

Abstract
The oncogene bcl-2 encodes a 26-kD protein localized to intracellular membranes, including the ER, mitochondria, and perinuclear membrane, but its mechanism of action is unknown. We have been investigating the hypothesis that Bcl-2 regulates the movement of calcium ions (Ca2+) through the ER membrane. Earlier findings in this laboratory indicated that Bcl-2 reduces Ca2+ efflux from the ER lumen in WEHI7.2 lymphoma cells treated with the Ca2+-ATPase inhibitor thapsigargin (TG) but does not prevent capacitative entry of extracellular calcium. In this report, we show that sustained elevation of cytosolic Ca2+ due to capacitative entry is not required for induction of apoptosis by TG, suggesting that ER calcium pool depletion may trigger apoptosis. Bcl-2 overexpression maintains Ca2+ uptake in the ER of TG-treated cells and prevents a TG-imposed delay in intralumenal processing of the endogenous glycoprotein cathepsin D. Also, Bcl-2 overexpression preserves the ER Ca2+ pool in untreated cells when extracellular Ca2+ is low. However, low extracellular Ca2+ reduces the antiapoptotic action of Bcl-2, suggesting that cytosolic Ca2+ elevation due to capacitative entry may be required for optimal ER pool filling and apoptosis inhibition by Bcl-2. In summary, the findings suggest that Bcl-2 maintains Ca2+ homeostasis within the ER, thereby inhibiting apoptosis induction by TG.
The oncogene bcl-2 encodes a 26-kD protein that localizes to the ER and perinuclear membrane, as well as the mitochondrial membrane (8, 48). bcl-2 is a member of a family of apoptosis regulators, some of which inhibit apoptosis, including bcl-2 and bcl-xL, and some of which promote apoptosis. The Bcl-2 protein anchors to intracellular membranes through a hydrophobic region located near its carboxy terminus, while the major portion of the molecule is in the cytoplasm. Bcl-2's mechanism of action is unknown. Bcl-2 overexpression is associated with decreased formation of reactive oxygen species (19) and decreased oxygen radical–mediated lipid peroxidation (16), suggesting that Bcl-2 works in an antioxidant pathway to inhibit apoptosis. However, it has been suggested that Bcl-2 can also inhibit apoptosis in the absence of oxygen radicals (17, 35, 43).
Another theory is that Bcl-2 and Bcl-xL may regulate ion fluxes. This concept is supported by evidence that bcl-2 overexpression prevents Ca2+ redistribution from the ER to mitochondria following growth factor withdrawal (1) and that bcl-2 overexpression inhibits apoptosis-associated Ca2+ waves (28) and nuclear Ca2+ uptake (29). Also, bcl-2 overexpression potentiates maximal uptake of Ca2+ by mitochondria (34) and preserves mitochondrial transmembrane potential (49). Two recent findings provide strong evidence that Bcl-2 family members regulate ion fluxes. First, the X-ray and NMR structure of Bcl-xL resembles the physical structure of ion channel–forming bacterial toxins (33). Second, Bcl-xL forms an ion channel in synthetic lipid membranes (32).
We have been investigating the hypothesis that Bcl-2 regulates the movement of Ca2+ through the ER membrane. A high concentration of Ca2+ is maintained in the ER lumen by a member of the sarcoplasmic endoplasmic reticulum calcium ATPase family of Ca2+-ATPases that pump Ca2+ into the ER, counterbalancing a leak of Ca2+ ions through the ER membrane into the cytoplasm (27). Thapsigargin (TG),1 a sesquiterpene lactone tumor promoter derived from the plant Thapsia garganica, is a selective inhibitor of the ER-associated Ca2+-ATPase (45). Because the concentration of Ca2+ in the ER lumen is several orders of magnitude higher than the concentration of Ca2+ in the cytoplasm, TG-mediated Ca2+-ATPase inhibition allows Ca2+ to flow from the ER lumen into the cytoplasm, producing a transient elevation of cytosolic Ca2+ concentration, followed by sustained elevation of cytosolic Ca2+ in response to capacitative entry (39, 45). We have shown that in TG-treated WEHI7.2 mouse lymphoma cells, which lack Bcl-2, these disturbances of intracellular Ca2+ homeostasis precede cell death accompanied by the typical hallmarks of apoptosis, including CASPASE activation, cleavage of poly(ADP-ribose) polymerase, chromatin condensation, and DNA fragmentation (20; unpublished data). In stable transfectants of the WEHI7.2 line that express high levels of Bcl-2, TG treatment inhibits cell growth but does not induce apoptosis (20). Bcl-2 overexpression is associated with a reduction in the transient elevation of cytosolic Ca2+ induced by TG-mediated ER Ca2+-ATPase inhibition (20). Based on this observation, we proposed that Bcl-2 inhibits apoptosis in TG-treated cells by inhibiting Ca2+ efflux from the ER. However, in subsequent studies we found that Bcl-2 does not prevent sustained cytosolic Ca2+ elevation due to capacitative entry of extracellular Ca2+; in fact, Bcl-2 prevents apoptosis even when cytosolic Ca2+ is markedly elevated following TG treatment (10). Therefore, based on the premise that sustained elevation of cytosolic Ca2+ triggers apoptosis in TG-treated cells (14, 18), we concluded that Bcl-2 acts downstream and independent of Ca2+ fluxes to inhibit apoptosis (10).
In the present report, we show that sustained elevation of cytosolic Ca2+ in TG-treated cells can be prevented by reducing extracellular Ca2+ concentration, which inhibits capacitative entry. These conditions do not prevent TG-induced apoptosis, indicating that sustained elevation of cytosolic Ca2+ secondary to capacitative entry is not required for induction of apoptosis by TG. Therefore, we focused on the hypothesis that Bcl-2 inhibits apoptosis by maintaining Ca2+ homeostasis in the ER. This hypothesis is supported by evidence that Bcl-2 maintains Ca2+ uptake and protein processing in the ER when the Ca2+-ATPase is inhibited by TG. Also, Bcl-2 preserves the ER Ca2+ pool and thereby prevents growth inhibition when extracellular Ca2+ is withdrawn. Finally, we show that the antiapoptotic action of Bcl-2 is abrogated by a reduction in extracellular Ca2+. Based on these findings, a model of how Bcl-2 preserves Ca2+ homeostasis in the ER and inhibits TG-induced apoptosis is proposed.
Materials and Methods
Cell Lines and Culture Conditions
W.Hb12 and W.Hb15 cell lines were derived from the WEHI7.2 line by stable transfection with a cDNA encoding Bcl-2 (20). WEHI7.2-neo cells were derived by stable transfection with pSFFV-neo vector. The CaBP3 and CaBP20 cell lines were derived from the WEHI7.2 line by stable transfection of a cDNA encoding calbindin D (12). The S49-Bcl-2 line was derived by stably transfecting the S49 line with a cDNA encoding Bcl-2 (2). Cell lines were routinely cultured in DME supplemented with 2 mM glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin, 0.4 mM nonessential amino acids, and 10% (vol/vol) heat-inactivated horse serum at 37°C in a 7% CO2 atmosphere. In certain experiments, cells were pelleted and resuspended in Ca2+-free DME supplemented with 10% horse serum.
Cell Counting and Apoptosis Assay
Viable cells, defined as cells that exclude trypan blue dye, were counted using a hemocytometer. The percentage of cells with an apoptotic nuclear morphology was assessed by fluorescence microscopy (10). Five million cells were gently pelleted and resuspended in 0.2 ml tissue culture medium, and both ethidium bromide and acridine orange were added from 100 μg/ml stock solutions to achieve 4 μg/ml final concentrations of each. Cells were examined under a glass coverslip with ultraviolet illumination using a Nikon Optiphot microscope (Melville, NY).
