

Abstract
We have isolated a fission yeast karyogamy mutant, tht1, in which nuclear congression and the association of two spindle pole bodies occurs but the subsequent fusion of nuclear envelopes is blocked. The tht1 mutation does not prevent meiosis, so cells execute meiosis with two unfused nuclei, leading to the production of aberrant asci. The tht1 + gene was cloned and sequenced. Predicted amino acid sequence has no significant homology to previously known proteins but strongly suggests that it is a type I membrane protein. The tht1 + gene is dispensable for vegetative growth and expressed only in conjugating cells. Tht1p is a glycoprotein susceptible to endoglycosilase H digestion. Site- directed mutagenesis showed that the N-glycosylation site, as well as the COOH-terminal region of Tht1p, is essential for its function. A protease protection assay indicated that the COOH terminus is cytoplasmic. Immunocytological analysis using a HA-tagged Tht1p suggested that the protein is localized in nuclear envelopes and in the ER during karyogamy and that its levels are reduced in cells containing fused nuclei.
Karyogamy, or nuclear fusion, is a process in which two haploid nuclei fuse to produce a diploid nucleus in yeast. Genetic and cytological analyses in Saccharomyces cerevisiae showed that karyogamy consists of at least two distinct processes: one is the congression of two nuclei and the other is the fusion of nuclear envelopes of juxtaposed nuclei (Rose, 1991; Kurihara et al., 1994). The mixing of chromosomes and nuclear matrices brought by the two nuclei must follow envelope fusion and results in the formation of a diploid nucleus, however, this aspect of karyogamy has received little attention to date. Genes identified in S. cerevisiae as required for the nuclear congression include KAR1, KAR3, CIK1, BIK1, and TUB2, which are all components of spindle pole body (SPB)1– microtubular system (Rose, 1991). These factors are also shown to be either essential or important for vegetative growth. KAR4 is involved in the expression of KAR3 and CIK1 and therefore also required for the nuclear congression (Kurihara et al., 1996). On the other hand, a class of endoplasmic reticulum proteins, such as Kar2p (BiP), Sec63p, Sec71p, Sec72p, and Jem1p (Normington et al., 1989; Rose et al., 1989; Ng and Walter, 1996; Nishikawa and Endo, 1997), were all shown to be required for the fusion of nuclear envelopes. BiP is know to be a ubiquitous ER lumenal protein which is a member of stress-inducible chaperones. BiP functions in modulating protein folding and protein translocation (Gething and Sambrook, 1992). Sec63p, Sec71p and Sec72p form a membrane-bound complex required for protein translocation (Deshaies et al., 1991; Green et al., 1992; Ng and Walter, 1996), and Kar2p has been shown to interact with Sec63p (Scidmore et al., 1993; Brodsky et al., 1995). However, it remains to be elucidated how these ER proteins are involved in the nuclear envelope fusion.
We have been interested in the karyogamy in Schizosaccharomyces pombe, because cytological studies have proven it to be an excellent model system to study the regulation of nuclear organization. Very dynamic yet genetically regulated rearrangement of chromosomes, as well as morphological changes in nuclear shape, can be observed upon entry into and during karyogamy and subsequent meiosis in the fission yeast. In vegetatively proliferating cells, centromeres are either located near the SPB as a cluster or linked to the SPB with microtubules. Thus, the SPB take a major role in the positioning and movement of chromosomes (Funabiki et al., 1993). In sharp contrast to the situation in the mitotic cell cycle, during karyogamy and meiotic prophase telomeres form a single cluster near the SPB and dominate in the chromosomal movement (Chikashige et al., 1994, 1997). The SPB appears to play a vital role not only in these chromosomal events but also in the movement of nucleus, that is, during both congression of two haploid nuclei and the oscillatory movement of a meiotic prophase nucleus along the long axis of the zygote, the SPB leads the nuclear movement (Chikashige et al., 1994). The nucleus in the meiotic prophase is also known to have characteristically elongated and ever changing morphology, often referred to as a “horse tail” nucleus (Robinow, 1977).
We have recently isolated a fission yeast mutant, kms1 (Shimanuki et al., 1997), which does not form the smoothly elongated horse tail nucleus in meiotic prophase but produces an aberrantly shaped nucleus in which the telomeres often fail to form a single cluster. Instead they tend to be distributed in multiple clusters. The kms1 mutant is also impaired in karyogamy and in the meiotic chromosome segregation and sporulation. These mutant phenotypes are most likely correlated with the failure of SPB function. In fact, we recently found that the kms1 + gene product may function as an indispensable component of the SPB when the telomere cluster associates with it (Shimanuki, M., unpublished observation). The observations of the kms1 phenotype further emphasized the importance of SPB function and chromosomal organization in sexual nuclear processes in fission yeast. In an effort to identify more factors involved in such a process, we previously made a collection of mutants that were defective in diploid formation after conjugation (Tange and Niwa, 1995). In this study we have isolated and characterized a mutant that is defective in the tht1 gene. Results described in this report indicate that the nuclear congression and the association of two SPBs takes place but that the subsequent fusion of nuclear envelopes is blocked in the mutant. The tht1 + gene encodes a novel glycoprotein transiently produced during conjugation. It is the first gene identified in the fission yeast that may be specifically required for the fusion of nuclear membranes during karyogamy.
Materials and Methods
Genetic Procedures
Standard genetic methods were followed (Gutz et al., 1974; Moreno et al., 1990). YEA and YPD were used as complete media, MEA for conjugation and sporulation, and EMM2 as a minimal medium. In some experiments conjugation was induced in liquid according to the procedure described by Miyata et al. (1997). In brief, heterothallic strains of different mating types were cultured separately at 30°C overnight to a concentration of 1 × 108 cells/ml in MEB-Gal medium (2% malt extract [Oxoid] and 1% d-galactose). For the nitrogen starvation, cells were washed once in 0.1% d-glucose and the two strains were mixed in MSM medium (1% d-galactose, 0.4% d-mannose, 0.15% KH2PO4 and four vitamins as in EMM2), each at a concentration of 3 × 107 cells/ml, followed by incubation at 30°C with vigorous shaking (140 rpm). For homothallic strains the final cell density in MSM was brought to 6 × 107 cells/ml. In the experiment for immunolocalization, we induced conjugation by the procedure described by Beach et al. (1985).
Mutant Isolation
We have made a mutant collection based on the genetic background of h 90 mei1-102 leu1 tsh1, aiming a class of mutants defective in the diploid formation after conjugation (Tange and Niwa, 1995). Individual strains were examined for the nuclear morphology in zygotes by 4,6-diamidino-2-phenylindole (DAPI) staining. Strain DF4-3 found in this screening was subjected to detailed analyses in this study.
Determination of Ploidy
Homothallic yeast strains were incubated on a MEA medium at 30°C overnight to induce zygote formation. Individual zygotes were transferred onto fresh YEA medium using a micromanipulator and incubated at 30°C for several days. Colonies produced from the separated zygotes were examined for their ploidy by the flow cytometric analysis as described in Tange and Niwa (1995).
Plasmids
The multicopy plasmid pKD10 (Shimanuki et al., 1997) was used for subcloning and complementation test. Fragments inserted into the vector are shown in Fig. 6 A. For the overexpression experiments, pREP1, pREP42 (gifts from Dr. K. Maundrell, Glaxo Institute for Molecular Biology, Geneva, Switzerland), and pAS248 (Toda et al., 1991) were used. pREP1 and pREP42 carry the nmt1-promoter (Maundrell, 1993), which is inducible by thiamine depletion. A DNA fragment covering the tht1 +-ORF was produced by PCR and inserted at the NdeI–BamHI site of pREP1 and pREP42 to make pNT45 and pNT97, respectively. pAS248 carries a constitutively active adh promoter. The BamHI–PstI fragment from pNT37 was blunt ended and inserted into the SmaI site in pAS248 to generate pNT79. The HA-tag sequence (AYPYDVPDYAGYPYDVPDYAMGY PYDVPDYA, repeated HA-epitopes are underlined) was amplified from a plasmid pHA41 (a gift from I. Hagan, University of Manchester, Manchester, UK) and inserted at the BsmI, HpaI, StuI, and NcoI sites in pNT28 to make pNT72, pNT73, pNT74, and pNT75, respectively. pNT72, pNT73, and pNT74 could not complement the tht1-1 mutation, whereas pNT75 could. A control plasmid, pNT77, containing the HA-tag sequence was constructed by replacing the NspV fragment of pNT28 with the HA-tag sequence. In this construct the HA-tag is fused to the NH2-terminal 38 amino acids of tht1 +. The plasmid pEB9 carries the BiP gene tagged with the myc epitope (Pidoux and Armstrong, 1992). The plasmid D817, which was employed for staining the nuclear envelope in this study, had been isolated from a GFP-fusion genomic library based on a multicopy vector with the LUE2 selection marker. Sequence determination of the D817 clone showed that the 276 amino terminal residues of the 678–amino acid cytochrome P-450 reductase (Miles, 1992) were fused with GFP in this plasmid (Ding, D.-Q., and Y. Hiraoka, manuscript in preparation). GFP-tagged Kms1p was used to stain the SPB, which was expressed from pGK77, a pREP1-based plasmid. The fusion protein produced under the repressive condition was shown to localize to the SPB. Details about the localization of Kms1p will be published elsewhere.
Figure 6.
