

Abstract
Previously we have demonstrated that focal adhesion kinase (FAK)-promoted migration on fibronectin (FN) by its overexpression in CHO cells is dependent on FAK autophosphorylation at Y397 and subsequent binding of Src to this site. In this report, we have examined the role of FAK association with Grb2 and p130Cas, two downstream events of the FAK/Src complex that could mediate integrin-stimulated activation of extracellular signal-regulated kinases (Erks). We show that a Y925F FAK mutant was able to promote cell migration as efficiently as FAK and that the transfected FAK demonstrated no detectable association with Grb2 in CHO cells. In contrast, cells expressing a FAK P712/715A mutant demonstrated a level of migration comparable to that of control cells. This mutation did not affect FAK kinase activity, autophosphorylation, or Src association but did significantly reduce p130Cas association with FAK. Furthermore, FAK expression in CHO cells increased tyrosine phosphorylation of p130Cas and its subsequent binding to several SH2 domains, which depended on both the p130Cas binding site and the Src binding site. However, we did not detect increased activation of Erks in cells expressing FAK, and the MEK inhibitor PD98059 did not decrease FAK-promoted cell migration. Finally, we show that coexpression of p130Cas further increased cell migration on FN and coexpression of the p130Cas SH3 domain alone functioned as a dominant negative mutant and decreased cell migration. Together, these results demonstrate that p130Cas, but not Grb2, is a mediator of FAK-promoted cell migration and suggest that FAK/ p130Cas complex targets downstream pathways other than Erks in mediating FAK-promoted cell migration.
Focal adhesion kinase (FAK)1 is a cytosolic protein tyrosine kinase that is critical in integrin-mediated signal transduction pathways (Clark and Brugge, 1995; Schwartz et al., 1995; Parsons, 1996). It is localized to focal contacts in many adherent cells in culture and becomes activated and tyrosine phosphorylated in response to cell adhesion to extracellular matrix (ECM) proteins in a variety of cell types (Schwartz et al., 1995). FAK associates with a number of signal transduction molecules, including Src and related kinases (Cobb et al., 1994; Xing et al., 1994), Grb2 (Schlaepfer et al., 1994), p130Cas (Polte and Hanks, 1995), and phosphatidylinositol 3-kinase (PI 3-kinase) (Chen and Guan, 1994). It also associates with the focal contact and cytoskeletal proteins paxillin (Turner and Miller, 1994; Schaller and Parsons, 1995) and talin (Chen et al., 1995) and is believed to associate with tensin (Miyamoto et al., 1995). Its interactions with these and potentially other proteins mediate FAK's functions in integrin-dependent signal transduction.
Upon activation, FAK becomes autophosphorylated with the major site being Y397, which serves as a binding site for the Src homology 2 (SH2) domain of Src and related kinases (Chan et al., 1994; Schaller et al., 1994) as well as the SH2 domains of the regulatory subunit (p85) of PI 3-kinase (Chen et al., 1996). Upon binding to Y397, Src further phosphorylates FAK at several sites including Y925, which serves as a binding site for the SH2 domain of the Grb2 adaptor protein (Schlaepfer et al., 1994; Schlaepfer and Hunter, 1996). The direct binding of Grb2 to FAK has been proposed to initiate downstream signaling pathways, resulting in activation of extracellular signal-regulated kinases (Erks) (Schlaepfer et al., 1994), which have been shown to be activated in cell adhesion (Chen et al., 1994; Schlaepfer et al., 1994; Zhu and Assoian, 1995).
p130Cas is another signal transduction protein that binds directly to FAK. It is an adaptor protein, containing an Src homology 3 (SH3) domain as well as multiple phosphotyrosine motifs for binding a variety of SH2 domains (Sakai et al., 1994). It is phosphorylated upon cell adhesion to ECM (Nojima et al., 1995; Petch et al., 1995; Vuori and Ruoslahti, 1995) and in a FAK- and Src-dependent manner (Vuori et al., 1996; Schalepfer et al., 1997). The proline-rich region of FAK spanning amino acids 712–718 has been mapped as a binding site for the SH3 domain of p130Cas (Polte and Hanks, 1995; Harte et al., 1996). FAK association with p130Cas has also been proposed to initiate a signal transduction pathway to activate Erks (Schlaepfer et al., 1997).
FAK is clearly an important regulator of integrin signal transduction, and recently its roles in specific cellular functions have begun to be elucidated. It has been proposed to regulate cell spreading because when it is inhibited by expression of its noncatalytic carboxy-terminal domain in chicken embryo fibroblasts, the rate of cell spreading is delayed (Richardson and Parsons, 1996). It has also been suggested to play a role in ECM-mediated apoptosis and cell proliferation (Frisch et al., 1996; Gilmore and Romer, 1996; Hungerford et al., 1996). However, more evidence exists to demonstrate that FAK plays a crucial and specific role in cell migration events. Inhibition of endogenous FAK by microinjection of its carboxy-terminal domain in endothelial cells resulted in decreased cell migration (Gilmore and Romer, 1996). Likewise, cultured fibroblasts isolated from FAK null mice, which demonstrated an embryonic lethal phenotype, showed markedly reduced cell motility in vitro (Ilic et al., 1995). Similarly, we have recently demonstrated that stable overexpression of FAK in CHO cells resulted in increased migration (Cary et al., 1996).
Using the FAK overexpression system in CHO cells has enabled us to examine the molecular mechanisms of FAK signal transduction. We have found that phosphorylation of Y397 of FAK and subsequent Src binding are crucial for its role in signal transduction resulting in cell migration. CHO cells expressing the FAK point mutant Y397F, which did not bind Src, demonstrated a basal level of cell migration, while a kinase-defective FAK mutant that was tyrosine phosphorylated and associated with Src was able to promote CHO cell migration (Cary et al., 1996). Interestingly, Klemke et al. (1997) have recently reported that activation of Erks regulates cell migration on ECM by directly phosphorylating a cytoplasmic target myosin light chain kinase. Because both Grb2 and p130Cas have been proposed to activate Erks in a manner dependent on the FAK/Src complex, we investigated the potential role of FAK association with Grb2 and/or p130Cas in mediating the FAK-promoted cell migration in the present study.
Materials and Methods
Antibodies
The mouse mAb KT3 (Cary et al., 1996), the mouse mAb 12CA5 (Chen et al., 1995), and the rabbit polyclonal anti-FAK serum (Chen and Guan, 1994) have been described previously. The following antibodies were purchased as indicated: rabbit anti-p130Cas (C-20) from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); anti-Src mouse mAb 2-17 from Quality Biotech (Camden, NJ); antiphosphotyrosine mouse mAb PY20 from Transduction Laboratories (Lexington, KY); rabbit anti–phospho-MAPK from New England Biolabs, Inc. (Beverly, MA); and mouse mAb 9E10, which recognizes the c-Myc epitope tag (EQKLISEEDL), from Calbiochem (La Jolla, CA).
Construction of Expression Plasmids
The expression plasmid pCDM8-FAK has been described previously (Cary et al., 1996). The FAK point mutant Y925F was generated using overlap extension by the polymerase chain reaction as described previously (Ho et al., 1989). The sense (5′-CCCACCCTCTTCAGCCTTATG-3′) and antisense (5′-CTTGTACTCCTGCTGCAGGCTGG-3′) oligonucleotides were used as amplification primers, and the mutation was introduced using the sense (5′-ATGACAAAGTCTTTGAGAATGTAAC-3′) and its complementary antisense oligonucleotide as internal mismatch primers. The product of the PCR mutagenesis was then ligated to pBluescript-FAK (a generous gift from Dr. J. Thomas Parsons, University of Virginia, Charlottesville, VA) after digestion of each with NdeI and NheI. Generation of the mutation was verified by dideoxy DNA sequencing (United States Biochemical Corp., Cleveland, OH). The MscI-NheI fragment containing the mutation was then subcloned into pCDM8-FAK, from which the MscI-NheI fragment had been removed, and the resulting plasmid was designated pCDM8-Y925F. The P712/715A double point mutant was similarly generated, using the sense (5′-CCCACCCTCTTCAGCCTTATG-3′) and antisense (5′-CTTGTACTCCTGCTGCAGGCTGG-3′) oligonucleotides as amplification primers, and the sense (5′-CAGATGAAGCTGCTCCCAAGGCCAGCAGGCC-3′) and its complementary antisense oligonucleotide as internal mismatch primers. The PCR product was ligated into pBluescript using EcoRV, the AccI-NheI fragment containing the mutation was subcloned to pBluescript-FAK, and finally the MscI-SalI fragment was ligated into pCDM8-FAK, from which this wild-type sequence had been removed. The resulting plasmid was designated pCDM8-P712/715A.
The pKH3 and pKH3-FAK vectors were described previously (Chen et al., 1996). The SH3 domain of p130Cas was PCR amplified from pVP16-cas 1a-2 (Polte and Hanks, 1995) using the forward and reverse primers 5′-GCGGATCCATGAAGTACCTGAACGTGC-3′ and 5′-GCGAATTCTCCTGCTGGCTTCTTATC-3′. The PCR reaction was carried out using Deep Vent DNA polymerase, digested with BamHI and EcoRI, and cloned into pKH3 to generate pKH3-CasSH3. The hygromycin-resistance vector pREP3 was a generous gift of Dr. Andrew Yen (Cornell University, Ithaca, NY).
