

Abstract
Phospholipase D (PLD) enzymes catalyze the hydrolysis of phosphatidylcholine and are involved in membrane trafficking and cytoskeletal reorganization. The Saccharomyces cerevisiae SPO14 gene encodes a PLD that is essential for meiosis. We have analyzed the role of PLD in meiosis by examining two mutant proteins, one with a point mutation in a conserved residue (Spo14pK→ H) and one with an amino-terminal deletion (Spo14pΔN), neither of which can restore meiosis in a spo14 deletion strain. Spo14pK→ H is enzymatically inactive, indicating that PLD activity is required, whereas Spo14pΔN retains PLD catalytic activity in vitro, indicating that PLD activity is not sufficient for meiosis. To explore other aspects of Spo14 function, we followed the localization of the enzyme during meiosis. Spo14p is initially distributed throughout the cell, becomes concentrated at the spindle pole bodies after the meiosis I division, and at meiosis II localizes to the new spore membrane as it surrounds the nuclei and then expands to encapsulate the associated cytoplasm during the formation of spores. The catalytically inactive protein also undergoes relocalization during meiosis; however, in the absence of PLD activity, no membrane is formed. In contrast, Spo14pΔN does not relocalize properly, indicating that the failure of this protein to complement a spo14 mutant is due to its inability to localize its PLD activity. Furthermore, we find that Spo14p movement is correlated with phosphorylation of the protein. These experiments indicate that PLD participates in regulated membrane formation during meiosis, and that both its catalytic activity and subcellular redistribution are essential for this function.
Phospholipases play a central role in cell signaling by generating lipid second messengers in response to a wide variety of stimuli. Phosphatidylcholine-specific phospholipase D (PLD)1 catalyzes the hydrolysis of phosphatidylcholine (PC) to produce phosphatidic acid (PA) and choline. PA can modulate the activity of a variety of regulatory proteins in vitro, including protein kinases (Bocckino et al., 1991), lipid kinases (Moritz et al., 1992), protein phosphatases (Zhao et al., 1993), and the neutrophil respiratory burst NADPH oxidase (Bellavite et al., 1988). Consequently, PA is believed to be the primary signaling molecule generated by PLD-catalyzed hydrolysis of PC in vivo. In addition, PA can be metabolized to form diacylglycerol (DAG), a well-characterized activator of protein kinase C (PKC) (Nishizuka, 1995), and lyso-phosphatidic acid, a potent mitogen that acts on specific cell surface receptors (Guo et al., 1996). The generation of these signaling molecules via PLD hydrolysis of PC may be important for sustained cellular responses (Exton, 1994). Since PC is the major phospholipid component of all cellular membranes, the intracellular location of PLD-generated second messengers may govern the nature of the response elicited by different stimuli.
Recent studies have suggested that PLD activation is directly involved in membrane trafficking (Ktistakis et al., 1996) and cytoskeletal reorganization (Cross et al., 1996). PLD is thought to function in regulated vesicular movement either by activating a downstream effector essential for trafficking and/or by altering the local structural characteristics of membranes (Liscovitch and Cantley, 1995). In support of this latter hypothesis, Ktistakis et al. (1996) have shown a direct requirement for the production of PA by PLD in the in vitro formation of coated vesicles from mammalian Golgi cisternae. In yeast, DAG appears to be the critical lipid for secretion through the Golgi complex (Kearns et al., 1997). PLD has also been proposed to regulate reorganization of the actin cytoskeleton by activating the small GTP-binding protein, Rho (Cross et al., 1996). However, the physiological role of PLD activation in these processes is unclear.
Sporulation in the yeast Saccharomyces cerevisiae is a program of cellular differentiation analogous to gametogenesis in vertebrates. Yeast cells induced to sporulate undergo meiosis, which consists of a single round of DNA replication followed by two successive rounds of chromosome segregation. The four haploid nuclei are then enveloped by an internal membrane followed by spore wall formation. This double-layered membrane is thought to arise from the fusion of vesicles near the meiosis II spindle pole bodies (Byers, 1981), the yeast equivalent of vertebrate centrosomes. Genetic and cytological analyses of secretory mutants in sporulation suggest that the vesicles that fuse to form the membrane are derived from the Golgi complex (Neiman, 1998). However, little is known about what distinguishes spore membrane formation from other membrane trafficking events and how the haploid nuclei and associated cytoplasm are encapsulated within the body of the mother cell.
The Saccharomyces cerevisiae SPO14 gene is essential for meiosis; spo14 mutants enter meiosis and complete meiotic prophase, but a large number of cells are unable to progress through both of the meiotic divisions, and none form spores (Honigberg et al., 1992; Rose et al., 1995). We have previously shown that SPO14 encodes a PLD (Rose et al., 1995; Ella et al., 1996; Waksman et al., 1996). Genetic analysis of a mutation that renders the protein catalytically inactive indicates that the essential function of Spo14p is the hydrolysis of PC (Sung et al., 1997). In this study we show that the NH2-terminal region of the protein is required for proper localization of Spo14p to the developing membrane, which forms around the haploid meiotic nuclei and is a substrate for phosphorylation. Furthermore, cells expressing a catalytically inactive protein fail to form the membrane. Taken together, our results indicate that localized PLD activity is essential for elaboration of this new internal membrane.
Materials and Methods
Strains and Genetic Procedures
Genotypes of yeast strains are listed in Table I. KR52-3C (Rose et al., 1995) and NH144 (Hollingsworth et al., 1995) have been described. Yeast manipulations were performed and media were prepared using standard procedures (Rose et al., 1990). Yeast transformations were carried out by the lithium acetate procedure (Ito et al., 1983).
Table I.
Yeast Strains
| Strain | Genotype | Source | ||
|---|---|---|---|---|
| KR52-3C | HOura3-1leu2arg4-8 thr1-4trp1-1ade2spo14::URA3 | Rose et al., 1995 | ||
| HO ura3-1 leu2 arg4-8 thr1-4 trp1-1 ade2 spo14::URA3 | ||||
| Y501 | KR52-3C, plus pME865 (SPO14 LEU2 CEN) | This study | ||
| Y568 | KR52-3C, plus pME940 (HA-SPO14 LEU2 CEN) | This study | ||
| Y951 | KR52-3C, plus pME1132 (HA-spo14ΔN LEU2 CEN) | This study | ||
| Y795 | KR52-3C, plus pME1043 (HA-spo14-K→ H LEU2 CEN) | This study | ||
| Y603 | KR52-3C, plus pME962 (SPO14 LEU2 2μ) | This study | ||
| Y602 | KR52-3C, plus pME957 (HA-SPO14 LEU2 2μ) | This study | ||
| Y1031 | KR52-3C, plus pME1121 (HA-spo14-ΔN LEU2 2μ) | This study | ||
| NH144 | MATa leu2-k HIS4 ho:: LYS2 ura3 lys2 arg4-Nsp | Hollingsworth et al., 1995 | ||
| MATα leu2::hisG his4-x ho::LYS2 ura3 lys2 ARG4 | ||||
| Y433 | NH144 but homozygous spo14::URA3 | This study | ||
| Y969 | Y433, plus pME1096 (GFP-SPO14 LEU2 2μ) | This study | ||
| Y988 | NH144, plus pME1124 (GFP-spo14-ΔN LEU2 2μ) | This study | ||
| Y989 | Y433, plus pME1124 (GFP-spo14-ΔN LEU2 2μ) | This study | ||
| Y1019 | NH144, plus pME1130 (GFP-spo14-K→ H LEU2 2μ) | This study | ||
| Y1020 | Y433, plus pME1130 (GFP-spo14-K→ H LEU2 2μ) | This study |
Plasmid Constructions
pME865 contains the 6-kb SPO14 complementing sequences derived from pKR325 (Rose et al., 1995) at the XbaI-ApaI sites of pUN105 (Elledge and Davis, 1988). Three copies of the hemagglutinin epitope (HA; Wilson et al., 1984) were introduced after amino acid 72 in the SPO14 open reading frame by inserting the SphI-digested product of PCR amplification (primers 5′HA: AAGCATGGCGAATTCCTGCAGCCCATCT and 3′HA: ATGCATGCAGAGCGTAATCTGGAACGT) using plasmid SK P/X HA (Neiman et al., 1997) as template, into the SphI site of pME865. The resulting plasmid, pME940, was sequenced to determine orientation and verify the open reading frame. Plasmid ME910 contains the lysine to histidine change at amino acid 1098 in Spo14p and was generated by site-directed mutagenesis of pME865 as described (Sung et al., 1997). A three-way ligation with the 2-kb XbaI-SacI fragment from pME940, the 4-kb SacI-ApaI fragment harboring the mutation from pME910, and the 6-kb XbaI-ApaI fragment of pUN105 was performed to create the HA-tagged version of this mutant protein in pME1043. The spo14-ΔN allele was constructed by removing the 0.45-kb EcoRI fragment from pKR325 to generate pME419. The PCR product described above was inserted into the unique SphI site of pME419 to generate pME1104. These sequences were moved into a LEU2 CEN plasmid by inserting the 6.0-kb XbaI-ApaI fragment from pME1104 into the corresponding sites of pUN105 to generate pME1131. Plasmids ME962 (SPO14 LEU2 2μ), ME957 (HA-SPO14 LEU2 2μ), and ME1121 (HA-spo14-ΔN LEU2 2μ) were constructed by inserting the XbaI-XhoI fragment from pME865, pME940, and pME1104, respectively, into the corresponding sites of YEp351 (Hill et al., 1986).
