

Abstract
It has been proposed that the urokinase receptor (u-PAR) is essential for the various biological roles of urokinase-type plasminogen activator (u-PA) in vivo, and that smooth muscle cells require u-PA for migration during arterial neointima formation. The present study was undertaken to evaluate the role of u-PAR during this process in mice with targeted disruption of the u-PAR gene (u-PAR−/−). Surprisingly, u-PAR deficiency did not affect arterial neointima formation, neointimal cell accumulation, or migration of smooth muscle cells. Indeed, topographic analysis of arterial wound healing after electric injury revealed that u-PAR−/− smooth muscle cells, originating from the uninjured borders, migrated over a similar distance and at a similar rate into the necrotic center of the wound as wild-type (u-PAR+/+) smooth muscle cells. In addition, u-PAR deficiency did not impair migration of wounded cultured smooth muscle cells in vitro. There were no genotypic differences in reendothelialization of the vascular wound. The minimal role of u-PAR in smooth muscle cell migration was not because of absent expression, since wild-type smooth muscle cells expressed u-PAR mRNA and functional receptor in vitro and in vivo. Pericellular plasmin proteolysis, evaluated by degradation of 125I-labeled fibrin and activation of zymogen matrix metalloproteinases, was similar for u-PAR−/− and u-PAR+/+ cells. Immunoelectron microscopy of injured arteries in vivo revealed that u-PA was bound on the cell surface of u-PAR+/+ cells, whereas it was present in the pericellular space around u-PAR−/− cells. Taken together, these results suggest that binding of u-PA to u-PAR is not required to provide sufficient pericellular u-PA–mediated plasmin proteolysis to allow cellular migration into a vascular wound.
Formation of blood vessels during embryogenesis involves proliferation and migration of endothelial and smooth muscle cells. During adulthood, these vascular cell types differentiate, are quiescent, and have a prolonged lifetime. However, after arterial injury such as during vascular interventions for the treatment of atherothrombosis in patients, smooth muscle cells of the medial layer accumulate into the intima and endothelial cells migrate into denuded wounds (15). Since smooth muscle cells are “encaged” by a basement membrane, they need to proteolytically degrade extracellular matrix components to migrate to distant sites. Another cell type frequently present in the injured vessel wall is the leukocyte, which participates in removal of necrotic material, orchestrating the inflammatory response, and assisting migration of other cells by clearing a path for them (31). Smooth muscle cells, endothelial cells, and leukocytes produce a variety of matrix-degrading proteinases, of which the plasminogen system and the matrix metalloproteinase system are generally believed to play an essential role in cellular migration; because they can, in concert, break down all different matrix components of the vessel wall (7, 24, 30, 56).
Matrix metalloproteinases (MMPs)1 constitute a family of proteinases able to degrade most extracellular matrix components in the vessel wall. In the mouse, MMP-13 (collagenase 3) appears to be the primary interstitial collagenase, whereas MMP-2 (gelatinase A) and MMP-9 (gelatinase B) degrade collagen type IV, V, VII, and X, elastin, and denatured collagens (40). Since MMPs are secreted as zymogens, they require extracellular activation. We recently demonstrated that urokinase-type plasminogen activator (u-PA)–generated plasmin is a likely pathological activator of several zymogen MMPs during atherosclerotic aneurysm formation in vivo (11). Increased levels of matrix metalloproteinases have been proposed to mediate cellular migration and vessel wall remodeling during arterial intimal thickening (3, 24).
The plasminogen system comprises two plasminogen activators (PAs), tissue-type PA (t-PA), and urokinase-type PA (u-PA), which convert the proenzyme plasminogen to its active derivative, plasmin (17). They are controlled by plasminogen activator inhibitors (PAIs), of which PAI-1 appears to be the predominant physiological inhibitor. Whereas t-PA is primarily involved in clot dissolution, u-PA binds to a membrane-anchored glycoprotein (u-PAR/ CD87), which has been implicated in pericellular proteolysis during cell migration or tissue remodeling (2, 4, 8, 57). It has been claimed that binding of u-PA to u-PAR is required for its role in pericellular proteolysis because it would accelerate plasminogen activation, delay inhibition by PAI-1, regulate clearance of u-PA, and localize plasmin proteolysis to the cell surface at the leading edge of the migrating cell (2, 4, 27, 57). Recent studies suggest that u-PAR may be a multifunctional receptor, not only promoting pericellular proteolysis but also involved in integrin-mediated cell adhesion and migration via interaction with vitronectin (22, 28, 58, 59, 64) or the complement receptor type-3/ Mac-1 receptor (CD11b/CD18) (53, 61, 63). In addition, u-PAR has been implicated in intracellular signaling, cellular differentiation, growth, and chemotaxis via a mechanism independent of the enzymatic activity of u-PA but dependent on receptor occupancy (1, 20, 28, 43, 44, 46, 58).
Although expression and manipulation of u-PAR gene function have been directly correlated with cellular migration in vitro, and with primary tumor growth, tumor-associated neovascularization and metastasis in vivo (18, 19, 38, 42, 50, 62, 66), it remains to be determined whether receptor binding of u-PA is required during other (patho) biological processes in vivo. In fact, binding of u-PA to u-PAR may not be always necessary since a membrane-anchored form of u-PA catalyzes plasminogen activation equally well as receptor-bound u-PA (29), and inhibition (60) or overexpression (65) of soluble u-PA also alters cellular invasiveness. In addition, loss of u-PAR gene function does not cause appreciable morphological or functional defects in gene-inactivated mice in vivo (5, 6, 23). Indeed, in contrast to the markedly impaired fibrin surveillance and poor general health in combined t-PA/u-PA deficient mice, mice with combined t-PA/u-PAR deficiency display only minimal fibrin deposits and a normal lifespan (6).
In an uninjured artery, endothelial cell–produced t-PA may promote vascular patency. After injury, expression of t-PA, u-PA, and u-PAR by smooth muscle cells, endothelial cells, and leukocytes is significantly induced (16, 41, 45); and despite induced PAI-1 expression, a significant hyperfibrinolytic response occurs. We recently demonstrated, using a perivascular electric injury model (14), that deficiency of plasminogen or u-PA but not of t-PA, or adenoviral PAI-1 gene transfer, suppresses arterial wound healing because of impaired migration of smooth muscle cells, whereas deficiency of PAI-1 enhances this process (9, 12, 13).
In the present study, the role of u-PAR in arterial wound healing was studied in mice with an inactivated gene encoding u-PAR (u-PAR−/−). The dependence of cellular migration during arterial wound healing on u-PA–mediated plasmin proteolysis (9, 13), allowed us to examine whether binding of u-PA to u-PAR is essential for its role in controlling directional plasmin proteolysis. Surprisingly, neointima formation and smooth muscle cell migration were not impaired by deficiency of u-PAR, indicating that u-PAR is not essential for the biological role of u-PA in vascular wound healing. In fact, the present analysis suggests that pericellular u-PA around vascular cells induces sufficient pericellular plasmin and MMP proteolysis to allow normal cellular migration. These findings may have implications for the design of therapeutic strategies to reduce u-PA–mediated arterial stenosis.
Materials and Methods
Arterial Injury Model, Morphometric Analysis, Histology and Immunocytochemistry
6–8-wk-old u-PAR+/+ and u-PAR−/− mice (23) of either sex with a mixed C57Bl6 × 129/SvPas genetic background were used. Groups of 8–14 animals were studied. Perivascular electric injury of left femoral arteries and analysis of neointima (morphometric measurements of cross-sectional areas, cell counts, and proliferation rates, reendothelialization, topographic analysis, and immunostainings) were performed as described elsewhere (14). t-PA, u-PA, and PAI-1, or MMP-9 and MMP-13 were immunostained as described, except that cultured cells were pretreated for 24 h with monensin (1 μM; Sigma Chemical Co., St. Louis, MO) (9, 14).
Smooth Muscle Cell Cultures and In Vitro Migration Assay
Smooth muscle cells were prepared (9) and wound assays were performed in vitro (49) as described. Briefly, smooth muscle cells were seeded in DME containing 10% FCS onto 35-mm dishes, precoated with human fibronectin (5 μg/ml during 24 h at 4°C) to prevent retraction of wounded smooth muscle cells. After 7 d, confluent smooth muscle cells were wounded with a razor blade, washed with PBS, and further incubated for 72 h at 37°C in DME containing 0.1% gelatin and 10% FCS. After fixation with absolute methanol, the cells were stained with Giemsa. Cells that had migrated from the edge of the wound into the denuded area were counted in seven successive 125-μm increments at ×100 magnification.
In Situ Hybridization and Electron Microscopy
In situ hybridization was performed using a 1,113-nucleotide human u-PAR, or a 159-nucleotide murine u-PAR 35S-labeled riboprobe, with similar results (35). Ultrastructural analysis was performed as described (33). For immunogold labeling, the tissues were perfusion fixed for 10 min, followed by immersion fixation at room temperature for a further 2 h using 3% formaldehyde in PBS. Thereafter, the samples were dehydrated with increasing concentrations of ethanol at progressively lower temperatures. Finally, the samples were embedded overnight in 100% Lowicryl K4M at −35°C. Polymerization was carried out by irradation of tissue in gelatin capsules by indirect UV light for 24 h at −35°C, followed by final hardening under UV light at room temperature for 1–2 d. The thin sections were placed on Formvar-coated 200-mesh nickel grids, and then immunogold labeled for u-PA using a postembedding staining protocol as previously described (34). A polyclonal rabbit anti-murine u-PA antiserum (10) or a monoclonal anti-murine u-PA antibody (clone No. H77A10; 30 μg/ml) (9) were used, with similar labeling characteristics.
Reverse Transcriptase (RT)-PCR Analysis
Expression of u-PAR mRNA was evaluated using RT-PCR analysis of total RNA prepared from cultured smooth muscle cells, uninjured arteries, or arteries at the indicated periods after injury. Preparation of RNA, synthesis of first strand cDNA and PCR amplification were carried out as described (23). The following primers were used: 5′-CAGAGCTTTCCACCGAATGGCTTCC-3′ sense; and 5′-CAGCAACAATATTATCTCAACTAAGAGG-3′ and 5′-GAAACCGGAGTTATACCGTAAGAGGC- 3′, antisense.
