

Abstract
Myf5 is the earliest-known muscle-specific factor to be expressed in vivo and its expression is associated with determination of the myoblast lineage. In C2 cells, we show by immunocytolocalization that Myf5 disappears rapidly from cells in which the differentiation program has been initiated. In proliferating myoblasts, the levels of Myf5 and MyoD detected from cell to cell are very heterogeneous. We find that some of the heterogeneity of Myf5 expression arises from a posttranscriptional regulation of Myf5 by the cell cycle. Immunoblotting of extracts from synchronized cultures reveals that Myf5 undergoes periodic fluctuations during the cell cycle and is absent from cells blocked early in mitosis by use of nocodazole. The disappearance of Myf5 from mitotic cells involves proteolytic degradation of a phosphorylated form of Myf5 specific to this phase of the cell cycle. In contrast, MyoD levels are not depleted in mitotic C2 cells. The mitotic destruction of Myf5 is the first example of a transcription factor showing cell cycle–regulated degradation. These results may be significant in view of the possible role of Myf5 in maintaining the determination of proliferating cells and in timing the onset of differentiation.
Terminal differentiation of muscle cells, both in vivo and ex vivo, is dependent upon the functions of the myogenic regulatory factors (MRFs)1 (for review see Yun and Wold, 1996). These include factors of the basic helix-loop-helix family MyoD, Myf5, and myogenin (Davis et al., 1987; Braun et al., 1989b ; Wright et al., 1989). Expression of any one of these MRFs is sufficient to convert a variety of cell types to the myogenic differentiation pathway (for review see Weintraub et al., 1991; Olson and Klein, 1994).
The mechanisms by which MyoD induces myogenesis (for review see Lassar et al., 1994; Molkentin and Olson, 1996) involve both the activation of muscle-specific gene expression and withdrawal from the cell cycle (Crescenzi et al., 1990; Sorrentino et al., 1990). MyoD-induced cell cycle arrest in G1 requires the retinoblastoma protein or its homologue p107 (Gu et al., 1993; Schneider et al., 1994; Zacksenhaus et al., 1996), and is maintained through accumulation of the cyclin-dependent kinase (cdk) inhibitor p21 (Guo et al., 1995; Halevy et al., 1995). The functions of MyoD are repressed in proliferating myoblasts. At least one such mechanism is mediated by cyclin D1 (Rao et al., 1994; Skapek et al., 1995, 1996).
MyoD and Myf5 are often classed as “determination” factors since they are expressed in proliferating myoblasts, in contrast to myogenin expression, which coincides with cell cycle arrest, and MRF4, which is found only in mature myotubes (Braun et al., 1989a ; Montarras et al., 1991; Andrés and Walsh, 1996). The functions of Myf5 are poorly understood. Myf5 is the earliest of the factors expressed in the mouse embryo and its expression is transient (between 8 and 12 d post coitum [p.c.]) (Ott et al., 1991). Downregulation of Myf5 transcripts precedes full muscle differentiation both in vivo (Ott et al., 1991) and ex vivo (Montarras et al., 1991). This transient expression of Myf5 is required to maintain the determined state of certain muscle precursor cells, since these abandon the myogenic lineage in myf5 −/− embryos (Tajbakhsh et al., 1996). The phenotypes of mice mutated in Myf5 and MyoD indicate that these two factors are capable of some functional redundancy (Braun et al., 1992; Rudnicki et al., 1992, 1993). It has been suggested that MyoD and Myf5 could determine parallel and equivalent myogenic lineages (Braun and Arnold, 1996).
Muscle cell lines such as C2 (Yaffé and Saxel, 1977; Pinset et al., 1988) provide strong models for the study of myoblasts and their terminal differentiation into multinucleated myotubes. In their proliferative state these cells express both MyoD and Myf5 (Braun et al., 1989a ; Montarras et al., 1991). To further clarify the role of Myf5 in determination and differentiation and its relationship to the proliferative state of myoblasts, we have examined the expression of Myf5 protein in C2 cells and in a C2-derived cell line incapable of autonomous differentiation (Montarras et al., 1996). We find that Myf5 is downregulated in cells undergoing differentiation and that in proliferative cells, Myf5 levels are strongly regulated by cell cycle–associated events that include the destruction of this factor at mitosis.
Materials and Methods
C2 Cell Culture and Differentiation
C2.7 and anti–insulin-like growth factor II (anti-IGFII) C2 cells have been previously described (Pinset et al., 1988; Montarras et al., 1996). Both cell lines are cultured in a 1:1 (vol/vol) mixture of MCDB 202 medium and DME (both from BICEF, l' Aigle, France) containing 20% (vol/vol) FCS (Jacques Boy, Reims, France). C2.7 cells are maintained in a proliferative state by replating at low cell density. To obtain autonomously differentiating C2.7 cells, these were plated at a density of either 2.5 × 103 cells/cm2 (see Figs. 1 and 2 B) or 2.5 × 102 cells/cm2 (see Fig. 2 A), and the cultures were allowed to age, without change of medium, for 6 d. Cells were prepared for synchronization or preparation of mitotic cell extracts (see below) by plating at an initial density of 2.5 × 103 cells/cm2 and 48 h of proliferation.
Figure 1.


Downregulation of Myf5 is a very early event in myogenic differentiation. C2.7 cells were cultured under conditions favorable to differentiation for 6 d and treated for indirect immunofluorescence. Cells were stained for Myf5 (red) and troponin T (green) (A), or Myf5 (green) and myogenin (red) (B). The resulting images were prepared by confocal microscopy to give a better estimation of coexpression of the markers examined. Bars, 10 μm.
Figure 2.


Expression patterns of MyoD and Myf5 in C2.7 cells are distinct. C2.7 cells in cultures undergoing autonomous differentiation were simultaneously stained for Myf5 expression (green) and MyoD expression (red). Cultures prepared (as described in Materials and Methods) such that cells were either still at the myoblast stage (A), or had undergone some fusion (B), were selected for analysis by confocal microscopy. Bars, 10 μm.
Synchronization of Anti-IGFII C2 Myoblasts, and Flow Cytometry
Synchronized populations of cells were obtained as described in figure legends. Aphidicolin (stored as 1 mg/ml stock in H2O), nocodazole (1 mg/ ml in DMSO), and N-acetyl-Leu-Leu-norleucinal (ALLN; 100 mM in DMSO) were obtained from Sigma Chemical Co. (St. Louis, MO).