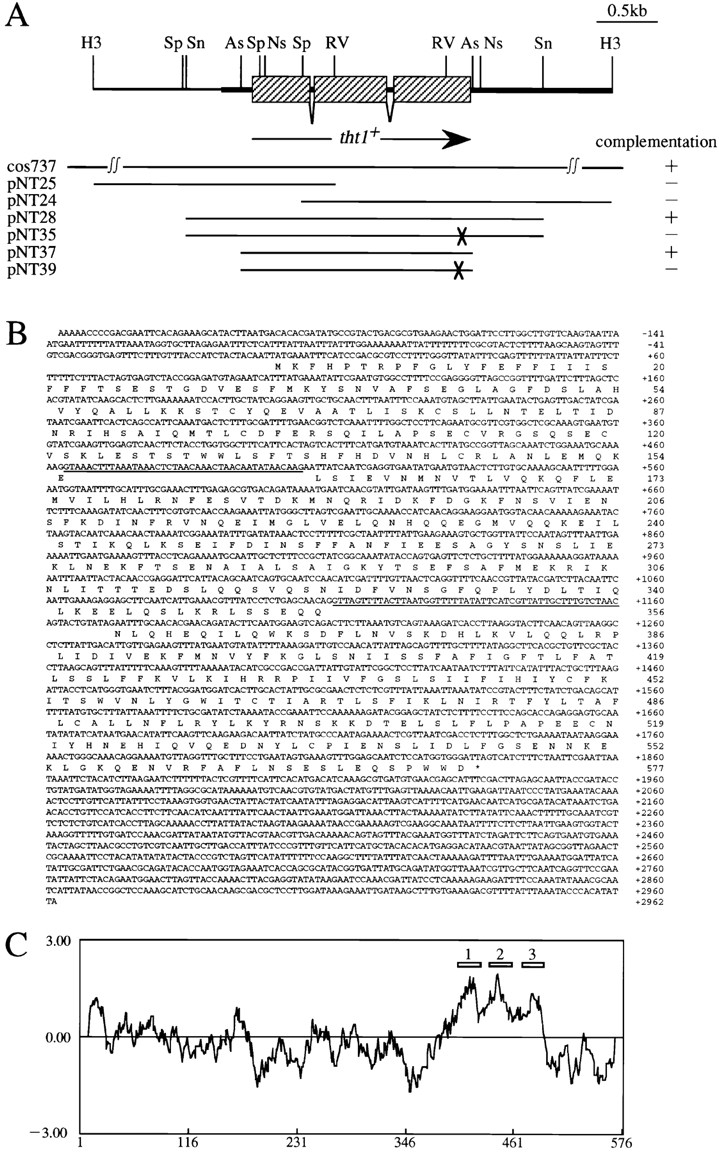
(A) Simplified restriction map around the tht1 + gene and the complementation activity of subclones. Shaded rectangles represent tht1 +-ORF interrupted with two introns. Each subclones contain indicated genomic segment. pNT35 and pNT39 carry tht1-1 mutant sequence as indicated by the crosses. Complementation test was carried out as in Fig. 10. (B) Genomic nucleotide sequence of the DNA segment shown in the thick line in A (this sequence has been deposited in the DDBJ database with the accession number D87337). Predicted amino acid sequence of Tht1p and two introns (underlined) are shown. (C) A hydropathy plot of Tht1 according to the Kite and Doolittle method (plotted as the average of 20 amino acids). The numbered rectangles indicate the position of postulated transmembrane segments.
Gene Cloning and Sequencing
tht1 + gene was cloned from cosmid libraries of S. pombe genomic DNA. One of the libraries had been made by using a cosmid vector sCos1-LEU2 (Murakami and Niwa, 1995). Another library was obtained from Dr. D. Beach via Dr. Y. Oshima (Kyushu University, Japan), which is based on the pSS10 vector (Nakaseko et al., 1986). h 90 leu1-32 tht1-1 was transformed by the lithium acetate method. Cosmid DNA was recovered from yeast transformants according to Moreno et al. (1991). One cosmid, cos737, was used for further study. The relevant DNA segments were sequenced by the dideoxy method. cDNA analysis was performed as described in Shimanuki et al. (1997).
One Step Gene Disruption
The 2.9-kb SnaBI fragment of the tht1 + gene was inserted in the SmaI site of Bluescript II (Stratagene, La Jolla, CA), and the ura4 + gene sequence was substituted with the 1.7-kb NspV fragment in the insert to generate pNT31. This substitution nearly completely disrupts the tht1 + gene, leaving only 38 amino acid residues at its NH2 terminus. The KpnI–SacI fragment of pNT31 was excised and transformed into a diploid cell to obtain stable transformants, for which Southern blot hybridization was performed to verify the correct integration. For the disruption of the 740-bp open reading frame (ORF), the 2.2-kb NspV fragment bearing the ORF was inserted into the EcoRV site in Bluescript II (pNT60). The EcoT14I– EcoRV fragment of pNT60 was replaced with the ura4 + sequence to completely remove the ORF sequence and then the XhoI–SpeI fragment was cut out of this plasmid for transformation.
Northern Blot Hybridization
Total RNA was extracted according to Alfa et al. (1993). Poly(A)+ mRNA was purified by using the oligotex-dT30 (super; Takeda Pharmaceutical Co., Osaka, Japan). RNA probes were used for hybridization. Each probe was transcribed from the T7 promoter using the DIG RNA labeling kit (Boehringer Mannheim Corp., Indianapolis, IN) with the following template plasmid DNAs. For probe 1 (see Fig. 8) the XhoI–RsaI fragment (from nucleotide −1202 to −189, numbering is from the initiation codon of the tht1 + gene) was ligated with the XhoI–EcoRV site of Bluescript II to make pNT46. XhoI-digested pNT46 DNA was used as the template. For probe 2 the HindIII–EcoRV fragment (from nucleotide −1271 to +670) was cloned into the HindIII–SmaI site of pKD10 (pNT25). pNT25 was digested at the SalI site (located at −39). For probes 3, 4, and 5 the EcoRV fragment (+670 to +1588), the MunI fragment (+2030 to +2482; blunt ended), and the DdeI fragment (+2781 to +3333; blunt ended) were inserted at the EcoRV site of Bluescript II in an appropriate orientation to make pNT42, pNT58, and pNT59, and then digested with HindIII, HindIII, and XhoI, respectively, before use. Another plasmid, pNT47, was made by ligating the RsaI–HindIII fragment (+2356 to +2922) with the HincII and HindIII sites in Bluescript II followed by digestion with XhoI. This probe was used for the analysis of the disruptant of the 740-bp ORF (see text). Hybridization and washing was conducted using the standard conditions recommended by Boehringer Mannheim Corp., with the exception of probe 4, which was hybridized in a high SDS buffer without formamide to reduce the stringency of hybridization (Church and Gilbert, 1984). Band detection was performed with the DIG luminescent detection system (Boehringer Mannheim Corp.).
Figure 8.

Analysis of tht1 + transcripts. At indicated times after transferring to the MSM medium, poly(A)+ RNA was extracted and probed with indicated sequences (shaded rectangles). The nature of the bands produced by probe 4 (solid arrowhead) is not known.
Production of Antibodies
A segment of the tht1 + gene corresponding to amino acids 42– 154 was inserted into the pGEX vector (Pharmacia Biotech, Inc., Piscataway, NJ) to produce a GST-fused protein. Bacterially produced fusion protein was purified and injected into rabbits to raise antibodies. Antibodies were affinity purified with the fusion protein according to Smith and Fisher (1984).
Preparation of Cell Extracts and Western Immunoblotting
Cell extracts for Western immunoblotting were prepared according to Moreno et al. (1991) and Funabiki et al. (1996) with modifications. 3 × 107 cells were harvested and washed once with ice-cold STOP buffer (150 mM NaCl, 50 mM NaF, 10 mM ethylenediaminitetraacetate [EDTA], 1 mM NaN3, pH 8.0). Cells were resuspended in 150 μl of disruption buffer (50 mM Tris-HCl, pH 7.5, 10 mM EDTA, 10% glycerol, 30 mM NaCl, 1 mM DTT, 1 mM PMSF, 2 μg/ml pepstatin A, 10μM E-64) and disrupted with 0.3g glass beads (425–600 μm; Sigma Chemical Co., St. Louis, MO) by vortexing four times each for 20 s. 200 μl of disruption buffer was added to the disrupted cell suspension and centrifuged at 1,500 g for 5 min. Supernatant fraction was then centrifuged at 57,000 g for 20 min. Resultant supernatant was referred as the soluble fraction and the pellet was suspended in 60 μl of disruption buffer and referred as the insoluble fraction. To each fraction, one-fourth volume of loading buffer (0.25 M Tris-HCl, pH 6.8, 4% SDS, 40% glycerol, 10% mercaptoethanol, 40 mg/ml bromophenol blue) was added and boiled for 5 min and then chilled on ice. In some cases this boiling step was omitted. Before loading on a gel, samples were centrifuged at 10,000 g for 5 min to remove insoluble materials. Alternatively, cell extracts were prepared according to the alkaline lysis method (Silve et al., 1991) with a slight modification. 1.5 × 107 cells were washed once in 0.5 ml of ice–cold STOP buffer and resuspended in a minimum amount of STOP buffer. 0.5 ml of 1.85 M NaOH 7.5% mercaptoethanol was added to the cell suspension and incubated for 10 min on ice. 0.5 ml of 50% TCA was mixed and kept on ice for 10 min, followed by a centrifugation at 15,000 g for 10 min. The pellet was rinsed with 0.5 ml of 1 M Tris-OH and suspended in 50 μl of disruption buffer plus 17 μl of loading buffer. This mixture was kept on ice for 20 min and then centrifuged at 10,000 g for 5 min to remove insoluble materials. Proteins were run on 12.5% PAGE and transferred to a nitrocellulose membrane. Protein detection was performed by a chemiluminescence detection system (ECL; Amersham Corp., Arlington Heights, IL) with horse radish peroxidase-conjugated protein A (Amersham Corp.).