Plasmid pRc/CMV-Cas(Myc) was constructed by inserting the full-length mouse Cas cDNA (GenBank/EMBL/DDBJ accession number U28151; Polte and Hanks, 1995) into the NotI site of eukaryotic expression vector pRc/CMV (Invitrogen, Carlsbad, CA). The Cas cDNA was engineered to encode an additional short sequence including the human c-Myc epitope, GREQKLISEEDLRGRLA, inserted two amino acid residues upstream of the Cas carboxyl terminus. A Myc-epitope cassette was generated by annealing partially complementary oligonucleotides (coding: 5′-CTAGCCGGCCGCGAGCAGAAGCTGATCTCCGAGGAGGACCTCCGCGGCCGG-3′; noncoding: 5′-CTAGCCGGCCGCGGAGGTCCTCCTCGGAGATCAGCTTCTGCTCGCGGCCGG-3′) to produce a double-stranded cDNA with overhanging NheI-compatible ends. The cassette was ligated into the NheI site located near the 3′-end of the Cas coding sequence before subcloning into pRc/CMV.
Cells and Transfections
CHO cells were maintained in F-12 medium with 10% FBS (both from GIBCO BRL, Gaithersburg, MD). Cells expressing wild-type (WT), kinase-defective (KD), or Y397F FAK as well as control cells (Neo) have been described previously (Cary et al., 1996). To generate cells stably expressing either the Y925F or P712/715A FAK mutant, CHO cells grown on 35-mm dishes were transfected essentially as described previously (Cary et al., 1996) using lipofectamine (GIBCO BRL) and following the manufacturer's instructions. Briefly, 1 μg of either pCDM8-Y925F or pCDM8-P712/715A and 0.1 μg of pSV2neo were transfected, and neomycin-resistant cells were selected using growth medium containing 0.5 mg/ ml G418 (GIBCO BRL). Clones were selected and screened for exogenous FAK expression by indirect immunofluorescence and Western blotting using the mAb KT3.
To generate cells stably expressing both FAK and wild-type p130Cas (WT/Cas cells), WT cells were transfected as described above using lipofectamine, 1 μg of pRc/CMV-Cas(Myc), and 0.1 μg of pREP3. Clones were selected in growth medium containing 0.5 mg/ml G418 and 0.8 mg/ml Hygromycin B (Sigma Chemical Co., St. Louis, MO) and screened for FAK and p130Cas expression by Western blotting with KT3 and 9E10, respectively. Likewise, cells stably expressing both FAK and CasSH3 (WT/ CasSH3 cells) were generated by transfection with lipofectamine, 1 μg of pKH3-CasSH3, and 0.1 μg of pREP3 and selected as above for WT/Cas cells. Cells were screened for FAK and CasSH3 expression by Western blotting with KT3 and 12CA5, respectively.
Human embryonic kidney 293 cells were maintained in DME with 10% FBS (both from GIBCO BRL). Transient transfection of 293 cells grown on 10-cm dishes was carried out similarly using lipofectamine and 9 μg of either pKH3-FAK or pCDM8-FAK. Lysates were generated from these cells 2 d after transfection.
Immunoprecipitations, Western Blots, and In Vitro Kinase Assays
For most experiments, attached cells were lysed in NP-40 lysis buffer containing protease inhibitors (Cary et al., 1996). For the Erk activation experiment, some cells were harvested by brief treatment with trypsin, which was inhibited by soybean trypsin inhibitor (0.5 mg/ml) and washed, followed by lysis as suspended cells in NP-40 lysis buffer. For coimmunoprecipitation of FAK with endogenous p130Cas, attached cells were lysed in modified RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% NP-40, 1 mM sodium vanadate, 0.5% sodium deoxycholate, and 0.05% SDS) containing protease inhibitors (1 mM PMSF, 0.1 trypsin inhibitory U/ml aprotinin, and 20 μg/ml leupeptin). For detection of FAK-promoted phosphorylation of p130Cas and binding to SH2 domains, cells were treated with 50 μM sodium vanadate (a tyrosine phosphatase inhibitor) for 16 h at 37°C as described previously (Schaller and Parsons, 1995) followed by lysis in NP-40 lysis buffer. Lysates were cleared by centrifugation, and total protein concentrations were determined using Bio-Rad Protein Assay (Hercules, CA).
Immunoprecipitations (Cary et al., 1996) and in vitro kinase assays (Guan and Shalloway, 1992) were carried out as described previously. For coimmunoprecipitation of the p130Cas SH3 domain with FAK, SDS-PAGE was performed under nonreducing conditions to eliminate comigration of the antibody light chain with the SH3 domain. Western blotting was also carried out as before (Guan et al., 1991) using dilutions of 1:2,000 for KT3 mouse ascites fluid, 1:1,000 for anti-p130Cas, 1:1,000 for PY20, 1:600 for 12CA5 mouse ascites fluid, 1 μg/ml for 9E10, 1:5,000 for anti-FAK, and 1:2,000 for anti–phospho-MAPK.
Cell Migration Assays
Migration assays in 48-well chemotaxis chambers (Neuro Probe, Inc., Cabin John, MD) were carried out as described previously (Cary et al., 1996). Briefly, human plasma fibronectin (FN) at various concentrations was added to the lower wells. Cells were harvested by brief trypsinization, washed, and added to the upper wells of the chamber, which were separated from the lower wells by a polycarbonate membrane. Cells were allowed to migrate for 6 h at 37°C. The membrane was fixed and stained, unmigrated cells were removed by wiping the upper side of the membrane, and the migrated cells were counted under a light microscope at 200× magnification. For experiments using the MEK inhibitor PD98059 (New England Biolabs, Inc.), cells were harvested and washed as above, resuspended in F-12 medium containing the indicated concentrations of PD98059, and pretreated for 1 h at 37°C. Without removing the PD98059, the cells were then added to the chemotaxis chamber and allowed to migrate to FN as above.
Expression of Glutathione S-Transferase Fusion Proteins and In Vitro Binding Assays
The pGEX-Src-SH2, pGEX-Grb2-SH2, and pGEX-p85-NSH2 plasmids were described previously (Chen et al., 1996). The pGEX-Nck-SH2 vector was kindly provided by Dr. Wei Li (University of Chicago, Chicago, IL). Expression and affinity purification of glutathione S-transferase (GST) fusion proteins were carried out essentially as described previously (Xing et al., 1994). Briefly, bacteria harboring the indicated expression vector were cultured at 37°C in medium containing 40 μg/ml ampicillin, isopropyl β-d-thiogalactopyranoside (IPTG) was then added to a concentration of 0.15 mM, and the bacteria were cultured for an additional 2 h. Cells were centrifuged, resuspended in PBS containing 1% Triton X-100, and lysed by sonication. The lysates were cleared by centrifugation, and GST fusion proteins were affinity purified using glutathione-Sepharose beads, which were then washed three times and resuspended in NP-40 lysis buffer. For in vitro binding assays, cell lysates in NP-40 lysis buffer were incubated with the indicated GST fusion protein beads for 2–3 h at 4°C, the beads were washed with NP-40 lysis buffer, and bound proteins were eluted by boiling in SDS sample buffer and then detected by Western blotting.
Results
Grb2 Binding to Y925 of FAK Is Not Necessary for FAK-promoted Cell Migration
To examine the potential role of FAK association with Grb2 in its ability to promote cell migration, we created a FAK mutant replacing Y925 with F using site-directed mutagenesis as described in Materials and Methods. Since Y925 of FAK has been mapped as the binding site for Grb2 in cell adhesion (Schlaepfer et al., 1994; Schlaepfer and Hunter, 1996), this mutation (designated Y925F) is predicted to disrupt FAK binding to Grb2. The expression vector pCDM8-Y925F with epitope tags was cotransfected with pSV2neo to CHO cells as described in Materials and Methods. Multiple clones were analyzed for expression of exogenous FAK by indirect immunofluorescent staining and Western blotting using KT3, a mAb against the epitope tag. Fig. 1 A shows expression of the mutant FAK in two independent Y925F cell clones as detected by Western blotting with KT3. The level of the mutant FAK expression in these two clones was comparable to that in a clone overexpressing wild-type FAK (WT), described previously (Cary et al., 1996). As expected, KT3 did not recognize the endogenous FAK in control CHO cells (Neo).
Figure 1.