Mitotic shake-off fractions were prepared by mechanical detachment of nocodazole-arrested cells. These were washed in PBS and used to make extracts, as was the adherent fraction of cells remaining attached to the culture dishes (see below).
In preparation for flow cytometric analysis of DNA content, synchronized cells (2–5 × 106) were harvested by trypsinization, washed twice in cold PBS, and then fixed by resuspension in cold 70% ethanol and incubation at 4°C overnight. Cells were then washed once in PBS and resuspended in 1 ml PBS, to which were added 50 μg RNase and propidium iodide to a final concentration of 10 μM. Propidium iodide fluorescence of 20,000 cells per sample was measured using a FACStar® Plus cytometer (Becton and Dickinson, Co., Mountain View, CA).
Indirect Immunofluorescence
Immunofluorescence was performed on cells grown on 35-mm plastic tissue culture plates (Falcon Plastics, Cockeysville, MD). PBS was used to wash cells extensively before fixation and after each step of the procedure described, which was carried out at room temperature. Cells were fixed in 4% (wt/vol) paraformaldehyde in PBS for 10 min, and then neutralized for 10 min in 50 mM NH4Cl in PBS. Permeabilization of cells was achieved with 0.2% Triton X-100 in PBS. Immunodetection involved three consecutive incubations with antibodies diluted in PBS containing 0.2% (wt/vol) gelatin (Merck, Darmstadt, Germany): (a) 1-h incubation with primary antibodies. These are described in detail below. One rabbit and one mouse antibody were used simultaneously for the codetections shown in Figs. 1 and 2. (b) 30-min incubation with secondary antibodies: goat anti–mouse immunoglobulins coupled to Texas red (1/100 dilution; Amersham International, Little Chalfont, UK), and goat anti–rabbit immunoglobulins coupled with biotin (1/200; Sigma Chemical Co.) (Fig. 1 A); or goat anti–rabbit coupled to FITC (1/200; Sigma Chemical Co.), and goat anti–mouse coupled with biotin (1/200; Sigma Chemical Co.) (Figs. 1 B and 2); or goat anti–rabbit antibody coupled with biotin (1/200; Sigma Chemical Co.) alone (see Fig. 3). (c) 20-min incubation with FITC-coupled streptavidin (FITC-Extravidin; 1/200; Sigma Chemical Co.) (Figs. 1 A and 3); or with Texas red–coupled streptavidin (1/100; Amersham International) (Figs. 1 B and 2). Cells were mounted in Mowiol (Calbiochem-Novabiochem, La Jolla, CA) under glass coverslips.
Figure 3.
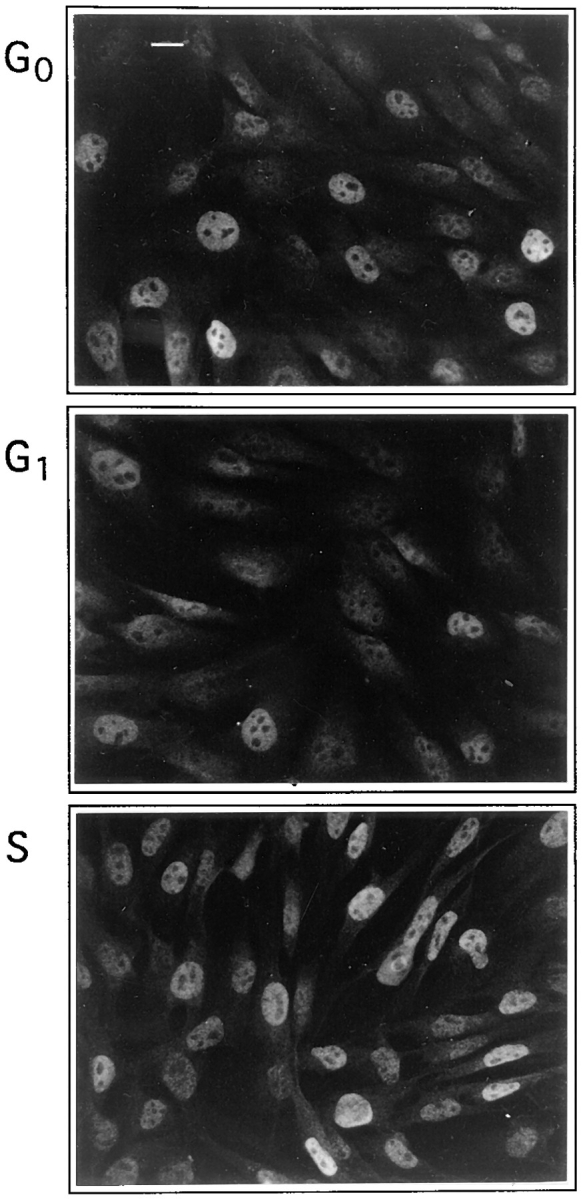
Myf5 expression varies with the cell cycle. Anti-IGFII C2 cells were synchronized by growth in 0.5% serum for 24 h (G0, top) followed by 6 h stimulation in 20% serum in the presence of 2 μg/ml aphidicolin (G1, center) followed by removal of aphidicolin for 6 h (S, bottom). Cells were fixed and stained for Myf5 expression. The designation of cell cycle phases in this figure is justified in Fig. 4 A. Bar, 10 μm.
Preparation of Protein Extracts and Western Blotting
Protein extracts were made by chilling plates on ice and washing with cold PBS followed by lysis into radioimmunoprecipitation assay (RIPA) buffer (50 mM NaCl, 25 mM Tris 8.2, 0.5% NP-40, 0.5% Na deoxycholate, 0.1% SDS, 1 mM DTT containing protease inhibitors as described in Mellits et al. [1993]). Lysates were collected, sheared by six passages through a 25-gauge needle, and debris removed by centrifugation at 12,000 g at 4°C. Approximately 20 μg extract per sample was analyzed by 9% SDS-PAGE, and transferred to nitrocellulose Hybond-C Extra filters (Amersham International) using a trans-blot semi-dry electrophoretic transfer cell (Bio-Rad Laboratories, Hercules, CA). Homogeneity of loading and transfer were checked by staining filters with Ponceau S dye (Sigma Chemical Co.). Filters were blocked for nonspecific binding by incubation for 2 h at room temperature in PBS containing 0.1% Tween-20 with 5% (wt/vol) powdered skim milk, which was also used to dilute all antibodies. Incubations with primary antibodies (described below) were for 90 min at room temperature. Filters were washed extensively in Tween-20–PBS before secondary incubation for 30 min with goat polyclonal antibodies against either rabbit or mouse immunoglobulins, coupled to horseradish peroxidase (diluted 1/104; Sigma Chemical Co.). Filters were washed and treated with ECL Western blotting detection reagents (Amersham International).
