

Abstract
ATP hydrolysis has been regarded as a general requirement for internalization processes in mammalian cells. We found, however, that treatment of ATP-depleted macrophages and fibroblasts with exogenous sphingomyelinase (SMase) rapidly induces formation of numerous vesicles that pinch off from the plasma membrane; the process is complete within 10 min after adding SMase. By electron microscopy, the SMase-induced vesicles are ∼400 nm in diameter and lack discernible coats. 15–30% of plasma membrane is internalized by SMase treatment, and there is no detectable enrichment of either clathrin or caveolin in these vesicles. When ATP is restored to the cells, the SMase-induced vesicles are able to deliver fluid-phase markers to late endosomes/lysosomes and return recycling receptors, such as transferrin receptors, back to the plasma membrane. We speculate that hydrolysis of sphingomyelin on the plasma membrane causes inward curvature and subsequent fusion to form sealed vesicles. Many cell types express a SMase that can be secreted or delivered to endosomes and lysosomes. The hydrolysis of sphingomyelin by these enzymes is activated by several signaling pathways, and this may lead to formation of vesicles by the process described here.
Mammalian cells use several mechanisms for internalizing fluids and membrane components. These include receptor-mediated endocytosis via clathrin-coated pits, actin-based phagocytosis, and the pinching off of various types of uncoated membranes (Mukherjee et al., 1997). ATP use has been considered to be a hallmark of all internalization processes (Silverstein et al., 1977). The mechanisms for budding and pinching off of endocytic vesicles from the plasma membrane are only partially understood. It has been shown that there are at least two ATP-dependent steps in the formation of clathrin-coated vesicles (Schmid and Smythe, 1991). One of these steps apparently involves the invagination of the membrane, and the second is the complete sealing of the vesicle, a process that requires fusion of the plasma membrane with itself. The general mechanism by which ATP catalyzes budding and fusion is not known at present.
The role of lipid components in vesicle budding and fusion in living cells is poorly understood. Recent studies have shown that phosphatidylinositol (PI)31 kinase is involved in several steps of vesicle traffic in mammalian cells (Shepherd et al., 1996). Inhibition of PI3 kinase by wortmannin and LY294002 was reported to prevent invaginated phagosomes from closure on the plasma membrane (Araki et al., 1996). The mechanisms by which PI3 kinase activity modulates vesicle traffic are not known. Treatment of Golgi membranes with bacterial phospholipase D stimulates coatomer binding (Ktistakis et al., 1996), and this may be part of a mechanism for controlling vesicle budding in the Golgi (Bednarek et al., 1996). Through studies in model membrane systems, it is known that budding can be facilitated by altering lipid composition to provide favorable membrane bending energy (Sackmann, 1994; Chernomordik et al., 1995). This favorable bending energy can arise through enrichment of certain types of lipids in an asymmetrical lipid bilayer. Alteration of lipid composition can also create a local phase separation so that fusion is facilitated after membrane bending. For instance, membrane fusion in dimyristoylphosphatidylcholine–cholesterol vesicles was attributed to the accumulation of cholesterol that formed a separate phase within the neck interconnecting the membranes (Knoll et al., 1985).
In an investigation of the ATP dependence of cholesterol esterification, we treated J774 macrophages with exogenous sphingomyelinase (SMase), which was shown previously to enhance delivery of plasma membrane cholesterol to a microsomal enzyme, acetyl-CoA-cholesterol acyl transferase (ACAT; Slotte et al., 1990). Since cholesterol preferentially associates with sphingomyelin in membranes (Gronberg et al., 1991), it was thought that depletion of sphingomyelin by SMase treatment would cause release of cholesterol from the plasma membrane, allowing its transport to ACAT. We found that enhanced cholesterol delivery because of SMase was not affected by ATP depletion but was associated with the formation of membrane vesicles (Skiba et al., 1996). We report here that treatment of cells with SMase rapidly induces formation of numerous vesicles that pinch off from the plasma membrane, and the formation of these endocytic compartments does not require cellular ATP. Electron microscopic visualization shows that the sphingomyelinase-induced vesicles are ∼400 nm in diameter and lack discernible coats.
Materials and Methods
Materials
SMase (Bacillus cereus), sodium azide, 2-deoxyglucose, Lucifer yellow, and ATP bioluminescent assay kit were purchased (Sigma Chemical Co., St. Louis, MO). FITC-dextran (70-kD), tetramethylrhodamine isothiocyanate (TRITC)-dextran (70-kD), rhodamine-dextran (70-kD), and 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene-c12-sphingomyelin (BODIPY-C12-SM) were from Molecular Probes (Eugene, OR). SMase (Staphylococcus aureus) was from ICN Biomedicals Inc. (Irvine, CA). Streptolysin O (SL-O) was obtained from Murex Diagnostics (Atlanta, GA). Human transferrin (Tf; Sigma Chemical Co.) was iron-loaded, purified by Sephacryl S-300 (Pharmacia Diagnostics AB, Uppsala, Sweden), and labeled with fluorophore Cy3 (Biological Detection Systems, Pittsburgh, PA) according to the manufacturer's instructions. Rabbit polyclonal antibody to caveolin/ VIP-2 was purchased from Transduction Laboratories (Lexington, KY), and mouse monoclonal antibodies against adaptor protein 2 (AP6) and clathrin (X22) were gifts from F. Brodsky (University of California, San Francisco, CA).
Cells
J774 cells (American Type Culture Collection, Rockville, MD) were maintained in spinner culture in DME (Gibco Laboratories, Grand Island, NY) with 10% FCS. TRVb-1 cells (McGraw et al., 1987), a CHO cell line lacking endogenous transferrin receptor and expressing transfected human Tf receptors, were cultured in HF-12 (Gibco Laboratories) with 5% FCS. 2 d before an experiment, cells were plated on polylysine-coated coverslip-bottom dishes (Salzman and Maxfield, 1988). All experiments were performed on day 3 unless otherwise mentioned. ATP measurements were performed on both J774 macrophages and TRVb-1 cells using an ATP bioluminescent assay kit (Sigma Chemical Co.). Cells grown on 12-well culture dishes were pretreated with sodium azide/2-deoxyglucose for 5 min to deplete ATP and then incubated with or without SMase for an additional 10 min at 37°C before ATP assay. To measure ATP in permeabilized cells, cells were preincubated with sodium azide/2-deoxyglucose for 5 min and cooled to 0°C. Cells were then treated with SL-O (0.4 U/ml) for 5 min on ice, rinsed three times with cold PBS, and transferred to 37°C for 10 min. After a 10-min incubation with SMase, cells were washed and assayed for ATP.
