

Abstract
kar9 was originally identified as a bilateral karyogamy mutant, in which the two zygotic nuclei remained widely separated and the cytoplasmic microtubules were misoriented (Kurihara, L.J., C.T. Beh, M. Latterich, R. Schekman, and M.D. Rose. 1994. J. Cell Biol. 126:911–923.). We now report a general defect in nuclear migration and microtubule orientation in kar9 mutants. KAR9 encodes a novel 74-kD protein that is not essential for life. The kar9 mitotic defect was similar to mutations in dhc1/dyn1 (dynein heavy chain gene), jnm1, and act5. kar9Δ dhc1Δ, kar9Δ jnm1Δ, and kar9Δ act5Δ double mutants were synthetically lethal, suggesting that these genes function in partially redundant pathways to carry out nuclear migration. A functional GFP-Kar9p fusion protein localized to a single dot at the tip of the shmoo projection. In mitotic cells, GFP-Kar9p localized to a cortical dot with both mother–daughter asymmetry and cell cycle dependence. In small-budded cells through anaphase, GFP-Kar9p was found at the tip of the growing bud. In telophase and G1 unbudded cells, no localization was observed. By indirect immunofluorescence, cytoplasmic microtubules intersected the GFP-Kar9p dot. Nocodazole experiments demonstrated that Kar9p's cortical localization was microtubule independent. We propose that Kar9p is a component of a cortical adaptor complex that orients cytoplasmic microtubules.
The nucleus in the yeast Saccharomyces cerevisiae migrates to distinct regions within the cell during different phases of the life cycle, mating, and mitosis. Each type of nuclear migration is dependent upon cytoplasmic microtubules. The cytoplasmic microtubules are attached to the nucleus at the spindle pole body (SPB),1 the microtubule organizing center in yeast. The SPB is embedded in the nuclear envelope, which remains intact at all stages of the yeast life cycle (Byers, 1981).
In preparation for mating, the yeast cell arrests in G1 and forms a projection—called a shmoo projection—in response to mating pheromone. The nucleus moves to the base of the shmoo neck and the cytoplasmic microtubules extend from the SPB to the tip of the shmoo (Byers and Goetsch, 1974; Rose and Fink, 1987; Read et al., 1992). Two shmoos of opposite mating type fuse to form a zygote and the intervening cell walls break down (Byers and Goetsch, 1975). The cytoplasmic microtubules can then interdigitate, and the nuclei are drawn together in a microtubule-dependent manner by the Kar3p kinesin-like motor protein. Karyogamy, or nuclear fusion, then ensues (Meluh and Rose, 1990). The process of nuclear and cell fusion has recently been reviewed (Rose, 1996; Marsh and Rose, 1997).
Nuclear migrations also occur during mitotic divisions in yeast. At the end of G2 phase the nucleus moves up to the neck, between the mother and bud (Pringle and Hartwell, 1981). The nucleus then elongates quickly, coincident with (Yeh et al., 1995), or just before (Kahana et al., 1995) translocation of the sausage-shaped nucleus into the neck. The sausage-shaped nucleus then undergoes a rapid set of oscillations across the bud neck (Yeh et al., 1995). The function of these oscillations remains unknown, but may correspond to the “DNA transits” observed by others (Palmer et al., 1989). The nucleus then undergoes a slower phase of elongation. It takes on an hour-glass shape until the two lobes of the nucleus are located at the distal poles of the mother and bud. After a brief pause, each of the nuclei returns to the center of its respective cell. Cytokinesis then follows (Yeh et al., 1995). In cells undergoing axial budding, the nucleus then reorients such that the SPB faces the site of new bud emergence (Byers and Goetsch, 1975; Snyder et al., 1991).
The cytoplasmic microtubules are required for most, if not all, nuclear migrations (Sullivan and Huffaker, 1992). Specific depolymerization of the cytoplasmic microtubules using a cold-sensitive allele of β-tubulin, TUB2-401, results in a massive failure of nuclear migration. The nuclear division cycle as well as the budding cycle continue in the absence of nuclear migration into the growing bud. This results in a multinucleated mother cell surrounded by a rosette of anucleate buds (Sullivan and Huffaker, 1992).
Another gene that plays a critical role in nuclear migration is the minus end–directed microtubule motor protein, cytoplasmic dynein, DHC1/DYN1 (Li et al., 1993; Eshel et al., 1993). Deletion of dynein from the yeast cell results in the failure of the nucleus to fully migrate to the bud neck, with spindle elongation occurring entirely within the mother cell (Li et al., 1993). Interestingly, the nuclear oscillations that occur in the neck of wild-type cells are absent in dynein mutant strains. One model for dynein function in yeast is that it exerts a pulling force on the cytoplasmic microtubules, perhaps through an attachment to the cell surface (Li et al., 1993; Eshel et al., 1993). Indeed, such a cortical localization is found for cytoplasmic dynein in the filamentous fungus, Aspergillus nidulans (Xiang et al., 1995b ).
Several other genes have been implicated in the process of nuclear migration, including ACT5 and JNM1. Whereas neither gene is essential, both mutants have nuclear migration phenotypes strikingly similar to that of dynein heavy chain mutants (Clark and Meyer, 1994; McMillan and Tatchell, 1994; Muhua et al., 1994). ACT5 is the yeast homologue of the Arp1 component of the vertebrate dynactin complex (Clark and Meyer, 1994; Muhua et al., 1994). Jnm1p has also been suggested to be an auxiliary subunit for cytoplasmic dynein (McMillan and Tatchell, 1994; Geiser et al., 1997). Consistent with this hypothesis, the double mutant, dhc1 act5, is not significantly worse for nuclear migration than either single mutant alone (Muhua et al., 1994). These data indicate that Dhc1p and Act5p function in the same pathway to promote nuclear migration (Muhua et al., 1994). Another protein, Num1p, also affects nuclear migration, possibly affecting cytoplasmic microtubule functions. It localizes primarily to the mother cell cortex and is expressed in a cell cycle–dependent manner (Farkasovsky and Kuntzel, 1995).
Nuclear migration has been extensively characterized in the filamentous fungi Aspergillus nidulans and Neurospora crassa. Nuclei migrate down the germ tube of these filamentous fungi before hyphal segmentation. As in Saccharomyces cerevisiae, the nuclear movements require microtubules, cytoplasmic dynein, and dynactin components (Plamann et al., 1994; Xiang et al., 1995b ). Three other Aspergillus genes have also been identified that are also required for nuclear migration: nudG (Willins et al., 1995); nudF (Xiang et al., 1995 a), a gene with homology to the human LIS-1 gene; and nudC (Osmani et al., 1990), which controls the level of the NUDF protein.
In S. cerevisiae, Kurihara et al. (1994) have shown that in kar9 mutant zygotes the cytoplasmic microtubules are misoriented and that the nuclei fail to migrate together. Thus the two haploid nuclei in kar9 zygotes remain widely separated (Kurihara et al., 1994). In this paper, we investigated the function of Kar9p during the mitotic and early mating stages of the yeast life cycle. In both instances, we found nuclear migration and microtubule orientation defects in kar9. Genetic interactions between KAR9 and DHC1/DYN1 mutations indicated that KAR9 functions in a nuclear migration pathway that is at least partially redundant with the dynein pathway. A fusion of green fluorescent protein (GFP) to Kar9p localized to a cortical region at the tip of the bud and at the tip of the shmoo projection in a microtubule-independent fashion. We propose that Kar9p functions as a cytoplasmic tether or adaptor linking the cytoplasmic microtubules to a specific site at the bud cortex.
Materials and Methods
Strains and Cell Culture
The yeast strains used in this study are listed in Table I. Bacterial strains and plasmids are listed in Table II. Yeast cells were cultured by standard methods as described previously (Rose et al., 1990). To prepare shmoos, cells were arrested with α factor (Syn/Seq facility; Princeton University, Princeton, NJ) at 10 μg/ml for 90 or 180 min in yeast peptone dextrose (YPD) media. The longer arrest time was used when induction of GFP-Kar9p was also required.
Table I.
List of Yeast Strains Used in This Study
| Strain | Genotype | Source | ||
|---|---|---|---|---|
| MS52 | MATα leu2-3 leu2-112 trp1-Δ1 ura3-52 | Rose | ||
| MS810 | MAT a/α ura3-52/ura3-52 ade2-101/+ leu2-3 leu2-112/leu2-3 leu2-112 trp1-Δ1/+ | Rose | ||
| MS1556 | MAT a ura3-53 leu2-3 leu2-112 ade-2-101 his3-Δ200 | Rose | ||
| MS1614 | MATα kar3-Δ::LEU2 ura3-52 leu2-3 leu2-112 his3-Δ200 ade2-101 {KAR3 URA3 CEN4 ARS1} | Rose | ||
| MS2305 | MAT a kip1-1::URA3 ura3-52 leu2-3 leu2-112 trp1-Δ1 | Rose | ||
| MS2684 | matΔ::LEU2 kar9-485 ura3-52 leu2-3 leu 2-112 trp1-Δ1 his3::TRP1 | Rose | ||
| MS3662 | MAT a kar9-485 leu2-3 leu2-112 ura3-52 | This study | ||
| MS4062 | MATα kar9-Δ1::LEU2 ura3-52 leu2-3 leu2-112 trp1-Δ1 | This study | ||
| MS4262 | MATα dhc1-Δ::URA leu2-3 leu2-112 trp1-Δ1 ura3-52 | This study | ||
| MS4263 | MAT a kar9-Δ1::LEU2 ura3-52 leu2-3 leu2-112 ade2-101 his3-Δ200 | This study | ||
| MS4306 | MAT a kar9-Δ2::HIS3 ura3-53 leu2-3 leu2-112 ade2-101 his3-Δ200 | This study | ||
| MS4313 | MAT a kar9-Δ1::LEU2 leu2-3 leu2-112 ura3-52 trp1-Δ1 | This study | ||
| MS4316 | MATα kar9-Δ1::LEU2 ura3-52 leu2-3 leu2-112 ade2-101 his3-Δ200 | This study | ||
| MS4321 | MAT a jnm1::LEU2 ura3-52 leu2-3 leu2-112 ade2-101 his3-Δ200 | This study | ||
| MS4382 | MAT a kar9-Δ2::HIS3 ura3-53 leu2-3, leu2-112 ade2-101 his3-Δ200 {pMR3465} | This study | ||
| MS4387 | MAT a ura3-52 leu2-3 leu2-112 ade2-101 his3-Δ200 {pMR3465} | This study | ||
| MS4516 | MAT a kip3-Δ::HIS3 ura3-52 leu2-3 leu2-112 ade2-101 his3-Δ200 | Rose | ||
| MS4586 | MATα act5-Δ1::HIS3 ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 | This study | ||
| MS4587 | MATα kar9-Δ2::HIS3 ura3-53 leu2-3 leu2-112 his3-Δ200 trp1-Δ1 | This study | ||
| MS4589 | MAT a kar9-Δ1::LEU2 ura3-52 leu2-3 leu2-112 ade2-101 his3-Δ200 | This study | ||
| MS4671 | MATα leu2-3 leu2-112 trp1-Δ1 ura3-52 {pMR3465} | This study | ||
| MS4683 | MAT a dhc1Δ::URA trp1-Δ1 leu2-3, leu2-112 ura3-52 | This study | ||
| MS4734 | MATα bik1::TRP leu2-3, leu2-112 trp1-Δ1 ura3-52 | This study | ||
| MS4903 | MAT a dhc1-Δ::LEU2 ura3-52 leu2-3 leu2-112 ade2-101 his3-Δ200 | This study | ||
| APY4ΔD5 | MAT a smy1-Δ::URA leu2-3 leu2-112 ura3-52 trp1 ade2 his6 | Lillie | ||
| MAY2058 | MATα cin8::LEU2 leu2-3 leu2-112 his3-Δ200 ura3-52 lys2-801 | Hoyt | ||
| YJC1159 | MATα act5-Δ1::HIS3 ura3-52 lys2-801 ade2-101 trp1-Δ63 his3-Δ200 leu2-Δ1 | Cooper |
Table II.