Ca2+ Flux Measurements
Methods for measuring 45Ca2+ uptake in the ER were described in detail (10, 20). Cells were pretreated with oxalate, which permeates the ER membrane and forms insoluble complexes with Ca2+, reducing Ca2+ efflux and permitting unidirectional measurement of 45Ca2+ uptake (6). Measurements were performed in the presence of azide at a free Ca2+ concentration buffered to 150 nM by EGTA, essentially eliminating the uptake of Ca2+ into low-affinity mitochondrial stores. Cells were washed twice with 0.9% NaCl, 50 μM EDTA solution at 4°C, and permeabilized by incubation for 5 min at 37°C in buffer containing 120 mM KCl, 30 mM imidazole, pH 6.8, 2 mM MgCl2, 1 mM ATP, 1 mM EGTA, and 0.0005% digitonin. Cells were then pelleted and resuspended at 4°C in buffer containing 140 mM KCl, 10 mM NaCl, 2.5 mM MgCl2, 0.0005% digitonin, 10 mM Hepes-KOH, pH 7.0, with 4 mM oxalic acid. To begin uptake measurements, 0.5-ml aliquots of the cell suspension were added at 37°C to 2.5 ml buffer containing 120 mM KCl, 30 mM imidazole, pH 6.8, 5 mM MgCl2, 5 mM ATP, 10 mM NaN3, 4 mM oxalic acid, and 45CaCl2 buffered to 150 nM free 45Ca2+ (8.9 mCi/mg) with EGTA as described (46). Immediately and at 1-min time intervals thereafter, 0.25-ml aliquots were removed from the continuously stirring cell suspension and placed into wash buffer containing 140 mM KCl, 10 mM NaCl, 2.5 mM MgCl2, 10 mM Hepes-KOH, pH 7.0, and 1 mM LaCl3. Cells were collected on glass fiber filters and washed three times, each with 4 ml of wash buffer. Filters were dried, dissolved in 10 ml 3a70 scintillation cocktail, and counted in a liquid scintillation counter (model LS7800; Beckman Instruments, Fullerton, CA).
Intracellular Ca2+ Measurements
Fluorometric measurements of intracellular Ca2+ were according to published methods (10, 20). Twenty million cells were pelleted by centrifugation at 150 g for 10 min and resuspended in 10 ml basal salt solution, CaBSS (130 mM NaCl, 5 mM KCl, 1.5 mM CaCl2, 1 mM MgCl2, 25 mM Hepes, pH 7.5, 5 mM glucose, and 1 mg/ml BSA). In certain experiments, CaCl2 was deleted, yielding a buffer referred to as BSS. Free Ca2+ concentration of buffers and tissue culture medium was measured with a Ca2+ electrode. Cell suspensions were incubated for 45 min at 25°C with 1 μM Fura-2 AM from a 1 mM stock solution dissolved in DMSO. Cells were then pelleted and resuspended in 10 ml fresh CaBSS or BSS and maintained at 25°C for up to 60 min before fluorometry. For each measurement, a 1.5-ml aliquot of Fura-2–loaded cells was equilibrated to 37°C in a stirred quartz cuvette. Fura-2 fluorescence was continuously monitored at 339 nm excitation and 500 nm emission. Ca2+-dependent Fura-2 fluorescence was calibrated after lysis of cells with 20 μg/ml digitonin, and cytosolic free Ca2+ concentration was calculated, assuming a Fura-2 K d of 220 nM.
Ca2+ Overlay and Western Blotting
Cells were lysed by suspension in lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 10 mM iodoacetamide, 1% Triton X-100) for 15 min at 4°C and paired samples of 30 μg cytosolic protein were separated by SDS–gel electrophoresis under reducing and denaturing conditions. One half of the membrane corresponding to one set of samples was used for 45Ca2+ overlay, and the other half of the membrane corresponding to the other identical set of samples was used for Western blotting. After electrophoresis, the proteins were transferred to a nitrocellulose membrane at a constant current of 100 mA for 1 h in Tris-glycine buffer. 45Ca2+ overlay was performed essentially as described by Maruyama et al. (30). After transfer, the membrane was washed in IKM buffer (10 mM imidazole-HCl, pH 6.8, 5 mM MgCl2, and 60 mM KCl) for 1 h with three changes of buffer. The membrane was then incubated in the same buffer with 1 mCi/liter 45Ca2+ for 10 min. After incubation, the membrane was rinsed with 50% ethanol for 5 min, three times. Excess ethanol was blotted away using Whatman filter paper No. 1, and the membrane was dried at room temperature for 3 h. After the membrane was completely dry, it was exposed to film for 2 d at −80°C.
For Western blotting, membranes were blocked by incubation for one h with 5% dry milk in TBS, pH 7.5, at 25°C or overnight at 4°C. After blocking, the membranes were overlaid with diluted primary antibody (monoclonal anti–calbindin D, Sigma Chemical Co. [St. Louis, MO]; or monoclonal anti–Bcl-2, PharMingen, San Diego, CA) and incubated for 1 h at 25°C. After washing, the membranes were incubated for 1 h at 25°C with horseradish peroxidase–conjugated goat anti–mouse whole immunoglobulin (Amersham Corp., Arlington Heights, IL) diluted 1:500 in TBS. After washing, the enhanced chemiluminescence Western blotting detection reagent (Amersham Corp.) was added, and the reaction was allowed to proceed according to the manufacturer. The membranes were exposed to film at room temperature for 30 s and then developed.
Cathepsin D Immunoprecipitation
Wild-type S49 cells and Bcl-2 transfectants (40 million cells each) were suspended in 2 ml methionine-free medium supplemented with 10% horse serum, 1 mM glutamine, and nonessential amino acids and incubated for 30 min before adding 150 μCi [35S]methionine. Either 200 nM TG or DMSO vehicle were added to duplicate sets of cells at the beginning of the pulse label. The cells were incubated for 2 h until the chase was initiated by adding 1 mg/ml unlabeled methionine. The chase was ended by washing cells twice with PBS at 4°C. Cell pellets were suspended in an equal volume of lysis buffer (25 mM Tris-HCl, pH 8.0, 50 mM NaCl, 1 mM EDTA, 10% glycerol, 0.8% deoxycholic acid, 0.8% NP-40). At 30–120 min into the chase, the cell suspensions were frozen and then thawed, followed by centrifugation at 14,000 g for 20 min at 4°C. The clear supernatant was collected and, based on TCA precipitation analysis, equal amounts of [35S]methionine-labeled protein were incubated with rabbit anti–human cathepsin D antiserum (1:30 dilution) (13) for 1 h at 4°C and then incubated with protein A–Sepharose for 30 min at 4°C. Protein A–Sepharose pellets were then washed four times with lysis buffer at 4°C. Bound proteins were extracted by resuspending protein A–Sepharose pellets in sample buffer for gel electrophoresis and analyzed by SDS-PAGE under reducing and denaturing conditions as described previously (21).
