

Abstract
It is not known how membrane fusion proteins that function at neutral pH, for example the human immunodeficiency virus envelope (Env) glycoprotein and intracellular fusion machines, are activated for target bilayer binding. We have addressed this question using a soluble oligomeric form of an avian retroviral Env glycoprotein (API) and soluble forms of its receptor. Binding of soluble receptor to API induces API to bind to liposomes composed of phosphatidylcholine and cholesterol at neutral pH. Liposome binding only occurs at fusion permissive temperatures (T > 20°C), is complete between 2 to 5 min at 37°C, and is stable to high salt, carbonate, and urea. Liposome binding is mediated by the ectodomain of the transmembrane subunit of API, and a mutant with a Val to Glu substitution in the Env fusion peptide (located in the ectodomain of the transmembrane subunit) shows significantly reduced liposome binding. Moreover, under conditions of equivalent binding to API, a mutant receptor that does not support infection (Zingler, K., and J.A.T. Young. 1996. J. Virol. 70:7510–7516) does not induce significant liposome binding. Our results indicate that a highly specific interaction between an avian retroviral Env and its receptor activates the retroviral glycoprotein for target bilayer binding at neutral pH in much the same way as low pH activates the influenza hemagglutinin. Our findings are discussed in terms of the mechanisms of viral and cellular fusion proteins that function at neutral pH.
Protein-mediated membrane fusion is a step in many important biological processes (Rothman and Warren, 1994; Hernandez et al., 1996). Due to its relative simplicity, virus–cell fusion is an attractive system with which to study the molecular basis of membrane fusion. Fusion between an enveloped virus and a target cell is mediated by viral surface glycoproteins. For many viruses, fusion takes place in the endosomal compartment where the mildly acidic pH triggers structural rearrangements that convert the fusion protein to a fusogenic form. The most significant consequence of the low pH–induced conformational change is exposure of the fusion peptide and concomitant conversion of the previously hydrophilic ectodomain of the fusion protein to a hydrophobic form capable of binding membranes.
Conversion to a hydrophobic form has been demonstrated for the ectodomains of several low pH–dependent viral fusion proteins including the hemagglutinin (HA)1 of influenza virus and the E1 protein of Semliki Forest virus (SFV; Harter et al., 1989; Bron et al., 1993; Klimjack et al., 1994; White, 1995). A water-soluble ectodomain of the influenza HA (BHA) can be prepared by treating influenza virus particles with bromelain. BHA aggregates if exposed to low pH in aqueous solution (Skehel et al., 1982). If, however, target membranes are present during the low pH treatment, BHA associates with membranes (Skehel et al., 1982; Doms et al., 1985). Photolabeling experiments have shown that this interaction is mediated by the fusion peptide (Harter et al., 1989). In addition, a mutant HA with a Gly to Glu substitution at position 1 of the fusion peptide, which displays no fusion activity, shows reduced (∼50%) liposome binding (Gething et al., 1986). The E1 ectodomain of SFV also binds to target membranes when exposed to low pH (Klimjack et al., 1994), and mutations in the SFV fusion peptide that impair fusion also affect liposome binding (Kielian et al., 1996).
Many viruses do not require low pH in order to fuse with host cells, and appear to be able to fuse directly at the plasma membrane. This includes members of approximately half of the known families of enveloped viruses including serious pathogens such as the human immunodeficiency virus (HIV) and respiratory syncytial virus (Hernandez et al., 1996). In contrast to what is known about the mechanisms of viral fusion proteins that function at low pH, little is known about the mechanisms of viral fusion proteins that function at neutral pH. Unlike viruses that fuse at low pH, those that fuse at neutral pH appear to require host cell receptors or additional factors (Weiss, 1992). Because of this we and others have proposed that interaction of the neutral pH viral fusion protein with its host cell receptor(s) triggers conformational changes in the viral fusion protein that activate it for fusion (White, 1990; Weiss, 1992). By analogy to the low pH–induced activation of the influenza HA and the SFV E1, the transition to a fusogenic state would include exposure of the previously buried fusion peptide whose interaction with the target membrane would initiate fusion. As far as we know, all cellular fusion reactions such as trafficking of endocytic and exocytic vesicles, egg fertilization, and myotube formation occur at neutral pH. Therefore cellular membrane fusion proteins/machines may use similar receptor-mediated strategies to initiate fusion.
A number of receptors for enveloped viruses that fuse at neutral pH have now been identified (Weiss, 1992; Brojatsch et al., 1996; Deng et al., 1996; Feng et al., 1996). Studies with the HIV envelope glycoprotein (Env) and its primary receptor, CD4, have provided preliminary support for the basic theory of “receptor-induced” activation. Upon binding to CD4, HIV Env undergoes a number of conformational changes, including exposure of the V3 loop as well as changes in sensitivity to proteolysis and antibody binding (Sattentau et al., 1993; Matthews et al., 1994; Stamatatos and Cheng-Mayer, 1995; Sullivan et al., 1995). In addition, structural data suggest that there may be similarities between fusion-related forms of the influenza HA (Bullough et al., 1994) and HIV Env (Chan et al., 1997; Weissenhorn et al., 1997). Attainment of the former structure does (White and Wilson, 1987; Bullough et al., 1995) and attainment of the latter structure most likely involves large conformational changes. Despite these noted changes, neither exposure of the fusion peptide nor changes in hydrophobicity have yet been demonstrated for HIV Env upon binding of either CD4 or a chemokine receptor, two host cell factors required for HIV entry.
Like most retroviruses, the avian leukosis and sarcoma virus (ALSV) fuses at neutral pH (Gilbert et al., 1990). Its Env is synthesized as an inactive precursor that is cleaved intracellularly by a host protease into a surface (SU) and a transmembrane (TM) subunit. The SU and TM subunits remain associated with each other through a disulfide bond(s) and form a trimer of SU/TM heterodimers (Einfeld and Hunter, 1988). The SU subunit has determinants for receptor binding whereas the TM subunit anchors the protein in the viral membrane and contains the fusion peptide. For most retroviruses the fusion peptide is located at the NH2 terminus of the TM protein, but ALSV is unique among known retroviruses in that its fusion peptide is internal to the NH2 terminus of the TM subunit (Hernandez, L.D., and J.M. White, manuscript submitted for publication). Structural models suggest a strong similarity between the ALSV TM and the Ebola virus glycoprotein implying that aspects of their fusion mechanisms may be similar (Gallaher, 1996).