List of Plasmids Used in This Study
| Strain | Genotype/Description | Source | ||
|---|---|---|---|---|
| pMR2254 | pUC18 carrying the LEU2 yeast selectable marker | Rose | ||
| pMR3143 | KAR9 HIS3 CEN6 ARS AMPR | Rose | ||
| pMR3381 | KAR9 AMPR URA3 integrating plasmid | This study | ||
| pMR3382 | kar9-Δ1::LEU2 URA3 AMPR | This study | ||
| pMR3386 | kar9-Δ2::HIS3 URA3 AMPR | This study | ||
| pMR3465 | PGAL1-GFP-KAR9 CEN4 ARS1 LEU2 AMPR | This study | ||
| pMR2886 | 2.1-kb SpeI-SpeI fragment cloned into pRS406 | This study | ||
| pB886 | plasmid carrying the HIS3 yeast-selectable marker | Broach | ||
| pB1893 | PGAL1-GFP CEN4 ARS1 LEU2 AMPR | Broach | ||
| pRS406 | URA3 AMPR integrating plasmid | Sikorski | ||
| pJM1432 | jnm1::LEU2 disruption plasmid | Tatchell | ||
| pBR2-1U | dhc1::URA3 disruption plasmid | Bloom | ||
| pMR3861 | dhc1::LEU2 disruption plasmid | Bloom | ||
| pVB17 | bik1::TRP disruption plasmid | Fink |
Fluorescence and Immunological Techniques
Morphological analysis of cells and shmoos by 4′,6-diamidino-2-phenylindole (DAPI) staining was performed as described (Rose et al., 1990). For nuclear migration analyses (see below), cells and shmoos were fixed with methanol/acetic acid (3:1) on ice for 30 min, washed twice in PBS, and then stained with DAPI (1 μg/ml). Indirect immunofluorescence of microtubules was performed as described (Rose et al., 1990). Cytoplasmic microtubules were stained with rabbit anti–β tubulin (a gift from F. Solomon, Massachusetts Institute of Technology, Cambridge, MA) at 1:500 dilution or rat anti–α tubulin YOL 1/34 neat (Accurate Biochemical, Westbury, NY). Double-label immunofluorescence was carried out with rabbit anti-GFP (CLONTECH, Palo Alto, CA) at a dilution of 1:30 and goat anti–rabbit conjugated with FITC (Boehringer Mannheim, Indianapolis, IN) at a 1:55 dilution. Each antibody was applied serially to the cells for 24 h at 4°C in the following order: anti-GFP, anti-tubulin YOL1/34, and mixed secondaries of goat anti-rabbit–FITC and goat anti–rat conjugated with Texas red (Organon Teknika Corp., West Chester, PA). Both anti-GFP and its secondary antibody were preabsorbed against a formaldehyde-fixed, zymolyase-digested wild-type strain lacking the GFP-KAR9 plasmid. Microscopy was done on an Axiophot microscope equipped with a ×100 Neofluor lens (1.4NA) (Carl Zeiss, Inc., Thornwood, NY).
Isolation and Genetic Mapping of the KAR9 Gene
The wild-type KAR9 gene was cloned using a yeast genomic centromere-based library (Rose et al., 1987). 100,000 transformants were screened for suppression of kar9-485's benomyl sensitivity using plates containing 20 μg/ml of benomyl incubated at 30°C. The plasmids that complemented were recovered from yeast using the procedure of Hoffman and Winston (1987) and retransformed into Escherichia coli. Candidate plasmids were mapped by restriction enzyme digest analysis. Three independently isolated plasmids were found to overlap by 5.8 kb. These plasmids were retested to determine whether they suppressed both the benomyl sensitivity and the mitotic defects of kar9-485. An internal 1.4-kD SpeI-SpeI fragment in the overlap region was used to carry out physical mapping. Pulse field gel electrophoresis and Southern blots were carried out by the methods of Rose et al. (1990), and graciously provided by L.J. Kurihara (Princeton University, Princeton, NJ). λ prime clone blots were used by the methods of Riles et al. (1993).
Sequencing
Subclones used for complementation analysis (see Fig. 1) and several ExoIII nuclease deletion constructs (data not shown) were sequenced using the Sequenase Version 2.0 kit (United States Biochemical Corp., Cleveland, Ohio), and double-stranded templates. This sequence was verified by the S. cerevisiae genome project. KAR9 corresponds to open reading frame (ORF) No. YPL269W on chromosome XVI. Analysis of the DNA sequence was carried out using the GCG program (Wisconsin software package; Oxford Molecular Group, Inc., Oxford, UK). Analysis of potential coiled-coils regions was by the Coils program (Lupas et al., 1991).
Figure 1.

Map of KAR9 clones and kar9 deletions. The region of overlap between the two original genomic library plasmids is shown by the hatched bar. Subclones were constructed and tested for their ability to suppress kar9's benomyl sensitivity (20 μg/ml at 30°C) and its nuclear migration defect, which was scored by DAPI. The integration plasmid (pMR2886), linearized with BglII, was used for linkage analysis. In the deletion vector (pMR3382), LEU2 replaces the KAR9 sequence from the BglII to the second SpeI site. In the deletion vector (pMR3386), HIS3 replaces the KAR9 sequence from the BglII sequence to BamHI. Numbering begins ∼100 bp upstream of the start of the MDL2-coding region. Restriction sites are represented as follows: S, SalI; BH, BamHI; Xh, XhoI; K, KpnI; Bg, BglII; P, PvuII; Sp, SpeI; Mu, MluI. Vector sequences are represented by thin lines.
Linkage Analysis
To construct an integration vector for use in linkage analysis, we cloned the 2.1-kb SpeI-SpeI fragment of this region into a URA3-containing integration vector pRS406 to create pMR2886 (see Fig. 1). This was cleaved with BglII, and integrated into the wild-type diploid, MS810, thus marking the wild-type KAR9 locus with URA3. After sporulation, a colony with the URA3-marked KAR9 locus was mated to a kar9-485 strain (MS3662), sporulated, and then tetrads were dissected. Of 32 tetrads analyzed, the URA3-marked wild-type KAR9 locus segregated together with benomyl resistance in 31 tetrads. The remaining tetrad segregated 2:2 for benomyl resistance, but contained three Ura− spores, two of which were benomyl sensitive, and one of which was benomyl resistant. The Ura− benomyl-resistant spore most likely represents a gene conversion event or a plasmid loop-out event leading to loss of the URA3 plasmid.
Construction of the KAR9 Deletion Strains
Two null alleles of KAR9 were constructed (see Fig. 1). For kar9-Δ1:: LEU2, amino acids 7–644 were deleted and replaced with LEU2. This nearly complete deletion of KAR9 was constructed by cutting the KAR9-containing plasmid, MS3381, with BglII and SpeI, and then replacing the deleted KAR9 fragment with an XbaI/BamHI fragment containing the LEU2 gene from the YIp vector, pMR2254. This yielded the kar9-Δ1:: LEU2 deletion plasmid, pMR3382. kar9-2Δ::HIS3, which replaces amino acids 7–574, was constructed by cutting pMR3381 with BglII and BamHI and replacing it with a BamHI/BamHI fragment containing the HIS3 gene from B886 (from J. Broach, Princeton University). This created the kar9-Δ2::HIS3 disruption plasmid, pMR3386. Both the kar9-Δ1::LEU2 and the kar9-Δ2::HIS3 deletion mutations were introduced into the genomic locus of the haploid strains by the one-step gene replacement method of Rothstein (1983). A 2.6-kb fragment liberated by SwaI digestion of pMR3382 and pMR3386 was gel purified and transformed into the wild-type strains, MS52 and MS1556, to create strains MS4062, MS4263, and MS4306. Whereas both deletions leave the NH2-terminal six amino acids of Kar9p intact, this small fragment is unlikely to be functionally significant. The kar9-Δ1::LEU2 and kar9-Δ2::HIS3 deletions were confirmed by Southern blot analysis and diagnostic PCR analysis, respectively. Both the kar9-Δ1:: LEU2 and the kar9-Δ2::HIS3 mutations in yeast result in benomyl sensitivity and mitotic defects identical to those described for the original point mutant, kar9-485. There was no difference in the growth rate between kar9Δ and wild-type strains.
Construction of Mutants
dhc1Δ.
The dhc1Δ::URA disruption (Li et al., 1993) was made in an isogenic strain background by the one-step gene replacement method (Rothstein, 1983). The plasmid pBR2-1U (Li et al., 1993) was cleaved with EcoRI, the 2.2-kb fragment was gel purified, and transformed into the wild-type strain, MS52, to create MS4262. Consistent with the creation of the proper disruption, the dhc1Δ::URA segregated 2:2 in crosses to wild type. The dhc1Δ::URA disruption strain was benomyl sensitive (20 μg/ml at 30°C), and also exhibited the same array of mitotic spindle orientation defects as expected from previous reports (Eshel et al., 1993; Li et al., 1993).
jnm1Δ.
The jnm1Δ disruption was made in a strain isogenic with kar9Δ by the one-step gene replacement method (Rothstein, 1983). pJM1432 (McMillan and Tatchell, 1994) was digested with XbaI and the 4.8-kb fragment was gel purified. Transformation of this fragment into MS1556 produced the jnm1 deletion strain, MS4321.
act5Δ.
The act5Δ::HIS3 disruption strain, YJC1159 (Muhua et al., 1994), was back-crossed to MS1556 yielding the act5Δ strain, MS4586.
bik1Δ.
pVB17 was digested with SnaBI and EcoRI to liberate a 3.3-kb fragment. This was transformed into MS52 to create the bik1::TRP1 strain, MS4734, by the one-step gene replacement method.
Construction of GFP-KAR9
To construct a GFP-Kar9p fusion protein, the KAR9 gene was synthesized with SalI ends by PCR using the following primers:
KAR9-NH2 5′-AC GCG TCG ACC ATG GAT AAT GAT GG-3′ and
KAR9-COOH 5′-GCG TGT CGA CTC AAT AAG TTG GGG-3′.
The resulting PCR product was cleaved with SalI and cloned in frame at the SalI site of the centromere based PGAL1-GFP vector, B1893, to create the PGAL1-GFP–KAR9 construct, pMR3465. The centromere based PGAL1-GFP–cloning vector, B1893, was constructed by C. Davis (Princeton University) by combining the No. 1 GFP mutant of B. Cormack and S. Falkow (Cormack, 1996) and the YCpIF1 vector of Foreman and Davis (Foreman and Davis, 1994).