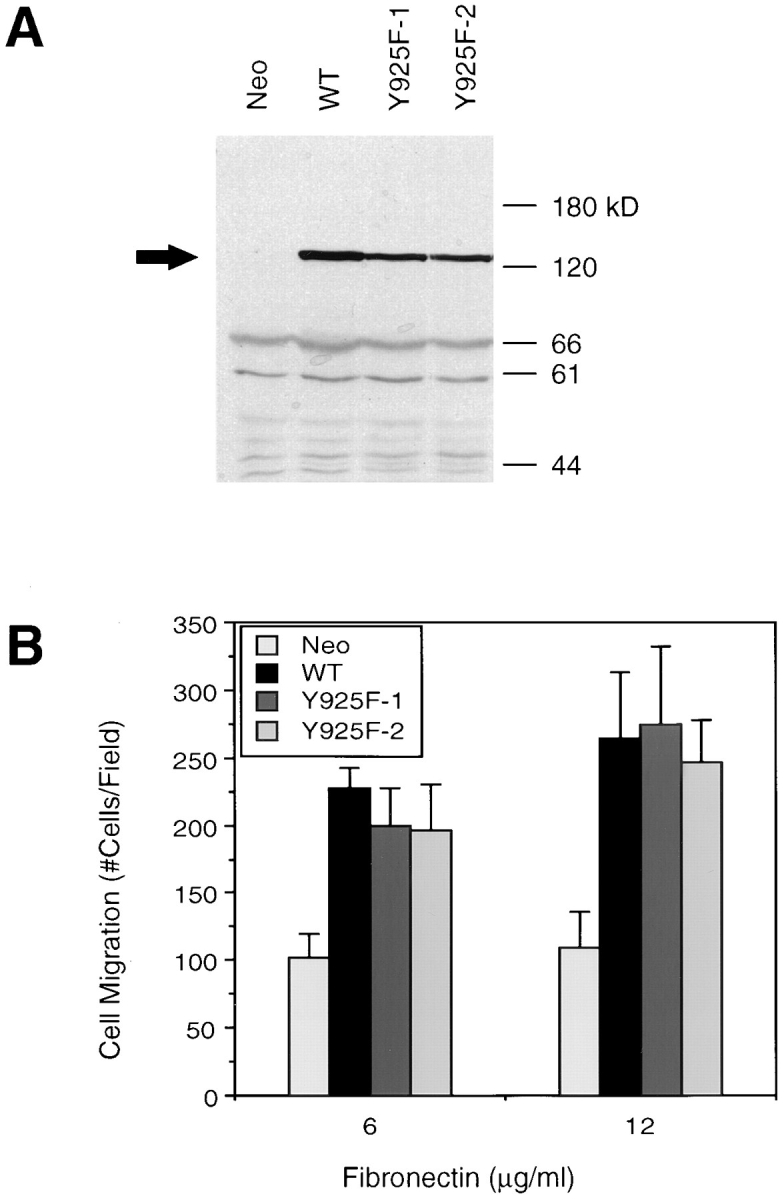
Grb2 binding to Y925 of FAK does not mediate FAK-promoted cell migration. (A) Cell lysates were generated from two CHO cell clones expressing the Y925F FAK mutant (Y925F-1 and Y925F-2), a clone overexpressing wild-type FAK (WT), and a control clone (Neo), as indicated. Total cell lysate proteins were Western blotted with the mAb KT3, which recognizes the epitope-tagged exogenous FAK, the position of which is indicated by an arrow. Molecular mass positions (kD) are shown on the right. (B) Fibronectin-mediated cell migration of the above clones was analyzed using a NeuroProbe chemotaxis chamber. Cells in the upper chamber were allowed to migrate through a porous membrane toward the lower chamber to which fibronectin had been added at either 6 or 12 μg/ml. Cells that migrated through the membrane were then fixed, stained, and counted using a light microscope. Mean cell counts from at least 10 fields and three experiments are shown. Error bars represent standard deviations. No significant migration was observed in the absence of fibronectin (not shown).
To determine the effects of this FAK mutation on its ability to promote cell migration, these two clones expressing Y925F FAK were analyzed by modified Boyden chamber assays using FN as an attractant (Fig. 1 B), as described previously (Cary et al., 1996). As expected, the WT cells exhibited an increased level of migration as compared with the control clone (Neo). At two different FN concentrations, both clones expressing the Y925F FAK mutant also demonstrated an increased level of migration that was comparable to that of WT cells. These results demonstrated that Y925 was not necessary for FAK-promoted cell migration and suggested that Grb2 binding to FAK and its downstream signaling pathways were dispensable for integrin-mediated cell migration.
To examine the ability of FAK to bind Grb2 in our CHO cell system, we carried out coimmunoprecipitation experiments, but we were unable to detect exogenous FAK association with Grb2 in these cells (data not shown). To rule out the possibility that there was a weak but undetectable interaction of Grb2 with FAK, we carried out in vitro binding experiments as shown in Fig. 2. Lysates prepared from cells overexpressing wild-type FAK, Y397F mutant FAK (described previously by Cary et al., 1996), Y925F mutant FAK, or control cells (Neo) were used for in vitro binding experiments with the indicated GST fusion proteins, and bound FAK was detected by Western blotting with KT3 (Fig. 2 A). There was no detectable binding of exogenous FAK in CHO cells with the Grb2-SH2 domain. As a control for this experiment, the same lysates were used for a binding assay with the Src-SH2 domain, demonstrating that the exogenous FAK was able to bind GST-Src-SH2 and furthermore that this binding was dependent on the Src-binding site of FAK (Y397) but not the Grb2 binding site (Y925). No binding of FAK to GST alone was detected.
Figure 2.

FAK expressed in CHO cells does not bind Grb2 in vitro. (A) Cell lysates were prepared from a CHO control clone (Neo) or clones expressing either wild-type (WT), Y397F, or Y925F mutant FAK, as indicated. Total cell lysate proteins were Western blotted with KT3 to demonstrate levels of FAK expression (Input). The lysates were used for an in vitro binding experiment with the SH2 domains of Grb2 or Src expressed as recombinant GST fusion proteins or with GST alone as a control. The GST fusion proteins were immobilized on glutathione-Sepharose beads with which the above lysates were incubated followed by several washes with lysis buffer. Bound FAK was detected by Western blotting with KT3. (B) Cell lysates were prepared from 293 cells that had been mock transfected or transiently transfected with either pKH3-FAK or pCDM8-FAK, as indicated. Total cell lysate proteins were Western blotted with anti-FAK to demonstrate levels of FAK expression (Input). The lysates were used for an in vitro binding assay with either GST-Grb2-SH2 or GST, as above, and bound FAK was detected by Western blotting with anti-FAK.
These results indicated that one or both of the epitope tags on the exogenous FAK may disrupt its binding to Grb2. To examine this possibility, lysates from 293 cells transiently transfected with the indicated FAK expression vectors were used for an in vitro binding assay with GST fusion proteins, as above, and the bound FAK was detected by Western blotting with anti-FAK (Fig. 2 B). The pCDM8-FAK construct, which was used to generate the CHO WT cells, results in a FAK protein tagged with one HA epitope at the amino terminus and one T-antigen epitope at the carboxy terminus. The pKH3-FAK construct results in a FAK protein tagged with three repeats of the HA epitope at the amino terminus and no epitope at the carboxy terminus. As shown in Fig. 2 B, FAK expressed by the pKH3-FAK construct was able to bind the Grb2-SH2 domain in vitro, while FAK expressed by the pCDM8-FAK construct was not. No significant binding of either FAK protein was detected to GST alone. These results demonstrated that the exogenous FAK expressed in CHO cells was not able to bind Grb2, and that this binding may have been disrupted by the carboxy-terminal epitope tag on FAK. These results further support that Grb2 binding to Y925 of FAK is not necessary for the ability of FAK to promote CHO cell migration.
P712 and P715 of FAK Are Necessary for Its Ability to Promote Cell Migration
The proline-rich region of FAK spanning amino acids 712–718 has been mapped as a binding site for the SH3 domain of p130Cas (Polte and Hanks, 1995; Harte et al., 1996). To examine the role of p130Cas binding to FAK in cell migration, a FAK mutant P712/715A was generated converting the two critical prolines P712 and P715 to alanines. The expression vector pCDM8-P712/715A was transfected to CHO cells, and multiple clones expressing this FAK mutant were selected as described in Materials and Methods. Western blotting with KT3 showed that two independent clones expressing this FAK mutant (P712/715A-1 and P712/715A-2) contained similar levels of exogenous FAK compared with cells expressing wild-type FAK (WT) (Fig. 3 A). These two clones were then subjected to cell migration assays along with a WT clone and a control clone (Neo), as shown in Fig. 3 B. Again, the WT cells demonstrated increased migration as compared with control cells. However, neither clone expressing the P712/715A FAK mutant exhibited increased migration on FN. These results demonstrated that this proline-rich region of FAK is necessary for its function and suggest that p130Cas binding to this site may mediate FAK-promoted cell migration.
Figure 3.