PCR amplification was performed with SK+GFP (Cormack et al., 1996; generously provided by C. DeMattie and J. Konopka, SUNY, Stony Brook) as template to generate a fragment containing the green fluorescent protein (GFP) (Chalfie et al., 1994) flanked with SphI sites (primers GFP5′: ACATGCATGCAAAAGGAGAAGAACTTTTCACT and GFP3′: ACATGCATGCTTGTATAGTTCATCCATGCC). The resulting 700-bp SphI fragment was inserted into the SphI site of pME865 (after amino acid 72 in the SPO14 open reading frame, identical to the position of the HA tag), creating pME1086. Sequence analysis verified orientation and reading frame; complementation analysis indicates that the GFP–Spo14 fusion is functional. This fusion was subcloned into the XbaI and SalI sites of the 2μ plasmid YEp351 on a 6-kb XbaI-XhoI fragment, generating pME1096 (GFP–SPO14 LEU2 2μ). Plasmid ME1124 (GFP–spo14-ΔN LEU2 2μ) was constructed by inserting the GFP amplification product described above into the SphI site of pME1121; the resulting product was sequenced to verify orientation and reading frame. A three-way ligation was performed with the 2.5-kb XbaI-SacI from pME1096, the 4-kb SacI-XhoI fragment from pME1043, and the 6-kb XbaI-SalI fragment of YEp351, generating pME1130 (GFP–spo14-K[H LEU2 2μ]).
Preparation of Particulate and Cytosol Fractions
15 h after transfer to sporulation medium, the time of the meiotic divisions in this strain background, yeast cells were collected by centrifugation at 1,000 g for 6 min. After a wash with distilled water, the cells were resuspended in 20 ml of spheroplast buffer A (200 mM Tris, pH 7.5, 20 mM DTT) and incubated for 15 min at room temperature with gentle agitation. The cells were pelleted by centrifugation at 1,000 g for 6 min and resuspended in 50 ml of spheroplast buffer B (50 mM Tris, pH 7.5, 500 mM potassium chloride, 10 mM DTT). Zymolylase 100T (United States Biological, Swampscott, MA) was added (20 μg/ml, final concentration), and the cells were incubated for 30 min at 30°C with gentle agitation. Spheroplasts were centrifuged at 1,000 g for 6 min, washed once with spheroplast buffer B, and suspended in 1.6 ml ice-cold lysis buffer (10 mM triethanolamine, pH 7.5, 300 mM sorbitol, 2 mM EDTA, 50 mM sodium fluoride, 40 mM β-glycerophosphate, 1 mM DTT, 2 mM PMSF, 2 mM benzamidine, 0.057 U/ml aprotinin, 2.5 μg/ml leupeptin). The cells were incubated for 40 min at 4°C with gentle agitation, and the lysate was adjusted to 1 M sorbitol in 2 ml. Unlysed cells were removed by centrifugation (1,000 g for 6 min at 4°C). The supernatant was centrifuged at 100,000 g for 1 h at 4°C to yield the particulate (pellet) and cytosolic (supernatant) cell fractions. The particulate fraction was washed twice with lysis buffer and suspended in a volume of lysis buffer equal to the volume of cytosol collected. Sample buffer was then added and the samples boiled for 5 min. Equal volumes of each fraction were used for immunoblot analysis.
Immunoprecipitations
Spheroplasts, prepared as described above, were suspended in 6 ml ice-cold immunoprecipitation (IP) lysis buffer (10 mM triethanolamine, pH 7.5, 150 mM sodium chloride, 5 mM EDTA, 5 mM EGTA, 50 mM sodium fluoride, 40 mM β-glycerophosphate, 10 mM sodium pyrophosphate, 1 mM DTT, 2 mM PMSF, 2 mM benzamidine 0.057 U/ml aprotinin, 2.5 μg/ml leupeptin) containing 1% Nonidet P-40 (wt/vol) (BDH Laboratory Supplies, Poole, England). The cells were incubated at 4°C for 40 min with gentle agitation. The lysate was centrifugation at 1,000 g for 6 min at 4°C to remove unlysed cells and large cellular debris. The supernatant was centrifuged at 15,000 g for 30 min at 4°C to yield the Nonidet P-40–soluble (supernatant) fraction. HA-Spo14 variants were immunoprecipitated directly from this fraction using the 12CA5 monoclonal antibody, which recognizes the HA epitope (BAbCO, Richmond, CA). 1-ml aliquots (1 mg total protein) of the Nonidet P-40–soluble fraction were incubated for 1.5 h at 4°C in tubes containing 3 μg of affinity-purified 12CA5. Protein A–agarose was then added (50 μl of a 50% suspension equilibrated in lysis buffer), followed by a further incubation for 1.5 h at 4°C.Immune complexes were washed three times in 1 ml IP lysis buffer (without PMSF, benzamidine, aprotinin, and leupeptin) containing 1% Nonidet P-40 (wt/ vol), three times in 1 ml of IP lysis buffer without detergent, and once in 1 ml of TBS. Protein concentrations were determined (Bradford, 1976) by using BSA as standard.
Phospholipase D Assays of Immunoprecipitated HA-Spo14p
HA-Spo14 variants were immunoprecipitated from Nonidet P-40–soluble fractions of meiotic yeast as described above. The immune complex was then split into two equal halves. One half was suspended in 20 μl 1 × sample buffer and boiled for 5 min, and the remaining half was washed once with 1 ml PLD assay buffer (25 mM Hepes, pH 7.0, 150 mM sodium chloride, 5 mM EGTA, 1 mM EDTA, 40 mM β-glycerophosphate, and 1 mM DTT) and suspended in 50 μl of 2× PLD assay buffer.
The PLD reaction was initiated by addition of 50 μl of lipid vesicles containing 20–200 μM 2-decanoyl-1-(O-[11-{4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionyl}amino]undecyl)-sn-glycero-3-phosphocholine (BODIPY-PC; Molecular Probes, Inc., Eugene, OR) and 10 μM PIP2. Lipid vesicles were prepared by bath sonication of dry lipid films. After incubation for 30 min at 30°C with occasional gentle agitation, the reaction was terminated with the addition of 375 μl of chloroform/ methanol (1:2 vol/vol). Chloroform (125 μl) and 1 M MgCl2 (100 μl ) were then added, and the lipid products of the lower phase were extracted and separated by TLC as described (Rose et al., 1995). The PLD reaction products were viewed by UV, and the bands corresponding to BODIPY-PC and 2-decanoyl-1-(O-[11-{4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionyl}amino]undecyl)-sn-glycero-3-phosphatidate (BODIPY-PA) were scraped from the plates and extracted with methanol. In all cases, BODIPY-PA was the only PLD reaction product observed. The fluorescence of the methanol extracts was determined using a Packard fluorometer (Meriden, CT) at 485 nm excitation and 530 nm emission. Fluorescence of BODIPY-PA was quantified as a percentage of BODIPY-PC. This value was then converted into nanomolar BODIPY-PA per relative protein.
