae, 65% identity; and M. musculus and S. cerevisiae, 64% identity.
That the rescuing gene encoded dim1 + and not a high copy suppressor was confirmed by integration mapping. pKG695, containing 3.7 kb of rescuing DNA and the selectable ura4 + marker was linearized within the dim1 5′ flanking sequence and integrated into the genome of the dim1-35 ura4-D18 strain KGY698 by homologous recombination. In outcrosses of the integrant strain to either wild type or dim1-35, the ura4 +marker segregated with a dim1 + phenotype in 26 out of 26 tetrads analyzed, confirming that the rescuing DNA had integrated adjacent to the dim1-35 locus.
That the rescuing gene consisted of the five identified exons and not another gene contained within the rescuing DNA fragment was confirmed by designing oligonucleotide primers for PCR-mediated amplification of the corresponding cDNA from an S. pombe cDNA library. Amplification generated a PCR product of predicted size. The PCR product was subcloned into the S. pombe expression vector pMNS21L under control of the thiamine-repressible nmt1 promoter. Two independent subclones were sequenced. Sequence of the cDNA confirmed the previously determined sequence of the genomic clone and verified the predicted intron/exon boundaries; no errors were found in the PCR amplification products. Plasmids carrying the dim1 cDNA were transformed into dim1-35 mutant cells and shown to rescue the mutant phenotype, thus confirming the identity of the rescuing gene as dim1 (data not shown).
Finally, genomic DNA was prepared from the dim1-35 mutant strain, and the primers described above were utilized to amplify the dim1-35 gene. The PCR product was subcloned, and two independent subclones were sequenced. Sequence analysis revealed a single mutation in nucleotide 644 (G to A), producing a single amino acid change (G126D; see Fig. 4 b). By all of the above criteria, we have cloned the dim1 gene.
A search of the databases revealed that S. pombe dim1 shares extensive homology with expressed sequence tags (ESTs) isolated from a number of other eukaryotic species, from budding yeast to human (Fig. 4 c). None of these ESTs has been characterized; as described in greater detail below, we show here that both S. pombe dim1 and the S. cerevisiae dim1 homolog (CDH1 for Saccharomyces cerevisiae dim1 homolog) are essential genes; that mouse dim1 (mdim1) can functionally complement S. pombe dim1; and that both S. pombe dim1 and mouse dim1 can functionally complement a null mutation in CDH1.
S. pombe dim1 Is an Essential Gene; the dim1 Deletion Mutant Displays Characteristics of G2 Arrest
To determine whether dim1 encodes an essential gene, we used the method of one step gene disruption to replace one copy of the dim1 coding region with either the ura4 + or his3 + selectable marker in diploids heterozygous for the dim1-35 mutation (Fig. 5 a). Replacement of one allele of dim1 with the ura4 + cassette or the his3 + cassette in putative deletion strains (putative ura4 + deletions: KGY 833 and KGY 841; putative his3 + deletions: KGY1087 and KGY 1088) was confirmed by Southern blot analysis (data not shown). Diploids were allowed to sporulate, and tetrads were dissected. In all cases, tetrads segregated two viable: two inviable progeny. In all tetrads, the inviable segregants germinated and underwent up to two residual cell divisions before arresting as slightly elongated cells (Fig. 5 b). For KGY 833, all viable segregants formed colonies of Ura−Dim1ts cells, and for KGY 1087, all viable segregants formed His−Dim1ts colonies, confirming replacement of the dim1 wild-type allele in KGY 833 and KGY 1087. For KGY 841, all viable segregants formed Ura−Dim1+ colonies, and for KGY 1088, all viable segregants formed His−Dim1+ colonies, verifying replacement of the dim135ts allele in KGY 841 and 1088. As final confirmation for deletion of the dim1 gene in KGY 833, 841, 1087, and 1088, various constructs carrying the dim1 + cDNA under control of the thiamine-repressible nmt1 promoter were shown to rescue the lethality of the putative dim1::ura4 + and dim1::his3 + mutants, in the presence or absence of thiamine, after transformation and sporulation of diploids (Fig. 5, c and d; and data not shown). Thus, the lethal deletions in KGY 833, 841, 1087, and 1088 had occurred at the dim1 locus; dim1 encodes an essential gene in S. pombe. These experiments further demonstrated that overexpression of dim1 + had no toxic or deleterious phenotypic consequences. As shown in Fig. 4 (c and d), cells overexpressing dim1 + from the full strength nmt1 promoter (KGY842) grew and divided normally. (Overproduction of Dim1p in these cells was verified by Western blot analysis using polyclonal α-Dim1p antibodies; data not shown.)
Figure 5.


Deletion of the dim1 coding region from the S. pombe genome. (a) Schematic showing replacement of entire dim1 coding region with the ura4 + or his3 + selectable marker. (b) Tetrad dissected from sporulated KGY833 diploid cell, germinated at 25°C. Viable colonies (first and third images) were Ura−Dim1ts. Inviable cells (presumed dim1::ura4 +; second and fourth images) divided once and then arrested. (c) Rescue of dim1::ura4 + by dim1 + and mdim1. Cells were streaked to minimal medium either lacking (middle) or containing (right) thiamine and then incubated at 32°C for 3 d. (Left) dim1::ura4 + carrying: (1) nmt1-T81:: dim1 + (KGY1091), (2) nmt1-T4::dim1 + (KGY858), (3) nmt1:: dim1 + (KGY842), and (4) nmt1::mdim1 (KGY1092). (d and e) Rescue of dim1::ura4 + by nmt1::dim1 + (d) or nmt1::mdim1 (e). KGY842 (d) or KGY1092 (e) was grown to midlog phase in minimal medium lacking thiamine, fixed with ethanol, and stained with DAPI. Bar, 10 μm.
The fact that dim1::ura4 + or dim1::his3 + spores germinated and underwent up to two residual cell divisions complicated analyses of the terminal null phenotype. Therefore, a dim1::his3 + strain carrying a single integrated copy of nmt1-T81 integrated at the leu1-32 locus (KGY1180) was generated. Although multicopy nmt1-T81::dim1 + rescued dim1::his3 + lethality in either the absence or presence of thiamine (Fig. 5), KGY1180 proved viable in the absence of thiamine but not in the presence of thiamine (data not shown). To examine the terminal phenotype of KGY1180, cells were grown to mid-log phase in minimal medium lacking thiamine and then shifted to rich medium containing thiamine. Samples were collected hourly after shift. In the first 6 to 7 h after shift, cell number continued to double approximately once every 3 h, with kinetics identical to those of the isogenic wild-type control KGY1216 (dim1 + nmt1-T81::dim1 +; Table I). By 8 to 10 h after shift, however, cell number began to level off, and by 13 h after shift, cells had ceased to divide, indicating that cells had reached a terminal phenotype (Fig. 6 a). At the 13-h time point and throughout the time course, cells maintained a 2C content of DNA (Fig. 6 b). Moreover, by 13 h after shift, cells had elongated moderately, contained interphase nuclei, and displayed an interphase array of microtubules, all suggestive of G2 arrest (Fig. 6 c). Biochemical data were consistent with the morphological evidence of G2 arrest; the KGY1180 mutant did not arrest with elevated levels of histone H1 kinase activity as compared to KGY1216. Indeed, histone H1 kinase activity in KGY1180 was approximately half that of KGY1216 both in the absence and presence of thiamine (data not shown). In short, the terminal phenotype of the dim1 deletion mutant gives no indication of entry into M phase, suggesting that dim1 + is required for entry into mitosis and that the mitotic phenotypes observed in dim1-35 result from the “leaky” nature of the dim1 allele (see Discussion).