Fluorescence Microscopy
In most experiments, cells were preincubated for 5 min in medium alone or containing 2-deoxyglucose and sodium azide. The cells were then incubated in the same medium containing 5 mg/ml FITC-dextran alone, or plus 50 mU/ml SMase for 10 min before being fixed with 4% paraformaldehyde for 5 min in PBS. Cells were immediately viewed using a microscope (model Diavert; Leitz, Wetzlar, Germany) with a 63×, NA 1.4 objective, and recorded with a cooled charge-coupled device (CCD) camera (KAF 1400; Photometrics, Tucson, AZ). To quantify the endocytosed FITC-dextran, fluorescence images of cells were taken using the same system as above but with a 25×, NA 0.8 objective. Total fluorescence intensity from a field of cells (∼70 cells) was measured and, after background correction, divided by the number of cells in the field. Each data point represents an average of four or five fields. For immunofluorescence experiments, cells were fixed with 4% paraformaldehyde in PBS for 10 min and then permeabilized with 0.1 mg/ml saponin before incubating with primary antibodies and a TRITC-conjugated secondary antibody. Confocal fluorescence images were obtained with a laser scanning confocal microscope (model MRC600; Bio-Rad Microscience, Cambridge, MA). FITC-dextran and Cy3-Tf images in Fig. 8 were obtained using an image-intensified video camera (model K51380; Videoscope, Washington, DC) on a fluorescence microscope (model DMIRB; Leica Inc., St. Gallen, Switzerland) using a 63×, NA 1.32 objective.
Figure 8.

Sorting of transferrin receptors in SMase-induced vesicles. J774 macrophages were preincubated for 15 min in 0.2% BSA/DME plus 100 mM 2-deoxyglucose/sodium azide, followed by a 10-min incubation in the same medium containing Cy3-Tf (20 μg/ml). Because of the energy depletion, Cy3-Tf was only bound to the cell surface at this stage. The cells were then rinsed and incubated in DME containing energy poisons, FITC-dextran, and SMase for 10 min. At the end of this 10-min incubation, a mild acid wash was performed to strip Cy3-Tf that still remained on the cell surface. Cells were then either viewed immediately by fluorescence microscopy (a and b) or chased in DME for 20 min (c and d). Without any chase, internalized Cy3-Tf (b) was colocalized with FITC-dextran (a) in the peripheral vesicles. After a 20-min chase in complete medium without energy poison, FITC-dextran–containing vesicles moved into the cells (c), and Tf was mostly returned to the cell surface (d). Bar, 20 μm.
Electron Microscopy
After incubating with HRP plus SMase for 10 min, ATP-depleted cells were fixed with 3.5% glutaraldehyde in 100 mM Hepes, pH 7.4, for 15 min at 22°C. After three rinses of 5 min each with TBS (50 mM Tris, pH 7.4, 100 mM NaCl), cells were incubated with 10 mg/ml diaminobenzidine in TBS with 0.3% H2O2 for 60 min at room temperature with agitation. Cells were rinsed with three washes of 100 mM sodium cacodylate, pH 6.8, and then postfixed with 1% osmium tetroxide and 1.25% potassium ferrocyanide in 100 mM sodium cacodylate for 4 h at room temperature. After two rinses with 100 mM sodium cacodylate, cells were dehydrated in a graded series of ethanol, embedded in Epon, and sectioned in the plane of the monolayer. Thin sections (80 nm) were counterstained with uranyl acetate and lead citrate before observation using a transmission electron microscope (model 1200EX II; JEOL USA, Peabody, MA) operating at 80 kV.
Quantification of Endocytosed Plasma Membrane
N-([6-{7-nitrobenz-2-oxa-1,3-diazol-4-yl} amino] hexanoyl) sphingosyl phosphocholine (C6-NBD-gal) was prepared as described previously (Mayor et al., 1993). C6-NBD-gal/lipid vesicles (100 μM total lipid) were prepared by injecting an ethanolic solution of a 1:1 mixture of C6-NBD-gal and dioleylphosphatidylcholine (DOPC; 2:5 mM total lipid; Avanti Polar Lipids, Inc., Albaster, AL) into 150 mM NaCl, 20 mM Hepes, pH 7.4. TRVb-1 cells were incubated on ice with 3 μM of this vesicle solution (<0.2% ethanol) in Hepes-buffered, glucose-free Hams HF-12 medium (HF-12; Specialty Media, Inc., Lavallette, NJ) for 30 min. Cells were washed extensively with ice-cold HF-12, incubated with 5 mM 2-deoxyglucose and 5 mM sodium azide for 10 min at 37°C, and then incubated for an additional 10 min at 37°C in the same medium with or without 50 mU/ml SMase. For some samples, the lipid label remaining on the plasma membrane was backexchanged by washing (in six rinses of 5 min each) with ice-cold HF-12 containing 5% BSA. All samples were washed with cold PBS before lysis with 1% Triton X-100, and then C6-NBD-gal was extracted from the samples by homogenizing the lysates with chloroform. The C6-NBD-gal content in the chloroform extracts was determined with an SLM 8000C spectrofluorometer (Aminco, Urbana, IL).
Cells were prepared for microscopy as above except that cells were labeled with 0.75 μM C6-NBD-gal. Cells were fixed with 3.3% paraformaldehyde after the backexchange step, and images were acquired with a cooled CCD camera (model 30427; Princeton Instruments, Princeton, NJ) on a fluorescence microscope (model DMIRB; Leica, Inc.). Because concentration-dependent self-quenching of C6-NBD-gal could lead to underestimation of total fluorescence, NBD fluorescence emission was integrated over time until photobleaching was complete. This method allows the accurate quantification of total fluorescence (Jovin and Arndt-Jovin, 1989).
SMase Treatment of BODIPY-C12-sphingomyelin–labeled, SL-O–permeabilized Cells
43 μg of BODIPY-C12- SM (1 mg/ml in DMSO) was mixed with 20 μl of 5 mM DOPC in ethanol. The mixture was evaporated for 30 min under argon to remove ethanol and then centrifuged for 20 min at 4°C and 100,000 g. The supernatant was then rapidly injected into 1.5 ml PBS to yield a BODIPY-C12-SM stock solution. To label the plasma membrane, TRVb-1 cells were chilled to 0°C for about 20 min and were then incubated with BODIPY-C12-SM (1:5 dilution of stock solution with HF-12 medium) for 30 min at 0°C. Cells were washed with cold PBS three times to remove unbound BODIPY-C12-SM. Some of the cells were further incubated with SL-O (0.4 U/ml) for 5 min at 0°C and then washed with cold PBS three times. Cells were then transferred to 37°C for 3 min, treated with SMase (50 mU/ml)/rhodamine-dextran for 5 min, washed with medium 1 and then immediately observed by fluorescence microscopy.
Results
SMase-induced, ATP-independent Vesicle Formation in J774 Macrophages
Fig. 1 a shows the uptake of a fluid endocytosis marker, FITC-dextran, by J774 cells during a 10-min incubation at 37°C. These control cells internalize FITC-dextran and deliver it to endosomes that are seen as bright fluorescent dots. As expected, this uptake is blocked when cellular ATP is depleted by preincubation with sodium azide and 2-deoxyglucose (Fig. 1 b). Under the conditions used for Fig. 1 b, cellular ATP is 2.4 ± 0.5% of control values, as determined by an ATP luminescence assay. When ATP-depleted cells are treated with SMase (50 mU/ml), there is substantial uptake of FITC-dextran into the cells (Fig. 1 c). SMase treatment had no effect on cellular ATP levels. To determine whether FITC-dextran is in sealed vesicles, these cells were either chased in dextran-free medium for long periods of time, or exposed to trypan blue, a membrane-impermeant quencher of FITC fluorescence (Hed et al., 1987). FITC-dextran labeling was not reduced after a 60-min chase, and the fluorescence was not quenched by 2 mg/ml trypan blue (data not shown). It has been shown previously that trypan blue is able to quench fluorescence even in deep surface invaginations (Myers et al., 1993), and the lack of quenching is strong evidence for the sealing of the SMase-induced vesicles. As seen in Fig. 1 c, these vesicles remain in the cell periphery, consistent with the requirement for ATP-dependent motor proteins such as dynein for translocation of vesicles along microtubules into the center of a cell (Holzbaur and Vallee, 1994). When ATP is restored to the cells, these vesicles move rapidly into the center (see below).