Results
Bcl-2 Mediates ER Ca2+ Uptake
We compared WEHI7.2 cells, which do not express Bcl-2, and W.Hb12 cells, which were derived from WEHI7.2 by stable transfection with the human Bcl-2 cDNA and therefore express a high level of Bcl-2 mRNA and protein (20). Findings have been confirmed in an additional high Bcl-2 clone and WEHI7.2 cells stably transfected with pSFFV-neo vector. Because TG is an irreversible Ca2+-ATPase inhibitor, TG treatment inhibits Ca2+ uptake in the ER, thereby producing sustained ER Ca2+ pool depletion (45). Based on this information, one would predict that TG treatment should cause sustained ER calcium pool depletion in both WEHI7.2 and W.Hb12 cells, unless Bcl-2 mediates ER Ca2+ uptake. To test this hypothesis, the uptake of 45Ca2+ in the ER of digitonin-permeabilized cells was measured in the presence of azide at a free Ca2+ concentration buffered to 150 nM to eliminate Ca2+ uptake into low-affinity mitochondrial stores. Uptake was measured in the presence and absence of oxalate, which enters the ER through nonselective anion channels and binds Ca2+ to form insoluble Ca2+–oxalate complexes, thus inhibiting Ca2+ efflux (47). Oxalate permeability is a property specific to the ER (47 and references therein). In the absence of oxalate, 45Ca2+ uptake was detected in W.Hb12 cells, but not in WEHI7.2 cells (Fig. 1 A). In the presence of oxalate, 45Ca2+ uptake was detected in WEHI7.2 cells, but the rate of uptake was consistently greater in W.Hb12 cells than in WEHI7.2 cells (Fig. 1 B). These findings indicate that Bcl-2 overexpression enhances calcium uptake and retention by the ER of untreated cells. 45Ca2+ uptake was markedly diminished in both WEHI7.2 and W.Hb12 cells when the ER calcium-ATPase was inhibited by 5–100 nM TG (Fig.1, C–F), indicating that 45Ca2+ uptake by the ER of untreated cells was primarily mediated by the ER-associated calcium-ATPase. However, residual 45Ca2+ uptake was detected in TG-treated W.Hb12 cells, whereas 45Ca2+ uptake was barely detected in TG-treated WEHI7.2 cells. Thus, Bcl-2 enhances Ca2+ accumulation in the ER of untreated cells and maintains a low but detectable level of Ca2+ accumulation in the ER of TG-treated cells.
Figure 1.

Bcl-2 mediates ER Ca2+ uptake. Uptake of 45Ca2+ was measured in digitonin-permeabilized cells in the absence (A) or presence of oxalate (B–F), and in the absence (A and B) or presence of 5 nM TG (C), 10 nM TG (D), 20 nM TG (E), or 100 nM TG (F). Open symbols, WEHI7.2 cells; closed symbols, W.Hb12 cells. Symbols represent the mean of duplicate determinations in a single experiment that was repeated at least twice with a similar result.
The increased accumulation of 45Ca2+ in the ER of W.Hb12 cells could be due to a direct effect of Bcl-2 on Ca2+ movement through the ER membrane or to an indirect effect of Bcl-2 on the Ca2+-binding capacity in the ER lumen. The levels of three major resident ER Ca2+-binding proteins, GRP78 (BiP), GRP94, and calreticulin, are unaffected by Bcl-2 overexpression (31; McColl, K., and C. Distelhorst, unpublished data). To exclude the possibility that Bcl-2 increases the expression of an unidentified Ca2+-binding protein, we compared Ca2+-binding proteins in WEHI7.2 and W.Hb12 cells by 45Ca2+ overlay (Fig. 2). Cellular proteins were separated by SDS-PAGE, transferred to a nitrocellulose membrane, renatured, and incubated with 45Ca2+. An autoradiogram reveals a number of radioactive bands corresponding to proteins that bound 45Ca2+. As a positive control, extracts from CaBP3 and CaBP20 cells were included. These cells were derived previously by stably transfecting WEHI7.2 cells with a cDNA encoding the Ca2+-binding protein, calbindin (12). As shown by the Western blot in Fig. 2 (lanes 3 and 4), calbindin protein is detected in CaBP20 but not in CaBP3 cells. A 45Ca2+-labeled protein corresponding to calbindin is detected in CaBP20 cells (Fig. 2, lane 5), documenting the detection of Ca2+-binding proteins by the 45Ca2+ overlay technique. Significantly, the pattern of 45Ca2+-binding proteins is the same in WEHI7.2 and W.Hb12 cells. Therefore, Bcl-2-mediated Ca2+ uptake in the ER does not appear to be secondary to altered expression of Ca2+-binding proteins.
Figure 2.

Bcl-2 does not increase expression of Ca2+-binding proteins. Extracts from untreated cells listed on the horizontal axis were resolved by SDS-PAGE and transferred to nitrocellulose membranes. Lanes 1–4 were subjected to Western blotting using anti-Bcl-2 antibody (lanes 1 and 2) and anticalbindin antibody (lanes 3 and 4) as probes. Lanes 5–8 were incubated with 45Ca2+ and then exposed to x-ray film. Molecular mass standards are in lane 9. An unidentified protein that immunoreacts with anticalbindin antibody is detected at ∼80-kD in lane 4, and an unidentified Ca2+-binding protein was detected in CaBP20 cells (lane 5) midway between the 49.5- and 80-kD markers. The appearance of these proteins was variable among experiments and therefore not considered significant.
BCL-2 Prevents a TG-imposed Delay in Protein Processing
Maintenance of Ca2+ homeostasis within the ER is required for normal protein processing and transport (15, 25, 42). Depletion of the ER Ca2+ pool by TG inhibits glycoprotein processing and maturation in the ER lumen (25). Therefore, to investigate the effect of Bcl-2 on Ca2+-dependent ER function, we examined the effect of TG treatment on the processing of cathepsin D, an endosomal cysteine protease that has been implicated as a mediator of apoptosis (9). The 48-kD cathepsin D protein is glycosylated within the ER, forming a 53-kD precursor that is modified to a 51-kD mature form after transfer to the Golgi (13). In earlier studies, we observed a similar pattern of cathepsin D maturation in the S49 mouse lymphoma cell line, a T-cell line similar to WEHI7.2 (21). Therefore, in the present study, the processing of cathepsin D was assessed by pulse–chase analysis in S49 cells, which do not express Bcl-2, and stable transfectants that are resistant to TG-induced apoptosis (2). The processing of cathepsin D is relatively rapid in untreated wild-type S49 cells and Bcl-2 transfectants, such that up to half of the cathepsin D synthesized during the 2 h pulse-labeling period was converted to the more mature 51-kD form by the beginning of the chase (Fig. 3). (Labeling of cathepsin D was suboptimal for immunoprecipitation analysis when the labeling period was shortened.) During the chase, the labeled 53-kD form matured to the 51-kD form almost completely in both wild-type S49 cells and Bcl-2 transfectants. Examination of the labeling pattern at 30 min into the chase suggests that processing of cathepsin D may be somewhat more rapid in the Bcl-2 transfectants compared to the wild-type cells. After TG treatment, however, there was a major difference between the wild-type cells and the Bcl-2 transfectants. In wild-type cells, TG treatment completely inhibited cathepsin D processing, whereas cathepsin D processing was unaffected by TG treatment in the Bcl-2 transfectants. Similar findings were observed in comparisons of WEHI7.2 and W.Hb12 cells (data not shown). In summary, these observations indicate that Bcl-2 overexpression maintains calcium-dependent protein processing, providing additional evidence that Bcl-2 preserves calcium homeostasis within the ER lumen when the ER Ca2+-ATPase is inhibited by TG.
Figure 3.

Bcl-2 prevents TG-induced delay in cathepsin D processing. Pulse–chase analysis of cathepsin D processing in wild-type S49 cells (−Bcl-2) and Bcl-2 transfectants (+Bcl-2) in the presence or absence of 200 nM TG. [35S]methionine-labeled cathepsin D was immunoprecipitated, resolved by PAGE under reducing and denaturing conditions, and analyzed by autoradiography.