Treatment of Immunoprecipitated Protein with Shrimp Alkaline Phosphatase
Immune complexes subjected to treatment with shrimp alkaline phosphatase were suspended in 15 μl phosphatase buffer (40 mM Hepes, pH 8.0, 10 mM magnesium chloride) and incubated for 30 min at 30°C with 1.5 U of shrimp alkaline phosphatase (United States Biochemical, Cleveland, OH) in the presence or absence of phosphatase inhibitors (10 mM sodium pyrophosphate, 5 mM EGTA, 5 mM EDTA). For immunoblot analysis, the phosphatase reaction was terminated with the addition of 5 μl of 4× sample buffer, and the beads were boiled for 5 min.
To determine if phosphorylation alters the in vitro catalytic activity of Spo14p, the phosphatase reactions were quenched with the addition of l ml ice-cold TBS containing phosphatase inhibitors. The immune complex was then split into two equal halves. One half was suspended in 20 μl 1× sample buffer and boiled for 5 min. The remaining half of the immunocomplex was washed once in 1 ml ice-cold PLD assay buffer supplemented with phosphatase inhibitors and suspended in 50 μl of 2× PLD assay buffer containing phosphatase inhibitors. The PLD reaction was initiated as described above.
In Vivo [32P]PO4 2− Labeling of HA-Spo14p
Sporulation cultures were washed twice with water and once in spheroplast labeling solution (200 mM MES, pH 6, 1 M sorbitol). Cells were then suspended in a total volume of 10 ml spheroplast labeling solution containing 20 mM DTT and 5 mCi [32P]PO4 2−. After 30 min at 30°C, 1 mg of zymolyase 100T was added, and the cells were incubated for a further 30 min at 30°C. [32P]PO4 2−-labeled spheroplasts were washed once in labeling solution and HA-Spo14p was immunoprecipitated directly from the Nonidet P-40–soluble fraction as described above. Immune complexes were boiled in sample buffer for immunoblot analysis.
Immunoblot Analysis
Cell extracts or immunoprecipitates prepared as described above were centrifuged at 16,000 g for 10 min before being subjected to SDS-PAGE on 5% SDS–polyacrylamide gels. Proteins were electrophoretically transferred onto nitrocellulose membranes (pore size: 0.45 mm; Bio-Rad Laboratories, Hercules, CA) for 22–24 h. Blots were blocked by incubation for 2 h at room temperature with 10% nonfat dry milk in TBS with 0.2% Tween-20 (vol/vol). Blots were then washed three times for 10 min with TBS-T (TBS with 0.1% Tween-20 [vol/vol]) and incubated with mAb 12CA5 diluted 1:3,000 in TBS-T with 1% fatty acid–free BSA (wt/vol) for 2 h. After three washes with TBS-T, blots were incubated for 2 h with horseradish peroxidase–conjugated anti–mouse antiserum (Amersham International, Buckinghamshire, England) diluted 1:5,000 in TBS-T with 1% fatty acid–free BSA (wt/vol). After three final washes in TBS-T, proteins on immunoblots were visualized by enhanced chemiluminescence detection on preflashed film. The resulting films were quantitated on an imaging densitometer (model GS-670; Bio-Rad Laboratories).
Cytology
Yeast strains used for cytology were derived from the rapidly sporulating strain SK1 (Kane and Roth, 1974; Hollingsworth et al., 1995) and were grown and sporulated as previously described (Krisak et al., 1994). Living cells were examined by fluorescence microscopy on the fluorescein channel. Double labeling experiments were performed by fixing cells with 3.7% formaldehyde for 10 min at room temperature. Immunofluorescence with antitubulin antibody (YOL1/34; Kilmartin et al., 1982; Accurate Chemical and Scientific Corp., Westbury, NY), and 4′-6′diaminophenylindole (DAPI) staining was performed as described (Pringle et al., 1991).
Cells from strains NH144 and Y433 were sporulated and, at various times, centrifuged, washed in H2O, and prepared for electron microscopy as described (Friesen et al., 1994).
Results
PLD Activity Is Required but Not Sufficient for Meiosis
SPO14 belongs to a gene family with orthologs in vertebrates, plants, and bacteria (Hammond et al., 1995; Rose et al., 1995). Sequence alignments have defined five conserved regions; regions II and IV contain triads of charged amino acids that putatively mediate catalysis (Morris et al., 1996), while the PX domain is postulated to mediate protein–protein interactions (Ponting, 1997; Fig. 1). Mutational analysis has demonstrated that the putative catalytic triads are essential for human PLD1 and Spo14p catalytic activity (Sung et al., 1997). Furthermore, spo14 deletion strains expressing a protein containing an amino acid change in one of these triads, Spo14pK→ H, are unable to sporulate (Sung et al., 1997; Fig. 1), indicating that PLD catalytic activity is essential for Spo14p function.
Figure 1.

Alterations in the Spo14 protein. Boxed regions denote conserved domains in PLD gene family members (PX, Ponting, 1997; I, II, III, and IV, Morris et al., 1996). The two putative catalytic triads and the single amino acid change in Spo14pK→ H are designated. ΔN refers to the NH2-terminal deletion in Spo14pΔN.
In addition to the conserved domains, Spo14p contains a large NH2-terminal extension that does not display similarity to other PLD family members (Fig. 1). To determine the function of this domain, a protein lacking 150 amino acids in the NH2-terminal portion of the protein, Spo14pΔN, was constructed and analyzed. spo14 deletion strains expressing spo14-ΔN are unable to sporulate, indicating that this region is also essential for Spo14p function.
To examine protein and catalytic activity, sequences encoding three epitopes from the influenza virus hemagglutinin protein (HA) were introduced into SPO14, spo14-ΔN and spo14-K→ H. The resulting products, HA-Spo14p, HA-Spo14pΔN, and HA-Spo14pK→ H, respectively, allowed for the specific detection of these proteins with the monoclonal antibody 12CA5. HA-SPO14 CEN (expressed from a low copy centromeric plasmid) enabled a yeast strain deleted for the SPO14 gene (Y568) to sporulate as well as wild-type SPO14 CEN (Y501) (59 vs. 58%, respectively), while neither HA-spo14-ΔN CEN nor HA-spo14K→ H CEN rescued the sporulation defect of the spo14 mutant (Y951, Y795; <0.01% sporulation; Fig. 2 B).
Figure 2.