Where filters were probed successively with several antibodies (see Fig. 4), previous antibodies were removed by stripping in PBS containing 2% SDS and 100 mM β-mercaptomethanol at 50°C for 20 min, after which the filters were washed and reblocked.
Figure 4.
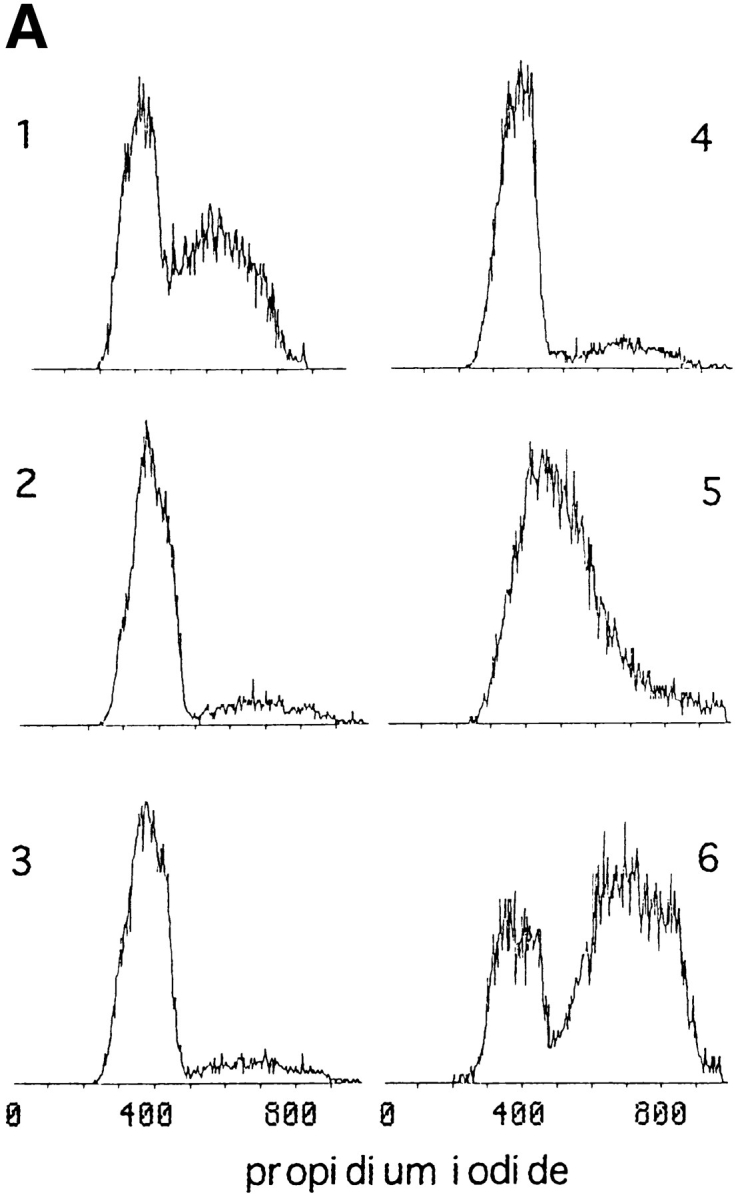


Myf5 is regulated by posttranscriptional mechanisms that are cell cycle– dependent. (A and B) Synchronized populations of anti-IGFII C2 cells were prepared either for propidium iodide labeling and flow cytometric analysis, or for protein analysis, or for RNA analysis. Nonsynchronized population (1). Cells grown in medium containing 0.5% FCS for 24 h (2); this was replaced with medium containing 20% FCS plus 1 μg/ml aphidicolin (Sigma Chemical Co.), for 6 h (3). Aphidicolin-containing medium was removed and replaced with medium containing 20% FCS, for 1 h (4), 6 h (5), and 16 h (8); or medium containing 20% FCS plus 200 ng/ml nocodazole for 6 h (9) and 16 h (6). Cells treated for 16 h with nocodazole, washed, and then replated in medium containing 20% FCS, for 6 h (7). (A) Analysis by flow cytometry of DNA content of samples 1–6. Propidium iodide content is in arbitrary units and the vertical axis indicates cell count (not to scale). The distribution of cell populations in these samples confirms the synchronization of cells from G1 (propidium iodide content equivalent to 2 N DNA content) through S phase to mitosis (4 N DNA content). A certain fraction of cells seem to be unable to exit G0, since the 2 N population seen in sample 6 is absent from cells that have been treated with nocodazole without prior serum withdrawal (not shown). (B) Analysis of Myf5 protein and mRNA in extracts from synchronized cells. Immunoblot analyses used rabbit anti-Myf5 NH2-terminal antiserum, and mouse mAb against cJun as a control. Myf5 mRNA levels measured by Northern blot analysis were quantitated by use of a PhosphorImager and corrected for the level of mRNA of ribosomal subunit S-26 (not shown) to confirm that the variation of Myf5 mRNA in these extracts is low. (C) Proliferating anti-IGFII C2 cells treated for 6 h with 200 ng/ml nocodazole were used to generate a mitotic fraction (SO, shake-off fraction) and a nonmitotic fraction (Ad, adherent). These were analyzed by immunoblotting for expression of Myf5 (anti–NH2-terminal antiserum), cJun, and Sp1.
Antibodies
The Myf5 antibody used to generate Fig. 3 is a polyclonal rabbit serum raised against the COOH-terminal peptide (237–255). Antibodies raised against the Myf5 NH2-terminal and COOH-terminal domains and used for all other figures in this paper were the gift of Dr. M. Primig (Unité de Génétique Moléculaire du Développement, Pasteur Institute, Paris, France). The NH2-terminal antibody used in Figs. 1 and 2 and the COOH-terminal antibody in Fig. 5, had been affinity purified. All Myf5 antibodies were used at 1/1,000 dilution for indirect immunofluorescence and 1/300 in Western blotting. Myogenin was detected using mouse monoclonal F5D (at 1/100 dilution), provided by Dr. W. Wright (University of Texas, Dallas, TX). Mouse monoclonal antibodies were used to detect MyoD (NCL-MyoD1; Novocastra, Burlingame, CA), troponin T (T6277; Sigma Chemical Co.), and cJun (J31920; Transduction Laboratories, Lexington, KY) at concentrations recommended by the suppliers. Sp1 was detected with a rabbit polyclonal antibody (sc-059; Santa Cruz Biotechnology, Santa Cruz, CA).