Protease Protection Assay
Methods described by Baker et al. (1990) and Garnier et al. (1996) were modified as follows. 5 × 109 conjugating cells were harvested after 5 h incubation in MSM medium, washed once with 50 mM Tris-HCl, pH 7.5, 50 mM EDTA and once again with the same buffer containing 1.2 M sorbitol (TES). Cells were digested at 30°C for 45 min with 0.5 mg/ml Zymolyase 100T (Seikagaku America, Inc., Rockville, MD) and 0.2 mg/ml lysing enzymes (Sigma Chemical Co.) in TES at a cell density of 5 × 108 cells/ml. The digested cells were washed once with solution A (50 mM Tris-HCl, pH 7.5, 10 mM EDTA, 30 mM NaCl, 0.5% 2-mercaptoethanol, 1 mM PMSF, 2 μg/ml pepstatin A, 10 μM E-64) containing 1.2 M sorbitol and suspended in 4 ml of lysis buffer (solution A containing 0.1 M sorbitol). The suspension was homogenized by a Dounce homogenizer (10 strokes), followed by centrifugation at 3,000 g for 10 min. The supernatant was centrifuged at 12,000 g for 10 min. The resultant pellet was suspended in 0.4 ml of lysis buffer, layered onto two tubes of 2.0-ml sucrose step gradient (1.0 ml each of 1.5 M sucrose and 1.2 M sucrose in lysis buffer), and then centrifuged at 100,000 g for 1 h. The microsomes at the 1.2/1.5 M interface were collected and washed once with lysis buffer and once with reaction buffer (lysis buffer without protease inhibitors), before being suspended in 1.0 ml of reaction buffer. 100-μl aliquots were incubated with TPCK-treated trypsin (0-5 μg/ml; Sigma Chemical Co.) at 25°C for 10 min. Reaction was terminated by adding 25 μl of 50% TCA. After 15 min incubation on ice, the pellet was collected (15,000 g for 10 min) and dissolved in 50 μl of solution A. Where indicated, Triton X-100 was added at a final concentration of 0.1% and the resulting solution was incubated at 25°C for 5 min before the addition of trypsin.
Overexpression of the tht1+ Gene
For the overexpression of the tht1 + gene in vegetative cells, HM123 (h − leu1) was transformed either with pNT45 or with pNT79. Transformants carrying pNT45 were transferred from EMM2 with thiamine to the thiamine-depleted medium at the cell density of 3 × 105 cells/ml at 30°C to induce the expression of the tht1 + gene.
Digestion with Endoglycosilase H
Cell extracts prepared by the alkaline lysis method were digested with endoglycosilase H (endoH; New England Biolabs Inc., Beverly, MA) according to the manufacturer's instructions.
Site-directed Mutagenesis
The Quikchange site-directed mutagenesis kit (Stratagene) was used to introduce desired mutations in pNT28. For each mutant gene the entire nucleotide sequence of the ORF was determined to confirm that only the desired changes had been introduced.
Fluorescent Staining
For indirect immunofluorescent staining, we followed the procedure described in Chappell and Warren (1989) but Novozyme was omitted from the digestion mixture. In this method, 4% formaldehyde was used for fixing cells but glutaraldehyde was not included. In some experiments, including GFP-DAPI double staining, the method described in Shimanuki et al. (1997) was followed. But for double staining, cell wall digestion was performed only briefly and the Triton X-100 treatment was omitted. Mouse monoclonal anti–HA antibody (12CA5; Boehringer Mannheim Corp.) was used in combination with a goat anti–mouse Cy3-conjugated antibody (The Jackson Laboratories, Bar Harbor, ME). We also used a goat anti–rabbit Cy3-conjugated antibody (Jackson Laboratories). For immunolocalization of the tagged BiP, mouse monoclonal anti–c-myc antibody (9E10; Sigma) was used (Pidoux and Armstrong, 1992). For membrane staining 0.25 or 0.125 μg/ml of DiOC6(3) (3,3′-dihexyloxacarboctanine iodide; LAMBDA, Graz, Austria) was added (Terasaki, 1994). Fluorescent in situ hybridization (FISH) was performed using a telomere specific probe cos212 according to Shimanuki et al. (1997). Microscopic observation was carried out with the Delta Vision system (Applied Precision Inc., Issaquah, WA). Coloring in all figures was arbitrary.
Results
tht1 Mutant Is Defective in the Nuclear Fusion
From a collection of S. pombe mutants that are defective in diploid formation after mating, we found one in which nuclear fusion was severely impaired. Tetrad analysis showed that this defect was due to a mutation in a single nuclear gene, which we have designated tht1 (twin horse tails, see below). Homothallic (self conjugating) strains with or without the mutation were compared for the efficiency of nuclear fusion by DAPI staining. These strains carried the mei1-102 mutation that blocks meiotic divisions after nuclear fusion (Egel, 1989), so that 99% of the tht1 + zygotes contained a single fused nucleus (Fig. 1 a). In sharp contrast, in the tht1 mutant 96% of conjugated cells contained two closely juxtaposed, but unfused, DAPI-stained bodies (Fig. 1, b and c, left). We then tried to examine whether the two nuclear envelopes also remained unfused. To this end, we used a green fluorescent protein (GFP)–fused hybrid protein that had been shown to stain the nuclear envelope in fission yeast (Ding, D.-Q., and Y. Hiraoka, unpublished data; see Materials and Methods). As shown in Fig. 1, b and c (right), the nuclear envelopes remained largely unfused in the mutant. We can not rule out the possibility that nuclear membranes were partially fused in the arrested zygotes. Electron microscopic observation will be needed to address this issue. However, it may be worth noting that the unfused nuclei persisted for at least 24 h while maintaining their individual identity, suggesting that the unfused state is fairly stable. Consistent with this, when we put individual zygotes of the tht1 mei1 double mutants back into nutrient-rich medium to let them reenter the cell cycle and form colonies, ∼85% were found to form haploid colonies. Under the same condition, tht1 + zygotes produced diploid colonies in >90% of the cases.
Figure 1.
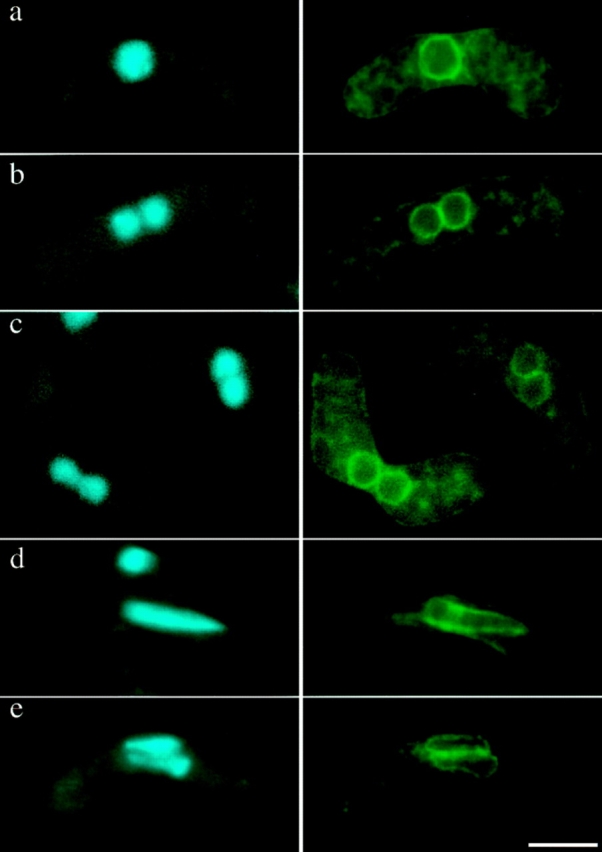
The terminal phenotype of the tht1-1 mutant. Strains carrying D817 plasmid were incubated on MEA at 18°C for 3 d. (Left) Stained with DAPI; (right) nuclear envelopes visualized with a GFP-fused hybrid protein produced from D817 (see text). (a–c) zygotes with mei1-102 mutation, blocking before entry into meiosis. (a) tht1 +; (b and c) tht1-1; (d and e) mei1 + zygotes in the horse tail stage; (d) tht1 +; (e) tht1-1. Bar, 5 μm.
Twin Horse Tails in the tht1 Mutant
Wild-type S. pombe cells usually proceed to meiosis immediately after nuclear fusion (zygotic meiosis), although meiosis can be induced directly from a diploid cell without intervening conjugation (azygotic meiosis). In both of these types of meiosis, prophase is accompanied by the presence of an oscillating horse tail nucleus (see Introduction, Fig. 1 d). When the tht1 mutant was induced to undergo a zygotic meiosis, we did not find a single horse tail nucleus in each cell, but instead we frequently found a pair of small horse tail nuclei lying side by side both by DAPI staining and by the nuclear envelope staining (Fig. 1 e). As anticipated, we found that telomere clusters were located near the tips of the paired nuclei (Fig. 2 A). In these twin horse tails the pointed ends were always on the same side, suggesting that these pairs of horse tail nuclei were moving in a coordinated fashion. In fact, when we observed the horse tail movement in live cells according to the procedures of Chikashige et al. (1994), twin horse tail nuclei always moved in such a way that the two pointed ends were dragged together from a single oscillating site where the SPB should reside, suggesting that in the conjugation of the tht1 mutant the pair of SPBs have bound to form a single diploid SPB after the nuclear congression, which can lead the movement of two unfused horse tail nuclei. To verify this, we performed an immunofluorescence staining of the SPB using the anti–Sad1 antibody (Hagan and Yanagida, 1995). In this analysis, we have carried out optical sectioning with 0.1-μm intervals to minimize the possibility of overlapped signals. In 24 out of 25 cases observed, only a single stained spot could be observed (Fig. 2 B). In the remaining one case, two distinct spots were observed, although they were very closely opposed. In wild-type control cells, in all of 25 cells single spot was stained. Similar results were obtained from analysis of a tht1 mutant strain with the mei1 mutation. In this analysis, GFP-tagged Kms1p was used for staining the SPB, because this fusion protein had been shown to localize to the SPB (Shimanuki, M., unpublished result). In 22 out of 25 cases, there was only one signal between juxtaposed nuclei (Fig. 3) and in the remaining three cases two adjoining spots were observed. In a tht1 + control strain, 24 out of 25 contained single spots. These observations not only confirmed the results on the twin horse tail nuclei, but also indicated that the binding or close association of SPBs was completed before the horse tail period, that is, meiotic prophase.