Figure 1.
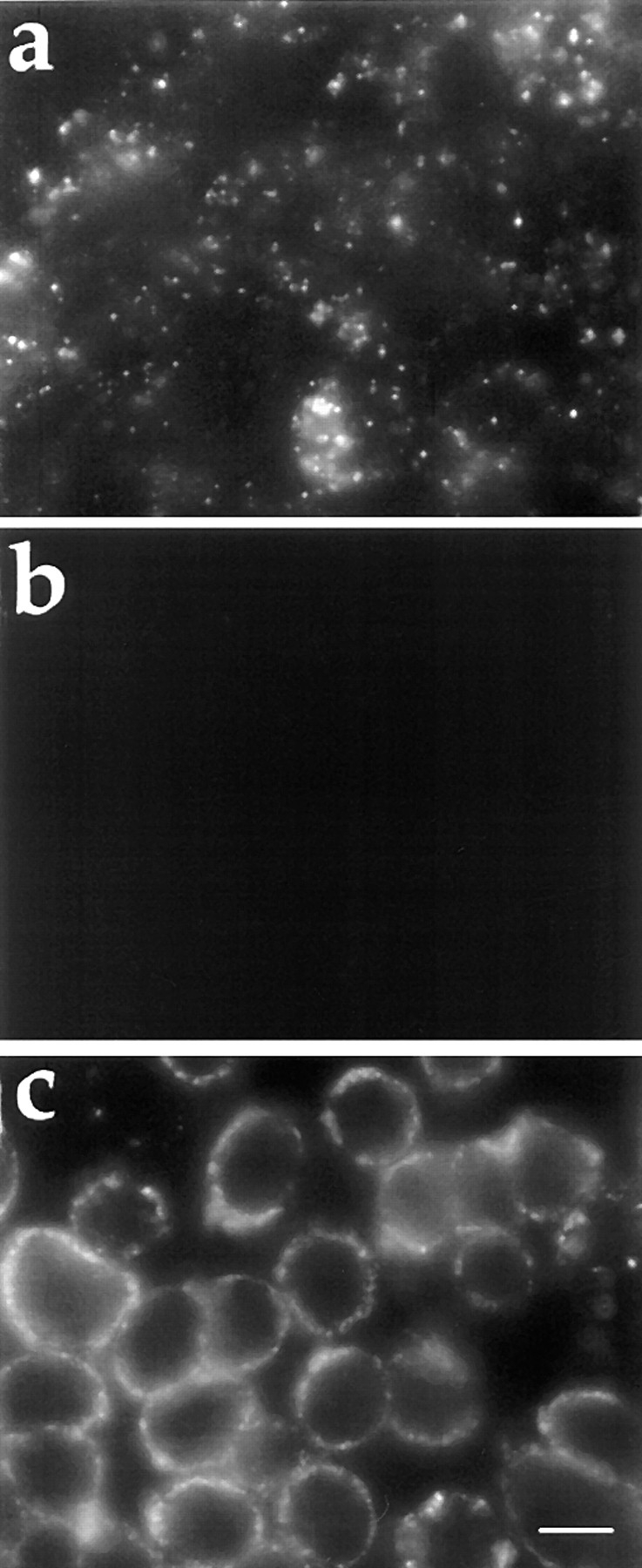
Fluorescence microscopy of FITC-dextran endocytosis in J774 macrophages. J774 macrophages were preincubated for 5 min in 0.2% BSA/DME alone (a), or containing 100 mM 2-deoxyglucose and 100 mM sodium azide (b and c). The cells were then incubated in the same medium containing 5 mg/ml FITC-dextran alone (a and b), or plus 50 mU/ml SMase (c) for 10 min. Cells were fixed and examined by fluorescence microscopy as described in Materials and Methods. Bar, 10 μm.
To visualize the SMase-induced vesicles by electron microscopy, ATP-depleted J774 cells were incubated for 10 min with HRP and 50 mU/ml SMase. Internalized HRP is visualized after diaminobenzidine treatment as a dark precipitate. As shown in Fig. 2, SMase treatment induces the formation of numerous vesicles that are typically 50–500 nm in diameter and marked by diaminobenzidine reaction product precipitate along the lumenal face of the vesicle membranes. There was not a significant difference in either the size distribution or number of vesicles when the incubation time was reduced from 10 to 2.5 min (data not shown). This suggests that the formation of vesicles is complete in a few min after exposure to SMase. The SMase- induced vesicles exhibit only peripheral staining, and no internal vesicles are seen. We did not observe a discernible coat on the vesicles.
Figure 2.

Electron microscopy of HRP endocytosis. J774 macrophages were preincubated with sodium azide/2-deoxyglucose (100 mM) for 5 min and then incubated in the same medium plus SMase (50 mU/ml) and HRP (10 mg/ml) for 10 min. The cells were fixed, incubated with diaminobenzidine and H2O2, and processed for electron microscopy as described in Materials and Methods. HRP-containing vesicles typically exhibited precipitated diaminobenzidine along the lumenal edge of the compartment (filled arrowheads). Lumenal diaminobenzidine precipitation often formed a horseshoe shape (open arrowheads). A lack of precipitated diaminobenzidine in the center of the compartment indicated a relatively low concentration of soluble protein. The SMase-induced compartments exhibited diameters ranging from 50 to 500 nm. In the presence of energy poisons, SMase-generated compartments exhibited a peripheral distribution in the cytoplasm. Limiting membranes of SMase-generated compartments did not contain discernible coat structures and were not continuous with membranes of any other organelles. Bar, 1 μm.
SMase-induced Vesicle Formation in Fibroblasts
The formation of vesicles in response to SMase is also observed in the TRVb-1 line derived from CHO fibroblasts. An advantage to using CHO cells is that they are less active in fluid uptake compared with macrophages, so the effect of SMase can be examined even in the absence of energy poisons. During a short incubation with FITC-dextran (2.5 min) little uptake is observed in control cells (Fig. 3 a). When SMase is added to these energy-replete cells, there is a burst of vesicle formation (Fig. 3 b), similar to the vesicles induced by SMase treatment of ATP-depleted cells (Fig. 3, c and d). Therefore, SMase-induced vesicle formation does not require ATP depletion, although we have shown that SMase-induced vesicle formation is an ATP-independent event. SMase-induced vesicles in energy-replete TRVb-1 cells (Fig. 3 b) were observed in the cell periphery because of the short incubation (2.5 min); vesicle formation because of SMase is completed within 5 min (earliest time point examined). Electron microscopy using HRP showed that the SMase-induced vesicles in TRVb-1 cells were structurally similar to those in J774 cells (data not shown).
Figure 3.