Bcl-2 Preserves the ER Ca2+ Pool
The preceding experiments examined the effect of Bcl-2 on ER Ca2+ homeostasis when the ER Ca2+-ATPase was inhibited by TG. Another condition that compromises ER Ca2+ pool filling is a reduction in extracellular Ca2+. Therefore, we compared the effect of low extracellular Ca2+ on the ER Ca2+ pool of WEHI7.2 and W.Hb12 cells. These cells are routinely cultured in DME supplemented with 10% serum, providing a physiologic extracellular Ca2+ concentration of 1.3 mM. In preliminary experiments, we incubated cells under completely Ca2+-free conditions by adding EDTA or EGTA to the tissue culture medium; however, rapid loss of viability was observed in both WEHI7.2 and W.Hb12 cells (Lam, M., and C. Distelhorst, unpublished observations). Moreover, incubating cells in Ca2+-free DME supplemented with either no serum or only 1% serum produced rapid loss of viability. Therefore, in the following experiments cells were incubated in Ca2+-free DME supplemented with 10% serum; under these conditions, the extracellular Ca2+ concentration, measured with a Ca2+ electrode, is 0.13 mM, ∼10-fold below the normal extracellular Ca2+ concentration.
The effect of low extracellular Ca2+ on free cytosolic Ca2+ concentration and the TG-mobilizable Ca2+ pool was measured using Fura-2 AM, a Ca2+-responsive fluorescent indicator that localizes to the cytoplasm of intact cells (Fig. 4). Cells were loaded with Fura-2 AM at 3 and 18 h after being placed in either normal or low extracellular Ca2+ medium. In the presence of a physiologic concentration of extracellular Ca2+, the free cytoplasmic Ca2+ concentration in WEHI7.2 and W.Hb12 cells is 184 ± 10 and 141 ± 8 nM, respectively, whereas the free cytoplasmic Ca2+ concentration decreases to 118 ± 4 and 111 ± 9 nM, respectively, when the extracellular Ca2+ concentration is decreased to 0.13 mM. The mobilizable ER Ca2+ pool was estimated by measuring the transient increase in cytosolic Ca2+ produced by adding 100 nM TG to Fura-2 AM– loaded cells. Note that EGTA was added immediately before adding TG to chelate extracellular Ca2+; hence, the increase in Fura-2 fluorescence is due to Ca2+ efflux from the ER, rather than uptake of extracellular Ca2+. The increase in cytosolic Ca2+ concentration induced by TG was markedly reduced when WEHI7.2 cells were incubated for either 3 or 18 h in low extracellular Ca2+. In contrast, the TG-mobilizable Ca2+ pool was essentially unchanged when W.Hb12 cells were incubated in low extracellular Ca2+. Thus, Bcl-2 overexpression preserves the ER Ca2+ pool when extracellular Ca2+ is withdrawn.
Figure 4.

Bcl-2 prevents ER Ca2+ pool depletion. WEHI7.2 cells (A–D) and W.Hb12 cells (E–H) were incubated for 3 h (A, B, E, and F) or 18 h (C, D, G, and H) in medium with a Ca2+ concentration of 1.3 mM (A, C, E, and G) or medium with a Ca2+ concentration of 0.13 mM (B, D, F, and H) and then loaded with Fura2-AM. Shown are continuous fluorometric tracings in which addition of 100 nM TG (right hand arrow) induces an increase in cytosolic Ca2+ concentration. Note that EGTA (left hand arrow) was added immediately before TG to chelate extracellular Ca2+; hence, the increase in cytosolic Ca2+ that follows TG addition represents release of Ca2+ from the ER pool. Large vertical oscillations in fluorescence near the beginning of each tracing correspond to brief light exposure during EGTA and TG additions.
Effect of Low Extracellular Ca2+ on Cell Growth and Viability
When extracellular Ca2+ is reduced, the growth of WEHI7.2 cells is inhibited (Fig. 5 A), whereas the growth of W.Hb12 cells is maintained (Fig. 5 C). These findings are consistent with earlier evidence that the ER Ca2+ pool is required for cell division and that a decrease in this pool induces growth arrest (44). It appears that Bcl-2 maintains cell growth by preserving the ER Ca2+ pool when extracellular Ca2+ is reduced. Apoptosis was not induced by incubating untreated WEHI7.2 cells in low extracellular Ca2+, indicating that incomplete reduction of the ER Ca2+ pool is compatible with survival (Fig. 5 B).
Figure 5.
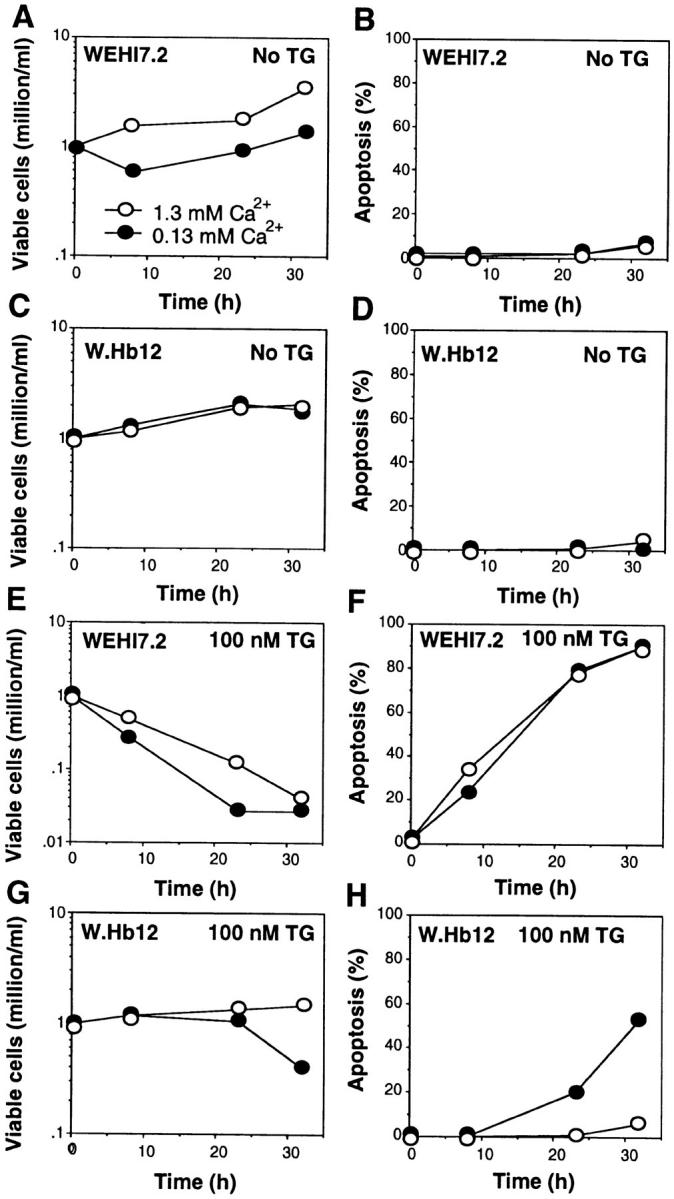
Effect of Bcl-2 on cell growth and viability. WEHI7.2 cells (A, B, E, and F) and W.Hb12 cells (C, D, G, and H) were incubated in medium containing 1.3 mM Ca2+ (open symbols) or 0.13 mM Ca2+ (closed symbols), in either the absence of TG (A–D) or presence of 100 nM TG (E–H), for 32 h. At various time points, the number of viable cells, defined as cells that exclude trypan blue dye, and the percentage of apoptotic cells, based on nuclear apoptotic changes detected on fluorescence microscopy of cells stained with acridine orange, were measured. Symbols represent the mean of duplicate determinations in a single experiment that was repeated three times with a similar result.
The alterations in intracellular Ca2+ homeostasis induced by TG are modified considerably by low extracellular Ca2+. Thus, the transient elevation of cytosolic Ca2+ induced by TG-mediated ER Ca2+-ATPase inhibition is reduced by incubating WEHI7.2 cells in low extracellular Ca2+ (Fig. 4, B and D). Also, the marked elevation of cytosolic Ca2+ secondary to capacitative entry is markedly reduced when either WEHI7.2 or W.Hb12 cells are treated with TG in low extracellular Ca2+ (Fig. 6). Nevertheless, low extracellular Ca2+ does not prevent the induction of apoptosis by TG in WEHI7.2 cells (Fig. 5 F), indicating that marked elevation of cytosolic Ca2+ due to capacitative entry is not necessary for apoptosis induction by TG.
