

Abstract
Sperm from nematodes use a major sperm protein (MSP) cytoskeleton in place of an actin cytoskeleton to drive their ameboid locomotion. Motility is coupled to the assembly of MSP fibers near the leading edge of the pseudopod plasma membrane. This unique motility system has been reconstituted in vitro in cell-free extracts of sperm from Ascaris suum: inside-out vesicles derived from the plasma membrane trigger assembly of meshworks of MSP filaments, called fibers, that push the vesicle forward as they grow (Italiano, J.E., Jr., T.M. Roberts, M. Stewart, and C.A. Fontana. 1996. Cell. 84:105–114). We used changes in hydrostatic pressure within a microscope optical chamber to investigate the mechanism of assembly of the motile apparatus. The effects of pressure on the MSP cytoskeleton in vivo and in vitro were similar: pressures >50 atm slowed and >300 atm stopped fiber growth. We focused on the in vitro system to show that filament assembly occurs in the immediate vicinity of the vesicle. At 300 atm, fibers were stable, but vesicles often detached from the ends of fibers. When the pressure was dropped, normal fiber growth occurred from detached vesicles but the ends of fibers without vesicles did not grow. Below 300 atm, pressure modulates both the number of filaments assembled at the vesicle (proportional to fiber optical density and filament nucleation rate), and their rate of assembly (proportional to the rates of fiber growth and filament elongation). Thus, fiber growth is not simply because of the addition of subunits onto the ends of existing filaments, but rather is regulated by pressure-sensitive factors at or near the vesicle surface. Once a filament is incorporated into a fiber, its rates of addition and loss of subunits are very slow and disassembly occurs by pathways distinct from assembly. The effects of pressure on fiber assembly are sensitive to dilution of the extract but largely independent of MSP concentration, indicating that a cytosolic component other than MSP is required for vesicle-association filament nucleation and elongation. Based on these data we present a model for the mechanism of locomotion-associated MSP polymerization the principles of which may apply generally to the way cells assemble filaments locally to drive protrusion of the leading edge.
Lammelipodial motility is essential to such wide-ranging processes as wound healing, inflammation, and metastatic invasion. This type of movement is thought to be based on forces derived from the actin cytoskeleton (for review see Condeelis, 1993; Stossel, 1993; Oliver et al., 1994; Mitchison and Cramer, 1996). However, in addition to locomotion, actin is also involved in other motile activities such as responses to external signals, uptake of nutrients, rearrangement of vesicles and organelles, and cell division. To carry out these multiple functions, a typical cell may contain 50–100 accessory proteins designed to orchestrate the dynamics of its actin filament system (Tilney and Tilney, 1993). As a consequence, it has been difficult to identify exactly which of these components are required for locomotion or precisely how they interact to produce the force for translocation.
The ameboid sperm of the nematode, Ascaris suum, have a specialized motile apparatus that offers distinct advantages for investigating the mechanism of lammelipodial cell motility (for review see Roberts and Stewart, 1995, 1997; Theriot, 1996). Like other ameboid cells, these sperm crawl by extending a pseudopod, and protrusive activity at the leading edge is tightly coupled to localized polymerization and centripetal flow of the cytoskeleton. However, these cells contain no F-actin; the polymerizing unit involved in their locomotion is the 14-kD major sperm protein (MSP)1 (Roberts and Stewart, 1995). MSP and actin have neither sequence nor structural homology, yet the patterns of motility the two proteins produce are so remarkably similar that the physical principles underlying MSP- and actin-based crawling movement must be shared (Theriot, 1996).
The simplicity of nematode sperm makes these cells particularly attractive for studying how cells crawl. At the end of meiosis, the endoplasmic reticulum, Golgi, protein synthesizing machinery, and conventional actin and microtubule cytoskeletons are cast aside so that the cell is reduced to the minimum components required to carry out its specialized function (for review see Ward, 1986; Roberts, 1987). The MSP cytoskeleton in these streamlined cells is used only for locomotion and, thus, its properties can be explored without the complications associated with a multifunctional actin-based motile apparatus.
The MSP filaments in Ascaris sperm are organized into long, branched meshworks called fiber complexes, which span 15–20 μm from the base of the pseudopod to its leading edge (Sepsenwol et al., 1989). As the cell crawls, new filaments are assembled along the pseudopodial membrane and become incorporated into the fiber complexes so that advance of the cell is accompanied by a continuous rearward flux of the filament system (Roberts and King, 1991; King et al., 1994). The rates of cytoskeletal assembly and sperm movement are tightly coupled, suggesting that localized polymerization and bundling of MSP filaments are key elements of locomotion.
An indication of the simplicity of the MSP motility system is the ease with which the events involved in pseudopodial protrusion can be reconstituted in vitro (Italiano et al., 1996). Addition of ATP to concentrated, clarified extracts of sperm results in the assembly of discrete filament meshworks, or fibers, similar to the fiber complexes observed in vivo. Each fiber has a vesicle derived from the pseudopodial plasma membrane at one end and fiber growth is because of assembly and bundling of MSP filaments at the vesicle-bearing end. Thus, elongation of the fiber, which can occur at rates approaching the average velocity of sperm locomotion, moves the associated vesicle forward in much the same way as growth of the fiber complexes appears to push the pseudopodial plasma membrane in vivo.
Fractionation of the sperm extract showed that fiber assembly requires at least four components: (a) MSP to build filaments, (b) membrane factors supplied by the vesicle, (c) ATP as an energy source, and (d) at least one additional cytosolic component. Dilution assays indicated that the activity of this cytosolic factor, and not the concentration of MSP, determines the rate of fiber growth (Italiano et al., 1996).
This in vitro motility system allows investigation of how the polymerization and bundling of MSP filaments moves membranes and how these processes are performed. In this study, we used hydrostatic pressure as a tool to study MSP cytoskeletal dynamics. Pressure perturbs reactions that involve volume changes; its effects are rapid and reversible, and it can be applied noninvasively (for review see Silva et al., 1996). Thus, pressure is well-suited for investigating processes that involve protein–protein interactions, such as cytoskeletal assembly. It has been known since 1936 that pressure blocks the movement of Amoeba proteus (Marsland and Brown, 1936), and more recent studies have documented that pressure alters assembly of actin filaments and microtubules both in vivo and in vitro (Salmon, 1975a ,b; Begg et al., 1983; Bourns et al., 1988).