Cell Culture, Fixation Methods and Drug Treatments of GFP-Kar9p–expressing Strains
For induction of GFP-Kar9p by galactose, cells were grown to early exponential phase in synthetic complete minus leucine media containing 2% raffinose, 2% galactose, and either no glucose or 0.1% glucose. Growth curve analysis demonstrated that induction of GFP-KAR9 by addition of 2% galactose did not inhibit growth in kar9Δ strains (MS4306) or wild-type strains (MS1556) for as long as 10 h of induction. However, to produce lower expression levels more like that found in wild type, 0.1% glucose was added to the culture media. This resulted in the reduction of GFP-Kar9p expression levels by approximately fivefold as determined by Western blot analysis.
For GFP-Kar9p localization studies, the following conditions were used. For vegetatively growing cells the wild-type strain containing a GFP-Kar9p plasmid, MS4387, was grown to early exponential phase in synthetic complete minus leucine media containing 2% raffinose, 2% galactose, and 0.1% glucose. To visualize the nuclear material in these cells, cells were fixed in 3.7% formaldehyde for 5–10 min, washed three times in PBS, and stained with DAPI (1 μg/ml). For shmoos the kar9 delete stain, MS4382, was grown to early exponential phase in synthetic complete minus leucine media containing 2% raffinose. GFP-Kar9p expression was induced for 4 h with 2% galactose. After 1–2.5 h, α factor (10 μg/ml) was added to arrest the cells for 1.5–3 h. For zygotes, a and α wild-type cells were grown to early log phase in synthetic complete minus leucine media containing 2% raffinose (MS4387 and MS4671). Cells were induced for GFP-Kar9p expression for 2.5–3 h by the addition of galactose (2%), and then allowed to mate on YPD for 2–3 h. Shmoos and zygotes were fixed with 3.7% formaldehyde for 1 h, lightly spheroplasted with glusulase NEE-154 (DuPont-NEN, Boston, MA) and stained with DAPI (1 μg/ml).
For nocodazole treatments, a wild-type strain containing the GFP-Kar9p plasmid (MS4387), was grown to early exponential phase in synthetic complete minus leucine media containing 2% raffinose at 30°C. Nocodazole (Sigma Chemical Co.) was added to 15 μg/ml from a 1.5 mg/ml stock in DMSO. α Factor was added to 10 μg/ml from a 1 mg/ml stock in methanol. PGAL1-GFP–Kar9p expression was induced by the addition of galactose to a concentration of 2%. At the time of fixation, each treated culture was split and prepared either for indirect immunofluorescence with anti-tubulin or analysis of GFP-Kar9p localization with DAPI staining.
For hydroxyurea treatment, cells were grown to early exponential phase in synthetic complete minus leucine media containing 2% raffinose, 2% galactose, and 0.1% glucose. Hydroxyurea (Sigma Chemical Co.) was added to 100 mM for 3 h at 30°C. Cells were washed twice in prewarmed media and returned to 30°C. After 1 h 25 min, cells were fixed in formaldehyde (5 min), washed twice in PBS, and then scored for GFP-Kar9p localization. For all induction protocols, we observed no GFP-Kar9p fluorescent dots when galactose was not added to the media.
Results
KAR9 Is a Novel Gene
In a collection of karyogamy mutants defective for nuclear congression, kar9 was unique in exhibiting misoriented cytoplasmic microtubules in the zygote (Kurihara et al., 1994). In addition, we found that the kar9 mutants were sensitive to the microtubule-destabilizing drug, benomyl. Growth was significantly suppressed at 15 μg/ml at 30°C (data not shown). In contrast, wild type was resistant to 25 μg/ml benomyl. As described below, kar9 mutants also showed mitotic defects consistent with a defect in nuclear migration. The karyogamy defect, the benomyl sensitivity, and the mitotic defect segregated together in five tetrads derived from meiotic crosses. Therefore, we considered it highly likely that each of these phenotypes was the result of the kar9 mutation. These phenotypes also suggested that Kar9p might play a broader role in microtubule function. To investigate further, the wild-type KAR9 gene was isolated by suppression of kar9's benomyl sensitivity. A kar9-485 strain (MS2684) was transformed with a YCp-based yeast genomic library (Rose et al., 1987) and screened for plasmid-dependent benomyl resistance (20 μg/ml at 30°C). Three independent plasmids were recovered that suppressed both the benomyl sensitivity and mitotic defects of kar9-485. These plasmids contained inserts that overlapped by 5.8 kb (Fig.1). To identify the minimal complementing region, additional subclones were constructed and tested for their ability to complement kar9-485's benomyl sensitivity and mitotic defects. Subclones containing all or almost all of the KAR9 ORF corrected the benomyl sensitivity defect (Fig. 1). In contrast, two subclones truncated at the NH2 terminus only partially suppressed the mitotic defect.
To determine that the region on the three overlapping plasmid inserts contained the authentic KAR9 gene rather than an extragenic suppressor, the 2.1-kb SpeI/SpeI fragment within this region was used for linkage analysis (Fig. 1). Integration of this fragment on pMR2886 was used to mark the wild-type KAR9 locus with URA3. When crossed to a kar9 mutant, the marked allele segregated together with the benomyl resistance in 31 out of 32 tetrads analyzed (see Materials and Methods). These results indicate that the sequences contained on pMR2886 were tightly linked to and therefore derived from the authentic KAR9 gene.
Hybridization methods demonstrated that KAR9 mapped to the distal end of the left arm of chromosome XVI. DNA sequence analysis showed that KAR9 was between MDL2 and PLC1 (Fig. 1). This sequence was subsequently confirmed by the S. cerevisiae genome sequencing project. KAR9 potentially encoded a 644–amino acid, 74-kD protein (Fig. 2) that showed no significant homology to any known proteins in available databases using BLAST or FASTA programs (Pearson and Lipman, 1988). Kar9p is predicted to be a basic protein with an overall calculated average isoelectric point (pI) of 8.9. However, the NH2-terminal 175 amino acids is predicted to be very acidic with an average calculated pI of 4.1. The COOH half of the protein contains three regions, which are predicted to be very basic, each with a calculated pI > 11.4 (Fig. 2). This bipolar distribution of electrostatic charges is similar to that of the microtubule-associated protein, MAPU (Aizawa et al., 1990). The COOH-terminal third of the protein is also predicted to be proline rich, containing ∼10% proline residues and three PXXP motifs, possibly indicating an interaction with SH3 proteins (Yu et al., 1994; Alexandropoulos et al., 1995). Alternatively, basic and proline-rich regions are also characteristic of microtubule-binding proteins (Meluh and Rose, 1990). However, the basic domain of Kar9p does not have sequence homology to other known microtubule-binding proteins, including TAU, MAPU, Kar3p, Mhp1p, Bik1p, and p150glued. Nevertheless, the possibility that Kar9p may possess a microtubule-binding domain is supported by the behavior of GFP-Kar9p described below. Together, these data indicate that KAR9 is a novel gene.
Figure 2.
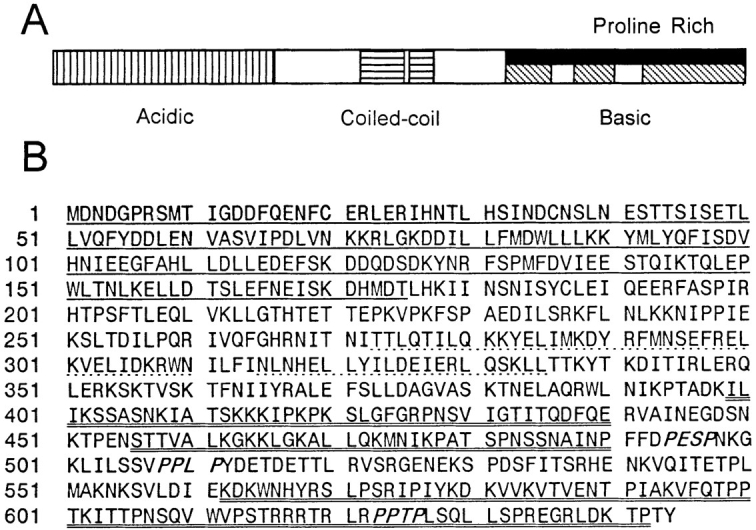
Kar9p sequence features. (A) Structural representation of Kar9p domains. Vertical-striped region represents the acidic domain of Kar9p. The horizontal-striped region represents the putative coiled-coil. The diagonally striped areas represent the highly basic region. The solid region represents the proline-rich domain, which roughly coincides with the basic domain. (B) The Kar9p protein sequence. Underlined region represents the acidic domain of Kar9p. Dotted line represents the putative, possible coiled-coil region of the protein. Double-underlined region represents the highly basic region. Three PXXP motifs are shown in italics. KAR9 corresponds to ORF YPL269W of the yeast genome sequencing project. These sequence data are available from GenBank/EMBL/DDBJ under accession number AF034763.
KAR9 Is Not Essential for Life, but Is Required for Cytoplasmic Microtubule Orientation and Nuclear Migration
To determine if the KAR9 gene product performs an essential function, we replaced the KAR9 sequence encoding amino acids 7–644 with LEU2 in haploid cells by homologous recombination using the one-step gene replacement method (Rothstein, 1983). The recovery of viable haploid colonies demonstrated that KAR9 is not essential for life. As described below, the kar9Δ mutants exhibited the same array of phenotypes as the kar9-485 point mutant, confirming the identification of the authentic KAR9 gene. Given the defects in cytoplasmic microtubule orientation in zygotes and the benomyl sensitivity of mitotic cells, we next examined the nuclear migration and microtubule morphology of the kar9Δ mutant in detail.
To determine how early in the mating process the defects were evident, we examined nuclear positioning and microtubule orientation in kar9Δ mutant shmoos. First, kar9Δ mutants were arrested with α factor and scored for nuclear position using DAPI stain. In wild-type shmoos, the nucleus normally moves up to the neck of the pear-shaped shmoo in preparation for mating (Byers and Goetsch, 1975; Rose, 1991; Read et al., 1992). Confirming this observation, we found that in 93% of wild-type shmoos with projections (n = 401), the nuclei were in or at the neck of the shmoo (Table III). Like wild type, the nuclei of dynein mutants were positioned correctly in 94% of shmoos (n = 400). In contrast, the nuclei of kar9Δ mutants were found in the center of the cell in ∼60% of shmoos (n = 300 and n = 324). We next used indirect immunofluorescence microscopy to examine the microtubules in the shmoo. In wild-type shmoos, 85% or more of the cells had microtubule bundles that extended to the shmoo tip (Fig. 3 A). In contrast, 73% of the kar9Δ mutant shmoos had misoriented microtubule bundles that did not extend into the shmoo tip (Fig. 3 D). Thus, the mating defect of the kar9 mutant is likely to be the result not only of the failure of cytoplasmic microtubules within the zygote to interdigitate, but also of an earlier defect in which cytoplasmic microtubules were misoriented in the shmoo.
Table III.
Nuclear Migration Defect in kar9Δ Shmoos

|
||||||||
|---|---|---|---|---|---|---|---|---|
| Nucleus: | In/at neck | In center | At bottom | n | ||||
| Wild type | 93% | 6% | 0.5% | 401 | ||||
| kar9Δ2::HIS3 | 37% | 60% | 3% | 324 | ||||
| kar9Δ1::LEU2 | 40% | 57% | 3% | 300 | ||||
| dhc1Δ | 94% | 6% | 0.5% | 400 | ||||
Wild type (MS1556), kar9Δ strains (MS4306 and MS4263), and dhc1Δ strain (MS4683) were arrested for 3 h with α factor. Cells were fixed, stained with DAPI, and then scored for nuclear position. Results are shown for a single experiment. Similar results were obtained for cells arrested for 90 min (data not shown).