Figure 6.

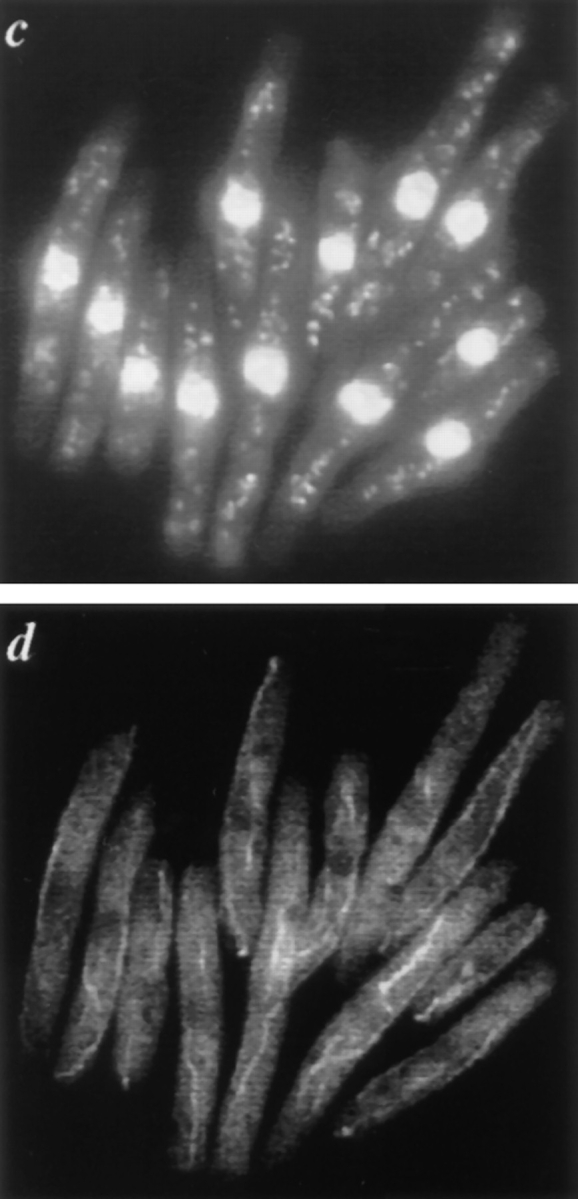
dim1::his3 + mutant cells arrest in G2. dim1::his3 + or dim1 + cells carrying a single integrated copy of nmt1−T81:: dim1 + (KGY1180 or KGY1216, respectively) were grown in minimal medium lacking thiamine and then shifted to rich medium containing thiamine to repress expression of the nmt1-T81:: dim1 + cassette. Samples were collected for analysis at 1-h intervals after shift. (a) KGY1180 (□) and KGY1216 (•) total cell number after shift to rich medium. (b) KGY1180 DNA content after shift to rich medium. (c and d) DAPI (c) and corresponding tubulin (d) staining of KGY1180 cells 13 h after shift.
Mouse dim1 (mdim1) Rescues Lethality of the dim1 Deletion Mutant
Given the high degree of sequence homology between dim1 and a number of ESTs listed in the databases, we wished to determine whether dim1 may be functionally conserved across the eukaryotic lineage. To this end, oligonucleotide primers were designed to PCR amplify the S. cerevisiae homolog of dim1 (CDH1) from S. cerevisiae genomic DNA. In addition, the mouse homolog of dim1 (mdim1) was obtained from Genome Systems Inc. (St. Louis, MO). Both CDH1 and mdim1 clones were sequenced to confirm their identities and then subcloned into the S. pombe pREP series of expression vectors to test for ability to rescue the ts defect of dim1-35 and/or the lethal phenotype of the dim1 deletion.
Driven by either the full strength nmt1 promoter or the partially attenuated nmt1-T4 promoter, mdim1 rescued dim1-35 in the absence of thiamine (promoter induced) but not in the presence of thiamine (promoter repressed). mdim1 under control of the fully attenuated nmt1-T81 promoter failed to rescue dim1-35 in the absence or presence of thiamine (data not shown). Requirements for rescue of a dim1 null allele appeared somewhat more stringent; mdim1 rescued dim1::ura4 + (or dim1::his3 +) only under control of the full strength nmt1 promoter and only in the absence of thiamine (Fig. 5, c and d).
In contrast to mdim1, CDH1 failed to rescue dim1-35 or dim1::ura4 + under control of any one of the three variable-strength nmt1 promoters, in the absence or presence of thiamine. Indeed, overexpression of CDH1 in the dim135 mutant proved toxic. dim1-35 mutant cells carrying either multi-copy nmt1::CDH1 or an integrated copy of nmt1::CDH1 were unable to form colonies on media lacking thiamine at either 29° or 25°C, temperatures fully permissive for growth of dim1-35 (data not shown). In a dim1 + background, overexpression of CDH1 was not lethal, although cells grew slowly and poorly, with a slightly elongated morphology (data not shown).
The S. cerevisiae Homolog of dim1 (CDH1) Is an Essential Gene; Both S. pombe dim1 and Mouse dim1 Rescue a Null Mutation in CDH1
To further investigate functional conservation among dim1 homologs, we chose to generate a null allele of S. cerevisiae CDH1. By the method of one step gene disruption, one copy of the CDH1 coding region was replaced with the selectable marker HIS3 in the diploid strain KGY823. Replacement of one allele of CDH1 in the putative deletion strain KGY937 was confirmed by Southern blot analysis (data not shown). KGY937 was induced to sporulate, and tetrads were dissected. 24 out of 24 tetrads segregated two viable to two inviable progeny. Inviable segregants germinated and formed microcolonies of ∼4 to 30 cells before arresting (data not shown). All viable colonies were His−. Thus, CDH1 encodes an essential gene in S. cerevisiae.
CDH1, dim1 +, or mdim1 cDNAs under control of the inducible MET25 or GALS promoters were tested for the ability to rescue the cdh1::HIS3 mutant when carried on multi-copy plasmids or integrated into the genome in single copy. Single integrated copies of MET25::CDH1, GALS::CDH1, MET25:: dim1 +, or GALS::dim1 + were capable of rescuing the cdh1::HIS3 mutant under either repressing or inducing conditions for the heterologous MET25 or GALS promoters (Fig. 7, a and b, and data not shown). In the case of mdim1, rescue of cdh1::HIS3 was achieved only when GALS::mdim1 was carried on a multicopy plasmid (pKG900) and only under inducing conditions for the GALS promoter (Fig. 7, a and b).
Figure 7.