Fluorescence microscopy of FITC-dextran uptake in TRVb-1 cells. Cells were preincubated for 5 min in 0.2% BSA/ HF-12 medium (a and b), or glucose-free 0.2% BSA/HF-12 plus 5 mM 2-deoxyglucose and 5 mM sodium azide (c and d). The cells were then incubated in the same medium with 5 mg/ml FITC-dextran (a and c) or FITC-dextran plus SMase (50 mU/ml; b and d) for 2.5 min. Cells were then fixed and viewed by fluorescence microscopy. Bar, 10 μm.
To further verify that SMase-induced vesicle formation is indeed ATP independent, TRVb-1 cells were incubated with sodium azide/2-deoxyglucose and then permeabilized with SL-O before being exposed to SMase. Cellular ATP level in these cells after SMase treatment was down to 0.07 ± 0.01% of the level in intact, untreated cells. In response to SMase treatment, the SL-O–permeabilized cells took up a fluid-phase marker, Lucifer yellow in this experiment, into peripheral vesicles (Fig. 4 a). SL-O permeabilization of ATP-depleted cells without SMase did not induce vesicle formation (data not shown). SMase-treated cells were able to retain Lucifer yellow (mol wt 457) after a 2-h chase in a dye-free medium (Fig. 4 b), indicating these vesicles are completely sealed off from the plasma membrane.
Figure 4.

Fluorescence microscopy of Lucifer yellow uptake and retention in SL-O–permeabilized TRVb-1 cells. Cells were preincubated for 10 min in 0.2% BSA/glucose-free HF-12 plus 10 mM 2-deoxyglucose and 10 mM sodium azide. The cells were then chilled to 0°C, treated with SL-O (0.4 U/ml) for 3 min, and followed by three washes with cold PBS. After returning to 37°C for 10 min, cells were exposed to Lucifer yellow (10 mg/ml) with SMase (50 mU/ml) for another 10 min. The cells were either viewed right away (a) or transferred into a medium free of Lucifer yellow for an additional 2 h before viewed by fluorescence microscopy (b). Bar, 10 μm.
To verify that the observed effects of SMase were because of SMase enzyme activity, we tested another SMase from a different bacterial source. SMase from Staphylococcus aureus had the same effect on TRVb-1 cells and J774 cells as the SMase from Bacillus cereus. Furthermore, when the SMase preparation was size-fractionated by fast protein liquid chromatography, the vesicle-forming activity copurified with the SMase activity (data not shown).
Kinetics of SMase-induced Vesicle Formation
To determine the kinetics of SMase-induced vesicle formation, J774 macrophages were incubated with FITC-dextran with or without SMase for different lengths of time, and total dextran uptake per cell was measured by quantitative fluorescence microscopy. Results are shown in Fig. 5. As expected, in energy-replete cells treated with or without SMase (inset), the uptake of dextran increased approximately linearly with time. ATP depletion completely blocked dextran internalization in control cells (closed squares), but SMase treatment of ATP-depleted cells caused a rapid burst of dextran uptake (closed circles) that is complete within 10 min (earliest time point examined). Cells apparently became insensitive to SMase after the initial burst since longer exposure (>10 min) to SMase did not result in additional internalization. FITC-dextran uptake kinetics in TRVb-1 cells were similar to those of J774 cells (data not shown). The amount of FITC-dextran uptake induced by SMase in TRVb-1 cells during a 5-min incubation was equivalent to the fluid uptake by energy- replete cells over a 20–30-min incubation.
Figure 5.

Kinetics of dextran uptake in J774 macrophages. Cells were preincubated for 5 min in 0.2% BSA/DME containing 100 mM azide and 100 mM 2-deoxyglucose, or in the same medium without energy poisons (inset). Cells were then incubated with 5 mg/ml FITC-dextran (▪, ♦) or FITC-dextran plus 50 mU/ml SMase (•, ▾) for times ⩽2 h. The uptake of FITC-dextran was then quantified by fluorescence microscopy as described previously (Dunn and Maxfield, 1990). Each data point presents an average fluorescence intensity from four or five fields of cells (50–70 cells/fields) ± SD. SMase treatment of energy-poisoned cells led to uptake of FITC-dextran that was comparable to the cumulative uptake in control cells after ∼20 min. In energy-replete cells (inset), SMase treatment caused an increase in the initial dextran uptake with a rate of sustained uptake similar to control cells.
Characterization of the Membrane Structures in SMase-induced Vesicles
To determine the extent of plasma membrane internalization caused by SMase treatment, a fluorescent membrane-lipid analogue, C6-NBD-gal, was used to label the plasma membrane of TRVb-1 cells. Cells were incubated on ice with C6-NBD-gal, depleted of ATP, and then treated with or without SMase. As shown in Fig. 6, in the absence of SMase the plasma membrane was relatively uniformly labeled by C6-NBD-gal (a), and a backexchange in BSA-containing medium extracted most of this surface label (b). SMase-treated cells (c) show more punctate labeling than untreated cells. After surface C6-NBD-gal was removed by backexchanging in BSA-containing medium, the peripheral punctate labeling of the SMase-treated cells is retained (d). This retained C6-NBD-gal label resembles the FITC-dextran uptake pattern in SMase-treated cells (Fig. 3 d). The fact that C6-NBD-gal in SMase-induced vesicles is not available for backexchange provides further evidence that these vesicles are completely sealed off from the extracellular environment. After quantifying C6-NBD-gal fluorescence images, we find that 30 ± 5% (average of five fields of cells ± SD) of the plasma membrane C6-NBD-gal is internalized in response to SMase. In parallel experiments using a spectrofluorometer to detect C6-NBD-gal extracted from solubilized cells, we find that 14 ± 7% of the C6-NBD-gal label is resistant to backexchange after treatment with SMase. The difference between the results obtained by fluorometry and digital microscopy may be explained by a small population of cells that take up a large amount of C6-NBD-gal throughout the cell even on ice (c). C6-NBD-gal in these cells can be extracted by backexchange. The fluorescence intensity of these cells, therefore, contributes to the total fluorescence intensity in the spectrophotometric analysis but not the microscopic analysis, and this may account for the difference in the values obtained from these two methods. Nevertheless, the results obtained by both methods show that the amount of membrane internalized is much greater than the area covered by clathrin-coated pits and/or caveolae, which typically comprise only 2–4% of the plasma membrane. The total intensities of the membrane C6-NBD-gal labeling of ATP-depleted cells with or without SMase treatment are not significantly different, indicating that SMase induces endo- but not exovesiculation.
Figure 6.

SMase-induced endovesiculation of the plasma membrane. The plasma membrane of TRVb-1 cells was labeled with the fluorescent lipid analogue C6-NBD-gal. Cells were then energy depleted by incubation with sodium azide/deoxyglucose (5 mM) for 10 min at 37°C. After a subsequent incubation in the same medium with (c and d) or without (a and b) SMase (50 mU/ml), the lipid label was backexchanged by washing with ice-cold medium containing 5% BSA (b and d) or washed with ice-cold medium alone (a and c). In the absence of SMase (a and b), almost all of the C6-NBD-gal is backexchanged, whereas in the presence of SMase, C6-NBD-gal located in peripheral vesicles is not available for backexchange. Bar, 10 μm.