Figure 3.
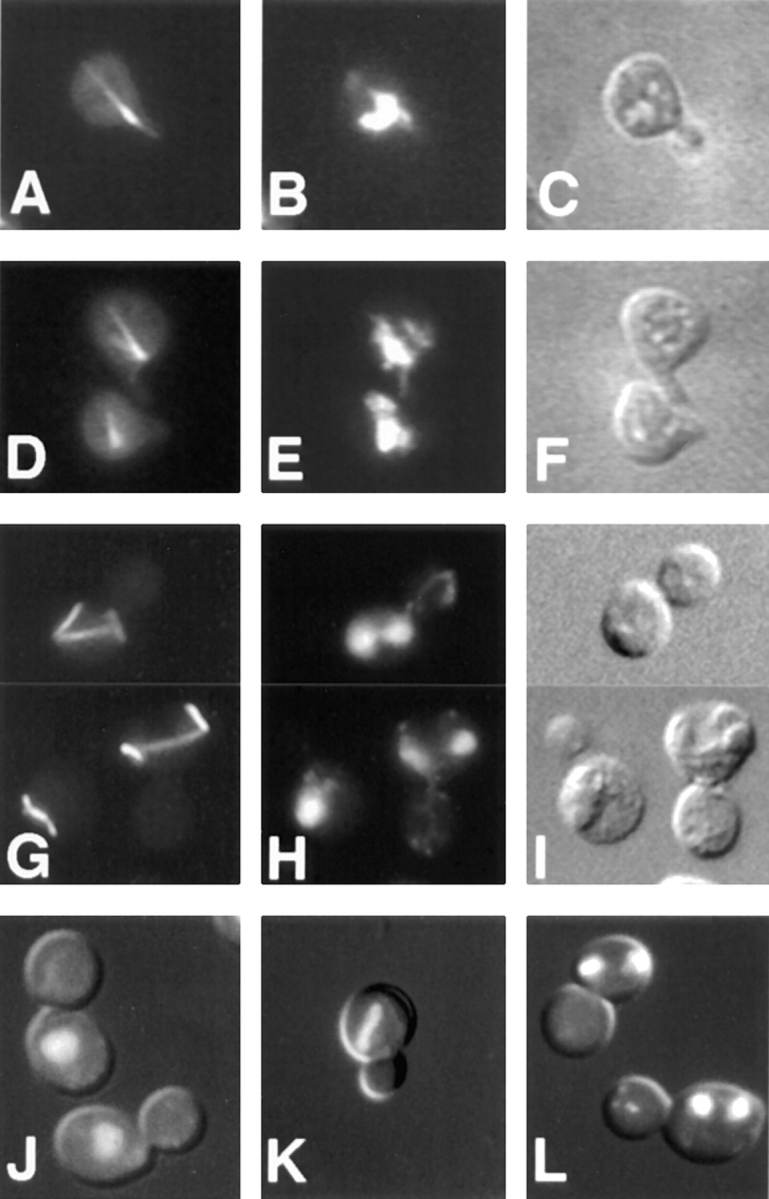
kar9 defects: aberrant cytoplasmic microtubules and nuclear migration. Wild-type (A–C) (MS1556) and kar9Δ (D–F) (MS4306) shmoos, as well as kar9-485 vegetatively growing cells (G–I) (MS2684) were fixed for indirect immunofluorescence using rabbit anti-tubulin (G) or rat anti-tubulin YOL1/34 (A and D). Examples two of vegetative kar9-485 cells with abnormal microtubules are shown in the composite panels, G–I. J–L are DAPI and Nomarski double-exposure images illustrating the array of abnormal mitotic phenotypes found in the kar9-485 stain, MS2684, grown to early logarithmic phase at 23°C. At this temperature, 15–20% of the cells exhibited such mitotic defects as large-budded cells with a single nucleus (J), mitosis occurring within the mother cell (K), and mother cells containing two nuclei (L).
Because kar9 mutants are sensitive to benomyl and because microtubule orientation is important during mitosis, we examined the kar9 mutants for nuclear positioning defects in mitosis. First, cultures of asynchronously growing kar9 cultures were stained with DAPI and scored for defects in nuclear location (Fig. 3, J–L). kar9 mutants exhibited significantly increased frequencies of abnormal mitotic phenotypes in large-budded cells. These included cells that contained a single nucleus that had failed to migrate to the bud neck (Fig. 3 J), cells with mitosis occurring within the mother cell (Fig. 3 K), and cells with two nuclei within the mother cell (Fig. 3 L). Anucleate cells were rarely seen (<0.5%) in kar9 mutant cultures. Cells exhibiting these three defects usually totaled between 10 and 15% of the culture at 30°C (Table IV). In contrast, wild-type cells showed only 2–3% of such abnormal cells. Because similar defects were reported for dhc1 mutants (Eshel et al., 1993; Li et al., 1993), we compared directly the nuclear migration defects observed in dhc1 with those of kar9 (Table IV). While the dhc1 mutant phenotypes were less severe at 30°C, the magnitude of the defects for both strains was aggravated by growth in the cold, with the sum of all defects increasing to >20% at 12°C. Thus, both dhc1 and kar9 exhibited strikingly similar defects in nuclear positioning (Table IV).
Table IV.
Nuclear Positioning Defects in kar9 Mutants
| A | B | C | D | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (°C) | Strain | n |

|
Σ | ||||||||||
| 30 | WT | 309 | 0.0% | 0.3% | 1.3% | 0.7% | 2.3% | |||||||
| 30 | dhc1Δ | 241 | 0.0% | 1.7% | 2.5% | 3.7% | 7.9% | |||||||
| 30 | kar9Δ | 256 | 0.4% | 4.3% | 4.3% | 3.9% | 13% | |||||||
| 12 | WT | 402 | 0.0% | 0.4% | 1.7% | 0.4% | 2.1% | |||||||
| 12 | dhc1Δ | 201 | 1.0% | 7.5% | 4% | 9.5% | 22% | |||||||
| 12 | kar9Δ | 278 | 0.0% | 2.9% | 5% | 13% | 21% | |||||||
The position of nuclear DNA was visualized by DAPI staining in asynchronously growing cultures of early exponential dhc1Δ (MS4262), kar9Δ (MS4589), and wildtype (MS52) strains at 308 and 128C. The percentage of abnormal nuclear position phenotypes are shown relative to the total cell culture. The remaining percents appeared wild type. A, anucleate cell. B, large-budded cell with a single nucleus in the mother cell. C, mitosis occurring within the mother cell. D, large-budded cell with two nuclei per mother cell. A representative experiment is shown. In repeat experiments, percentage vary by about 5%. n, the total cells counted. Σ, the sum of nuclear positioning defects.
Indirect immunofluorescence was carried out to determine if the kar9Δ mutant exhibited mitotic nuclear migration defects that correlated with microtubule abnormalities. Exponentially growing cultures of kar9 mutants contained many cells with abnormal microtubule morphologies for both the mitotic spindle and the cytoplasmic microtubules (Fig. 3 G). The mitotic spindle was often rotated away from the long axis of the mother–bud. Occasionally, the spindle appeared bent along the inner surface of the mother cell (data not shown), similar to that observed in other nuclear migration mutants, DHC1/DYN1 (Eshel et al., 1993; Li et al., 1993), ACT5 (Clark and Meyer, 1994; Muhua et al., 1994), and JNM1 (McMillan and Tatchell, 1994). The cytoplasmic microtubules were also frequently positioned aberrantly in kar9 mutants (Fig. 3 G). In contrast to both wild-type and dhc1Δ cells, 30% of kar9Δ cells carrying out anaphase within the mother cell had cytoplasmic microtubules that failed to extend into the bud (Fig. 4 A). In those kar9Δ cells that contained two nuclei within the mother cell, 70% exhibited cytoplasmic microtubules that did not extend into the bud (Fig. 4 B). This is in sharp contrast to the phenotype of equivalent dhc1Δ mutant cells, in which 83% of the cytoplasmic microtubule bundles were observed to extend into the bud (Fig. 4 B; Li et al., 1993).
Figure 4.


Quantitation of cytoplasmic microtubule orientation defect. The percentage of cells with cytoplasmic microtubule orientation defects was determined in A (cells carrying out anaphase within the mother cell), and B (cells with two nuclei within the mother cell by indirect immunofluorescence). The kar9Δ strain (MS4306), the dhc1Δ strain (MS4903), and the wild-type strain (MS1554), were grown to early exponential phase at 14°C, and then fixed for indirect immunofluorescence using the tubulin antibody, YOL1/34.
The kar9Δ mutants displayed no obvious actin defect by rhodamine-phalloidin staining. In kar9, actin patches localized to the growing bud in small-budded cells and to the cleavage furrow in cells undergoing cytokinesis as they normally do in wild type (data not shown). From this, together with the benomyl sensitivity, we conclude that the kar9 mutation affects the functions of cytoplasmic microtubules.
kar9Δ and dhc1Δ Mutants Are Synthetically Lethal
Because kar9 and dhc1 display similar phenotypes in nuclear migration, we wanted to determine whether KAR9 functions in the same or different pathways as other genes involved in this process. To carry out the analysis, marked deletions of KAR9 and several other nuclear migration genes were crossed together.
The first nuclear migration protein tested was the microtubule motor protein, dynein, encoded by DHC1/DYN1. To make the kar9Δ dhc1Δ mutant, the dhc1Δ strain (MS4262) was crossed to kar9-Δ1::LEU2 (MS4589). Of the 35 segregants predicted to be double mutants on the basis of segregation of markers in sister spores, 9% failed to germinate and 91% formed microcolonies (Table V; Fig. 5, A and B) at 30°C. In contrast, wild-type and single mutant colonies showed 97% spore viability and no apparent growth defects (data not shown). The microcolonies exhibited heterogeneity in size, but by 3–4 d after germination at 30°C they usually attained the size of an 18–24-h-old wild-type colony (e.g., <1,000 cells). Examination of the cells within these microcolonies by DAPI staining and Nomarski optics revealed a wide range of defects. Most cells were multinucleate, anucleate, deformed in cell shape, or lysed. These defects are consistent with a severe defect in nuclear migration (Fig. 5 B).
Table V.
