

Abstract
We have monitored fusion between cell pairs consisting of a single human immunodeficiency virus–1 (HIV-1) envelope glycoprotein–expressing cell and a CD4+ target cell, which had been labeled with both a fluorescent lipid in the membrane and a fluorescent solute in the cytosol. We developed a new three-color assay to keep track of the cell into which fluorescent lipids and/or solutes are redistributed. Lipid and solute redistribution occur as a result of opening a lipid-permissive fusion pore and a solute-permissive fusion pore (FPS), respectively. A synthetic peptide (DP178) corresponding to residues 643–678 of the HIV-1LAI gp120-gp41 sequence (Wild, C.T., D.C. Shugars, T.K. Greenwell, C.B. McDanal, and T.J. Matthews. 1994. Proc. Natl. Acad. Sci. USA. 91:12676–12680) completely inhibited FPS at 50 ng/ml, whereas at that concentration there was 20–30% fusion activity measured by the lipid redistribution. The differences detected in lipid mixing versus contents mixing are maintained up to 6 h of coculture of gp120-41–expressing cells with target cells, indicating that DP178 can “clamp” the fusion complex in the lipid mixing intermediate for very long time periods. A peptide from the NH2-terminal of gp41, DP107, inhibited HIV-1LAI gp120-gp41–mediated cell fusion at higher concentrations, but with no differences between lipid and aqueous dye redistribution at the different inhibitor concentrations. The inhibition of solute redistribution by DP178 was complete when the peptide was added to the fusion reaction mixture during the first 15 min of coculture. We have analyzed the inhibition data in terms of a fusion pore dilation model that incorporates the recently determined high resolution structure of the gp41 core.
The human immunodeficiency virus–1 (HIV-1)1 envelope glycoprotein is synthesized as a gp160 precursor, which is cleaved by a cellular proteinase at a characteristic sequence to create the mature glycoproteins gp120 and gp41 (23). Gp120 is noncovalently attached to the extracellular domain of gp41 and mediates the binding of the virus to appropriate host cells through interactions with the receptor CD4 (26) and coreceptors (18). The gp41 plays an important role in fusion between virus and cell membranes (30). The three-dimensional structure of the ectodomain of gp41 has been modeled using the crystal structure of influenza hemagglutinin (HA) as a conceptual framework (20). The model predicted two regions of the gp41 ectodomain as extended α helices. One of these, on the NH2-terminal side of the molecule, was predicted to contain a leucine zipperlike motif, whereas the other is located in the COOH-terminal end of the gp41 ectodomain (see Fig. 1). In the absence of gp120 and the NH2-terminal fusion peptide, the ectodomain of gp41 forms a soluble α-helical rodlike oligomer (40). The core structure of gp41 has recently been determined by x-ray crystallography (10, 41).
Figure 1.

Amino acid sequence and schematic representation of the localization of DP178 in the gp41 protein (46). Sequence (36 amino acids):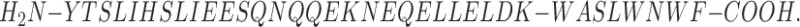
A synthetic peptide (DP178) corresponding to the COOH-terminal ectodomain sequence (residues 643–678 of the HIV-1LAI isolate) blocked 100% of virus-mediated cell–cell fusion at <5 ng/ml (measured in a syncytium formation assay), and reduced infectious titer of cell-free virus 10 times at ∼80 ng/ml (46). The mechanism of its potent inhibitory effect remains to be elucidated.
The viral fusion reaction proceeds along a series of discrete steps before the final event occurs that results in delivery of the nucleocapsid into the cell (3, 42, 43). In previous studies of cell fusion mediated by influenza HA an important new intermediate, the fusion pore, was revealed by the differential dispersion of lipid molecules, small aqueous molecules, and large molecules (34), and by capacitance patch clamping studies (37, 48). Using analytical and quantitative video microscopy we have established the sequence of these events during HA-induced fusion with fluorescently labeled red blood cells (RBCs) (6). Our results indicate that lipid mixing and transfer of large solutes are sequential events occurring with different probabilities (6, 36). We have analyzed the kinetics of events that occur as a result of transitions between a lipid-permissive fusion pore (FPL) and a solute-permissive fusion pore (FPS) (6). Based on the hypothesis that HIV-1 envelope glycoprotein–induced fusion pore dilation follows a similar pattern as that seen with HA, we reasoned that DP178 might be used to dissect these various stages of fusion pore dilation.
In this report, we evaluate the ability of DP178 to block HIV-1 envelope glycoprotein-mediated cell–cell fusion by two different methods, the gene reporter fusion assay (32) and fluorescent dye transfer analyzed by video microscopy (33, 39). We developed a new three-color assay to assess lipid and solute movement from the same target cell into an envelope-expressing cell. We find that DP178 blocks movement of large molecules at a 10-fold lower concentration than lipid mixing. We analyze these findings in terms of a model for gp120-gp41–mediated fusion pore dilation.
Materials and Methods
Cells
The human lymphoid cell line TF228.1.16 (19) (a gift from Z.L. Jonak, SmithKline and Beecham, Philadelphia, PA), which constitutively expresses HIV-1 envelope glycoprotein, GP4F cells (a gift from J.M. White, University of Virginia, Charlottesville, VA), mouse 3T3 fibroblasts, which constitutively express influenza HA (16, 31), and the CD4+ lymphocyte cell line SupT1 (obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD) were used. The cells were propagated in RPMI 1640 (for human cell lines) or DME (for GP4F) media supplemented with 10% FCS and antibiotics (Gibco BRL, Life Technologies Inc., Gaithersburg, MD). Cell cultures were maintained at 37°C in a humidified 5% CO2 atmosphere.
Reagents
The fluorescent probes DiI (1,1′-dioctadecyl-3,3,3′3′-tetramethylindocarbocyanine perchlorate; excitation/emission [ex/em] 550/565 nm), DiO (3,3′-dioctadecyloxaocarbocyanine perchlorate; ex/em 484/501 nm), calcein-AM (ex/em 496/517 nm), and CMAC (7-amino-4-chloromethylcoumarin; ex/em 354/469 nm) were all products from Molecular Probes (Eugene, OR). NP-40, the substrate CPRG (chlorophenol red–β-galactopyranoside), X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside), and β-galactosidase from Escherichia coli (600 U/mg) were obtained from Boehrringer Mannheim (Indianapolis, IN). Diluent C and 1-β-d-arabinofuranosylcytosin (Ara-C) were purchased from Sigma Chemical Co. (St. Louis, MO).