Binding of 125I-Labeled u-PA to Cultured Smooth Muscle Cells
Binding of u-PA to smooth muscle cells was evaluated by iodination of a recombinant murine u-PA polypeptide, containing the first 48 amino acid residues (mu-PA1–48) (38), as previously described (23). Smooth muscle cells were plated at a density of 2 × 105 cells per well in 96-well plates. After overnight culture, the cells were acid-treated to dissociate endogenous u-PA from u-PAR by adding 100 μl of a 50 mM glycine-HCl buffer, pH 3.0, containing 100 mM NaCl for 3 min, which was then neutralized by addition of 20 μl of 500 mM Hepes buffer, pH 7.5, containing 100 mM NaCl. After a wash with binding medium (DME containing 0.1% sterile BSA), the cells were incubated for 1 h at room temperature in 100 μl binding medium containing 125I-labeled mu-PA1–48, with or without cold mu-PA–48 or human u-PA at the indicated concentrations. After three washes, the cells were lysed with 100 μl of 1 M NaOH, and the lysate counted for cell-associated radioactivity. These values were corrected for background radioactivity in the absence of cells. Cell loss was evaluated in separate wells, treated in an identical manner as those used for counting of radioactivity.
In Situ Autoradiography of 125I–u-PA Binding
For in situ autoradiography, cultured smooth muscle cells were plated at a density of 2–20 × 103 cells per 8-well chamber of the Lab-Tek slides (Chamber slidesTM; Nunc Inc., Naperville, IL) and cultured overnight. The cells were then washed, acid treated, and incubated in binding medium containing 125I-labeled with or without cold mu-PA1–48 as described above. After incubation, the cells were washed three times. For light microscopy, the cells were fixed with 2% glutaraldehyde in 0.1 M sodium- cacodylate buffer, pH 7.6, for 1 h, washed three times for 10 min with cacodylate buffer, dried, dipped in photographic emulsion (LM-1 emulsion; Amersham, Brussels, Belgium), and then developed after 3–14 d. For electron microscopy, the cells were fixed with 2.5% glutaraldehyde in 0.1 M HCl-sodium-cacodylate buffer, postfixed in 1% osmiumtetroxide in the same buffer, dehydrated in graded ethanol series, and then embedded in epoxy resin. Ultrathin sections were placed on Formvar-coated grids, coated in the dark by dipping in Ilford L4 radioautographic emulsion (Ilford Ltd., Mobberley Knutsford, Cheshire, UK), exposed for 14 d, and then developed in Kodak D19 and Kodak Ectaflow (Eastman Kodak, Rochester, NY) fixer before examination.
Fibrin Zymography, Plasminogen Activation, and 125I-Fibrin Degradation
Zymographic analysis of plasminogen activator activities in arterial extracts or cell-conditioned media (9, 13), in situ zymography of plasminogen activators on 7-μm arterial cryosections (9), plasminogen activation by cell-associated endogenous u-PA (in the presence of the chromogenic plasmin substrate S2403) (23), 125I-fibrin degradation (9), and quantitation of murine t-PA and u-PA antigen in cell-conditioned media by ELISA (21) were performed as described.
Macrophage Cell Culture and Metalloproteinase Assays
Peritoneal macrophages were harvested 3–5 d after intraperitoneal injection of 0.5 ml 4% Brewers thioglycollate medium (Difco Laboratories Inc., Detroit, MI) and cultured overnight with serum-free DME supplemented with 2 mM glutamine, 0.02% lactalbumine hydrolyzate, and 300 nM phorbol myristate acetate (PMA). The following day, the cells were starved with methionine/cysteine-free, serum-free DME supplemented with 2 mM glutamine, 0.02% lactalbumine hydrolyzate, and 300 nM PMA for 60 min, after which 100 μCi/ml of 35S-labeled methionine/cysteine (Amersham) and plasminogen (10 μg/ml final concentration as indicated) were added. After 8 h, the conditioned medium was immunoprecipitated with rabbit anti-murine MMP-9 or MMP-13 antibodies, and subjected to zymographic gelatin gel electrophoresis (MMP-9) or gel electrophoresis and autoradiography (MMP-9, MMP-13) (11).
Statistical Analysis
Experimental values were expressed as the mean ± SEM. Statistically significant differences between groups were calculated by analysis of variance followed by Bonferroni correction.
Results
Vascular Wound Healing in u-PAR+ /+ Mice
Electric injury denuded the endothelium and destroyed all cells in the media (<1% surviving medial cells), resulting in a wound healing response characterized by transient thrombosis, removal of necrotic debris, reendothelialization, repopulation of the media by smooth muscle cells, and the formation of a neointima with multiple layers of smooth muscle cells within 3–4 wk after injury (Fig. 1 and 2, a and b) (12). A neoadventitia developed that was rich in leukocytes and fibroblastlike cells. Wound healing and the associated arterial neointima formation, and reendothelialization initiated at the uninjured borders with subsequent progression into the necrotic center.
Figure 1.

Schematic representation of the wound healing response to perivascular electric injury and the topographic pattern of neointima formation in u-PAR+/+ and u-PAR−/− arteries. The media of an uninjured mouse femoral artery consists of two to three layers of smooth muscle cells. There are no smooth muscle cells in the intima. 2 d after injury, the injured segment in the media is depleted of smooth muscle cells. Within 1 wk after injury, cells begin to repopulate the media and a small neointima is formed at the borders of the injury. The insert shows the presumed migration of smooth muscle cells across the internal elastic lamina, within the media, and alongside the lumen. Within 3–4 wk after injury, the media is repopulated and the neointima has uniformly developed throughout the whole injured region. The vertical lines below each artery denote the equally spaced locations that were used for the topographic analysis in Tables II, III and Fig. 4.
Vascular Wound Healing in u-PAR− /− Mice
After perivascular injury, removal of the necrotic debris, repopulation of the media by smooth muscle cells, and accumulation of neointimal cells appeared to be not affected by u-PAR deficiency, as evidenced by a normal neointima at 3 wk after injury (Fig. 2 c). This was not anticipated since deficiency of u-PA significantly impairs wound healing and arterial neointima formation (Fig. 2 d displays a representative example of impaired wound healing and reduced neointima formation in u-PA−/− arteries, as previously reported [9]). Quantitative measurements of the neointimal cross-sectional area (Fig. 3 a), the intima to media ratio (Fig. 3 b), the percent luminal stenosis (Fig. 3 c), and the number of neointimal cells (Fig. 3 d) revealed that neointima formation in u-PAR−/− mice was not statistically different from that in u-PAR+/+ mice.
Figure 2.

Light microscopic analysis of vascular wound healing and neointima formation after electric injury. All sections were stained with hematoxylin-eosin. (a) uninjured u-PAR+/+ artery, revealing the presence of smooth muscle cells in the media. (b–d) artery from a u-PAR+/+ (b) and a u-PAR−/− (c) mouse (at location 5 as defined in Fig. 1) 3 wk after injury, revealing a similar neointima in u-PAR−/− as in u-PAR+/+ arteries. For comparison, an electrically injured artery from a u-PA−/− mouse (d), revealing impaired wound healing and a much smaller neointima is displayed (reproduced from reference 9). The arrows indicate the internal elastic lamina whereas the arrowheads indicate the external elastic lamina. Bar, 50 μm.
Figure 3.

Morphometric analysis in electrically injured arteries, revealing similar cross-sectional neointimal area (a), intima to media ratio (b), percent luminal stenosis (c), and neointimal cell counts (d) in u-PAR−/− and in u-PAR+/+ mice after electric injury. The data represent the mean ± SEM of measurements in 8–14 arteries. u-PAR +/+, wild-type; u-PAR −/−, u-PAR deficient.
Proliferation of Smooth Muscle Cells
To evaluate whether deficiency of u-PAR affects cellular proliferation, incorporation of 5′-bromo-2′-deoxyuridine into replicating cells was determined (Table I). Proliferation of smooth muscle cells in electrically injured arteries is best evaluated beyond 1 wk after injury, since the media and neointima are then largely devoid of leukocytes (14). As shown in Table I, medial and neointimal cell proliferation were not significantly different between u-PAR+/+ and u-PAR−/− arteries, except for an increased proliferation rate of u-PAR−/− intimal smooth muscle cells within 2 wk after injury.
Table I.
Proliferation (in Percent) of Medial and Neointimal Cells in u-PAR+ /+ and u-PAR− /− Arteries After Electric Injury
| Time after injury | ||||||||
|---|---|---|---|---|---|---|---|---|
| Week 1 | Week 2 | Week 3 | Week 4 | |||||
| Neointimal cells | ||||||||
| u-PAR+/+ | 11 ± 2 | 8 ± 2 | 5 ± 2 | 0.5 ± 0.3 | ||||
| u-PAR−/− | 8 ± 2 | 15 ± 3 | 4 ± 1 | 0.3 ± 0.3 | ||||
| Medial cells | ||||||||
| u-PAR+/+ | 29 ± 4 | 15 ± 4 | 12 ± 3 | 0.8 ± 0.5 | ||||
| u-PAR−/− | 21 ± 4 | 37 ± 4* | 23 ± 10 | 0.4 ± 0.3 | ||||
Proliferation of medial and neointimal cells was quantified by counting the number of 5′-bromo-2′-deoxyuridine–positive nuclei on five equally spaced sections per artery across the injured segment, and were averaged per artery. The values represent the mean ± SEM of these averages in groups containing between 8 and 14 arteries. u-PAR +/+, wild type; u-PAR −/−, u-PAR deficient.
P < 0.05 vs. wild type by analysis of variance.
Migration of Smooth Muscle Cells
In Vivo Migration.