Differing host range, receptor binding, and interference patterns have divided ALSV into five major subgroups designated A–E. The host cell receptor for ALSV-A is a small transmembrane protein termed Tva (Bates et al., 1993; Young et al., 1993); Tva binds specifically to the subgroup A envelope glycoprotein (Connolly et al., 1994; Gilbert et al., 1994). The extracellular domain of Tva has a single copy of a cysteine-rich motif, seven of which are present in the low density lipoprotein (LDL) receptor. This 40–amino acid repeat motif is sufficient to mediate virus entry (Rong and Bates, 1995). We have previously shown that upon interaction with sTva, a soluble form of the Tva ectodomain, Env A undergoes a temperature- dependent conformational change in the SU subunit that correlates with the activation of its fusion function (Gilbert et al., 1995). Here we show that binding of Tva at fusion-permissive temperatures renders the Env A ectodomain hydrophobic as measured by a liposome binding assay. Liposome binding is mediated by the TM subunit and, at least in part, by the fusion peptide. Liposome binding also appears to require a specific sequence in the receptor. This is the first example of receptor-induced conversion of the ectodomain of a fusion protein that functions at neutral pH from a hydrophilic to a hydrophobic entity.
Materials and Methods
Construction of API-Glu2
The KpnI-AflII fragment from pCB6-Env-API (GPI-linked Env A; Gilbert et al., 1993) was replaced with the corresponding KpnI-AflII fragment of pCB6-Glu2 yielding pCB6-Glu2-PI. The resulting Env gene has the single amino acid substitution Val to Glu at residue 30 of the TM subunit (in the fusion peptide) as well as the signal for glycosylphosphatidylinositol (GPI) addition in place of the transmembrane domain and cytoplasmic tail.
Tissue Culture
The pCB6-Glu2-PI plasmid was transfected into NIH 3T3 cells by the calcium phosphate precipitation method (Wigler et al., 1977). 24 h after transfection, cells were placed in selective medium (DME, 10% supplemented calf serum; [Hyclone, Logan, UT], and 500 mg/Liter geneticin GIBCO BRL, Gaithersburg, MD) for 14 d. Geneticin-resistant clones were picked and screened for expression of envelope protein after treatment for 16–18 h with 10 mM sodium butyrate (Sigma Chemical Co., St. Louis, MO), and the highest expressing clones were amplified. The NIH 3T3 cell line expressing Env-API was maintained in the above medium and has been described previously (Gilbert et al., 1993). Env-API cells were treated for 16–18 h with 5 mM sodium butyrate to induce higher Env-API expression levels. The secreted form of Tva (sTva) was a gift of P. Bates (University of Pennsylvania, Philadelphia, PA)
Antibodies and Reagents
The anti-Tva antiserum used was that described previously (Bates et al., 1993) and was a gift of P. Bates (University of PA, Philadelphia, PA). Polyclonal antisera were raised in rabbits against peptides corresponding to part of the host range 2 (hr2) domain of the SU subunit of Env A (amino acids 206–220), and the decay-accelerating factor glycosylphosphatidylinositol (DAF-GPI) signal peptide. These latter antibodies were affinity purified by chromatography over their respective peptides coupled to SulfoLink resin (Pierce, Rockford, IL) according to the manufacturer's instructions. Phosphatidylinositol-specific phospholipase-C (PI-PLC) was purified from an overexpressing bacterial strain (Koke et al., 1991). A gift of P. Bjorkman (California Institute of Technology, Pasadena, CA) and O. Griffith Hayes (University of Oregon, Eugene, OR). Two additional soluble forms of Tva, s47 and s47WA, were purified and refolded from overexpressing bacterial strains.
Biotinylation and Coimmunoprecipitation Binding Assay
After induction with sodium butyrate (5 mM for API and 10 mM for Glu2-PI) for 16–18 h, cells expressing GPI-anchored proteins were labeled with the membrane-impermeant reagent NHS-LC-biotin (Pierce) and treated with PI-PLC as described previously (Gilbert et al., 1993). sTva, s47, and s47WA proteins were biotinylated on ice for 45 min in PBS (pH 7.8) containing 0.5 mM MgCl2 and 1 mg/ml NHS-LC-biotin. The excess biotin was quenched by the addition of 50 mM glycine for 1–18 h at 4°C. For coimmunoprecipitation of s47 and API, the indicated amount of biotinylated s47 or s47WA protein was incubated with unlabeled PI-PLC– released API for 1 h on ice in a total volume of 200 μl. Anti-DAF antibody was added and incubated for 1 h at 4°C followed by another 1-h incubation after the addition of protein A–agarose (Boehringer Mannheim Biochemicals, Indianapolis, IN). Samples were washed four times in lysis buffer (Hepes-buffered saline with 1% Nonidet P-40), boiled, and reduced in Laemmli sample buffer, and separated by SDS-PAGE. Proteins were transferred to nitrocellulose and probed with streptavidin coupled to HRP (strep-HRP; Pierce) to detect biotinylated proteins. The HRP signal was detected by enhanced chemiluminescence (ECL; Amersham Corp., Arlington Heights, IL).
For measuring sTva dissociation from API, unlabeled PI-PLC–released API was immunoprecipitated with anti-DAF/protein A–agarose as described above. Samples were washed four times in calcium- and magnesium-free PBS (PBS−−). Biotinylated sTvA was added to the immunoprecipitates on ice for 20 min. Liposomes or PBS−− was added and samples were incubated for 5 min at 37°C. An additional 100 μl PBS−− was added and the samples were centrifuged at 14,000 rpm in a microcentrifuge for 10 s at 4°C. Supernatants were chloroform–methanol precipitated as described below. The immunecomplexes and the supernatant proteins were analyzed by SDS-PAGE and blotted with strep-HRP.