Peptide Synthesis
The DP178 peptide (Fig. 1) was synthesized by the solid phase method with Fmoc chemistry using a peptide synthesizer (430A; Applied Biosystems, Inc., Foster City, CA). Cleavage of the peptide from the resin and removal of the side chain–protecting groups were carried out using reagent K. The peptide was purified by HPLC on a Vydac C-8 column with 0.05% TFA/water-acetonitrile. The mass of the peptide (M r = 4,451) was confirmed by electrospray ionization mass spectrometry on a Finnigan MAT SSQ 7000 (Finnigan MAT, San Jose, CA).
Gene Reporter Fusion Assay
Recombinant vaccinia (vCB21R and vTF7.3, respectively, provided by Dr. E.A. Berger, National Institutes of Allergy and Infectious Diseases, Bethesda, MD) was used to express β-galactosidase under the control of the T7 promoter in TF228 cells, and T7 polymerase in SupT1 cells (18). Cells were infected at a multiplicity of infection of 10 for 1.5 h at 37°C, followed by incubation at 31°C overnight. For the fusion assay, 2 × 105 cells of each type were mixed in 96-well plates for 2 h at 37°C in presence of Ara-C to reduce background. To quantitate fusion, the cells were lysed with NP-40 to a final concentration of 0.5%, and aliquots of this lysate were monitored for β-galactosidase activity at 37°C using the colorimetric assay (32).
Dye Transfer Fusion Assay
The second method used for detecting cell–cell fusion was based on the redistribution of fluorescent probes between effector and target cells upon fusion (33, 39). The application of three different probes was used to monitor lipid versus cytosolic mixing in the same cell population.
Labeling with the Lipophilic Probes.
DiI and DiO are the most widely used lipophilic carbocyanine membrane probes. Their spectral properties are independent of the alkyl chains, but are determined by the heteroatoms in the terminal ring systems and the length of the connecting bridge. DiI has excitation and emission spectra compatible with rhodamine filter sets, whereas the DiO analogue can be used with the FITC optical filter set. This enables the study of lipid mixing between pairs of cells labeled differently with either dye. Cells were incubated for 15 min with the probe in RPMI/Diluent C 1:0.6 (final concentration = 34 μM) at room temperature, shaking in the dark. The cells were then washed three times with RPMI, changing tubes for each wash, and then resuspended at 106 cells/ml in RPMI.
Calcein-AM Labeling.
Cells were incubated with 0.5 μM of calcein-AM for 45 min at 37°C, washed, incubated in fresh medium for 30 min at 37°C, washed twice, and then resuspended at 106 cells/ml in RPMI.
CMAC Loading.
This blue fluorescent dye freely passes through cell membranes and then reacts with free sulfhydryls, producing a cell-impermeant reaction product. Cells were loaded with 20 μM of CMAC in RPMI for 45 min at 37°C, washed, incubated in fresh medium for 30 min at 37°C, washed twice, and then resuspended at 106 cells/ml in RPMI.
Fusion Assay.
Fluorescently labeled gp120-41–expressing cells and CD4+ cells were cocultured at 1:1 ratio (105 cells of each type) for 2 h at 37°C in coated microwells (MatTek Corp., Asland, MA), with different concentrations of peptide. Bright field and fluorescent images were acquired using an Olympus IX70 microscope coupled to a CCD camera (Princeton Instruments, Trenton, NJ) with ×40 UplanApo oil immersion objective. “FITC” (exciter: BP470-490; beamsplitter: DM505; emitter BA515-550), “rhodamine” (exciter: BP530-550; beamsplitter: DM570; emitter BA590), and “DAPI” (exciter: D360/40; beamsplitter: 400DCLP; emitter: D450/60) optical filter cubes were carefully chosen to avoid spillover when observing fluorescence of the three dyes. For each sample, three different fields were collected, and data were analyzed by overlaying the images using Metamorph software (Universal Imaging Corporation, West Chester, PA). When using two dyes, effector and target cells were labeled with different probes. The total number of cells positive for one or two dyes was scored. Bright field images were used to eliminate false overlaying positives.
The percentage of fusion was calculated as the mean of a and b: a = 100 × (number of cells positive for two dyes/number of target cells positive for one dye); b = 100 × (number of cells positive for two dyes/number of effector cells positive for one dye). If three probes were used, TF228 cells were labeled with the cytoplasmic probe CMAC and the target cells, SupT1, with both calcein (cytoplasmic) and DiI (lipophilic). The total number of cells positive for one, two, or three dyes was scored. The percentage of lipid mixing was calculated as the mean of c and d: c = 100 × (number of cells positive for DiI and CMAC/number of target cells positive for DiI and calcein); d = 100 × (number of cells positive for DiI and CMAC/number of effector cells positive for CMAC). The percentage of cytoplasm mixing was calculated as the mean of e and f: e = 100 × (number of cells positive for three dyes/number of target cells positive for DiI and calcein); f = 100 × (number of cells positive for three dyes/number of effector cells positive for CMAC).
Dye transfer between GP4F cells and labeled RBCs was monitored as described previously (6, 31, 34, 37, 48).
Results
Inhibition of Cell–Cell Fusion by DP178
In the first set of experiments, we assayed the concentration-dependent inhibitory effect of DP178 on cell–cell fusion using the gene reporter assay (32). TF228 cells (infected with the vaccinia recombinant vCB21R that expresses β-galactosidase under the control of the T7 promoter) and SupT1 cells (infected with vTF7.3 vaccinia with T7 polymerase gene) were incubated with different concentrations of peptide as described in Materials and Methods. After 2 h of incubation, the extent of cell–cell fusion was calculated as percentage of β-galactosidase activity compared with the control without peptide. The results are summarized in Table I A. As shown, 2.5 ng/ml of peptide reduced >50% of cell–cell fusion as measured by β-galactosidase activity, and 50 ng/ml blocked the activity completely.
Table I.
Inhibition of HIV-1–mediated Cell–Cell Fusion by DP-178 Peptide
| Peptide concentration | (A) β-Galactosidase activity | (B) DiI/DiO transfer | ||
|---|---|---|---|---|
| ng/ml | % | % | ||
| 0 | 100 | 100 | ||
| 0.5 | 109 ± 12.7 | 104.5 ± 4.5 | ||
| 2.5 | 49.05 ± 1.3 | 79.6 ± 6.4 | ||
| 5 | 24.4 ± 3.4 | 41.5 ± 16.4 | ||
| 25 | 12.5 ± 2.7 | 34.9 ± 9.6 | ||
| 50 | 1.15 ± 0.8 | 31.7 ± 9.1 | ||
| 100 | N.D. | 20.5 ± 4.0 | ||
| 500 | 2.5 ± 3.6 | N.D. |
Values were calculated as mean ± SD of two independent experiments in A, and of three experiments in B. For data in A, β-galactosidase activity was detected according to the gene reporter assay. After 2 h of incubation in 96-microwell plates, the percentage of cell–cell fusion was calculated from the enzymatic activity of the control (0 ng/ ml of peptide). For data in B, TF228 cells were labeled with DiI, and SupT1 cells with DiO; percentage of cell–cell fusion was calculated from the control as described in the text. N.D., not determined.