Viability of kar9Δ Double Mutants
| Viability with kar9Δ | Tetrads (PD:TT:NPDs) | Predicted double mutants | Percent of double mutants dead or forming microcolonies | |||||
|---|---|---|---|---|---|---|---|---|
| dhc1Δ | not viable | 32 (2:25:5) | 35 | 100 | ||||
| jnm1Δ | not viable | 17 (4:12:1) | 14 | 100 | ||||
| act5Δ | not viable | 23 (7:12:4) | 20 | 100 | ||||
| bik1Δ | not viable | 16 (3:10:3) | 16 | 94 | ||||
| smy1Δ | viable | 8 (0:6:2) | 10 | 0 | ||||
| cin8Δ | viable | 15 (2:8:5) | 18 | 33* | ||||
| kip1Δ | viable | 7 (0:7:0) | 7 | 0 | ||||
| kar3Δ | viable | 22 (6:13:3) | 15‡ | NA§ |
Double mutants were created by crosses. 2–3 d after germination, plates were scored for colony size and the presence of microcolonies. The following crosses were carried out to create the corresponding kar9Δ double mutants. kar9Δ dhc1Δ MS4262 × MS4589. kar9Δ act5Δ MS4586 × MS4263. kar9Δ jnm1Δ MS4321 × MS4587. kar9Δ bik1Δ, MS4734 × MS4313. kar9Δsmy1Δ, APY4ΔD5 × MS4062. kar9Δ cin8Δ, MAY2058 × MS4306. kar9Δ kip1Δ, MS2305 × MS4263. For the analysis of kar9Δ kar3Δ double mutants, MS1614, which carries KAR3 on a URA3-marked CEN plasmid was crossed to the KAR9 delete strain (MS4306). This yielded 15 double mutants carrying the KAR3 URA3 CEN plasmid. The ability of the double mutant to lose the KAR3 plasmid and grow was assayed on plates containing 5-fluoro-orotic acid plates. The ability of the double mutant to lose the KAR3 plasmid was not significantly different in comparison to that of the kar3Δ single mutants. NA, not applicable.
This is the same rate found for single cin8Δ mutants.
Plasmid loss assay on media containing 5-fluoro-orotic acid was used for the analysis of KAR3.
This represents the number of kar9Δ kar3Δ double mutants that carried the KAR3 URA3 plasmid.
Figure 5.

Synthetic lethality. kar9-Δ1::LEU2 (MS4589) was crossed to dhc1Δ::URA (MS4262). Spores were allowed to germinate 4 d on YPD and photographed (A). Arrows show double-mutant microcolonies. 15–20 microcolonies were removed from the dissection plate, fixed, and then stained with DAPI. Examples of the kar9Δ dhc1Δ double mutant (B), and kar9Δ bik1-518 double mutant (C) are shown using DAPI and Nomarski double-exposure images.
Several genes, including ACT5 and JNM1, have been suggested to operate in nuclear migration in concert with the dynein heavy chain as part of the dynactin complex (McMillan and Tatchell, 1994; Muhua et al., 1994). If such a model is correct, then deletions in these genes would be predicted also to result in synthetic lethality when combined with the KAR9 deletion. To test this possibility, the jnmΔ strain (MS4321) was crossed to the kar9-Δ2::HIS3 strain (MS4587) to create kar9Δ jnm1Δ mutants. Seventeen tetrads were examined, yielding 14 predicted double mutants. Of these, 100% formed microcolonies (Table V). To create kar9Δ act5Δ double mutants, the ACT5 delete strain (MS4586) was crossed to the kar9-Δ1::LEU2 strain (MS4263). From this cross, 23 tetrads were dissected, producing 20 predicted double mutants. Of these double mutants, 5% were dead and 95% formed microcolonies (Table V). Like the kar9Δ dhc1Δ double mutant, analysis of the cells within these microcolonies also revealed lysed cells, anucleate cells, and multinucleate cells. In contrast, dhc1 act5, dhc1 jnm1, and act5 jnm1 double mutants are viable and show no more severe defect than any single mutant (Tatchell, K., personal communication; Muhua et al., 1994; Geiser et al., 1997). We conclude therefore that Kar9p acts in a nuclear migration pathway that is separate from and partially redundant with that of the dynein/dynactin complex.
Several additional genes were also tested for the possibility of genetic interactions with kar9Δ. A bilateral karyogamy mutation, bik1 (Berlin et al., 1990), with additional functions during mitosis, also exhibited a microcolony phenotype in combination with KAR9 deletions (Table V). Cells found within the microcolonies of the kar9Δ bik1-518 (Trueheart et al., 1987) double mutant (Fig. 5 C) exhibited defects similar to those of the kar9Δ dhc1Δ mutant.
To test the specificity of the synthetic lethality, crosses to mutants in other microtubule motor proteins were performed. KIP1 (Roof et al., 1992) and CIN8 (Hoyt et al., 1992) are required for establishment of a bipolar mitotic spindle. kar9Δ cin8Δ and kar9Δ kip1Δ double mutants resulted in no detectable growth defect (Table V). KAR3 is required for mitosis and mating, possibly by mediating the sliding of microtubules past each other (Meluh and Rose, 1990). The kar9Δ kar3Δ mutant spores exhibited no obvious growth defect worse than that of the kar3Δ single mutant alone. Deletions in SMY1, a kinesin involved in secretion (Lillie and Brown, 1992), also exhibited no apparent growth defect when combined with the KAR9 deletion (Table V).
GFP-Kar9p Localizes to the Tip of the Shmoo
To better understand how Kar9p might be functioning, its localization was determined inside living yeast cells. A GFP-Kar9p fusion protein was constructed on a centromere-based plasmid with its expression under the control of the GAL1 promoter (pMR3465). This construct fully suppressed the microtubule orientation and nuclear migration defects in kar9Δ shmoos (data not shown). The localization of GFP-Kar9p was examined in shmoos and zygotes. In each instance, GFP-Kar9p fluorescence was observed primarily as a single small dot. In 73% of kar9Δ shmoos, the dot was located at the tip of the shmoo projection (n = 152) (Fig. 6 A). In an additional 12% of the shmoos, a thin line extended from the single dot toward the nucleus (data not shown). Identical results were observed in wild-type cells expressing GFP-Kar9p. In zygotes, GFP-Kar9p was located as an elongated dot at the future site of cell fusion of the pre-zygote (100%; n = 15) (Fig. 6 D), as if two dots had fused. In budded zygotes, it was also found at the tip of the emerging bud (Fig. 6 G).
Figure 6.

Localization of GFP-Kar9p in shmoos and zygotes. kar9Δ shmoos (MS4382) (A–C), pre-zygotes (MS4671X MS4387) (D–F), and budding zygotes (MS4671X MS4387) (G–I), each expressing GFP-Kar9p (pMR3465) were fixed and stained with DAPI. In each instance, >90% of all cells showed GFP localization. GFP: A, D, and G. DAPI: B, E, and H. Nomarski: C, F, and I.
GFP-Kar9p Localization to the Tip of the Bud Is Cell-Cycle Dependent
To gain an understanding of how Kar9p might be functioning to orient microtubules in mitotically growing cells, the localization of GFP-Kar9p was scored throughout the cell cycle. First, asynchronously growing cultures were examined using bud size and nuclear position as an indicator of the cell cycle stage. In unbudded cells, little or no GFP localization was observed (76%; n = 51) (Figs. 7 A and 8). In small–medium-budded cells, the majority of cells showed GFP-Kar9p localization as a single dot at the tip of the growing bud (58%; n = 57) (Figs. 7 D and 8). An additional 14% of small-budded to medium-budded cells also had the dot at the tip of the bud with a second dot between the first cortical dot and the nucleus (Fig. 8). At anaphase, the major localization pattern (49% of all anaphase cells; n = 57) was a single dot of GFP at the tip of the bud (Figs. 7 G and 8). An additional 14% had the dot at the tip of the bud, but also had a second dot between the cortical dot and the nucleus (Fig. 8). For the small-budded and anaphase stages, the percentage of cells that showed no localization was relatively low, 19 and 30%, respectively (Fig. 8). However at telophase, the majority of cells exhibited no localization (58%; n = 80) (Figs. 7 J and 8). To confirm the latter stages of localization, cells were first synchronized with hydroxyurea, and then released from the block. Under these conditions, >85% of telophase cells showed no localization (n = 70). In comparison, only 17% of anaphase cells exhibited no localization at the corresponding time point (n = 88) (Fig. 8). In addition to the single dot in the bud and the “two dots in a line” patterns (Fig. 8), some cells also exhibited an additional spot. These cells were classified as “other” (Fig. 8). The additional spot was located at one of the six locations depicted in Fig. 7 M in >90% of cells in the “other” category. In a few rare examples, three spots per cell were observed. We conclude that GFP-Kar9p shows both mother–daughter asymmetry and cell cycle dependence for its localization.
Figure 7.
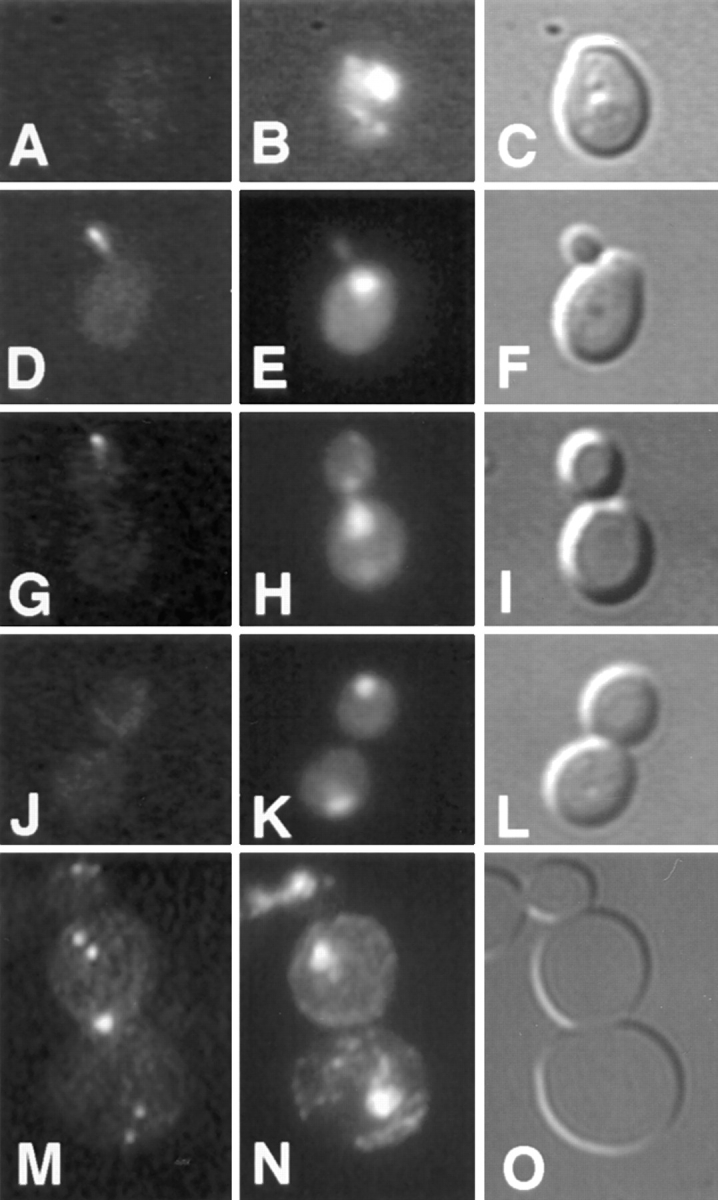
Cell cycle–dependent localization of GFP-Kar9p. The cell cycle position of a wild-type strain expressing GFP-Kar9p (MS4387) was estimated based on bud size and nuclear morphology. The GFP localization pattern was then determined in unbudded or G1 cells (A–C), small-budded to medium-budded cells or S to M phase cells (D–F), anaphase (G–I), and telophase (J–L) cells. A rare telophase cell with an abnormal localization pattern is shown in panel M. Minor localization patterns were found to be a subset of these six dots. The largest subsets of minor localization patterns were those of two and three dots. GFP: A, D, G, J, and M. DAPI: B, E, H, K, and N. Nomarski: C, F, I, L, and O.
Figure 8.