Deletion of CDH1 from the S. cerevisiae genome. (a) Cells were streaked to synthetic complete medium containing 1% galactose/1% raffinose (middle) or 2% glucose (right) and then incubated at 32°C for 2 d. (Left) cdh1::HIS 3 carrying: (1) MET25::CDH1 (KGY 572); (2) GALS::CDH1 (KGY965); (3) GALS::UBCDH1 (KGY1026); (4) MET25::dim1 + (KGY966); (5) GALS::dim1 + (KGY311); (6) GALS::UBdim1 +(KGY1023); (7) GALS::mdim1 (KGY1093). (b–d) Rescue of cdh1::HIS3 by GALS::CDH1 (b), GALS::dim1+ (c), or GALS::mdim1 (d). KGY 965 (b), KGY311 (c), or KGY1093 (d) was grown to midlog phase in SGR, fixed with ethanol, and stained with DAPI. Bar, 10 μM.
cdh1::HIS3 Cells Accumulate and Arrest with a G2/M Content of DNA
To examine the phenotype of the cdh1::HIS3 mutant in greater detail, we wished to create a system for conditional expression of CDH1 function. As described above, repression of the heterologous MET25 or GALS promoters was not sufficient to abrogate rescue of cdh1::HIS3 by single integrated copies of MET25::CDH1, GALS::CDH1, MET25::dim1 +, or GALS:: dim1 +. (At the time of these experiments, the mdim1 clone was unavailable.) Therefore, we made use of the Ubiquitin/N-degron tagging strategy described by Althoeffer et al. (1995). Under repressing conditions, a single integrated copy of a cDNA encoding a Ubiquitin/N-degron–CDH1 fusion protein driven by the GALS promoter (GALS::UBCDH1) still allowed growth, albeit poor growth, of the cdh1::HIS3 mutant (Fig. 7 a). In contrast, a single integrated copy of GALS:: UBdim1 + rescued under inducing conditions but failed to rescue under repressing conditions for the GALS promoter (Fig. 7 a). Therefore, we utilized strain KGY1023 (cdh1::HIS3 carrying GALS::UBdim1 + integrated in single copy; Table II) to examine the null phenotype of cdh1.
KGY1023 or wild-type strain KGY820 was grown to mid-log phase at 32°C in synthetic complete medium containing 1% galactose/1% raffinose as the sole carbon source (SGR medium) to induce expression of GALS:: UBdim1 +. At time 0, cells were washed three times in synthetic complete medium containing 2% glucose (SD medium) to repress expression of pGALS::UBdim1 + and then resuspended in SD medium and incubated at 32°C. At hourly intervals, samples were collected for determination of total cell number, viable cell number, DNA content, and cell and nuclear morphology. The results of these analyses are summarized in Fig. 6. While wild-type cells continued to double approximately every 2 h and maintained ∼90% viability throughout the time course of the experiment, KGY1023 underwent about two and a half viable cell doublings during the first 5 h of the time course and then ceased to divide (Fig. 8 a). Viability was maintained at ∼85% during the first 6 h of the time course and then began to decline. By 10 h after shift from SGR to SD, viability had fallen to 43% (Fig. 8 b). The loss in viability was not associated with a homogenous arrest morphology. Even at the 10-h time point, KGY1023 consisted of a mixed population of unbudded cells, budded cells with an undivided nucleus, and budded cells with DNA divided between mother and daughter cells. Beginnning at the 4-h time point and throughout the remainder of the time course, however, a unique phenotype was observed. Approximately 10% of cells displayed a large budded phenotype with DNA positioned not at the mother bud neck but rather adjacent to the mother cell wall most distal to the daughter cell (data not shown). In addition, throughout the time course, the percentage of budded cells in KGY1023 was greater than that in KGY820 (data not shown). Consistent with the differential ratio of budded cells in the two populations, flow cytometric analysis revealed that, throughout the time course, the cdh1::HIS3 population contained 50 to 60% more cells with 2N DNA content than did the wild-type population (Fig. 8, b and c), suggesting that loss of CDH1 function resulted in accumulation of cells in the G2 and/or M phases of the cell cycle.
Figure 8.
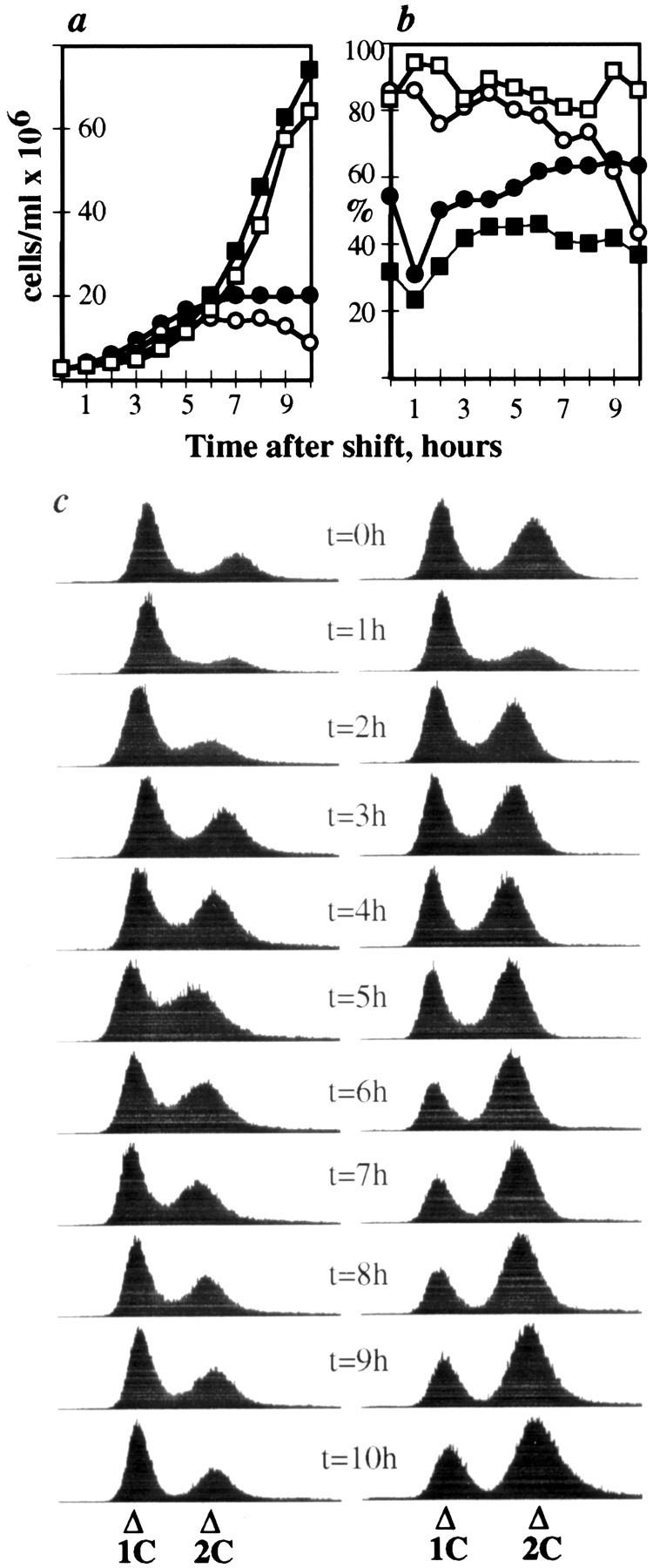
Phenotype of the cdh1::HIS3 mutant. (a) Growth of cdh1:: HIS3 GALS::UBdim1 + (KGY1023), and wild type (KGY820) after shift from SGR to SD. ▪, KGY820 total cell number; □, KGY820 viable cell number; •, KGY1023 total cell number; ○, KGY1023 viable cell number. (b) KGY1023 and KGY820 percent viability and percent of cells containing 2C DNA after shift from SGR to SD. □, KGY820 viability; ○, KGY1023 viability; ▪, KGY820 2C DNA; •, KGY1023 2C DNA. (c) Flow cytometric analysis showing DNA content of KGY820 (left) and KGY1023 (right) after shift from SGR to SD.