We used an optical pressure chamber (Salmon and Ellis, 1975) to visualize directly the influence of pressures ⩽500 atm on the motility of live sperm and growth of fibers in vitro. We found that MSP cytoskeletal dynamics in whole cells and in cell-free extracts are affected by similar pressures. Analysis of the in vitro assembly system showed that membrane and cytosolic factors operate in concert to determine the rates of both nucleation and elongation of filaments, and appear to function by converting MSP transiently into a form that polymerizes readily under physiological conditions. Moreover, these events occur at or very near the membrane surface, so that once an MSP filament is assembled and becomes part of a fiber, its rates of subunit addition and loss are very slow. The physical events involved in MSP-based membrane movement are very similar to those proposed for motile systems associated with actin filament assembly and may, therefore, reveal general properties of polymerization-driven protrusion that occurs at the leading edge of crawling cells.
Materials and Methods
Isolation of Sperm and Preparation of Cell Extracts
Males of A. suum were collected from the small intestine of infected hogs at a slaughterhouse (Lowell Packing Co., Fitzgerald, GA), and then transported to the lab in phosphate-buffered saline containing 10 mM NaHCO3 at 37–39°C. Spermatids were obtained by draining the contents of the seminal vesicle (∼5 × 107 cells/male) into tubes containing 50 mM Hepes, 65 mM KCl, 10 mM Na HCO3, pH 6.7, (HKB buffer), at 39°C. These cells were activated to complete development into motile spermatozoa by treatment with a homogenate of the vas deferens (Sepsenwol et al., 1986) and then used immediately for experiments requiring live, motile cells.
To prepare extracts of sperm for in vitro assembly assays, activated sperm were pelleted by centrifugation for 5 s at 5,000 g. The supernatant was removed by aspiration, and then the pellet of packed cells was frozen in liquid nitrogen and stored at −70°C. These cells were lysed via two cycles of freeze-thaw and then centrifuged (model TL-100 Ultracentrifuge; Beckman Instruments, Inc., Palo Alto, CA) at 100,000 g for 60 min at 4°C. The resulting supernatant (S100), which contained the soluble sperm proteins plus a population of small membrane-bound vesicles (Italiano et al., 1996), was divided into aliquots and stored at −70°C.
Examination of Pressure Effects by Light Microscopy
All assays of the effects of pressure were conducted in a small chamber designed to allow application of pressures ⩽600 atm while continuously viewing specimens by light microscopy (Salmon and Ellis, 1975; modified as described in Crenshaw and Salmon, 1996). This stainless steel chamber uses 1-mm-thick strain-free optical glass (Edmunds Scientific, Barrington, NJ) for viewing windows and is connected via thin steel tubing to a pressure valve. The tubing is filled with low viscosity silicone oil (Crenshaw and Salmon, 1996), and the pressure adjusted by using a pressure valve for volume displacement. Pressure is measured with a low displacement electrical pressure gauge and can be altered as desired over the range of 1–500 atm within 3 to 7 s (Salmon and Ellis, 1975).
For examination of live cells, sperm were pipetted onto 4 × 4-mm fragments of ethanol-washed No. 1 coverslips that were placed horizontally on top of the lower window in the pressure chamber filled with HKB at 39°C. The chamber was sealed and mounted on the stage of a microscope (model Axiovert; Carl Zeiss, Inc., Thornwood, NY) so that the optical axis of the chamber was aligned with the light path of the microscope. An airstream incubator (model ASI400; Nicholson Precision Instruments, Bethesda, MD) was used to maintain the chamber at 39°C. Images were obtained with a 40× long-working distance differential interference contrast objective (Carl Zeiss, Inc.) with a correction collar adjusted to optimize focus on the cells through the 1-mm-thick glass window in the lower surface of the pressure chamber.
To examine fibers assembled in vitro, aliquots of S100 were diluted as required with buffer containing 8 mM KH2PO4, 2 mM K2HPO4, and 5 mM MgCl2 (KPM buffer), and then ATP was added to 1 mM. A 0.5-μl drop of this material was pipetted onto the center of a 4 × 4-mm coverslip fragment lined on each edge with a thin strip of silicon grease. A second 4 × 4-mm piece of coverslip was lowered onto the drop and pressed gently to create a sealed chamber containing a thin layer of S100. These preparations were then placed into the pressure chamber filled with KPM buffer, sealed, and then examined as described above using a phase-contrast objective with a correction collar.
Images obtained using a charge-coupled device video camera (model TI-24A; NEC Corp., Elk Grove Village, IL), were digitized and processed by background subtraction and contrast enhancement using Image-1AT software and hardware (Universal Imaging, West Chester, PA), and then recorded on super VHS VCR (JVC model HR-S5200U; Victor Company of Japan, Ltd., Elmwood Park, NJ). The images shown in Figs. 1–3 were prepared using Adobe Photoshop software (Adobe Systems Inc., San Jose, CA). Fiber growth rates were determined using an Image-1AT measurement subroutine.
Figure 1.

Effects of pressure on the MSP cytoskeleton in vivo. (a) Video-enhanced differential interference contrast micrograph of two Ascaris sperm at 1 atm. In each cell, a pseudopod with its characteristic array of fiber complexes extends forward from a hemispherical, organelle-packed cell body. Raising the pressure to 75 atm (b) causes the cytoskeleton to disassemble and the pseudopod to round up within 5 to 10 s. After 2 min at 75 atm (c) the cytoskeleton reforms. Increasing the pressure to 200 atm causes the cytoskeleton to disassemble again. After 4 min at this pressure (d), partial cytoskeletal reassembly occurs as shown by the formation of protrusions (arrow) at the leading edge. Only when the pressure is lowered to 75 atm for 1 min (e) are these cells able to rebuild their complete filament system and resume motility. Bar, 5 μm.
Figure 3.

Variation in optical density of fibers as a function of pressure. Growth of the two fibers shown in this video-enhanced phase-contrast micrograph was initiated at 150 atm. At the point indicated by the black arrow the pressure was raised to 275 atm for 2 min, resulting in formation of a short segment with reduced optical density. When the pressure was lowered to 125 atm (white arrow), both the optical density and the growth rate increased. Note the abrupt change in optical density at the points of pressure shift. The arrowheads indicate the vesicle associated with each fiber. Bar, 5 μm.