After electric injury, smooth muscle cells migrate within the media and alongside the lumen from the uninjured borders into the center of the necrotic wound (14). Progression of the neointima from the borders to the center was quantitated by measuring the luminal narrowing (percent stenosis) at equally spaced positions across the injured segment. Within 1 wk after injury, a significant neointima was initiated at the borders of the injury in both u-PAR−/− arteries and in u-PAR+/+ arteries (P = NS versus u-PAR+/+ by analysis of variance) (Fig. 4 a). Within 3 wk after injury, the neointima had progressed equally far into the center of the injury in u-PAR−/− and in u-PAR+/+ arteries (P = NS) (Fig. 4 b).
Figure 4.
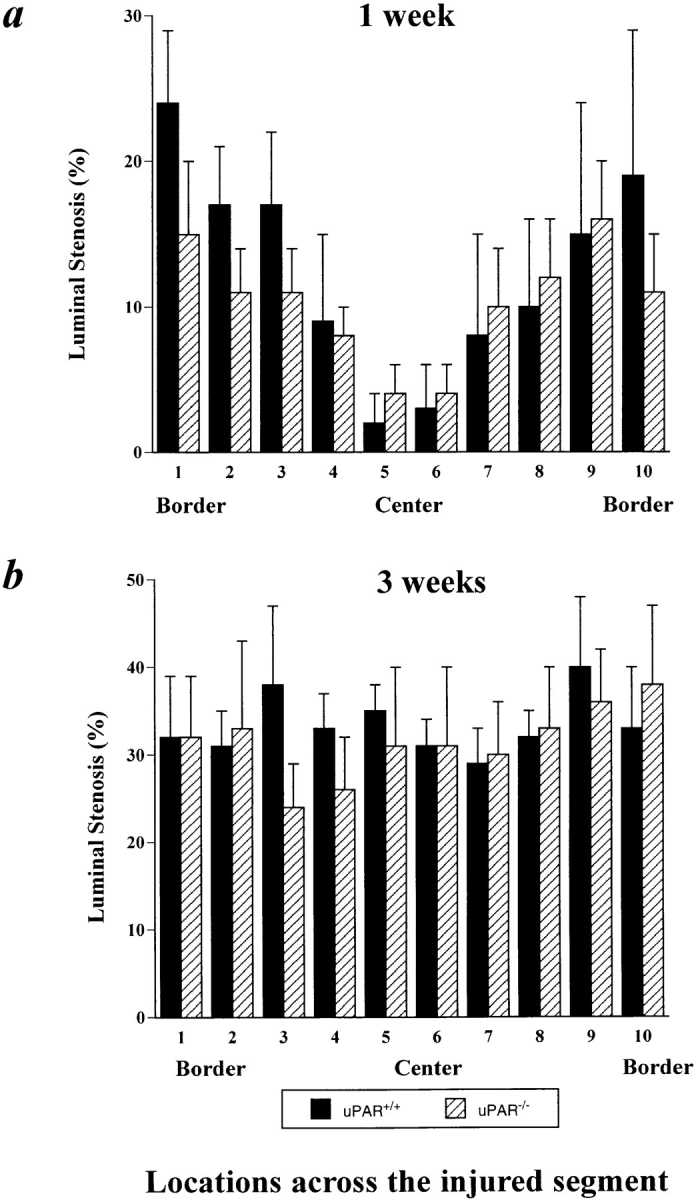
Topographic analysis of the arterial neointima formation in u-PAR+/+ and u-PAR−/− arteries within 1 (a) and 3 (b) wk after injury. Within 1 wk after injury, neointima formation initiates from the uninjured borders, whereas within 3 wk after injury, a significant neointima has formed across the entire injured segment in both u-PAR−/− and u-PAR+/+ arteries (P = NS versus u-PAR−/−). The data represent the mean ± SEM of the morphometric measurements in at least seven arteries, determined at similar relative topographic locations throughout the injured segment. The number of the relative locations throughout the injured segment refers to those defined in Fig. 1 beneath each schematically represented artery.
These findings were extended by counting the medial and neointimal cell nuclei at the left border, center, and right border of the injured segment (Fig. 1, locations 1, 5, and 10 in the arteries), revealing that the cells migrated over a similar distance and at a similar rate into the center of the wound in u-PAR−/− as in u-PAR+/+ arteries (Tables II and III). This process is schematically represented in Fig. 1. The lack of an effect of u-PAR deficiency on smooth muscle cell migration in vivo contrasts with the impaired smooth muscle cell migration in mice lacking u-PA or plasminogen (9, 13).
Table II.
Topographic Pattern of Medial Cell Repopulation
| Time after injury | Genotype | Topographic location across injury | ||||||
|---|---|---|---|---|---|---|---|---|
| Border (position 1) | Center (position 5) | Border (position 10) | ||||||
| 1 week | u-PAR+/+ | 53 ± 7 | 8 ± 3* | 58 ± 5 | ||||
| u-PAR−/− | 99 ± 12 | 22 ± 8* | 94 ± 10 | |||||
| 3 weeks | u-PAR+/+ | 62 ± 7 | 47 ± 6 | 72 ± 5 | ||||
| u-PAR−/− | 63 ± 8 | 46 ± 11 | 55 ± 13 | |||||
The data represent the mean ± SEM of the number of cells in at least seven arteries, determined at similar relative topographic locations throughout the injured arterial segment. The topographic locations are indicated on the schematically represented arteries in Fig. 1.
P < 0.05 versus uninjured border (positions 1 or 10) by analysis of variance with Bonferroni correction.
Table III.
Topographic Pattern of Neointimal Cell Accumulation
| Time after injury | Genotype | Topographic location across injury | ||||||
|---|---|---|---|---|---|---|---|---|
| Border (position 1) | Center (position 5) | Border (position 10) | ||||||
| 1 week | u-PAR+/+ | 124 ± 29 | 47 ± 14* | 117 ± 25 | ||||
| u-PAR−/− | 121 ± 31 | 30 ± 10* | 166 ± 23 | |||||
| 3 weeks | u-PAR+/+ | 184 ± 47 | 153 ± 28 | 219 ± 32 | ||||
| u-PAR−/− | 201 ± 36 | 158 ± 40 | 159 ± 33 | |||||
The data represent the mean ± SEM of the number of neointimal cells in at least seven arteries, determined at similar relative topographic locations at the uninjured borders or throughout the injured arterial segment. The topographic positions are indicated on the schematically represented arteries in Fig. 1.
P < 0.05 versus uninjured border (locations 1 or 10) by analysis of variance with Bonferroni correction.
In vitro Migration.
To confirm the lack of an effect of u-PAR in smooth muscle cell migration, cultured smooth muscle cells from wild-type and u-PAR−/− mice were wounded by scraping them from one part of the culture dish and quantifying their accumulation into the denuded area. Similar numbers of u-PAR+/+ smooth muscle cells (165 ± 12) and u-PAR−/− smooth muscle cells (163 ± 8; P = NS) accumulated into the denuded area within 72 h. However, migration of smooth muscle cells was dependent on u-PA since only 35 ± 2 u-PA−/− smooth muscle cells or 43 ± 7 u-PAR+/+ smooth muscle cells in the presence of neutralizing u-PA antibodies (50 μg/ml) accumulated in the wound. Although accumulation of cells into the denuded area could also depend on proliferation, the following observations indicate that migration was the principal (if not the only) mechanism responsible for cell accumulation. Indeed, similar numbers of u-PAR+/+ cells (25 ± 4) and u-PAR−/− cells (26 ± 2) accumulated within 6 h after scraping, i.e., before the cells had divided (the doubling time of cultured murine smooth muscle cells was estimated to be ∼10–12 h). In addition, γ-irradiation of smooth muscle cells (which blocked proliferation of wild-type and u-PAR−/− cells by >98% without cytotoxic effects on lactate dehydrogenase release) did not affect cellular accumulation. Indeed, 187 ± 13 non–γ-irradiated vs. 176 ± 19 γ-irradiated wild-type cells (P = NS), and 178 ± 21 non–γ-irradiated vs. 181 ± 18 γ-irradiated u-PAR−/−/ cells (P = NS) accumulated into the wound within 72 h after wounding. Moreover, proliferation (quantitated by [3H]thymidine incorporation) of smooth muscle cells during the 72-h migration period was similar in wild-type cells (7,830 ± 1,200 cpm/ 105 cells) as in u-PAR−/− cells (7,740 ± 1,000 cpm/105 cells; P = NS). Taken together, deficiency of u-PA but not of u-PAR impairs smooth muscle cell migration under the present experimental conditions.
Reendothelialization
Electric injury completely denuded the injured segment of intact endothelium as revealed by Evans blue staining immediately after injury (2.8 ± 0.2 mm in six u-PAR+/+ arteries versus 2.9 ± 0.3 mm in five u-PAR−/− arteries). Reendothelialization initiated from the uninjured borders into the necrotic center and was almost complete within 1 wk after injury in u-PAR+/+ mice (0.7 ± 0.2 mm; n = 7 arteries). u-PAR deficiency did not affect endothelial regrowth within 7 d after injury (0.8 ± 0.4 mm; n = 5 arteries; P = NS).
Expression of a Functional u-PAR by Smooth Muscle Cells
To exclude that u-PAR gene inactivation failed to affect smooth muscle cell migration because u-PAR would not be expressed by wild-type smooth muscle cells, expression of the u-PAR gene, and protein function by wild-type cells were analyzed.
In Situ Hybridization.
A low level of basal u-PAR mRNA expression was detectable in uninjured arteries (Fig. 5 a). Beyond 1 wk after injury, when smooth muscle cells migrate into the necrotic wound, u-PAR mRNA levels were significantly increased in cells that repopulated the media, accumulated in the neointima, and infiltrated in the adventitia (Fig. 5 c). u-PAR mRNA expression appeared to be maximal at the leading edge of the cellular migration front. Only background hybridization was detected using a control sense probe (Fig. 5, b and d). RT-PCR, followed by sequencing of cloned RT-PCR amplicons, confirmed the presence of specific u-PAR mRNA transcripts in wild-type arteries before and 1–2 wk after injury, as well as in wild-type cultured smooth muscle cells (not shown). Taken together, these data suggest that u-PAR is induced in migratory cells during wound healing, and indicate that the lack of an effect on neointima formation by u-PAR deficiency is not because of absent u-PAR expression in wild-type arteries.