SMase-induced vesicles are formed from part of the plasma membrane, raising the question as to whether they are from some specialized or invaginated plasma membrane regions. To address this, we examined if clathrin or caveolae were preferentially associated with these vesicles. TRVb-1 fibroblasts were treated with SMase in the presence of FITC-dextran, fixed, permeabilized, and stained with antibodies against clathrin or adaptor protein 2 (AP2). We did not find any significant enrichment of either clathrin or AP2 in FITC-dextran–containing vesicles by immunofluorescence microscopy (data not shown). When SMase-treated cells were colabeled with antibody to caveolin/ VIP-21, the major protein in caveolae, there was no significant colocalization between caveolin and FITC-dextran (data not shown). SMase-induced vesicles, therefore, appear to not be enriched in either clathrin-coated pits or caveolae.
Sorting of Membrane and Fluid Content in SMase-induced Vesicles
We next investigated the fate of internalized receptors and the fluid contents of SMase-induced vesicles. J774 cells were preincubated with TRITC-dextran to label the late endosomes and lysosomes. The cells were then energy-poisoned and incubated with SMase and FITC-dextran. After 10 min, cells were rinsed and chased for an additional 60 min either in complete medium that allows restoration of ATP, or in the continued presence of energy poisons. In the continuous presence of energy poisons, SMase-induced vesicles (FITC-dextran–containing vesicles) remained in the cell periphery (Fig. 7 a), well separated from the TRITC-dextran–labeled late endosomes and lysosomes (Fig. 7 b). When ATP was restored, however, the FITC dextran in the SMase-induced vesicles moved into the cell and merged with TRITC-dextran–containing compartments (Fig. 7, c and d), indicating that these vesicles are able to deliver FITC-dextran to the previously formed late endosomes and lysosomes.
Figure 7.

Fusion of SMase-induced vesicles with late endosomes/ lysosomes. J774 macrophages were incubated with TRITC-dextran for 1 h and chased in dextran-free medium for 30 min. The cells were then incubated for 5 min in 0.2% BSA/DME plus 100 mM 2-deoxyglucose/sodium azide, followed by a 10-min incubation in the same medium containing FITC-dextran (5 mg/ml) and SMase (50 mU/ml). After rinsing, the cells were chased for 1 h either in the same medium containing energy poisons (a and b) or in complete medium without energy poisons (c and d). The cells were fixed, and then a pair of fluorescence images of FITC-dextran (a and c) and TRITC-dextran (b and d) were acquired from each field of cells by confocal microscopy. In the continuous presence of energy poisons, the FITC-containing compartments remain in the periphery (a) and do not fuse with the TRITC-dextran– labeled endosomes and lysosomes (b; arrows). In cells chased in complete medium that allows restoration of ATP, there is extensive colocalization between FITC-dextran–containing compartments (c) and TRITC-dextran compartments (d; arrows). Bar, 10 μm.
To test if the SMase-induced vesicles are also able to sort receptors for recycling, Cy3-transferrin was bound onto the surface of ATP-depleted J774 cells. The cells were then treated with SMase in the presence of FITC-dextran. Transferrin that remained on the cell surface was stripped off with a mild acid wash (Salzman and Maxfield, 1988). As shown in Fig. 8, Tf and dextran were initially internalized into the same compartments after SMase treatment (Fig. 8, a and b). When the cells were returned to complete medium in the absence of energy poisons, the Tf (Fig. 8 d) was rapidly sorted away from the FITC-dextran compartments (Fig. 8 c) and returned to the cell surface. Thus, the contents of the SMase-induced vesicles can be sorted with recycling receptors returned to the cell surface and the remaining contents being delivered to late endosomes.
Ceramide Is Included in SMase-induced Vesicles
To understand the mechanism of SMase-induced vesicle formation, it would be useful to know the lipid composition of these vesicles. As a start on these studies, we have used a fluorescent sphingomyelin to determine if ceramide formed from it enters the induced vesicles. We incorporated BODIPY-C12-SM into the plasma membrane of TRVb-1 cells. When cells were exposed to SMase, a significant fraction of the BODIPY fluorescence translocated to the Golgi region (Fig. 9 a). This is consistent with the behavior of fluorescent acyl chain derivatives of ceramide that have been shown to be transported to the Golgi apparatus by a nonvesicular mechanism (Martin and Pagano, 1994). The sphingomyelin analogue itself does not translocate to the Golgi as demonstrated by the cells in Fig. 9 b, which were incubated at 37°C for the same time as the cells in Fig. 9 a, but without SMase exposure. It is evident that under our experimental conditions, SMase hydrolyzed the majority of BODIPY-C12-SM and turned it into BODIPY-C12-ceramide that could translocate to the Golgi. Since the transport of fluorescent ceramides is thought to occur via flipping to the cytoplasmic leaflet and binding to cytoplasmic carrier proteins, it should be blocked by removal of such proteins. This would allow us to observe the presence of the ceramide in SMase-induced vesicles. We permeabilized BODIPY-C12-SM–labeled cells with SL-O to remove cytosol proteins. As shown in Fig. 9 d, SMase treatment in these cells caused BODIPY fluorescence to leave the plasma membrane and, significantly, to concentrate in SMase-induced vesicles that were labeled by rhodamine-dextran (Fig 9 c). Taken together, these studies indicate that a substantial amount of the BODIPY ceramide formed by SMase enters the induced vesicles, and this BODIPY-C12-ceramide can flip to the cytoplasmic leaflet.
Figure 9.

SMase treatment of BODIPY-C12-SM–labeled cells. TRVb-1 cells were labeled with BODIPY-C12-SM on ice for 30 min. Cells were incubated with SMase (50 mU/ml) (A) or without (B) for 5 min. Some of the cells were incubated with SL-O (0.4 U/ml) for 5 min on ice (C and D), washed with cold PBS and transferred to 37°C for 3 min, and then incubated with SMase (50 mU/ml)/ rhodamine-dextran (5 mg/ml) for 5 min. Cells were washed and transferred into HF-12 medium for ∼20 min at room temperature before being viewed by fluorescence microscopy. Rhodamine-dextran–containing vesicles (C) colocalized with vesicles labeled with BODIPY fluorescence (D). Bar, 10 μm.
Discussion
We have shown here that treatment of energy-depleted macrophages and CHO cells with exogenous SMase induces vesicle formation from the plasma membrane. To our knowledge, this is the first instance of an internalization process in living cells that does not require ATP. SMase-induced vesiculation was reported in human erythrocytes, although ATP dependence of the process was not addressed (Allan and Walklin, 1988).
The mechanism by which SMase causes vesicle formation in our system is unclear. One possibility is that the SMase causes an inward deformation of the lipid bilayer. It is known that plasma membrane lipids are asymmetrically distributed with sphingomyelin located mainly in the outer leaflet of the bilayer (Koval and Pagano, 1991). Removal of the head group from sphingomyelin would reduce crowding in the outer leaflet, and the ceramide produced by SMase could flip across the bilayer because of its hydrophobicity (Pagano, 1990). Both of these processes would promote inward curvature (Sheetz and Singer, 1974). Ceramide may form droplets in the hydrophobic core of the plasma membrane in human erythrocytes after SMase treatment (Verkleij et al., 1973), and it can promote lipid fusion in model systems (van Meer, 1993). Contact of ceramide-rich membranes at the neck of an invagination might promote formation of sealed vesicles. Furthermore, it is believed that sphingomyelin preferentially associates with cholesterol, and removal of sphingomyelin from the plasma membrane by SMase may result in the destabilization and subsequent transport of cholesterol to intracellular locations (Slotte and Bierman, 1988). However, it is possible that a substantial component of intracellular cholesterol transport after SMase treatment may also occur by the vesicular carriers described here (Skiba et al., 1996).