Quantification of GFP-Kar9p localization. The cell cycle position of cells expressing GFP-Kar9p was estimated as described in Fig. 7. Cells in the Other category had one or more dots in the locations depicted in Fig. 7 M. Subsets of two or three dots were the most common minor localization patterns.
Cytoplasmic Microtubules Intersect the GFP-Kar9p Dot
To determine the relationship between cytoplasmic microtubules and the GFP-Kar9p dot, double-label indirect immunofluorescence was conducted using antibodies specific for tubulin and GFP. In shmoos, the cytoplasmic microtubule bundle terminated at the dot of anti-GFP staining in all cases (n = 100) (Fig. 9, A–D). In vegetative cells, microtubule staining intersected the anti-GFP staining dot in 85% of the large-budded cells examined (n = 20; Fig. 9, E–H). In a small percentage of both shmoos and large-budded cells, a line of anti-GFP immunofluorescence extended away from the anti-GFP dot toward the nucleus. In all cases, the line of staining colocalized with the cytoplasmic microtubules.
Figure 9.

Cytoplasmic microtubules intersect the GFP-Kar9p dot. Double-label indirect immunofluorescence was carried out on wild-type shmoos (A–D) and cells (E–H) expressing GFP-Kar9p using anti-GFP and anti-tubulin (YOL 1/34). Two shmoos are shown in A–D and three cells are shown in E–H. Anti-tubulin: A and E. GFP: B and F. DAPI: C and G. Nomarski: D and H.
GFP-Kar9p Localization at the Cortex Is Independent of Microtubules
Kar9p might function in microtubule orientation by two different mechanisms. In the first model, Kar9p might function as a microtubule-associated protein which stabilizes cytoplasmic microtubules. Localization of Kar9p at the cortex would therefore be dependent solely on the cytoplasmic microtubules. Alternatively, Kar9p might instead serve as a target for the cytoplasmic microtubules at the cortex. In this second model, Kar9p localization would be solely dependent on cortical information and independent of microtubules.
To examine these possibilities and to ascertain whether microtubules are required for the maintenance of Kar9p localization, wild-type shmoos already expressing GFP-Kar9p were treated with nocodazole to depolymerize preexisting microtubules (Fig. 10 B) or mock-treated with DMSO (Fig. 10 A). 74% of shmoos treated with nocodazole under these conditions had a GFP-Kar9p dot at the tip of the shmoo, as compared to 90% of the mock-treated control shmoos. Thus, Kar9p remained at the tip of these shmoos and microtubules were not required to maintain GFP-Kar9p at its cortical location. Alternatively, microtubules might play a role in establishing the localization of GFP-Kar9p at its cortical site. To investigate this possibility, cells were induced to form shmoos either in the presence (Fig. 10 C) or absence of microtubules (Fig. 10 D) and the expression of GFP-Kar9p was then induced. GFP-Kar9p localization was found at the tip of 72% of shmoos treated with nocodazole versus 85% of shmoos mock treated with DMSO. Thus, under microtubule-depolymerizing conditions, GFP-Kar9p was still able to localize to the tip of the shmoo. Identical results were obtained using the GFP-KAR9 plasmid in a kar9 deletion strain.
Figure 10.


Kar9p localization at the cortex is independent of microtubules. The wild-type strain (MS1556) containing the GFP-KAR9 plasmid (pMR3465), was grown to early exponential phase. Representative examples of each phenotype scored are shown. 100 cells were counted for each analysis of GFP-Kar9p localization and anti-tubulin. Shmoos displaying both a dot and a line emanating from the dot were scored in the cortical dot category. Under conditions A and B, GFP-Kar9p expression was induced for 1.5 h. Microtubules were then depolymerized by the addition of nocodazole (see Materials and Methods) or mock treated with DMSO. Under conditions C and D, cells were treated simultaneously with α factor and nocodazole or DMSO for 1.5 h to create shmoos devoid of microtubules. GFP-Kar9p was then induced for an additional 1.75 h by the addition of galactose. Whereas this treatment also resulted in some cells with a large-budded arrest phenotype, only cells with a distinct shmoo morphology were counted in this analysis. Under conditions E and F, GFP-Kar9p expression was induced for 2 h. Microtubules were then depolymerized by nocodazole or mock treated with DMSO. Under conditions G and H, microtubules were depolymerized with nocodazole or mock treated with DMSO for 1.5 h. GFP-Kar9p expression was then induced by the addition of galactose. Because of the cell cycle constraints of GFP-Kar9p localization in cells, only large-budded cells not yet in anaphase were scored. Cells with two GFP-Kar9p dots in a line were scored in the cortical dot category if one of the dots was at the bud cortex. Cells with a cortical dot and a distinct line emanating from it were scored in the line category. αF, α factor.
A similar analysis was carried out to determine if GFP-Kar9p localization was also independent of microtubules in vegetative cells. When microtubules were depolymerized after induction of GFP-Kar9p, localization was found at the tip of the bud in 89% of large-budded cells (Fig. 10 F). In comparison, in the mock-treated controls, 72% had the GFP-Kar9p dot at the tip of the large bud (Fig. 10 E). Similarly, when microtubules were depolymerized before the induction of GFP-Kar9p, 79% of large-budded cells contained a dot of GFP-Kar9p localization at the tip of the bud (Fig. 10 H). Thus, without microtubules present, GFP-Kar9p could still localize at the tip of the bud. Therefore, we conclude that like shmoos, Kar9p localization at the cortex was independent of microtubules in vegetative cells.
In 11 and 12% of the mock-treated control cells, a line of GFP-Kar9p localization extending from the cortex was also observed (Fig. 10, E and G). When cells were treated with nocodazole, none of the cells displayed the line of GFP-Kar9p fluorescence (Fig. 10, F and H). In the experiment with shmoos, a GFP-Kar9p dot with a line of fluorescence was scored in the “cortical dot” category. When the shmoos were treated with nocodazole, the lines of GFP-Kar9p fluorescence were not observed (data not shown). Therefore, localization along microtubules was dependent on their polymerization. These data support the finding that the line of GFP-Kar9p fluorescence colocalized with microtubules (Fig. 9) and are consistent with the idea that Kar9p may contain a microtubule-binding domain.
Discussion
In this paper we have shown that KAR9 encodes a novel protein that is required for nuclear migration during mitosis and mating. Loss of Kar9p results in misoriented cytoplasmic microtubules, which appears to be the primary cause of the nuclear migration defect. Genetic analysis revealed that deletion mutations in KAR9 and genes encoding components of the dynactin complex are synthetically lethal. GFP-Kar9p localized to cortical sites in buds and shmoos. Consistent with a role for Kar9p in orienting the microtubules, the cytoplasmic microtubules intersected the GFP-Kar9p dot. The localization of GFP-Kar9p to the bud tip was cell cycle dependent, indicating that localization was both spatially and cell cycle regulated. Finally, the microtubule independence of the cortical localization of Kar9p indicates that it can respond directly to the spatial information present in the growing cell.
Nuclear Migration Defects and Misoriented Microtubules in kar9 Mutants during Mating
kar9 was previously identified as a karyogamy mutant with a class I nuclear fusion defect characterized by widely separated nuclei in the zygote (Kurihara et al., 1994). However, the kar9 mutant was unlike other class I karyogamy mutants in that its cytoplasmic microtubules were grossly misoriented. We have now examined kar9 mutants at the shmoo stage and found that the defect occurs before the time of cell fusion. Unlike wild type or DHC1 mutants, a significant number of kar9 nuclei did not move to the neck of the shmoo in response to pheromone treatment (Table III). Examination of the microtubules in kar9 shmoos revealed that the microtubules were misoriented (Fig. 3). It seems reasonable to conclude that the defect in nuclear fusion arises from the earlier defect in nuclear migration. Presumably the cytoplasmic microtubules from misoriented nuclei are unable to intersect and pull the nuclei together in the limited time available for nuclear fusion in the zygote.
Mating cells represent a simpler system than mitotically growing cells in which to identify cellular components. For instance, the kinesin-like motor, Kar3p, is one example of a nonredundant gene required for functions in the shmoo and zygote and deletion of KAR3 results in complete blockage of nuclear migration in the zygote. Hence, it is likely to be the sole motor responsible for nuclear migration in zygotes. The very high incidence of misoriented microtubules in kar9Δ shmoos suggests that Kar9p is the major, if not the only, mechanism involved in orienting cytoplasmic microtubules toward the tip of the shmoo projection.
The Role Played by KAR9 in Nuclear Migration in Mitosis Is Different Than the Role Played by the Dynein Heavy Chain Motor
Mitotically growing kar9 mutants also displayed severe nuclear migration defects. The cytoplasmic microtubules were also misoriented in the mitotic example of nuclear migration defect. Because the cytoplasmic microtubules are required for nuclear migration in mitotic cells (Sullivan and Huffaker, 1992), the most likely explanation for the defect in kar9 mutants is the abnormally oriented cytoplasmic microtubules. Our results indicate that Kar9p function is not specific to either the mating or mitotic processes. Rather, Kar9p must function in microtubule orientation in general and that microtubule orientation is Kar9p's primary function.
Several pieces of evidence strongly suggest that Kar9p plays a very different role in nuclear migration than Dhc1p. First, Kar9p functions both during mating and mitotic cell divisions. The dhc1 mutants do not exhibit a mating defect (Li et al., 1993), and dhc1 mutants do not display nuclear migration defects in the shmoo (Table III). These results are consistent with the earlier view that Kar3p is the minus end–directed microtubule motor responsible for nuclear migration during mating. Second, in kar9 mutants the cytoplasmic microtubules do not enter the bud, whereas in dhc1 cells the cytoplasmic microtubules consistently extend into the bud (Muhua et al., 1993; Geiser et al., 1997). This result is consistent with the view that Kar9p is responsible for orientation, whereas dynein is required for movement.
Genetic interactions between kar9Δ and dhc1Δ suggest that KAR9 functions in a pathway that is separate and partially redundant with DHC1. The double mutants between kar9 and dhc1 were severely handicapped for growth in comparison to the minor growth defects of either single mutant alone (Fig. 5). Furthermore, kar9 was synthetically lethal with deletions in two other components postulated to act with dynein, ACT5 and JNM1 (McMillan and Tatchell, 1994; Muhua et al., 1994). Importantly, double mutants between deletions in DYN1, ACT5, and JNM1 have been reported to be no worse than the single mutants for nuclear migration defects. These data are consistent with the model that DHC1/DYN1, ACT5, and JNM1 function in the same pathway (Tatchell, K., personal communication; Muhua et al., 1994; Geiser et al., 1997).
Synthetic lethality can be observed in two very different kinds of genetic interactions. On the one hand, two mutations might cause the disruption of one essential complex. For example, each point mutation might only reduce the stability of the complex. However together the two mutations could greatly reduce the stability such that the complex never forms. In this case, one might expect complete deletions of proteins to be lethal. On the other hand, two mutations might disrupt the function of two redundant pathways required for an essential process. In this case neither gene would be essential.
The use of deletion strains in these studies demonstrated that neither KAR9 nor DHC1/DYN1 are essential. We therefore interpret the synthetic lethality between kar9 and mutants in the dynein pathway as a reflection of their respective functions in separate pathways required for the essential process of nuclear migration. The converse side of this argument suggests that kar9 deletions are not disrupting the dynein complex, as would be expected if a dynein intermediate chain were removed. If this were the case, the expected phenotype would be the same as if the dynein heavy chain itself had been removed and the phenotype of double mutant would be no worse than that of either single deletion. Therefore, we conclude that the synthetic lethality exhibited between kar9Δ and dhc1Δ cannot be the result of Kar9p functioning as an intermediate chain or similar accessory subunit to the dynein heavy chain. Indeed, Kar9p does not have homology to any known dynein intermediate chains. Therefore, Kar9p must be functioning in a manner that is distinct from that of the pulling force of the dynein heavy chain.