Liposome Binding Assay
Liposomes were freshly prepared before each experiment as described previously (White and Helenius, 1980) and contained phosphatidylcholine and cholesterol (Sigma Chemical Co.) in a 4:1 molar ratio. Biotinylated PI-PLC released GPI-linked envelope glycoproteins were incubated with soluble receptor (either sTva, s47, or s47WA) or diluted fetal calf serum on ice for 15 min in a total volume of 60 μl. PI-PLC released material from ∼2 × 106 cells was used for each sample. 40 μl of liposomes were then added and the samples were incubated at the indicated temperature for the indicated amount of time. 300 μl of 67% sucrose was added to bring the final concentration of sucrose to 50%. This solution was overlaid with 300 μl of 25% sucrose and 200 μl of 10% sucrose in a clear polycarbonate ultracentrifuge tube (Beckman Instruments Inc., Fullerton, CA). After centrifugation at 200,000 g for 3 h at 4°C, 9 × 100 μl fractions were collected from the top of the gradient, brought up to a final volume of 400 μl with lysis buffer, immunoprecipitated with the anti-DAF antibody, separated by SDS-12.5% PAGE and processed for the detection of biotinylated proteins as described above. For quantitation of liposome binding, blots were probed with 125I-labeled streptavidin, dried, and exposed to a phosphorimaging screen. Exposed phosphorimaging screens were scanned into a PhosphorImager workstation using the ImageQuant program (Molecular Dynamics, Inc., Sunnyvale, CA). Protein band intensities were determined by summing the pixels in a constant volume rectangle. The top three fractions are defined as liposome bound. For 6 M urea, 1 M NaCl, and 15 mM carbonate (pH 11.0) treatments, liposomes with bound API were incubated with the appropriate solution for 10 min at room temperature (RT) before the addition of the sucrose solution. For the detection of sTva, fractions were chloroform-methanol precipitated: 10 μl of salmon sperm DNA (1 mg/ml) was added to each 100 μl fraction. 550 μl methanol, 150 μl chloroform, and 550 μl H2O were added. Samples were vortexed and centrifuged at 14,000 rpm in a microcentrifuge for 3–5 min at RT. The top layer was aspirated and 550 μl of methanol was added to the bottom layer. Samples were again vortexed and centrifuged at 14,000 rpm in a microcentrifuge. Pellets were resuspended in Laemmli sample buffer, boiled, and then processed by SDS-PAGE and Western blotting with the anti-Tva antibody.
Dissociation of the SU and TM Subunits
Biotinylated PI-PLC–released API was incubated with or without s47 on ice as described above. Liposomes were added and the mixture was incubated for 5 min at 37°C. 75 μl of 8 M urea and 17.5 μl of 1 M DL-dithiothreitol (DTT) were then added and the solution was incubated at RT for an additional 20 min before being placed in the bottom of a sucrose step gradient and processed as described above. Fractions were immunoprecipitated with either anti-DAF or a mixture of both the anti-DAF and anti-hr2 antibodies.
Results
sTva Induces API Liposome Binding
Exposure of influenza HA or SFV E1 to acidic conditions triggers the conversion of each ectodomain to a hydrophobic form capable of binding to liposomes (Skehel et al., 1982; Doms et al., 1985; Gething et al., 1986; Harter et al., 1989; Klimjack et al., 1994). We performed a liposome binding assay using the water-soluble Env A ectodomain, API, to test whether binding of sTva to Env A causes an analogous change in the hydrophobicity of the Env A ectodomain. API is a GPI anchored form of Env A that contains the signal for GPI addition from DAF in place of the Env A transmembrane and cytoplasmic tail domains. It is expressed, oligomerized, and transported to the cell surface in a manner similar to full-length Env A. It can be removed from the cell surface with PI-PLC yielding a water-soluble oligomer (Gilbert et al., 1993). The soluble oligomer binds specifically to the subgroup A receptor (Gilbert et al., 1994), and undergoes a temperature-dependent conformational change in the SU subunit upon this interaction in a manner similar to full-length Env A (Gilbert, 1995; Hernandez, L.D., unpublished results). In this paper, the term API refers to the PI-PLC–released soluble oligomeric form of API.
For the first liposome binding assays, biotinylated API was incubated with sTva on ice to allow formation of the sTva–API complex (Gilbert et al., 1994). After 15 min, liposomes composed of phosphatidylcholine and cholesterol were added and the samples were incubated at 37°C for 30 min, returned to 4°C, placed in the bottom of a centrifuge tube, overlaid with a sucrose step gradient, and subjected to ultracentrifugation. The gradient was fractionated and the position of API was determined by immunoprecipitation with an anti-DAF antibody that recognizes the first nine residues at the COOH terminus of the engineered TM subunit. In the absence of sTva, API did not bind liposomes and remained at the bottom of the gradient (Fig. 1, top). In the presence of sTva, however, API was found at the top of the gradient indicating it had associated with the liposomes (Fig. 1, bottom).
Figure 1.

Receptor-induced liposome binding. Biotinylated PI-PLC–released API was incubated with or without sTva for 15 min at 4°C incubated with liposomes at 37°C for 30 min and then analyzed on flotation gradients as described in Materials and Methods. Gradients were fractionated, and samples were immunoprecipitated with anti-DAF antibody, boiled and reduced, separated by SDS-12.5% PAGE, transferred to nitrocellulose, probed with streptavidin-HRP, and visualized by ECL to identify API.
Because Tva is related to the LDL receptor (Bates et al., 1993), it was possible that sTva itself has an affinity for liposomes. We therefore asked which component of the sTva–API complex is responsible for liposome binding. sTva alone or the sTva–API complex was incubated with liposomes at 37°C for 30 min and analyzed for liposome binding by a sucrose flotation gradient as described above. Fractions were chloroform–methanol precipitated and Western blotted with anti-Tva serum. sTva that had not been incubated with API remained at the bottom of the gradient showing that sTva itself does not have strong affinity for liposomes (Fig. 2 A, top). sTva that had been prebound to API and incubated with liposomes also remained at the bottom of the gradient (Fig. 2 A, bottom) indicating that it had dissociated from API, which floats to the top of the gradient (Fig. 1). We used a more sensitive method for detecting receptor by using biotinylated sTva in the liposome binding assay and determining where it ran in the gradient. Again sTva was found only at the bottom of the gradient (data not shown). This was somewhat unexpected since sTva binds to Env A with a K d of 1.5 nM (Zingler and Young, 1996; Rong et al., 1997) and remains bound to Env A through sucrose density centrifugation and coimmunoprecipitation in detergent-containing solutions (Gilbert et al., 1994). As shown in Fig. 2 B by a coimmunoprecipitation assay, a significant fraction of sTva dissociates from API in the presence, but not in the absence, of liposomes.
Figure 2.


sTva dissociates from API. (A) sTva or sTva-API was incubated with liposomes and subjected to centrifugation as in Fig. 1. Gradient fractions were chloroform-methanol precipitated, separated by SDS-12.5% PAGE, transferred to nitrocellulose, and Western blotted with anti-Tva serum. (B) Unlabeled API was immunoprecipitated with the anti-DAF antibody and protein A agarose. Biotinylated sTva was added to the immunoprecipitates on ice for 20 min. Liposomes or PBS were added and samples were incubated for 5 min at 37°C. Beads were centrifuged and the supernatants collected and chloroform-methanol precipitated. Beads and supernatant proteins were reduced, boiled, separated by SDS-12.5% PAGE, transferred to nitrocellulose, and blotted with streptavidin-HRP.
sTva contains the entire 83–amino acid extracellular domain of Tva. Within this domain the 40–amino acid LDL receptor repeat binding motif suffices to mediate virus entry (Rong and Bates, 1995). We therefore generated in Escherichia coli and refolded a 47–amino acid fragment of Tva that contains the 40-residue LDL receptor repeat motif (Peters, R., and D. Agard, unpublished results). s47 binds to Env A and induces the same structural changes in Env A that sTva does (Hernandez, L., and J.M. White, unpublished results). Since we found s47 to be a more stable reagent than sTva, we used s47 as the “receptor” for all subsequent experiments.