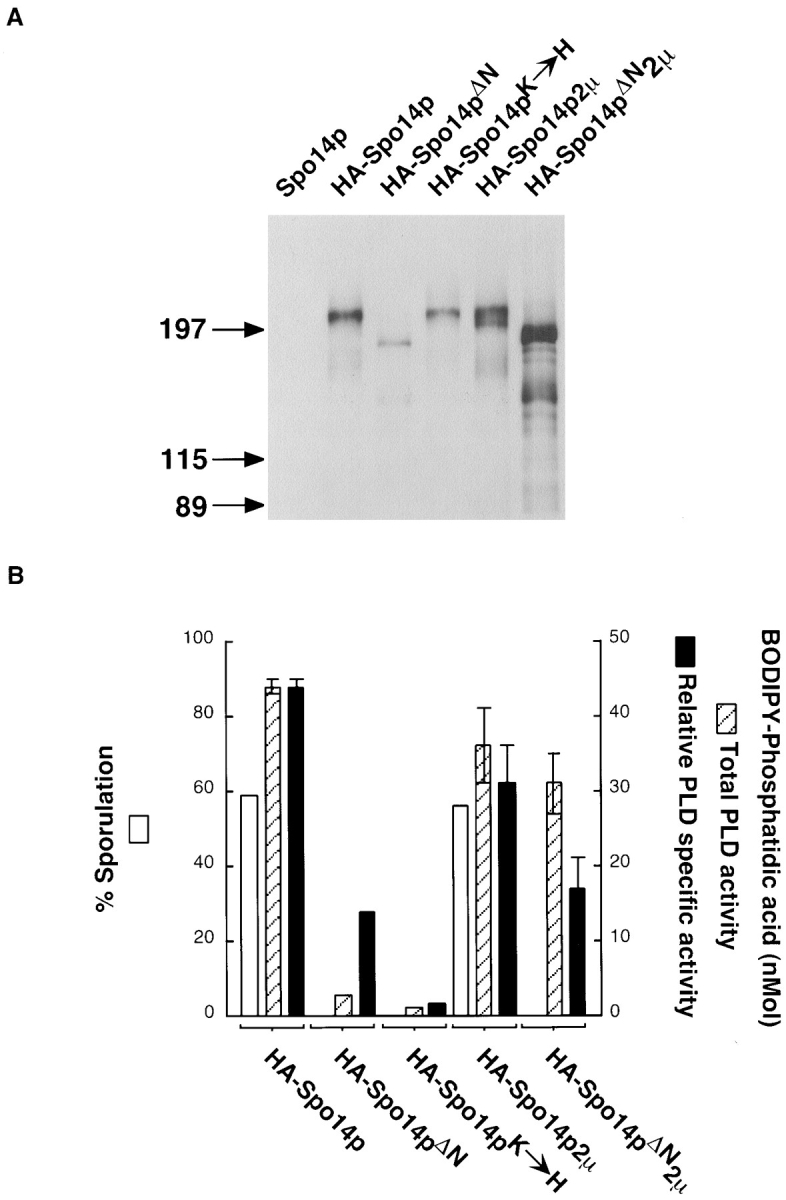
Protein and PLD activity of Spo14 variants. (A) Immunoblot blot analysis of immunoprecipitates from cells harboring SPO14 CEN (lane Spo14p; Y501), HA-SPO14 CEN (lane HA-Spo14p; Y568), HA-spo14-ΔN CEN (lane HA-Spo14pΔN; Y951), HA-spo14-K→ H CEN (lane HA-Spo14pK→ H; Y795), HA-SPO14 2μ (lane HA-Spo14p 2μ; Y602), and HA-spo14-ΔN 2μ (lane HA-Spo14pΔN 2μ; Y1031). The numbers on the left of the immunoblot indicate the positions of molecular mass standards (kD). (B) Sporulation (white bars) and total PLD activity (hatched bars) and relative PLD specific activity (black bars) of the corresponding proteins. PLD assays were performed with vesicles containing 100 μM BODIPY-PC and 5 μM PIP2.
Immunoprecipitations were performed from cells harboring the different Spo14 proteins induced to undergo meiosis. The amount of HA-Spo14p immunoprecipitated represents ∼95% of the total protein expressed. The immunoprecipitates were examined by immunoblot analysis and assayed for PLD activity. As shown in Fig. 2 A, a protein of ∼210 kD is specifically detected in immunoprecipitates from cells expressing HA-SPO14 or HA-spo14-K→ H but not from cells expressing SPO14 without the HA sequences. A protein of ∼190 kD is immunoprecipitated from cells expressing HA-spo14-ΔN. The predicted molecular masses of the proteins are 195 kD (Spo14p, Spo14pK→ H; Rose et al., 1995) and 180 kD (Spo14pΔN). While all three proteins are synthesized, the amount of protein and PLD activity immunoprecipitated from the different strains was not equal. There was approximately fivefold less protein and 15-fold less PLD activity immunoprecipitated from cells harboring HA-Spo14pΔN compared with HA-Spo14p (Fig. 2). When protein amounts are taken into account, the specific PLD activity of HA-Spo14pΔN is decreased approximately threefold compared with the wild-type protein. In contrast, the relative specific PLD activity of HA-Spo14pK→ H is 30-fold less than the wild-type protein (Fig. 2; Sung et al., 1997).
To determine if HA-spo14-ΔN is unable to rescue the sporulation defect of spo14 null mutants because there is less protein and PLD activity, HA-spo14-ΔN was expressed from a 2μ plasmid, which is present at high copy number within yeast cells (for review see Broach and Volkert, 1991). Cells expressing HA-spo14-ΔN from a 2μ plasmid were unable to sporulate (Y1031; <0.01% sporulation), while cells expressing HA-SPO14 from a 2μ plasmid sporulated as efficiently as when these sequences were expressed on a centromere plasmid (56 vs. 59% sporulation for strain Y602 and Y568, respectively; Fig. 2 B). The catalytically inactive protein expressed from a 2μ plasmid also failed to complement the sporulation defect of spo14 null mutants (data not shown). Immunoblot analysis and activity assays revealed that approximately nine times more full-length protein and ten times more PLD activity were immunoprecipitated from strains harboring HA-spo14-ΔN 2μ compared with strains harboring HA-spo14-ΔN CEN (Fig. 2). A faster-migrating species was also observed in immunoprecipitates from cells expressing this protein from a 2μ plasmid and is probably a degradative product of the full-length HA-Spo14pΔN. Thus, the total amount of PLD activity is similar to the activity immunoprecipitated from cells expressing HA-SPO14 from either a centromere or 2μ plasmid. These results indicate that the inability of spo14-ΔN to rescue the sporulation defect of spo14 mutants is not caused by decreased PLD activity.
Spo14p catalytic activity in vitro is dependent on the lipid cofactor, phosphatidylinositol 4,5-bisphosphate (PIP2; Rose et al., 1995). In this respect, Spo14p is similar to the mammalian orthologs (Hammond et al., 1995; Colley et al., 1997). No PLD activity was detected in the absence of PIP2, indicating that like Spo14p, Spo14pΔN is PIP2 dependent (data not shown). Therefore, the inability of cells expressing HA-spo14-ΔN to sporulate is unlikely to be a consequence of a change in the protein's ability to respond to its putative in vivo activator, PIP2.
Cell fractionation experiments were performed to determine the subcellular location of Spo14p in meiosis. HA-Spo14p is found almost exclusively in the particulate fraction (96%; Fig. 3), suggesting that Spo14p is associated with membranes or exists in a large protein complex. HA-Spo14pK→ H also partitioned almost exclusively to the particulate fraction (92%; Fig. 3). In contrast, Spo14pΔN is found in nearly equivalent amounts in the cytosol (42%) and the particulate fractions (58%; Fig. 3), raising the possibility that the failure of this protein to rescue a spo14 null allele is due to mislocalization.
Figure 3.

Biochemical fractionation of Spo14 proteins. Immunoblot of particulate (P) and cytosolic (C) fractions prepared from yeast cells harboring HA-SPO14 CEN (Y568), HA-spo14-ΔN CEN (Y951), and HA-spo14-K→ H CEN (Y795). The numbers on the left of the immunoblot indicate the positions of molecular mass standards (kD).
Spo14p Relocalizes during Meiosis
A GFP–Spo14 fusion was constructed, and its localization was examined in living yeast cells. GFP–Spo14p is fully functional; this fusion protein expressed from either a centromere or a 2μ plasmid enables a spo14 null mutant to sporulate as efficiently as wild type. Although GFP–Spo14p expressed from a centromere plasmid rescued the sporulation defect of spo14 null mutants, we were unable to detect a fluorescent signal; however, a specific signal was detected when this fusion was expressed from a 2μ plasmid. The top panel of Fig. 4 shows representative cells expressing GFP–SPO14 before induction of meiosis (0 hr) and throughout meiosis (6–10 hr). In vegetative cells and early in meiosis, GFP–Spo14p appears to be dispersed in the cytoplasm (Fig. 4, 0 hr). As meiosis progresses, GFP–Spo14p converges into discrete foci (Fig. 4, 6 hr), expands into ringlike structures (Fig. 4, 8 hr), and eventually enlarges to outline the mature spore (Fig. 4, 10 hr).
Figure 4.


Relocalization of GFP–Spo14p during meiosis. (Top) Micrographs of living yeast cells harboring GFP–SPO14 2μ (Y969) at the indicated times after induction of meiosis. (Middle) A fixed cell after the meiosis I division showing GFP–Spo14p (Spo14p), antitubulin antibody (Tubulin) and DAPI (DNA) labeling. A schematic representation of the cell is on the right. The circles represent Spo14p, lines represent the spindles, and the ovals represent DNA. The dashed line is the nuclear envelope. (Bottom) A cell expressing GFP–Spo14p (Spo14p) after the meiosis II division stained with DAPI (DNA).