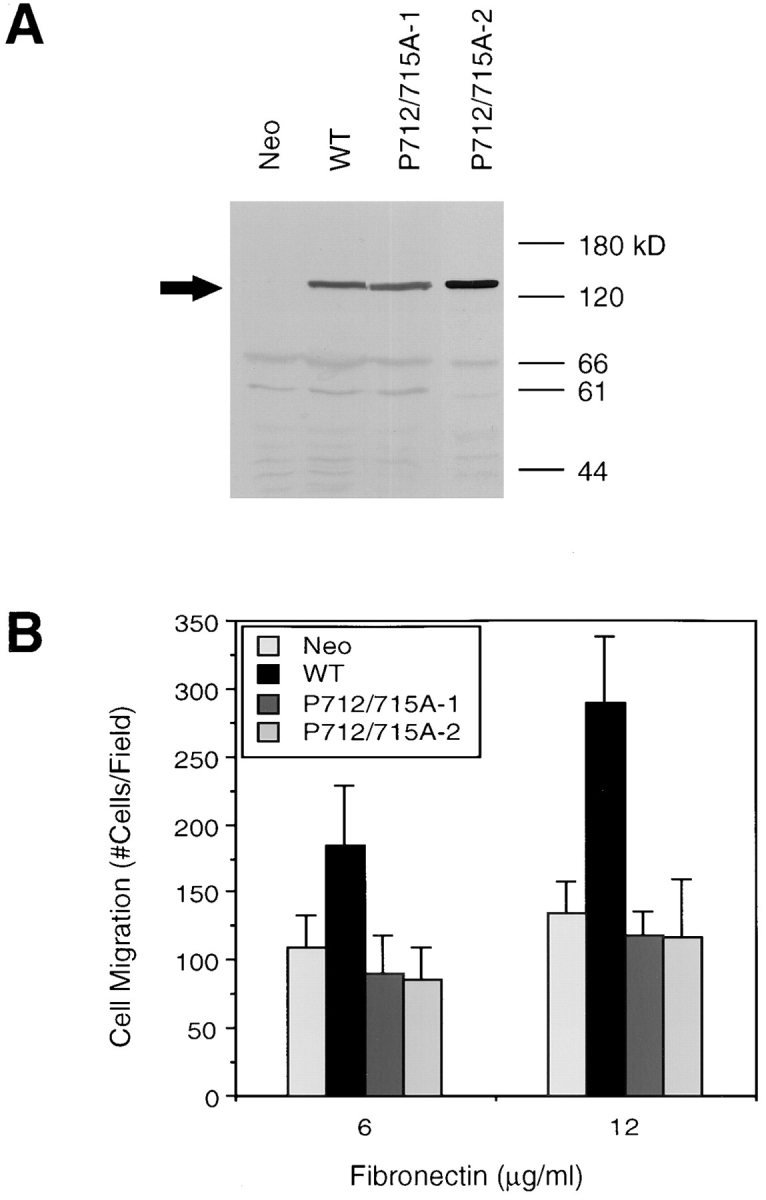
The proline-rich p130Cas binding site on FAK is necessary for FAK-promoted cell migration. (A) Cell lysates were generated from a control clone (Neo), a clone expressing wild-type FAK (WT), or two independent clones expressing the P712/715A FAK mutant (P712/715A-1 and P712/715A-2), as indicated. Total cell lysate proteins were Western blotted with KT3 to demonstrate comparable levels of FAK overexpression, the position of which is indicated on the left by an arrow. Molecular mass positions (kD) are shown on the right. (B) Fibronectin-mediated cell migration of the above clones was analyzed using a NeuroProbe chemotaxis chamber as in Fig. 1. Mean cell counts from at least nine fields and two experiments are shown. Error bars represent standard deviations. No significant migration was observed in the absence of FN (not shown).
To exclude the possibility that the lack of function of the P712/715A FAK mutant was a result of gross alterations of structure and/or kinase activity, the phosphorylation and activity of this mutant were examined, as shown in Fig. 4. Cell lysates were prepared from a representative clone expressing the P712/715A FAK mutant as well as a WT clone and a control clone (Neo). The exogenous FAK was immunoprecipitated from the lysates using KT3, and the immune complexes were Western blotted with KT3 (Fig. 4 A) or the antiphosphotyrosine mAb PY20 (Fig. 4 B). Alternatively, the KT3 immune complexes were subjected to in vitro kinase assays either with (Fig. 4 D) or without (Fig. 4 C) poly(Glu,Tyr) as an exogenous substrate. The P712/ 715A FAK mutant demonstrated levels of in vivo tyrosine phosphorylation (Fig. 4 B) and in vitro kinase activity to the exogenous substrate (Fig. 4 D) similar to those of the wild-type FAK, with a slightly higher level of in vitro autophosphorylation (Fig. 4 C). The inability of the P712/715A FAK mutant to promote cell migration was therefore not a result of reduced kinase activity or phosphorylation.
Figure 4.
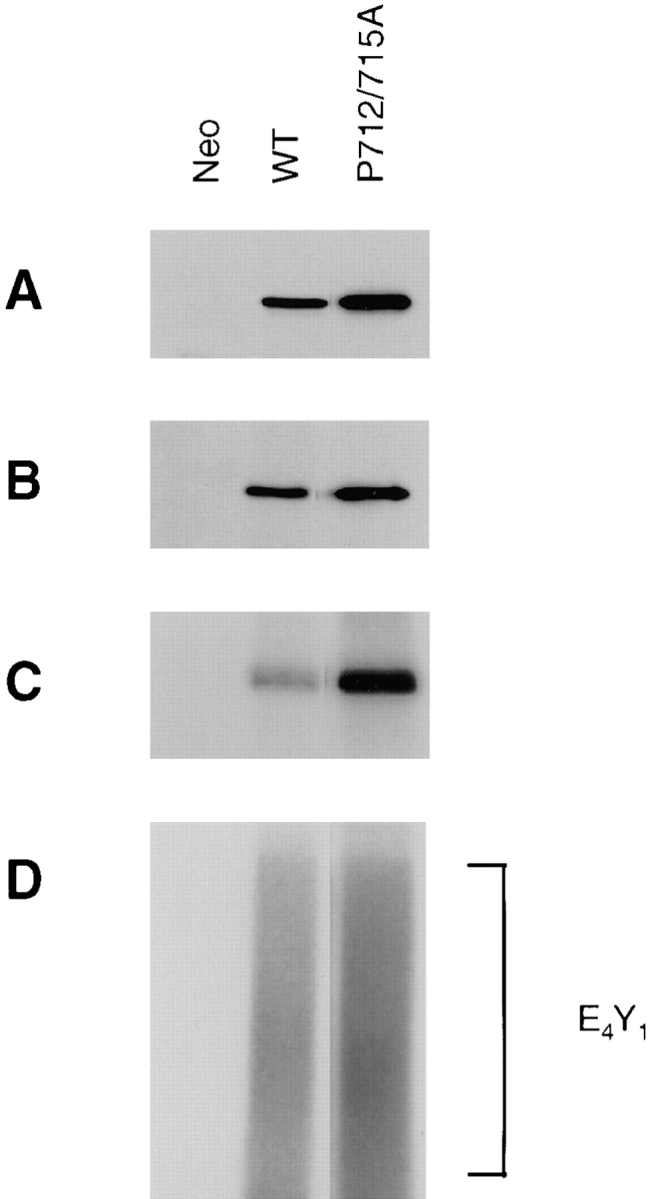
The P712/715A FAK mutant demonstrates efficient kinase activity and tyrosine phosphorylation. Cell lysates were generated from a control clone (Neo) or clones expressing either wild-type (WT) or P712/715A FAK, as indicated. The exogenous FAK was immunoprecipitated using KT3, and the immune complexes were Western blotted with either KT3 (A) or the antiphosphotyrosine mAb PY20 (B). Alternatively, the immune complexes were subjected to in vitro kinase assays using [γ-32P]ATP in the absence (C) or presence (D) of poly(Glu,Tyr) (E4Y1) as an exogenous substrate, the position of which is indicated on the right.
We have previously demonstrated that Src binding to Y397 of FAK is critical for FAK signaling in cell migration processes (Cary et al., 1996). To exclude the possibility that the mutation of P712/715A in FAK affected its ability to associate with Src, we performed the following coimmunoprecipitation experiment. Cell lysates were prepared from a WT, Y397F, P712/715A, or control clone. The lysates were Western blotted with KT3 to demonstrate relative expression levels of exogenous FAK, which varied somewhat in this particular experiment (Fig. 5 A). Alternatively, Src was immunoprecipitated from the lysates, and the immune complexes were Western blotted with KT3 to show exogenous FAK association with Src (Fig. 5 B). As shown previously (Chan et al., 1994; Schaller et al., 1994; Cary et al., 1996), the Y397F mutation resulted in significantly reduced association of FAK with Src. However, densitometric analysis showed that the P712/715A mutant demonstrated a level of in vivo Src association comparable to that of wild-type FAK and significantly higher than that of the Y397F mutant, indicating that the inability of the P712/715A FAK mutant to promote cell migration was not a result of a secondary effect on Src association.
Figure 5.

A mutation of the p130Cas binding site on FAK does not affect its association with Src. Cell lysates were prepared from a control clone (Neo) or from clones expressing either wild-type (WT), Y397F, or P712/715A FAK, as indicated. Total cell lysate proteins were Western blotted with KT3 to demonstrate relative levels of exogenous FAK expression (A). Alternatively, Src was immunoprecipitated from the lysates using the anti-Src mAb 2-17, and the immune complexes were Western blotted with KT3 to demonstrate Src-associated FAK (B).
p130Cas Phosphorylation Is Correlated with FAK-promoted Cell Migration
The lack of function of the P712/715A mutant in promoting cell migration suggested that FAK binding to p130Cas may be important in triggering downstream signaling pathways resulting in cell migration. To test this hypothesis, we first examined association of exogenous FAK and its mutants with p130Cas in CHO cells. Cell lysates were prepared from a control clone or clones expressing either WT, KD, Y397F, or P712/715A mutant FAK. A fraction of each lysate was Western blotted with KT3 to verify similar expression levels of exogenous FAK in these clones (Fig. 6 A). The remaining lysates were used for immunoprecipitations with anti-p130Cas, and the immune complexes were Western blotted with KT3 to detect p130Cas-associated FAK (Fig. 6 B). Consistent with previous reports (Polte and Hanks, 1995, 1997; Harte et al., 1996), the wild-type FAK was associated with p130Cas. The kinase-defective and Y397F FAK mutants were found to associate with p130Cas as efficiently as wild-type FAK. In contrast, the P712/715A FAK mutant showed a decreased association with p130Cas, as observed previously (Polte and Hanks, 1997). Densitometric analysis of four such experiments indicated an ∼50% reduction in P712/715A mutant association with p130Cas relative to wild-type FAK.
Figure 6.