The gene reporter assay monitors transfer of large molecules (e.g., RNA polymerase) between cells. We were also interested in examining the effect of the peptide on earlier stages of fusion pore dilation. This can be done by monitoring transfer of lipid fluorophores incorporated into effector and target cell membranes. For convenience we will designate the fusion pore that allows lipid transfer as FPL and the fusion pore that allows transfer of large solutes as FPS (6). vCB21R-infected TF228 cells were labeled with the lipophilic probe DiI, and vTF7.3-infected SupT1 with DiO, and upon dye redistribution cells were stained with both dyes. As DiI and DiO are both lipophilic probes, dye transfer will reflect lipid mixing. Table I B shows the lipid redistribution at different concentrations of peptide. The results indicate a significantly different effect of the peptide on lipid mixing as compared to transfer of large molecules. At 50 ng/ml peptide, the inhibition of β-galactosidase activity was almost complete, whereas there was 30% of fusion activity measured by the lipid redistribution assay.
Lipid Mixing Versus Cytoplasm Mixing
To examine whether the differences in inhibition by DP178 of lipid mixing and transfer of large molecules (Table I) were because of the different methods used, we monitored lipid mixing and cytoplasmic mixing in the same cells. Effector cells were labeled with CMAC, a dye that reacts with free sulfhydryl groups inside the cell, and the target cells were labeled with both calcein and DiI. The lipophilic probe DiI was chosen instead of DiO because it exhibits red fluorescence, whereas DiO is green like calcein. When only DiI is redistributed to the CMAC-labeled cells, these cells are scored as positive for lipid mixing; cells labeled with all three dyes were positive for both lipid and cytoplasmic mixing. Fig. 2 shows a montage of video images from a typical redistribution experiment using three dyes. Images were taken after 2 h of incubation in presence of 50 ng/ml DP178 (Fig. 2 a) or in absence of peptide (Fig. 2 b), and are shown in bright field and in fluorescence, using the DAPI (for CMAC), rhodamine (for DiI), and FITC (for calcein) optical filter cubes, respectively. The characteristics of the filter cubes are listed in the Materials and Methods section. In Fig. 2 a (50 ng/ml peptide), two cells in contact are presented in bright field. The one in the left (TF228) was originally labeled with CMAC; the cell in the right was labeled with DiI in the membrane and calcein in the cytoplasm. It is clear from the DiI image that the lipophilic probe has been transferred from the upper cell to the lower, whereas the hydrophilic probes have not moved, indicating lipid redistribution only. In the sequence presented in Fig. 2 b (no peptide) one single cell is shown labeled with the three dyes as result of complete fusion of two cells.
Figure 2.

Inhibitory effect of DP178 peptide detected by the three-color assay. TF228 cells were labeled with the cytosolic probe CMAC, and SupT1 with both the lipophilic probe DiI and the cytosolic dye calcein, and cocultured at 37°C for 2 h. Images were acquired as described in the text with an Olympus IX70 microscope. The images are acquired in bright field and in fluorescence, using the “DAPI” (CMAC), “rhodamine” (DiI), and “FITC” (Calcein) optical filter cubes, respectively. The characteristics of the filter cubes are listed in the Materials and Methods section. The figure shows fields of a representative experiment. (a) 50 ng/ml peptide. Two cells in contact are presented in bright field. The one in the left (TF228) was originally labeled with CMAC; the cell in the right (SupT1) was labeled with DiI in the membrane and calcein in the cytoplasm. The DiI image shows the transfer of the lipophilic probe from the upper cell to the lower while the hydrophilic probes have not moved. (b) No peptide. This cell has the three dyes indicating complete fusion.
We averaged data from hundreds of cell pairs similar to those shown in Fig. 2 and plotted the average percentage cell fusion based on redistribution of lipid and cytoplasmic fluorophores, respectively. Fig. 3 a shows that DP178 inhibits cytoplasmic mixing nearly completely at 50 ng/ml peptide at which concentration there is still ∼30% lipid mixing. However, lipid mixing could be completely inhibited at 1,000 ng/ml peptide (Fig. 3 a, inset) indicating that the peptide does not exhibit nonspecific lipid mixing– inducing characterisitics. To exclude the possibility that the effects seen with DP178 might be due to the scoring procedure in our assays, or that they might have a trivial explanation such as a change in surface density of active gp120-gp41, we have performed the same experiments with the synthetic peptide DP107 corresponding to the leucine zipper motif of gp41 (44). This peptide has been shown to inhibit HIV-1 envelope glycoprotein fusion, albeit at higher concentrations as compared to DP178 (44). Fig. 3 b shows the inhibition, with no differences between lipid and aqueous dye redistribution at the different inhibitor concentrations. This experiment clearly demonstrates that the difference we see in lipid and contents mixing is specific for DP178. Moreover, we have confirmed the observation made by Lu and coworkers (25) with peptides equivalent to DP178 and DP107, that the inhibitory effect of DP178 is markedly reduced in the presence of stoichiometric amounts of DP107 (data not shown).
Figure 3.


Comparison of lipid mixing versus cytoplasmic mixing. TF228 cells were labeled with the cytosolic probe CMAC, and SupT1 with both the lipophilic probe DiI and the cytosolic dye calcein. After 2 h of incubation at 37°C, cells positive for the three dyes were scored as positive for cytoplasm mixing, and cells positive for DiI and CMAC as positive for lipid mixing only. The percentage of cell–cell fusion in both cases is relative to the control without peptide. For every sample, three different fields were acquired. Data are the mean ± SD of two or three independent experiments. •, lipid mixing; ○, cytoplasm mixing. (a) Plot for DP178. Solid line, curve-fit to the lipid mixing data according to Eq. 1 with ε = 1.35 and K I = 14.4 ng/ml; dashed line, curve-fit to the cytoplasm mixing data according to Eq. 1 with ε = 2.06 and K I = 0.96 ng/ml. Inset, same plot for DP178 at concentrations up to 1,000 ng/ml. (b) Plot for DP107. Solid line, curve-fit to the lipid and aqueous mixing data according to Eq. 1 with ε = 8.5 and K I = 185 ng/ml.