Figure 2.
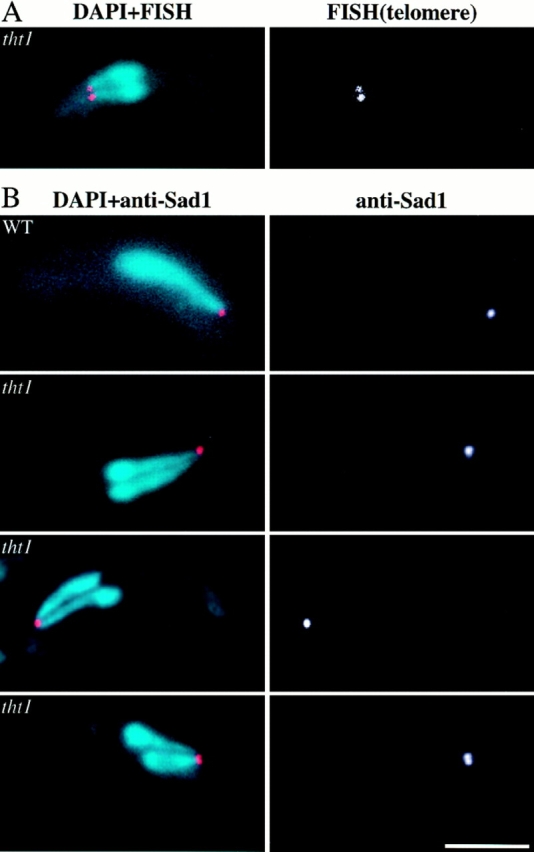
(A) FISH analysis of the twin horse tails with a telomere probe (right). (Left) Merged image of DAPI (light blue) and telomere-FISH (red). (B) SPBs stained with the anti–Sad1 antibody (right). Merged images of (light blue) DAPI stains and (red) anti–Sad1 (left). Bar, 5 μm.
Figure 3.

SPB staining with GFP-fused Kms1p (green). DAPI images are in red. Bar, 5 μm.
Aberrant Sporulation in tht1 Zygotes
Zygotic asci produced by the tht1 mutant were abnormal, containing an irregular number of spores with aberrant morphology (Fig. 4). DAPI-stained bodies observed in these mutant asci were also irregular in number and intensity. Furthermore, they often contained nuclei that were not encapsulated with spore walls (Fig. 5, b, e, f, arrows). The viability of the spores, which were released by enzymatic treatment of the asci, was 28%, while for wild-type control spores it was 70%. Moreover, as many as 35% of the viable mutant spores were diploid. These results suggested that chromosomes were not correctly transmitted into the mutant spores. When diploid cells homozygous for the tht1 mutation were induced to carry out meiosis/ sporulation, however, resultant azygotic asci appeared completely normal and contained four viable haploid spores (Fig. 4). The recombination frequency as well as the morphology of horse tail nucleus was also normal in the azygotic meiosis (data not shown), indicating that the tht1 mutation itself does not interfere with meiosis and sporulation. Therefore, it is likely that the aberrant mutant asci were produced via the inability to fuse the nuclei resulting in the twin horse tail stage.
Figure 4.
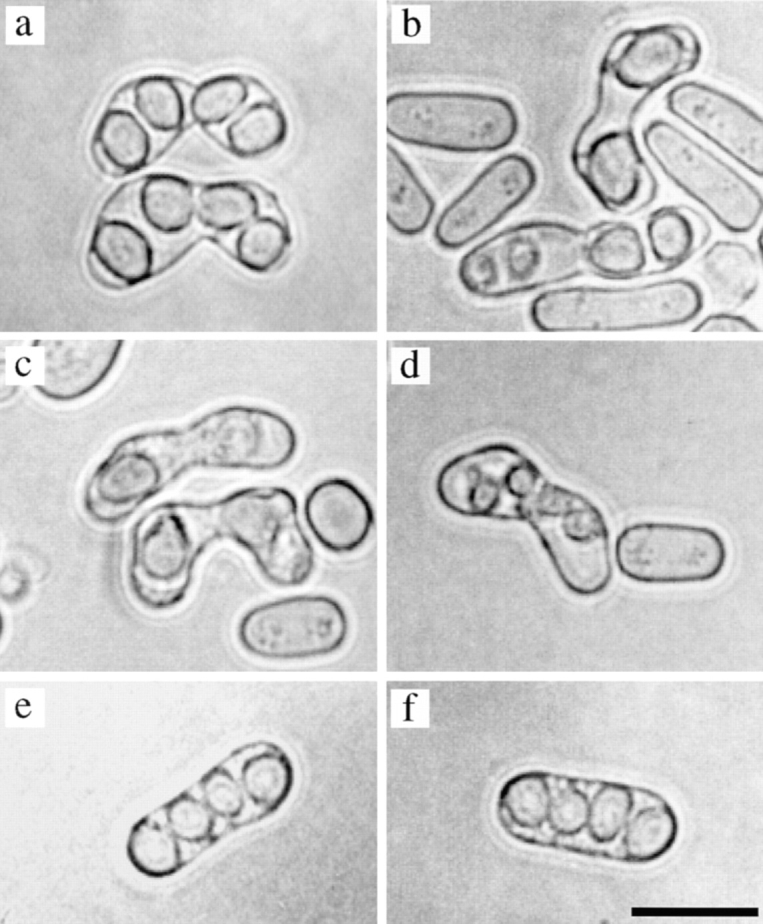
Aberrant sporulation in the tht1 mutant. Zygotic asci are a–d. Azygotic asci are shown in e and f. (a and e) Wild type; (b–d and f) tht1. Bar, 10 μm.
Figure 5.

Irregular distribution of chromosomes in the tht1 mutant. All are zygotic asci. a is wild type and the others are tht1 mutant. Arrows indicate nuclei that are not encapsulated with spore wall. Bar, 10 μm.
Cloning of the tht1+ Gene
Since the tht1 mutation was recessive to wild type regarding the abnormal ascus formation, the tht1 + gene was cloned by using cosmid libraries. Two overlapping cosmids derived from the same chromosomal region were isolated. Integration mapping indicated that the cosmid clones contained the tht1 + gene. By subcloning and sequencing segments of the cosmids we found that the tht1 + gene contained an ORF of ∼1.8 kb that was interrupted with two introns. The presence of these introns has been confirmed by a PCR based cDNA analysis. We found that a cosmid sequence that had been previously deposited in the EMBL/ GenBank/DDBJ database (available under accession number Z50112) contained the tht1 + sequence. This cosmid (c13C5) had been mapped to the distal portion of the left arm of chromosome I (Hoheisel et al., 1993). We then made a complete loss-of-function mutant allele at the tht1 locus by a one-step gene disruption method (Materials and Methods). Strains carrying the disruptant allele were viable and did not show any defective phenotype in vegetative growth, but, as anticipated, were as defective in karyogamy as the original tht1-1 mutant.
The predicted amino acid sequence of Tht1p along with a hydropathy plot is shown in Fig. 6. The calculated mol wt of the 577–amino acid protein is 66.9 kD. Computational searches have failed to find out any known proteins with significant sequence homology, indicating that Tht1p represents a novel class of proteins. There are three potential transmembrane segments in the COOH-terminal half of the protein, with positively charged amino acid residues enriched between segments 1 and 2 and near the COOH-terminal side of segment 3 (Fig. 6). There is also a typical signal peptide sequence at the NH2 terminus. These features of the protein may indicate that the tht1 + gene product is a type I membrane protein, with the COOH-terminal portion being in the cytoplasm (High and Dobberstein, 1992).
The Expression of tht1+ Gene Is Confined to Conjugating Cells
Northern blot hybridization analysis was performed using probes specific for the tht1 + gene (Fig. 7). About 3 h after the induction of conjugation by nitrogen starvation, two kinds of poly(A)+ transcripts of ∼1.8 and 3.8 kb appeared and the amount of these transcripts peaked at 5 h and then decreased (Fig. 7 A). The timing of the induction was roughly coincident with the appearance of copulating cells. Nitrogen starvation and the presence of two different mating type genes were not sufficient for the induction, since neither heterothallic haploid cells nor diploid cells heterozygous for the mating type gene could produce the transcripts after nitrogen starvation (Fig. 7 B). These results indicated that tht1 gene is only expressed in conjugating cells.
Figure 7.

Northern hybridization analysis of the expression of the tht1 + gene. (A) Poly(A)+ mRNA were extracted at the times indicated after transferring to MSM from MEB-gal medium (Materials and Methods). Probe 3 (see Fig. 8 for its position) was used. (B) Lack of gene expression in diploid and heterothallic haploid cells. A h +/h − diploid strain was transferred to nitrogen-depleted medium. Poly(A)+ RNA was extracted at 0 (a and d), 3 (b and e), and 6 h (c and f) and then hybridized with probe 3 (a–c) and probe 5 (d– f). In g–i, total RNA was extracted at 5 h after transferring to the MSM medium from h − wild-type haploid cells (g), h + (h) and mixture of h − and h + cells (i) and hybridized with probe 3.