Figure 6.

Low extracellular Ca2+ inhibits cytosolic Ca2+ elevation in TG-treated cells. WEHI7.2 cells (A) and W.Hb12 cells (B) were incubated in medium containing 1.3 mM Ca2+ or 0.13 mM Ca2+, in the presence or absence of 100 nM TG, for 4 h before loading with Fura-2 AM. Cytosolic Ca2+ concentration was measured according to Fura-2 fluorescence. Symbols represent the mean ± SE of multiple determinations in three separate experiments.
When W.Hb12 cells are suspended in medium containing a physiologic concentration of Ca2+, TG treatment inhibits cell growth (Fig. 5 G) but does not induce apoptosis (Fig. 5 H). However, when W.Hb12 cells are suspended in low Ca2+ medium, TG induces not only growth arrest (Fig. 5 G), but also apoptosis (Fig. 5 H). These observations suggest that TG-induced ER Ca2+ pool depletion is sufficient to inhibit cell growth, but not to induce apoptosis, when W.Hb12 cells are suspended in medium containing a physiologic concentration of Ca2+. Low extracellular Ca2+ may reduce the ability of Bcl-2 to maintain ER Ca2+ uptake when the ER Ca2+-ATPase is inhibited by TG, allowing the concentration of Ca2+ within the ER to fall below a critical level necessary for cell survival.
Discussion
Evidence that ER Ca2+ Pool Depletion Induces Apoptosis
Inhibition of the ER Ca2+-ATPase by TG induces three well-defined changes in intracellular Ca2+ homeostasis: transient elevation of cytosolic Ca2+ due to efflux of Ca2+ through the ER membrane, ER Ca2+ pool depletion, and sustained elevation of cytosolic Ca2+ secondary to capacitative entry of extracellular Ca2+. In this report, we show that both transient and sustained elevations of cytosolic Ca2+ are inhibited when WEHI7.2 cells are treated with TG in the presence of low extracellular Ca2+ (Figs. 4 and 6), without inhibiting TG-induced apoptosis (Fig. 5). Thus, it appears that ER Ca2+ pool depletion can trigger apoptosis in TG-treated WEHI7.2 cells. The concept that ER Ca2+ pool depletion induces cell death is consistent with the findings of others (2, 22, 23, 41). However, in thymocytes and prostate cancer cells, TG-induced apoptosis is inhibited by preincubating cells with the intracellular Ca2+ indicator, BAPTA-AM, or overexpressing the cytosolic Ca2+-binding protein, calbindin, suggesting that cytosolic Ca2+ elevation may also be a factor in inducing apoptosis (14, 18). However, TG-induced apoptosis in WEHI7.2 cells is not inhibited by pretreatment with BAPTA-AM or overexpression of calbindin (Lam, M., and C. Distelhorst, unpublished observations).
Maintenance of Ca2+ homeostasis within the ER is essential for a number of vital cellular functions, including signal transduction (7), translation (4), protein processing and transport (15, 25, 42), and cell division (44). Therefore, ER Ca2+ pool depletion might affect cells adversely in multiple ways. Three potential mechanisms by which ER Ca2+ pool depletion might contribute to apoptosis induction are considered here. First, depletion of the ER Ca2+ pool might destabilize the Ca2+–protein gel and its associated membrane (3, 42), leading to vesiculation and formation of apoptotic blebs. This concept is intriguing in view of evidence that apoptotic blebs are composed of ER membrane and resident ER Ca2+-binding proteins, including calreticulin, GRP78 (BiP), and GRP94 (5). Second, disruption of protein processing and transport within the ER may contribute to TG-induced apoptosis. This concept is consistent with evidence that induction of the genes encoding the ER proteins GRP78, GRP94, and calreticulin, in response to ER Ca2+ pool depletion, protects against TG-induced cell death (24). These proteins bind Ca2+ with low affinity and, therefore, facilitate Ca2+ sequestration in the ER lumen (3, 42). Moreover, these proteins function as chaperones, aiding in the folding and processing of nascent proteins in the ER lumen. Accordingly, the failure of WEHI7.2 cells to induce the transcription of these stress genes may account for the marked susceptibility of WEHI7.2 cells to TG-induced apoptosis (31). Disruption of normal protein processing secondary to TG-induced ER Ca2+ pool depletion and failure to induce GRP78 (BiP), GRP94, and calreticulin may cause a proteolytically active cathepsin D precursor, or other protease precursor, to accumulate within the ER lumen, initiating a proteolytic cascade that leads to apoptosis. This theory is based on recent evidence implicating increased cathepsin D expression and delayed processing in apoptosis induction by tumor necrosis factor-α, interferon-γ, and Fas/ APO-1 (9). A third theory is that TG-induced ER Ca2+ pool depletion releases an endonuclease into the nucleus responsible for DNA fragmentation. This theory is based on evidence that a deoxyribonuclease involved in nuclear DNA fragmentation is confined to the ER until it gains access to the nucleus during apoptosis (37). Although these hypotheses have not been formally tested in TG-treated cells, they illustrate potential mechanisms by which TG-induced ER Ca2+ pool depletion might trigger apoptosis.
Evidence that Bcl-2 Maintains Ca2+ Homeostasis in the ER
The findings of this report, summarized in the form of a model (Fig. 7), suggest that Bcl-2 maintains ER Ca2+ homeostasis when the Ca2+-ATPase is inhibited by TG or extracellular Ca2+ is withdrawn. In untreated WEHI7.2 cells cultured in the presence of a physiologic extracellular Ca2+ concentration, ER Ca2+ pool filling is maintained by the ER-associated Ca2+-ATPase, which pumps Ca2+ from the cytoplasm into the ER against a steep concentration gradient (Fig. 7, A and B). When the ER Ca2+-ATPase is inhibited by TG, the ER Ca2+ pool is reduced, signaling capacitative entry and thereby producing sustained elevation of cytosolic Ca2+ (Fig. 7, C and D). In cells that lack Bcl-2, TG treatment inhibits Ca2+ uptake, and the ER Ca2+ pool is severely depleted (Fig. 7 C). In the model, we propose that Bcl-2 overexpression maintains partial ER pool filling by mediating Ca2+ uptake when the Ca2+-ATPase is inhibited by TG (Fig. 7 D). However, Bcl-2 does not appear to completely fill the ER Ca2+ pool under these conditions; this conclusion is based on evidence that TG treatment induces capacitative entry of extracellular Ca2+, the signal for which is a reduction in the ER calcium pool (40). The concept that Bcl-2 prevents complete ER Ca2+ pool depletion is based primarily on 45Ca2+ uptake studies (Fig. 1) and evidence that Bcl-2 overexpression prevents a TG-imposed delay in protein processing (Fig. 3).
Figure 7.