dim1 Displays only Weak Genetic Interactions with Mitotic Control Genes cdc2, cdc13, and cdr1
dim1-35 was isolated as a second-site mutation capable of lowering the restrictive temperature of cdc2-D217N from 36° to 32°C. Furthermore, the dim1-35 and cdh1::HIS3 mutant phenotypes described above suggested a role for dim1 in G2/M progression. Therefore, we wished to determine whether dim1-35 would show genetic interactions with various G2/M mutants, such as cdc2-22, cdc13-117, cdc13A382V, cdc25-22, and cdr1-76 (Table I). When double mutants were constructed, a lowering of restrictive temperature from 36° to 32°C again was observed. Upon more careful analysis, however, 32°C proved a semipermissive temperature for dim1-35 itself; the dim1-35 mutant streaked to 32°C formed small colonies of slow growing cells. At 29°C, a temperature fully permissive for dim1-35, all double mutants also proved viable (data not shown). Thus, dim1-35 did not display strong genetic interactions with other G2/M mutants. In addition to G2/M defects, dim1-35 also displayed characteristics reminiscent of mitotic mutants, including cut mutants, dis mutants, and nda mutants (see Introduction). As in the case of the G2/M mutants, however, double mutant construction and analysis revealed that dim1-35 exhibited no significant genetic interactions with cut1-205, cut1-RB5, cut2-364, cut3-447, dis1-288, or nda3km311 (data not shown; interaction with nda2-km52 could not be analyzed, as no temperature examined proved fully permissive for both the cold-sensitive nda2-km52 mutant and the temperature-sensitive dim1-35 mutant).
The dim1-35 Mutant Arrests with Low Histone H1 Kinase Activity
Because genetic analyses did not prove informative in probing the function of Dim1p, we decided to take a biochemical approach. We considered the possibility that the failure of dim1-35 to complete mitosis successfully may reflect an inability to attain or maintain levels of Cdc2p/ Cdc13p kinase activity sufficient to drive productive entry into and successful passage through mitosis. Indeed, as described above, the dim1::his3 + mutant displayed reduced levels of histone H1 kinase activity as compared to a wildtype control. To determine whether Cdc13p-associated Cdc2p kinase activity was compromised in dim1-35 as well, protein lysates prepared from dim1-35 as well as from wild-type, cdc25-22, and nuc2-663 cells incubated at 36.5°C were subjected to immunoprecipitation using the polyclonal anti-Cdc13p antibody GJG56. Immunoprecipitates were assayed for histone H1 kinase activity. As illustrated in Fig. 9, H1 kinase activity in the dim1-35 mutant was comparable to that of cdc25-22, a temperature-sensitive mutant that arrests in G2, before entry into mitosis (Cdc2p/Cdc13p kinase activity low). The kinase activity of dim1-35 or of cdc25-22 was ∼50% that of wild-type cells and ∼25% that observed in the temperature-sensitive mutant nuc2-663, which arrests in mid-mitosis (Cdc2p/Cdc13p activity high). Thus, the dim1-35 mutant incubated at restrictive temperature contained low levels of Cdc13p-associated histone H1 kinase activity, although the level of Cdc2p/Cdc13p complex was not detectably lower (data not shown).
Figure 9.

Relative histone H1 kinase activity in wild-type or temperature-sensitive mutant strains. Strains were grown to midlog phase at 25°C in YE medium and then shifted to 36.5°C for 4.5 h. Cells were collected and lysed. Lysates were subjected to immunoprecipitation using α-Cdc13p antibodies. Immunoprecipitates were assayed for histone H1 kinase activity and Cdc2 protein levels. Relative histone H1 kinase activity is expressed in arbitrary units as detectable histone H1 phosphorylation normalized to corresponding Cdc2p level.
The dim1-35 Mutant Displays Sensitivity to the Microtubule Destabilizing Drug TBZ
To investigate further the function of Dim1p, we chose to examine the sensitivity of dim1-35 to various drugs. In particular, the failure of nuclear division in those dim1-35 cells that leak into M phase suggested that the dim1-35 mutation may affect spindle function. Note that spindle formation and elongation in the dim1-35 mutant appeared normal (see above). Nevertheless, we considered the possibility that the dim1-35 mutation may affect spindle function indirectly, or in such a way as to prove undetectable at the level of resolution of light microscopy. Therefore, we tested dim1-35 for sensitivity to the microtubule destabilizing drug TBZ. The dim1-35 mutant did in fact prove sensitive to TBZ. At the permissive temperature of 29°C, dim1-35 cells were unable to form single colonies on YE agar containing 9 μg/ml TBZ, whereas wild-type cells were uninhibited in single colony formation by TBZ at this concentration (Fig. 10 a). To determine the terminal phenotype of the dim1-35 mutant exposed to TBZ, wild-type or dim1-35 mutant cells were incubated at 29°C in YE medium containing 10 μg/ml TBZ. TAT1 immunofluorescence performed on fixed cells revealed that spindle formation and elongation did occur in dim1-35 under these conditions (data not shown). Nevertheless, 40% of dim135 cells displayed a cut phenotype or gross missegregation of DNA; only 4% of wild-type cells displayed such phenotypes (Fig. 10, b and c).
Figure 10.

TBZ sensitivity of dim1-35. (a) Wild-type (KGY28) or dim1-35 (KGY392) cells were streaked to YE agar containing 9 μg/ml TBZ dissolved in DMSO (right) or an equivalent amount of DMSO alone (left) and then incubated at 29°C for 3 d. (b and c) Phenotype of wild type cells (b) or dim1-35 cells (c) grown at 29°C for 6 h in YE medium containing 10 μg/ml TBZ. Cells were fixed with ethanol and stained with DAPI. Bar, 5 μm.
Discussion
We have identified a fission yeast gene, dim1 +, essential for entry into and progression through mitosis. Mutant cells deleted for the dim1 coding region arrest in G2 with cytoplasmic microtubules and single interphase nuclei of 2C DNA content. The terminal phenotype of the dim1 deletion mutant shows no evidence of entry into mitosis. In contrast, in the temperature-sensitive dim1-35 mutant, cells may enter mitosis at restrictive temperature. Specifically, in a synchronous population of dim1-35 cells shifted to restrictive temperature, 80% of cells divide at the first mitosis without significant loss of viability; 20% fail to divide. At the second mitosis, ∼50% of cells fail to divide. Of the 50% of cells that do progress through mitosis and cytokinesis, 70% cut or missegregate their DNA, as judged by DAPI staining. Consistent with the DAPI phenotype, cells of greater or less than 2C DNA content accumulate, and viability decreases significantly at the second round of septation. Loss of 2C DNA occurs subsequent to loss of viability, suggesting that cutting per se is not the lethal event. Rather, based upon the terminal phenotype of the dim1 deletion mutant in conjunction with that of dim1-35, we conclude that (a) dim1 gene function is required in late G2 for successful entry into and progression through M phase; and (b) dim1-35 represents a hypomorphic allele that blocks some cells at the dim1 execution point before entry into mitosis but allows other cells to leak into M phase. Those cells that leak past the dim1 block, however, are unable to complete mitosis successfully, due to previous failure of dim1 function.
Not only is dim1 gene function required for proper G2/M progression, but in the dim1-35 mutant at restrictive temperature, faithful DNA segregation is uncoupled from subsequent cell cycle events. That is to say, septation occurs in the absence of nuclear division. Moreover, after missegregation of DNA and subsequent septation, daughter cells that receive greater or less than a full chromosomal complement appear to enter S phase of the next cell cycle, resulting in accumulation of cells of greater than or less than 2C DNA content.