Stability of Liposome Binding
We examined the stability of the receptor-induced API– liposome interaction by treating API–liposome complexes with 6 M urea, high salt (1 M sodium chloride) or carbonate, pH 11.0 ,before centrifugation. As seen in Fig. 3, none of these treatments affected the attachment of API to liposomes. The interaction of API with liposomes is therefore nonionic, hydrophobic, and stable.
Figure 3.

Stability of the API–liposome interaction. API–liposome complexes were incubated with either 1 M NaCl, 6 M urea, or 15 mM carbonate, pH 11.0, for 10 min at RT. Samples were processed as described in the legend to Fig. 1.
Time and Temperature Dependence of Liposome Binding
We next examined the time course of liposome binding by varying the length of the 37°C incubation with liposomes from 1 to 30 min. Samples were subjected to sucrose flotation gradients and analyzed as described above. API binding to liposomes occurs rapidly and is complete between 2 to 5 min at 37°C (Fig. 4, A and B). This is similar to BHA which binds to liposomes within 1 to 2 min after exposure to low pH (Doms et al., 1985; Gething et al., 1986).
Figure 4.


Time course of liposome binding. The API–s47 complex was incubated with liposomes at 37°C for varying amounts of time and analyzed on sucrose flotation gradients. (A) Sucrose gradient fractions were immunoprecipitated and processed as in Fig. 1. (B) Sucrose gradient fractions were immunoprecipitated and processed as in Fig. 1 with the exception that the blots were probed with 125I-labeled streptavidin and subjected to quantitation by phosphorimager analysis as described in Materials and Methods. (% Bound) The percent API in the top three fractions.
ALSV binds to chicken embryo fibroblasts at 4°C but only fuses with them at temperatures >22°C (Gilbert et al., 1990). We therefore proposed that activation of the ALSV fusion protein must be temperature-dependent. Our previous studies on Tva–Env interactions are consistent with this hypothesis; sTva causes a change in the sensitivity of the SU subunit of Env A to thermolysin at 37°C but not at 4°C (Gilbert et al., 1995). We therefore asked if receptor-induced liposome binding is temperature-dependent. To do this API–s47 complexes were incubated with liposomes for 5 min at 4°, 10°, 22°, or 37°C, returned to 4°C, and analyzed for liposome binding (Fig. 5). Little liposome binding was detected at 4° or 10°C. Liposome binding then increased as the temperature was further increased with a high degree of binding (72%) occurring at 37°C. These results show that liposome binding is temperature-dependent similar to the sTva-induced conformational change in SU and ALSV fusion with chicken embryo fibroblast cells.
Figure 5.

Temperature dependence of liposome binding. The API–s47 complex was incubated with liposomes for 5 min at the indicated temperatures and analyzed on sucrose flotation gradients. Sucrose gradient fractions were immunoprecipitated, processed, and quantitated as described in the legend to Fig. 4 B.
TM Subunit Binds Liposomes
We next asked which subunit of API (SU or TM) mediates liposome binding. SU and TM are associated through a disulfide bond(s). We found that they could be dissociated but only by treatment with 100 mM DTT and 6 M urea. Under these conditions the anti-DAF antibody efficiently immunoprecipitates TM and an antibody against the hr2 region of Env efficiently immunoprecipitates SU (Fig. 6 A). It is interesting that the anti-hr2 antibody only recognized SU after treatment with urea/DTT, suggesting that the hr2 epitope is obstructed or in a different conformation in native API. Similar results were obtained with full length Env A (Delos, S., and J. White, unpublished results).
Figure 6.


API binds liposomes through the TM subunit. (A) Biotinylated API was incubated with either PBS, 6 M urea, or 6 M urea/100 mM DTT for 20 min at RT in a total volume of 100 μl. Samples were brought up to 400 μl with PBS and immunoprecipitated with anti-DAF or anti-hr2 antibody and processed as described in the legend to Fig. 1. (B) The s47-induced API–liposome complex was treated with 6 M urea and 100 mM DTT before centrifugation to dissociate the TM and SU subunits. Each sucrose gradient fraction was immunoprecipitated with anti-DAF or a mixture of anti-DAF and anti-SU antibodies and processed as described in the legend to Fig. 1.
Biotinylated API bound to receptor (s47) was incubated with liposomes for 5 min at 37°C. The sample was then treated with urea and DTT for 20 min at room temperature, and analyzed by a flotation gradient. Gradient fractions were immunoprecipitated with either anti-DAF (Fig. 6 B, top) or a combination of anti-DAF and anti-hr2 antibodies (Fig. 6 B, bottom). Immunoprecipitation with anti-DAF (Fig. 6 B, top) showed that TM and SU are efficiently dissociated from each other by the urea/DTT treatment; only TM was detected in the immunoprecipitates. Despite this, with s47-pretreated API the majority of TM remained bound to the liposomes (migrated to the top of the gradient). The TM subunit is more spread out in the gradient (Fig. 6 B) than in samples that had not been treated with urea/DTT (e.g., Fig. 1). This is likely due to the combined action of urea and DTT, since treatment with urea alone did not cause TM to spread out on the gradient (Fig. 3, middle). When samples of the API–liposome complexes were incubated with urea/DTT, SU stayed at the bottom of the gradient (Fig. 6 B, bottom). Conversely TM from the same sample migrated to the top of the gradient (Fig. 6 B, bottom). These results show that TM, and not SU, mediates API binding to liposomes.