We measured changes in fiber contrast by measuring changes in the difference in pixel grayscale value for the middle of a fiber and a background region just outside the fiber for images recorded by phase contrast. In phase-contrast imaging, the difference in intensity between a fiber and the surrounding medium depends on the phase retardation of light produced by the fiber relative to the surrounding medium, (nf − nm)d, where (nf − nm) is the difference in refractive index between the fiber and the medium with d as the fiber thickness (Martin, 1966; Pluta, 1989). The refractive index of the fiber is proportional to the density of protein in the fiber. As density increases, nf increases, and the contrast measurement is initially a sine function of the phase difference. For small values of phase difference, contrast is proportional to (nf − nm)d. When we pressurized MSP fibers, the optical density of the fibers decreased, but the diameter remained constant. To test if contrast was proportional to fiber density in our studies, we examined how contrast changed when one region of a fiber crossed over another region of the same fiber or another fiber during spiral growth at atm pressure. We measured 10 fiber crossovers and generally the relationship we were looking for was maintained (crossover contrast = fiber 1 contrast + fiber 2 contrast). The crossover value was a little lower (6– 14%) than the sum of the two fibers, indicating that doubling normal fiber contrast moves the measurement slightly out of the linear range of the method. However, one of the fibers was often slightly out of focus at the crossover point, which could have reduced the contrast measurement at the crossover. Nevertheless, this test demonstrates that our measurements of contrast are within 15%, and more likely, close to proportional to the density of MSP protein within the fibers. Thus, we defined the optical density of a fiber as its grayscale value measured in the middle of the fiber minus the grayscale value obtained for nearby background.
Results
Effects of Pressure on Sperm Motility
Application of pressures of 50–300 atm caused disruption of the MSP cytoskeleton in crawling Ascaris sperm so that the pseudopod rounded up and the cell stopped moving. At these pressures, the cells adapted and reassembled their cytoskeleton while the pressure remained constant (Fig. 1). The extent of this recovery varied with pressure. For example, at 50–150 atm, the cells completely rebuilt their cytoskeleton and resumed locomotion. At 150–300 atm, fiber complexes started to reassemble at the leading edge and their associated protrusions reformed, but the cell failed to complete cytoskeletal reconstruction or translocate. At pressures >300 atm, the cytoskeleton only reassembled when the pressure was lowered. Adaptation to pressure has not been observed in other motile cells, and we have not determined the mechanism of recovery of the MSP cytoskeleton from the initial effects of pressure. The transient response of sperm to pressures <300 atm made it difficult to discriminate direct effects of pressure on the cytoskeleton from indirect effects resulting from pressure-induced modulation of other physiological parameters. Moreover, although the effects of pressure on the MSP filament system appeared to occur primarily at the pseudopodial membrane, we were unable to determine if alteration of cytoskeletal dynamics in intact cells was because of modification of filament assembly, disassembly, or both.
Effects of Pressure on Cytoskeletal Assembly and Disassembly Reactions Can Be Assessed Independently in the In Vitro Motility System
We found that fibers assembled in vitro in the S100 fraction of sperm extracts provided a simpler system for evaluation of the effects of pressure on MSP cytoskeletal dynamics. The rate of fiber assembly was affected by pressures of about the same magnitude as those that modulated the MSP filament system in vivo but, unlike cells, fibers failed to adapt to pressure. We were able to image in phase contrast both the fiber and the vesicle attached at its growing end within the pressure chamber and pressure-induced changes in fiber dynamics and anatomy could be examined in real time. This property allowed us to distinguish pressure regimes that influenced fiber assembly from those that induced fiber disassembly. Pressures ⩽300 atm had rapid (within several seconds), reversible effects on fiber assembly without inducing detectable disassembly (loss of fiber phase contrast). Pressures >300 atm were required to disassemble fibers and the effects were not reversible. Thus, fiber assembly and disassembly are separable, distinct processes. We focused on assembly and its role in vesicle motility in the in vitro system.
MSP Assembly Occurs Only in the Immediate Vicinity of the Vesicle
The sequence shown in Fig. 2 illustrates the effects of rapid, large-scale changes in pressure on fibers assembled in vitro. In this case, a fiber growing at 75 atm was rapidly pressurized to 450 atm. This caused the fiber to stop growing, release the vesicle from its growing end, and start to disassemble along its entire length (as evidenced by the decrease in optical density). This pressure-induced separation of the vesicle from the fiber allowed us to pinpoint the site of MSP polymerization associated with fiber growth. If, for example, filament assembly was because of the addition of subunits onto the end of existing filaments, then when we reduced the pressure we would have expected to observe an increase in optical density of the partially disassembled fiber. Instead, when the pressure was returned to 75 atm, the fiber shown in Fig. 2 failed to regain mass. By contrast, the vesicle immediately began to assemble a new fiber. We observed this pattern for several fibers; regardless of the extent of disassembly, fibers that lost their vesicles were unable to reassemble filaments when the pressure was reduced, but each of the released vesicles built new fibers. Thus, in this in vitro system, the MSP filaments in the fiber do not nucleate polymerization since no new growth occurs in fibers detached from a vesicle by pressurization. Likewise, vesicle-associated fiber growth does not require the end of a fiber because pressure-detached vesicles immediately began normal fiber growth when pressure was released. Thus, MSP polymerization is nucleated by factors at or very near the vesicle surface. The activity of these factors withstands pressure sufficient to disassemble the filaments that they produce.
Figure 2.

Effects of high pressure on filament assembly in vitro. (a) Video-enhanced phase-contrast micrograph of a fiber with its associated vesicle (arrow) grown at 75 atm. As the fiber continues to grow at this pressure for 20 s (b), it pushes its vesicle forward. Raising the pressure to 450 atm (c) causes the vesicle to release from the end of the fiber. At this pressure, the filaments in the original fiber disassemble so that by 60 s (d), its optical density is reduced substantially. When the pressure is returned to 75 atm (e), the vesicle (arrow) immediately starts to build a new fiber but the original fiber fails to regain its optical density even as the new fiber continues to grow for 1 min (f). Bar, 2 μm.