Figure 5.

Myf5 undergoes destruction at mitosis after mitosis-specific phosphorylation. (A) Mitotic and nonmitotic fractions were prepared as described in Fig. 4 C but with the following additions to cultures 2 h before shake-off: ALLN was added to a final concentration of 0.1 mM from a stock in DMSO (+ALLN); controls received an equivalent volume of DMSO (−ALLN). S0, shake-off fraction (mitotic cells); Ad, adherent fraction (nonmitotic cells). (B) Three of the extracts analyzed in A (lanes 6, 7, and 8) were incubated for 1 h at 37°C with (phosphatase-treated) or without (mock-treated) calf intestinal phosphatase (5 U/100 μl extract; from Pharmacia Biotechnology Inc.) and analyzed by immunoblotting with the COOH-terminal antibody used in A (right-hand panel).
Preparation of RNA and Northern Blotting
Preparation, electrophoresis, and blotting of total RNA from synchronized cell cultures was as previously described (Pinset et al., 1988). 10 μg of each sample was analyzed. Homogeneity of loading was monitored by staining of gels with ethidium bromide before transfer, and measured by probing for the ribosomal protein S26 (Vincent et al., 1993). The Myf5 probe used has been described in Montarras et al. (1996).
Results
Myf5 Is Absent from Myoblasts at the Onset of Differentiation
The recent availability of antibodies against Myf5 (Primig, M., manuscript in preparation; and Auradé et al., 1997) enabled us to investigate the expression of this factor during the autonomous differentiation of C2 myoblasts (Pinset et al., 1988). These antibodies specifically recognize Myf5 by immunocytochemical techniques and show no cross-reaction with other MRFs in immunoblot analyses (not shown). Previous studies have shown that the level of Myf5 mRNA in various myogenic cell lines is downregulated under conditions favorable to differentiation (Montarras et al., 1991, 1993; Mangiacapra et al., 1992). Codetection by indirect immunofluorescence of Myf5 with troponin T, a marker for myogenic differentiation (Pinset and Montarras, 1994) shows that Myf5 protein is not detected in cells that express troponin T (Fig. 1 A). This finding is in agreement with previous expression data, and suggests that the Myf5 mRNA detected in extracts from myotube cultures (Montarras et al., 1991) reflects the presence of residual myoblasts. The half-life of Myf5 protein is very short—not >30 min in C2 cells (Lindon, C., unpublished observations)—thus the disappearance of the protein must follow closely the downregulation of the Myf5 message.
To establish more precisely the timing of the downregulation of Myf5, we examined its codetection with myogenin (Fig. 1 B). Myogenin is the earliest known marker for myoblasts committed to the differentiation pathway (Wright et al., 1989), and in a recent study has been shown to be expressed before the establishment of the postmitotic state (Andrés and Walsh, 1996). We find that, in general, detection of Myf5 in myogenin-positive nuclei is reduced compared to the levels detected in myogenin-negative nuclei. We propose that the downregulation of Myf5 occurs either before or simultaneously with the appearance of myogenin in these cells. The disappearance of Myf5 protein is thus a very early event in the exit of C2 myoblasts from a determined state to one in which they are committed to differentiate.
Fig. 2 A shows both Myf5 and MyoD expressed in proliferating C2 myoblasts. The levels of these factors in the myoblast population are heterogeneous, but there is no clear relationship between the relative levels of the two factors in these cells. In a differentiating culture, by contrast, the codetection of Myf5 with MyoD is much reduced. The myoblasts resolve almost completely into two populations distinguished by the predominant expression of either one or the other of the two factors (Fig. 2 B). At this stage in the differentiation of the culture, the MyoD-expressing population of cells has undergone some fusion into myotubes, whereas the Myf5-expressing cells have mostly failed to differentiate.
Thus the pattern of expression in differentiating myoblasts of the determination factor Myf5 contrasts strongly with that of MyoD. Whereas MyoD is upregulated in differentiating cells, Myf5 is downregulated early in differentiation. Our data suggest that this downregulation of Myf5 may coincide with the commitment of myoblasts to the differentiation pathway.
Myf5 Protein Levels Are Regulated by the Cell Cycle
The considerable heterogeneity of the levels of Myf5 and MyoD detected in proliferating myoblasts led us to examine the possibility of cell cycle–dependent effects on the expression of these factors. We chose to study Myf5 expression in a C2-derived cell line, in which expression of IGFII and MyoD are eliminated by the expression of an antisense-IGFII construct (anti-IGFII C2 myoblasts; Montarras et al., 1996). Since these myoblasts are unable to differentiate autonomously (requiring treatment with IGFs to do so), they can be synchronized in G0 and G1 phases of the cell cycle without undergoing terminal differentiation. Previous detection of Myf5 in these cells by immunofluorescence has shown that the basal level of expression is as heterogeneous as in the parental C2 cells (Auradé et al., 1997).
We investigated the cell cycle–associated changes in the level of Myf5 by synchronization of myoblasts through the cell cycle (Figs. 3 and 4). This was achieved by sequential use of serum deprivation, aphidicolin, and nocodazole, to arrest the myoblasts in G0, at the G1/S boundary, and in early M, respectively. Aphidicolin is an inhibitor of DNA polymerase α (Spadari et al., 1982), and nocodazole is an inhibitor of microtubule depolymerization used at weak doses to specifically block the progress of cells through metaphase (Rieder and Palazzo, 1992). The effects of both cell cycle blockers are fully reversible.
Our initial observations are illustrated in Fig. 3 and reveal that synchronization of myoblast populations affects the levels of Myf5 detected. We find that Myf5 levels are generally lower in cultures synchronized in G1 (Fig. 3, middle), than in those progressing through S phase (Fig. 3, bottom). In serum-deprived cells (G0; Fig. 3, top), Myf5 is strongly heterogeneous, thus in noncycling cells there are clearly additional mechanisms that generate variations in the expression of Myf5. It was not possible to investigate the presence of Myf5 in mitotic cells by immunofluorescence because of nonspecific staining of these cells (not shown).