Ceramide can also function as a second messenger, triggering a cascade of intracellular events (Obeid and Hannun, 1995). However, it is unlikely that the SMase-induced vesicle formations in our system are related to the signaling functions of ceramide. Ceramide was shown to inhibit both fluid-phase and receptor-mediated endocytosis in a dose-dependent manner (Chen et al., 1995). Interestingly, in the same study SMase was also shown to inhibit endocytosis. This is most likely because of experimental procedures used in this study. The uptake of HRP as an endocytosis marker was measured after a 20-min preincubation with SMase. From our results, we know now that endocytosis should actually be increased within the first 20 min in the presence of SMase and could well be decreased after that because of depletion of sphingomyelin from the plasma membrane.
Endogenous sphingomyelin is believed to be synthesized in the Golgi and transported to the plasma membrane (Mandon et al., 1992). Ceramide produced by exogenous SMase is converted back to sphingomyelin, possibly in the recycling pathway (Kallen et al., 1994). Both processes require ATP. After treating ATP-depleted cells with SMase, sphingomyelin on the plasma membrane would eventually be depleted because of either hydrolysis or internalization. This may explain our observation that uptake of dextran is a time-limited process. Endocytosis reaches its maximum within 10 min after exposure to the enzyme, and further exposure did not generate additional vesiculation (Fig. 3, closed circles). This suggests that the formation of the vesicles after 10 min may be limited by the availability of sphingomyelin on the plasma membrane.
The characterization of endocytosis activated by SMase has several implications. First, our results demonstrate that cellular energy is not directly required for the formation of vesicles that invaginate from the plasma membrane. Since these vesicles apparently have incorporated nonspecific membrane, our results imply that much of the plasma membrane has the information required to recycle receptors and deliver fluid contents to late endosomes. This is consistent with observations that macropinosomes initially contain receptors that are removed from them (Racoosin and Swanson, 1993). The mechanisms for the sorting of the membrane and fluid contents of the SMase-induced vesicles are not known. Secondly, our results indicate that budding and fusion of mammalian cell membranes could occur as a result of altering the lipid composition of the membrane. While budding and fusion in living cells are known to be assisted by proteins such as clathrin and dynamin (Schmid, 1992; Hinshaw and Schmid, 1995), there is increasing evidence that these processes can also be governed by the properties of the lipids. Several diverse biological fusion processes, from sea urchin egg cortical exocytosis to mast cell degranulation, have been shown to be inhibited or enhanced by adding exogenous lipids into the membrane (Chernomordik et al., 1995). It was reported that Semliki Forest virus requires sphingomyelin in the target membrane to fuse (Nieva et al., 1994). It is possible that mammalian cells may use some lipid-modifying enzymes, such as SMase, to change lipid composition locally and, therefore, promote membrane budding and fusion.
The effect of SMase on cholesterol trafficking must now be reexamined. It is well known that SMase treatment can lead to stimulation of cholesterol esterification by transport of free cholesterol from the plasma membrane to intracellular ACAT locations (Slotte and Bierman, 1988). ACAT is thought to be primarily localized in the endoplasmic reticulum, although other intracellular locations have not been excluded. The mechanism by which cholesterol is transported from the plasma membrane to ACAT in response to SMase treatment is not known at present. It is known, however, that energy depletion does not impair this transport (Skiba et al., 1996). How the SMase-induced vesicles are related to the cholesterol translocation is not clear. It is possible that these vesicles are enriched with cholesterol relative to sphingomyelin. This relative enrichment could make cholesterol on these vesicles much more mobile than cholesterol in conventional endosomes, so that it can be easily transferred to cholesterol-poor membranes, such as the endoplasmic reticulum. Thus, the vesicles formed by SMase may act as efficient cholesterol transporters.
Although we used a nonphysiological addition of bacterial enzyme to hydrolyze plasma membrane sphingomyelin, our results may be relevant to several important biological processes. Mammalian cells express acidic and neutral SMases (Das et al., 1984; Merrill and Jones, 1990; Chatterjee, 1993; Hannun, 1994), and activation of mammalian SMases and ceramide production have been implicated in many signal transduction cascades (Hannun, 1994). These regulated SMases were found to be located in either the cytosol or light membrane fraction, possibly the plasma membrane (Liu and Anderson, 1995). Furthermore, a Zn-dependent SMase was recently found to be secreted by several cell lines (Schissel et al., 1996), and it has been found that this secreted Zn-dependent SMase is encoded by the same gene, mRNA, and protein precursor as acid lysosomal SMase (Schissel et al., 1996; Schissel, S., E. Schuchman, K. Williams, and I. Tabas, manuscript submitted for publication). With lipoproteins, treatments such as oxidation or preexposure to phospholipase A2 cause the sphingomyelin to become a good substrate for secreted SMase at pH 7.4 (Callahan et al., 1983; Schissel et al., 1998). Therefore, even though secreted SMase does not cause extensive ceramide production when added to mammalian cells (our unpublished data), a change in sphingomyelin accessibility to the enzyme may occur under some conditions at the plasma membrane. In summary, there are various forms of SMase in the extracellular medium and in the secretory and endocytic compartments of many cells. Although the mechanisms of activation remain to be worked out, it is likely that one or more of these forms of SMase cause significant hydrolysis of sphingomyelin at the plasma membrane. Our data suggest that among other effects, activation of SMase would be expected to cause endovesiculation.
Recent data point to a specific example where this occurs (Grassme et al., 1997). Infection by Neisseria gonorrhoeae requires entry into mucosal epithelial cells (McGee et al., 1983). It was found that invasion of human epithelial cells and primary fibroblasts was accompanied by hydrolysis of phosphatidyl choline (PC) by a PC-specific phospholipase C, and by hydrolysis of sphingomyelin by SMase (Grassme et al., 1997). Approximately 35% of metabolically labeled sphingomyelin was hydrolyzed within 5 min of exposure to the bacteria, and the sphingomyelin hydrolysis did not occur in fibroblasts from Niemann-Pick Disease type A patients. These fibroblasts lack both the lysosomal and secreted forms of the (acid) SMase. The Niemann-Pick A fibroblasts did not internalize Neisseria gonorrhoeae in contrast to normal human fibroblasts. Transient transfection with the SMase gene restored the SMase activity after contact with bacteria, and it also restored the ability to internalize the bacteria in the transfected cells. Inhibition of PC-specific phospholipase C also reversibly blocked bacterially induced SMase activity and uptake of the bacteria. These data suggest that Neisseria gonorrhoeae can activate a signaling cascade through PC-specific phospholipase C that activates the target cell's SMase, resulting in very extensive and rapid conversion of sphingomyelin to ceramide. This SMase activity is required for internalization of the bacteria (Grassme et al., 1997).