To exclude the possibility that the mixing in the presence of high concentrations of DP178 is because of nonspecific transfer of the membrane dyes, we expressed a construct in which amino acids at the gp120-gp41 junction were deleted, and proteolytic cleavage of gp160 did not occur. The uncleaved gp160 is inserted into the plasma membrane and binds soluble CD4, but cannot trigger fusion (15). BJAB cells (an Epstein-Barr virus–immortalized B lymphocytic cell line used to construct TF228 cells [19]) were infected with vaccinia recombinant vCB16 (10 multiplicity of infection), which expresses the uncleaved gp160 (7). These cells were labeled with CMAC and mixed with calcein-DiI–labeled SupT1. Experiments were performed in parallel with BJAB cells infected at the same multiplicity of infection with the vaccinia recombinant vSC60, which expresses the mature envelope glycoprotein from HIV-1 IIIB strain (7). After 2 h of coculture at 37°C, the samples were analyzed for dye transfer by video microscopy. The percentage of DiI transfer was 2.32 ± 5.2 (mean ± SD of five data sets) of control, indicating that the 20–30% dye transfer in the presence of 100 ng/ml peptide (Fig. 3 a) is not because of nonspecific effects. As an additional control for nonspecific dye transfer mediated by DP178 we have examined the effect of the peptide on dye transfer between GPF4 cells (HA-expressing cells) and RBCs (6, 31, 34, 37, 48). Table II clearly shows that the peptide does not induce nonspecific lipid transfer in this system. Neither does the peptide inhibit HA-induced fusion.
Table II.
Effects of DP178 in Fusion between GP4F Cells and RBCs*
| GP4F cells | pH | DP178 | Fluorescent GP4F** | |||
|---|---|---|---|---|---|---|
| ng/ml | % | |||||
| HA0 | 7.4 | 0 | 0 | |||
| HA0 | 7.4 | 50 | 0 | |||
| HA | 7.4 | 0 | 3.1 ± 4.38 | |||
| HA | 7.4 | 50 | 0 | |||
| HA | 5.0 | 0 | 74.4 ± 9.5 | |||
| HA | 5.0 | 50 | 76.9 ± 8.77 |
DiI-labeled RBCs were added to plated GP4F cells (16, 31). For cleavage of HA0, neuraminidase (1 U/ml) and trypsin (5 μg/ml) were added to the HA-expressing cells (31). Samples at pH 7.4 were incubated for 2 h at 37°C; samples at pH 5.0 were activated for 1 min at low pH, and then incubated for 30 min at 37°C.
Percent of GP4F cells bound to at least one RBC, which have been stained with DiI. Data are mean ± SD of two independent experiments.
Kinetics of DP178 Peptide Inhibition
To study the kinetics of the inhibitory effect of DP178 on HIV-1 envelope glycoprotein-mediated cell–cell fusion, vCB21R-infected TF228 cells and vTF7.3-infected SupT1 cells were cocultured at 1:1 ratios in 96-microwell plates. 50 ng/ml of peptide was added to effector/target mixtures at different times after the initiation of coculture. After 2 h of incubation at 37°C, the extent of cell–cell fusion was calculated as percentage of β-galactosidase activity of control. Fig. 4 shows that the fusion was completely inhibited when the peptide was added to the fusion reaction mixture during the first 15 min of coculture. When the peptide was added at later time points, the inhibitory effect of DP178 was reduced, and no inhibitory response was detected if the peptide was added >75 min after mixing.
Figure 4.

Time dependence of the inhibitory effect of DP178 peptide. vCB21R-infected TF228 cells and vTF7.3-infected SupT1 were cocultured at 1:1 ratios in 96-wells plates (4 × 105, total number of cells by well). 50 ng/ml of peptide was added to effector/target mixtures at t = 0, and at different times after the initiation of coculture. After 2 h of total incubation at 37°C, the percentage of cell–cell fusion was calculated according to the gene reporter fusion assay. Data are mean ± SD of three independent experiments.
We have studied the kinetics of fusion in absence and presence of DP178 for up to 6 h of incubation of TF228 cells with SupT1 cells using the three color assay (see Materials and Methods). Cells expressing HIV-1 envelope glycoprotein were labeled with CMAC, and CD4 expressing cells were labeled with both DiI and calcein. The same number of cells were cocultured in absence or presence of three different concentrations of DP178: 2.5, 25, and 50 ng/ ml, for 1 to 6 h. Cell fusion was calculated as the percent of control without peptide. As can be seen in Fig. 5, the differences detected in lipid mixing versus contents mixing are maintained in the frame time studied. This indicates that the peptide can “clamp” the fusion complex in the intermediate (i.e., lipid mixing) state for very long time periods.
Figure 5.

Kinetics of fusion in presence of DP178. TF228 cells were labeled with the cytosolic probe CMAC, and SupT1 with both the lipophilic probe DiI in the membrane and the cytosolic dye calcein. The cells were cocultured at 37°C for 1–6 h in absence (control) or presence of of 2.5, 25, or 50 ng/ml of DP178. Cell fusion was calculated as a percent of control as described in Fig. 3 using the three-color assay method.
Discussion
The first step in the HIV-1 fusion reaction is a conformational change in the envelope glycoprotein that results in exposure of specific epitopes in gp41 (27, 35). Synthetic peptides corresponding to sequences of specific domains on gp41 (Fig. 1) have been used to examine fusion mechanisms (27). This paper describes experiments in which we have studied the effects of the synthetic peptides DP178 and DP107 in HIV-1 envelope glycoprotein–mediated cell membrane fusion. Our measurements of the inhibitory effect of the peptides on redistribution of lipids and of large solutes inform us about the various stages of the fusion cascade (4).
In the case of influenza HA–catalyzed fusion, a sequence of events has been resolved after the conformational changes. These include transfer of fluorescent-labeled lipid molecules because of the merging of the outer membrane monolayers (hemifusion) (22), and transfer of large, aqueous fluorescent dye molecules because of formation and expansion of a fusion pore (6, 42, 43, 48). We have denoted these events as the opening of a FPL and a FPS, respectively (6). Hemifusion is predicted by the “stalk-pore” model (12) to involve intermixing of lipids between contacting leaflets but not between noncontacting membrane leaflets. The task of the viral envelope glycoprotein is to produce sufficient curvature in the lipid bilayer to create the hemifusion and fusion pore structures. To complete fusion a lipidic fusion pore of critical radius has to form and expand in the contact bilayer. Mutations in various domains of influenza HA were found to have a profound effect on the outcome of the fusion cascade. Replacement of the membrane-spanning domain of HA with a glycosylphosphatidylinositol anchor results in a very stable hemifusion intermediate (22, 28). Furthermore, specific single site mutations in the fusion peptide of HA were found not to affect lipid redistribution but significantly impaired the ability of HA to mediate delivery of large aqueous molecules from RBCs to HA-expressing cells (36).