To investigate in a more quantitative fashion the cell cycle regulation of Myf5, we prepared extracts from synchronized cell populations for Northern and Western blot assays (Fig. 4). The distribution of cells in G0/G1, S, and G2/M phases contributing to the extracts prepared for analysis was measured by flow cytometric analysis of parallel samples stained with propidium iodide (Fig. 4 A). Western blot analysis confirmed that Myf5 protein levels are regulated by progression through the cell cycle (Fig. 4 B, middle). Moreover the observed variations in protein level are largely independent of the level of Myf5 mRNA measured (Fig. 4 B, bottom), implying that the cell cycle–associated fluctuations of Myf5 protein result from posttranscriptional mechanisms. The level of transcription factor cJun (Fig. 4 B, top) does not show cell cycle regulation. The variations seen in the level of Myf5 mRNA are slight: quantification of the Northern blot shown in Fig. 4 B, after PhosphorImager analysis (not shown), reveals a two-fold difference between the highest level, in M phase cell extracts, and that in the unsynchronized population.
We find that whereas Myf5 mRNA is downregulated in G0 (Fig. 4 B, lane 2; and Montarras et al., 1996), the protein accumulates to a higher level in G0 than in the unsynchronized population, but that this level falls as cells pass through G1 (Fig. 4 B, lane 3). Myf5 protein accumulates during S phase (Fig. 4 B, lanes 4 and 5). The most striking discrepancy between the levels of message and protein occurs in the population that is largely synchronized in M phase (Fig. 4 B, lane 6). Here the message is most abundant, but the Myf5 protein appears to be almost completely absent. Release of cells from the nocodazole block allows completion of mitosis and resynthesis of Myf5 (Fig. 4 B, lane 7).
There is a population of cells that does not exit G0/G1 after serum deprivation (Fig. 4 A, sample 6), thus the fluctuations in level of Myf5 may be underestimated by this experiment.
Attempts to synchronize cells through mitosis without use of nocodazole (i.e., by release of cells arrested at the G1/S transition) were not successful. Thus it was necessary to consider the possibility that nocodazole regulates the level of Myf5 (but not those of cJun, cFos, Sp1; Fig. 4, B and C; and data not shown) by a mechanism that is not related to cell cycle arrest. We prepared separate fractions of mitotic and nonmitotic cells from plates treated with nocodazole for 6 h (time of treatment that had no effect on the level of Myf5 in nonmitotic cells; Fig. 4 B, compare lanes 5 and 9). In a typical experiment, the mitotic fraction consisted of 90–95% cells containing condensed chromatin as judged by propidium iodide labeling of ethanol-fixed cells (not shown). Replated cells from the mitotic fraction completed mitosis and showed a growth rate and differentiation capacity identical to that of the nonmitotic fraction (not shown). Extracts prepared from these fractions were analyzed by immunoblot (Fig. 4 C). It was found that Myf5 is specifically absent from the mitotic fraction comprising cells arrested in pro-metaphase. Since treatment of cells with nocodazole has no effect on the level of Myf5 in nonmitotic cells, it can be concluded that mitotic arrest is responsible for the disappearance of Myf5.
The observed appearance of a more slowly migrating form of Sp1 in the mitotic fraction (Fig. 4 C, top) is consistent with the previously described phosphorylation of Sp1 in nocodazole-blocked HeLa cells (Martinez-Balbas et al., 1995).
Myf5 Is Phosphorylated and Degraded at Mitosis
We investigated the possibility that the disappearance of Myf5 is because of an increase in its rate of degradation as cells pass into mitosis. ALLN is commonly used as an inhibitor of proteasome-mediated proteolysis, and was used to treat cells prepared for mitotic shake-off for 2 h before preparation of extracts. Extracts were analyzed by immunoblotting using antibodies raised against both the NH2-terminal and COOH-terminal portions of Myf5 to confirm that lack of detection of Myf5 in mitotic extracts was not because of epitope masking or partial degradation of the protein. The results are shown in Fig. 5 A. The disappearance of Myf5 from mitotic cell extracts, observed with both of the antibodies tested (Fig. 5 A, lanes 2 and 6), is blocked in the presence of ALLN (Fig. 5 A, lanes 4 and 8). Thus proteolysis of Myf5 is responsible for its disappearance at mitosis, and this probably occurs via the ubiquitin– proteasome pathway. However the specificity of ALLN also extends to nonproteasome proteases such as calpain; one of these could be involved in the degradation of Myf5.
The Myf5 protein detected in the presence of ALLN exists as a more slowly migrating form (Fig. 5 A, lanes 4 and 8), which is detected only in mitotic extracts. By treating extracts shown in Fig. 5 A with alkaline phosphatase, we investigated whether the shift in mobility of Myf5 in mitotic cells was because of phosphorylation of the protein (Fig. 5 B). Phosphatase treatment of the ALLN-treated mitotic shake-off extract did indeed reverse the shift in mobility of Myf5 (Fig. 5 B, extract 8). Parallel treatment of a mitotic shake-off extract confirms that there is no epitope masking through phosphorylation of Myf5 in mitotic cells (Fig. 5 B, extract 6). Both mitotic and nonmitotic forms of Myf5 appear to be stable in the extracts used, even at 37°C (Fig. 5 B, panels 7 and 8).
These data strongly suggest that Myf5 undergoes phosphorylation by a mitosis-specific kinase(s), and that this modified Myf5 is highly unstable in mitotic cells and is rapidly degraded, probably by a 26-S proteasome-dependent mechanism.
MyoD Does Not Undergo Cell Cycle–dependent Variation in Expression
To investigate whether MyoD might be subject to similar cell cycle regulation we have synchronized C2.7 cells by suspension in methyl cellulose–containing medium for 48 h as described in Milasincic et al. (1996). This leads to reversible arrest in G0 and extinction of MyoD expression (Milasincic et al., 1996). MyoD is resynthesized on reattachment of these cells but we have been unable to detect any variation in the level of MyoD during the subsequent cell cycle (data not shown). Moreover, analysis of the mitotic fraction from nocodazole-treated cultures of C2.7 cells reveals that MyoD is present in these cells (Fig. 6). The migration of MyoD as a doublet in SDS–polyacrylamide gels has previously been ascribed to phosphorylation of the slowly migrating form (Skapek et al., 1995). There is an increase in the relative amount of the slowly migrating form of MyoD in mitotic cells (Fig. 6, top right), this could be the result of new phosphorylation of the protein, or of preferential degradation of the faster migrating form during mitosis. However, the total quantity of MyoD in mitotic cells does not appear to be significantly diminished, whereas Myf5 has undergone complete destruction (Fig. 6, bottom).