Figure 5.
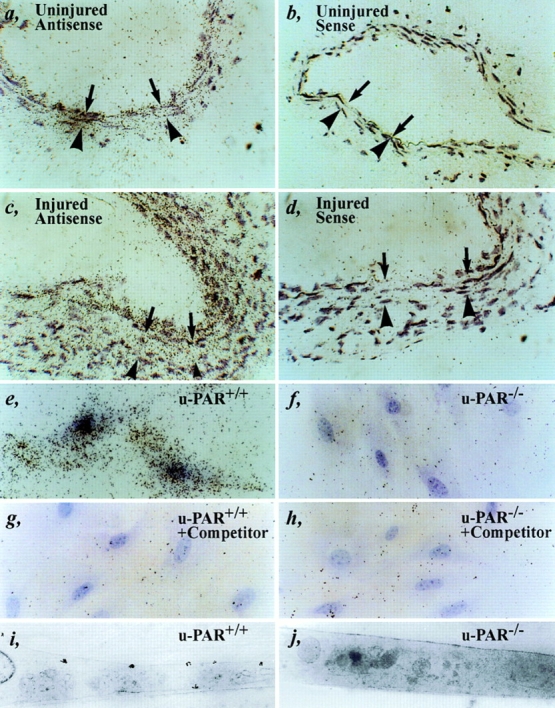
Expression of u-PAR. (a–d) In situ hybridization of an uninjured (a and b) or injured (c and d) wild-type artery within 1 wk after injury (at locations 1–3 or 8–10 as defined in Fig. 1) with an antisense (a and c) or sense (b and d) murine u-PAR probe, revealing a low basal level of u-PAR mRNA expression in a quiescent artery and significantly induced u-PAR mRNA levels at the leading front of cellular migration. The arrows indicate the internal elastic lamina; the arrowheads indicate the external elastic lamina. (e–j) Autoradiography (light microscopy: e–h, or electron microscopy: i and j) of cultured u-PAR+/+ (e, g, and i) and u-PAR−/− (f, h, and j) smooth muscle cells, incubated with 2 nM murine 1–48u-PA alone (e, f, i, and j) or together with a 100-fold excess of unlabeled competitor (g and h), revealing significant binding to u-PAR+/+ but not to u-PAR−/− smooth muscle cells. Bar, 25 μm.
Binding of 125I–mu-PA1–48 to Smooth Muscle Cells.
To provide direct proof for the presence of a functional u-PAR, binding of 125I–mu-PA1–48 to cultured smooth muscle cells was evaluated. u-PAR+/+ cells incubated with 2 nM 125I–mu-PA1–48 bound 420 ± 11 cpm 125I–mu-PA1–48. A 10- or 100-fold molar excess of unlabeled mu-PA1–48 reduced binding to 8 ± 4 cpm and 0 ± 0 cpm, respectively (P < 0.05; n = 3). In contrast, a 100-fold molar excess of human single-chain u-PA was less effective (265 ± 21 cpm; P < 0.05; n = 3), which is in line with the observations that u-PA binding to u-PAR is species-specific (26). In contrast, u-PAR−/− cells bound only 10 ± 9 cpm when incubated with 2 nM 125I–mu-PA1–48. Similar binding characteristics were observed with other murine cell types (fibroblasts and macrophages) (23).
These quantitative data were extended by in situ autoradiography to visualize binding of 125I–mu-PA1–48 on the surface of cultured smooth muscle cells. As can be seen from the light microscopic analysis, 125I–mu-PA1–48 (2 nM) bound to u-PAR+/+ cells (Fig. 5 e) but not in the presence of 100-fold molar excess of unlabeled competitor (Fig. 5 g). Background binding of 125I–mu-PA1–48 to u-PAR−/− cells (Fig. 5 f) was not affected by excess unlabeled competitor (Fig. 5 h). Electron microscopy confirmed that 125I–mu-PA1–48 was associated with the plasma membrane of u-PAR+/+ cells but not of u-PAR−/− cells (Fig. 5, i and j).
Taken together, smooth muscle cells express u-PAR during wound healing in vivo and in vitro, enabling them to bind u-PA on their cellular surface with high affinity. In addition, since binding on u-PAR+/+ cells was completely inhibited by a 100-fold excess of competitor murine u-PA, and residual binding of u-PA on u-PAR−/− cells was minimal, it is unlikely that smooth muscle cells express another high affinity u-PA–binding receptor.
Pericellular u-PA–mediated Plasmin Proteolysis
A possible explanation for the similar migration rate of u-PAR−/− versus u-PAR+/+ cells may relate to the presence of sufficient pericellular u-PA–mediated plasmin proteolysis around mutant cells. This was analyzed by evaluating the presence of u-PA antigen and activity around u-PAR−/− cells.
Immunoelectron Microscopy of u-PA in Injured Arteries In Vivo.
The localization of u-PA around migrating cells in injured arteries in vivo was ultrastructurally examined by immunogold labeling of u-PA in the injured arteries within 1 wk after injury i.e., when cell migration is maximal and u-PA is readily detectable in the injured arteries. Two antisera (a polyclonal and monoclonal) with similar labeling characteristics were used, which were previously shown to specifically detect murine u-PA, as indicated by the lack of staining in u-PA−/− arteries, by the colocalization of u-PA immunoreactivity with u-PA mRNA hybridization signals and with in situ zymographic u-PA activity, by their potential to immunoneutralize u-PA activity in murine tissue extracts or cell-conditioned media (10), and by their recognition of purified murine u-PA antigen by Western blot analysis (not shown).
In u-PAR+/+ cells, u-PA gold particles were detected in close association with the cell surface, specifically on cell projections (Fig. 6, a–c, arrows) as well as in the extracellular matrix (Fig. 6, b and c, arrowheads). In contrast, although u-PA gold particles were present in secretory granules inside u-PAR−/− cells (presumably indicating biosynthesis of u-PA) (Fig. 6, d and e, SG), they were predominantly observed in the extracellular matrix in the vicinity of u-PA producing cells (Fig. 6, d and e, arrowheads). Overall, comparable numbers of u-PA gold particles appeared to be present in the extracellular matrix around u-PAR−/− cells as bound on the surface of, and associated with the extracellular matrix around u-PAR+/+ cells (although a quantitative analysis was not performed). The various components of the extracellular matrix were not individually characterized, but u-PA gold particles were detected on collagen type I and III fibers, which can be identified because of their characteristic structural appearance (Fig. 6 b, arrowheads). In addition, u-PA gold particles appeared scattered throughout the extracellular matrix, which is known to contain laminin and fibronectin, but were only rarely detected on elastin fibers (Fig. 6 e, IEL), indicating that binding occurred preferentially to some but not to all extracellular matrix components.
Figure 6.
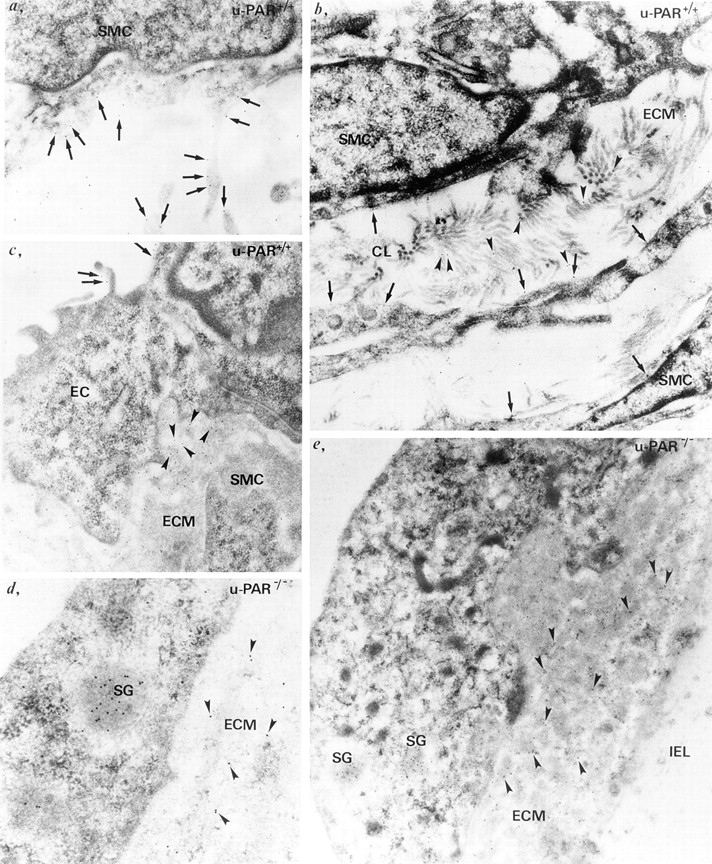
Ultrastructural localization of u-PA in vivo. Immunogold labeling of u-PA in u-PAR+/+ (a–c) and u-PAR−/− (d and e) arteries, revealing u-PA bound to the cell surface (a–c, arrows) on endothelial cells (EC) or smooth muscle cells (SMC), or associated with the extracellular matrix (ECM) (b and c, arrowheads) in u-PAR+/+ arteries. Note the presence of u-PA gold particles on cell projections (a), and on collagen (CL) fibers in the extracellular matrix (b). In contrast, in u-PAR−/− arteries, u-PA gold particles were predominantly detected in the pericellular milieu associated with extracellular matrix components (d and e, arrowheads). Note the paucity of u-PA gold particles on the elastin fibers of the internal elastic lamina (IEL) (e). Gold particles labeling u-PA were also found in secretory granules inside u-PAR−/− cells (d and e, SG).
Accumulation of u-PA in the Pericellular Milieu.
Because u-PAR has been involved in internalization of u-PA, deficiency of u-PAR may result in increased extracellular accumulation of u-PA. Therefore, smooth muscle cells, fibroblasts, and macrophages were cultured in vitro and the amount of u-PA accumulated in their conditioned medium quantitated by ELISA. As shown in Table IV, slightly higher amounts of u-PA accumulated around u-PAR−/− than u-PAR+/+ cells. Quantitation of u-PA by the more sensitive zymography revealed that u-PA activity levels (expressed as units/106 cells/48 h) were 1,600 ± 70 for u-PAR+/+ smooth muscle cells and 3,600 ± 400 for u-PAR−/− smooth muscle cells (P < 0.05 vs. wild-type).