The P712/715A FAK mutant demonstrates reduced association with p130Cas. Cell lysates from a control clone (Neo), a clone expressing wild-type FAK (WT), or clones expressing the indicated FAK mutants (kinase-defective [KD], Y397F, or P712/715A) were prepared using modified RIPA lysis buffer. Total cell lysate proteins were Western blotted with KT3 to demonstrate relative levels of exogenous FAK expression (A). The lysates were used for immunoprecipitations using anti-p130Cas, followed by Western blotting with KT3 to demonstrate p130Cas-associated FAK (B).
Besides directly binding to FAK, p130Cas has also been demonstrated to be tyrosine phosphorylated in response to cell adhesion in a FAK- and Src-dependent manner (Vuori et al., 1996). However, it is not clear if p130Cas binding to FAK is required for its phosphorylation. We therefore examined whether FAK overexpression in CHO cells could promote p130Cas phosphorylation and whether this was dependent on either the p130Cas binding site or the Src binding site. Cell lysates were prepared from cells overexpressing FAK (WT, KD, Y397F, or P712/715A) or a control clone (Neo) as described in Materials and Methods. Lysates were immunoprecipitated with anti-p130Cas, and the immune complexes were Western blotted with anti-p130Cas to verify similar expression levels (Fig. 7 B) or with PY20 to determine the in vivo tyrosine phosphorylation levels of p130Cas (Fig. 7 A). Overexpression of wild-type FAK resulted in increased tyrosine phosphorylation of p130Cas in CHO cells, and this was dependent on the p130Cas binding site since it was abolished by the P712/ 715A FAK mutation. Furthermore, the Y397F FAK mutant did not promote p130Cas phosphorylation, suggesting a role for Src binding to FAK in the increased p130Cas phosphorylation. Consistent with this, p130Cas phosphorylation was also elevated by the KD FAK mutant, which has been shown to associate with Src and promote CHO cell migration (Cary et al., 1996). Therefore, increased p130Cas phosphorylation required FAK binding to both Src and p130Cas, and it was correlated with increased cell migration in FAK-overexpressing CHO cells.
Figure 7.

FAK-promoted tyrosine phosphorylation of p130Cas and its subsequent binding to SH2 domain–containing proteins may mediate FAK-promoted cell migration. Cells overexpressing FAK (wild-type [WT] or the mutants kinase-defective [KD], Y397F, or P712/715A) or a control clone (Neo) were treated with sodium vanadate and lysed. Lysates were used for immunoprecipitations with anti-p130Cas, and the immune complexes were Western blotted with PY20 (A) or anti-p130Cas (B). The SH2 domains of Src (D), Nck (E), or the p85 subunit of phosphatidylinositol 3-kinase (F) were expressed as recombinant bacterial GST fusion proteins, or GST alone (C) was expressed as a control. The GST fusion proteins were immobilized on glutathione-Sepharose beads, with which the above cell lysates were incubated followed by several washes with lysis buffer. Bound p130Cas was detected by Western blotting with anti-p130Cas.
p130Cas is an adaptor protein containing multiple tyrosine phosphorylation sites (Sakai et al., 1994; Schlaepfer et al., 1997); therefore, we examined the interaction of tyrosine-phosphorylated p130Cas with its potential downstream effectors. The same lysates described above were used for in vitro binding experiments with various SH2 domains expressed as GST fusion proteins or with GST alone as a control, and the associated p130Cas was detected by Western blotting (Fig. 7, C–F). The increased tyrosine phosphorylation of p130Cas in cells expressing wild-type or kinase-defective FAK correlated with its increased binding to the SH2 domains of Src, Nck, or the p85 subunit of phosphatidylinositol 3-kinase, whereas in the absence of increased p130Cas phosphorylation (in cells expressing the Y397F or P712/715A FAK mutant), there was a basal level of p130Cas binding to these SH2 domains. These results suggested that increased association of p130Cas with SH2 domain–containing intracellular signaling molecules may be responsible for triggering downstream signaling pathways in cell migration.
The Erk Signaling Pathway Does Not Mediate FAK-promoted Cell Migration
Activation of Erks has been suggested to be one of the possible downstream pathways of p130Cas (Schlaepfer et al., 1997). To investigate the potential role of Erks in FAK-promoted cell migration, we first examined whether stable overexpression of FAK in CHO cells could activate Erks, as shown in Fig. 8 A. A clone expressing wild-type FAK (WT) or a control clone (Neo) were lysed as either a monolayer of attached cells (Att) or in suspension (Sus). As a control for Erk activation, NIH 3T3 cells were lysed after treatment with PDGF. Lysates were Western blotted with anti–phospho-MAPK antibody, which recognizes the phosphorylated and activated forms of Erk1 and Erk2. PDGF treatment of NIH 3T3 cells resulted in strong activation of both Erk1 and Erk2, as indicated. While attached CHO cells demonstrated increased Erk activation compared with cells in suspension, there was no detectable Erk activation as a result of FAK overexpression. We were unable to detect activation of Erks in CHO cells replated on fibronectin, regardless of FAK overexpression (data not shown). These results suggested that Erks are not important mediators of FAK signaling resulting in cell migration.
Figure 8.

The Erk signaling pathway does not mediate FAK-promoted cell migration. (A) Cells overexpressing wild-type FAK (WT) or control cells (Neo) were lysed as either a monolayer of attached cells (Att) or in suspension (Sus). As a control for Erk activation, NIH 3T3 cells were serum-starved and treated with or without PDGF (25 ng/ml for 10 min) followed by lysis. Total cell lysate proteins were Western blotted with anti–phospho-MAPK antibody. Arrows indicate the position of activated Erk1 and Erk2. (B) WT or Neo cells were pretreated for 1 h with the indicated concentrations of the MEK inhibitor PD98059, and their migration on 12 μg/ml FN was then analyzed using a chemotaxis chamber. Mean cell counts from at least five fields and two experiments are shown. Error bars represent standard errors.
Although no FAK-dependent Erk activation was detected, it was possible that Erks were activated to a low and undetectable level by FAK overexpression and therefore could mediate FAK signal transduction resulting in cell migration. To rule out this possibility, we examined the ability of CHO cells to migrate in the presence of the MEK inhibitor PD98059, which subsequently inhibits both Erk1 and Erk2. CHO cells overexpressing wild-type FAK (WT) or control cells (Neo) were subjected to FN-stimulated cell migration assays in the presence of PD98059 at the indicated concentrations (Fig. 8 B). This inhibitor did not affect FAK-promoted cell migration at concentrations necessary for inhibition of MEK1 and MEK2 (Alessi et al., 1995; Dudley et al., 1995; Pang et al., 1995). These results suggest that p130Cas may target signaling pathways other than those involving Erks in mediating FAK-promoted cell migration.
p130Cas Is an Important Mediator of FAK-promoted Cell Migration
While the above results demonstrating the lack of migration by the P712/715A FAK mutant and the correlation of FAK-promoted p130Cas phosphorylation with migration strongly suggested that p130Cas was a crucial downstream mediator of FAK in cell migration, the possibility existed that another protein that could bind FAK at this site could be mediating this signaling. To confirm the role of p130Cas in FAK-promoted cell migration, we expressed wild-type p130Cas in cells overexpressing FAK (WT cells) to determine if this would further enhance cell migration. The expression vector pRc/CMV-Cas(Myc) was cotransfected with pREP3 to WT cells, and stable hygromycin-resistant clones were selected and screened for exogenous p130Cas expression using the mAb 9E10, which recognizes the epitope-tagged p130Cas. Three such clones (WT/Cas-1, 2, and 3) demonstrated similar levels of exogenous FAK expression (Fig. 9 A) and exogenous p130Cas expression (Fig. 9 B). The ability of these clones to migrate on FN was examined as shown in Fig. 9 C. At 3 μg/ml FN, all three WT/Cas clones exhibited an increased level of migration as compared with the WT clone. At higher FN concentrations, these WT/Cas clones exhibited levels of migration comparable to that of the WT clone (data not shown). These results showed that FAK and p130Cas can cooperate to promote cell migration and suggested that this may be a result of direct interaction between the two proteins. However, it was possible that this effect was due to a second p130Cas-initiated pathway that was independent of FAK.
Figure 9.