Increased Pressure Causes Vesicles to Assemble Fewer Filaments More Slowly
To investigate the mechanism of vesicle-associated filament assembly, we examined the effects of pressure on fiber growth systematically. We found that alteration of pressure affected both the rate of fiber growth and the optical density at the growing end. In the example shown in Fig. 3, when the fibers growing at 150 atm were pressurized to 275 atm, their rate of elongation decreased from 13 to 2 μm/min. In addition, the optical density of the segment grown at the higher pressure was fourfold lower than that of the adjacent segment constructed at the lower pressure. Conversely, when the pressure was lowered to 125 atm, the rate of fiber elongation increased to 15 μm/min, and the optical density of the newly formed segment was fivefold greater than that of the segment grown at 275 atm. Alteration of the pressure resulted in abrupt changes in both growth rate and optical density so that fibers treated in this way exhibited sharp boundaries between segments grown at different pressures. Because the optical density of a fiber is correlated with its filament mass per unit volume, the decrease in optical density that we observed at increased pressure could have been because of reduction in the number of filaments per unit volume of the fiber or to alignment of the filaments with the axis of fiber growth. A change in filament orientation from perpendicular to parallel to the fiber axis should increase the rate of fiber growth. We found, instead, that reduction in optical density correlated with reduced growth rate. Thus, decreased optical density is because of a reduction in filament number. This indicates that pressure influences both the number of filaments assembled at the vesicle surface per unit time and the rate at which those filaments polymerize.
We measured the growth rate and optical density of fibers grown in S100 diluted 1:4 with KPM buffer at pressures ranging from 1 to 350 atm. Pressures <75 atm had little effect on the rate of fiber growth, but at pressures ⩾75 atm we observed a roughly linear reduction in growth rate with increased pressure (Fig. 4 a). Thus, at 175 atm the growth rate (9 μm/min) was ∼50% of that observed at 1 atm, and at ∼325 atm fiber growth could not be detected in these preparations. Likewise, the optical density of fibers, normalized to that of fibers grown at 1 atm, decreased linearly with the pressure (Fig. 4 b). Comparison of these two measures of fiber dynamics revealed a direct correlation between normalized growth rate and normalized optical density over the range of pressures tested (Fig. 4 c).
Figure 4.
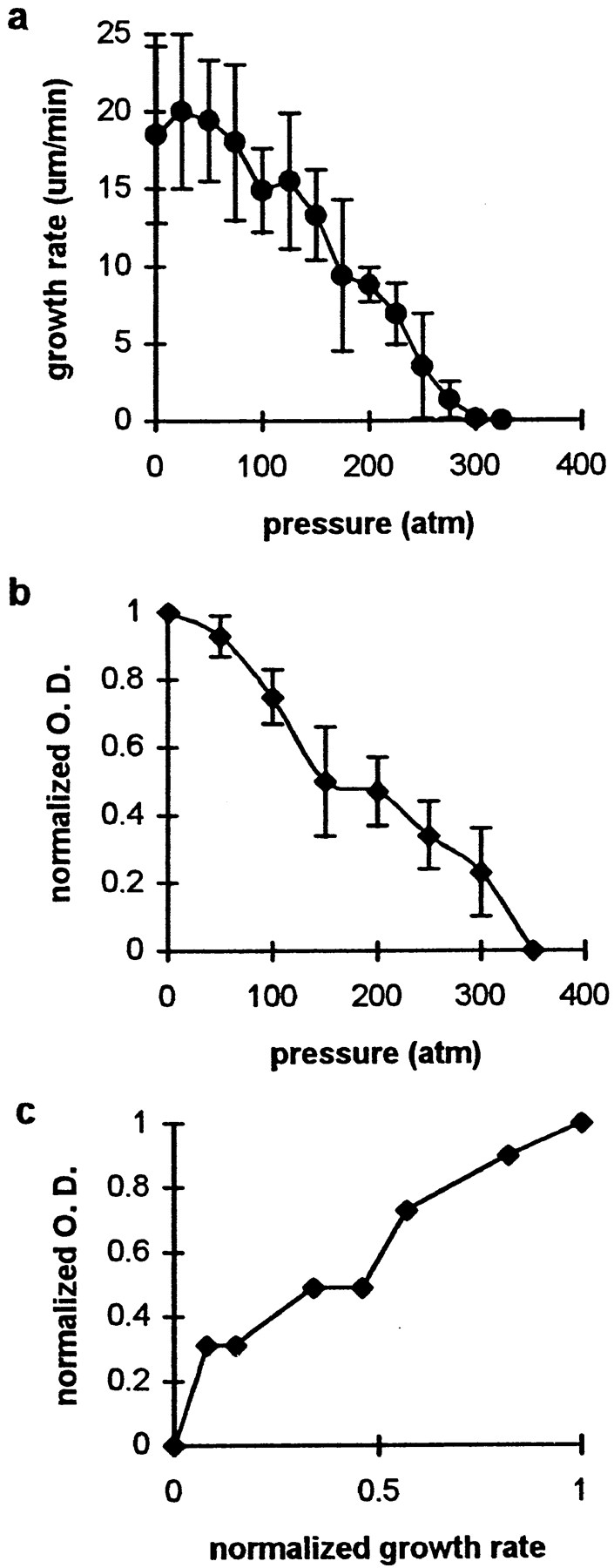
Relationship of fiber growth properties to pressure in S100 diluted 1:4 with KPM buffer. (a) Effect of pressure on the rate of fiber growth. Each point represents the mean of three trials, each of which included measurement of ⩾10 fibers for 45 s or more of growth. (b) Variation in optical density at the growing end of fibers with pressure. Normalized optical density was determined by measuring the grayscale value of a fiber, subtracting the background, and normalizing to the value obtained for a fiber grown in the same batch of S100 at 1 atm. Points represent the mean of three trials, each including measurements from ⩾12 fibers. The bars in a and b are standard errors of the mean. (c) Data in a and b replotted to show the relationship of growth rate to optical density. Growth rates were normalized to the mean rate at 1 atm.
Effects of Pressure and Dilution on Fiber Assembly Are Synergistic
Previous studies at atmospheric pressure demonstrated that dilution of S100 slows the rate of fiber growth (Italiano et al., 1996). We compared the effects of dilution and pressure on fiber assembly by examining the relationship of fiber growth rate to pressure at two identical dilutions of two batches of S100 (Fig. 5). We found that increased dilution of S100 not only reduced the rate of fiber growth, but also decreased the pressure required to stop fiber assembly. As a result, the slopes of the fiber growth rate vs. pressure curves for the two dilutions were similar. We also measured the pressure required to stop fiber growth over a range of dilutions of S100 and found that the stall pressure (i.e., the pressure at which growth ceased) decreased approximately linearly with S100 concentration (Fig. 6). These observations indicate that the effects of dilution and pressure on fiber growth are synergistic. Thus, dilution, like pressure, modulates both the nucleation activity at the vesicle surface and the rate of filament elongation.
Figure 5.

Effect of dilution of S100 on the relationship of fiber growth rate to pressure. Points represent the means from two trials, each including measurement of ⩾10 fibers for ⩾30 s.
Figure 6.