Table IV.
Accumulation of u-PA Around uPAR+ /+and u-PAR− /−Cells
| u-PA | t-PA | |||
|---|---|---|---|---|
| Macrophages | ||||
| u-PAR+/+ | 3.6 ± 0.2 | 0.04 ± 0.02 | ||
| u-PAR−/− | 7.3 ± 0.5* | 0.10 ± 0.01 | ||
| Smooth muscle cells | ||||
| u-PAR+/+ | ⩽50 | 40 ± 4 | ||
| u-PAR−/− | 55 ± 6 | 78 ± 8* | ||
| Fibroblasts | ||||
| u-PAR+/+ | 26 ± 18 | 50 ± 10 | ||
| u-PAR−/− | 47 ± 23 | 43 ± 1 |
The data represent the mean ± SEM of the measurements (n = 2 or 3). u-PA and t-PA concentrations in the conditioned media were measured via ELISA expressed as ng/ 106 cells/48 h culture. Thioglycollate-stimulated peritoneal macrophages were freshly harvested, whereas smooth muscle cells and fibroblasts were cultured.
P < 0.05 versus wild type by analysis of variance.
125I-Fibrin Degradation and Plasminogen Activation by Endogenous u-PA.
Pericellular plasmin proteolysis by intact u-PAR−/− cells was evaluated by degradation of 125I-fibrin. Initial experiments revealed that cultured smooth muscle cells could not reliably be used for studying degradation of 125I-fibrin since they produced significant amounts of t-PA, which lysed the underlying fibrin and caused cells to detach. Therefore, thioglycollate-stimulated peritoneal macrophages were used because they produce significant amounts of u-PA but not of t-PA (10). Furthermore, macrophages significantly participate in the repair of the vascular wound after electric injury (14). Plasminogen-dependent degradation of 125I-labeled fibrin after 8 h by u-PAR+/+ and u-PAR−/− macrophages was 9,500 ± 3,500 cpm (n = 6) and 12,400 ± 2,400 cpm (n = 8), respectively (P = NS), but only 1,000 ± 300 cpm (n = 6) by u-PA−/− cells (P < 0.05 vs. wild-type and u-PAR−/− by analysis of variance). The latter was not significantly different from the plasminogen-independent degradation by wild-type cells (600 ± 200 cpm; n = 6), u-PAR−/− cells (800 ± 100 cpm; n = 9), or u-PA−/− cells (500 ± 160 cpm; n = 5). Thus, u-PAR−/− cells can degrade 125I-labeled fibrin equally well as wild-type cells, whereas u-PA−/− cells are defective in matrix degradation.
Plasminogen activation by thioglycollate-stimulated macrophages (quantitated by conversion of the chromogenic plasmin substrate S2403 and expressed in milliabsorbance at 405 nm; n = 3) was threefold lower with u-PAR−/− cells (28 ± 5) than with u-PAR+/+ cells (95 ± 19; P < 0.05) within 3 h, but comparable with u-PAR−/− cells (480 ± 120) as with u-PAR+/+ cells (590 ± 100; P = NS) within 7 h, indicating that plasminogen activation by u-PAR−/− cells was only transiently reduced (23). Taken together, the ultrastructural, fibrin degradation, and plasminogen activation data suggest that u-PA is present in the extracellular matrix around u-PA–producing cells, providing them with sufficient pericellular plasmin proteolysis to migrate.
Role of t-PA in u-PAR− /− Arteries
t-PA plays a neglible role in smooth muscle cell migration in vitro and in vivo (9). To examine whether deficiency of u-PAR would compensatorily upregulate expression of t-PA, which could rescue impaired migration of u-PAR−/− cells, extracts from uninjured arteries were zymographically analyzed for t-PA activity. u-PAR+/+ arteries contained 1.14 ± 0.19 units t-PA (n = 10), which was not different from u-PAR−/− arteries (which contained 1.37 ± 0.19 units t-PA [n = 8; P = NS versus u-PAR+/+]). Thus, it is unlikely that t-PA plays a more important role in u-PAR−/− than in u-PAR+/+ arteries.
Role of u-PAR in Activation of MMPs
Expression of MMPs during Arterial Wound Healing.
In a recent study, u-PA–generated plasmin was demonstrated to activate the proform of several MMPs, including proMMP-9 and proMMP-13 (11). Therefore, the role of u-PAR in this process was evaluated. Immunostaining revealed absent expression of MMP-9 and MMP-13 in uninjured control arteries, but significantly induced expression of MMP-9 and MMP-13 in wild-type arteries within 1 and 2 wk after injury (e.g., when cells migrate most actively) (Fig. 7). Since double-labeling studies demonstrated that MMP-9 and MMP-13 are expressed by macrophages (not shown) and macrophages play a central role in arterial wound healing in vivo, macrophages were used to study the role of u-PAR in activation of proMMP-9 and proMMP-13.
Figure 7.

Immunostaining of MMP-9 and MMP-13. MMP-9 (a and b) and MMP-13 (c and d) immunoreactivity were absent in uninjured wild-type arteries (a and c), but were markedly upregulated throughout the entire vessel wall after injury (b and d). The arrows indicate the internal elastic lamina whereas the arrowheads indicate the external elastic lamina.
Activation of proMMPs.
To study activation of proMMPs by plasmin (which occurs extracellularly), and to avoid possible contamination by artefactual activation of intracellularly stored proMMPs upon tissue extraction, thioglycollate-stimulated peritoneal macrophages were cultured in the absence or presence of plasminogen; and activation of proMMP-9 and proMMP-13 assayed by gelatin zymography of their conditioned medium (MMP-9), or by autoradiography of conditioned medium from metabolically labeled macrophages after immunoprecipitation (MMP-9, MMP-13). As shown in Fig. 8, conditioned medium from wild-type and u-PAR−/− macrophages, cultured without plasminogen, contained the inactive form of MMP-9 (∼92 kD) and MMP-13 (∼57 kD). When the macrophages were, however, cultured in the presence of plasminogen, their conditioned medium contained in addition the active cleavage product of MMP-9 (∼82 kD) and MMP-13 (∼45-kD doublet and an intermediately processed higher molecular mass form). In contrast, medium from u-PA−/− macrophages only contained the inactive MMP-9 and MMP-13 zymogens, irrespective of the presence of plasminogen in the culture medium. Taken together, these results indicate that u-PAR−/− cells produce sufficient pericellular u-PA– generated plasmin to activate proMMPs.
Figure 8.
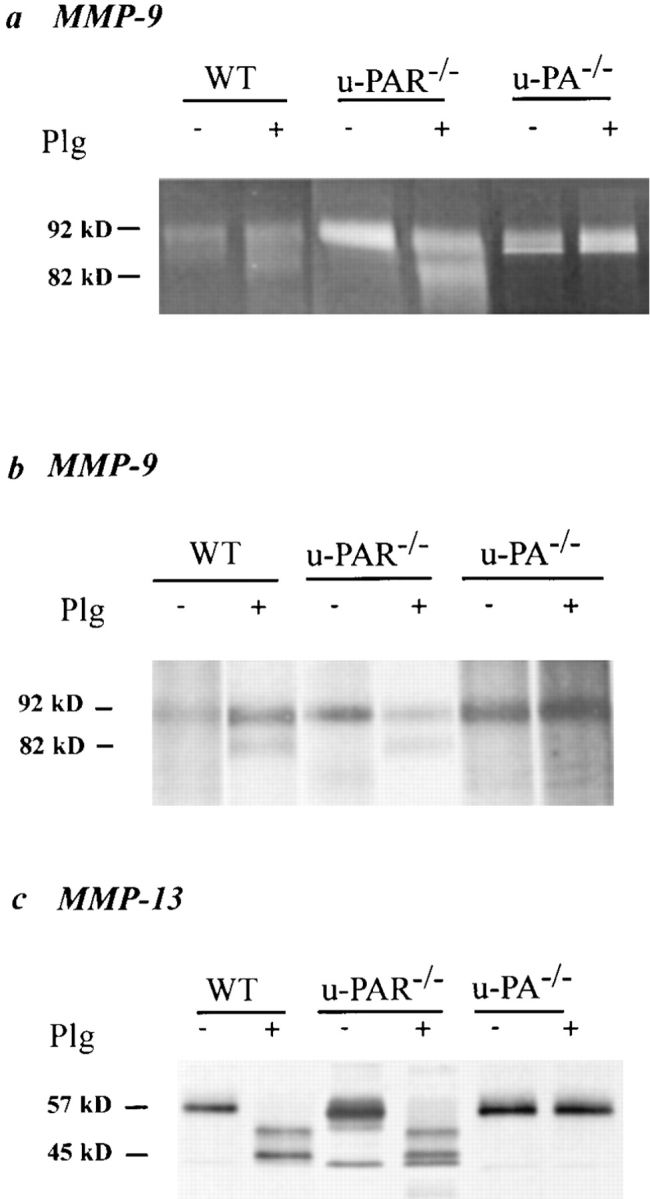
Plasmin(ogen) dependent activation of matrix metalloproteinases. (a) gelatin zymography after MMP-9 immunoprecipitation; (b and c) autoradiograph from metabolically labeled macrophage-conditioned medium after MMP-9 (b) or MMP-13 (c) immunoprecipitation. In the absence of plasminogen (−Plg), only the zymogen proMMP-9 (∼92 kD, a and b) and proMMP-13 (∼57 kD, c) are present in the conditioned medium from macrophages of all genotypes. In the presence of plasminogen (+Plg; 10 μg/ml), these proforms are processed to the active MMP-9 (∼82 kD) and MMP-13 (∼45-kD doublet and partially processed forms) by wild-type (WT) and u-PAR–deficient (u-PAR −/−) macrophages but not by u-PA– deficient (u-PA −/−) macrophages.