Even in the case of fusion of wild-type HA-expressing cells with labeled RBCs at optimal pH and temperature only ∼40% of fusion events that cause lipid mixing result in redistribution of large solutes (36, 37) a phenomenon dubbed by Melikyan et al. as “stunted fusion” (28a).
We have not observed stunted fusion in the case of the gp120-gp41–expressing TF228 cells incubated with SupT1 cells at 37°C for up to 6 h. However, addition of 50 ng/ml of a COOH-terminal peptide from the gp41 ectodomain (DP178 [46]) fully blocks complete fusion (contents mixing), at which concentration 30% of lipid mixing was still in effect. Lipid mixing was completely blocked at 1,000 ng/ ml peptide. Interestingly, the NH2-terminal peptide DP107, which inhibits at a higher dissociation constant K i, showed no distinction between the effects on lipid and contents mixing. This indicates that inhibition by DP178 is not because of some trivial effect, such as reducing the surface density of active gp120-gp41. Previous studies on inhibition of syncytia by DP178 using the same isolate (HIV-1LAI) (46) yielded 50% inhibitory concentration (IC50) values in the range of 0.5–1.7 ng/ml. Since this is in the range of inhibition of FPS reported here, it appears that there are no substantial barriers between opening of the large fusion pore and completing the fusion process (14). Interestingly, cell fusion escapes inhibition by the peptide when it is added up to 15 min after incubation of the cells (Fig. 4). We have recently shown that conformational changes in cell surface–expressed gp120-gp41 occur within 1 min of interaction of envelope-expressing cells with appropriate target cells (21). These observations taken together indicate that the peptide acts after a conformational change of the envelope glycoprotein.
We have interpreted our observations on inhibition of gp120-gp41–mediated fusion on the basis of recently determined high resolution crystal structures of the HIV-1 gp41 core (10, 41), which is a bent-in-half, antiparallel, heterotrimeric coiled-coil structure. Interestingly, a trimeric envelope glycoprotein structure had previously been proposed for HIV-1 on the basis of biochemical cross-linking and sedimentation studies (38). Comparison with the crystal structures of the influenza HA2 subunits in a low pH– induced conformation (8) and of fragments of the Moloney murine leukemia virus fusion protein (17) reveals common structural motifs, which provide growing support for the “spring-loaded” type of mechanistic models (9). In this scenario, activation of the fusion protein results in release of the fusion peptide and extension of the central coiled-coil structure. The new positioning of the fusion peptides at the tip of the stalk provides for easy contact with the target cell membrane. A small group of proximal fusion proteins that are simultaneously inserted into both the viral and target membranes would constitute a potential fusion site. A concerted collapse of this protein complex, actuated by the bending-in-half of the stalks at a central hinge region and formation of the antiparallel six-helical bundle seen in the crystal structures, would position the COOH-terminal transmembrane anchors and NH2-terminal fusion peptides on top of each other in the center. This would bring the two membranes into contact, and thus allow for formation of the hemifusion intermediate and eventually the fusion pore (Fig. 6). The DP178 inhibitor, which is a peptide fragment of the COOH-terminal helix seen in the crystal structures (10, 41), presumably acts by binding to the triple-stranded coiled-coil of NH2-terminal helices. If it displaces the COOH-terminal helices and prevents formation of a sufficiently bent-in-half structure (as described above), then the membranes would never get close enough for fusion to commence.
Figure 6.

Schematic structural model of the effect of DP178 on the membrane fusion process. (a) Top view of six, semi-aggregated gp41 trimers sticking straight up, in the initial stage of forming a potential fusion site (FS) complex. (b) Top view of lipid-mixing fusion site (FPL). The six gp41 proteins have undergone the bending-in-half transition, and are in the six-helical bundle, heterotrimeric coiled-coil conformation observed in the crystal. The NH2-terminal regions of the gp41 ectodomain are in magenta, and the COOH-terminal regions are in blue. The aggregate of magenta helices at the center are the NH2-terminal fusion peptides inserted into the target cell membrane (note that the target membrane has been removed to reveal the protein complex). A side view of this structure is shown in the insert (f). This includes the target cell membrane (top), and shows the initial contact of the alkyl cores of the two membranes (which allows for interchange of the lipids and leads to hemifusion). Also seen in the side view are the COOH-terminal transmembrane domains in the viral membrane. (c) Top view of the disrupted protein scaffold, which allows for expansion of the pore and contents mixing (FPS). The transition from a to d represents binding of DP178 (green) to the low affinity site on gp41, which prevents formation of the heterotrimeric coiled-coil structure, and thus all subsequent stages of fusion. The transition from b to e represents binding of DP178 to the high affinity site, which clamps the protein subunits together, and prevents the transition from the lipid-mixing (FPL) to the solute-mixing (FPS) stages. The protein models shown in b, c, e, and f were developed from a recently determined crystal structure of gp41 fragments in the heterotrimeric coiled-coil conformation (10) (1AIK.pdb). The lengths of the NH2- and COOH-terminal helices were expanded as suggested by experimental studies of the complex in solution (25). The loops in the structures are simply placed in arbitrary positions, and were included solely to test the steric feasibility of the models.
It has been proposed that an assembly of viral envelope glycoprotein oligomers could form a molecular scaffold responsible for bringing the viral membrane close to the target cell membrane, and create the architecture that enables lipid bilayers to merge (1, 2, 5, 42). Such scaffolds have also been proposed for exocytotic fusion pores (29). Movement of the viral glycoproteins away from the FPL scaffold would break the hemifusion diaphragm, and allow for the flow of solutes between aqueous compartments (6). An analysis of data relating the lag phase for the onset of HA-mediated fusion to the density of HA in the donor membrane suggests that a minimum of 3–4 HA trimers is required to induce a fusion event (13). Our kinetic data on influenza HA fusion pore dilation indicate that six HA trimers need to move to form a FPS (6). The putative transitions between fusion site, FPL and FPS are diagramed in Fig. 6.