Liposome Binding Properties of a Fusion Peptide Mutant
Studies with the influenza HA have demonstrated that liposome binding (at low pH) is mediated by the fusion peptide (Harter et al., 1989). We therefore hypothesized that the ALSV fusion peptide mediates liposome binding. We have recently shown that mutations in the candidate fusion peptide of ALSV Env A (residues 21–42 of TM) significantly impair the ability of Env A to mediate both virus– cell and cell–cell fusion without affecting Env processing, folding, oligomerization, cell surface expression, receptor binding, or ability to undergo a receptor-induced conformational change (Hernandez, L.D., and J.M. White, manuscript submitted for publication). We therefore constructed a GPI-linked form of one of these mutants (Val30Glu) and tested it in the liposome binding assay. The GPI form of this mutant is referred to as Glu2-PI. Glu2-PI was impaired in receptor-induced liposome binding (Fig. 7 A, middle) when compared to wt API (Fig. 7 A, bottom). The Glu2-PI protein was spread out in the gradient suggesting that its interaction with liposomes was relatively weak. Quantitation of the data indicated that Glu2-PI binds to liposomes with ∼50% the efficiency of wt API (Fig. 7 B). This phenotype is similar to the behavior of the Gly1 to Glu HA fusion peptide mutant (Gething et al., 1986). These results support our hypothesis that the fusion peptide mediates liposome binding (see Discussion).
Figure 7.
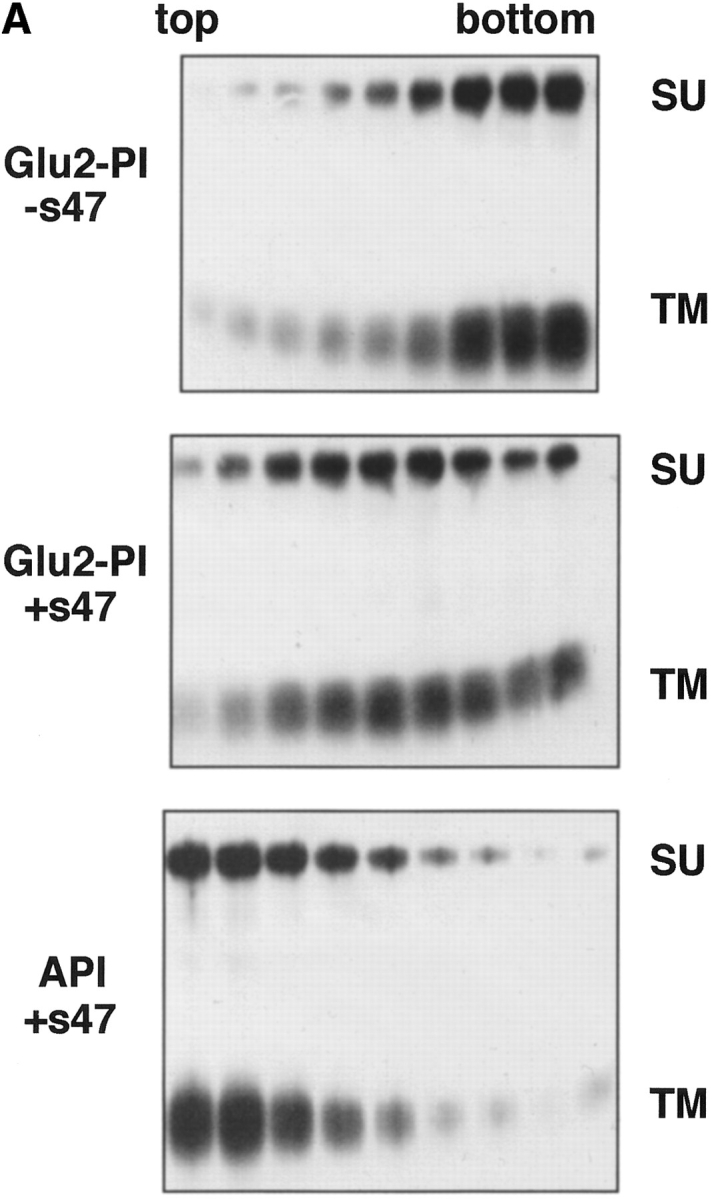

Liposome binding capability of Glu2-PI. The position 2 glutamate mutant protein (Glu2-PI) was assayed for liposome binding as in Fig. 4. Fractions were immunoprecipitated with anti-DAF, processed by SDS-12.5% PAGE and transferred to nitrocellulose. Blots were probed with (A) streptavidin-HRP (visualized by enhanced chemiluminescence), or (B) 125I-labeled streptavidin (quantitated by phosphorimager analysis as described in Materials and Methods). (% Bound) Percent API or Glu2-PI in the top three fractions.
Mutant Tva Does Not Induce Significant Liposome Binding
Residues 46–48 in the Tva ectodomain are crucial for virus entry (Zingler et al., 1995; Zingler and Young, 1996). A substitution that replaced the tryptophan at position 48 with alanine reduced infectivity titers approximately three to four orders of magnitude, but only decreased cell surface Tva binding affinity for an ALSV–A SU–Ig fusion protein approximately sevenfold (Zingler and Young, 1996). These findings suggested that the W48A mutant has a defect in a postbinding step(s) necessary for virus entry. We therefore asked if W48A could induce liposome binding.
s47WA, a soluble form of the W48A Tva mutant analogous to s47, was expressed in E. coli, purified, and refolded (Peters, R., and D. Agard, unpublished results). By our coimmunoprecipitation assay, s47WA appeared to bind to API approximately 100-fold less well than wild-type (wt) s47 (Fig. 8 A). This apparent 100-fold reduction in binding differs from the sevenfold reduction reported by Zingler and Young (1996), and may be due to differences in the systems used. Nonetheless equal binding to API could be achieved using 100 times more s47WA protein than wt s47 (Fig. 8 A). Under conditions of comparable binding, the s47WA receptor (100×) did not induce significant liposome binding when compared to (1×) wt s47 (Fig. 8 B). With 100× mutant receptor the majority of TM stays at the bottom of the gradient. If the amount of mutant receptor is further increased (250×) to binding levels that exceed the wt (Fig. 8 A), API now appears to spread on the gradient. Nonetheless, even using 250× mutant protein, API does not display a clear cut liposome binding phenotype as occurs with 1× of the wt receptor (Fig. 8 B).
Figure 8.

Ability of a mutant receptor, s47WA, to (A) bind to API and (B) induce liposome binding. (A) Indicated amounts of biotinylated s47 or s47WA protein were incubated with equal amounts of PI-PLC–released API protein in a total volume of 200 μl for 1 h on ice. Samples were immunoprecipitated with the anti-DAF antibody, separated by SDS-15% PAGE and transferred to nitrocellulose. Blots were probed with streptavidin-HRP and visualized by enhanced chemiluminescence. 1× = 0.36 μg protein. (B) Biotinylated PI-PLC–released API was incubated with 0.36 μg (1×) s47 or 3.6 μg (10×), 36 μg (100×), or 90 μg (250×) s47WA protein at 4°C for 20 min. Liposomes were added and samples were incubated at 37°C for 5 min before centrifugation. Fractions were analyzed as described in the legend to Fig. 1. Similar results were obtained with the s47WA mutant (100×) when the 4°C incubation step was increased to 90 min.