Figure 6.

MyoD is present in mitotic C2 cells. Mitotic and nonmitotic fractions were prepared from proliferating C2.7 cells as described in Fig. 4 C. These were immunoblotted with antibodies against MyoD and Myf5 (NH2 terminus). The right-hand panel shows a shorter exposure of MyoD.
Discussion
The results presented in this paper show that expression of the muscle determination factor Myf5 is associated with proliferating myoblasts and tightly regulated by the cell cycle.
We show that downregulation of Myf5 in C2 cells can occur as early as the onset of differentiation, when cells become myogenin-positive. Previous results obtained using primary muscle cell cultures (Smith et al., 1993) have described the codetection of Myf5 with a differentiation marker in myotubes and these authors conclude that the different MRFs define different types of myoblast rather than different stages of determination and differentiation. We have examined Myf5 expression in mouse and human primary myoblast cultures and found that the level of Myf5 expression in differentiating cells is low compared to that in surrounding myoblasts, although we can detect Myf5 in some young myotubes (Pinset, C., D. Montarras, and C. Lindon, unpublished observations).
It is possible that the sequence of events that occurs in differentiating C2 cells represents the differentiation of a subset of myoblasts in vivo. However our observations in primary cultures are consistent with the conclusion that strong expression of Myf5 (in contrast to that of MyoD) is restricted to myoblasts in the proliferative state. This conclusion raises the possibility that the downregulation of Myf5 could be requisite for the progress of differentiation. In support of this idea, we have previously observed that in myoblasts unable to undergo differentiation Myf5 expression is upregulated (Montarras et al., 1996).
We also show that Myf5 accumulates in S phase, and is depleted in mitotic cells. Since Myf5 mRNA levels are not affected by the cell cycle, the loss of Myf5 protein expression must be a posttranscriptional event. This is not a general block in translation since the level of cJun (t1/2 = 90 min) is not affected even after prolonged nocodazole treatment (24 h; data not shown). We have shown that the proteolytic degradation of Myf5, most probably by the 26-S proteasome, is responsible for its disappearance. However, we have not excluded the possibility that a specific arrest in translation of the Myf5 message contributes to the downregulation of this factor at mitosis.
Although nocodazole artificially prolongs the time cells spend in mitosis, the absence of any trace of Myf5 from mitotic extracts leads us to conclude that this factor undergoes complete destruction either at, or just before, entry into mitosis. Proteolytic degradation follows a phosphorylation(s) specific to this phase of the cell cycle. Phosphorylation at mitosis has been documented for several transcription factors. In some cases this has been described as a mechanism for preventing DNA binding (Roberts et al., 1991; Caelles et al., 1995), and in other cases has been shown to correlate with dissociation of transcription factors from condensed chromatin (Martinez-Balbas et al., 1995; Muchardt et al., 1996). However, to our knowledge this is the first description of the destruction of a transcription factor at mitosis.
We have previously observed that high level expression of Myf5 is incompatible with cellular proliferation in nontransformed cells: Muscle cell lines derived from embryonic 10T1/2 fibroblasts transfected with Myf5 are found to express the gene at a level severalfold lower than that seen for any of the other MRF factors, and detectable only by RT-PCR analysis (Auradé et al., 1994). In contrast, we find that HeLa cells (which are transformed and not convertible to the myogenic lineage) are able to express Myf5 in a stable fashion at high levels (Lindon, C., unpublished observation). In addition, we note that the only established muscle cell lines described that express high levels of Myf5 are those that have been chemically transformed (Braun et al., 1989a ). We propose that nontransformed, cycling cells need to degrade Myf5 during each cell cycle to overcome a negative effect of Myf5 on cell cycle progression, and that this is the basis for the apparent toxicity of Myf5 overexpression.
Do Myf5 and MyoD play distinct roles in proliferating myoblasts? The cell cycle–associated variation in Myf5 that we have described contributes to the heterogeneity of Myf5 in proliferating myoblasts observed by immunofluorescence. There is no clear correlation between the levels of Myf5 and MyoD from cell to cell and we have been unable to detect cell cycle effects on MyoD levels present in C2 cell extracts. Given the upregulation of MyoD observed in differentiating myoblasts, it seems likely that the accumulation of MyoD is incremental rather than cyclic, and that above a certain threshold level cells become committed to differentiate in response to favorable conditions (such as serum depletion).
Mechanisms by which cyclin D1 suppresses MyoD activity during the cell cycle have been described (Rao et al., 1994; Skapek et al., 1995, 1996). Clearly there are additional mechanisms by which the cell cycle regulates the activity of the myogenic bHLH factor family and it seems likely that Myf5 functions in response to conditions distinct from those that induce the myogenic activity of MyoD.
We have previously observed that transcription of myf5 is highly sensitive to culture conditions and rapidly upregulated by agonists of the stress-activated protein kinases (Auradé et al., 1997). Our finding that basal Myf5 levels are very low or nonexistent in G1 and M phases suggests that the induction of Myf5 during these phases of the cycle could influence progress through the cell cycle and the commitment to differentiation. We believe that the further study of Myf5 functions and of its posttranscriptional regulation will advance our understanding of the mechanisms involved in determination.
Acknowledgments
We thank F. Auradé, M. Primig, and A. Garcia for useful discussions during the course of this work; and F. Auradé and S. Whiteside for their comments on the manuscript. Confocal microscopy was carried out with the help of R. Helio, and flow cytometric analysis with the help of H. Kiefer.
Abbreviations used in this paper
- ALLN
N-acetyl-Leu-Leu-norleucinal
- cdk
cyclin-dependent kinase
- IGF
insulin-like growth factor
- MRF
myogenic regulatory factors
- RIPA
radioimmunoprecipitation assay
Footnotes
C. Lindon is the recipient of a bourse d'étude from the Fondation Simone et Cino del Duca. The Groupe de Développement Cellulaire is funded by the Institut Pasteur, the Centre National de la Recherche Scientifique, and the Association Française contre les Myopathies.