The behavior of the dim1-35 mutation in a cdc11-119 background supports our interpretation of data obtained in synchronous shift experiments. As in the dim1-35 single mutant, 20% of cdc11-119 dim1-35 double mutants shifted to restrictive temperature fail to undergo a first mitosis; 20% of cells remain uninucleate throughout the time course of the temperature shift experiment. The remaining 80% of cells complete mitosis of a first cell cycle and then arrest predominantly as binucleates. Although these cells do not block with features characteristic of midmitotic arrest (i.e., condensed chromosomes and/or presence of a mitotic spindle), evidence suggests mitotic defects in at least a subpopulation of cells. First, 20% of cells leak through the binucleate block to accumulate greater than two nuclei per cell by 6 to 7 h after shift. Of these, 50% contain three rather than four nuclei. In contrast, cdc11119 single mutant cells undergo synchronous rounds of mitosis such that cells always contain 2, 4, 8, or 16 nuclei. We hypothesize that cdc11-119 dim1-35 trinucleate cells arise when a binucleate leaks into M phase, but due to inadequate Dim1p function, only one of the two nuclei is able to complete mitosis successfully.
Additional evidence for “leaky” passage through M phase and subsequent progression through the cell cycle in a subpopulation of cdc11-119 dim1-35 double mutant cells lies in the accumulation of cells of 8C DNA content, despite almost complete stabilization of nuclear content at two nuclei per cell throughout the last 3 h of the time course. Such accumulation of DNA without a corresponding increase in number of nuclei per cell (i.e., without chromosomal segregation) has been observed previously in both cdc11-119 top2 and cdc11-119 cut1 double mutants (Uemura and Yanagida, 1986; Uzawa et al., 1990). After shift to restrictive temperature, these mutants undergo multiple rounds of the cell cycle without nuclear division, such that the single nucleus in a cdc11-119 cut1 double mutant cell accumulates multiple aberrant spindle pole bodies, as well as polyploid DNA content. The single nucleus in a cdc11119 top2 double mutant cell accumulates greater than 2C DNA, but multiple SPBs are not observed.
The second cell cycle effect as well as the “leaky” nature of the dim1-35 mutation in either a cdc11 + or cdc11-119 background suggests that dim1-35 represents a hypomorphic allele. An alternative explanation for the second cell cycle arrest observed in dim1-35 is that loss of dim1 function in G1 or S results in failure to complete the subsequent mitosis. Thus, cells synchronized in early G2 and shifted to restrictive temperature would progress through a first round of mitosis and would arrest only at the second round of mitosis, after passing through G1/S in the absence of dim1 function. Such a phenomenon is observed in the S. cerevisiae mutant pds1-1, in which an anaphase defect occurs as the result of a temperature-sensitive step in cell cycle progression which occurs between G1 and S (Yamamoto et al., 1996). In an attempt to address the possibility of a role for dim1 in G1 or S, we performed nitrogen starvation/release experiments and spore germination experiments. In these experiments, a first cell cycle arrest was observed. However, these experiments do not necessarily indicate a role for dim1 in G1/S. Rather, given the hypomorphic nature of the dim1-35 allele, we consider it more likely that the first cell cycle arrest observed in the nitrogen starvation/release and spore germination experiments reflects the length of time spent at restrictive temperature before entry into M phase in these experiments: cells synchronized in early G2 enter a first round of M phase 85 min after shift to restrictive temperature; nitrogen-starved cells, 3.5 h after shift; and germinating spores, 7 h after shift (data not shown). Thus, we hypothesize that the weakly temperature-sensitive Dim1-35 protein remains functional through the first mitosis in synchronous shift experiments but has lost function by the time of the first mitosis in nitrogen starvation/release and spore germination experiments. This results in an apparent second cell cycle arrest in synchronous cultures but a first cell cycle arrest in nitrogen starvation/release and spore germination experiments.
Although a G1/S role for dim1 remains a remote possibility, our results demonstrate that neither the dim1-35 mutation nor deletion of dim1 from the genome blocks progression through S phase per se. In the synchronous shift experiments, cells continue to accumulate DNA despite cutting and/or missegregation of chromosomes; in the cdc11-119 dim1-35 double mutant, DNA replication is not impaired, despite failure of chromosome segregation; in both nitrogen starvation/release and spore germination experiments, cells accumulate 2C DNA at restrictive temperature; and, finally, both chd1::HIS3 and dim1::his3 + mutants arrest with 2C DNA. It should be noted that certain S phase mutants are capable of bulk DNA replication, such that accumulation of a G2 content of DNA is not blocked at restrictive temperature (Nasmyth and Nurse, 1981; Coxon et al., 1992; Kelly et al., 1993; MacNeill et al., 1996). Because DNA replication is defective, however, these mutants arrest in G2 by virtue of invoking the DNA replication checkpoint. In a genetic background in which the checkpoint gene rad1 is deleted, the DNA replication checkpoint is lost, and S phase mutants enter mitosis without having properly replicated their DNA, resulting in dramatic loss of viability (Al-Khodairy and Carr, 1992). In contrast, dim1-35 in a rad1::ura4 + background exhibits no increased frequency of cutting or loss of viability at restrictive temperature, as compared to dim1-35 in a rad1 + background (data not shown). Therefore, dim1-35 mutant cells are capable not only of bulk DNA replication but of proper DNA replication, further supporting our conclusion that dim1 does not play a role in S phase but rather functions in late G2 or early M.
Molecular cloning has provided little insight into the nature of Dim1p function. However, cloning has revealed a remarkable evolutionary conservation of Dim1p at the sequence level. Our studies further demonstrate conservation at the level of function: Both S. pombe dim1 and mouse dim1 are capable of rescuing a null allele of S. cerevisiae CDH1; mdim1 functionally complements a null allele of S. pombe dim1. Analysis of the S. cerevisiae cdh1:: HIS3 mutant phenotype also suggests functional conservation of Dim1p, in that the accumulation of cells with 2C DNA content in cdh1::HIS3 indicates a possible role for Cdh1 protein in the G2/M transition.
The accumulation of cdh1::HIS3 cells with a G2/M content of DNA, taken together with our analyses of the dim1::his3 + and dim1-35 mutant phenotypes, suggest a model for Dim1p function. We propose that Dim1p plays an essential role in reorganization of microtubules at mitosis and/or in reorganization of chromatin for attachment to the mitotic spindle. For proper segregation of DNA during mitosis, the cytoplasmic microtubule network must disassemble, the microtubule-based mitotic spindle must form, and condensed chromosomes must attach to the spindle. Both TBZ sensitivity and the DNA missegregation/cut phenotype of dim1-35 are consistent with defects in one or more of the above processes. Thus, dim1::his3 + mutant cells arrest before entry into mitosis, in the absence of spindle formation or changes in chromatin structure. In contrast, in the hypomorphic dim1-35 mutant, some cells may block tightly before entry into mitosis, with an interphase array of microtubules, a single interphase nucleus (i.e., uncondensed chromosomes), and low histone H1 kinase activity. dim1-35 cells that leak into M phase form spindles, but spindle function and/or mitotic chromosome dynamics are compromised. Cells then proceed through mitosis in the absence of chromosome segregation, resulting in development of a cut phenotype or segregation of DNA to only one daughter cell.
Although septation in the absence of chromosome segregation in dim1-35 could suggest loss of a mitotic checkpoint, we do not imagine that dim1 plays a role in checkpoint control. As discussed above, many fission yeast mitotic mutants uncouple mitotic processes. For example, the cut mutants undergo septation and cytokinesis without nuclear division; septation mutants undergo repeated rounds of S phase and mitosis without septation and cytokinesis; cdc11 top2 and cdc11 cut1 double mutants undergo repeated rounds of S phase and mitosis without nuclear division or septation and cytokinesis. It seems unlikely that all of these mutants define genes whose products function in checkpoint pathways. Rather, like dim1-35, these mutants demonstrate the separability of mitotic processes, both from each other and from subsequent cell cycle events.