Discussion
Receptor-independent Role of u-PA in Smooth Muscle Cell Migration
It has been proposed that binding of u-PA to u-PAR is required for its proteolytic activity, since this might accelerate plasminogen activation, concentrate u-PA onto the cell surface, and possibly protect u-PA from inactivation by PAI-1 (2, 4, 27, 57). We previously demonstrated that smooth muscle cells produce and require u-PA for migration and formation of a neointima. Since smooth muscle cell migration was impaired by u-PA deficiency (9) as well as by plasminogen deficiency (13), the biological role of u-PA during vascular wound healing appears to depend primarily on generation of plasmin. The latter mediates degradation of the extracellular matrix components either directly or indirectly via activation of zymogen MMPs. In the present study, evidence is provided for a receptor- independent role of u-PA in smooth muscle cell migration. Indeed, the observations that u-PAR deficiency did not affect the initiation, formation, and progression of the neointima from the uninjured borders into the necrotic center of the wound, and that migration of smooth muscle cells in vivo and in vitro was unaffected by u-PAR deficiency, indicates that u-PAR is not essential for u-PA–dependent plasmin proteolysis during smooth muscle cell migration (see also Fig. 1 for schematic representation). The present study does, however, not exclude that u-PA may also affect cellular migration via nonproteolytic mechanisms, e.g., via binding of the NH2-terminal fragment of u-PA to u-PAR with subsequent signaling (1, 20, 28, 43, 44, 46, 58). Elucidation of such a mechanism in vivo may, however, have to await the generation of transgenic mice selectively expressing mutant u-PA that is catalytically inactive but still able to bind u-PAR.
Lack of an Effect of u-PAR Is Not Because of Lack of u-PAR Expression by Wild-Type Cells or to Compensatory t-PA Overexpression
There are several possibilities why loss of u-PAR gene function might not result in impairment of neointima formation, as observed in u-PA–deficient arteries. One reason is that u-PAR may not be expressed by smooth muscle cells during vascular wound healing. However, the observations that smooth muscle cells express u-PAR mRNA in vitro and in vivo, and that they possess a functional u-PAR, able to bind u-PA with similar affinity as observed for other cell types (41, 45), excludes this possibility and makes it unlikely that u-PAR has modified binding properties. In addition, there was no evidence for another u-PA–binding receptor with a similar high affinity on smooth muscle cells, consistent with previous observations (5, 23).
A second possible reason is that compensatorily upregulated levels of t-PA in u-PAR−/− arteries could rescue migration of u-PAR−/− smooth muscle cells. However, t-PA activity levels were similar in wild-type and in mutant arteries. In addition, t-PA plays a neglible role in smooth muscle cell migration in vitro and in vivo (9). Furthermore, a compensatory upregulation of t-PA expression in u-PA−/− or u-PAR−/− mice was never observed. In fact, it might have been expected that deficiency of u-PA rather than of u-PAR would compensatorily increase t-PA, since total plasminogen activator activity is lower in u-PA−/− than in u-PAR−/− mice. Nevertheless, t-PA activity was not compensatorily increased in u-PA−/− mice (9).
Pericellular Localization of u-PA and u-PA–mediated Plasmin and MMP Proteolysis Around u-PAR− /− Cells
Another explanation for the negligible role of u-PAR in smooth muscle cell migration is that u-PA would not need to be bound to u-PAR but could be present in the pericellular environment to provide sufficient u-PA–mediated plasmin proteolysis to u-PAR−/− cells. This hypothesis is substantiated by our findings that u-PA accumulates in slightly increased amounts in the conditioned medium around u-PAR−/− cells in vitro, that u-PA is detected ultrastructurally in the surrounding extracellular matrix in u-PAR−/− arteries in vivo, and that pericellular plasmin proteolysis (evidenced by 125I-fibrin degradation) or matrix metalloproteinase proteolysis (evidenced by proMMP-9 and proMMP-13 activation) by u-PAR−/− cells is similar to that produced by u-PAR+/+ cells. Such a mechanism is further supported by previous observations that membrane-anchored u-PA (29), or soluble u-PA (60, 65), is biologically active.
The role of u-PAR in plasminogen activation remains controversial. u-PAR on whole cells accelerates plasminogen activation by u-PA within a period of <1 h, as the addition of human U937 cells to mixtures of plasminogen and u-PA resulted in a significant increase in plasmin generation (25). Since nM concentrations of u-PA (e.g., within the range of the Kd for u-PA-binding to u-PAR) were added to cells in suspension, binding of u-PA to u-PAR (and not to other binding sites, such as those in the extracellular matrix) occurred. This increase was abolished by agents blocking the cellular binding site of either u-PA or plasminogen, indicating the dependence of the accelerating effect on the presence of both types of binding sites (4). In contrast, acceleration of plasminogen activation was not observed in a similar assay using murine u-PA–deficient endothelioma cells, in spite of the presence of functional u-PAR on these cells (32). These conflicting results might relate to differences between the human and murine system, or to the characteristics of the cell lines used.
These assays may, however, not reliably reflect the in vivo situation in which not only cellular u-PAR but also alternative u-PA–binding sites in the pericellular milieu may contribute to the biological role of u-PA. Indeed, migrating cells in the electrically injured arteries produce >100-fold increased u-PA levels during prolonged periods (days to weeks). Consequently, the pericellular u-PA levels might raise above those needed to saturate binding to u-PAR and allow its binding to other sites in the pericellular milieu (cf the ultrastructural analysis in vivo). Consistent with such a hypothesis, adherent u-PAR–deficient macrophages generated less plasmin upon short incubation (acceleration of plasminogen activation by u-PAR) (4), whereas they generated comparable plasmin levels to wild-type cells upon prolonged incubation (e.g., >4 h, a time period sufficient for accumulation of u-PA in the pericellular milieu and supernatant) (23). Similarly, the lack of effect of u-PAR deficiency on 125I-fibrin degradation or cell migration, both processes requiring prolonged assay times (hours to days), might be explained by the presence of sufficient u-PA in the pericellular milieu. Taken together, although u-PAR may accelerate plasmin generation over a period of minutes to hours, this kinetic advantage may be less significant for slow processes (requiring days to weeks to develop) than the total amount of plasmin generated.
To what components in the extracellular matrix u-PA binds, remains to be determined, but previous studies have demonstrated that u-PA can bind fibrin, heparin, laminin, thrombospondin, and fibronectin (17, 36, 37, 39, 48, 51, 52, 54, 55). Furthermore, since plasminogen also binds some of the above-mentioned matrix components, u-PA (through binding with plasminogen) can be closely associated with the extracellular matrix. The present experiments do not exclude that u-PA may bind to another low affinity cellular or soluble receptor, although no evidence for such a molecule has been provided to date.
Cellular Proliferation
Cellular proliferation was not affected by deficiency of u-PAR, except for a transient increase of neointimal cell proliferation within 2 wk after injury. It is unclear why deficiency of u-PAR would induce cell proliferation. u-PAR has been implicated in cell growth and other cellular processes via mediating intracellular signaling (20, 43), but to date, an inhibitory role for u-PAR in cell growth has not been documented. Smooth muscle cells dedifferentiate when they proliferate and migrate into the intima (as evidenced by their reduced α-actin expression) (14) but redifferentiate after their arrival in the intima (increased α-actin expression) (14). Whether u-PAR deficiency would prevent/delay smooth muscle cell differentiation, as in macrophages (44), remains to be determined.
Role of u-PAR in Endothelial Cell Regeneration
Endothelial cell regrowth was not affected by u-PAR deficiency. However, reendothelialization was also not affected by deficiency of u-PA, t-PA, PAI-1, or plasminogen (9, 13), indicating that regenerating endothelial cells may not require plasmin to migrate alongside the denuded internal elastic lamina. This is somewhat surprising in view of the induced expression of plasminogen activators in regenerating endothelium in vivo and in vitro after injury (45, 57). The plasminogen system plays, however, a more essential role during pathological blood vessel formation when endothelial cells migrate through anatomic barriers (47).
Requirement of u-PAR in Other Biological Processes
Previous studies have demonstrated that inhibition of u-PAR gene function, either by antisense to u-PAR or by inhibition of the u-PA/u-PAR interaction, suppresses primary tumor growth, tumor-associated neovascularization, and metastasis (19, 38, 42, 66). Apparently, u-PAR plays a more essential role in these processes than during arterial neointima formation. This may, however, be related to differences in the level or the temporospatial pattern of u-PAR expression, or the contextual requirement for u-PA–mediated plasmin proteolysis. In addition, u-PAR is a multifunctional receptor involved in vitro in pericellular proteolysis, cell proliferation, differentiation, and adhesion, and the contribution of each of these mechanisms may determine the requirement of u-PAR in different biological processes. Unresolved issues include whether u-PAR may have other biological functions during vascular wound healing, for example on cellular differentiation or adhesion, which were not revealed by our analysis, or whether receptor binding is essential during other u-PA–dependent biological processes in vivo. The growing list of phenotypes resulting from loss of u-PA gene function in gene-inactivated mice, which include fibrin deposition (10), impaired myocardial healing (unpublished data), and atherosclerotic aneurysm formation (11) may aid in addressing this issue in the future. In particular, since u-PAR has been implicated in cancer, appropriate models in the gene-inactivated mice may be useful to unravel its possible role in this process.
In conclusion, deficiency of u-PAR does not affect arterial neointima formation, most likely because accumulation of u-PA in the extracellular environment compensates for binding on its cellular receptor. These data suggest that antifibrinolytic therapy for arterial stenosis should be targeted at inhibiting u-PA activity rather than its binding on u-PAR.
Acknowledgments
The authors thank Dr. R. Lijnen for the MMP-9 antiserum, Dr. Y. Eeckhout for the MMP-13 antiserum, V. Attenburrow, A. Bouché, I. Cornelissen, M. De Mol, B. Hermans, S. Janssen, A. Manderveld, A. Van den Bomen, S. Wyns (CTG, Leuven, Belgium), A. Westmuckett, D. Goulding, J. Crawley (Thrombosis Research Institute, London, UK), and Jennifer Stratton-Thomas (Chiron, Emeryville, CA) for their help; and M. Deprez for artwork.