Our results present an obvious mechanism for the SMase-dependent uptake of the Neisseria gonorrhoeae bacteria by the nonphagocytic cells. The massive SMase activity would drive endovesiculation that could engulf the bacteria. Further work will be required to determine if the activation of SMase is localized near the site of bacterial uptake or if there is a widespread activation.
Acknowledgments
The authors are grateful to T. McGraw (Cornell University Medical College) for critical reading of the manuscript.
This work was supported by National Institutes of Health (NIH) grants to F.R. Maxfield (DK27083) and to I. Tabas (HL39703 and HL21006). X. Zha was supported by a postdoctoral fellowship from the American Heart Association, New York City Affiliate, and P. Skiba was supported in part by an NIH postdoctoral training grant (5T32HL-07343). P.L. Leopold was supported by an Aaron Diamond Foundation postdoctoral fellowship.
Abbreviations used in this paper
- ACAT
acetyl-CoA-cholesterol acyl transferase
- BODIPY-C12-SM
4,4-difluoro-4-bora-3a,4a-diaza-s-indacene- C12-sphingomyelin
- C6-NBD-gal
N-([6-{7-nitrobenz-2-oxa-1,3-diazol-4-yl} amino] hexanoyl) sphingosyl phosphocholine
- PC
phosphatidyl choline
- SL-O
streptolysin O
- SMase
sphingomyelinase
- Tf
transferrin
- TRITC
tetramethylrhodamine isothiocyanate
Footnotes
Address all correspondence to Frederick R. Maxfield, Department of Biochemistry, Cornell University Medical College, 1300 York Ave., New York, NY 10021. Tel.: (212) 746-6405. Fax: (212) 746-8875. E-mail: frmaxfie@mail.med.cornell.edu
References
- Allan D, Walklin CM. Endovesiculation of human erythrocytes exposed to sphingomyelinase C: a possible explanation for the enzyme-resistant pool of sphingomyelin. Biochim Biophys Acta. 1988;938:403–410. doi: 10.1016/0005-2736(88)90138-1. [DOI] [PubMed] [Google Scholar]
- Araki N, Johnson MT, Swanson JA. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J Cell Biol. 1996;135:1249–1260. doi: 10.1083/jcb.135.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarek SY, Orci L, Schekman R. Traffic COPs and the formation of vesicle coats. Trends Cell Biol. 1996;6:468–473. doi: 10.1016/0962-8924(96)84943-9. [DOI] [PubMed] [Google Scholar]
- Callahan JW, Jones CS, Davidson DJ, Shankaran P. The active site of lysosomal sphingomyelinase: evidence for the involvement of hydrophobic and ionic groups. J Neurosci Res. 1983;10:151–163. doi: 10.1002/jnr.490100205. [DOI] [PubMed] [Google Scholar]
- Chatterjee S. Neutral sphingomyelinase. Adv Lipid Res. 1993;26:25–48. [PubMed] [Google Scholar]
- Chen CS, Rosenwald AG, Pagano RE. Ceramide as a modulator of endocytosis. J Biol Chem. 1995;270:13291–13297. doi: 10.1074/jbc.270.22.13291. [DOI] [PubMed] [Google Scholar]
- Chernomordik LV, Kozlov MM, Zimmerberg J. Lipid in biological membrane fusion. J Membr Biol. 1995;146:1–14. doi: 10.1007/BF00232676. [DOI] [PubMed] [Google Scholar]
- Das DVM, Cook HW, Spence MW. Evidence that neutral sphingomyelinase of cultured neuroblastoma cells is oriented externally on the plasma membrane. Biochim Biophys Acta. 1984;777:339–342. doi: 10.1016/0005-2736(84)90437-1. [DOI] [PubMed] [Google Scholar]
- Dunn, K.W., and F.R. Maxfield. 1990. Use of fluorescence microscopy in the study of receptor-mediated endocytosis. In Noninvasive Techniques in Cell Biology. J.K. Foskett, and S. Grinstein, editors. Wiley-Liss, Inc., New York. 153–176.
- Grassme H, Gulbins E, Brenner B, Ferlinz K, Sandhoff K, Harzer K, Lang F, Meyer TF. Acidic sphingomyelinase mediates entry of Neisseria gonorrhoeae into non-phagocytic cells. Cell. 1997;91:605–615. doi: 10.1016/s0092-8674(00)80448-1. [DOI] [PubMed] [Google Scholar]
- Gronberg L, Ruan ZS, Bittman R, Slotte JP. Interaction of cholesterol with synthetic sphingomyelin derivatives in mixed monolayers. Biochemistry. 1991;30:10746–10754. doi: 10.1021/bi00108a020. [DOI] [PubMed] [Google Scholar]
- Hannun YA. The sphingomyelin cycle and the second messenger function of ceramide. J Biol Chem. 1994;269:3125–3128. [PubMed] [Google Scholar]
- Hed J, Haliden G, Johanson SGO, Lerssen F. The use of fluorescence quenching in flow cytometry to measure the attachment and ingestion phases in phagocytosis in peripheral blood without prior cell separation. J Immunol Methods. 1987;101:119–125. doi: 10.1016/0022-1759(87)90224-9. [DOI] [PubMed] [Google Scholar]
- Hinshaw JE, Schmid SL. Dynamin self-assembles into rings suggesting a mechanism for coated vesicle budding. Nature. 1995;374:190–192. doi: 10.1038/374190a0. [DOI] [PubMed] [Google Scholar]
- Holzbaur EL, Vallee RB. Dyneins: molecular structure and cellular function. Annu Rev Cell Biol. 1994;10:339–372. doi: 10.1146/annurev.cb.10.110194.002011. [DOI] [PubMed] [Google Scholar]
- Jovin, T.M., and D.J. Arndt-Jovin. 1989. FRET microscopy: digital imaging of fluorescence resonance energy transfer. Application in cell biology. In Cell Structure and Function by Microspectrofluorometry. E. Kohen and J.G. Hirschberg, editors. Academic Press, San Diego, CA. 99–117.