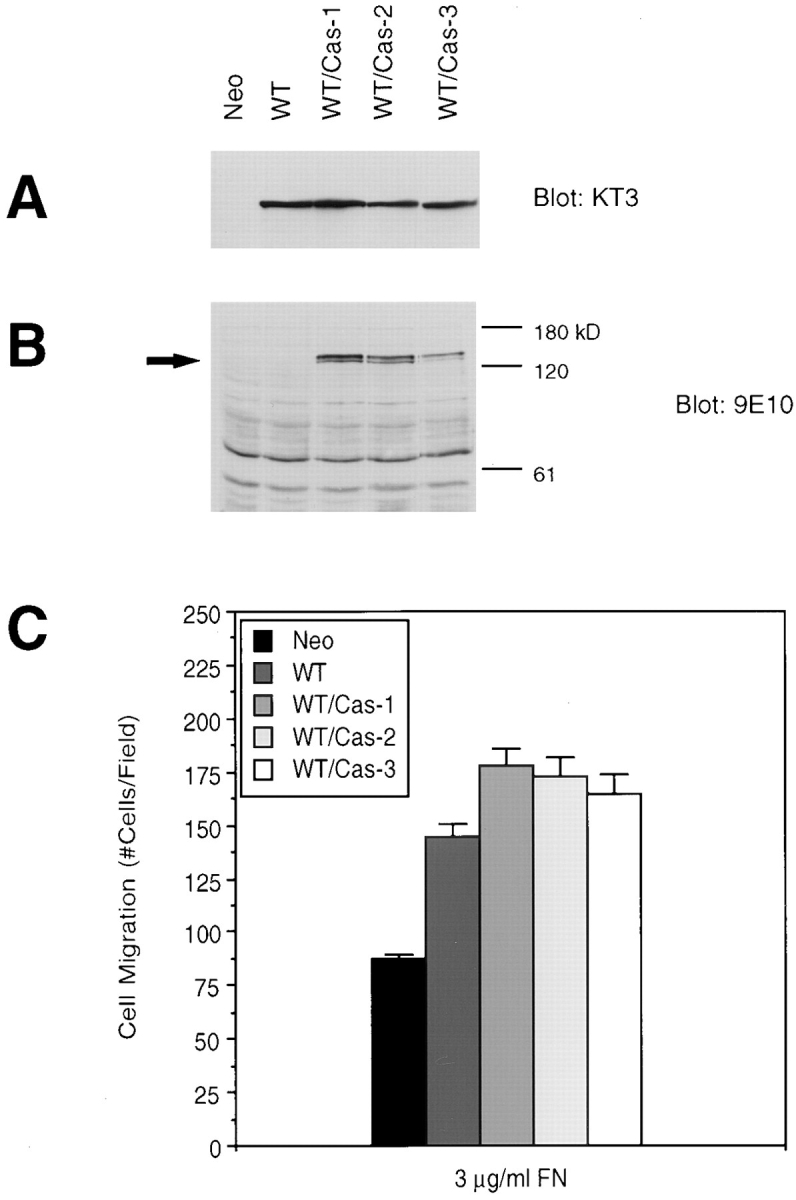
Expression of p130Cas enhances FAK-promoted cell migration. Cell lysates were prepared from a clone expressing FAK (WT), three clones expressing both FAK and wild-type p130Cas (WT/Cas-1, 2, and 3), or a control clone (Neo), as indicated. Total cell lysate proteins were Western blotted with mAb KT3 to demonstrate exogenous FAK expression (A) or mAb 9E10 to demonstrate exogenous p130Cas expression, which is indicated by an arrow (B). Molecular mass positions (kD) are shown on the right (B). Migration of the above clones was analyzed using a chemotaxis chamber and 3 μg/ml FN as an attractant (C). Mean cell counts from at least 20 fields and three experiments are shown. Error bars represent standard errors.
To show that p130Cas is an important mediator of FAK-promoted cell migration as a result of its direct association with FAK, we expressed the SH3 domain of p130Cas (CasSH3) in cells overexpressing FAK. CasSH3 was expected to bind to FAK and inhibit endogenous p130Cas binding, thus acting in a dominant-negative manner. The expression vector pKH3-CasSH3 was cotransfected with pREP3 to WT cells. Stable hygromycin-resistant clones were selected and screened for CasSH3 expression using the mAb 12CA5, which recognizes the epitope-tagged CasSH3. Two such clones (WT/CasSH3-1 and 2) demonstrated similar levels of exogenous FAK expression (Fig. 10 A) and CasSH3 expression (Fig. 10 B). The association of exogenous FAK with CasSH3 in these cells was confirmed by coimmunoprecipitation (Fig. 10 C). As shown in Fig. 10 D, both WT/CasSH3 clones exhibited a reduced level of migration as compared with the WT clone. These results showed that p130Cas is an important downstream mediator of FAK-promoted cell migration as a result of its SH3 domain–dependent binding to FAK.
Figure 10.
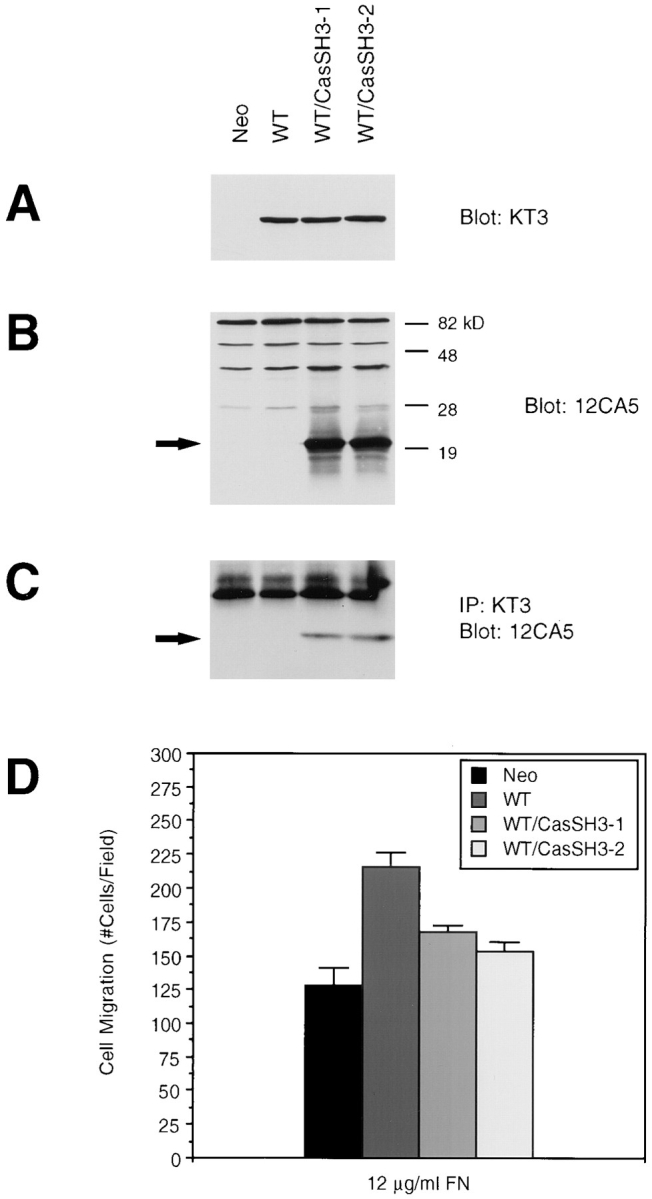
Expression of the p130Cas SH3 domain and its association with FAK inhibits FAK-promoted cell migration. Cell lysates were prepared from a clone expressing FAK (WT), two clones expressing both FAK and CasSH3 (WT/CasSH3-1 and 2), or a control clone (Neo), as indicated. Total cell lysate proteins were Western blotted with the mAb KT3 to demonstrate exogenous FAK expression (A) or mAb 12CA5 to demonstrate expression of the SH3 domain of p130Cas (B). Lysates were immunoprecipitated with KT3 and Western blotted with 12CA5 to show association of exogenous FAK with CasSH3 (C). The position of CasSH3 is indicated by arrows (B and C). Molecular mass positions (kD) are shown on the right (B). Migration of the above clones was analyzed using a chemotaxis chamber and 12 μg/ml FN as an attractant (D). Mean cell counts from at least 21 fields and three experiments are shown. Error bars represent standard errors.
Discussion
FAK is a key player in integrin-mediated signal transduction pathways; consistent with this it has been shown to associate with a number of important signaling molecules (Clark and Brugge, 1995; Schwartz et al., 1995). However, the functional significance of these interactions is largely unknown in the context of appropriate cellular processes. Recent studies have demonstrated that FAK plays a crucial role in mediating integrin signal transduction resulting in cell migration (Ilic et al., 1995; Cary et al., 1996; Gilmore and Romer, 1996). Using a system of FAK overexpression in CHO cells, we are beginning to elucidate the molecular mechanisms of FAK signaling in these pathways. We have previously shown that Y397 is necessary for FAK's promotion of cell migration, implicating a role for Src and/or PI 3-kinase downstream of FAK in these pathways (Cary et al., 1996). In this report, we have analyzed the role of FAK interaction with Grb2 and p130Cas, two signaling events downstream of the FAK/Src complex. Our data excluded a role for Grb2 binding to Y925 of FAK in FAK-promoted cell migration. Conversely, we have identified p130Cas as a mediator of FAK-promoted CHO cell migration in this system. Unlike cells expressing wild-type FAK, cells expressing a FAK P712/715A mutant with reduced p130Cas association showed a level of migration comparable to that of control cells. Coexpression of p130Cas with FAK further increased cell migration, and coexpression of the p130Cas SH3 domain alone functioned as a dominant negative mutant and decreased cell migration. Biochemical analysis indicated that FAK expression in CHO cells increased tyrosine phosphorylation of p130Cas and its subsequent binding to several SH2 domains, which could stimulate downstream signaling pathways other than the Erk pathway to mediate FAK-promoted cell migration.
One of the downstream events of the FAK/Src complex formation is Src phosphorylation of Y925 of FAK and Grb2 binding to this site (Schlaepfer et al., 1994; Schlaepfer and Hunter, 1996). FAK association with Grb2 is believed to mediate activation of Erks in response to FN, which has been well documented (Chen et al., 1994; Schlaepfer et al., 1994; Zhu and Assoian, 1995). However, the putative role of FAK in this pathway remains controversial. One report has demonstrated that various FN fragments and integrin-activating antibodies could activate Erk independently of FAK tyrosine phosphorylation, suggesting that FAK does not play a role in FN-stimulated Erk activation (Lin et al., 1997). Conversely, FAK overexpression in human 293 cells significantly enhanced FN-stimulated Erk2 activation (Schlaepfer and Hunter, 1997). In any case, our data here demonstrated that Grb2 binding to Y925 of FAK was not responsible for FAK-promoted cell migration. The Y925F FAK mutant was able to promote cell migration as efficiently as wild-type FAK (Fig. 1), and there was no detectable association of Grb2 with the transfected FAK in these cells, which exhibit FAK-dependent increased cell migration (Fig. 2). Nevertheless, the binding of Grb2 to FAK, which could result in activation of Erks, might play a role in alternative integrin-mediated signal transduction events, such as cell proliferation or cell survival (Frisch et al., 1996; Gilmore and Romer, 1996; Hungerford et al., 1996).