Abbreviations used in this paper
- MMP
matrix metalloproteinase
- PA
plasminogen activator
- PAI
PA inhibitors
- PMA
phorbol myristate acetate
- RT
reverse transcriptase
- t-PA
tissue-type PA
- u-PA
urokinase-type PA
Footnotes
The work of the London group was supported by the British Heart Foundation.
Address all correspondence to P. Carmeliet, Center for Transgene Technology and Gene Therapy, Flanders Interuniversity Institute for Biotechnology, Campus Gathuisberg, KU Leuven, Herestraat 49, B-3000 Leuven, Belgium. Tel.: 32-16-345772. Fax: 32-16-345990. E-mail: peter.carmeliet@med.kuleuven.ac.be
References
- 1.Anichini E, Fibbi G, Pucci M, Caldini R, Chevanne M, Del Rosso M. Production of second messengers following chemotactic and mitogenic urokinase-receptor interaction in human fibroblasts and mouse fibroblasts transfected with human urokinase receptor. Exp Cell Res. 1994;213:438–448. doi: 10.1006/excr.1994.1221. [DOI] [PubMed] [Google Scholar]
- 2.Behrendt N, Ronne E, Dano K. The structure and function of the urokinase receptor, a membrane protein governing plasminogen activation on the cell surface. Biol Chem Hoppe Seyler. 1995;376:269–279. [PubMed] [Google Scholar]
- 3.Bendeck MP, Zempo N, Clowes AW, Galardy RE, Reidy MA. Smooth muscle cell migration and matrix metalloproteinase expression after arterial injury in the rat. Circ Res. 1994;75:539–545. doi: 10.1161/01.res.75.3.539. [DOI] [PubMed] [Google Scholar]
- 4.Blasi F, Conese M, Moller LB, Pedersen N, Cavallaro U, Cubellis MV, Fazioli F, Hernandez-Marrero L, Limongi P, Munoz-Canoves P, et al. The urokinase receptor: structure, regulation and inhibitor- mediated internalization. Fibrinolysis. 1994;8:182–188. [Google Scholar]
- 5.Bugge TH, Suh TT, Flick MJ, Daugherty CC, Romer J, Solberg H, Ellis V, Dano K, Degen JL. The receptor for urokinase-type plasminogen activator is not essential for mouse development or fertility. J Biol Chem. 1995;270:16886–16894. doi: 10.1074/jbc.270.28.16886. [DOI] [PubMed] [Google Scholar]
- 6.Bugge TH, Flick MJ, Danton MJ, Daugherty CC, Romer J, Dano K, Carmeliet P, Collen D, Degen JL. Urokinase-type plasminogen activator is effective in fibrin clearance in the absence of its receptor or tissue-type plasminogen activator. Proc Natl Acad Sci USA. 1996;93:5899–5904. doi: 10.1073/pnas.93.12.5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carmeliet P, Collen D. Role of the plasminogen/plasmin system in thrombosis, hemostasis, restenosis and atherosclerosis. Evaluation in transgenic animals. Trends Cardiovasc Med. 1995;5:117–122. doi: 10.1016/1050-1738(95)00050-J. [DOI] [PubMed] [Google Scholar]
- 8.Carmeliet P, Collen D. Gene manipulation and transfer of the plasminogen system and coagulation system in mice. Semin Thromb Hemostasis. 1996;22:525–542. doi: 10.1055/s-2007-999055. [DOI] [PubMed] [Google Scholar]
- 9.Carmeliet P, Moons L, Herbert J-M, Crawley J, Lupu F, Lijnen R, Collen D. Urokinase-type but not tissue-type plasminogen activator mediates arterial neointima formation in mice. Circ Res. 1997;81:829–839. doi: 10.1161/01.res.81.5.829. [DOI] [PubMed] [Google Scholar]
- 10.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord JJ, Collen D, Mulligan RC. Physiological consequences of loss of plasminogen activator gene function in mice. Nature. 1994;368:419–424. doi: 10.1038/368419a0. [DOI] [PubMed] [Google Scholar]
- 11.Carmeliet P, Moons L, Lijnen R, Baes M, Lemaître V, Tipping P, Drew A, Eeckhout Y, Shapiro S, Lupu F, Collen D. Urokinase-generated plasmin is a candidate activator of matrix metalloproteinases during atherosclerotic aneurysm formation. Nat Genet. 1997;17:439–444. doi: 10.1038/ng1297-439. [DOI] [PubMed] [Google Scholar]
- 12.Carmeliet P, Moons L, Lijnen R, Janssens S, Lupu F, Collen D, Gerard RD. Inhibitory role of plasminogen activator inhibitor-1 in arterial wound healing and neointima formation. A gene targeting and gene transfer study in mice. Circulation. 1997;96:3180–3191. doi: 10.1161/01.cir.96.9.3180. [DOI] [PubMed] [Google Scholar]
- 13.Carmeliet P, Moons L, Ploplis V, Plow EF, Collen D. Impaired arterial neointima formation in mice with disruption of the plasminogen gene. J Clin Invest. 1997;99:200–208. doi: 10.1172/JCI119148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carmeliet P, Moons L, Stassen JM, van den Oord JJ, Declercq C, Kockx M, Collen D. A model for arterial neointima formation using perivascular electric injury in mice. Am J Pathol. 1997;150:761–777. [PMC free article] [PubMed] [Google Scholar]
- 15.Clowes AW, Reidy MA, Clowes MM. Mechanisms of stenosis after arterial injury. Lab Invest. 1983;49:208–215. [PubMed] [Google Scholar]
- 16.Clowes AW, Clowes MM, Au YP, Reidy MA, Belin D. Smooth muscle cells express urokinase during mitogenesis and tissue-type plasminogen activator during migration in injured rat carotid artery. Circ Res. 1990;67:61–67. doi: 10.1161/01.res.67.1.61. [DOI] [PubMed] [Google Scholar]
- 17.Collen D, Lijnen HR. Basic and clinical aspects of fibrinolysis and thrombolysis. Blood. 1991;78:3114–3124. [PubMed] [Google Scholar]
- 18.Conese M, Blasi F. The urokinase/urokinase-receptor system and cancer invasion. Baillieres Clin Haemotol. 1995;8:365–389. doi: 10.1016/s0950-3536(05)80273-2. [DOI] [PubMed] [Google Scholar]
- 19.Crowley CW, Cohen RL, Lucas BK, Liu G, Shuman MA, Levinson AD. Prevention of metastasis by inhibition of the urokinase receptor. Proc Natl Acad Sci USA. 1993;90:5021–5025. doi: 10.1073/pnas.90.11.5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Petro G, Copeta A, Barlati S. Urokinase-type and tissue-type plasminogen activators as growth factors of human fibroblasts. Exp Cell Res. 1994;213:286–294. doi: 10.1006/excr.1994.1200. [DOI] [PubMed] [Google Scholar]
- 21.Declerck PJ, Verstreken M, De Cock F, Carmeliet P, Collen D. Generation of monoclonal antibodies against autologous proteins in gene-inactivated mice. J Biol Chem. 1995;270:8397–8400. doi: 10.1074/jbc.270.15.8397. [DOI] [PubMed] [Google Scholar]
- 22.Deng G, Curriden SA, Wang S, Rosenberg S, Loskutoff DJ. Is plasminogen activator inhibitor–1 the molecular switch that governs urokinase receptor-mediated adhesion and release? . J Cell Biol. 1996;134:1563–1571. doi: 10.1083/jcb.134.6.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dewerchin M, Van Nuffelen A, Wallays G, Bouche A, Moons L, Carmeliet P, Mulligan RC, Collen D. Generation and characterization of urokinase receptor-deficient mice. J Clin Invest. 1996;97:870–878. doi: 10.1172/JCI118489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dollery CM, McEwan JR, Henney AM. Matrix metalloproteinases and cardiovascular disease. Circ Res. 1995;77:863–868. doi: 10.1161/01.res.77.5.863. [DOI] [PubMed] [Google Scholar]
- 25.Ellis V, Scully MF, Kakkar VV. Plasminogen activation initiated by single-chain urokinase-type plasminogen activator. Potentiation by U937 monocytes. J Biol Chem. 1989;264:2185–2188. [PubMed] [Google Scholar]
- 26.Estreicher A, Wohlwend A, Belin D, Schleuning WD, Vassalli JD. Characterization of the cellular binding site of the urokinase-type plasminogen activator. J Biol Chem. 1989;264:1180–1189. [PubMed] [Google Scholar]
- 27.Higazi AA, Mazar A, Wang J, Reilly R, Henkin J, Kniss D, Cines D. Single-chain urokinase type plasminogen activator bound to its receptor is relatively resistant to plasminogen activator inhibitor type 1. Blood. 1996;87:3545–3549. [PubMed] [Google Scholar]
- 28.Kanse SM, Kost C, Wilhelm OG, Andreasen PA, Preissner KT. The urokinase receptor is a major vitronectin-binding protein on endothelial cells. Exp Cell Res. 1996;224:344–353. doi: 10.1006/excr.1996.0144. [DOI] [PubMed] [Google Scholar]
- 29.Lee SW, Kahn ML, Dichek DA. Expression of an anchored urokinase in the apical endothelial cell membrane. Preservation of enzymatic activity and enhancement of cell surface plasminogen activation. J Biol Chem. 1992;267:13020–13027. [PubMed] [Google Scholar]
- 30.Libby P. Molecular basis of the acute coronary syndromes. Circulation. 1995;91:2844–2850. doi: 10.1161/01.cir.91.11.2844. [DOI] [PubMed] [Google Scholar]
- 31.Libby, P., D. Schwartz, E. Brogi, H. Tanaka, and S.K. Clinton. 1992. A cascade model for restenosis. A special case of atherosclerosis progression. Circulation. 86(Suppl.):47–52. [PubMed]
- 32.Lijnen HR, Van Hoef B, Collen D. Characterization of the murine plasminogen/urokinase-type plasminogen activator system. Eur J Biochem. 1996;241:840–848. doi: 10.1111/j.1432-1033.1996.00840.x. [DOI] [PubMed] [Google Scholar]