Using this concept we have developed a model for inhibition of FPL and FPS formation by DP178 (see Appendix 1). Assuming a Poissonian distribution of fusion pores over the contact area between cells, the fraction of FPL or FPs is then given by:
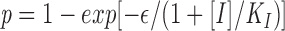 |
1 |
where [I] is the concentration of inhibitor, and K I is the concentration of I where half of the fusion sites are inhibited and ε reflects the efficiency of conversion from fusion site to fusion pore. Fitting the data from Fig. 3 a (DP178) to Eq. 1 yields ε = 1.35, K I = 14.4 ng/ml, and ε = 2.06, K I = 0.96 ng/ml for FPL and FPS, respectively. Fitting the data from Fig. 3 b (DP107) to Eq. 1 yields ε = 8.5 and K I = 185 ng/ml for both FPL and FPS.
The large difference in inhibition constants in the case of DP178 is consistent with the notion that there are different binding sites for this peptide on gp41 with different values of K I (Fig. 6). Since the inhibitory effect of DP178 works against different isolates of HIV-1 but not of HIV-2 (45, 46), it is not likely that the low affinity site represents nonspecific binding to the cell membrane leading to inhibition of cell fusion. Moreover, using labeled DP178 we have only observed binding to HIV-1 envelope expressing cells (unpublished results). Based on the structural information about the gp41 core, an obvious choice for the low affinity site (affecting both FPL and FPS formation) is the leucine zipper domain (10, 11, 24, 25, 41, 47); with helical DP178 peptides binding in the same way as the corresponding amino acid sequence regions of the three protein COOH-terminal helices in the crystal structures. At this position the peptides would sterically block the regular binding of the COOH-terminal helices to the inner core of NH2-terminal helices, and thus prevent formation of the bent-in-half, antiparallel, heterotrimeric coiled-coil structure presumably required to bring the viral and target cell membranes into contact for fusion (as described above). Similarly, the lower-affinity binding of DP107 to the COOH-terminal domains would inhibit formation of this structure in an inverse manner, thus also precluding all stages of membrane fusion.
However, at certain intermediary concentrations of DP178, we find that it is possible to fully block the exchange of solutes (FPS), while still having significant exchange of lipids (FPL). Thus, binding of the peptide to the high affinity site is taken to be associated with the transition from the FPL to FPS states (Fig. 6). Since this transition (which constitutes breaking of the hemifusion diaphragm and expansion of the fusion pore) is believed to result from disruption of the oligomeric fusion protein scaffolding, the DP178 inhibitor is envisioned forming links between adjacent subunits that stabilize the complex. This concept is speculatively illustrated in Fig. 6 e, in which the NH2- and COOH-terminal ends (as short helices) of six DP178 peptides (green) simultaneously bind to all the adjacent gp41 proteins in the complex. In this working model the small helices of DP178 displace the ends of the outer gp41 COOH-terminal helices that are in the coiled-coil complex (blue), and bind in the grooves between the inner NH2-terminal helices (magenta). It should be noted, however, that there is no physical evidence for the depicted conformation of the DP178 peptides, nor for the binding site on gp41; rather, this simply illustrates the steric feasibility of a single peptide linking adjacent protein oligomers. Nevertheless, this model is consistent with the experimental results of Wild and coworkers (47), in which deletion of only one to four residues from either the NH2 or COOH-terminal ends of DP178 significantly reduced the fusion inhibition activity. As seen in Fig. 6 e, this is because such deletions would diminish the interactions (and thus the binding constant) between the inhibitor and gp41. Additionally, having two regions of the peptide (i.e., the NH2 and COOH-terminal ends) bind the two putative sites on the heterotrimer would make for a larger effective binding affinity than for each region individually. This could explain the higher observed binding constant for blocking formation of FPS than for FPL. This model also illustrates the hypothesis that, in contrast to the low affinity site, the bound DP178 does not prevent the close contact of the fusion peptides and COOH-terminal viral membrane anchors presumed to be required for merging of the membranes. It should be emphasized that the high affinity binding site model shown in Fig. 6 e is only one of many possibilities. For example, an alternative class of models would have the link between adjacent gp41 stalks made by an aggregate of DP178 peptides. Obviously, more detailed experimental studies are needed to identify the sites of inhibitor peptide binding to the fusion proteins.
These data and analysis also form the basis for an explanation why fusion of intact virus (infectivity) is inhibited by DP178 at a concentration >10-fold higher than needed for inhibition of cell–cell fusion (46). Since the envelope glycoproteins in the intact virus are tightly packed, DP178 might not be able to access the interoligomer space in the FPL site to form the clamp. However, binding of DP178 to the leucine zipper domain, which according to our data occurs with similar affinity to that seen for inhibition of viral infectivity, will have a profound effect on fusion of intact virus.
Acknowledgments
We thank Dr. E. Berger for vaccinia recombinants, Dr. Z. Jonak for the TF228 line, and Dr. J.M. White for GP4F cells. We are also grateful to Dr. D. Dimitrov, A. Puri, P. Hug, T. Korte, and P. Jones for careful reading of the manuscript and helpful suggestions.
Abbreviations used in this paper
- CMAC
7-amino-4-chloromethylcoumarin
- DiI
1,1′-dioctadecyl-3,3,3′3′-tetramethylindocarbocyanine perchlorate
- DiO
3,3′-dioctadecyloxaocarbocyanine perchlorate
- FPL
lipid-permissive fusion pore
- FPS
solute permissive fusion pore
- HA
hemagglutinin
- HIV-1
human immunodeficiency virus type–1
- IC50
50% effective inhibitory concentration
- RBCs
red blood cells
Appendix
An FS (see Fig. 6) is assembled according to
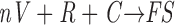 |
A1 |
,
where V is the concentration of envelope glycoprotein oligomer on the surface, n is the number of Vs per fusion site. Assuming that the concentrations of receptor (R) and coreceptor (C) on the target cell are in excess of V, then FS =V/n. If the inhibitor (I) interacts with the oligomer according to V + I ⇔ VI, with an equilibrium constant K I, the concentration of free envelope glycoprotein (V) is given by:
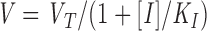 |
A2 |
,
where V T is the total concentration of envelope glycoprotein.
Assuming a Poissonian distribution of fusion pores over a contact area between cells, the probability p(i) of i fusion pores in the contact area is given by:
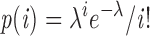 |
A3 |
,
where λ = αFS is the average number of fusion pores in the contact area, FS is the number of fusion sites in the contact area, and α the efficiency of conversion from fusion site to fusion pore. Redistribution of lipid and/or large solute will occur if at least one fusion pore opens. The probability of such an event is given by (see reference 36):
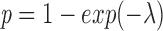 |
A4 |
.
Substituting αFS for λ, V/n for FS, and Eq. A2 for V yields:
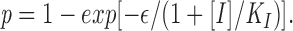 |
A5 |
where ε = αV T/n. Eq. A5 is the same as Eq. 1 in the main text.