Double labeling experiments with the DNA-specific dye DAPI indicate that Spo14p begins to converge into specific foci at the meiosis I division (Fig. 4, middle). At meiosis II, these foci expand into circles that encompass the four-lobed nucleus, which appear as elongated rings (Fig. 4, bottom). The spindle pole bodies are the sites of membrane formation (Byers, 1981); therefore, we labeled yeast cells harboring GFP–Spo14p with a monoclonal antibody directed against tubulin. The discrete foci of GFP–Spo14p after the meiosis I division colocalize with the ends of the spindle, presumably at the spindle pole bodies (Fig. 4, middle).
Localized PLD Activity Is Required for Membrane Formation
GFP derivatives of Spo14pΔN and Spo14pK→ H were constructed and examined in wild-type and spo14 deletion mutants. In contrast to the wild-type protein, GFP– Spo14pΔN remains dispersed in the cytoplasm throughout meiosis when expressed in either wild-type or mutant cells (Fig. 5, top). Thus, consistent with the biochemical fractionation, the failure of this protein to complement a spo14 deletion strain is most likely due to its inability to relocalize during meiosis.
Figure 5.


Visualization of GFP–Spo14pΔN and GFP–Spo14pK→ H during meiosis. (Top) Live cells expressing GFP–Spo14pΔN (live; Y988) and a fixed cell expressing GFP–Spo14pΔN (Spo14pΔN), labeled with antitubulin antibodies (Tubulin) and DAPI (DNA). (Middle) spo14 mutant cells expressing GFP–Spo14pK→ H (Y1020) showing GFP (Spo14pK→ H ), antitubulin antibody (Tubulin) and DAPI (DNA) labeling. (Bottom) Wild-type cells expressing GFP–Spo14pK→ H (Spo14pK→ H; Y1019) late in meiosis.
Examination of GFP–Spo14pK→ H in wild-type and spo14 deletion cells revealed that this protein relocalized to the spindle pole bodies after the meiosis I division in the majority of the cells examined (Fig. 5, middle). However, while membranes were observed in wild-type cells expressing GFP–Spo14pK→ H (Fig. 5, bottom), no membrane structures were observed in spo14 deletion cells expressing GFP–Spo14pK→ H.
To confirm that in the absence of PLD activity no spore membrane is formed, we examined thin sections of yeast cells induced to sporulate by electron microscopy. In wild-type cells, the spore membrane and spore wall layers were observed readily as discrete compartmentalized entities within the body of the mother cell (Fig. 6 A). In contrast, in greater than 100 cells examined, no compartmentalization was observed in spo14 null mutants, indicating that no membrane nor spore wall is formed (Fig. 6 B). Taken together, these results indicate that relocalization of PLD activity is essential for the formation of the spore membrane that encompasses the haploid meiotic nuclei.
Figure 6.

Electron microscopy of sporulated SPO14/ SPO14 (NH144) and spo14/ spo14 (Y433) cells. (A) A typical wild-type cell late in sporulation (12 h) showing three spore-like compartments. (B) A typical spo14 cell at the same time showing three nuclei but no compartmentalization. SC, spore compartment; N, nucleus. Bars, 500 nm.
Phosphorylation of Spo14p Correlates with Relocalization
Immunoblot analysis of whole cell extracts derived from cells harboring HA-Spo14p revealed that Spo14p changes electrophoretic mobility during meiosis (Fig. 7 A). The shift in apparent molecular mass of HA-Spo14p during meiosis suggests that the protein is posttranslationally modified. Phosphorylation is a common protein modification and has been shown to affect enzymatic activity (Liu and Simon, 1996) and protein localization (Keranen et al., 1995). To determine if Spo14p is a phosphoprotein, immunoprecipitates of HA-Spo14p were performed from meiotic cells that were labeled in vivo with [32P]orthophosphate. Label was specifically incorporated into HA-Spo14p (Fig. 7 B), indicating that Spo14p is phosphorylated during meiosis.
Figure 7.
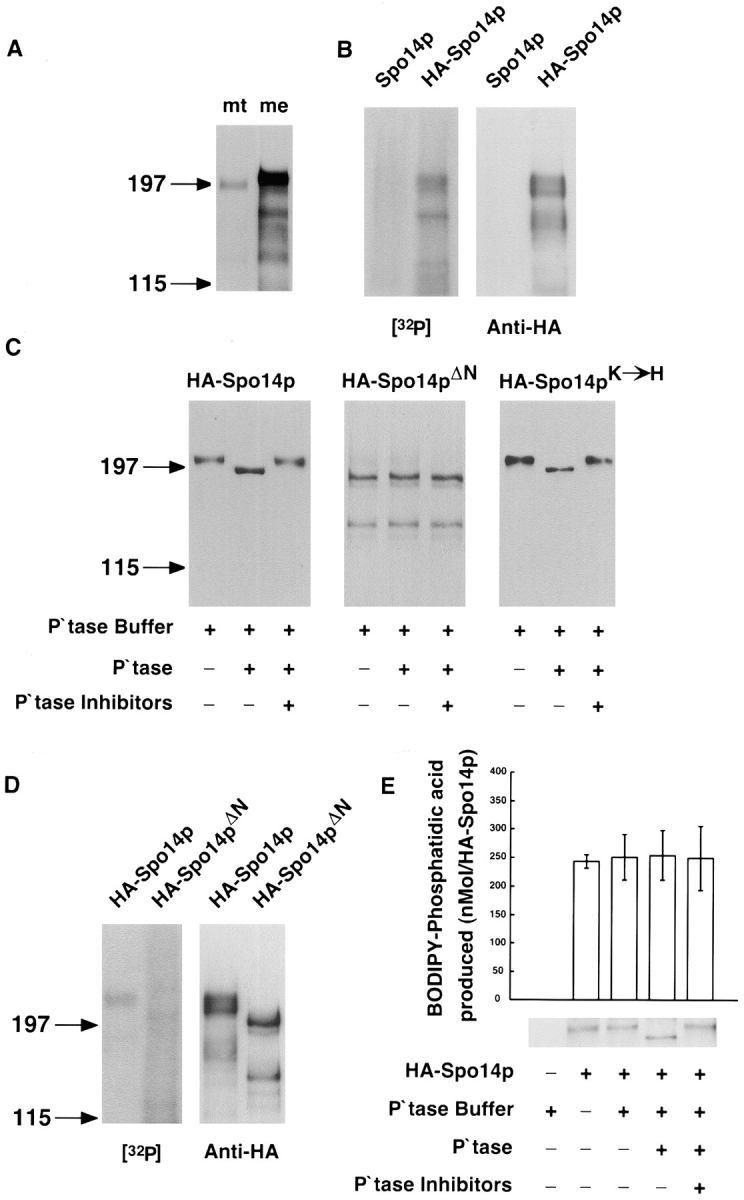
Phosphorylation of Spo14 proteins during meiosis. (A) Immunoblot of Y568 whole cell protein extracts containing HA-Spo14p prepared from mitotically dividing cells (lane mt) and midway through meiosis (lane me). (B) Autoradiograph of immunoprecipitates derived from meiotic yeast cells harboring either Spo14p 2μ (Y603) or HA-Spo14p 2μ (Y602) labeled with 32P ([32P]) and the corresponding immunoblot (Anti-HA). (C) Immunoblot of immunoprecipitated HA-Spo14p (Y568), HA-Spo14pΔN (Y951), and HA-Spo14pK→ H (Y795) from meiotic yeast cells treated with either shrimp alkaline phosphatase buffer (P\Qtase buffer) or shrimp alkaline phosphatase (P\Qtase) with or without phosphatase inhibitors (P\Qtase Inhibitors). (D) Autoradiograph of immunoprecipitates derived from 32P-labeled meiotic yeast cells harboring either HA-Spo14p 2μ (Y602) or HA-Spo14pΔN 2μ (Y1031) ([32P]) and the corresponding immunoblot (Anti-HA). (E) PLD activity and immunoblot of immunoprecipitated meiotic HA-Spo14p (Y568) treated with shrimp alkaline phosphatase in the presence and absence of phosphatase inhibitors. PLD assays were performed with vesicles containing 10 μM BODIPY-PC and 5 μM PIP2. The numbers on the left of the immunoblot indicate the positions of molecular mass standards (kD).