We then examined the origins of the two different transcripts. Southern hybridization analysis indicated that there is only one copy of the tht1 + gene in the genome. We performed Northern hybridization using several different probes specific for either the tht1 +-ORF sequence or neighboring sequences (Fig. 8). Both 1.8- and 3.8-kb mRNAs were hybridized to probes 2 and 3, which were specific for 5′- and 3′-half of the ORF, respectively. However, with probes 4 and 5, only a 3.8-kb band was detected. Interestingly, probe 5 also hybridized to a 0.8-kb mRNA, which was probably corresponded to the 740-bp ORF. Probe 1, an upstream specific probe, did not hybridize to the tht1 + transcripts, but instead hybridized to a 1-kb mRNA covering the 1 kb-ORF. A PCR-based analysis suggested that both of the two transcripts start at about the same site (∼50 bp upstream from the initiation codon) and that the 1.8-kb mRNA terminates ∼100 bp downstream from the termination codon. These results indicated that the tht1-ORF is transcribed with two different mRNAs. Since it was possible that the 740-bp ORF has some functional relation to the tht1 gene, we disrupted this ORF by a ura4 + substitution method (Materials and Methods). The disruptant was viable and displayed no karyogamy related phenotype. In this disruptant, the 0.8-kb mRNA was no longer transcribed and the 3.8-kb mRNA became shorter. The amount of the 1.8-kb mRNA in the disruptant was comparable to its level in wild-type controls (data not shown).
Identification of the tht1+ Gene Product
We prepared extracts from cells carrying a multicopy plasmid with the tht1 + gene (pNT28) and performed a Western immunoblot analysis using anti–Tht1p antibodies (1-3-2). We found a 67-kD protein specifically expressed in conjugating cells (Fig. 9 A). The temporal expression profile was very similar to the mRNA accumulation profiles detected by Northern hybridization analysis. Moreover, the protein levels were very low in strains without the plasmid or with the vector alone (Fig. 9 A), suggesting that the 67-kD band represented the tht1 + gene product. This protein partitioned to the insoluble fraction (Fig. 9 B). It could be barely solubilized with 1% Triton X-100 or with 2 M NaCl (data not shown). The same 67-kD insoluble protein was produced when we forced gene expression in mitotic cells by using the adh-promoter (see Fig. 9 C).
Figure 9.

Western immunoblot analyses of the tht1 gene product. (A) Heterothallic strains with opposite mating types each carried pNT28 or pKD10 (vector) were mixed in the MSM medium and incubated for the times indicated. (B) Extracts from conjugating cells prepared as in A at 5 h were divided into the soluble (S) and the insoluble (P) fractions. (C) Cell extracts made from h − cells overproducing Tht1p from pNT79 and from conjugating homothallic cells containing pNT28 were treated with or without endoH. (D) Homothallic strains carrying pNT28 or its derivative plasmid bearing mutations at the potential N-linked glycosylation sites were brought to conjugation by 4 h incubation in the MSM medium. Extracts were prepared by the alkali method (in A, C, and D) or by the glass beads method (as in B; see Materials and Methods). anti–Tht1p antibody 1-3-2 was used for band detection in these experiments. The bands indicated by the asterisks were nonspecific because it appeared even from cells with disrupted tht1 gene (data not shown).
Tht1p Is a Glycoprotein
Since there are two potential N-linked glycosylation sites in the tht1 + protein sequence, we examined whether Tht1p is actually glycosylated. To this end we took two approaches. First, we examined whether the protein is a substrate for endoH, an N-glycosylation–specific glycosilase. Results shown in Fig. 9 C indicated that endoH digestion could make the 67-kD band shift down to 64 kD. This 3-kD difference roughly corresponds to the removal of a single oligosaccharide. Second, we performed site-directed mutagenesis to change the acceptor Asn residues to Asp. Mutant genes carrying N163D, N372D, or doubly mutated sequence were expressed in conjugating cells. Western immunoblot analysis clearly indicated Asn163 is glycosylated, but that Asn372 is not a target of N-glycosylation (Fig. 9 D). Functional complementation analysis showed that neither the N163D mutant nor the double mutant were able to rescue the tht1-1 mutation, while N372D was as active as wild type (Fig. 10). From these results we concluded that the Asp residue at 163 is glycosylated and that this modification is likely to be essential for function.
Figure 10.

Complementation activity of various tht1 mutants produced by site-directed mutagenesis. h 90 leu1 tht1-1 and h 90 mei1-102 leu1 tht1-1 were transformed with respective plasmid and each transformant was incubated on MEA at 30°C for 2 d to score the frequency of normal asci (with the mei1 + strain) and of zygotes with fused nuclei (with the mei1-102 strain). The left-most shaded rectangle represents the signal sequence and the gray rectangle represents the postulated membrane spanning domain. The two dots show approximate positions of potential N-glycosylation sites.
The COOH-terminal End of Tht1p Is Essential and Cytoplasmic
In the original tht1-1 mutant gene we found a one-base substitution near the 3′-end of the ORF (Fig. 10). It was a nonsense mutation that would produce a truncated protein missing three amino acids (WWD) from the COOH terminus. This suggested the COOH-terminal end of the protein performs an essential functional role. To test this possibility we created several kinds of mutations near the COOH terminus. For each of these mutant genes we examined the ability to rescue the defects both in nuclear fusion and ascus formation of a strain carrying the tht1 disruption allele. Results summarized in Fig. 10 indicated that both of the two consecutive tryptophane residues are essential for function. Alterations in the surrounding amino acids had very little effect except for the deletion of the most terminal Asp residue which led to a partial loss of function. Even the addition of an extra sequence of four amino acids to the COOH terminus, as well as the insertion of the HA-tag sequence before the PWWD sequence, did not impair the activity. It should be noted that Western blot analysis showed that the amount of protein produced from each of these mutant genes was comparable to that produced from the wild-type gene (data not shown).
As noted above, it was predicted from the amino acid sequence that the tht1 + gene product is a type I membrane protein, with its COOH terminus in the cytoplasm. To test this prediction, we prepared microsomes from conjugating cells expressing HA-tagged or wild-type Tht1p. One of the tagged proteins, Tht1-NcoI-HA, bears the HA epitope near the COOH terminus, while the other one, Tht1-StuI-HA, contained it on the NH2-terminal side of the presumed membrane-spanning domain. When the microsome samples were incubated with trypsin and subjected to Western immunoblot analysis using anti–HA antibodies, the band intensity of Tht1-NcoI-HA fusion decreased as the amount of trypsin increased (Fig. 11 A, lanes 1– 5). However, when Tht1-StuI-HA was examined there was a shift to a band that migrated with a mobility equivalent to a reduction in size of 3 kD and there was only a partial decrease in band intensity. (Fig. 11 A, lanes 11– 15), indicating that the HA sequence was more resistant to trypsin in Tht1-StuI-HA than in Tht1-NcoI-HA. The 55-kD band that appeared in the preparation of Tht1-StuI-HA was also resistant to trypsin digestion. This band probably corresponded to a polypeptide truncated near the COOH-terminal end of the third transmembrane segment. When trypsin digestion was performed in the presence of Triton X-100, which can permeabilize microsomes, the bands from Tht1-StuI-HA were no longer resistant to trypsin (Fig. 11 A, lanes 16–20). We then used anti–Tht1p antibody 1-3-2, which was raised against an segment from the NH2-terminal part of Tht1p, to probe the tryptic digests. Upon digestion of Tht1-NcoI-HA by trypsin, as the intensity of the 76-kD band decreased, a lower 64-kD band became more intense (Fig. 11 B, lanes 1– 3). The same 64-kD band was produced from Tht1p in the absence of Triton X-100 (Fig. 11 B, lanes 13– 15). In the presence of the detergent, this 64-kD band was not stable in either Tht1-NcoI-HA or Tht1p (Fig. 11 B, lanes 4–6 and 16–18). These results were all consistent with the notion that the COOH terminus of Tht1p is in the cytoplasm and the NH2-terminal portion is in the lumen so that it is protected from attack by trypsin in these assays.
Figure 11.

Protease protection assay. Microsome fractions prepared from conjugating cells carrying the plasmids pNT75 (Tht1-NcoI-HA), pNT74 (Tht1-StuI-HA), pNT28 (Tht1p), or pKD10 (vector) were treated with trypsin either in the absence or presence of Triton X-100 and subjected to Western immunoblot analysis. (A) Western blots probed with anti–HA antibodies. The amount of trypsin added was 0 μg/ml (lanes 1, 6, 11, and 16), 0.5 μg/ml (lanes 2, 7, 12, and 17), 1.5 μg/ ml (lanes 3, 8, 13, and 18), 3 μg/ml (lanes 4, 9, 14, and 19), and 5 μg/ml (lanes 5, 10, 15, and 20). (B) Western blots probed with anti–Tht1 antibody, 1-3-2. The amount of trypsin added was 0 μg/ml (lanes 1, 4, 7, 10, 13, 16, and 19), 0.5 μg/ml (lanes 2, 5, 8, 11, 14, and 17), and 5 μg/ml (lanes 3, 6, 9, 12, 15, and 18). The open arrow indicates a nonspecific band.