Footnotes
The work was supported by the AIDS Intramural Targeted Antiviral Program. I. Muñoz-Barroso is a postdoctoral fellow of the Direccion General de Investigacion Cientifica y Enseñanza Superior, Spain.
Address all correspondence to Robert Blumenthal, Miller Drive, P.O. Box B, Building 469, Room 213, Frederick, MD 21702-1201. Tel.: (301) 846-1446. Fax: (301) 846-6192. E-mail: blumen@helix.nih.gov
References
- 1.Bentz J, Ellens H, Alford D. An architecture for the fusion site of influenza hemagglutinin. FEBS (Fed Eur Biochem Soc) Lett. 1990;276:1–5. doi: 10.1016/0014-5793(90)80492-2. [DOI] [PubMed] [Google Scholar]
- 2.Blumenthal R. Cooperativity in viral fusion. Cell Biophys. 1988;12:1–12. doi: 10.1007/BF02918347. [DOI] [PubMed] [Google Scholar]
- 3.Blumenthal, R., and D.S. Dimitrov. 1997. Membrane fusion. In Handbook of Physiology. J.F. Hoffman, and J.C. Jamieson, editors. Oxford University Press, New York. 569–604.
- 4.Blumenthal R, Schoch C, Puri A, Clague MJ. A dissection of steps leading to viral envelope protein-mediated membrane fusion. Ann NY Acad Sci. 1991;635:285–296. doi: 10.1111/j.1749-6632.1991.tb36499.x. [DOI] [PubMed] [Google Scholar]
- 5.Blumenthal R, Pak CC, Raviv Y, Krumbiegel M, Bergelson LD, Morris SJ, Lowy RJ. Transient domains induced by influenza haemagglutinin during membrane fusion. Mol Membr Biol. 1995;12:135–142. doi: 10.3109/09687689509038509. [DOI] [PubMed] [Google Scholar]
- 6.Blumenthal R, Sarkar DP, Durell S, Howard DE, Morris SJ. Dilation of the influenza hemagglutinin fusion pore revealed by the kinetics of individual cell–cell fusion events. J Cell Biol. 1996;135:63–71. doi: 10.1083/jcb.135.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Broder CC, Berger EA. Fusogenic selectivity of the envelope glycoprotein is a major determinant of human immunodeficiency virus type 1 tropism for CD4+ T-cell lines vs. primary macrophages. Proc Natl Acad Sci USA. 1995;92:9004–9008. doi: 10.1073/pnas.92.19.9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 9.Carr CM, Kim PS. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell. 1993;73:823–832. doi: 10.1016/0092-8674(93)90260-w. [DOI] [PubMed] [Google Scholar]
- 10.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 11.Chen CH, Matthews TJ, McDanal CB, Bolognesi DP, Greenberg ML. A molecular clasp in the human immunodeficiency virus (HIV) type 1 TM protein determines the anti-HIV activity of gp41 derivatives: implication for viral fusion. JVirol. 1995;69:3771–3777. doi: 10.1128/jvi.69.6.3771-3777.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chernomordik LV, Melikyan GB, Chizmadzhev YA. Biomembrane fusion: a new concept derived from model studies using two interacting planar lipid bilayers. Biochim Biophys Acta. 1987;906:309–352. doi: 10.1016/0304-4157(87)90016-5. [DOI] [PubMed] [Google Scholar]
- 13.Danieli T, Pelletier SL, Henis YI, White J M. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J Cell Biol. 1996;133:559–569. doi: 10.1083/jcb.133.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dimitrov DS, Blumenthal R. Photoinactivation and kinetics of membrane fusion mediated by the human immunodeficiency type 1 envelope glycoprotein. JVirol. 1994;68:1956–1961. doi: 10.1128/jvi.68.3.1956-1961.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Earl PL, Koenig S, Moss B. Biological and immunological properties of human immunodeficiency virus type 1 envelope glycoprotein: analysis of proteins with truncations and deletions expressed by recombinant vaccinia viruses. JVirol. 1991;65:31–41. doi: 10.1128/jvi.65.1.31-41.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellens H, Bentz J, Mason D, Zhang F, White JM. Fusion of influenza hemagglutinin-expressing fibroblasts with glycophorin-bearing liposomes: role of hemagglutinin surface density. Biochemistry. 1990;29:9697–9707. doi: 10.1021/bi00493a027. [DOI] [PubMed] [Google Scholar]
- 17.Fass D, Harrison SC, Kim PS. Retrovirus envelope domain at 1.7 angstrom resolution. Nat Struct Biol. 1996;3:465–469. doi: 10.1038/nsb0596-465. [DOI] [PubMed] [Google Scholar]
- 18.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 19.Fu YK, Hart TK, Jonak ZL, Bugelski PJ. Physicochemical dissociation of CD4-mediated syncytium formation and shedding of human immunodeficiency virus type 1 gp120. JVirol. 1993;67:3818–3825. doi: 10.1128/jvi.67.7.3818-3825.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallaher WR, Ball JM, Garry RF, Griffin MC, Montelaro RC. A general model for the transmembrane proteins of HIV and other retroviruses. AIDS Res Hum Retroviruses. 1989;5:431–440. doi: 10.1089/aid.1989.5.431. [DOI] [PubMed] [Google Scholar]
- 21.Jones P, Korte T, Blumenthal R. Conformational changes in cell surface HIV-1 envelope glycoproteins are triggered by cooperation between cell surface CD4 and coreceptors. J Biol Chem. 1998;273:404–409. doi: 10.1074/jbc.273.1.404. [DOI] [PubMed] [Google Scholar]
- 22.Kemble GW, Danieli T, White JM. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell. 1994;76:383–391. doi: 10.1016/0092-8674(94)90344-1. [DOI] [PubMed] [Google Scholar]
- 23.Kowalski M, Potz J, Basiripour L, Dorfman T, Goh WC, Terwilliger E, Dayton A, Rosen C, Haseltine W, Sodroski J. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science. 1987;237:1351–1355. doi: 10.1126/science.3629244. [DOI] [PubMed] [Google Scholar]
- 24.Lawless MK, Barney S, Guthrie KI, Bucy TB, Petteway SR, Jr, Merutka G. HIV-1 membrane fusion mechanism: structural studies of the interactions between biologically-active peptides from gp41. Biochemistry. 1996;35:13697–13708. doi: 10.1021/bi9606962. [DOI] [PubMed] [Google Scholar]
- 25.Lu M, Blacklow SC, Kim PS. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat Struct Biol. 1995;2:1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- 26.Maddon PJ, Dalgleish AG, McDougal JS, Clapham PR, Weiss RA, Axel R. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell. 1986;47:333–348. doi: 10.1016/0092-8674(86)90590-8. [DOI] [PubMed] [Google Scholar]
- 27.Matthews TJ, Wild C, Chen CH, Bolognesi DP, Greenberg ML. Structural rearrangements in the transmembrane glycoprotein after receptor binding. Immunol Rev. 1994;140:93–104. doi: 10.1111/j.1600-065x.1994.tb00866.x. [DOI] [PubMed] [Google Scholar]
- 28.Melikyan GB, White JM, Cohen FS. GPI-anchored influenza hemagglutinin induces hemifusion to both red blood cell and planar bilayer membranes. J Cell Biol. 1995;131:679–691. doi: 10.1083/jcb.131.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28a.Melikyan GB, Brener SA, Ok DC, Cohen FS. Inner but not outer membrane leaflets control the transition from glycosylphosphatidylinositol-anchored influenza hemagglutinin-induced hemifusion to full fusion. J Cell Biol. 1997;136:995–1005. doi: 10.1083/jcb.136.5.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monck JR, Fernandez JM. The fusion pore and mechanisms of biological membrane fusion. Curr Opin Cell Biol. 1996;8:524–533. doi: 10.1016/s0955-0674(96)80031-7. [DOI] [PubMed] [Google Scholar]
- 30.Moore, J.P., B.A. Jameson, R.A. Weiss, and Q.J. Sattentau. 1993. The HIV-cell fusion reaction. In Viral Fusion Mechanisms. J. Bentz, editor. CRC Press. Boca Raton. 233–289.