To date, biochemical analyses have not proven informative in examining dim1 function, primarily due to lack of effective reagents. Although polyclonal antibodies generated against recombinant Dim1p as well as anti-epitope antibodies used to analyze epitope-tagged versions of Dim1p, are adequate for Western blotting applications (data not shown), these antibodies are unable to immunoprecipitate Dim1p or to detect Dim1p by immunofluorescence. Thus, extensive biochemical analyses of Dim1p have not been possible. The generation of more effective reagents in addition to the isolation of new alleles of dim1 as well as suppressors and enhancers of the dim1-35 mutant phenotype may allow us to examine Dim1p function in greater detail. Regardless of the precise molecular nature of Dim1p function, however, the work presented here demonstrates a fundamental, evolutionarily conserved role for dim1 in G2/M progression as well as in chromosome segregation during mitosis.
Acknowledgments
We wish to thank all members of the Gould laboratory for helpful advice and discussion; Dr. J. Price for processing and analysis of flow cytometric samples; Dr. I. Hagan for providing the cut1-RB5 strain and α-Sad1p antibodies; Dr. K. Gull for providing TAT1 antibodies; Dr. J.R. McIntosh for providing the cut1-205, cut2-364, and cut3-447 strains; Dr. T. Graham for providing the S. cerevisiae strains SEY6210, SEY6211, and SEY6210/6211 (KGY820, KGY821, and KGY823, respectively); and Dr. P.A. Weil for providing the S. cerevisiae vectors pRS405, pRS415GALS, and pRS415MET25.
This work was supported by National Institutes of Health grant GM47728. L.D. Berry was supported by a National Science Foundation Graduate Research Fellowship. K.L. Gould is an assistant investigator of the Howard Hughes Medical Institute.
Footnotes
1. Abbreviations used in this paper: cut, cell untimely torn; EST, expressed sequence tag; SPB, spindle pole body; TBZ, thiabendazole; ts, temperature sensitive; YE, yeast extract.
Please address all correspondence to Lynne D. Berry, Howard Hughes Medical Institute, Department of Cell Biology, Vanderbilt University, Nashville, TN 37212. Tel.: (615) 343-9500; Fax: (615) 343-4539; E-mail: GouldK@ctrvax.vanderbilt.edu
References
- Al-Khodairy F, Carr AM. DNA repair mutants defining G2 checkpoint pathways in Schizosaccharomyces pombe. . EMBO (Eur Mol Biol Organ) J. 1992;11:1343–1350. doi: 10.1002/j.1460-2075.1992.tb05179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Althoefer H, Schleiffer A, Wassmann K, Nordheim A, Ammerer G. Mcm1 is required to coordinate G2-specific transcription in Saccharomyces cerevisiae. . Mol Cell Biol. 1995;15:5917–5928. doi: 10.1128/mcb.15.11.5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbet N, Muriel WJ, Carr AM. Versatile shuttle vectors and genomic libraries for use with Schizosaccharomyces pombe. . Gene (Amst) 1992;114:59–66. doi: 10.1016/0378-1119(92)90707-v. [DOI] [PubMed] [Google Scholar]
- Basi G, Schmid E, Maundrell K. TATA box mutations in the Schizosaccharomyces pombe nmt1promoter affect transcription efficiency but not the transcription start point or thiamine repressibility. Gene (Amst) 1993;123:131–136. doi: 10.1016/0378-1119(93)90552-e. [DOI] [PubMed] [Google Scholar]
- Baum P, Yip C, Goetsch L, Byers B. A yeast gene essential for regulation of spindle pole duplication. Mol Cell Biol. 1988;8:5386–5397. doi: 10.1128/mcb.8.12.5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, D.M., and V. Lundblad. 1994. Introduction of DNA into yeast cells. In Current Protocols in Molecular Biology. F.A. Ausubel, R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl, editors. John Wiley and Sons, New York. 13.7.1–13.7.10.
- Berry LD, Gould KL. Novel alleles of cdc13 and cdc2 isolated as suppressors of mitotic catastrophe in Schizosaccharomyces pombe. . Mol Gen Genet. 1996;251:635–646. doi: 10.1007/BF02174112. [DOI] [PubMed] [Google Scholar]
- Booher R, Beach D. Interaction between cdc13 + and cdc2 + in the control of mitosis in fission yeast: dissociation of the G1 and G2 roles of the cdc2 +protein kinase. EMBO (Eur Mol Biol Organ) J. 1987;6:3441–3447. doi: 10.1002/j.1460-2075.1987.tb02667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booher R, Beach D. Involvement of cdc13 + in mitotic control in Schizosaccharomyces pombe: possible interaction of the gene product with microtubules. EMBO (Eur Mol Biol Organ) J. 1988;7:2321–2327. doi: 10.1002/j.1460-2075.1988.tb03075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booher RN, Alfa CA, Hyams JS, Beach DH. The fission yeast cdc2/cdc13/suc1 protein kinase: regulation of catalytic activity and nuclear localization. Cell. 1989;58:485–497. doi: 10.1016/0092-8674(89)90429-7. [DOI] [PubMed] [Google Scholar]
- Coleman TR, Dunphy WG. Cdc2 regulatory factors. Curr Opin Cell Biol. 1994;6:877–882. doi: 10.1016/0955-0674(94)90060-4. [DOI] [PubMed] [Google Scholar]
- Coxon A, Maundrell K, Kearsey SE. Fission yeast cdc21 +belongs to a family of proteins involved in an early step of chromosome replication. Nucleic Acids Res. 1992;20:5571–5577. doi: 10.1093/nar/20.21.5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantes PA. Isolation of cell size mutants of a fission yeast by a new selective method: characterization of mutants and implications for division control mechanisms. J Bacteriol. 1981;146:746–754. doi: 10.1128/jb.146.2.746-754.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone C, Russell P. Fission yeast p107wee1mitotic inhibitor is a tyrosine/serine kinase. Nature (Lond) 1991;349:808–811. doi: 10.1038/349808a0. [DOI] [PubMed] [Google Scholar]
- Feilotter H, Nurse P, Young PG. Genetic and molecular analysis of cdr1/nim1 in Schizosaccharomyces pombe. . Genetics. 1991;127:1–10. doi: 10.1093/genetics/127.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fikes JD, Becker DM, Winston F, Guarente L. Striking conservation of TFIID in Schizosaccharomyces pombe and Saccharomyces cerevisiae. . Nature (Lond) 1990;346:291–294. doi: 10.1038/346291a0. [DOI] [PubMed] [Google Scholar]
- Funabiki H, Yamano H, Kumada K, Nagao K, Hunt T, Yanagida M. Cut2 proteolysis required for sister-chromatid separation in fission yeast. Nature (Lond) 1996a;381:438–441. doi: 10.1038/381438a0. [DOI] [PubMed] [Google Scholar]
- Funabiki, H., Kumada, K., and M. Yanagida. 1996b. Fission yeast Cut1 and Cut2 are essential for sister chromatid separation, concentrate along the metaphase spindle and form large complexes. EMBO (Eur. Mol. Biol. Organ.) J. 15:6617–6628. [PMC free article] [PubMed]
- Gould KL, Nurse P. Tyrosine phosphorylation of the fission yeast cdc2 +protein kinase regulates entry into mitosis. Nature (Lond) 1989;342:39–42. doi: 10.1038/342039a0. [DOI] [PubMed] [Google Scholar]
- Gould KL, Moreno S, Owen DJ, Sazer S, Nurse P. Phosphorylation of Thr 167 is required for Schizosaccharomyes pombe p34cdc2function. EMBO (Eur Mol Biol Organ) J. 1991;10:3297–3307. doi: 10.1002/j.1460-2075.1991.tb04894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan I, Yanagida M. Novel potential mitotic motor protein encoded by the fission yeast cut7 +gene. Nature (Lond) 1990;347:563–566. doi: 10.1038/347563a0. [DOI] [PubMed] [Google Scholar]
- Hagan I, Yanagida M. Kinesin-related Cut7 protein associates with mitotic and meiotic spindles in fission yeast. Nature (Lond) 1992;356:74–76. doi: 10.1038/356074a0. [DOI] [PubMed] [Google Scholar]