- 33.Lupu F, Danricu I, Simionescu N. The development of intracellular lipid deposits in the lipid laden cells of the atherosclerotic lesions. A cytochemical and ultrastructural study. Atherosclerosis. 1987;67:127–142. doi: 10.1016/0021-9150(87)90273-5. [DOI] [PubMed] [Google Scholar]
- 34.Lupu F, Bergonzelli GE, Heim DA, Cousin E, Genton CY, Bachmann F, Kruithof EKO. Localization and production of plasminogen activator inhibitor-1 in human healthy and atherosclerotic arteries. Arterioscler Thromb. 1993;13:1090–1100. doi: 10.1161/01.atv.13.7.1090. [DOI] [PubMed] [Google Scholar]
- 35.Lupu F, Heim DA, Bachmann F, Hurni M, Kakkar VV, Kruithof EK. Plasminogen activator expression in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 1995;15:1444–1455. doi: 10.1161/01.atv.15.9.1444. [DOI] [PubMed] [Google Scholar]
- 36.Martinez J, Santibanez JF. Surface-associated plasminogen activation in leukemic cells: interaction with extracellular matrix. Eur J Cell Biol. 1994;64:257–263. [PubMed] [Google Scholar]
- 37.McGuire PG, Seeds NW. The interaction of plasminogen activator with a reconstituted basement membrane matrix and extracellular macromolecules produced by cultured epithelial cells. J Cell Biochem. 1989;40:215–227. doi: 10.1002/jcb.240400210. [DOI] [PubMed] [Google Scholar]
- 38.Min HY, Doyle LV, Vitt CR, Zandonella CL, Stratton-Thomas JR, Shuman MA, Rosenberg S. Urokinase receptor antagonists inhibit angiogenesis and primary tumor growth in syngenic mice. Cancer Res. 1996;56:2428–2433. [PubMed] [Google Scholar]
- 39.Moser TL, Enghild JJ, Pizzo SV, Stack MS. Specific binding of urinary-type plasminogen activator (u-PA) to vitronectin and its role in mediating u-PA-dependent adhesion of U937 cells. Biochem J. 1995;307:867–873. doi: 10.1042/bj3070867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murphy G. Matrix metalloproteinases and their inhibitors. Acta Orthop Scand Suppl. 1995;66:55–60. [PubMed] [Google Scholar]
- 41.Okada SS, Tomaszewski JE, Barnathan ES. Migrating vascular smooth muscle cells polarize cell surface urokinase receptor after injury in vitro. Exp Cell Res. 1995;217:180–187. doi: 10.1006/excr.1995.1077. [DOI] [PubMed] [Google Scholar]
- 42.Ossowski L. Effect of antisense inhibition of urokinase receptor on malignancy. Curr Top Microbiol Immunol. 1996;213:101–112. doi: 10.1007/978-3-642-80071-9_7. [DOI] [PubMed] [Google Scholar]
- 43.Rabbani SA, Mazar AP, Bernier SM, Haq M, Bolivar I, Henkin J, Goltzman D. Structural requirements for the growth factor activity of the amino-terminal domain of urokinase. J Biol Chem. 1992;267:14151–14156. [PubMed] [Google Scholar]
- 44.Rao NK, Shi GP, Chapman HA. Urokinase receptor is a multifunctional protein: influence of receptor occupancy on macrophage gene expression. J Clin Invest. 1995;96:464–474. doi: 10.1172/JCI118057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reidy MA, Irvin C, Lindner V. Migration of arterial wall cells. Expression of plasminogen activators and inhibitors in injured rat arteries. Circ Res. 1996;78:405–414. doi: 10.1161/01.res.78.3.405. [DOI] [PubMed] [Google Scholar]
- 46.Resnati M, Guttinger M, Valcamonica S, Sidenius N, Blasi F, Fazioli F. Proteolytic cleavage of the urokinase receptor substitutes for the agonist-induced chemotactic effect. EMBO (Eur Mol Biol Organ) J. 1996;15:1572–1582. [PMC free article] [PubMed] [Google Scholar]
- 47.Sabapathy KT, Pepper MS, Keifer F, Möhel-Steinlein U, Tacchini-Cottier F, Fetka I, Breier G, Risau W, Carmeliet P, Montesano R, Wagner EF. Polyoma middle T-induced vascular tumor formation: the role of the plasminogen activator/plasmin system. J Cell Biol. 1997;137:953–963. doi: 10.1083/jcb.137.4.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salonen EM, Zitting A, Vaheri A. Laminin interacts with plasminogen and its tissue-type activator. FEBS (Fed Eur Biochem Soc) Lett. 1984;172:29–32. doi: 10.1016/0014-5793(84)80866-2. [DOI] [PubMed] [Google Scholar]
- 49.Sato K, Rifkin D. Autocrine activities of basic fibroblast growth factor: Regulation of endothelial cell movement, plasminogen activator synthesis, and DNA synthesis. J Cell Biol. 1988;107:1199–1205. doi: 10.1083/jcb.107.3.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmitt M, Wilhelm O, Janicke F, Magdolen V, Reuning U, Ohi H, Moniwa N, Kobayashi H, Weidle U, Graeff H. Urokinase-type plasminogen activator (u-PA) and its receptor (CD87): a new target in tumor invasion and metastasis. J Obstet Gynaecol. 1995;21:151–165. doi: 10.1111/j.1447-0756.1995.tb01089.x. [DOI] [PubMed] [Google Scholar]
- 51.Silverstein RL, Harpel PC, Nachman RL. Tissue plasminogen activator and urokinase enhance the binding of plasminogen to thrombospondin. J Biol Chem. 1986;261:9959–9965. [PubMed] [Google Scholar]
- 52.Silverstein RL, Nachman RL, Pannell R, Gurewich V, Harpel PC. Thrombospondin forms complexes with single-chain and two-chain forms of urokinase. [published erratum appears in J. Biol. Chem. 1990. 265:16025.] . J Biol Chem. 1990;265:11289–11294. [PubMed] [Google Scholar]
- 53.Sitrin RG, Todd RF, Albrecht E, Gyetko MR. The urokinase receptor (CD87) facilitates CD11b/CD18-mediated adhesion of human monocytes. J Clin Invest. 1996;97:1942–1951. doi: 10.1172/JCI118626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stephens RW, Bokman AM, Myohanen HT, Reisberg T, Tapiovaara H, Pedersen N, Grondahl J, Hansen, Llinas M, Vaheri A. Heparin binding to the urokinase kringle domain. Biochemistry. 1992;31:7572–7579. doi: 10.1021/bi00148a019. [DOI] [PubMed] [Google Scholar]
- 55.Stephens RW, Aumailley M, Timpl R, Reisberg T, Tapiovaara H, Myohanen H, Murphy-Ullrich J, Vaheri A. Urokinase binding to laminin-nidogen. Structual requirements and interactions with heparin. Eur J Biochem. 1992;207:937–942. doi: 10.1111/j.1432-1033.1992.tb17127.x. [DOI] [PubMed] [Google Scholar]
- 56.van Leeuwen RT. Extracellular proteolysis and the migrating vascular smooth muscle cell. Fibrinolysis. 1996;10:59–74. [Google Scholar]
- 57.Vassalli JD. The urokinase receptor. Fibrinolysis. 1994;8:172–181. [Google Scholar]
- 58.Waltz DA, Chapman HA. Reversible cellular adhesion to vitronectin linked to urokinase receptor occupancy. J Biol Chem. 1994;269:14746–14750. [PubMed] [Google Scholar]
- 59.Wei Y, Lukashev M, Simon DI, Bodary SC, Rosenberg S, Doyle MV, Chapman HA. Regulation of intergrin function by the urokinase receptor. Science. 1996;273:1551–1555. doi: 10.1126/science.273.5281.1551. [DOI] [PubMed] [Google Scholar]
- 60.Wilhelm O, Weidle U, Hohl S, Rettenberger P, Schmitt M, Graeff H. Recombinant soluble urokinase receptor as a scavenger for urokinase-type plasminogen activator (u-PA). Inhibition of proliferation and invasion of human ovarian cancer cells. FEBS (Fed Eur Biochem Soc) Lett. 1994;337:131–134. doi: 10.1016/0014-5793(94)80259-9. [DOI] [PubMed] [Google Scholar]
- 61.Wong WS, Simon DI, Rosoff PM, Rao NK, Chapman HA. Mechanisms of pertussis toxin-induced myelomonocytic cell adhesion: role of Mac-1 (CD11B/CD18) and urokinase receptor (CD87) . Immunology. 1996;88:90–97. doi: 10.1046/j.1365-2567.1996.d01-646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xing RH, Rabbani SA. Overexpression of urokinase receptor in breast cancer cells results in increased tumor invasion, growth and metastasis. Int J Cancer. 1996;67:423–429. doi: 10.1002/(SICI)1097-0215(19960729)67:3<423::AID-IJC18>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 63.Xue W, Kindzelskii AL, Todd RFR, Petty HR. Physical association of complement receptor type 3 and urokinase-type plasminogen activator receptor in neutrophil membranes. J Immunol. 1994;152:4630–4640. [PubMed] [Google Scholar]
- 64.Yebra M, Parry GCN, Stromblad S, Mackman N, Rosenberg S, Mueller BM, Cheresh DA. Requirement of receptor-bound urokinase-type plasminogen activator for integrin αvβ-5-directed cell migration. J Biol Chem. 1996;271:29393–29399. doi: 10.1074/jbc.271.46.29393. [DOI] [PubMed] [Google Scholar]
- 65.Yu HR, Schultz RM. Relationship between secreted urokinase plasminogen activator activity and metastatic potential in murine B16 cells transfected with human urokinase sense and antisense genes. Cancer Res. 1990;50:7623–7633. [PubMed] [Google Scholar]
- 66.Yu W, Kin J, Ossowski L. Reduction in surface urokinase receptor forces malignant cells into a protracted state of dormancy. J Cell Biol. 1997;137:767–777. doi: 10.1083/jcb.137.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]