In this report, we provide several lines of evidence that p130Cas is a downstream mediator of FAK-promoted cell migration. First, a mutation of P712/715A in FAK abolished its ability to promote CHO cell migration (Fig. 3). The proline-rich region on FAK spanning amino acids 712–718 has been mapped as the primary binding site for p130Cas through its SH3 domain (Polte and Hanks, 1995; Harte et al., 1996), although a second proline-rich region (amino acids 875–884) can also mediate FAK binding to p130Cas (Harte et al., 1996; Polte and Hanks, 1997). In vivo coimmunoprecipitation experiments showed that FAK/ p130Cas association was reduced but not abolished by the P712/715A mutation (Fig. 6), indicating that the second proline-rich region on FAK may mediate some binding to p130Cas, as demonstrated previously (Harte et al., 1996; Polte and Hanks, 1997). However, binding of p130Cas to an alternative site clearly does not allow for FAK-promoted migration. It has also been suggested that p130Cas may associate with FAK indirectly through Src (Schlaepfer et al., 1997), which can bind FAK via its SH2 domain (Cobb et al., 1994; Xing et al., 1994) and p130Cas via its SH3 domain (Nakamoto et al., 1996). This is not the case in our CHO cell system since a mutation of the Src binding site (Y397F) did not affect FAK association with p130Cas (Fig. 6). Since we have previously demonstrated that Src binding to FAK is necessary for FAK-promoted migration (Cary et al., 1996), we verified that the P712/715A FAK mutant did not indirectly reduce Src association with FAK, thereby abolishing FAK-promoted migration (Fig. 5). These results therefore suggested that binding of p130Cas to the first proline-rich region of FAK was necessary for FAK-promoted cell migration. Second, coexpression of wild-type p130Cas with FAK in CHO cells further enhanced the migration promoted by FAK alone, indicating that these two proteins can cooperate to promote cell migration (Fig. 9). These results strongly suggested that the inability of the P712/ 715A FAK mutant to stimulate cell migration was due to its reduced association with p130Cas rather than some other proteins that may bind to the same site on FAK. Finally, coexpression of CasSH3 in CHO cells expressing FAK decreased cell migration (Fig. 10). The SH3 domain of p130Cas has been mapped as the binding site for FAK previously (Polte and Hanks, 1995; Harte et al., 1996). Consistent with this, we have found stable association of CasSH3 with exogenous FAK in vivo (Fig. 10 C). Furthermore, Nakamoto et al. (1997) have shown recently that truncation of the SH3 domain of p130Cas abolished its localization to focal contacts. It is therefore conceivable that CasSH3 bound to FAK was localized to focal contacts and could function as a dominant-negative mutant by competing with endogenous p130Cas for association with FAK and/or localization to focal contacts. Together, these findings identify p130Cas as a downstream mediator of FAK-promoted cell migration as a result of its SH3 domain–dependent association with FAK.
Tyrosine phosphorylation of p130Cas as a result of its binding to FAK is likely to play a role in its initiation of signaling pathways resulting in cell migration. p130Cas has been shown to be regulated by a number of different tyrosine kinases and phosphatases (Sakai et al., 1994, 1997; Garton et al., 1996; Liu et al., 1996), and its phosphorylation in cell adhesion is believed to be dependent on both FAK and Src (Vuori et al., 1996; Schlaepfer et al., 1997). It has been shown that FAK expression can promote p130Cas tyrosine phosphorylation (Vuori et al., 1996), which we have confirmed in this study. Furthermore, we now demonstrate that this phosphorylation is dependent on direct binding of p130Cas to prolines 712 and 715 of FAK (Fig. 7). While its binding to FAK is a direct interaction and not dependent on Src, efficient tyrosine phosphorylation of p130Cas in response to integrin activation is believed to require Src in this complex. Tyrosine phosphorylation of p130Cas was decreased in cultured fibroblasts from Src− mice and increased in Csk− cells in which Src activity was increased. In addition, it was shown that FAK-promoted p130Cas phosphorylation was dependent on the Src binding site (Y397) of FAK (Vuori et al., 1996). Both FAK and Src could phosphorylate p130Cas in vitro at similar sites (Schlaepfer et al., 1997), leading us to question which kinase mediates p130Cas phosphorylation in the FAK-overexpressing CHO cells. We now demonstrate that FAK-promoted p130Cas phosphorylation occurred when a kinase-defective but not Y397F FAK mutant was expressed (Fig. 7), indicating that Src may be the kinase that mediates phosphorylation of p130Cas in this system, as observed previously in other cell types (Vuori et al., 1996). This result also suggests that the inability of the Y397F FAK mutant to promote cell migration may be due to inefficient p130Cas phosphorylation and recruitment of signaling molecules.
The downstream pathway of p130Cas binding to FAK in cell migration is unknown, although the activation of Erks has been proposed as a possible signaling event downstream of p130Cas (Schlaepfer et al., 1997). Activation of Erks could promote cell motility via regulation of gene expression, such as that described for the transcriptional regulator NF-κB, the inhibition of which resulted in inhibition of carcinoma cell migration on vitronectin (Yebra et al., 1995). Additionally, recent evidence demonstrating that Erk2 could directly phosphorylate and activate myosin light chain kinase indicates a more direct regulation of actin cytoskeletal proteins as a mechanism for Erk-dependent regulation of cell motility (Klemke et al., 1997). However, our data showed that Erks are not involved in FAK-promoted cell migration in our CHO cell system. We have been unable to detect an increase in Erk activity by FAK overexpression (Fig. 8 A). Furthermore, studies using the MEK inhibitor PD98059 have shown that inhibition of MEK kinases and presumably Erks did not inhibit FAK-promoted CHO cell migration on FN (Fig. 8 B). It is therefore possible that the Erk signaling pathway plays a role in cell migration in some cell types (Klemke et al., 1997) but not in others (this study). These results also suggested that in CHO cells, p130Cas is likely to target alternative downstream signaling pathways as a result of its FAK-promoted tyrosine phosphorylation and subsequent recruitment of other signaling molecules.
In conclusion, stable overexpression of FAK in CHO cells has enabled us to dissect the molecular mechanisms of FAK signal transduction and to analyze the putative roles of several FAK-binding proteins and their downstream pathways in cell migration. We now demonstrate that p130Cas, but not Grb2, is a mediator of FAK-promoted cell migration. Our data also suggest that the FAK/ p130Cas complex targets downstream pathways other than Erks in mediating FAK signaling in cell migration. Further work will examine the importance of other protein associations with FAK as well as downstream signals from the FAK/Src/p130Cas complex in integrin-mediated cell migration.
Acknowledgments
We are grateful to Dr. Gernot Walter for mAb KT3, Dr. Tom Parsons for cDNA encoding chicken FAK, Dr. Wei Li for the GST-Nck-SH2 construct, and Dr. Andy Yen for the pREP3 vector. We thank Laurie Warner and Frances Trasti for technical assistance and Mossaad Abdel-Ghany, Heinz Reiske, Kristin Wilson, Wannian Yang and Chuanhai Zheng for critical reading of the manuscript.
Abbreviations used in this paper
- CasSH3
SH3 domain of p130Cas
- ECM
extracellular matrix
- Erks
extracellular signal-regulated kinases
- FAK
focal adhesion kinase
- FN
fibronectin
- GST
glutathione S-transferase
- PI 3-kinase
phosphatidylinositol 3-kinase
- SH2 and SH3
Src homology 2 and 3
Footnotes
This research was supported by National Institutes of Health (NIH) grant GM48050 and The Council for Tobacco Research, USA, to J.-L. Guan, and NIH grant GM49882 to S. Hanks.
Address all correspondence to Jun-Lin Guan, Department of Pathology, College of Veterinary Medicine, Cornell University, Ithaca, NY 14853. Tel.: (607) 253-3586. Fax: (607) 253-3708.
Thomas Polte's current address is Children's Hospital/Harvard Medical School, Department of Surgical Research, 300 Longwood Ave., Boston, MA 02115.