Tht1p Is Localized in Nuclear Envelope and ER during Karyogamy
To determine the location of Tht1 protein, we first attempted to immunostain wild-type cells or those carrying integrated HA-tagged tht1 gene, but we failed to observe significant signals from these cells. We then expressed an HA-tagged Tht1 protein (Tht1-NcoI-HA) from a multicopy plasmid. This construct (pNT75) was capable of complementing the tht1 deficiency (see Fig. 10). Staining with anti–HA antibodies revealed signals from the nuclear peripheries in some fraction of the conjugating cells. In these cells there were always some aggregate-like bright signals in cytoplasm (Fig. 12, a and b). In other words, we encountered very few cells in which only the nuclear peripheries were stained. When the same cells were simultaneously stained with a general membrane dye, DiOC6(3), we found that the regions where the anti–HA antibodies stained were also stained by the dye. With DiOC6(3), however, more cytoplasmic components were stained and brighter signals were produced. In general DiOC6(3) staining was not coincident with the anti–HA signals (see Fig. 12). Interestingly, when cells had formed single fused nuclei, we could no longer see the anti–HA signal from the nuclear peripheries and the signals in cytoplasm were also diminished (Fig. 12 c). Another HA-tagged protein (Tht1-StuI-HA; pNT74) was examined for its localization. This protein was also localized around the nuclei and in the amorphous cytoplasmic components, however, the signal intensity was more concentrated around the nuclear peripheries and was at a much-reduced level in the cytoplasm when compared to Tht1-NcoI-HA (Fig. 12 d). It should be noted that pNT74 was unable to complement the tht1 mutation (Fig. 10). At the present, we do not understand the apparent difference in the signal distribution. We also do not know which cytoplasmic components were stained with the anti–HA probe, although they are likely to represent ER. Control cells carrying pNT77 (Nter-Tht1-HA) displayed no specifically localized anti–HA signals, although the DiOC6(3)-staining images were indistinguishable from the experimental cells (Fig. 12 e). It should be noted that we first attempted to use glutaraldehyde to fix cells but we found this method produced very high background. Therefore we employed formaldehyde for subsequent fixations, although this fixative may not be suited for the preservation of membranous structure (Terasaki, 1994). However, the glutaraldehyde fixation method could be applied to vegetative cells. We have overproduced Tht1p by using the nmt1 + promoter, which can be induced by thiamine depletion (Maundrell, 1993), and stained the cells with anti–Tht1p antibodies 1-3-2. As shown in Fig. 13 b, 14.5 h after the induction, we could find bright signals around the nucleus as well as in cytoplasmic reticular structures and the periphery of cytoplasm. While in cells harvested 10 h after induction, when only low levels of Tht1p could be detected by Western immunoblot analysis, very few such bright signals could be observed (Fig. 13 a). When we overproduced Tht1p by using a constitutively active promoter, the adh promoter, which has been shown to be less active than the nmt1 + promoter (Basi et al., 1993), we could observe bright signals only from the nuclear peripheries, and signals in the cytoplasm were weaker than the cases when the nmt1 + promoter was used (Fig. 13 c). The location of Tht1p in these vegetative cells was reminiscent of the distribution of BiP, an authentic ER protein, in S. pombe (Pidoux and Armstrong, 1992, 1993). Consistently the distribution of BiP and Tht1p in cells cooverexpressing both proteins was virtually identical (Fig. 13, d and e).
Figure 12.
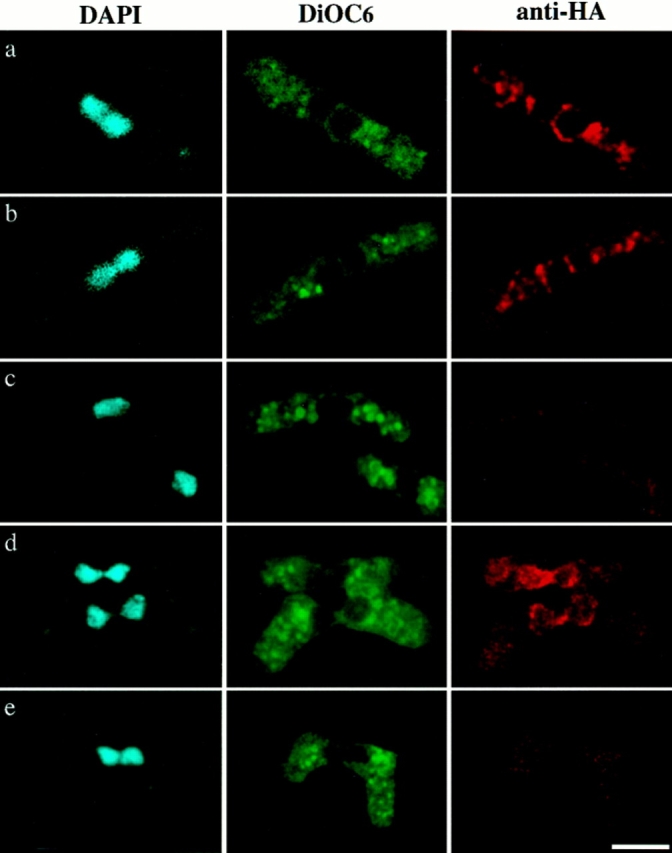
Localization of HA-tagged Tht1p. A homothallic strain with disrupted tht1 gene was transformed with pNT75 (a– c), pNT74 (d), and pNT77 (e) and conjugation was induced (see Materials and Methods). a and b represent the images before fusion and c represents a cell after nuclear fusion. In c there is also a vegetative cell at the bottom of right side. Note that pNT75 was capable of complementing the fusion defect but pNT74 and pNT77 were not. Bar, 5 μm.
Figure 13.
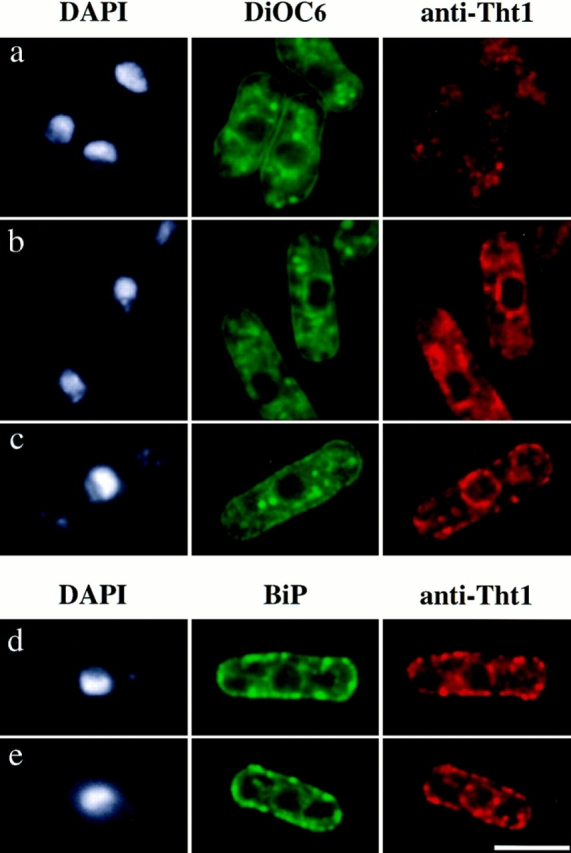
Localization of overproduced Tht1p. (a and b) HM123 (h − wild type) transformed with pNT45 was induced by thiamine depletion for 10 h (a) and 14.5 h (b). (c) HM123 was transformed with pNT79. Anti–Tht1 antibody 1-3-2 was used to stain for Tht1p. (d and e) HM180 (h − leu1 ura4) was transformed with both pEB9 and pNT97 and incubated for 14 h in the absence of thiamine. Cells were doubly stained with anti–c-myc 10E9 and anti–Tht1p antibody 1-3-2. Bar, 5 μm.
Discussion
The tht1 + gene reported in this study is the first karyogamy-specific gene identified in S. pombe. The kms1 + gene, which we identified recently, also plays an important role in karyogamy but it is required for progression through meiosis as well, particularly in the formation of the meiotic prophase-specific nuclear architecture (Shimanuki et al., 1997). The tht1 mutant severely blocks nuclear fusion, but does not affect meiosis at all when meiosis is induced directly from a diploid cell. We also showed that tht1 gene is dispensable for mitotic growth by completely disrupting the gene. In accordance with these observations, the expression of tht1 + gene is strictly confined to conjugating cells, where the gene product is required. The fission yeast gene fus1 + is also specifically required for conjugation but at an earlier step before karyogamy, that is, the degradation of cell walls leading to cytoplasmic fusion after the cell contact. It has been reported that the expression of fus1 + gene is dependent upon nitrogen starvation and the presence of cells of the opposite mating type (Petersen et al., 1995), a condition similar to that for the tht1 gene expression. It will be interesting to see if there are a class of genes specifically involved in conjugation and/or karyogamy that are under the same genetic control.