Discussion
In this study we present the first evidence that receptor binding to a neutral pH fusion protein transforms its ectodomain into a hydrophobic entity capable of binding to target membranes. Moreover, we show that soluble forms of the receptor are fully competent to elicit this response. This is the first demonstration of its kind for any fusion protein that functions at neutral pH. After interaction with soluble receptor, the ALSV Env A ectodomain binds to liposomes composed solely of phosphatidylcholine and cholesterol. Binding is stable and nonionic in nature, temperature dependent (T > 20°C), and it is complete between 2 to 5 min at 37°C. Somewhat surprisingly, the Env A-receptor complex appears to dissociate after Env A binds to liposomes. Liposome binding is mediated by the TM subunit of Env A and, at least in part, by the fusion peptide; a mutant in the fusion peptide region is significantly impaired in its ability to bind to liposomes. Furthermore, a mutant receptor that is competent to bind Env A but is unable to support virus infection (Zingler and Young, 1996) is compromised in its ability to induce liposome binding. We propose that this receptor-induced change in hydrophobicity of a neutral pH fusion protein ectodomain is analogous to the low pH–induced activation of fusion proteins such as the influenza HA (Doms et al., 1985; Gething et al., 1986) and the SFV E1 protein (Klimjack et al., 1994).
HA, the most extensively characterized viral fusion protein, is a homotrimer composed of two disulfide bonded subunits: HA1, which confers receptor binding activity, and HA2 which contains the fusion peptide. When exposed to low pH, the previously buried fusion peptide of HA2 is revealed and repositioned to interact with membranes, and the HA1 globular head domains separate from each other and from HA2 (White and Wilson, 1987; Bullough et al., 1994; Hernandez, 1996). Along with our previous findings, the results presented here suggest that ALSV Env A may undergo similar changes during fusion activation.
As for HA, a drastic change in the hydrophobicity of the ALSV-A ectodomain occurs in response to a trigger. In the case of HA the trigger is low pH; in the case of Env A the trigger is a temperature-dependent sequence-specific interaction with the viral receptor, Tva. Because Tva is sufficient to induce a change in the hydrophobicity of Env A, it appears highly likely that Tva is the only host factor required for fusion. This differs from HIV, which needs at least two receptors, CD4 and a chemokine receptor, to mediate virus entry (reviewed in Bates, 1996). Whereas CD4 binding has been shown to induce some structural changes in HIV Env, interaction of HIV Env with a chemokine receptor may be necessary to induce changes that expose the HIV fusion peptide and transforms its ectodomain into a hydrophobic form. Because soluble forms of Tva are able to induce Env A binding to liposomes, it is clear that, at least in the ALSV system, the proteinaceous fusion trigger does not need to be embedded in the target membrane to elicit the first steps of the fusion process. In this respect, it will be interesting to see if soluble Tva can induce virus– liposome fusion.
Previous studies suggested that Trp-48 of Tva may be an important determinant for triggering postbinding events necessary for fusion (Zingler et al., 1995; Zingler and Young, 1996). Our results are consistent with this prediction; the W48A Tva mutant, s47WA, is impaired in its ability to induce liposome binding even under conditions in which its binding to the Env A ectodomain is comparable to or exceeds that of wt Tva. Our results therefore show that binding of Tva to Env is necessary, but not sufficient, to effect a change in the hydrophobicity of the Env ectodomain; the two proteins must interact in a highly specific and temperature-dependent manner in order to induce the structural changes necessary for target bilayer binding. Trp-48 appears to be an important contact point in this interaction.
In response to its trigger, low pH, the fusion peptide of HA is exposed and the ectodomain becomes hydrophobic. Here we have clearly shown that in response to its trigger—specific Tva binding—the Env A ectodomain becomes hydrophobic. We propose that the increase in hydrophobicity is due at least in part to exposure of the fusion peptide. In previous work we have shown that an antibody against the Env A fusion peptide reacts preferentially with Env A following binding to Tva at T > 22°C (Gilbert et al., 1995). Here we show that a mutant in the Env A fusion peptide (Val30 to Glu) that is abolished in fusion activity (Hernandez, L.D., and J.M. White, manuscript submitted for publication) is significantly compromised in its ability to bind to liposomes: binding of the Glu-substituted mutant to liposomes was ∼50% and appeared unstable compared to that of the wt Env ectodomain. We believe that the phenotype of Glu-substituted mutant is caused by the presence of a charged residue, glutamate, in the hydrophobic surface of the Env A fusion peptide. In this respect the phenotype of the Glu-substituted Env is highly reminiscent of the first fusion peptide mutant: Gly1→ Glu of the HA fusion peptide (Gething et al., 1986). This original HA fusion peptide mutant binds to liposomes less well (∼50%) than wt HA (see Fig. 8 in Gething et al., 1986). Future studies using techniques such as labeling with photoactivatable phospholipids (Harter et al., 1989; Durrer et al., 1995) are necessary to test whether the hydrophobic interaction between the Env A ectodomain and target bilayers is an exclusive property of the fusion peptide. Computer modeling has predicted that the Ebola virus glycoprotein is structurally very similar to the ALSV TM subunit (Gallaher, 1996) with an analogous region postulated to act as the fusion peptide. Our results suggest that the corresponding region in the Ebola glycoprotein may act in a similar manner to attach the fusion protein to target membranes under fusion-inducing conditions.
In the case of HA, exposure to low pH also causes the globular head domains (HA1) to separate from each other, and this dissociation is necessary for fusion (Godley et al., 1992; Kemble et al., 1992). We have observed two structural changes in the receptor binding subunit (SU) of Env in response to its fusion trigger: SU becomes sensitive to thermolysin (Gilbert et al., 1995) and it dissociates from sTva in the presence of liposomes (Fig. 2). Analogous to HA, the structural changes in the Env SU subunit may reflect separation of the receptor binding domains of Env and they may be necessary for fusion.
Recently, CAR1 has been identified as the receptor for members of two cytopathic subgroups of ALSV, ALSV-B and ALSV-D (Brojatsch et al., 1996). CAR1 is a member of the tumor necrosis factor receptor family and contains two extracellular cysteine-rich domains and a death domain in its cytoplasmic tail that has been implicated in virus-induced cell death. CAR1 is not related to Tva or to any other known retroviral receptor. It will now be possible to determine if different ALSV subgroups, cytopathic and noncytopathic, share a common mechanism for activation of their fusion proteins despite using highly unrelated receptors. For example, it will be interesting to determine if CAR1 binding to Env B or D induces analogous structural changes to those seen in Env A upon Tva binding, particularly conversion of the Env B or D ectodomain to a hydrophobic entity.