Address all correspondence to C. Lindon, Groupe de Développement Cellulaire, Institut Pasteur, Département de Biologie Moléculaire, 25 rue du Dr Roux, 75724 Paris Cedex 15. Tel.: (33) 1-45-68-84-76. Fax: (33) 1-45-68-89-63. E-mail: clindon@pasteur.fr
References
- Andrés V, Walsh K. Myogenin expression, cell cycle withdrawal, and phenotypic differentiation are temporally separable events that precede cell fusion upon myogenesis. J Cell Biol. 1996;132:657–666. doi: 10.1083/jcb.132.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auradé F, Pinset C, Chaffey P, Gros F, Montarras D. Myf5, MyoD, Myogenin and MRF4myogenic derivatives of the embryonic mesenchymal cell line 10T1/2 exhibit the same adult phenotype. Differentiation. 1994;55:185–192. doi: 10.1046/j.1432-0436.1994.5530185.x. [DOI] [PubMed] [Google Scholar]
- Auradé F, Pfarr C, Lindon C, Primig M, Montarras D, Pinset C. The glucocorticoid receptor and c-jun are involved in a positive regulation of the muscle regulatory gene myf5 in cultured myoblasts. J Cell Sci. 1997;110:2771–2779. doi: 10.1242/jcs.110.22.2771. [DOI] [PubMed] [Google Scholar]
- Braun T, Arnold H-H. Myf-5 and MyoD genes are activated in distinct mesenchymal stem cells and determine different skeletal muscle cell lineages. EMBO (Eur Mol Biol Organ) J. 1996;15:310–318. [PMC free article] [PubMed] [Google Scholar]
- Braun T, Bober E, Buschhausen-Denker G, Kotz S, Grzeschik K-H, Arnold H. Differential expression of myogenic determination genes in muscle cells: possible autoactivation by the Myfgene products. EMBO (Eur Mol Biol Organ) J. 1989a;8:3617–3625. doi: 10.1002/j.1460-2075.1989.tb08535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun T, Buschhausen-Denker G, Bober E, Tannich E, Arnold H. A novel human muscle factor related but distinct from MyoD1 induces myogenic conversion in 10T1/2 fibroblasts. EMBO (Eur Mol Biol Organ) J. 1989b;8:701–709. doi: 10.1002/j.1460-2075.1989.tb03429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun T, Rudnicki MA, Arnold H-H, Jaenisch R. Targeted inactivation of the muscle regulatory gene Myf-5results in abnormal rib development and perinatal death. Cell. 1992;71:369–382. doi: 10.1016/0092-8674(92)90507-9. [DOI] [PubMed] [Google Scholar]
- Caelles C, Henneman H, Karin M. M-phase-specific phosphorylation of the POU transcription factor GHF-1 by a cell cycle-regulated protein kinase inhibits DNA binding. Mol Cell Biol. 1995;15:6694–6701. doi: 10.1128/mcb.15.12.6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crescenzi M, Fleming T, Lassar A, Weintraub H. MyoD induces growth arrest independent of differentiation in normal and transformed cells. Proc Natl Acad Sci USA. 1990;87:8442–8446. doi: 10.1073/pnas.87.21.8442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblast to myoblasts. Cell. 1987;51:987–1000. doi: 10.1016/0092-8674(87)90585-x. [DOI] [PubMed] [Google Scholar]
- Gu W, Schneider J, Condorelli G, Kaushal S, Mahdavi V, Nadal-Ginard B. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell. 1993;72:309–324. doi: 10.1016/0092-8674(93)90110-c. [DOI] [PubMed] [Google Scholar]
- Guo K, Wang J, Andrés V, Smith R, Walsh K. MyoD-induced expression of p21 inhibits cyclin-dependent kinase activity upon myocytes terminal differentiation. Mol Cell Biol. 1995;15:3823–3829. doi: 10.1128/mcb.15.7.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halevy O, Novitch B, Spicer D, Skapek S, Rhee J, Hannon G, Beach D, Lassar A. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- Lassar A, Skapek S, Novitch B. Regulatory mechanisms that coordinate skeletal muscle differentiation and cell cycle withdrawal. Curr Opin Cell Biol. 1994;6:788–794. doi: 10.1016/0955-0674(94)90046-9. [DOI] [PubMed] [Google Scholar]
- Mangiacapra F, Roof S, Ewton D, Florini J. Paradoxical decrease in Myf-5 messenger RNA levels during induction of myogenic differentiation by insulin-like growth factors. Mol Endocrinol. 1992;6:2038–2044. doi: 10.1210/mend.6.12.1337140. [DOI] [PubMed] [Google Scholar]
- Martinez-Balbas MA, Dey A, Rabindran SK, Ozato K, Wu C. Displacement of sequence-specific transcription factors from mitotic chromatin. Cell. 1995;83:29–38. doi: 10.1016/0092-8674(95)90231-7. [DOI] [PubMed] [Google Scholar]
- Mellits KH, Hay RT, Goodbourn S. Proteolytic degradation of MAD3 (IκBα) and enhanced processing of the NF-κB precursor p105 are obligatory steps in the activation of NF-κB. Nucl Acids Res. 1993;21:5059–5066. doi: 10.1093/nar/21.22.5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milasincic DJ, Dhawan J, Farmer SR. Anchorage-dependent control of muscle-specific gene expression in C2C12 mouse myoblasts. In Vitro Cell Dev Biol Anim. 1996;32:90–99. doi: 10.1007/BF02723040. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Olson E. Defining the regulatory networks for muscle development. Curr Opin Genet Dev. 1996;6:445–453. doi: 10.1016/s0959-437x(96)80066-9. [DOI] [PubMed] [Google Scholar]
- Montarras D, Chelly J, Bober E, Arnold H, Ott M-O, Gros F, Pinset C. Developmental patterns in the expression of myf5, MyoD, myogenin, and MRF4 during myogenesis. New Biol. 1991;3:592–600. [PubMed] [Google Scholar]
- Montarras D, Pinset C, Perez M-C, Ilan J, Gros F. Muscle differentiation: insulin-like growth factors as positive modulators of myogenic regulatory genes? . CR Acad Sci III. 1993;316:1029–1031. [PubMed] [Google Scholar]
- Montarras D, Auradé F, Johnson T, Ilan J, Gros F, Pinset C. Autonomous differentiation in the mouse myogenic cell line, C2, involves a mutual positive control between insulin like growth factor II and MyoD, operating as early as the myoblast stage. J Cell Sci. 1996;109:551–560. doi: 10.1242/jcs.109.3.551. [DOI] [PubMed] [Google Scholar]
- Muchardt C, Reyes J-C, Bourachot B, Legouy E, Yaniv M. The hbrm and BRG-1 proteins, components of the human SNF/SWI complex, are phosphorylated and excluded from the condensed chromosomes during mitosis. EMBO (Eur Mol Biol Organ) J. 1996;15:3394–3402. [PMC free article] [PubMed] [Google Scholar]
- Olson EN, Klein WH. bHLH factors in muscle development: dead lines and commitments, what to leave in and what to leave out. Genes Dev. 1994;8:1–8. doi: 10.1101/gad.8.1.1. [DOI] [PubMed] [Google Scholar]
- Ott M, Bober E, Lyons G, Arnold H, Buckingham M. Early expression of the myogenic regulatory gene, myf-5, in precursor cells of skeletal muscle in the mouse embryo. Development (Camb) 1991;111:1097–1107. doi: 10.1242/dev.111.4.1097. [DOI] [PubMed] [Google Scholar]
- Pinset, C., and D. Montarras. 1994. Cell systems for ex vivo studies of myogenesis: A protocol for the isolation of stable muscle cell populations from newborn to adult mice. In Cell Biology: A Laboratory Handbook. J.E. Celis, editor. Academic Press, Inc., San Diego. 199–206.