If Kar9p's microtubule orientation function is separate from dynein's microtubule pulling function, why then are KAR9 and DHC1 not essential? That is, what functions do each provide in the absence of the other protein? We postulate that Kar9p stably orients the cytoplasmic microtubules while dynein pulls. In Kar9p's absence and without proper microtubule orientation, dynein and/or other motors hypothesized to be located on the cortical surface would not pull efficiently. However, when microtubules transiently enter into the bud, then the motors could provide some force and orientation, which over time would provide sufficient nuclear migration for most cells. In Dhc1p's absence, Kar9p would still provide stable microtubule orientation. Without Dhc1p, it is possible that additional motor systems such as Kip2p and Kip3p may be involved in moving the nucleus into the bud and thus may compensate for the loss of dynein's pulling force (Miller, R., K. Heller, D. Wallack, L. Frisén, D. Loayza, and M.D. Rose, manuscript in preparation; Miller, R.K., and M.D. Rose. 1995. Mol. Biol. Cell. 6:256a. 1995. Cottingham and Hoyt, 1997; DeZwaan et al., 1997). Alternatively, the intrinsic dynamic instability of microtubules might provide sufficient force for the nuclear migration, much as depolymerization can move chromosomes on microtubules in vitro (Koshland et al., 1988; Lombillo et al., 1995). However, the cumulative effect of the loss of both Kar9p and Dhc1p is that without the correct orientation of cytoplasmic microtubules, any additional force that might be provided by auxiliary motors or by microtubule depolymerization is ineffectual. The cell then loses both the pulling force and correct microtubule orientation, resulting in a massive failure of nuclear migration.
GFP-Kar9p Localization
Using a GFP-Kar9p fusion, we found Kar9p localized to a distinct dot at the tip of the shmoo and at the tip of the growing bud. We never found GFP-Kar9p localization to be diffuse or spread out in a patch over the cortical surface. However, in minor localization patterns we did see a line of localization extending proximally toward the nucleus. Unlike the GFP-Kar9p cortical dot, the line of localization was dependent on microtubules. This may reflect an association of Kar9p with cytoplasmic microtubules, perhaps through the basic domain of Kar9p. Furthermore, since cytoplasmic microtubules terminated with the GFP-Kar9p dot, Kar9p is positioned at the predicted cellular location to orient cytoplasmic microtubules.
In addition to the strong mother–bud asymmetry, GFP-Kar9p also showed cell cycle variation. In unbudded cells when microtubule orientation is presumably not needed, localization was not observed. Kar9p was found at the tip of small-budded cells and remained at the bud tip through anaphase. At telophase, GFP-Kar9p localization was no longer observed. Why might it no longer be necessary for Kar9p to be localized at the tip of the bud after anaphase? At anaphase, the orientation of the nucleus through the neck of the bud has been clearly established. Using live cell observations with differential interference contrast microscopy, Yeh et al. (1995) demonstrated that after migration of the two nuclei to the distal ends of the cell and bud (and extension of the mitotic spindle to its maximal length), the nucleus then returns to the center of the cell. It could be possible that this “rebound” to the center of the cell requires that the Kar9p tether release the microtubule from the cortex.
How does GFP-Kar9p localization disappear after anaphase? One possibility is that Kar9p is degraded, perhaps through the anaphase promoting complex ubiquitination system. Alternatively, delocalization of GFP-Kar9p away from the spot might prevent its visualization. Either mechanism would have to overcome the continuous galactose-induced expression from the GAL1 promoter used in these experiments. Nevertheless, Kar9p is localized both when and where it would be predicted to function in orienting microtubules into the bud.
In a subset of cells additional spots of GFP localization were observed. All of these can be represented by subsets of the six GFP-Kar9p dots shown in Fig. 7 M, with two to three dots being the most prevalent minor class (Fig. 8). However, because of the impracticality of showing all combinations of six dots scored three ways, we show this very rare cell as an example. One site of minor localization was to the nuclear periphery, most likely at the SPB. GFP-Kar9p localization at the nucleus is likely to represent an overexpression phenotype, since this localization was greatly reduced when GFP-Kar9p expression levels were downregulated by the addition of glucose to the media and through the use of a gal2 mutant (data not shown). GFP-Kar9p localization as two dots at each side of the neck in large-budded cells could represent an involvement of Kar9p at sites required for the microtubule-dependent reorientation of the nucleus to face the new bud site for the next cycle of axial budding (Byers, 1981; Snyder et al., 1991). Alternatively, localization at the neck might represent premature association with the new bud site, before Kar9p's normal association at the tip of the new bud. Interestingly, the presence of these minor localization patterns did not appear to be affected by the depolymerization of microtubules by nocodazole (data not shown). Thus, whereas we interpret the significance of these minor localization sites with caution, they may represent the normal association of Kar9p with proteins of lower affinity. It is significant that each of these minor localization patterns are at locations within the cell that are important for microtubule function (Fig. 7 M).
We were somewhat surprised to find that Kar9p did not normally localize to a mother cell cortical site as well; cytoplasmic microtubules are predicted to be anchored to the mother cell cortex as well as to the bud cortex. Without a mother cell cortical attachment, the nucleus might be pulled completely into the bud, leaving the mother cell without a nucleus. Num1p has been proposed to be involved in this function (Farkasovsky and Kuntzel, 1995). Num1p localizes as many punctate spots widely distributed primarily over the mother cell's cortical surface (Farkasovsky and Kuntzel, 1995). In contrast, Kar9p localizes as a single dot at the tip of the bud. However like kar9 mutants, num1 mutants do not always orient cytoplasmic microtubules into the bud correctly (Farkasovsky and Kuntzel, 1995).
Nocodazole Depolymerization of Microtubules
By treating GFP-Kar9p–expressing cells with nocodazole, we have shown that the localization of Kar9p at the cortex is largely independent of microtubule polymerization. Microtubules are not required either for the establishment or the maintenance of GFP-Kar9p at the cortex. Therefore, Kar9p appears to carry out its microtubule orientation function via its own interaction with the cortex. This suggests that Kar9p does not function as a microtubule-associated “capping” protein. Rather it suggests that Kar9p provides an orientation function for cytoplasmic microtubules as part of the cortical cytoskeleton.
Since actin is an intrinsic element for many polarized functions in yeast, it is possible that Kar9p localization occurs through an association with the actin cytoskeleton. This hypothesis is consistent with previous observations that the orientation of the cytoplasmic microtubules occurs in response to cues from sites of polarized growth, and that microtubules are not involved in establishing this polarity (Jacobs et al., 1988; Read et al., 1992; Yang et al., 1997).
Previous studies in a variety of organisms have proposed models in which the actin cytoskeleton and cortical structures in general serve to anchor cytoplasmic microtubules and orient the mitotic spindle (Hyman, 1989; Dan and Tanaka, 1990; Palmer et al., 1992; Sullivan and Huffaker, 1992). In Caenorhabditis elegans embryos, irradiation of a putative microtubule attachment point at the cortex disrupted the rotation of the mitotic apparatus and centrosomes, the functional equivalent of the yeast SPB (Hyman, 1989). Micromanipulation of Chaetopterus oocytes demonstrated the existence of a single, localized spindle attachment point on the oocyte cortex (Lutz et al., 1988). In S. cerevisiae, disruption of the actin network of cables and cortical actin patches at the nonpermissive temperature of ACT1-4 results in a rapid deorientation of previously oriented pre-anaphase spindles (Palmer et al., 1992). However, the nature of the postulated microtubule attachment complex to the cortex remains elusive. The results reported here indicate that Kar9p should be considered for such a role. Whether Kar9p interacts directly with the actin cytoskeleton, indirectly through an actin-binding protein, or interacts with the bud cortex through some other attachment mechanism remains an interesting avenue of future investigation.
Mitosis comprises many critical steps. Universal in all systems is the establishment of the mitotic spindle in the correct orientation relative to the plane of cell division. In S. cerevisiae, spindle orientation is coordinated with the migration of the nucleus to the bud neck. We report here that Kar9p functions both in vegetatively growing cells and in shmoos as a part of an orientation mechanism for cytoplasmic microtubules. In both instances of polarized growth, Kar9p orients cytoplasmic microtubules toward the cell cortex. Therefore, we propose that the novel cortical protein, Kar9p (either directly or as part of a complex) is involved in anchoring cytoplasmic microtubules to the cell cortex and thus provides a mechanism for spindle orientation. We consider it likely that Kar9p-like adaptors will be found in other systems.
Acknowledgments
We thank C. Davis for re-engineering F. Cormack's GFP plasmid. A. Gammie, D. Sullivan, and L.J. Kurihara provided many helpful suggestions.
This work was supported by National Institutes of Health grant GM-37739 (to M.D. Rose). R.K. Miller was supported by a National Institutes of Health postdoctoral fellowship.
Abbreviations used in this paper
- GFP
green fluorescent protein
- DAPI
4′,6-diamidino-2-phenylindole
- pI
isoelectric point
- SPB
spindle pole body
- YPD
yeast peptone dextrose
Footnotes
Address all correspondence to M.D. Rose, Department of Molecular Biology, Princeton University, Princeton, NJ 08544. Tel.: (609) 258-2804. Fax: (609) 258-6175. E-mail: mrose@molecular.princeton.edu
References
- Aizawa H, Emori Y, Murofushi H, Kawasaki H, Sakai H, Szuki K. Molecular cloning of a ubiquitously distributed microtubule-associated protein with Mr190,000. J Biol Chem. 1990;265:13849–13855. [PubMed] [Google Scholar]
- Alexandropoulos K, Cheng G, Baltimore D. Proline-rich sequences that bind to Src homology 3 domains with individual specificities. Proc Natl Acad Sci USA. 1995;92:3110–3114. doi: 10.1073/pnas.92.8.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin V, Styles CA, Fink GR. BIK1, a protein required for microtubule function during mating and mitosis in Saccharomyces cerevisiaecolocalizes with tubulin. J Cell Biol. 1990;111:2573–2586. doi: 10.1083/jcb.111.6.2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers, B. 1981. Cytology of the Yeast Life Cycle. In The Molecular Biology of the Yeast Saccharomyces: Life Cycle and Inheritance. J. Strathern, E.W. Jones, and J.R. Broach, editors. Cold Spring Harbor Laboratory. Cold Spring Harbor, NY. 59–96.