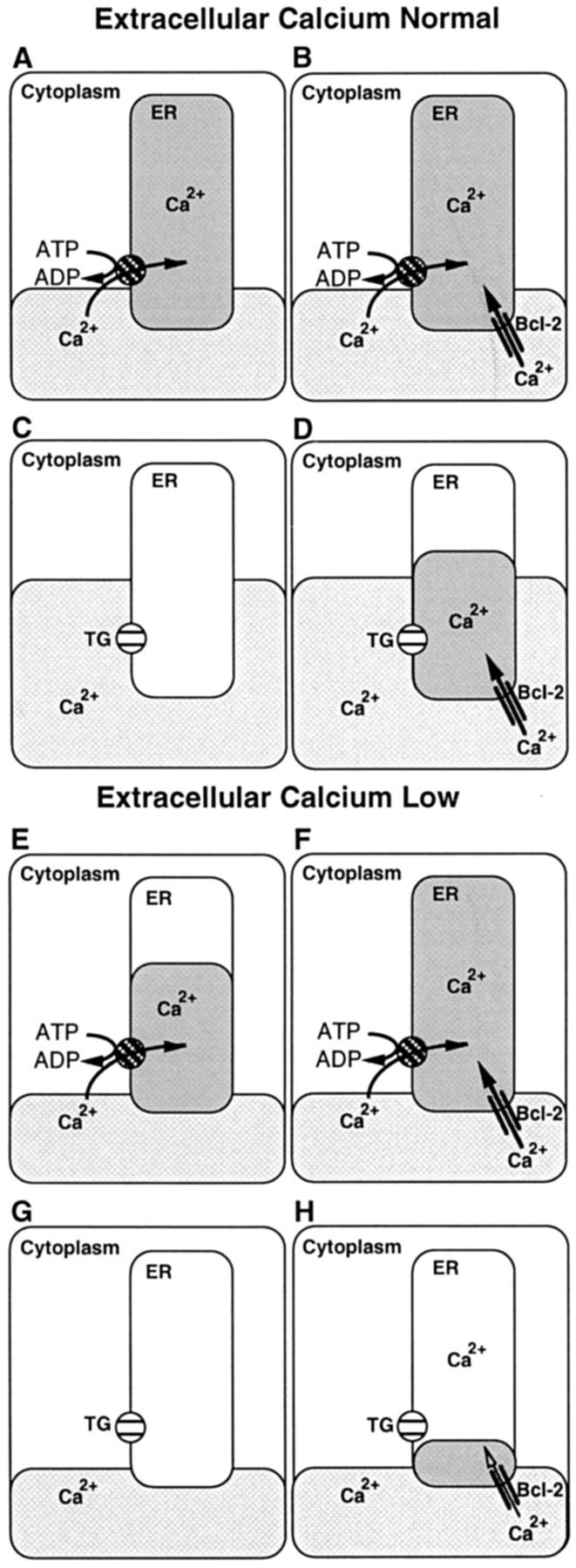
Model: Bcl-2 regulation of Ca2+ homeostasis in the ER. The relationship between Ca2+ pools in the cytoplasm and ER is illustrated. A–D represent cells cultured in medium containing a physiologic concentration of Ca2+ (1.3 mM), and E–H represent cells cultured in medium containing a low concentration of Ca2+ (0.13 mM). A, C, E, and G represent cells that lack Bcl-2, and B, D, F, and H represent cells that express Bcl-2. A, B, E, and F represent untreated cells in which Ca2+ is pumped into the ER by the Ca2+-ATPase, and C, D, G, and H represent cells in which the Ca2+-ATPase pump is inhibited by TG. Bcl-2 is represented by two parallel lines anchored in the ER membrane.
It should be noted that 45Ca2+ uptake measurements may underestimate Bcl-2–mediated Ca2+ entry because in the assay, free Ca2+ concentration is buffered to 150 nM to prevent uptake of Ca2+ by low-affinity pools (i.e., mitochondria). When cells are treated with TG in the presence of a physiologic concentration of extracellular Ca2+, cytosolic concentration is much higher than 150 nM because of capacitative entry of extracellular Ca2+ (Fig. 6). An elevated concentration of Ca2+ in the cytoplasm appears to be required for inhibition of TG-induced apoptosis by Bcl-2. This conclusion is based on evidence that TG induces apoptosis in cells that overexpress Bcl-2 if extracellular calcium is low, thereby preventing sustained elevation of cytosolic Ca2+ in response to capacitative entry. Thus, in the model Bcl-2 maintains at least partial ER Ca2+ pool filling when cells are treated with TG in the presence of a physiologic extracellular calcium concentration (Fig. 7 D) but is unable to maintain a level of Ca2+ in the ER necessary to inhibit apoptosis when cells are treated with TG in the presence of low extracellular calcium (Fig. 7 H).
The concentration of Ca2+ in the ER is difficult to quantitate directly. We have attempted to measure the ER Ca2+ pool with high-affinity fluorescent dyes (i.e., Mag-Fura-2 AM) and by electron probe microanalysis without success. Therefore, in the absence of a facile method for directly quantitating ER Ca2+ concentration, we have resorted to a functional assay to indirectly assess ER Ca2+ pool filling, based on previous evidence that protein processing within the ER lumen is inhibited by TG-induced ER Ca2+ pool depletion (25). Consistent with the hypothesis that Bcl-2 maintains Ca2+ homeostasis in the ER, we find that Bcl-2 overexpression prevents the TG-imposed delay in cathepsin D processing in the ER (Fig. 3).
ER Ca2+ pool filling is often estimated according to the ability of TG to dump Ca2+ from the ER lumen into the cytoplasm, producing a measurable elevation of cytosolic Ca2+. This method was used in Fig. 4 to measure the effect of extracellular Ca2+ withdrawal on the ER Ca2+ pool of untreated cells, indicating that the ER Ca2+ pool is diminished in cells that lack Bcl-2 (Fig. 7 E) but maintained in cells that overexpress Bcl-2 (Fig. 7 F). A recent study has reported that TG fails to induce a detectable elevation of cytosolic Ca2+ in Bcl-2–expressing cells that were treated with the nonselective Ca2+ ionophore ionomycin in the absence of extracellular Ca2+ (41). Based on this observation, the authors proposed that intracellular Ca2+ stores are not required for inhibition of apoptosis by Bcl-2 (41). However, ionomycin does not inhibit the Ca2+-ATPase pump and the failure to detect a mobilizable Ca2+ pool does not exclude the possibility that Ca2+-ATPase–mediated and/or Bcl-2–mediated Ca2+ uptake maintains a concentration of Ca2+ within the ER lumen necessary for cell survival. Moreover, the concept that Bcl-2 maintains cell survival in the absence of intracellular Ca2+ stores is improbable, in view of the requirement for Ca2+ to maintain the structural and functional integrity of the ER (3, 42).
The proposed effect of Bcl-2 on ER Ca2+ homeostasis, as conceptualized in Fig. 7, appears to correlate with maintenance of cell growth and viability in cells that overexpress Bcl-2. For example, the diminution in the ER Ca2+ pool induced by extracellular Ca2+ withdrawal (Fig. 4) is associated with growth inhibition in cells that lack Bcl-2 (Fig. 5 A), consistent with evidence that ER Ca2+ pool depletion inhibits cell division (44). In cells that overexpress Bcl-2, the ER Ca2+ pool is preserved when extracellular Ca2+ is withdrawn (Fig. 4), consistent with evidence that cell growth is maintained under these conditions (Fig. 5 C). In cells that overexpress Bcl-2, TG treatment in the presence of a physiologic extracellular Ca2+ concentration induces growth inhibition (Fig. 5 G), consistent with the hypothesis that Bcl-2 maintains partial filling of the ER Ca2+ pool (Fig. 7 G). However, as discussed above, TG treatment induces apoptosis when cells that overexpress Bcl-2 are suspended in low Ca2+ medium (Fig. 5 H), suggesting that extracellular Ca2+ is required for maintenance of ER Ca2+ homeostasis by Bcl-2 when the ER Ca2+-ATPase is inhibited by TG.