To determine whether phosphorylation is altered in the mutant proteins, HA-Spo14p, HA-Spo14pΔN, and HA-Spo14pK→ H were immunoprecipitated from yeast cells induced in meiosis. The immunoprecipitates were incubated with alkaline phosphatase and analyzed by immunoblotting. Treatment of the immunoprecipitated products with alkaline phosphatase converted HA-Spo14p and HA-Spo14pK→ H to faster migrating species; the presence of phosphatase inhibitors prevented the shift in mobility (Fig. 7 C). The dephosphorylated species migrated faster than the proteins derived from mitotic cells (data not shown); these results suggest that more than one residue on Spo14p is phosphorylated, and at least one of these sites is specifically modified during meiosis. In contrast to HA-Spo14p and HA-Spo14pK→ H, treatment of immunoprecipitated HA-Spo14pΔN with alkaline phosphatase did not result in a change in electrophoretic mobility (Fig. 7 C). To confirm that phosphorylation is altered in Spo14pΔN, we labeled HA-Spo14p and HA-Spo14pΔN in vivo with [32P]orthophosphate. Consistent with the analysis of phosphatase sensitivity, approximately seven-fold less label was incorporated into HA-Spo14pΔN, compared with HA-Spo14p (Fig. 7 D). In addition, analysis of the phosphorylation state of LexA–Spo14 fusion proteins in meiosis indicates that the majority of phosphorylated residues map to the NH2-terminal region defined by the Spo14pΔN deletion (data not shown).
To determine if phosphorylation alters the in vitro catalytic activity of Spo14p, we performed PLD assays on the immunoprecipitated protein before and after treatment with alkaline phosphatase. As shown in Fig. 7 E, in vitro PLD activity was unaffected by the phosphorylation state of the protein. Taken together, these results suggest that phosphorylated residues in the 150 amino acids defined by the deletion are important for relocalization.
Discussion
In this paper, we have shown that the localization of Spo14p is specifically altered during meiosis and that both proper redistribution and catalytic activity are required for the formation of the internal spore membrane. The meiosis-specific relocalization of Spo14p appears to be mediated by the nonconserved NH2-terminal portion of the protein and correlates with phosphorylation. However, while this region of the protein is necessary for relocalization, we have not demonstrated sufficiency. These results emphasize the importance of subcellular localization and posttranslational modification of PLD in regulating cellular differentiation.
Spo14p is present and active in mitotically dividing cells as measured by an in vitro assay; however, no effect on vegetative growth has been detected in spo14 deletion strains (Rose et al., 1995). The diffuse staining observed in mitotically dividing cells suggests that while active, the protein does not have access to its lipid cofactor, PIP2, or substrate, PC, and consequently does not generate PA or other biologically active lipids. Consistent with this idea, Spo14p isolated from mitotically dividing cells and early in meiosis is not solubilized with nonionic detergent at concentrations that are known to release membrane-bound proteins, suggesting that the particulate nature of Spo14p is due to association with a large protein complex such as the cytoskeleton. However, at the time of the meiotic divisions, Spo14p is readily solubilized by such treatment, indicating that it becomes associated with cellular membranes (Rudge, S.A., and J. Engebrecht, unpublished data). Thus, it seems likely that meiotic development triggers activation of Spo14p by relocalizing it to membranes.
Recent work has suggested a role for PLD activity in yeast Golgi function during mitotic growth (Patton-Vogt et al., 1997). Such a role was uncovered in a multigenic mutant and raises the possibility that Spo14p can be localized to Golgi membranes during mitotic growth. In fact, it is possible that Spo14p initially moves to the Golgi before movement to the spindle pole bodies during meiosis.
The relocalization of proteins to their lipid substrate is a common theme for the activation of proteins involved in signal transduction. For example, thrombin provokes the translocation of p110 phosphatidylinositol-3 kinase (Zhang et al., 1992) and phosphatidylinositol 4-phosphate 5-kinase (Hinchliffe et al., 1996) to the membrane cytoskeleton of platelets. Moreover, phospholipase Cγ translocates from the cytosol to the membrane fraction of HER14 cells in response to epidermal growth factor and platelet-derived growth factor, and this relocalization appears to be promoted by tyrosine phosphorylation (Kim et al., 1990).
Protein kinase C βII also undergoes translocation from a detergent-insoluble to a detergent-soluble cell fraction that contains its lipid cofactors, phosphatidylserine and diacylglycerol (Keranen et al., 1995). Recently, at least two classes of PKC-binding proteins have been identified that are not substrates for the kinase (receptors for activated C-kinase and proteins that interact with C-kinase; for review see Faux and Scott, 1996). These proteins are believed to participate in PKC targeting. Therefore, PKC translocation involves not only protein–lipid interactions but also protein–protein interactions. During meiosis, Spo14p relocalization to its membrane target might also involve binding to both its phospholipid substrate and cofactor, and to a putative Spo14p receptor protein.
What kinase(s) are responsible for phosphorylating Spo14p? Phosphatase sensitivity and in vivo labeling experiments suggest that there is more than one residue that is modified by phosphorylation. Whether a single kinase or multiple kinases are responsible for these phosphorylation events remains to be determined. Preliminary data suggest that phosphorylation occurs predominantly on serine and/or threonine residues (Rudge, S.A., and J. Engebrecht, unpublished data). In the region defined by Spo14pΔN, two of the serine and threonine residues conform to the consensus sequence for PKC, a known activator of mammalian PLD (Conricode et al., 1992; Lopez et al., 1995; Singer et al., 1996; Hammond et al., 1997). As stated above, mammalian PKC translocates from a detergent- insoluble cell fraction to a detergent-soluble fraction upon activation (Keranen et al., 1995). Therefore, yeast Pkc1p might phosphorylate Spo14p and translocate with it upon activation. Expression of an activated allele of PCK1 (PCK1-R398P; Nonaka et al., 1995) in mitotically dividing cells did not result in phosphorylation or relocalization of Spo14p, indicating that Pkc1p is not the kinase responsible for these events (Rudge, S.A., and J. Engebrecht, unpublished). Moreover, expression of PCK1-R398P in vegetative cells did not alter Spo14p catalytic activity. However, we cannot rule out the possibility that Pkc1p itself is specifically activated during meiosis and consequently promotes the phosphorylation and relocalization of Spo14p.
The localization pattern of Spo14p is similar to that of Spr3p, Cdc3p, and Cdc10p, yeast septins that play partially redundant roles during the process of spore formation (Fares et al., 1996). However, while spo14 mutants are defective in meiosis and do not synthesize the spore membrane, septin mutants are only partially defective in spore formation. Furthermore, Spo14p is found uniformly around the growing membrane, while the septins appear to define the leading edge of the spore membrane.
Examination of GFP–Spo14 fusions was only possible when these sequences were expressed on 2μ plasmids, which exist in multiple copies per cell (Broach and Volkert, 1991). Expression of the wild-type protein on a 2μ plasmid is not deleterious, indicating that there is no gross effect of overexpression. While we can not eliminate the possibility that overexpression alters the localization pattern, the relocalization of GFP–Spo14p is consistent with biochemical fractionation studies performed on cells containing endogenous levels of protein.
The results presented here suggest that PLD participates in regulated membrane formation during meiosis. This could occur in several ways. First, Spo14p may generate a lipid required for the formation of the internal spore membrane. Generation of PA or DAG could alter the characteristics of membranes to promote membrane curvature, similar to what is postulated to underlie the generation of Golgi vesicles (Ktistakis et al., 1996; Kearns et al., 1997). This could be important in the process of encapsulation of the nuclei and perhaps also for the generation of the vesicles that fuse to form the spore membrane. Alternatively, Spo14p could synthesize another lipid important for membrane formation. PLD has been proposed to synthesize bisphosphatidic acid, which is formed through transphosphatidylation when DAG is used as a nucleophile donor instead of water (van Blitterswijk and Hilkmann, 1993). A derivative of this lipid, semilysobisphosphatidic acid, has been shown to be a component of Golgi membranes and is also predicted to induce membrane curvature (Cluett and Machamer, 1996). Cluett and Machamer (1996) hypothesize that this lipid may be important for stabilizing membranes during vesicle budding and fusion events, both of which are likely to be essential for spore membrane formation.