While relatively little is known about viral fusion proteins that function at neutral pH, even less is known about endogenous proteins that mediate cell–cell fusion and intracellular fusion events, all of which are thought to occur at neutral pH. Several candidate cell–cell fusion proteins have been identified, including fertilin (Blobel et al., 1992) and meltrin (Yagami-Hiromasa et al., 1995), members of the ADAM gene family (Wolfsberg and White, 1996) involved in sperm–egg and myoblast fusion, respectively. N-ethylmaleimide–sensitive factor (NSF), soluble NSF- attachment proteins, and soluble NSF-attachment proteins receptors, are involved in intracellular fusion reactions such as vesicular transport between Golgi compartments (for reviews see Rothman and Warren, 1994; Denesvre and Malhotra, 1996; Monck and Fernandez, 1996). The large number of proteins involved in cell–cell and intracellular fusion events suggests that these latter fusion events are more complex than the apparent two component ALSV system. Nonetheless, the overall general fusion mechanism may be similar: specific protein–protein interactions cause conformational changes that expose a hydrophobic region(s) in one or more of the members of the fusion protein complex. This conformational change would, in turn, enable the fusion protein(s) to bind hydrophobically to target membranes and initiate membrane merger (Hernandez et al., 1996). In view of the findings presented here, it is now reasonable to consider, for example in the case of intracellular fusion events (Rothman and Warren, 1994; Denesvre and Malhotra, 1996; Monck and Fernandez, 1996), that a soluble protein may act as the fusion trigger.
Acknowledgments
We thank Paul Bates for baculovirus produced sTva and the anti-Tva antiserum; Pamela Bjorkman and O. Griffith Hayes for providing the PI-PLC overexpressing bacterial strain; and Joanna Gilbert for generating the anti-DAF antibody.
Abbreviations used in this paper
- ALSV
avian leukosis and sarcoma virus
- API
GPI-linked ALSV subgroup A Env glycoprotein
- BHA
bromelain-released hemagglutinin
- DAF
decay-accelerating factor
- DTT
dithiothreitol
- ECL
enhanced chemiluminescence
- Env
envelope glycoprotein
- GPI
glycosylphosphatidylinositol
- HA
hemagglutinin
- HIV
human immunodeficiency virus
- LDL
low density lipoprotein
- NSF
N-ethylmaleimide-sensitive factor
- PI-PLC
phosphatidylinositol-specific phospholipase-C
- RT
room temperature
- SFV
Semliki Forest virus
- sTva
soluble ALSV subgroup A receptor
- SU
surface
- TM
transmembrane
- Tva
ALSV subgroup A receptor
- wt
wild-type
Footnotes
Work in Dr. White's laboratory was supported by a grant from the National Institutes of Health (A122470). L.D. Hernandez was supported in part by a predoctoral fellowship from the Howard Hughes Medical Institute. D.A. Agard is an Investigator of the Howard Hughes Medical Institute.
Address all correspondence to Judith M. White, Department of Cell Biology, University of Virginia, Health Sciences Center, Box 439, Charlottesville, VA 22908. Tel.: (804) 924-2593. Fax: (804) 982-3912. e-mail: jw7g@virginia.edu
References
- Bates P. Chemokine receptors and HIV-1: an attractive pair? . Cell. 1996;86:1–3. doi: 10.1016/s0092-8674(00)80070-7. [DOI] [PubMed] [Google Scholar]
- Bates P, Young JAT, Varmus HE. A receptor for subgroup A Rous sarcoma virus is related to the low density lipoprotein receptor. Cell. 1993;74:1043–1051. doi: 10.1016/0092-8674(93)90726-7. [DOI] [PubMed] [Google Scholar]
- Bennett MK, Scheller RH. A molecular description of synaptic vesicle membrane trafficking. Annu Rev Biochem. 1994;63:63–100. doi: 10.1146/annurev.bi.63.070194.000431. [DOI] [PubMed] [Google Scholar]
- Blobel CP, Wolfsberg TG, Turck CW, Myles DG, Primakoff P, White JM. A potential fusion peptide and an integrin ligand domain in a protein active in sperm-egg fusion. Nature. 1992;356:248–252. doi: 10.1038/356248a0. [DOI] [PubMed] [Google Scholar]
- Brojatsch J, Naughton J, Rolls MM, Zingler K, Young JAT. CAR1, a TNFR-related protein, is a cellular receptor for cytopathic avian leukosis-sarcoma viruses and mediates apoptosis. Cell. 1996;87:845–855. doi: 10.1016/s0092-8674(00)81992-3. [DOI] [PubMed] [Google Scholar]
- Bron R, Wahlberg JM, Garoff H, Wilschut J. Membrane fusion of Semliki forest virus in a model system: correlation between fusion kinetics and structural changes in the envelope glycoprotein. EMBO J. 1993;12:693–701. doi: 10.1002/j.1460-2075.1993.tb05703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- Connolly L, Zingler K, Young JA. A soluble form of a receptor for subgroup A avian leukosis and sarcoma viruses (ALSV-A) blocks infections and binds directly to ALSV-A. J Virol. 1994;68:2760–2764. doi: 10.1128/jvi.68.4.2760-2764.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denesvre C, Malhotra V. Membrane fusion in organelle biogenesis. Curr Opin Cell Biol. 1996;8:519–523. doi: 10.1016/s0955-0674(96)80030-5. [DOI] [PubMed] [Google Scholar]
- Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- Doms RW, Helenius A, White J. Membrane fusion activity of the influenza virus hemagglutinin. J Biol Chem. 1985;260:2973–2981. [PubMed] [Google Scholar]
- Durrer P, Gaudin Y, Ruigrok RWH, Graf R, Brunner J. Photolabeling identifies a putative fusion domain in the envelope glycoprotein of rabies and vesicular stomatitis viruses. J Biol Chem. 1995;270:17575–17581. doi: 10.1074/jbc.270.29.17575. [DOI] [PubMed] [Google Scholar]
- Einfeld D, Hunter E. Oligomeric structure of a prototypic retrovirus glycoprotein. Proc Natl Acad Sci USA. 1988;85:8688–8692. doi: 10.1073/pnas.85.22.8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: Functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- Gallaher WR. Similar structural models of the transmembrane proteins of Ebola and avian sarcoma viruses. Cell. 1996;85:477–478. doi: 10.1016/s0092-8674(00)81248-9. [DOI] [PubMed] [Google Scholar]
- Gething M-J, Doms RW, York D, White JM. Studies on the mechanism of membrane fusion: site-specific mutagenesis of the hemagglutinin of influenza virus. J Cell Biol. 1986;102:11–23. doi: 10.1083/jcb.102.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J, Mason D, White J. Fusion of Rous sarcoma virus with host cells does not require low pH. J Virol. 1990;64:5106–5113. doi: 10.1128/jvi.64.10.5106-5113.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, J.M. 1995. Studies on the fusion mechanism of the rous sarcoma virus envelope glycoprotein. Ph.D. Thesis. University of California, San Francisco, CA. 207 pp.