Pressure required to stall fiber growth as a function of dilution of S100 with KPM buffer. At each dilution pressure was increased until fiber growth could not be detected by video-enhanced phase-contrast microscopy. No variation in stall pressure was observed among three trials at each dilution.
Growth Rate and Optical Density of Fibers Are Independent of MSP Concentration Except at High Pressure
The rate of fiber growth at 1 atm is not affected by MSP concentration and is affected primarily by the concentration of an additional soluble cytosolic factor (Italiano et al., 1996). To determine if the influence of pressure on either the rate of fiber growth or the number of filaments assembled at the vesicle surface was related to MSP concentration, we compared the effects of pressure on fiber assembly in S100 diluted fourfold either with buffer alone or with buffer containing 4 mM β-MSP (the MSP concentration in undiluted S100). MSP concentration had no effect on the rate of fiber assembly at pressures ⩽250 atm (Fig. 7 a). However, when the pressure was raised to 300 atm, fiber growth stalled in S100 diluted with buffer. Fiber growth in S100 diluted with MSP continued until the pressure was raised to 450 atm, where growth stalled. Dilution with MSP also had no effect on the pressure-induced reduction in optical density of fibers with the exception that, at pressures greater than required to stall assembly in buffer-diluted S100, fibers grown in S100 diluted with 4 mM MSP continued to grow slowly at low optical density (Fig. 7 b). Thus, MSP concentration had negligible effect on the rates of filament nucleation or elongation except at high pressures, where added MSP decreased the sensitivity of fiber growth to pressure, and so increased the stall pressure.
Figure 7.

Comparison of the effects of pressure on fiber growth (a) and OD (b) in S100 diluted with KPM buffer versus dilution with KPM buffer containing 4 mM β-MSP. Each point represents the mean of ⩾15 fibers. Normalized optical density values in b were calculated as described in Fig. 4.
Discussion
In vitro reconstitution of MSP-based motility provides a powerful assay for evaluating the mechanism of localized filament polymerization and its role in ameboid movement (Theriot, 1996). By using pressure to alter the assembly properties of this system, we have more precisely characterized how fibers form and shown that their assembly requires both nucleation and elongation of MSP filaments in the immediate vicinity of the vesicle surface. Surprisingly, the rates of nucleation and elongation near the vesicle surface are largely independent of MSP concentration, and instead are governed primarily by a soluble factor (SF) that activates MSP polymerization.
Model for the Mechanism of Membrane-associated MSP Polymerization
The results from both pressure and dilution studies suggest the model for fiber assembly shown in Fig. 8. In addition to MSP and ATP as an energy source (Italiano et al., 1996), vesicle-associated fiber assembly requires SF and an integral vesicle protein (VP). SF in combination with VP converts MSP to an activated form, MSP*, which polymerizes rapidly under physiological conditions to form filaments and ultimately fibers. This model accounts for the key features we have established for the effect of pressure, and MSP concentration on fiber generation in vitro in terms of a nucleation–elongation polymerization mechanism. Biological assemblies such as MSP filaments are invariably generated by nucleation–elongation polymerization (Oosawa and Asakura, 1975), in which a small number of subunits first come together to form a nucleus, after which the filament elongates by addition of subunits at one or both ends. The nucleation rate determines the number of filaments assembled and the elongation rate defines how rapidly those filaments grow.
Figure 8.

Mechanism of vesicle-associated MSP filament polymerization suggested by the pressure and dilution data. Because polymerization occurs primarily in the vicinity of the vesicle, we propose that fiber growth involves conversion of MSP into a short-lived, activated form (MSP*) that polymerizes much more rapidly under physiological conditions than MSP itself. Conversion of MSP to MSP* requires an integral membrane protein (VP) in the vesicle that recruits a soluble cytosolic protein (SF) to the vesicle surface. The active VP/SF component then converts MSP to MSP* that polymerizes to form fibers. Although the diagram shows SF bound to VP, SF could bind transiently to and activate VP, or be activated by VP and release. Because MSP* is short lived, its concentration would fall rapidly away from the vesicle, and the filaments in the bulk of the fiber would contain primarily MSP rather than MSP*. Therefore, this model accounts for the formation of MSP fibers only in the immediate vicinity of the vesicle and the effects of pressure and dilution on the rate of fiber formation.
Because pressure does not alter the refractive index of the medium and its effects are rapidly reversible, it provides an effective way to assess the contributions of nucleation of new filaments and elongation of existing filaments to polymerization. We measured changes in filament number (assessed by fiber optical density) and elongation rate (assessed by fiber growth rate) simultaneously, and showed that in the in vitro system increasing pressure resulted in fewer MSP filaments growing more slowly (Fig. 3). Because pressure modulated the rates of both nucleation and elongation of MSP filaments, they probably share the same pressure-sensitive mechanism. The pressures that influence fiber assembly are well below the level required to induce conformational changes in proteins, but are of the order that modulate protein–protein interactions (Mozhaev et al., 1996). Such interactions generate a net volume increase, probably by releasing bound water (Silva et al., 1996). By Le Chatelier's principle, increasing pressure should favor the lower volume state, making nucleation and elongation more difficult.
Significant MSP polymerization only occurred in the vicinity of the vesicle. Assembled fibers were stable at pressures that blocked growth. Moreover, when the pressure was reduced after a vesicle had been separated from its fiber by high pressure, the vesicle grew a new fiber at the same rate, diameter, and density as its original fiber, whereas the separated fiber did not. Furthermore, at <300 atm, the abrupt change in both density and growth rate seen at the vesicle–fiber interface when pressure was altered, indicated that filaments in the bulk of the fiber made a negligible contribution to assembly. Therefore, once filaments were incorporated into the fiber and moved away from the vesicle, the rates of subunit addition and loss fell below levels we could detect by optical methods. Therefore, fiber assembly is not a simple equilibrium process in which the polymerizing and depolymerizing MSP subunits at the vesicle and in the bulk of the fiber are the same; polymerization and depolymerization near the vesicle primarily involves MSP*, whereas in the bulk of the fiber it involves MSP. Moreover, the strict spatial control of polymerization in this system indicates that the lifetime of MSP* outside the fiber must be short. Because fiber formation occurs only at the vesicle surface and not spontaneously, both filament nucleation and elongation depend on the concentration of MSP*, and thus on the activity of VP/SF, rather than on the MSP concentration itself. Therefore, the requirement VP/SF to generate MSP* explains why polymerization only occurs close to the vesicle surface. The number of VP molecules on the vesicle would determine the number of SF molecules recruited and, in turn, the amount of MSP polymerized. Larger vesicles would have more VP and, as we observed, would grow thicker fibers.