- Byers B, Goetsch L. Duplication of spindle plaques and integration of the yeast cell cycle. Cold Spring Harbor Symp Quant Biol. 1974;38:123–131. doi: 10.1101/sqb.1974.038.01.016. [DOI] [PubMed] [Google Scholar]
- Byers B, Goetsch L. Behavior of spindles and spindle plaques in the cell cycle and conjugation of Saccharomyces cerevisiae. . J Bacteriol. 1975;124:511–523. doi: 10.1128/jb.124.1.511-523.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SW, Meyer DI. ACT3: a putative centractin homologue in S. cerevisiaeis required for proper orientation of the mitotic spindle. J Cell Biol. 1994;127:129–138. doi: 10.1083/jcb.127.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene (Amst) 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- Cottingham FR, Hoyt MA. Mitotic spindle positioning in Saccharomyces cerevisiaeis accomplished by antagonistically acting microtubule motor proteins. J Cell Biol. 1997;138:1041–1053. doi: 10.1083/jcb.138.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan, K., and Y. Tanaka. 1990. Attachment of one spindle pole to the cortex in unequal cleavage. In Cytokinesis: Mechanisms of Furrow Formation during Cell Division. G.W. Conrad and T.E. Schroeder, editors. New York. 108–119. [DOI] [PubMed]
- DeZwaan TM, Ellington E, Pellman D, Roof DM. Kinesin- related KIP3 of Saccharomyces cerevisiaeis required for a distinct step in nuclear migration. J Cell Biol. 1997;138:1023–1040. doi: 10.1083/jcb.138.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshel D, Urrestarazu LA, Vissers S, Jauniaux J-C, van Vliet-Reedijk JC, Planta RJ, Gibbons IR. Cytoplasmic dynein is required for normal nuclear segregation in yeast. Proc Natl Acad Sci USA. 1993;90:11172–11176. doi: 10.1073/pnas.90.23.11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkasovsky M, Küntzel H. Yeast Num1p associates with the mother cell cortex during S/G2 phase and affects microtubular functions. J Cell Biol. 1995;131:1003–1014. doi: 10.1083/jcb.131.4.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foreman PK, Davis RW. Cloning vectors for the synthesis of epitope-tagged, truncated and chimeric proteins in Saccharomyces cerevisiae. . Gene (Amst) 1994;144:63–68. doi: 10.1016/0378-1119(94)90204-6. [DOI] [PubMed] [Google Scholar]
- Geiser JR, Schott EJ, Kingsbury TJ, Cole NB, Totis LJ, Bhattacharyya G, He L, Hoyt MA. Saccharomyces cerevisiae genes required in the absence of the CIN8-encoded spindle motor act in functionally diverse mitotic pathways. Mol Biol Cell. 1997;8:1035–1050. doi: 10.1091/mbc.8.6.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DS, Winston F. A ten min DNA preparation from yeast efficiently releases autonomous plasmids for transformation of E. coli. . Gene (Amst) 1987;57:267–272. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]
- Hoyt MA, He L, Loo KK, Saunders WS. Two Saccharomyces cerevisiaekinesin-related gene products required for mitotic spindle assembly. J Cell Biol. 1992;118:109–120. doi: 10.1083/jcb.118.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA. Centrosome movement in the early divisions of Caenorhabditis elegans: A cortical site determining centrosome position. J Cell Biol. 1989;109:1185–1193. doi: 10.1083/jcb.109.3.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs CW, Adams AEM, Szaniszlo PJ, Pringle JR. Functions of microtubules in the Saccharomyces cerevisiaecell cycle. J Cell Biol. 1988;107:1409–1426. doi: 10.1083/jcb.107.4.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahana JA, Schnapp BJ, Silver PA. Kinetics of spindle pole body separation in budding yeast. Proc Natl Acad Sci USA. 1995;92:9707–9711. doi: 10.1073/pnas.92.21.9707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland DE, Mitchison TJ, Kirschner MW. Polewards chromosome movement driven by microtubule depolymerization in vitro. . Nature. 1988;331:499–504. doi: 10.1038/331499a0. [DOI] [PubMed] [Google Scholar]
- Kurihara LJ, Beh CT, Latterich M, Schekman R, Rose MD. Nuclear congression and membrane fusion: Two distinct events in the yeast karyogamy pathway. J Cell Biol. 1994;126:911–923. doi: 10.1083/jcb.126.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y-Y, Yeh E, Hays T, Bloom K. Disruption of mitotic spindle orientation in a yeast dynein mutant. Proc Natl Acad Sci USA. 1993;90:10096–10100. doi: 10.1073/pnas.90.21.10096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillie SH, Brown SS. Suppression of a myosin defect by a kinesin-related gene. Nature. 1992;356:358–361. doi: 10.1038/356358a0. [DOI] [PubMed] [Google Scholar]
- Lombillo VA, Nislow C, Yen TJ, Gelfand VI, McIntosh JR. Antibodies to the kinesin motor domain and CENP-E inhibit microtubule depolymerization-dependent motion of chromosomes in vitro. . J Cell Biol. 1995;128:107–115. doi: 10.1083/jcb.128.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Lutz DA, Hamaguchi Y, Inoue S. Micromanipulation studies of the asymmetric positioning of the maturation spindle in Chaetopterus sp.oocytes: I. anchorage of the spindle to the cortex and migration of a displaced spindle. Cell Motil Cytoskeleton. 1988;11:83–96. doi: 10.1002/cm.970110202. [DOI] [PubMed] [Google Scholar]
- Marsh, L., and M.D. Rose. 1997. The pathway of cell and nuclear fusion during mating in Saccharomyces cerevisiae. In Cell Cycle and Cell Biology. J.R. Pringle, J.R. Broach, and E.W. Jones, editors. Cold Spring Harbor Laboratory. Cold Spring Harbor, New York. 827–888.
- McMillan JN, Tatchell K. The JNM1 gene in the yeast Saccharomyces cerevisiaeis required for nuclear migration and spindle orientation during the mitotic cell cycle. J Cell Biol. 1994;125:143–158. doi: 10.1083/jcb.125.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meluh PB, Rose MD. KAR3, a kinesin-related gene required for yeast nuclear fusion. Cell. 1990;60:1029–1041. doi: 10.1016/0092-8674(90)90351-e. [DOI] [PubMed] [Google Scholar]
- Muhua L, Karpova TS, Cooper JA. A yeast actin-related protein homologous to that in vertebrate dynactin complex is important for spindle orientation and nuclear migration. Cell. 1994;78:669–679. doi: 10.1016/0092-8674(94)90531-2. [DOI] [PubMed] [Google Scholar]
- Osmani AH, Osmani SA, Morris NR. The molecular cloning and identification of a gene product specifically required for nuclear movement in Aspergillus nidulans. . J Cell Biol. 1990;111:543–551. doi: 10.1083/jcb.111.2.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer RE, Koval M, Koshland D. The dynamics of chromosome movement in the budding yeast Saccharomyces cerevisiae. . J Cell Biol. 1989;109:3355–3366. doi: 10.1083/jcb.109.6.3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer RE, Sullivan SD, Huffaker T, Koshland D. Role of astral microtubules and actin in spindle orientation and migration in the budding yeast, Saccharomyces cerevisiae. . J Cell Biol. 1992;119:583–593. doi: 10.1083/jcb.119.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plamann M, Minke PF, Tinsley JH, Bruno KS. Cytoplasmic dynein and actin-related protein Arp1 are required for normal nuclear distribution in filamentous fungi. J Cell Biol. 1994;127:139–149. doi: 10.1083/jcb.127.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle, J.R., and L.H. Hartwell. 1981. The Saccharomyces cerevisiae cell cycle. In The Molecular Biology of theYeast Saccharomyces: Life Cycle and Inheritance. J.N. Strathern, E.W. Jones, and J.R. Broach, editors. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 97–142.
- Read EB, Okamura HH, Drubin DG. Actin- and tubulin-dependent functions during Saccharomyces cerevisiaemating projection formation. Mol Biol Cell. 1992;3:429–444. doi: 10.1091/mbc.3.4.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riles L, Dutchik JE, Baktha A, McCauley BK, Thayer EC, Leckie MP, Braden VV, Depke JE, Olson MV. Physical maps of the six smallest chromosomes of Saccharomyces cerevisiaeat a resolution of 2.6 kilobase pairs. Genetics. 1993;134:81–150. doi: 10.1093/genetics/134.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roof DM, Meluh PB, Rose MD. Kinesin-related proteins required for assembly of the mitotic spindle. J Cell Biol. 1992;118:95–108. doi: 10.1083/jcb.118.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose MD. Nuclear fusion in yeast. Annu Rev Microbiol. 1991;45:539–567. doi: 10.1146/annurev.mi.45.100191.002543. [DOI] [PubMed] [Google Scholar]
- Rose MD. Nuclear fusion in the yeast Saccharomyces cerevisiae. . Annu Rev Cell Dev Biol. 1996;12:663–695. doi: 10.1146/annurev.cellbio.12.1.663. [DOI] [PubMed] [Google Scholar]
- Rose MD, Fink GR. KAR1, a gene required for function of both intranuclear microtubules in yeast. Cell. 1987;48:1047–1060. doi: 10.1016/0092-8674(87)90712-4. [DOI] [PubMed] [Google Scholar]
- Rose MD, Novick P, Thomas JH, Botstein D, Fink GR. Saccharomyces cerevisiaegenomic plasmid bank based on a centromere-containing shuttle vector. Gene (Amst) 1987;60:237–243. doi: 10.1016/0378-1119(87)90232-0. [DOI] [PubMed] [Google Scholar]
- Rose, M.D., F. Winston, and P. Hieter. 1990. Methods of Yeast Genetics. Cold Spring Harbor Laboratory. Cold Spring Harbor, New York. 198 pp.
- Rothstein RJ. One-step gene disruption in yeast. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- Snyder M, Gehrung S, Page BD. Studies concerning the temporal and genetic control of cell polarity in Saccharomyces cerevisiae. . J Cell Biol. 1991;114:515–532. doi: 10.1083/jcb.114.3.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan DS, Huffaker TC. Astral microtubules are not required for anaphase B in Saccharomyces cerevisiae. . J Cell Biol. 1992;119:379–388. doi: 10.1083/jcb.119.2.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trueheart J, Boeke JD, Fink GR. Two genes required for cell fusion during yeast conjugation: evidence for a pheromone-induced surface protein. Mol Cell Biol. 1987;7:2316–2328. doi: 10.1128/mcb.7.7.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willins DA, Xiang X, Morris NR. An alpha tubulin mutation suppresses nuclear migration mutations in Aspergillus nidulans. . Genetics. 1995;141:1287–1298. doi: 10.1093/genetics/141.4.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang S, Osmani AH, Osmani SA, Xin M, Morris NR. a. NudF, a nuclear migration gene in Aspergillus nidulans, is similar to the human LIS-1gene required for neuronal migration. Mol Biol Cell. 1995;6:297–310. doi: 10.1091/mbc.6.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang X, Roghi C, Morris N. Characterization and localization of the cytoplasmic dynein heavy chain in Aspergillus nidulans. . Proc Natl Acad Sci USA. 1995b;92:9890–9894. doi: 10.1073/pnas.92.21.9890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Ayscough KR, Drubin DG. A role for the actin cytoskeleton of Saccharomyces cerevisiaein bipolar bud-site selection. J Cell Biol. 1997;136:111–123. doi: 10.1083/jcb.136.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E, Skibbens RV, Cheng JW, Salmon ED, Bloom K. Spindle dynamics and cell cycle regulation of dynein in the budding yeast, Saccharomyces cerevisiae. . J Cell Biol. 1995;130:687–700. doi: 10.1083/jcb.130.3.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Chen JK, Feng S, Dalgarno DC, Brauer AW, Schreiber SL. Structural basis for the binding of proline-rich peptides to SH3 domains. Cell. 1994;76:933–945. doi: 10.1016/0092-8674(94)90367-0. [DOI] [PubMed] [Google Scholar]