- Hagan I, Yanagida M. The product of the spindle formation gene sad1 +associates with the fission yeast spindle pole body and is essential for viability. J Cell Biol. 1995;129:1033–1047. doi: 10.1083/jcb.129.4.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan IH, Hayles J, Nurse P. Cloning and sequencing of the cyclin related cdc13 +gene and a cytological study of its role in fission yeast mitosis. J Cell Sci. 1988;91:587–595. doi: 10.1242/jcs.91.4.587. [DOI] [PubMed] [Google Scholar]
- Hindley J, Phear GA. Sequence of the cell division gene cdc2 from Schizosaccharomyces pombe: patterns of splicing and homology to protein kinases. Gene (Amst) 1984;31:129–134. doi: 10.1016/0378-1119(84)90203-8. [DOI] [PubMed] [Google Scholar]
- Hirano T, Funahashi S, Uemura T, Yanagida M. Isolation and characterization of Schizosaccharomyces pombe cutmutants that block nuclear division but not cytokinesis. EMBO (Eur Mol Biol Organ) J. 1986;5:2973–2979. doi: 10.1002/j.1460-2075.1986.tb04594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T, Hiraoka Y, Yanagida M. Temperature-sensitive mutation of the Schizosaccharomyces pombe gene nuc2 +that encodes a nuclear scaffold-like protein blocks spindle elongation in mitotic anaphase. J Cell Biol. 1988;106:1171–1183. doi: 10.1083/jcb.106.4.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, C.S. 1993. Preparation of yeast DNA. In Current Protocols in Molecular Biology. F.A. Ausubel, R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl, editors. John Wiley and Sons, New York. 13.11.1–13.11.4.
- Keeney JB, Boeke JD. Efficient targeted integration at leu1-32 and ura4-294 in Schizosaccharomyces pombe. . Genetics. 1994;136:849–856. doi: 10.1093/genetics/136.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly TJ, Martin GS, Forsburg SL, Stephen RJ, Russo A, Nurse P. The fission yeast cdc18 +gene product couples S phase to START and mitosis. Cell. 1993;74:371–382. doi: 10.1016/0092-8674(93)90427-r. [DOI] [PubMed] [Google Scholar]
- Kentaro N, Kurooka H, Takeuchi M, Kinoshita K, Nakaseko Y, Yanagida M. p93dis1, which is required for sister chromatid separation, is a novel microtubule and spindle pole body-associating protein phosphorylated at the Cdc2 target sites. Genes Dev. 1995;9:1572–1585. doi: 10.1101/gad.9.13.1572. [DOI] [PubMed] [Google Scholar]
- Lee MS, Enoch T, Piwnica-Worms H. mik1 + encodes a tyrosine kinase that phosphorylates p34cdc2on tyrsoine 15. J Biol Chem. 1994;269:30530–30537. [PubMed] [Google Scholar]
- Lundgren K, Walworth N, Booher R, Dembski M, Kirschner M, Beach D. Mik1 and Wee1 cooperate in the inhibitory tyrosine phosphorylation of Cdc2. Cell. 1991;64:1111–1122. doi: 10.1016/0092-8674(91)90266-2. [DOI] [PubMed] [Google Scholar]
- Maundrell K. nmt1of fission yeast. J Biol Chem. 1991;265:10857–10864. [PubMed] [Google Scholar]
- Maundrell K. Thiamine-repressible vectors pREP and pRIP for fission yeast. Gene (Amst) 1993;123:127–130. doi: 10.1016/0378-1119(93)90551-d. [DOI] [PubMed] [Google Scholar]
- MacNeill SA, Moreno S, Reynolds N, Nurse P, Fantes PA. The fission yeast Cdc1 protein, a homologue of the small subunit of DNA polymerase δ, binds to Pol3 and Cdc27. EMBO (Eur Mol Biol Organ) J. 1996;15:4613–4628. [PMC free article] [PubMed] [Google Scholar]
- Millar JBA, Russell P. The cdc25M-phase inducer: an unconventional protein phosphatase. Cell. 1992;68:407–410. doi: 10.1016/0092-8674(92)90177-e. [DOI] [PubMed] [Google Scholar]
- Moreno S, Hayles J, Nurse P. Regulation of p34cdc2protein kinase during mitosis. Cell. 1989;58:361–372. doi: 10.1016/0092-8674(89)90850-7. [DOI] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. . Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M. Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 1994;22:5767–5768. doi: 10.1093/nar/22.25.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabeshima K, Kurooka H, Takeuchi M, Kinoshita K, Nakaseko Y, Yanagida M. p93disl, which is required for sister chromatid separation, is a novel microtubule and spindle pole body-associating protein phosphorylated at the Cdc2 target sites. Genes Dev. 1995;9:1572–1585. doi: 10.1101/gad.9.13.1572. [DOI] [PubMed] [Google Scholar]
- Nasmyth K, Nurse P. Cell division cycle mutants altered in DNA replication and mitosis in the fission yeast Schizosaccharomyces pombe. . Mol Gen Genet. 1981;182:119–124. doi: 10.1007/BF00422777. [DOI] [PubMed] [Google Scholar]
- Nurse P. Genetic control of cell size at cell division in yeast. Nature (Lond) 1975;256:547–551. doi: 10.1038/256547a0. [DOI] [PubMed] [Google Scholar]
- Nurse P, Thuriaux P. Controls over the timing of DNA replication during the cell cycle of fission yeast. Exp Cell Res. 1977;107:365–375. doi: 10.1016/0014-4827(77)90358-5. [DOI] [PubMed] [Google Scholar]
- Nurse P, Thuriaux P. Regulatory genes controlling mitosis in the fission yeast Schizosaccharomyces pombe. . Genetics. 1980;96:627–637. doi: 10.1093/genetics/96.3.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse P, Bissett Y. Gene required in G1 for commitment to the cell cycle and G2 for control of mitosis in the fission yeast. Nature (Lond) 1981;292:558–560. doi: 10.1038/292558a0. [DOI] [PubMed] [Google Scholar]
- Nurse P, Thuriaux P, Nasmyth K. Genetic control of the cell division cycle in the fission yeast Schizosaccharomyces pombe. . Mol Gen Genet. 1976;146:167–178. doi: 10.1007/BF00268085. [DOI] [PubMed] [Google Scholar]
- Ohi R, Feoktistova A, Gould KL. Construction of vectors and a genomic library for use with his3-deficient strains of Schizosaccharomyces pombe. . Gene (Amst) 1996;174:315–318. doi: 10.1016/0378-1119(96)00085-6. [DOI] [PubMed] [Google Scholar]
- Okhura H, Adachi N, Kinosha O, Niwa T, Toda T, Yanagida M. Cold-sensitive and caffeine supersensitive mutants of the Schizosaccharomyces pombe disgenes implicated in sister chromatid separation during mitosis. EMBO (Eur Mol Biol Organ) J. 1988;7:1465–1473. doi: 10.1002/j.1460-2075.1988.tb02964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okhura H, Hagan I, Glover D. The conserved Schizosaccharomyces pombe kinase plo1, required to form a bipolar spindle, the actin ring, and septum, can drive septum formation in G1 and G2 cells. Genes Dev. 1995;9:1059–1073. doi: 10.1101/gad.9.9.1059. [DOI] [PubMed] [Google Scholar]
- Prentice HL. High efficiency transformation of Schizosaccharomyces pombeby electroporation. Nucleic Acids Res. 1991;20:621. doi: 10.1093/nar/20.3.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell P, Nurse P. cdc25 +functions as an inducer in the mitotic control of fission yeast. Cell. 1986;45:145–153. doi: 10.1016/0092-8674(86)90546-5. [DOI] [PubMed] [Google Scholar]
- Russell P, Nurse P. Negative regulation of mitosis by wee1 +, a gene encoding a protein kinase homolog. Cell. 1987;49:559–567. doi: 10.1016/0092-8674(87)90458-2. [DOI] [PubMed] [Google Scholar]
- Saka Y, Sutani T, Yamashita Y, Saitoh S, Takeuchi M, Nakaseko Y, Yanagida M. Fission yeast cut3 and cut14, members of a ubiquitous protein family, are required for chromosome condensation and segregation in mitosis. EMBO (Eur Mol Biol Organ) J. 1994;13:4938–4952. doi: 10.1002/j.1460-2075.1994.tb06821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual, second edition. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York.