Our current data do not define the molecular nature of MSP* or precisely how it is generated by VP/SF. No proteins in S100 approach the molar concentration of MSP, so it is unlikely that VP/SF creates MSP* by removing a sequestering protein analogous to profilin (Machesky and Pollard, 1993) or β-thymosin (Nachmias, 1993) in actin-based systems. MSP does not appear to bind nucleotides, and so it is unlikely that VP/SF functions through nucleotide exchange analogous to the ATP–ADP exchange profilin promotes on actin (Sohn and Goldschmidt-Clermont, 1994). Extensive biochemical analysis of MSP has failed to detect a covalent posttranslational modification, such as phosphorylation, that could account for VP/SF-induced polymerization, but the amount of MSP modified at any time would probably be small and could be below the detection level of our assays. Although we cannot rule out the possibility that VP/SF removes a small molecule that prevents MSP polymerization, perhaps the most likely explanation is that VP/SF induces a conformational change in MSP that facilitates polymerization. We cannot eliminate the possibility that different activated forms of MSP are required for nucleation and elongation, but the rates and locations of these two processes are so tightly coupled that it seems more likely that both use the same activated MSP*.
Although vesicles (and thus VP) are clearly necessary for fiber formation, SF is also required and normally limits the rate of MSP polymerization. Isolated vesicles cannot induce detectable polymerization of purified MSP, and maintaining constant MSP concentration does not prevent the decrease in fiber growth rate resulting from dilution of S100 (Italiano et al., 1996). Moreover, as pressure increased, the rates of filament nucleation and elongation were independent of MSP concentration except when approaching the stall pressure (Fig. 7). Thus, SF and not MSP, must govern both the number of filaments assembled at the vesicle and how rapidly they elongate. There are indications that both SF and VP are proteins. Isolated vesicles recombined with cytosol still grow fibers (Italiano et al., 1996), although when isolated vesicles were treated with proteases (0.1 mg/ ml trypsin or 10 U/ml pronase, for 100 min), washed, and then recombined with cytosol, fibers did not assemble (our unpublished observations). VP is the likely target of these proteases. The cytosolic components (the fraction of S100 containing SF) required for fiber assembly failed to pass through a 5,000-M r cutoff filter (Italiano et al., 1996), suggesting that SF (or at least one of its constituents) is a macromolecule, probably a protein.
VP and SF could function as a complex or, alternatively, one could activate the other. Although our current data do not allow discrimination between these possibilities (thus, we refer to the active component as VP/SF), they do reveal important features of the filament production mechanism. The MSP concentration is high compared to other proteins in the cytoplasm, suggesting that VP/SF must function cyclically or catalytically to activate MSP to MSP*. Indeed, VP/SF appears analogous to an enzyme that follows Michaelis–Menton kinetics. At high substrate (MSP) concentration, the rate of MSP* production and fiber growth approaches v max and depends primarily on the concentration of VP/SF rather than MSP. Lowering the concentration of VP/SF by dilution would reduce the rate of MSP* production and, as we observed, lower the fiber growth rate even if MSP concentration remained constant. Thinking of VP/SF as an “enzyme” with MSP as its “substrate” can also account for the effect of added MSP seen near the stall pressure. The pressures that modulate fiber assembly are likely to alter the interaction of VP/SF with its substrate but not its conformation. Thus, pressure should alter the K m of VP/SF without significantly changing v max. If between 1–250 atm the K m of VP/SF was much lower than the MSP concentration (∼1 mM in S100 diluted 1:4), increasing MSP concentration fourfold would have little effect on the rate of MSP* formation and so, as we observed, would not produce a significant increase in fiber growth. MSP concentration would only influence the rate of MSP polymerization when it approached K m. This would explain the effect of added MSP above 250 atm, where we found that a fourfold increase in MSP concentration produced a small increase in the rate of fiber growth. If K m approached the MSP concentration at these high pressures, raising the MSP concentration would increase the reaction velocity, enabling VP/SF to generate more MSP*, and thereby sustaining fiber growth and increasing the stall pressure. The Michaelis–Menton behavior of VP/SF with MSP also provides compelling evidence for a direct interaction between the molecules analogous to an enzyme–substrate complex. Naturally, energy would be required to produce MSP*, and could result from ATP hydrolysis either when VP/SF was formed or when it generated MSP*.
Implications for Ameboid Movement
Although we focused on the effects of pressure on in vitro motility, we also showed that pressure modulates cytoskeletal dynamics in intact sperm. The morphology and dynamics of fibers assembled in vitro and the fiber complexes in vivo are similar (Italiano et al., 1996), and the shared response of these filament arrays to pressure suggests that mechanistic conclusions derived from studying the in vitro system are likely to also apply to the cytoskeletal dynamics associated with cell locomotion. The pseudopod of Ascaris sperm contains ∼4 mM MSP, but polymerization occurs almost exclusively along the membrane at their leading edge (Roberts and King, 1991; King et al., 1994) The vesicles that build fibers in vitro are derived from the plasma membrane at the front of the pseudopod, and so this is where VP is located in the cell. Recruitment of SF to this site would account for the localized polymerization of MSP observed in vivo, and the relationship of polymerization to vesicle movement in vitro would explain the tight coupling between filament assembly and protrusion of the leading edge of the pseudopod.
Relationship to Actin-based Locomotory Systems
Although different components are used in the MSP- and actin-based machinery, the mechanisms by which the two systems generate motility appear to be conserved. Our pressure studies may provide general insights about how cells assemble filaments at the membrane and determine sites of protrusive activity. For example, two models, nucleation of assembly of new filaments and addition of subunits onto the ends of existing filaments, have been proposed to account for actin polymerization associated with membrane protrusion (for review see Welch et al., 1997a ). Lamellipodial protrusion in fish epithelial keratocytes is thought to involve nucleation of filaments along the leading edge, followed by release to become incorporated into the dynamic lamellipodial cytoskeleton (Theriot and Mitchison, 1992). By contrast, rapid shape changes in activated platelets appear to be because of the addition of subunits onto barbed ends exposed by the severing and uncapping of preexisting actin filaments (Hartwig, 1992; Barkalow et al., 1996; Nachmias et al., 1996 ). Discrimination between these mechanisms has often relied on indirect methods, such as monitoring the dynamics of fluorescent actin in live cells (Theriot and Mitchison, 1991), or measuring filament length distributions (Small et al., 1995; Sechi et al., 1997), and has sometimes led to conflicting interpretations (for review see Mitchison and Cramer, 1996). Our pressure assays showed that MSP-based fiber assembly and vesicle movement require continuous nucleation of new MSP filaments, and that filament elongation is completed while the filament is near the vesicle.