Aberrant Meiotic Divisions from Unfused Nuclei
In fission yeast meiosis, like in mitosis, the spindle is formed between separated SPBs in the nucleus while keeping nuclear envelope intact (Hirata and Tanaka, 1982). Before the SPB proceeds into the duplication/separation cycle in meiosis, the two haploid SPBs must fuse to make a single unified SPB in karyogamy. A couple of pieces of evidence shown in this study suggest that the SPBs have completed this binding process before the tht1 arrest point. First, in almost all of the cases, only one SPB signal could be observed between two juxtaposed nuclei using the SPB-specific probes, anti–Sad1 antibodies and GFP-fused Kms1p. A second piece of evidence is provided by the coordinated movement of the twin horse tail nuclei, where only a single SPB seems to lead the movement. However, we have not ruled out the possibility that the binding of the SPBs is incomplete in the mutant. Rather, we think it could be a considerable possibility, because there could be strong tension produced by such unusual configuration of unfused and bulky nuclei against the SPB. In any case, there must be a topological problem in the formation of the spindle in a mutant situation such as that seen in tht1 where one unified SPB or two separating SPBs have to be stuck to the two unfused nuclear envelopes. In fact, we could find very few normal spindles and DAPI-stained images of separating nuclei in the mutant zygotes (data not shown). Although we could not strictly rule out the possibility that meiotic division is prohibited from the twin horse tail state, we feel that it is unlikely judging from the efficiency of sporulation. Moreover, the tws1 mutation that blocks meiosis II (Nakaseko et al., 1984), when combined with the tht1 mutation, reduced the number of spores (data not shown), indicating that even in the aberrant meiosis/ sporulation in the tht1 mutant, meiosis II, and therefore meiosis I as well, does take place. In this regard we thought it interesting that a considerable number of mutant asci contained a large nucleus that was not encapsulated with the spore wall (Fig. 5, arrows). Since it is known that spore wall formation initiates from a modified SPB (Tanaka and Hirata, 1982), we first speculated that this type of nucleus might have arisen from an event where the SPB had been stripped off from one of the unfused nuclei. However, to our surprise, such nuclei in fact bore two or three anti–Sad1 stains (data not shown). This phenotype was very unusual and we have currently no plausible explanation for it. Nevertheless, it seems apparent that the SPB-spindle cycle became catastrophic in the mutant zygotic meiosis. It may be also worth considering here that like the “twin meiosis” described by Gutz (1967) where two unfused nuclei go into meiosis separately, the two haploid nuclei might undergo independent meiosis and sporulation (Iino and Yamamoto, 1985; Nurse 1985) in the tht1 mutant, thereby producing aberrant asci. However, if this were the case a large fraction of the mutant asci should contain eight spores, but only very few asci actually contained eight spores. This argument further supports the notion that the SPBs have unified in the tht1 mutant.
The Role of Tht1p in Karyogamy
During conjugation in the tht1 mutant, the juxtaposition of two nuclei is not followed by the fusion of nuclear envelopes. Mutants showing similar defective phenotype have been isolated in S. cerevisiae, those include kar2, kar5, kar7, kar8, sec63, sec71, sec72, and jem1 (Kurihara et al., 1994; Ng and Walter, 1996; Nishikawa and Endo, 1997). For some of these genes their products have been identified, but very little is known about how these genes are involved in nuclear fusion. For instance, Sec63p, Sec71p, Sec72p, and also Kar2p (BiP) are involved as a complex in protein translocation across the ER membrane (Deshaies et al., 1991; Green et al., 1992; Brodsky et al., 1995; Ng and Walter, 1996), but it has been shown that the activity required for protein translocation is not directly required for the fusion of nuclear envelopes (Ng and Walter, 1996).
The ER is contiguous with the nuclear membrane and so it has not been determined from which site(s) membrane fusion starts in karyogamy. However, it was demonstrated that nuclear fusion is roughly paralleled with the homotypic fusion of ER membranes (Latterich and Schekman, 1994). Mutants defective in the fusion of nuclear envelopes were also defective in the ER membrane fusion both in vivo and in vitro (Kurihara et al., 1994; Latterich and Schekman, 1994). In this respect it is interesting that Cdc48p, an ER protein with homology to Sec18p, has been shown to participate in the fusion of ER membranes (Latterich et al., 1995). Sec18p is a yeast homologue of N-ethylmaleimide–sensitive factor (NSF), which together with Sec17p (a homologue of soluble NSF attachment protein α [α-SNAP]) is required for vesicular docking. Thus, it appears that Cdc48p is a component of fusion/docking machinery, mainly functioning in the fusion of ER membranes. Whether Cdc48p is required for nuclear fusion is yet to be examined experimentally. Furthermore, it has been shown that some novel proteins which are tightly bound to ER membrane are required for the membrane fusion and that Kar2p may participate in the activation of these proteins (Latterich and Schekman, 1994).
At the present we can not speculate as to the function of Tht1p from its amino acid sequence because of the lack of homology to known proteins. We cannot strictly rule out the possibility that the absence of the fusion of nuclear envelopes is a secondary effect of incomplete SPB binding, although it is unlikely as discussed above. However, several lines of evidence described in this study strongly suggest that Tht1p is a novel ER protein. The primary amino acid sequence indicates that it is a type I membrane protein. Tht1p is actually modified with N-linked glycosylation, and partitions to the insoluble fraction. Finally, immunofluorescence microscopy localized the protein in nuclear envelope and some portion of cytoplasmic membranous structures. In vegetative cells containing overproduced Tht1p it could localize to ER. Taking this fact into consideration together with the observation that the tht1 + gene is transiently expressed only in conjugating cells, we suggest that Tht1p may be directly and specifically involved in the fusion of nuclear envelopes and the ER membranes during karyogamy. One of the key issues in further studies for elucidating the role of Tht1p in karyogamy may be the role of the COOH-terminal end of Tht1p, because this end of the protein was shown to reside in the cytoplasm and could therefore be interacting with some other factors or perhaps with itself to perform an essential function to achieve the membrane fusion. Searches for such factors are in progress and may help to reveal a novel class of molecules involved in membrane fusion events. Another important issue addressed in this study is the temporal expression of the gene. Particularly interesting is the rapid disappearance of Tht1p from the membranes after the completion of karyogamy. Elucidation of mechanism involved in such elaborate regulation should be an important step toward understanding the membrane dynamics in the cell.
Acknowledgments
We are grateful to John Armstrong for the BiP plasmid. We thank Iain Hagan for critical reading of the manuscript and valuable comments. We also wish to thank Dr. N. Miyajima for his help in computational analyses.
This work was supported by Grant-in-Aid for Scientific Research on Priority Areas (to Y. Hiraoka and O. Niwa) and by the Kazusa DNA Research Institute Foundation.
Abbreviations used in this paper
- DAPI
4,6-diamidino-2-phenylindole
- EDTA
ethylenediaminitetraacetate
- endoH
endoglycosilase H
- FISH
fluorescent in situ hybridization
- GFP
green fluorescent protein
- NSF
N-ethylmaleimide-sensitive factor
- ORF
open reading frame
- SPB
spindle pole body
Footnotes
Dr. Horio present address is School of Medicine, Tokushima University, 3-18-15 Kuramoto-cho, Tokushima, Tokushima 770, Japan.
References
- Alfa, C., P. Fantes, J. Hyams, M. McLeod, and E. Warbrick. 1993. Experiments with fission yeast: a laboratory course manual. Cold Spring Harbor Press, New York. 186 pp.
- Baker D, Wuestehube L, Schekman R, Botstein D, Segev N. GTP-binding Ypt1 protein and Ca2+function independently in a cell-free protein transport reaction. Proc Natl Acad Sci USA. 1990;87:355–359. doi: 10.1073/pnas.87.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basi G, Schmid E, Maundrell K. TATA box mutations in the Schizosaccharomyces pombe nmt1promoter affect transcription efficiency but not the transcription start point or thiamine repressibility. Gene (Amst) 1993;123:131–136. doi: 10.1016/0378-1119(93)90552-e. [DOI] [PubMed] [Google Scholar]
- Beach D, Rodgers L, Gould J. RAN1 +controls the transition from mitotic division to meiosis in fission yeast. Curr Genet. 1985;10:297–311. doi: 10.1007/BF00365626. [DOI] [PubMed] [Google Scholar]
- Brodsky JL, Goeckeler J, Schekman R. BiP and Sec63p are required for both co- and posttranslational protein translocation into the yeast endoplasmic reticulum. Proc Natl Acad Sci USA. 1995;92:9643–9646. doi: 10.1073/pnas.92.21.9643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell TG, Warren G. A galactosyltransferase from the fission yeast Schizosaccharomyces pombe. J Cell Biol. 1989;109:2693–2702. doi: 10.1083/jcb.109.6.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikashige Y, Ding D-Q, Funabiki H, Haraguchi T, Mashiko S, Yanagida M, Hiraoka Y. Telomere-led premeiotic chromosome movement in fission yeast. Science. 1994;264:270–273. doi: 10.1126/science.8146661. [DOI] [PubMed] [Google Scholar]
- Chikashige Y, Ding D-Q, Imai Y, Yamamoto M, Haraguchi T, Hiraoka Y. Meiotic nuclear reorganization: switching the position of centromeres and telomeres in the fission yeast Schizosaccharomyces pombe. . EMBO (Eur Mol Biol Organ) J. 1997;16:193–202. doi: 10.1093/emboj/16.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church GM, Gilbert W. Genomic sequencing. Proc Natl Acad Sci USA. 1984;81:1991–1995. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Sanders SL, Feldheim DA, Schekman R. Assembly of yeast Sec proteins involved in translocation into the endoplasmic reticulum into a membrane-bound multisubunit complex. Nature. 1991;349:806–808. doi: 10.1038/349806a0. [DOI] [PubMed] [Google Scholar]
- Egel, R. 1989. Mating-type genes, meiosis, and sporulation. In Molecular Biology of the Fission Yeast. A. Nasim, P. Young, and B.F. Johnson, editors. Academic Press, San Diego. 37–73.