Mechanism of Bcl-2–mediated Ca2+ Uptake
The mechanism by which Bcl-2 mediates Ca2+ uptake in the ER is unknown. Bcl-2 does not appear to prevent Ca2+-ATPase inhibition (Fig. 1; reference 20) or to increase the level of Ca2+-binding proteins in the ER (Fig. 2). One theory is that Bcl-2 decreases oxidative damage to the ER membrane, thereby decreasing the leak of Ca2+ through the ER and increasing the Ca2+-sequestering capacity of the ER. This theory is consistent with evidence that the ER is a prime target of oxidative damage that depletes the ER Ca2+ pool (36) and with evidence that Bcl-2 overexpression reduces oxidative damage to membranes (16). Also, this hypothesis is consistent with evidence that Bcl-2 overexpression preserves ER Ca2+ pool filling when cells are treated with hydrogen peroxide (11). A second theory is that Bcl-2 functions as a cation channel that transports Ca2+ into the ER lumen, or alternatively as an anion channel that draws Ca2+ into the ER by producing an electrochemical potential across the ER membrane. This concept is supported by recent evidence of a structural similarity between Bcl-XL and bacterial toxins that form ion channels in cell membranes (33). One such toxin, the diphtheria toxin, appears to undergo a pH- and temperature-dependent conformational change by which it becomes hydrophobic and inserts into the cell membrane (26). It is possible that Bcl-XL might undergo a similar conformational change and insert through the ER membrane, the perinuclear membrane, or the mitochondrial membrane, thereby regulating ion homeostasis within these organelles. A recent study indicates that Bcl-XL forms an ion channel in synthetic lipid membranes (32), providing direct evidence that Bcl-2 family members are indeed involved in ion homeostasis.
General Conclusion
Bcl-2 inhibits apoptosis in response to a wide variety of signals. Furthermore, Bcl-2 is localized not only to the ER but also to the perinuclear membrane and the outer mitochondrial membrane. Each of these locations plays a role in inhibiting apoptosis, depending on the apoptosis-inducing signal (50). By regulating ion fluxes, Bcl-2 could act at different locations to regulate diverse cell death signals. Thus, while an effect of Bcl-2 on Ca2+ uptake at the level of the ER might inhibit TG-induced apoptosis, regulating ion fluxes in mitochondria, or both the ER and mitochondria, might inhibit apoptosis in response to other signals. The possibility that Bcl-2 might affect diverse cellular functions by regulating ion fluxes is illustrated by evidence that perinuclear Ca2+ concentration regulates nuclear pore activity (38). Hence, the ability of Bcl-2 to regulate ion fluxes through the ER membrane and contiguous perinuclear membrane might regulate nuclear translocation of transcription factors that control cell growth and viability.
Acknowledgments
We thank Gabriel Nunez for the pSFFV-neo vector; Diane Dowd (St. Louis University, St. Louis, MO), Roger Miesfeld, David Askew (both from University of Arizona, Tucson, AZ), and John Cidlowski (National Institute of Environmental Health Sciences, Bethesda, MD), for cell lines; and Richard Eckert for Ca2+-free medium. We are grateful to Stuart Kornfeld (Washington University, St. Louis, MO) for providing cathepsin D antibodies. We thank Manjunatha Bhat for Ca2+ electrode measurements. We are grateful to Michael Simonson and Edmunds Reineks for critically reading the manuscript, and George Dubyak for helpful suggestions regarding calcium measurements.
Footnotes
Address all correspondence to Clark W. Distelhorst, Department of Medicine, Biomedical Research Building Room 329, Case Western Reserve University School of Medicine, 10900 Euclid Avenue, Cleveland, OH 44106-4937. Tel.: (216) 368-1176. Fax: (216) 368-1166. e-mail: cwd@po.cwru.edu
1. Abbreviation used in this paper: TG, thapsigargin.
References
- 1.Baffy G, Miyashita T, Williamson JR, Reed JC. Apoptosis induced by withdrawal of interleukin-3 (IL-3) from an IL-3-dependent hematopoietic cell line is associated with repartitioning of intracellular calcium and is blocked by enforced Bcl-2 oncoprotein production. J Biol Chem. 1993;268:6511–6519. [PubMed] [Google Scholar]
- 2.Bian X, Hughes FM, Huang Y, Cidlowski JA, Putney JW. Roles of cytoplasmic Ca2+ and intracellular Ca2+stores in induction and suppression of apoptosis in S49 cells. Am J Physiol. 1997;272:C1241–C1249. doi: 10.1152/ajpcell.1997.272.4.C1241. [DOI] [PubMed] [Google Scholar]
- 3.Booth C, Koch GLE. Perturbation of cellular calcium induces secretion of luminal ER proteins. Cell. 1989;59:729–737. doi: 10.1016/0092-8674(89)90019-6. [DOI] [PubMed] [Google Scholar]
- 4.Brostrom CO, Brostrom MA. Calcium-dependent regulation of protein synthesis in intact mammalian cells. Annu Rev Physiol. 1990;52:577–590. doi: 10.1146/annurev.ph.52.030190.003045. [DOI] [PubMed] [Google Scholar]
- 5.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chueh S-H, Mullaney JM, Ghosh TK, Zachary AL, Gill DL. GTP- and inositol 1,4,5-trisphosphate-activated intracellular calcium movements in neuronal and smooth muscle cell lines. J Biol Chem. 1987;262:13857–13864. [PubMed] [Google Scholar]
- 7.Clapham DE. Calcium signaling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- 8.Cory S. Regulation of lymphocyte survival by the Bcl-2 gene family. Annu Rev Immunol. 1995;13:513–543. doi: 10.1146/annurev.iy.13.040195.002501. [DOI] [PubMed] [Google Scholar]
- 9.Deiss LP, Galinka H, Berissi H, Cohen O, Kimchi A. Cathepsin D protease mediates programmed cell death induced by interferon-γ, Fas/APO-1 and TNF-α. EMBO (Eur Mol Biol Organ) J. 1996;15:3861–3870. [PMC free article] [PubMed] [Google Scholar]
- 10.Distelhorst CW, McCormick TS. Bcl-2 acts subsequent to and independent of calcium fluxes to inhibit apoptosis in thapsigargin- and glucocorticoid-treated mouse lymphoma cells. Cell Calcium. 1996;19:473–483. doi: 10.1016/s0143-4160(96)90056-1. [DOI] [PubMed] [Google Scholar]
- 11.Distelhorst CW, Lam M, McCormick TS. Bcl-2 inhibits hydrogen peroxide-induced ER Ca2+pool depletion. Oncogene. 1996;12:2051–2055. [PubMed] [Google Scholar]
- 12.Dowd DR, MacDonald PN, Komm BS, Haussler MR, Miesfeld RL. Stable expression of the calbindin-D28K complementary DNA interferes with the apoptotic pathway in lymphocytes. Mol Endocrinol. 1992;6:1843–1848. doi: 10.1210/mend.6.11.1336124. [DOI] [PubMed] [Google Scholar]
- 13.Faust PL, Wall DA, Perara E, Lingappa VR, Kornfeld S. Expression of human cathepsin D in Xenopusoocytes: phosphorylation and intracellular targeting. J Cell Biol. 1987;105:1937–1945. doi: 10.1083/jcb.105.5.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furuya Y, Lundmo P, Short AD, Gill DL, Isaacs JT. The role of calcium, pH, and cell proliferation in the programmed (apoptotic) death of androgen-independent prostatic cancer cells induced by thapsigargin. Cancer Res. 1994;54:6167–6175. [PubMed] [Google Scholar]
- 15.Gething M-J, Sambrook J. Protein folding in the cell. Nature (Lond) 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- 16.Hockenbery DM, Oltvai ZN, Yin X-M, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 17.Jacobson MD, Raff MC. Programmed cell death and Bcl-2 protection in very low oxygen. Nature (Lond) 1995;374:814–816. doi: 10.1038/374814a0. [DOI] [PubMed] [Google Scholar]