Second, localized PLD activity may generate PA or DAG as a signal to activate downstream effectors important for membrane formation. This may lead to cytoskeletal reorganization, similar to what is observed in the formation of actin stress fibers (Cross et al., 1996). Cytoskeletal components are likely to mediate alignment of vesicles for directing membrane formation around the nuclear envelope. Third, PLD may generate lipids for both activating downstream effectors and membrane formation. Future studies should define which of the putative lipid products of PLD mediate the formation of this internal membrane and coordinate its synthesis to the nuclear events of meiosis.
Acknowledgments
We thank L. Davis, M. Frohman, A. Nieman, S. Strickland, and J. Trimmer (SUNY, Stony Brook) and C. Machamer (Johns Hopkins University, Baltimore, MD) for helpful discussions and comments on the manuscript.
This work was supported by National Institutes of Health Grants GM4863903 (J. Engebrecht), GM50388 and GM54641 (A.J. Morris), and a Catacosinos Young Investigator Award to J. Engebrecht. S.A. Rudge had an Affiliate Fellowship from New York State Heart Association (Grant 950209).
Abbreviations used in this paper
- BODIPY-PA
2-decanoyl-1-(O-[11-{4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3- propionyl}amino] undecyl]-sn-glycero-3-phosphatidate
- BODIPY-PC
2-decanoyl-1-(O-[11-{4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3- propionyl}amino] undecyl)-sn-glycero-3-phosphocholine
- DAG
diacylglycerol
- DAPI
4′-6′diaminophenylindole
- GFP
green fluorescent protein
- HA
hemagglutinin
- PA
phosphatidic acid
- PC
phosphatidylcholine
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PKC
protein kinase C
- PLD
phospholipase D
Footnotes
Address all correspondence to JoAnne Engebrecht, Department of Pharmacological Sciences, State University of New York, Stony Brook, Stony Brook, NY 11794-8651. Tel.: (516) 444-7815. Fax: (516) 444-3218. E-mail: joanne@pharm.sunysb.edu
References
- Bellavite P, Corso F, Dusi S, Grzeskowiakk M, Bella-Bianca V, Rossi F. Activation of NADPH-dependent superoxide production in plasma membrane extracts of pig neutrophils by phosphatidic acid. J Biol Chem. 1988;263:8210–8214. [PubMed] [Google Scholar]
- Bocckino SB, Wilson PB, Exton JH. Phosphatidate-dependent protein phosphorylation. Proc Natl Acad Sci USA. 1991;88:6210–6213. doi: 10.1073/pnas.88.14.6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilising the principle of protein-dye binding. Anal Biochem. 1976;76:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Broach, J.R., and F.C. Volkert. 1991. Circular DNA plasmids of yeasts. In The Molecular and Cellular Biology of the Yeast Saccharomyces. Genome Dynamics, Protein Synthesis, and Energetics. J.R. Broach, J.R. Pringle, and E.W. Jones, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 297–331.
- Byers, B. 1981. Cytology of the yeast life cycle. In The Molecular Biology of the Yeast Saccharomyces. Life Cycle and Inheritance. J.N. Strathern, E.W. Jones, and J.R. Broach, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 59–96.
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- Cluett EB, Machamer CE. The envelope of vaccinia virus reveals an unusual phospholipid in Golgi complex membranes. J Cell Sci. 1996;109:2121–2131. doi: 10.1242/jcs.109.8.2121. [DOI] [PubMed] [Google Scholar]
- Colley WC, Sung T-C, Roll R, Jenco J, Hammond SM, Altshuller Y, Bar-Sagi D, Morris AJ, Frohman MA. Phospholipase D2, a PLD1-related isoform with novel regulatory properties and discrete subcellular localization that provokes cytoskeletal reorganization. Curr Biol. 1997;7:191–201. doi: 10.1016/s0960-9822(97)70090-3. [DOI] [PubMed] [Google Scholar]
- Conricode KM, Brewer KA, Exton JH. Activation of phospholipase D by protein kinase C. Evidence for a phosphorylation-independent mechanism. J Biol Chem. 1992;267:7199–7202. [PubMed] [Google Scholar]
- Cormack BP, Valdivis RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- Cross MJ, Roberts S, Ridley AJ, Hodgkin MN, Stewart A, Claesson-Welsh L, Wakelam MJO. Stimulation of actin stress fibre formation mediated by activation of phospholipase D. Curr Biol. 1996;6:588–597. doi: 10.1016/s0960-9822(02)00545-6. [DOI] [PubMed] [Google Scholar]
- Ella KM, Dolan JW, Qi C, Meier KE. Characterization of Saccharomyces cerevisiaedeficient in expression of phospholipase D. Biochem J. 1996;314:15–19. doi: 10.1042/bj3140015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge SJ, Davis RW. A family of versatile centromeric vectors designed for use in the sectoring-shuffle mutagenesis assay in Saccharomyces cerevisiae. . Gene. 1988;70:303–312. doi: 10.1016/0378-1119(88)90202-8. [DOI] [PubMed] [Google Scholar]
- Exton JH. Phosphatidylcholine breakdown and signal transduction. Biochim Biophys Acta. 1994;1212:26–42. doi: 10.1016/0005-2760(94)90186-4. [DOI] [PubMed] [Google Scholar]
- Fares H, Goetsch L, Pringle JR. Identification of a developmentally regulated septin and involvement of the septins in spore formation in Saccharomyces cerevisiae. . J Cell Biol. 1996;132:399–411. doi: 10.1083/jcb.132.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faux MC, Scott JD. More on target with protein phosphorylation: conferring specificity by location. Trends Biol Sci. 1996;4:312–315. [PubMed] [Google Scholar]
- Friesen H, Lunz R, Doyle S, Segall J. Mutation of the SPS1- encoded protein kinase of Saccharomyces cerevisiaeleads to defects in transcription and morphology during spore formation. Genes Dev. 1994;8:2162–2175. doi: 10.1101/gad.8.18.2162. [DOI] [PubMed] [Google Scholar]
- Guo Z, Liliom K, Fischer DJ, Bathurst IC, Tomei LD, Kiefer MC, Tigyi G. Molecular cloning of a high-affinity receptor for the growth factor-like lipid mediator lysophosphatidic acid from Xenopusoocytes. Proc Natl Acad Sci USA. 1996;93:14367–14372. doi: 10.1073/pnas.93.25.14367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond SM, Altshuller YM, Sung T-C, Rudge SA, Rose K, Engebrecht J, Morris AJ, Frohman MA. Cloning of mammalian ARF-activated phosphatidylcholine-specific phospholipase D define a new and highly conserved family of genes. J Biol Chem. 1995;270:29640–29643. doi: 10.1074/jbc.270.50.29640. [DOI] [PubMed] [Google Scholar]
- Hammond SM, Jenco JM, Nakashima S, Cadwallader K, Gu Q, Cook S, Nozawa Y, Prestwich GD, Frohman MA, Morris AJ. Characterization of two alternatively spliced forms of phospholipase D1. J Biol Chem. 1997;272:3860–3868. doi: 10.1074/jbc.272.6.3860. [DOI] [PubMed] [Google Scholar]
- Hill JE, Myers AM, Koerner TJ, Tzagoloff A. Yeast/E. colishuttle vectors with multiple unique restriction sites. Yeast. 1986;2:163–167. doi: 10.1002/yea.320020304. [DOI] [PubMed] [Google Scholar]
- Hinchliffe KA, Irvine RF, Divecha N. Aggregation-dependent, integrin-mediated increases in cytoskeletally associated PtdIns (4,5)P2levels in human platelets are controlled by translocation of PtdIns 4-P 5-kinase C to the cytoskeleton. EMBO (Eur Mol Biol Organ) J. 1996;15:6516–6524. [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth NM, Ponte L, Halsey C. MSH5, a novel MutS homolog, facilitates meiotic reciprocal recombination between homologs in Saccharomyces cerevisiaebut not mismatch repair. Genes Dev. 1995;9:1728–1739. doi: 10.1101/gad.9.14.1728. [DOI] [PubMed] [Google Scholar]