- Funabiki H, Hagan I, Uzawa S, Yanagida M. Cell cycle-dependent positioning and clustering of centromeres and telomeres in fission yeast. J Cell Biol. 1993;121:961–976. doi: 10.1083/jcb.121.5.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funabiki H, Yamano H, Kumada K, Nagao K, Hunt T, Yanagida M. Cut2 proteolysis required for sister-chromatid separation in fission yeast. Nature. 1996;381:438–441. doi: 10.1038/381438a0. [DOI] [PubMed] [Google Scholar]
- Garnier C, Blondel M-O, Haguenauer-Tsapis R. Membrane topology of the yeast uracil permease. Mol Microbiol. 1996;21:1061–1073. doi: 10.1046/j.1365-2958.1996.621430.x. [DOI] [PubMed] [Google Scholar]
- Gething M-J, Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- Green N, Fang H, Walter P. Mutants in three novel complementation groups inhibit membrane protein insertion into and soluble protein translocation across the endoplasmic reticulum membrane of Saccharomyces cerevisiae. . J Cell Biol. 1992;116:597–604. doi: 10.1083/jcb.116.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutz H. “Twin meiosis” and other ambivalences in the life cycle of Schizosaccharomyces pombe. . Science. 1967;158:796–798. doi: 10.1126/science.158.3802.796. [DOI] [PubMed] [Google Scholar]
- Gutz, H., H. Heslot, U. Leupold, and N. Loprieno. 1974. Schizosaccharomyces pombe. In Handbook of Genetics. R.C. King, editor. Plenum Press, New York. 395–446.
- Hagan I, Yanagida M. The product of the spindle formation gene sad1 +associates with the fission yeast spindle pole body and is essential for viability. J Cell Biol. 1995;129:1033–1047. doi: 10.1083/jcb.129.4.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- High S, Dobberstein B. Mechanisms that determine the transmembrane disposition of proteins. Curr Opin Cell Biol. 1992;4:581–586. doi: 10.1016/0955-0674(92)90075-n. [DOI] [PubMed] [Google Scholar]
- Hirata A, Tanaka K. Nuclear behavior during conjugation and meiosis in the fission yeast Schizosaccharomyces pombe. . J Gen Appl Microbiol. 1982;28:263–274. [Google Scholar]
- Hoheisel JD, Maier E, Mott R, McCarthy L, Grigoriev AV, Schalkwyk LC, Nizetic D, Francis F, Lehrach H. High resolution cosmid and P1 maps spanning the 14 Mb genome of the fission yeast S. pombe. . Cell. 1993;73:109–120. doi: 10.1016/0092-8674(93)90164-l. [DOI] [PubMed] [Google Scholar]
- Iino Y, Yamamoto M. Mutants of Schizosaccharomyces pombe which sporulate in the haploid state. Mol Gen Genet. 1985;198:416–421. doi: 10.1007/BF00332932. [DOI] [PubMed] [Google Scholar]
- Kurihara LJ, Beh CT, Latterich M, Schekman R, Rose MD. Nuclear congression and membrane fusion: two distinct events in the yeast karyogamy pathway. J Cell Biol. 1994;126:911–923. doi: 10.1083/jcb.126.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara LJ, Stewart BG, Gammie AE, Rose MD. Kar4p, a karyogamy-specific component of the yeast pheromone response pathway. Mol Cell Biol. 1996;16:3990–4002. doi: 10.1128/mcb.16.8.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latterich M, Schekman R. The karyogamy gene KAR2 and novel proteins are required for ER-membrane fusion. Cell. 1994;78:87–98. doi: 10.1016/0092-8674(94)90575-4. [DOI] [PubMed] [Google Scholar]
- Latterich M, Frohlich KU, Schekman R. Membrane fusion and the cell cycle: Cdc48p participates in the fusion of ER membranes. Cell. 1995;82:885–893. doi: 10.1016/0092-8674(95)90268-6. [DOI] [PubMed] [Google Scholar]
- Maundrell K. Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene. 1993;123:127–130. doi: 10.1016/0378-1119(93)90551-d. [DOI] [PubMed] [Google Scholar]
- Miles JS. Structurally and functionally conserved regions of cytochrome P-450 reductase as targets for DNA amplification by the polymerase chain reaction: cloning and nucleotide sequence of the Schizosaccharomyces pombe cDNA. Biochem J. 1992;287:195–200. doi: 10.1042/bj2870195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata M, Doi H, Miyata H, Johnson BF. Sexual coflocculation by heterothallic cells of the fission yeast Schizosaccharomyces pombemodulated by medium constituents. Antonie Leeuwenhoek. 1997;71:207–215. doi: 10.1023/a:1000166426509. [DOI] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. . Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- Murakami S, Niwa O. Fission yeast sta mutations that stabilize an unstable minichromosome are novel cdc2-interacting suppressors and are involved in regulation of spindle dynamics. Mol Gen Genet. 1995;249:391–399. doi: 10.1007/BF00287100. [DOI] [PubMed] [Google Scholar]
- Nakaseko Y, Niwa O, Yanagida M. A meiotic mutant of the fission yeast Schizosaccharomyces pombethat produces mature asci containing two diploid spores. J Bacteriol. 1984;157:334–336. doi: 10.1128/jb.157.1.334-336.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaseko Y, Adachi Y, Funahashi S, Niwa O, Yanagida M. Chromosome walking shows a highly homologous repetitive sequence present in all the centromere regions of fission yeast. EMBO (Eur Mol Biol Organ) J. 1986;5:1011–1021. doi: 10.1002/j.1460-2075.1986.tb04316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng DTW, Walter P. ER membrane protein complex required for nuclear fusion. J Cell Biol. 1996;132:499–509. doi: 10.1083/jcb.132.4.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa S, Endo T. The yeast JEM1p is a DnaJ-like protein of the endoplasmic reticulum membrane required for nuclear fusion. J Biol Chem. 1997;272:12889–12892. doi: 10.1074/jbc.272.20.12889. [DOI] [PubMed] [Google Scholar]
- Normington K, Kohno K, Kozutsumi Y, Gething M-J, Sambrook J. S. cerevisiae encodes an essential protein homologous in sequence and function to mammalian BiP. Cell. 1989;57:1223–1236. doi: 10.1016/0092-8674(89)90059-7. [DOI] [PubMed] [Google Scholar]
- Nurse P. Mutants of fission yeast Schizosaccharomyces pombe which alter the shift between cell proliferation and sporulation. Mol Gen Genet. 1985;198:497–502. [Google Scholar]
- Petersen J, Weilguny D, Egel R, Nielsen O. Characterization of fus1 of Schizosaccharomyces pombe:a developmentally controlled function needed for conjugation. Mol Cell Biol. 1995;15:3697–3707. doi: 10.1128/mcb.15.7.3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidoux AL, Armstrong J. Analysis of the BiP gene and identification of an ER retention signal in Schizosaccharomyces pombe. . EMBO (Eur Mol Biol Organ) J. 1992;11:1583–1591. doi: 10.1002/j.1460-2075.1992.tb05203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidoux AL, Armstrong J. The BiP protein and the endoplasmic reticulum of Schizosaccharomyces pombe: fate of the nuclear envelope during cell division. J Cell Sci. 1993;105:1115–1120. doi: 10.1242/jcs.105.4.1115. [DOI] [PubMed] [Google Scholar]
- Robinow CF. The number of chromosomes in Schizosaccharomyces pombe: light microscopy of stained preparations. Genetics. 1977;87:491–497. doi: 10.1093/genetics/87.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose MD, Misra LM, Vogel JP. KAR2, a karyogamy gene, is the yeast homolog of the mammalian Bip/GRP78 gene. Cell. 1989;57:1211–1221. doi: 10.1016/0092-8674(89)90058-5. [DOI] [PubMed] [Google Scholar]
- Rose MD. Nuclear fusion in yeast. Annu Rev Microbiol. 1991;45:539–567. doi: 10.1146/annurev.mi.45.100191.002543. [DOI] [PubMed] [Google Scholar]
- Scidmore MA, Okamura HH, Rose MD. Genetic interaction between KAR2 and SEC63, encoding eukaryotic homologues of DnaK and DnaJ in the endoplasmic reticulum. Mol Biol Cell. 1993;4:1145–1159. doi: 10.1091/mbc.4.11.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimanuki M, Miki F, Ding D-Q, Chikashige Y, Hiraoka Y, Horio T, Niwa O. A novel fission yeast gene, kms1 +, is required for the formation of meiotic prophase specific nuclear architecture. Mol Gen Genet. 1997;254:238–249. doi: 10.1007/s004380050412. [DOI] [PubMed] [Google Scholar]
- Silve S, Volland C, Garnier C, Jund R, Chevallier MR, Haguenauer-Tsapis R. Membrane insertion of uracil permease, a polytopic yeast plasma membrane protein. Mol Cell Biol. 1991;11:1114–1124. doi: 10.1128/mcb.11.2.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DE, Fisher PA. Identification, developmental regulation, and response to heat shock of two antigenically related forms of a major nuclear envelope protein in Drosophilaembryo; an application of an improved method for affinity purification of antibodies using polypeptides immobilized on nitrocellulose blots. J Cell Biol. 1984;99:20–28. doi: 10.1083/jcb.99.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Hirata A. Ascospore development in the fission yeasts Schizosaccharomyces pombe and S. japonicus. . J Cell Sci. 1982;56:263–279. doi: 10.1242/jcs.56.1.263. [DOI] [PubMed] [Google Scholar]
- Tange Y, Niwa O. A selection system for diploid and against haploid cells in Schizosaccharomyces pombe. . Mol Gen Genet. 1995;248:644–648. doi: 10.1007/BF02191703. [DOI] [PubMed] [Google Scholar]
- Terasaki, M. 1994. Labeling of the endoplasmic reticulum with DiOC6(3). In Cell Biology: A Laboratory Handbook. Vol. 2. J.E. Celis, editor. Academic Press, San Diego. 381–386.
- Toda T, Shimanuki M, Yanagida M. Fission yeast genes that confer resistance to staurosporine encode an AP-1like transcription factor and a protein kinase related to the mammalian ERK1/MAP2 and budding yeast FUS3 and KSS1kinases. Genes Dev. 1991;5:60–73. doi: 10.1101/gad.5.1.60. [DOI] [PubMed] [Google Scholar]