- Kallen KJ, Allan D, Whatmore J, Quinn P. Synthesis of surface sphingomyelin in the plasma membrane recycling pathway of BHK cells. Biochim Biophys Acta. 1994;1191:52–58. doi: 10.1016/0005-2736(94)90232-1. [DOI] [PubMed] [Google Scholar]
- Knoll W, Schmidt G, Ibel K, Sackmann E. Small-angle neutron scattering study of lateral phase separation in dimyristoylphosphatidylcholine-cholesterol mixed membranes. Biochemistry. 1985;24:5240–5246. doi: 10.1021/bi00340a043. [DOI] [PubMed] [Google Scholar]
- Koval M, Pagano RE. Intracellular transport and metabolism of sphingomyelin. Biochim Biophys Acta. 1991;1082:113–125. doi: 10.1016/0005-2760(91)90184-j. [DOI] [PubMed] [Google Scholar]
- Ktistakis NT, Brown HA, Waters MG, Sternweis PC, Roth MG. Evidence that phospholipase D mediates ADP ribosylation factor- dependent formation of Golgi membrane vesicles. J Cell Biol. 1996;134:295–306. doi: 10.1083/jcb.134.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Anderson RG. Compartmentalized production of ceramide at the cell surface. J Biol Chem. 1995;270:27179–27185. doi: 10.1074/jbc.270.45.27179. [DOI] [PubMed] [Google Scholar]
- Mandon EC, Ehses I, Rother J, van Echten G, Sandhoff K. Subcellular location and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase, and sphinganine N-acyltransferase in mouse liver. J Biol Chem. 1992;267:11144–11148. [PubMed] [Google Scholar]
- Martin OC, Pagano RE. Internalization and sorting of a fluorescent analogue of glucosylceramide to the Golgi apparatus of human skin fibroblasts: utilization of endocytic and nonendocytic transport mechanisms. J Cell Biol. 1994;125:769–81. doi: 10.1083/jcb.125.4.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor S, Presley JF, Maxfield FR. Sorting of membrane components from endosomes and subsequent recycling to the cell surface occurs by a bulk flow process. J Cell Biol. 1993;121:1257–1269. doi: 10.1083/jcb.121.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee, Z.A., D.S. Stephens, L.H. Hoffman, W.F.R. Schiech, and R.G. Hom. 1983. Mechanism of mucosal invasion by pathogenic Neisseria. Rev. Infect. Dis. 5 (Suppl.):708–714. [DOI] [PubMed]
- McGraw TE, Greenfield L, Maxfield FR. Functional expression of the human transferrin receptor cDNA in Chinese hamster ovary cells deficient in endogenous transferrin receptor. J Cell Biol. 1987;105:207–214. doi: 10.1083/jcb.105.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill AH, Jones DD. An update of the enzymology and regulation of sphingomyelin metabolism. Biochim Biophys Acta. 1990;1044:1–12. doi: 10.1016/0005-2760(90)90211-f. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Ghosh RN, Maxfield FR. Endocytosis. Physiol Rev. 1997;77:759–803. doi: 10.1152/physrev.1997.77.3.759. [DOI] [PubMed] [Google Scholar]
- Myers JN, Tabas I, Jones NL, Maxfield FR. β-very low density lipoprotein is sequestered in surface-connected tubules in mouse peritoneal macrophages. J Cell Biol. 1993;123:1398–1402. doi: 10.1083/jcb.123.6.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieva JL, Bron R, Corver J, Wilschut J. Membrane fusion of Semliki Forest virus requires sphingolipids in the target membrane. EMBO (Eur Mol Biol Organ) J. 1994;13:2797–2804. doi: 10.1002/j.1460-2075.1994.tb06573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeid LM, Hannun YA. Ceramide: a stress signal and mediator of growth suppression and apoptosis. J Cell Biochem. 1995;50:191–198. doi: 10.1002/jcb.240580208. [DOI] [PubMed] [Google Scholar]
- Pagano RE. Lipid trafficking in eukaryotic cells: mechanisms for intracellular transport and organell-specific enrichment of lipids. Curr Opin Cell Biol. 1990;2:652–663. doi: 10.1016/0955-0674(90)90107-p. [DOI] [PubMed] [Google Scholar]
- Racoosin EL, Swanson JA. Macropinosome maturation and fusion with tubular lysosomes in macrophages. J Cell Biol. 1993;121:1011–1020. doi: 10.1083/jcb.121.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackmann E. Membrane bending energy concept of vesicle- and cell-shapes and shape transitions. FEBS (Fed Eur Biochem Sci) Lett. 1994;346:3–16. doi: 10.1016/0014-5793(94)00484-6. [DOI] [PubMed] [Google Scholar]
- Salzman NH, Maxfield FR. Intracellular fusion of sequentially formed endocytic compartments. J Cell Biol. 1988;104:1083–1091. doi: 10.1083/jcb.106.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schissel SL, Schuchman EH, Williams KJ, Tabas I. Zn2+-stimulated sphingomyelinase is secreted by many cell types and is a product of the acid sphingomyelinase gene. J Biol Chem. 1996;271:18431–18436. doi: 10.1074/jbc.271.31.18431. [DOI] [PubMed] [Google Scholar]
- Schissel, S., X. Jiang, J. Tweedie-Hardman, T. Jeong, E. Hurt Camejo, J. Najib, J. Rapp, K. Williams, and I. Tabas. 1998. Secretory sphingomyelinase, a product of the acid sphingomyelinase gene, can hydrolyze atherogenic lipoproteins at neutral pH. Implications for atherosclerotic lesion development. J. Biol. Chem. 273:In press. [DOI] [PubMed]
- Schmid SL. The mechanism of receptor-mediated endocytosis: more questions than answers. Bioessays. 1992;14:589–596. doi: 10.1002/bies.950140903. [DOI] [PubMed] [Google Scholar]
- Schmid SL, Smythe E. Stage-specific assays for coated pit formation and coated vesicles budding in vitro. J Cell Biol. 1991;114:869–880. doi: 10.1083/jcb.114.5.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheetz MP, Singer SJ. Biological membranes as bilayer couples. A molecular mechanism of drug-erythrocyte interactions. Proc Natl Acad Sci USA. 1974;71:4457–4461. doi: 10.1073/pnas.71.11.4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd PR, Reaves BJ, Davidson HW. Phosphoinositide 3-kinases and membrane traffic. Trends Cell Biol. 1996;6:92–97. doi: 10.1016/0962-8924(96)80998-6. [DOI] [PubMed] [Google Scholar]
- Silverstein SC, Steinman RM, Cohn ZA. Endocytosis. Annu Rev Biochem. 1977;46:669–722. doi: 10.1146/annurev.bi.46.070177.003321. [DOI] [PubMed] [Google Scholar]
- Skiba PJ, Zha X, Maxfield FR, Schissel SL, Tabas I. The distal pathway of lipoprotein-induced cholesterol esterification, but not sphingomyelinase-induced cholesterol esterification, is energy-dependent. J Biol Chem. 1996;271:13392–13400. doi: 10.1074/jbc.271.23.13392. [DOI] [PubMed] [Google Scholar]
- Slotte JP, Bierman EL. Depletion of plasma membrane sphingomyelin rapidly alters the distribution of cholesterol biosynthesis in cell cultures. Biochem J. 1988;250:650–658. doi: 10.1042/bj2500653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotte JP, Harmala AS, Jansson C, Porn MI. Rapid turn-over of plasma membrane sphingomyelin and cholesterol in baby hamster kidney cells after exposure to sphingomyelinase. Biochim Biophys Acta. 1990;1030:251–257. doi: 10.1016/0005-2736(90)90301-4. [DOI] [PubMed] [Google Scholar]
- van Meer G. Transport and sorting of membrane lipids. Curr Opin Cell Biol. 1993;5:661–673. doi: 10.1016/0955-0674(93)90137-f. [DOI] [PubMed] [Google Scholar]
- Verkleij AJ, Zwaal RFA, Roelofsen B, Comfurius P, Kastelijn D, van Deenen LLM. The assymmetric distribution of phospholipids in the human red cell membrane. A combined study using phospholipases and freeze-etch electron microscopy. Biochim Biophys Acta. 1973;323:178–193. doi: 10.1016/0005-2736(73)90143-0. [DOI] [PubMed] [Google Scholar]