References
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Cary L, Chang J, Guan J-L. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with Src and Fyn. J Cell Sci. 1996;109:1787–1794. doi: 10.1242/jcs.109.7.1787. [DOI] [PubMed] [Google Scholar]
- Chan P-Y, Kanner S B, Whitney G, Aruffo A. A transmembrane-anchored chimeric focal adhesion kinase is constitutively activated and phosphorylated at tyrosine residues identical to pp125FAK . J Biol Chem. 1994;269:20567–20574. [PubMed] [Google Scholar]
- Chen H-C, Guan J-L. Association of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1994;91:10148–10152. doi: 10.1073/pnas.91.21.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-C, Appeddu PA, Parsons JT, Hildebrand JD, Schaller MD, Guan J-L. Interaction of focal adhesion kinase with cytoskeletal protein talin. J Biol Chem. 1995;270:16995–16999. doi: 10.1074/jbc.270.28.16995. [DOI] [PubMed] [Google Scholar]
- Chen H-C, Appeddu PA, Isoda H, Guan J-L. Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J Biol Chem. 1996;271:26329–26334. doi: 10.1074/jbc.271.42.26329. [DOI] [PubMed] [Google Scholar]
- Chen Q, Kinch MS, Lin TH, Burridge K, Juliano RL. Integrin-mediated cell adhesion activates mitogen-activated protein kinases. J Biol Chem. 1994;269:26602–26605. [PubMed] [Google Scholar]
- Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- Cobb BS, Schaller MD, Leu T-H, Parsons JT. Stable association of pp60src and pp59fyn with the focal adhesion-associated protein kinase, pp125FAK . Mol Cell Biol. 1994;14:147–155. doi: 10.1128/mcb.14.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein tyrosine kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garton AJ, Flint AJ, Tonks NK. Identification of p130Casas a substrate for the cytosolic protein tyrosine phosphatase PTP-PEST. Mol Cell Biol. 1996;16:6408–6418. doi: 10.1128/mcb.16.11.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore AP, Romer LH. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol Biol Cell. 1996;7:1209–1224. doi: 10.1091/mbc.7.8.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J-L, Shalloway D. Regulation of focal adhesion-associated protein tyrosine kinase by both cellular adhesion and oncogenic transformation. Nature. 1992;358:690–692. doi: 10.1038/358690a0. [DOI] [PubMed] [Google Scholar]
- Guan J-L, Trevithick JE, Hynes RO. Fibronectin/integrin interaction induces tyrosine phosphorylation of a 120-kDa protein. Cell Regul. 1991;2:951–964. doi: 10.1091/mbc.2.11.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harte MT, Hildebrand JD, Burnham MR, Bouton AH, Parsons JT. p130Cas, a substrate associated with v-Src and v-Crk, localizes to focal adhesions and binds to focal adhesion kinase. J Biol Chem. 1996;271:13649–13655. doi: 10.1074/jbc.271.23.13649. [DOI] [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site- directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Hungerford JE, Compton MT, Matter ML, Hoffstrom BG, Otey CA. Inhibition of pp125FAKin cultured fibroblasts results in apoptosis. J Cell Biol. 1996;135:1383–1390. doi: 10.1083/jcb.135.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T, Aizawa S. Reduced cell motility and enhanced focal contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Klemke RL, Cai S, Giannini AL, Gallagher PJ, Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997;137:481–492. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TH, Aplin AE, Shen Y, Chen Q, Schaller M, Romer L, Aukhil I, Juliano RL. Integrin-mediated activation of MAP kinase is independent of FAK: evidence for dual integrin signaling pathways in fibroblasts. J Cell Biol. 1997;136:1385–1395. doi: 10.1083/jcb.136.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Hill DE, Chernoff J. Direct binding of the proline-rich region of protein tyrosine phosphatase 1B to the src homology 3 domain of p130Cas . J Biol Chem. 1996;271:31290–31295. doi: 10.1074/jbc.271.49.31290. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Akiyama S, Yamada KM. Synergistic roles for receptor occupancy and aggregation in integrin transmembrane function. Science. 1995;267:883–885. doi: 10.1126/science.7846531. [DOI] [PubMed] [Google Scholar]
- Nakamoto T, Sakai R, Ozawa K, Yazaki Y, Hirai H. Direct binding of C-terminal region of p130Casto SH2 and SH3 domains of Src kinase. J Biol Chem. 1996;271:8959–8965. doi: 10.1074/jbc.271.15.8959. [DOI] [PubMed] [Google Scholar]
- Nakamoto T, Sakai R, Honda H, Ogawa S, Ueno H, Suzuki T, Aizawa S, Yazaki Y, Hirai H. Requirements for localization of p130Casto focal adhesions. Mol Cell Biol. 1997;17:3884–3897. doi: 10.1128/mcb.17.7.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima Y, Morino N, Mimura T, Hamasaki K, Furuya H, Sakai R, Sato T, Tachibana K, Morimoto C, Yazaki Y, Hirai H. Integrin-mediated cell adhesion promotes tyrosine phosphorylation of p130Cas, a Src homology 3-containing molecule having multiple Src homology 2-binding motifs. J Biol Chem. 1995;270:15398–15402. doi: 10.1074/jbc.270.25.15398. [DOI] [PubMed] [Google Scholar]
- Pang L, Sawada T, Decker SJ, Saltiel AR. Inhibition of Map kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- Parsons JT. Integrin-mediated signalling:regulation by protein tyrosine kinases and small GTP-binding proteins. Curr Opin Cell Biol. 1996;8:146–152. doi: 10.1016/s0955-0674(96)80059-7. [DOI] [PubMed] [Google Scholar]
- Petch LA, Bockholt SM, Bouton A, Parsons JT, Burridge K. Adhesion-induced tyrosine phosphorylation of the p130 src substrate. J Cell Sci. 1995;108:1371–1379. doi: 10.1242/jcs.108.4.1371. [DOI] [PubMed] [Google Scholar]
- Polte TR, Hanks SK. Interaction between focal adhesion kinase and Crk-associated tyrosine kinase substrate p130Cas . Proc Natl Acad Sci USA. 1995;92:10678–10682. doi: 10.1073/pnas.92.23.10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polte TR, Hanks SK. Complexes of focal adhesion kinase (FAK) and Crk-associated substrate (p130Cas) are elevated in cytoskeleton-associated fractions following adhesion and Src transformation. Requirements for Src kinase activity and FAK proline-rich motifs. J Biol Chem. 1997;272:5501–5509. doi: 10.1074/jbc.272.9.5501. [DOI] [PubMed] [Google Scholar]
- Richardson A, Parsons JT. A mechanism for regulation of the adhesion-associated protein tyrosine kinase pp125FAK . Nature. 1996;380:538–540. doi: 10.1038/380538a0. [DOI] [PubMed] [Google Scholar]
- Sakai R, Iwamatsu A, Hirano N, Ogawa S, Tanaka T, Mano H, Yazaki Y, Hirai H. A novel signaling molecule, p130, forms stable complexes in vivo with v-Crk and v-Src in a tyrosine phosphorylation-dependent manner. EMBO (Eur Mol Biol Organ) J. 1994;13:3748–3756. doi: 10.1002/j.1460-2075.1994.tb06684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai R, Nakamoto T, Ozawa K, Aizawa S, Hirai H. Characterization of the kinase activity essential for tyrosine phosphorylation of p130Casin fibroblasts. Oncogene. 1997;14:1419–1426. doi: 10.1038/sj.onc.1200954. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Parsons JT. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol Cell Biol. 1995;15:2635–2645. doi: 10.1128/mcb.15.5.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller MD, Hildebrand JD, Shannon JD, Fox JX, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src . Mol Cell Biol. 1994;14:1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hunter T. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol Cell Biol. 1996;16:5623–5633. doi: 10.1128/mcb.16.10.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Hunter T. Focal adhesion kinase overexpression enhances Ras-dependent integrin signaling to ERK2/mitogen-activated protein kinase through interaction with and activation of c-Src. J Biol Chem. 1997;272:13189–13195. doi: 10.1074/jbc.272.20.13189. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin-mediated signal transduction linked to ras pathway by Grb2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Broome MA, Hunter T. Fibronectin-stimulated signaling from a focal adhesion kinase-c-Src complex: involvement of the Grb2, p130Cas, and Nck adaptor proteins. Mol Cell Biol. 1997;17:1702–1713. doi: 10.1128/mcb.17.3.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA, Schaller MD, Ginsberg MH. Integrins: emerging paradigms of signal transduction. Annu Rev Cell Dev Biol. 1995;11:549–599. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- Turner CE, Miller J. Primary sequence of paxillin contains putative SH2 and SH3 domain binding motif and multiple LIM domains: identification of a vinculin and pp125FAKbinding region. J Cell Sci. 1994;107:1583–1591. doi: 10.1242/jcs.107.6.1583. [DOI] [PubMed] [Google Scholar]
- Vuori K, Ruoslahti E. Tyrosine phosphorylation of p130Casand cortactin accompanies integrin-mediated cell adhesion to extracellular matrix. J Biol Chem. 1995;270:22259–22262. doi: 10.1074/jbc.270.38.22259. [DOI] [PubMed] [Google Scholar]
- Vuori K, Hirai H, Aizawa S, Ruoslahti E. Introduction of p130Cassignaling complex formation upon integrin-mediated cell adhesion: a role for Src family kinases. Mol Cell Biol. 1996;16:2606–2613. doi: 10.1128/mcb.16.6.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Z, Chen H-C, Nowlen JK, Taylor SJ, Shalloway D, Guan J-L. Direct interaction of v-Src with the focal adhesion kinase mediated by the Src SH2 domain. Mol Biol Cell. 1994;5:413–421. doi: 10.1091/mbc.5.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yebra M, Filardo EJ, Bayna EM, Kawahara E, Becker JC, Cheresh DA. Induction of carcinoma cell migration on vitronectin by NF-κB-dependent gene expression. Mol Biol Cell. 1995;6:841–850. doi: 10.1091/mbc.6.7.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Assoian RK. Integrin-dependent activation of MAP kinase: a link to shape-dependent cell proliferation. Mol Biol Cell. 1995;6:273–282. doi: 10.1091/mbc.6.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]