The molecular components that determine the site of MSP assembly may be analogous to those in actin-based motile systems. For example, intracellular movement of bacteria such as Listeria is driven by formation of an actin tail and resembles lamellipodial protrusion in many respects (for review see Theriot et al., 1995). Assembly of the actin filaments that compose the tail requires a bacterial surface protein, ActA, that recruits host cytosolic proteins, such as vasodilator-stimulated phosphoprotein, profilin (Chakraborty et al., 1995; Reinhard et al., 1995), and Arp 2/3 complex (Welch et al., 1997b ) to the site of polymerization. Therefore, in the MSP system, VP operates like ActA by recruiting components to the membrane to trigger filament formation. There are also several examples of actin-based protrusions in eukaryotic cells stimulated by interaction of an external signal with a membrane receptor and transduced by local activation of cytosolic components, such as members of the rho family of GTPases (Hall, 1994; Zigmond, 1996), and so the interaction between a cytosolic factor and membrane protein that defines the site of MSP polymerization may be a general mechanism by which cells specify the location of protrusive activity and determine their direction of locomotion.
Acknowledgments
We thank K. Riddle, A. Davis, and G. Roberts (all from Florida State University, Tallahassee, FL) for expert technical assistance, H. Crenshaw (Duke University, Durham, NC) for the use of his microscope objective, and J. Italiano, R. Hammell, and L. LeClaire (all from Florida State University) for valuable discussions and ideas.
This work was supported by National Institutes of Health grants (GM-29994) to T.M. Roberts and M. Stewart and (GM-24364) to E.D. Salmon.
Abbreviations used in this paper
- atm
atmospheres
- MSP
major sperm protein
- MSP*
activated major sperm protein
- SF
soluble factor
- VP
vesicle protein
Footnotes
Address all correspondence to T.M. Roberts, Department of Biological Science, 336 Bio Unit I, Florida State University, Tallahassee, FL 32306-3050. Tel.: (904) 644-3237. Fax: (904) 644-0481. E-mail: roberts@bio.fsu.edu
References
- Barkalow K, Witke W, Kwiatkowski DJ, Hartwig JH. Coordinated regulation of platelet actin filament barbed ends by gelsolin and capping protein. J Cell Biol. 1996;134:389–399. doi: 10.1083/jcb.134.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begg DA, Salmon ED, Hyatt HA. The changes in structural organization of actin in sea urchin egg cortex in response to hydrostatic pressure. J Cell Biol. 1983;97:1795–1805. doi: 10.1083/jcb.97.6.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourns B, Franklin S, Cassimeris L, Salmon ED. High hydrostatic pressure effects in vitro: changes in cell morphology, microtubule assembly, and actin organization. Cell Motil Cytoskel. 1988;10:380–390. doi: 10.1002/cm.970100305. [DOI] [PubMed] [Google Scholar]
- Chakraborty T, Ebel F, Domann E, Niebuhr K, Gerstel B, Pistor S, Temm-Grove CJ, Jockusch BM, Reinhard M, Walter U, Wehland J. A focal adhesion factor directly linking intracellularly motile Listeria monocytogenes and Listeria invanoviito the actin-based cytoskeleton of mammalian cells. EMBO (Eur Mol Biol Organ) J. 1995;14:1314–1321. doi: 10.1002/j.1460-2075.1995.tb07117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condeelis J. Life at the leading edge: the formation of cell protrusions. Annu Rev Cell Biol. 1993;9:411–444. doi: 10.1146/annurev.cb.09.110193.002211. [DOI] [PubMed] [Google Scholar]
- Crenshaw HC, Salmon ED. Hydrostatic pressure to 400 atm does not induce changes in the cytosolic concentration of Ca2+in mouse fibroblasts: measurements using fura-2 fluorescence. Exp Cell Res. 1996;227:277–284. doi: 10.1006/excr.1996.0277. [DOI] [PubMed] [Google Scholar]
- Hall A. Small GTP-binding proteins and the regulation of the actin cytoskeleton. Annu Rev Cell Biol. 1994;10:31–54. doi: 10.1146/annurev.cb.10.110194.000335. [DOI] [PubMed] [Google Scholar]
- Hartwig JH. Mechanism of actin rearrangements mediating platelet activation. J Cell Biol. 1992;118:1421–1442. doi: 10.1083/jcb.118.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Italiano JE, Jr, Roberts TM, Stewart M, Fontana CA. Reconstitution in vitro of the motile apparatus from the ameboid sperm of Ascarisshows that filament assembly and bundling move membranes. Cell. 1996;84:105–114. doi: 10.1016/s0092-8674(00)80997-6. [DOI] [PubMed] [Google Scholar]
- King KL, Essig J, Roberts TM, Moerland TS. Regulation of the Ascarismajor sperm protein (MSP) cytoskeleton by intracellular pH. Cell Motil Cytoskel. 1994;27:193–205. doi: 10.1002/cm.970270302. [DOI] [PubMed] [Google Scholar]
- Machesky LM, Pollard TD. Profilin as a potential mediator of membrane-cytoskeleton communication. Trends Cell Biol. 1993;3:381–385. doi: 10.1016/0962-8924(93)90087-h. [DOI] [PubMed] [Google Scholar]
- Marsland D, Brown DES. Amoeboid movement at high hydrostatic pressure. J Cell Comp Physiol. 1936;8:167–178. [Google Scholar]
- Martin, L.C. 1966. The Theory of the Microscope. American Elsevier Publishing Co., Inc. New York. 488 pp.
- Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- Mozhaev VV, Heremans K, Frank J, Masson P, Balny C. High pressure effects on protein structure and function. Proteins Struct Funct Genet. 1996;24:81–91. doi: 10.1002/(SICI)1097-0134(199601)24:1<81::AID-PROT6>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Nachmias VT. Small actin-binding proteins: the β-thymosin family. Curr Opin Cell Biol. 1993;5:56–62. doi: 10.1016/s0955-0674(05)80008-0. [DOI] [PubMed] [Google Scholar]
- Nachmias VT, Golla R, Casella JF, Barron-Casella E. CapZ, a calcium insensitive capping protein in resting and activated platelets. FEBS (Fed Eur Biochem Soc) Lett. 1996;378:258–262. doi: 10.1016/0014-5793(95)01474-8. [DOI] [PubMed] [Google Scholar]
- Oliver T, Lee J, Jacobson K. How do cells move along surfaces? . Semin Cell Biol. 1993;5:139–147. doi: 10.1006/scel.1994.1018. [DOI] [PubMed] [Google Scholar]
- Oosawa, F., and S. Asakura. 1975. Thermodynamics of the Polymerization of Protein. Academic Press, London. 204 pp.