- 18.Jiang S, Chow SC, Nicotera P, Orrenius S. Intracellular Ca2+ signals activate apoptosis in thymocytes: studies using the Ca2+-ATPase inhibitor thapsigargin. Exp Cell Res. 1994;212:84–92. doi: 10.1006/excr.1994.1121. [DOI] [PubMed] [Google Scholar]
- 19.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Örd T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science (Wash DC) 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 20.Lam M, Dubyak G, Chen L, Nuñez G, Miesfeld RL, Distelhorst CW. Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+fluxes. Proc Natl Acad Sci USA. 1994;91:6569–6573. doi: 10.1073/pnas.91.14.6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lam M, Vimmerstedt LJ, Schlatter LK, Hensold JO, Distelhorst CW. Preferential synthesis of the 78-Kd glucose-regulated protein in glucocorticoid-treated S49 mouse lymphoma cells. Blood. 1992;79:3285–3292. [PubMed] [Google Scholar]
- 22.Li WW, Alexandre S, Cao X, Lee AS. Transcription of the grp78 promoter by Ca2+ depletion: a comparative analysis with A23187 and the endoplasmic reticulum Ca2+-ATPase inhibitor thapsigargin. J Biol Chem. 1993;268:12003–12009. [PubMed] [Google Scholar]
- 23.Li X, Lee AS. Competitive inhibition of a set of endoplasmic reticulum protein genes (GRP78, GRP94, and ERp72) retards cell growth and lowers viability after ionophore treatment. Mol Cell Biol. 1991;11:3446–3453. doi: 10.1128/mcb.11.7.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Little E, Lee AS. Generation of a mammalian cell line deficient in glucose-regulated protein stress induction through targeted ribozyme driven by a stress-inducible promoter. J Biol Chem. 1995;270:9526–9534. [PubMed] [Google Scholar]
- 25.Lodish HF, Kong N, Wikstrom L. Calcium is required for folding of newly made subunits of the asialoglycoprotein receptor within the endoplasmic reticulum. J Biol Chem. 1992;267:12753–12760. [PubMed] [Google Scholar]
- 26.London E. Diptheria toxin: membrane interaction and membrane translocation. Biochim Biophys Acta. 1992;1113:25–51. doi: 10.1016/0304-4157(92)90033-7. [DOI] [PubMed] [Google Scholar]
- 27.Lytton J, Westlin M, Hanley MR. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem. 1991;266:17067–17071. [PubMed] [Google Scholar]
- 28.Magnelli L, Cinelli M, Turchetti A, Chiarugi VP. Bcl-2 overexpression abolishes early calcium waving preceding apoptosis in NIH-3T3 murine fibroblasts. Biochem Biophys Res Commun. 1994;204:84–90. doi: 10.1006/bbrc.1994.2429. [DOI] [PubMed] [Google Scholar]
- 29.Marin MC, Fernandez A, Bick RJ, Brisbay S, Buja LM, Snuggs M, McConkey DJ, von Eschenbach AC, Keating MJ, McDonnell TJ. Apoptosis suppression by bcl-2 is correlated with the regulation of nuclear and cytosolic Ca2+ . Oncogene. 1996;12:2259–2266. [PubMed] [Google Scholar]
- 30.Maruyama K, Mikawa T, Ebashi S. Detection of calcium binding proteins by 45Ca autoradiography on nitrocellulose membrane after sodium dodecyl sulfate gel electrophoresis. J Biochem. 1984;95:511–519. doi: 10.1093/oxfordjournals.jbchem.a134633. [DOI] [PubMed] [Google Scholar]
- 31.McCormick TS, McColl KS, Distelhorst CW. Mouse lymphoma cells destined to undergo apoptosis in response to thapsigargin treatment fail to generate a calcium mediated grp78/grp94 stress response. J Biol Chem. 1997;272:6087–6092. doi: 10.1074/jbc.272.9.6087. [DOI] [PubMed] [Google Scholar]
- 32.Minn AJ, Velez P, Schendel SL, Liang H, Muchmore SW, Fesik SW, Fill M, Thompson CB. Bcl-xL forms an ion channel in synthetic lipid membranes. Nature (Lond) 1997;385:353. doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- 33.Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong S-L, Ng S-C, Fesik SW. X-ray and NMR structure of human Bcl-XL, an inhibitor of programmed cell death. Nature (Lond) 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- 34.Murphy AN, Bredesen DE, Cortopassi G, Wang E, Fiskum G. Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc Natl Acad Sci USA. 1996;93:9893–9898. doi: 10.1073/pnas.93.18.9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muschel RJ, Bernhard EJ, Garza L, McKenna WG, Koch CJ. Induction of apoptosis at different oxygen tensions: evidence that oxygen radicals do not mediate apoptotic signaling. Cancer Res. 1995;55:995–998. [PubMed] [Google Scholar]
- 36.Orrenius S, Burkitt MJ, Kass GEN, Dypbukt JM, Nicotera P. Calcium ions and oxidative cell injury. Ann Neurol. 1992;32:S33–S42. doi: 10.1002/ana.410320708. [DOI] [PubMed] [Google Scholar]
- 37.Peitsch MC, Polzar B, Stephan H, Crompton T, MacDonald HR, Mannherz HG, Tschopp J. Characterization of the endogenous deoxyribonuclease involved in nuclear DNA degradation during apoptosis (programmed cell death) EMBO (Eur Mol Biol Organ) J. 1993;12:371–377. doi: 10.1002/j.1460-2075.1993.tb05666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez-Terzic C, Pyle J, Jaconi M, Stehno-Bittel L, Clapham DE. Conformational states of the nuclear pore complex induced by depletion of nuclear Ca2+stores. Science (Wash DC) 1996;273:1875–1877. doi: 10.1126/science.273.5283.1875. [DOI] [PubMed] [Google Scholar]
- 39.Putney JW. Capacitative calcium entry revisited. Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- 40.Putney JW, Bird GSJ. The signal for capacitative calcium entry. Cell. 1993;75:199–201. doi: 10.1016/0092-8674(93)80061-i. [DOI] [PubMed] [Google Scholar]
- 41.Reynolds JE, Eastman A. Intracellular calcium stores are not required for Bcl-2-mediated protection from apoptosis. J Biol Chem. 1996;271:27739–27743. doi: 10.1074/jbc.271.44.27739. [DOI] [PubMed] [Google Scholar]
- 42.Sambrook JF. The involvement of calcium in transport of secretory proteins from the endoplasmic reticulum. Cell. 1990;61:197–199. doi: 10.1016/0092-8674(90)90798-j. [DOI] [PubMed] [Google Scholar]
- 43.Shimizu S, Eguchi Y, Kosaka H, Kamiike W, Matsuda H, Tsujimoto Y. Prevention of hypoxia-induced cell death by Bcl-2 and Bcl-xL. Nature (Lond) 1995;374:811–813. doi: 10.1038/374811a0. [DOI] [PubMed] [Google Scholar]
- 44.Short AD, Bian J, Ghosh TK, Waldron RT, Rybak SL. Intracellular Ca2+pool content is linked to control of cell growth. Proc Natl Acad Sci USA. 1993;90:4986–4990. doi: 10.1073/pnas.90.11.4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsien R, Pozzan R. Measurement of cytosolic free Ca2+with Quin2. Methods Enzymol. 1989;172:230–262. doi: 10.1016/s0076-6879(89)72017-6. [DOI] [PubMed] [Google Scholar]
- 47.Waldron RT, Short AD, Gill DL. Thapsigargin-resistant intracellular calcium pumps: role in calcium pool function and growth of thapsigargin-resistant cells. J Biol Chem. 1995;270:11955–11961. doi: 10.1074/jbc.270.20.11955. [DOI] [PubMed] [Google Scholar]
- 48.Yang E, Korsmeyer SJ. Molecular thanatopsis: a discourse on the BCL2 family and cell death. Blood. 1996;88:386–401. [PubMed] [Google Scholar]
- 49.Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Migotte B, Kroemer G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182:367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, Andrews DW. Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO (Eur Mol Biol Organ) J. 1996;15:4130–4141. [PMC free article] [PubMed] [Google Scholar]