- Honigberg SM, Conicella C, Esposito RE. Commitment to meiosis in Saccharomyces cerevisiae: involvement of the SPO14gene. Genetics. 1992;130:703–716. doi: 10.1093/genetics/130.4.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Fukada Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane S, Roth J. Carbohydrate metabolism during ascospore development in yeast. J Bacteriol. 1974;118:8–14. doi: 10.1128/jb.118.1.8-14.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns BG, McGee TP, Mayinger P, Gedvilaite A, Phillips SE, Kagiwada S, Bankaitis VA. Essential role for diacylglycerol in protein transport from the yeast Golgi complex. Nature. 1997;387:101–105. doi: 10.1038/387101a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keranen LM, Dutil EM, Newton AC. Protein kinase C is regulated in vivoby three functionally distinct phosphorylations. Curr Biol. 1995;5:1394–1403. doi: 10.1016/s0960-9822(95)00277-6. [DOI] [PubMed] [Google Scholar]
- Kilmartin JV, Wright B, Milstein C. Rat monoclonal antitubulin antibodies derived by using a new nonsecreting rat cell line. J Cell Biol. 1982;93:576–582. doi: 10.1083/jcb.93.3.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim UH, Kim HS, Rhee SG. Epidermal growth factor and platelet-derived growth factor promote translocation of phospholipase C-γ from cytosol to membrane. FEBS Lett. 1990;270:33–36. doi: 10.1016/0014-5793(90)81228-g. [DOI] [PubMed] [Google Scholar]
- Krisak L, Strich R, Winters RS, Hall JP, Mallory MJ, Kreitzer D, Tuan RS, Winters E. SMK1, a developmentally regulated MAP kinase, is required for spore wall assembly in Saccharomyces cerevisiae. . Genes Dev. 1994;10:2151–2161. doi: 10.1101/gad.8.18.2151. [DOI] [PubMed] [Google Scholar]
- Ktistakis NT, Brown HA, Waters MG, Sternweis PC, Roth MG. Evidence that phospholipase D mediates ADP ribosylation factor– dependent formation of Golgi coated vesicles. J Cell Biol. 1996;134:295–306. doi: 10.1083/jcb.134.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscovitch M, Cantley LC. Signal transduction and membrane traffic: the PITP/phosphoinositide connection. Cell. 1995;81:659–662. doi: 10.1016/0092-8674(95)90525-1. [DOI] [PubMed] [Google Scholar]
- Liu M, Simon M. Regulation of cAMP-dependent protein kinase of a G-protein–mediated phospholipase C. Nature. 1996;382:83–87. doi: 10.1038/382083a0. [DOI] [PubMed] [Google Scholar]
- Lopez I, Burns DJ, Lambeth JD. Regulation of phospholipase D by protein kinase C in human neutrophils. J Biol Chem. 1995;270:19465–19472. doi: 10.1074/jbc.270.33.19465. [DOI] [PubMed] [Google Scholar]
- Moritz A, Graan PND, Gispen WH, Wirtz KW. Phosphatidic acid is a specific activator of phosphatidylinositol-4-phosphate kinase. J Biol Chem. 1992;267:7207–7210. [PubMed] [Google Scholar]
- Morris AJ, Engebrecht J, Frohman MA. Structure and regulation of phospholipase D. Trends Pharm Sci. 1996;17:182–185. doi: 10.1016/0165-6147(96)10016-x. [DOI] [PubMed] [Google Scholar]
- Neiman AM. Prospore membrane formation defines a developmentally regulated branch of the secretory pathway in yeast. J Cell Biol. 1998;140:29–37. doi: 10.1083/jcb.140.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neiman AM, Mhaiskar V, Manus V, Galibert F, Dean N. Saccharomyces cerevisiae HOC1, a suppressor of pkc1, encodes a putative glycosyltransferase. Genetics. 1997;145:637–645. doi: 10.1093/genetics/145.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB (Fed Eur Soc Exp Biol) J. 1995;9:484–496. [PubMed] [Google Scholar]
- Nonaka H, Tanaka K, Hirano H, Fujiwara T, Kohno H, Umikawa M, Mino A, Takai Y. A downstream target of RHO1 small GTP-binding protein is PKC1, a homolog of protein kinase C, which leads to activation of the MAP kinase cascade in Saccharomyces cerevisiae. . EMBO (Eur Mol Biol Organ) J. 1995;14:5931–5938. doi: 10.1002/j.1460-2075.1995.tb00281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton-Vogt JL, Griac R, Sreenivas A, Bruno V, Dowd S, Swede MJ, Henry SA. Role of the yeast phosphatidylinositol/phosphatidylcholine transfer protein (Sec14p) in phosphatidylcholine turnover and INO1regulation. J Biol Chem. 1997;272:20873–20883. doi: 10.1074/jbc.272.33.20873. [DOI] [PubMed] [Google Scholar]
- Ponting CP. Novel domains in NADPH oxidase subunits, sorting nexins, and PtdIns 3-kinases: binding partners of SH3 domains? . Protein Sci. 1997;5:2353–2357. doi: 10.1002/pro.5560051122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle JR, Adams AEM, Drubin DG, Haarer BK. Immunofluorescence methods for yeast. Methods Enzymol. 1991;194:565–602. doi: 10.1016/0076-6879(91)94043-c. [DOI] [PubMed] [Google Scholar]
- Rose K, Rudge SA, Frohman MA, Morris AJ, Engebrecht J. Phospholipase D signaling is essential for meiosis. Proc Natl Acad Sci USA. 1995;92:12151–12155. doi: 10.1073/pnas.92.26.12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, M.D., F. Winston, and P. Hieter. 1990. Methods in Yeast Genetics: Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory, NY. 198 pp.
- Singer WD, Brown HA, Bokoch GM, Sternweis PC. Resolved phospholipase D activity is modulated by cytosolic factors other than Arf. J Biol Chem. 1996;270:14944–14950. doi: 10.1074/jbc.270.25.14944. [DOI] [PubMed] [Google Scholar]
- Sung T-C, Roper K, Zhang Y, Rudge S, Temel R, Hammond SM, Morris AJ, Moss B, Engebrecht J, Frohman MA. Mutagenesis of phospholipase D defines a superfamily including a trans-Golgi viral protein required for poxvirus pathogenicity. EMBO (Eur Mol Biol Organ) J. 1997;16:4519–4530. doi: 10.1093/emboj/16.15.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Blitterswijk WJ, Hilkmann H. Rapid attenuation of receptor-induced diacylglycerol and phosphatidic acid by phospholipase D–mediated transphosphatidylation: formation of bisphosphatidic acid. EMBO (Eur Mol Biol Organ) J. 1993;12:2655–2662. doi: 10.1002/j.1460-2075.1993.tb05926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waksman M, Eli Y, Liscovitch M, Gerst JE. Identification and characterization of a gene encoding phospholipase D activity in yeast. J Biol Chem. 1996;271:2361–2364. doi: 10.1074/jbc.271.5.2361. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Niman HL, Houghten RA, Cherenson AR, Connolly ML, Lerner RA. The structure of an antigenic determinant in a protein. Cell. 1984;37:767–778. doi: 10.1016/0092-8674(84)90412-4. [DOI] [PubMed] [Google Scholar]
- Zhang J, Fry MJ, Waterfield MD, Jaken S, Liano L, Fox JEB, Rittenhouse SE. Activated phosphoinositide 3-kinase associates with membrane skeleton in thrombin-exposed platelets. J Biol Chem. 1992;267:4686–4692. [PubMed] [Google Scholar]
- Zhao Z, Shen SH, Fischer EH. Stimulation by phospholipids of a protein-tyrosine-phosphatase containing two src homology 2 domains. Proc Natl Acad Sci USA. 1993;90:4251–4255. doi: 10.1073/pnas.90.9.4251. [DOI] [PMC free article] [PubMed] [Google Scholar]