- Pluta, M. 1989. Advanced Light Microscopy. Vol. 2. Specialized Methods. Elsevier Publishing Co., Inc., Amsterdam, The Netherlands. 494 pp.
- Reinhard M, Giehl K, Abel K, Haffner C, Jarchau T, Hoppe V, Jockusch BM, Walter U. The proline-rich focal adhesion and microfilament protein VASP is a ligand for profilins. EMBO (Eur Mol Biol Organ) J. 1995;14:1583–1589. doi: 10.1002/j.1460-2075.1995.tb07146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, T.M. 1987. Nematode sperm as models for studying cell motility. In Vistas in Nematology. J. Veech and D. Dickson, eds. Williams and Wilkins, Baltimore, MD. 440–447.
- Roberts TM, King KL. Centripetal flow and directed reassembly of the major sperm protein (MSP) cytoskeleton in the amoeboid sperm of the nematode, Ascaris suum. . Cell Motil Cytoskel. 1991;20:228–241. doi: 10.1002/cm.970200306. [DOI] [PubMed] [Google Scholar]
- Roberts TM, Stewart M. Nematode sperm locomotion. Curr Opin Cell Biol. 1995;7:13–17. doi: 10.1016/0955-0674(95)80039-5. [DOI] [PubMed] [Google Scholar]
- Roberts TM, Stewart M. Nematode sperm: amoeboid movement without actin . Trends Cell Biol. 1997;7:368–373. doi: 10.1016/S0962-8924(97)01113-6. [DOI] [PubMed] [Google Scholar]
- Salmon ED. Pressure-induced depolymerization of brain microtubules in vitro. Science. 1975a;189:884–886. doi: 10.1126/science.1171523. [DOI] [PubMed] [Google Scholar]
- Salmon ED. Spindle microtubules: thermodynamics of in vivo assembly and role in chromosome movement. Ann NY Acad Sci. 1975b;253:383–406. doi: 10.1111/j.1749-6632.1975.tb19216.x. [DOI] [PubMed] [Google Scholar]
- Salmon ED, Ellis GW. A new miniature hydrostatic pressure chamber for microscopy. J Cell Biol. 1975;65:587–602. doi: 10.1083/jcb.65.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechi AS, Wehland J, Small JV. The isolated comet tail pseudopodium of Listeria monocytogenes: a tail of two actin filament populations, long and axial and short and random. J Cell Biol. 1997;137:155–167. doi: 10.1083/jcb.137.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepsenwol S, Nguyen M, Braun T. Adenylate cyclase activity is absent in inactive and motile sperm of the nematode parasite, Ascaris suum. . J Parasitol. 1986;72:962–964. [PubMed] [Google Scholar]
- Sepsenwol S, Ris H, Roberts TM. A unique cytoskeleton associated with crawling in the amoeboid sperm of the nematode, Ascaris suum. . J Cell Biol. 1989;108:55–66. doi: 10.1083/jcb.108.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JL, Foguel D, Da Poian AT, Prevelige PE. The use of hydrostatic pressure as a tool to study viruses and other macromolecular assemblages. Curr Opin Struct Biol. 1996;6:166–175. doi: 10.1016/s0959-440x(96)80071-6. [DOI] [PubMed] [Google Scholar]
- Sohn RH, Goldschmidt-Clermont PJ. Profilin: at the crossroads of signal transduction and the actin cytoskeleton. Bioessays. 1994;16:465–472. doi: 10.1002/bies.950160705. [DOI] [PubMed] [Google Scholar]
- Small JV, Herzog M, Anderson K. Actin filament organization in the fish keratocyte lamellipodium. J Cell Biol. 1995;129:1275–1286. doi: 10.1083/jcb.129.5.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stossel TP. On the crawling of animal cell. Science. 1993;260:1086–1094. doi: 10.1126/science.8493552. [DOI] [PubMed] [Google Scholar]
- Theriot JA. The cell biology of infection by intracellular bacterial pathogens. Annu Rev Cell Dev Biol. 1995;11:213–239. doi: 10.1146/annurev.cb.11.110195.001241. [DOI] [PubMed] [Google Scholar]
- Theriot JA. Worm sperm and advances in cell locomotion. Cell. 1996;84:1–4. doi: 10.1016/s0092-8674(00)80068-9. [DOI] [PubMed] [Google Scholar]
- Theriot JA, Mitchison TJ. Actin microfilament dynamics in locomoting cells. Nature. 1991;352:126–131. doi: 10.1038/352126a0. [DOI] [PubMed] [Google Scholar]
- Theriot JA, Mitchison TJ. Nucleation-release model of actin filament dynamics in cell motility. Trends Cell Biol. 1992;2:219–222. doi: 10.1016/0962-8924(92)90298-2. [DOI] [PubMed] [Google Scholar]
- Tilney LG, Tilney MS. The wiley ways of a parasite: induction of actin assembly by Listeria. . Trends Microbiol. 1993;1:25–31. doi: 10.1016/0966-842x(93)90021-i. [DOI] [PubMed] [Google Scholar]
- Ward, S. 1986. Asymmetric localization of gene products during development of Caenorhabditis elegans spermatozoa. In Gametogenesis and the Early Embryo. Gall, J.G., ed. Alan R. Liss, Inc. New York. 55–75.
- Welch MD, Mallavarapu A, Rosenblatt J, Mitchison TJ. Actin dynamics in vivo. . Curr Opin Cell Biol. 1997a;9:54–61. doi: 10.1016/s0955-0674(97)80152-4. [DOI] [PubMed] [Google Scholar]
- Welch MD, Iwamatsu A, Mitchison TJ. Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. . Nature. 1997b;385:265–269. doi: 10.1038/385265a0. [DOI] [PubMed] [Google Scholar]
- Zigmond SH. Signal transduction and actin filament organization. Curr Opin Cell Biol. 1996;8:66–73. doi: 10.1016/s0955-0674(96)80050-0. [DOI] [PubMed] [Google Scholar]