- 31.Morris SJ, Sarkar DP, White JM, Blumenthal R. Kinetics of pH-dependent fusion between 3T3 fibroblasts expressing influenza hemagglutinin and red blood cells. Measurement by dequenching of fluorescence. J Biol Chem. 1989;264:3972–3978. [PubMed] [Google Scholar]
- 32.Nussbaum O, Broder CC, Berger EA. Fusogenic mechanisms of enveloped-virus glycoproteins analyzed by a novel recombinant vaccinia virus-based assay quantitating cell fusion-dependent reporter gene activation. JVirol. 1994;68:5411–5422. doi: 10.1128/jvi.68.9.5411-5422.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Puri A, Morris SJ, Jones P, Ryan M, Blumenthal R. Heat-resistant factors in human erythrocyte membranes mediate CD4-dependent fusion with cells expressing HIV-1 envelope glycoproteins. Virology. 1996;219:262–267. doi: 10.1006/viro.1996.0244. [DOI] [PubMed] [Google Scholar]
- 34.Sarkar DP, Morris SJ, Eidelman O, Zimmerberg J, Blumenthal R. Initial stages of influenza hemagglutinin-induced cell fusion monitored simultaneously by two fluorescent events: Cytoplasmic continuity and lipid mixing. J Cell Biol. 1989;109:113–122. doi: 10.1083/jcb.109.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sattentau QJ, Moore JP. Conformational changes induced in the human immunodeficiency virus envelope glycoprotein by soluble CD4 binding. J Exp Med. 1991;174:407–415. doi: 10.1084/jem.174.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schoch C, Blumenthal R. Role of the fusion peptide sequence in initial stages of influenza hemagglutinin-induced cell fusion. J Biol Chem. 1993;268:9267–9274. [PubMed] [Google Scholar]
- 37.Spruce AE, Iwata A, White JM, Almers W. Patch clamp studies of single cell-fusion events mediated by a viral fusion protein. Nature. 1989;342:555–558. doi: 10.1038/342555a0. [DOI] [PubMed] [Google Scholar]
- 38.Weiss CD, Levy JA, White JM. Oligomeric organization of gp120 on infectious human immunodeficiency virus type 1 particles. JVirol. 1990;64:5674–5677. doi: 10.1128/jvi.64.11.5674-5677.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weiss CD, Barnett SW, Cacalano N, Killeen N, Littman DR, White JM. Studies of HIV-1 envelope glycoprotein-mediated fusion using a simple fluorescence assay. AIDS. 1996;10:241–246. doi: 10.1097/00002030-199603000-00001. [DOI] [PubMed] [Google Scholar]
- 40.Weissenhorn W, Wharton SA, Calder LJ, Earl PL, Moss B, Aliprandis E, Skehel JJ, Wiley DC. The ectodomain of HIV-1 env subunit gp41 forms a soluble, alpha-helical, rod-like oligomer in the absence of gp120 and the N-terminal fusion peptide. EMBO (Eur Mol Biol Organ) J. 1996;15:1507–1514. [PMC free article] [PubMed] [Google Scholar]
- 41.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic Structure of the Ectodomain from HIV-1 gp41. Nature. 1997;387:426–428. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 42.White JM. Membrane fusion. Science. 1992;258:917–924. doi: 10.1126/science.1439803. [DOI] [PubMed] [Google Scholar]
- 43.White JM. Membrane fusion: the influenza paradigm. Cold Spring Harbor Symp Quant Biol. 1995;60:581–588. doi: 10.1101/sqb.1995.060.01.062. [DOI] [PubMed] [Google Scholar]
- 44.Wild C, Oas T, McDanal C, Bolognesi D, Matthews T. A synthetic peptide inhibitor of human immunodeficiency virus replication: correlation between solution structure and viral inhibition. Proc Natl Acad Sci USA. 1992;89:10537–10541. doi: 10.1073/pnas.89.21.10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wild C, Greenwell T, Matthews T. A synthetic peptide from HIV-1 gp41 is a potent inhibitor of virus-mediated cell-cell fusion. AIDS Res Hum Retroviruses. 1993;9:1051–1053. doi: 10.1089/aid.1993.9.1051. [DOI] [PubMed] [Google Scholar]
- 46.Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci USA. 1994;91:9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wild C, Greenwell T, Shugars D, Rimsky-Clarke L, Matthews T. The inhibitory activity of an HIV type 1 peptide correlates with its ability to interact with a leucine zipper structure. AIDS Res Hum Retroviruses. 1995;11:323–325. doi: 10.1089/aid.1995.11.323. [DOI] [PubMed] [Google Scholar]
- 48.Zimmerberg J, Blumenthal R, Sarkar DP, Curran M, Morris SJ. Restricted movement of lipid and aqueous dyes through pores formed by influenza hemagglutinin during cell fusion. J Cell Biol. 1994;127:1885–1894. doi: 10.1083/jcb.127.6.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]