- Pinset C, Montarras D, Chenevert J, Minty A, Barton P, Laurent C, Gros F. Control of myogenesis in the mouse myogenic C2 cell line by medium composition and by insulin: characterization of permissive and inducible C2 myoblasts. Differentiation. 1988;38:28–34. doi: 10.1111/j.1432-0436.1988.tb00588.x. [DOI] [PubMed] [Google Scholar]
- Rao S, Chu C, Khotz S. Ectopic expression of cyclin D1 prevents activation of gene transcription by myogenic basic helix-loop-helix regulators. Mol Cell Biol. 1994;14:5259–5267. doi: 10.1128/mcb.14.8.5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL, Palazzo RE. Colcemid and the mitotic cycle. J Cell Sci. 1992;102:387–392. doi: 10.1242/jcs.102.3.387. [DOI] [PubMed] [Google Scholar]
- Roberts SB, Segil N, Heintz N. Differential phosphorylation of the transcription factor Oct1 during the cell cycle. Science. 1991;253:1022–1026. doi: 10.1126/science.1887216. [DOI] [PubMed] [Google Scholar]
- Rudnicki M, Braun T, Hinuma S, Jaenisch R. Inactivation of MyoD in mice leads to up-regulation of the myogenic HLH gene Myf-5and results in apparently normal muscle development. Cell. 1992;71:383–390. doi: 10.1016/0092-8674(92)90508-a. [DOI] [PubMed] [Google Scholar]
- Rudnicki MA, Schnegelsberg P, Stead R, Braun T, Arnold H, Jaenisch R. MyoD or Myf5 is required for the formation of skeletal muscle. Cell. 1993;75:1351–1359. doi: 10.1016/0092-8674(93)90621-v. [DOI] [PubMed] [Google Scholar]
- Schneider J, Gu W, Zhu L, Mahdavi V, Nadal-Ginard B. Reversal of terminal differentiation mediated by p107 in Rb-/- muscle cells. Science. 1994;264:1467–1471. doi: 10.1126/science.8197461. [DOI] [PubMed] [Google Scholar]
- Skapek S, Rhee J, Spicer D, Lassar A. Inhibition of myogenic differentiation in proliferating myoblasts by cyclin D1-Dependent kinase. Science. 1995;267:1022–1024. doi: 10.1126/science.7863328. [DOI] [PubMed] [Google Scholar]
- Skapek SX, Rhee J, Kim PS, Novitch BG, Lassar AB. Cyclin-mediated inhibition of muscle gene expression via a mechanism that is independent of pRB hyperphosphorylation. Mol Cell Biol. 1996;16:7043–7053. doi: 10.1128/mcb.16.12.7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TH, Block NE, Rhodes SJ, Konieczny SF, Miller JB. A unique pattern of expression of the four muscle regulatory factor proteins distinguishes somitic from embryonic, fetal and newborn mouse myogenic cells. Development (Camb) 1993;117:1125–1133. doi: 10.1242/dev.117.3.1125. [DOI] [PubMed] [Google Scholar]
- Sorrentino V, Pepperkok R, Davis R, Ansorge W, Philipson L. Cell proliferation inhibited by MyoD1 independently of myogenic differentiation. Nature. 1990;345:813–815. doi: 10.1038/345813a0. [DOI] [PubMed] [Google Scholar]
- Spadari S, Sala F, Pedrali-Noy G. Aphidicolin: a specific inhibitor of nuclear DNA replication in eukaryotes. Trends Biochem Sci. 1982;7:29–32. [Google Scholar]
- Tajbakhsh S, Rocancourt D, Buckingham M. Muscle progenitor cells failing to respond to positional cues adopt non-myogenic fates in myf-5 null mice. Nature. 1996;384:266–270. doi: 10.1038/384266a0. [DOI] [PubMed] [Google Scholar]
- Vincent S, Marty L, Fort P. S26 ribosomal protein RNA: an invariant control for gene regulation experiments in eucaryotic cells and tissues. Nucl Acids Res. 1993;21:1498. doi: 10.1093/nar/21.6.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H, Davis R, Tapscott S, Thayer M, Krause M, Benezra R, Blackwell TK, Turner D, Rupp R, Hollenberg S, et al. The myoD gene family: nodal point during specification of the muscle cell lineage. Science. 1991;251:761–766. doi: 10.1126/science.1846704. [DOI] [PubMed] [Google Scholar]
- Wright WE, Sassoon D, Lin VK. Myogenin, a factor regulating myogenesis, has a domain homologous to MyoD. Cell. 1989;56:607–617. doi: 10.1016/0092-8674(89)90583-7. [DOI] [PubMed] [Google Scholar]
- Yaffé D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1977;270:725–727. doi: 10.1038/270725a0. [DOI] [PubMed] [Google Scholar]
- Yun K, Wold B. Skeletal muscle determination and differentiation: story of a core regulatory network and its context. Curr Opin Cell Biol. 1996;8:877–889. doi: 10.1016/s0955-0674(96)80091-3. [DOI] [PubMed] [Google Scholar]
- Zacksenhaus E, Jiang Z, Chung D, Marth J, Phillips R, Gallie B. pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes Dev. 1996;10:3051–3064. doi: 10.1101/gad.10.23.3051. [DOI] [PubMed] [Google Scholar]