- Samejima I, Yanagida M. Bypassing anaphase by fission yeast cut9 mutation: requirement of cut9+to initiate anaphase. J Cell Biol. 1994;127:1655–1670. doi: 10.1083/jcb.127.6.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samejima I, Matsumoto T, Nakaseko Y, Beach D, Yanagida M. Identification of seven new cut genes involved in Schizosaccharomyces pombemitosis. J Cell Sci. 1993;105:135–143. doi: 10.1242/jcs.105.1.135. [DOI] [PubMed] [Google Scholar]
- Sazer S, Sherwood SW. Mitochondrial growth and DNA synthesis occur in the absence of nuclear DNA replication in fission yeast. J Cell Sci. 1990;97:509–516. doi: 10.1242/jcs.97.3.509. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Boguski MS, Goebl M, Hieter P. A repeating amino acid motif in CDC23 defines a family of proteins and a new relationship among genes required for mitosis and RNA synthesis. Cell. 1990;60:307–317. doi: 10.1016/0092-8674(90)90745-z. [DOI] [PubMed] [Google Scholar]
- Simanis V, Nurse P. The cell cycle control gene cdc2 +of fission yeast encodes a protein kinase potentially regulated by phosphorylation. Cell. 1986;45:261–268. doi: 10.1016/0092-8674(86)90390-9. [DOI] [PubMed] [Google Scholar]
- Thuriaux P, Nurse P, Carter B. Mutants altered in the control coordinating cell division with cell growth in the fission yeast Schizosaccharomyces pombe. . Mol Gen Genet. 1978;161:215–220. doi: 10.1007/BF00274190. [DOI] [PubMed] [Google Scholar]
- Toda T, Umesono K, Hirata A, Yanagida M. Cold-sensitive nuclear division arrest mutants of the fission yeast Schizosaccharomyces pombe. . J Mol Biol. 1983;168:251–270. doi: 10.1016/s0022-2836(83)80017-5. [DOI] [PubMed] [Google Scholar]
- Uemura T, Yanagida M. Isolation of type I and type II DNA topoisomerase mutants from fission yeast: single and double mutants show different phanotypes in cell growth and chromatin organization. EMBO (Eur Mol Biol Organ) J. 1984;3:1737–1744. doi: 10.1002/j.1460-2075.1984.tb02040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Yanagida M. Mitotic spindle pulls but fails to separate chromosomes in type II DNA topoisomerase mutants: uncoordinated mitosis. EMBO (Eur Mol Biol Organ) J. 1986;5:1003–1010. doi: 10.1002/j.1460-2075.1986.tb04315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Morikawa K, Yanagida M. The nucleotide sequence of the fission yeast DNA topoisomerase II gene: structural and functional relationships to other DNA topoisomerases. EMBO (Eur Mol Biol Organ) J. 1986;5:2355–2361. doi: 10.1002/j.1460-2075.1986.tb04504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Okhura H, Adachi Y, Morino K, Shiozaki K, Yanagida M. DNA topoisomerase II is required for condensation and separation of mitotic chromosomes in S. pombe. . Cell. 1987;50:917–925. doi: 10.1016/0092-8674(87)90518-6. [DOI] [PubMed] [Google Scholar]
- Umesono K, Toda T, Hayashi S, Yanagida M. Two cell division cycle genes NDA2 and NDA3 of the fission yeast Schizosaccharomyces pombecontrol microtubular organization and sensitivity to anti-mitotic benzimidazole compounds. J Mol Biol. 1983;168:271–284. doi: 10.1016/s0022-2836(83)80018-7. [DOI] [PubMed] [Google Scholar]
- Uzawa S, Samejima I, Hirano T, Tanaka K, Yanagida M. The fission yeast cut1 +gene regulates spindle pole body duplication and has homology to the budding yeast ESP1 gene. Cell. 1990;62:913–925. doi: 10.1016/0092-8674(90)90266-h. [DOI] [PubMed] [Google Scholar]
- Woods A, Sherwin T, Sasse R, McRae TH, Baines AJ, Gull K. Definition of individual components within the cytoskeleton of Trypanosoma bruceiby a library of monoclonal antibodies. J Cell Sci. 1989;93:491–500. doi: 10.1242/jcs.93.3.491. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Guacci V, Koshland D. Pds1p is required for faithful execution of anaphase in the fission yeast, Saccharomyces cerevisiae. . J Cell Biol. 1996;133:85–97. doi: 10.1083/jcb.133.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita YM, Nakaseko Y, Samejima I, Kumada K, Yamada H, Michaelson D, Yanagida M. 20S cyclosome complex formation and proteolytic activity inhibited by the cAMP/PKA pathway. Nature (Lond) 1996;384:276–279. doi: 10.1038/384276a0. [DOI] [PubMed] [Google Scholar]
- Yanagida M. Gene products required for chromosome separation. J Cell Sci. 1989;12:213–229. doi: 10.1242/jcs.1989.supplement_12.18. [DOI] [PubMed] [Google Scholar]
- Young PG, Fantes P. Schizosaccharomyces pombemutants affected in their division response to starvation. J Cell Sci. 1987;88:295–304. doi: 10.1242/jcs.88.3.295. [DOI] [PubMed] [Google Scholar]
- Zachariae W, Shin TH, Galova M, Obermaier B, Nasmyth K. Identification of subunits of the anaphase-promoting complex of Saccharomyces cerevisiae. . Science (Wash DC) 1996;274:1201–1204. doi: 10.1126/science.274.5290.1201. [DOI] [PubMed] [Google Scholar]
- Zhang MQ, Marr TG. Fission yeast gene structure and recognition. Nucleic Acids Res. 1994;22:1750–1759. doi: 10.1093/nar/22.9.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]