- Gilbert JM, Hernandez LD, Chernov-Rogan T, White JM. Generation of a water soluble oligomeric ectodomain of the Rous sarcoma virus envelope glycoprotein. J Virol. 1993;67:6889–6892. doi: 10.1128/jvi.67.11.6889-6892.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert JM, Bates P, Varmus HE, White JM. The receptor for the subgroup A avian leukosis sarcoma virus binds to subgroup A but not to subgroup C envelope glycoprotein. J Virol. 1994;68:5623–5628. doi: 10.1128/jvi.68.9.5623-5628.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert JM, Hernandez LD, Balliet JW, Bates P, White JM. Receptor-induced conformational changes in the subgroup A avian leukosis and sarcoma virus envelope glycoprotein. J Virol. 1995;69:7410–7415. doi: 10.1128/jvi.69.12.7410-7415.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godley L, Pfeifer J, Steinhauer D, Ely B, Shaw G, Kaufmann R, Suchanek E, Pabo C, Skehel JJ, Wiley DC, et al. Introduction of intersubunit disulfide bonds in the membrane-distal region of the influenza hemagglutinin abolishes membrane fusion activity. Cell. 1992;68:635–645. doi: 10.1016/0092-8674(92)90140-8. [DOI] [PubMed] [Google Scholar]
- Harter C, James P, Bächi T, Semenza G, Brunner J. Hydrophobic binding of the ectodomain of influenza hemagglutinin to membranes occurs through the “fusion peptide.” . J Biol Chem. 1989;264:6459–6464. [PubMed] [Google Scholar]
- Hernandez LD, Hoffman LR, Wolfsberg TG, White JM. Virus-cell and cell-cell fusion. Annu Rev Cell Dev Biol. 1996;12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- Kemble GW, Bodian DL, Rosé J, Wilson IA, White JM. Intermonomer disulfide bonds impair the fusion activity of influenza virus hemagglutinin. J Virol. 1992;66:4940–4950. doi: 10.1128/jvi.66.8.4940-4950.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian M, Klimjack MR, Ghosh S, Duffus WA. Mechanisms of mutations inhibiting fusion and infection by semliki forest virus. J Cell Biol. 1996;134:863–872. doi: 10.1083/jcb.134.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimjack MR, Jeffrey S, Kielian M. Membrane and protein interactions of a soluble form of the Semliki Forest virus fusion protein. J Virol. 1994;68:6940–6946. doi: 10.1128/jvi.68.11.6940-6946.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koke JA, Yang M, Henner DJ, Volwerk JJ, Griffith OH. High-level expression in Escherichia coli and rapid purification of phosphatidylinositol-specific phospholipase C from Bacillus cereus and Bacillus thuringiensis. Protein Expr Purif. 1991;2:51–58. doi: 10.1016/1046-5928(91)90009-8. [DOI] [PubMed] [Google Scholar]
- Matthews TJ, Wild C, Chen CH, Bolognesi DP, Greenberg ML. Structural rearrangements in the transmembrane glycoprotein after receptor binding. Immunol Rev. 1994;140:93–104. doi: 10.1111/j.1600-065x.1994.tb00866.x. [DOI] [PubMed] [Google Scholar]
- Monck JR, Fernandez JM. The fusion pore and mechanisms of biological membrane fusion. Curr Opin Cell Biol. 1996;8:524–533. doi: 10.1016/s0955-0674(96)80031-7. [DOI] [PubMed] [Google Scholar]
- Rong L, Bates P. Analysis of the subgroup A avian sarcoma and leukosis virus receptor: the 40-residue, cysteine-rich, low-density lipoprotein receptor repeat motif of Tva is sufficient to mediate viral entry. J Virol. 1995;69:4847–4853. doi: 10.1128/jvi.69.8.4847-4853.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong L, Edinger A, Bates P. Role of basic residues in the subgroup-determining region of the subgroup A avian sarcoma and leukosis virus envelope in receptor binding and infection. J Virol. 1997;71:3458–3465. doi: 10.1128/jvi.71.5.3458-3465.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JE, Warren G. Implications of the SNARE hypothesis for intracellular membrane topology and dynamics. Curr Biol. 1994;4:220–233. doi: 10.1016/s0960-9822(00)00051-8. [DOI] [PubMed] [Google Scholar]
- Sattentau QJ, Moore JP, Vignaux F, Traincard F, Poignard P. Conformational changes induced in the envelope glycoproteins of the human and simian immunodeficiency viruses by soluble receptor binding. J Virol. 1993;67:7383–93. doi: 10.1128/jvi.67.12.7383-7393.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel JJ, Bayley PM, Brown EB, Martin SR, Waterfield MD, White JM, Wilson IA, Wiley DCJ. Changes in the conformation of influenza virus hemagglutinin at the pH optimum of virus-mediated membrane fusion. Proc Natl Acad Sci USA. 1982;79:968–972. doi: 10.1073/pnas.79.4.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatatos L, Cheng-Mayer C. Structural modulations of the envelope gp120 glycoprotein of human immunodeficiency virus type 1 upon oligomerization and differential V3 loop epitope exposure of isolates displaying distinct tropism upon virion-soluble receptor binding. J Virol. 1995;69:6191–6198. doi: 10.1128/jvi.69.10.6191-6198.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan N, Sun Y, Li J, Hofmann W, Sodroski J. Replicative function and neutralization sensitivity of envelope glycoproteins from primary and T-cell line-passaged human immunodeficiency virus type 1 isolates. J Virol. 1995;69:4413–4422. doi: 10.1128/jvi.69.7.4413-4422.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, R. 1992. Cellular receptors and viral glycoproteins involved in retrovirus entry. In The Retroviruses. J. Levy, editor. Plenum Press, New York. Vol. 2: 1–108.
- Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- White J, Helenius A. pH-dependent fusion between the Semliki Forest virus membrane and liposomes. Proc Natl Acad Sci USA. 1980;77:3273–3277. doi: 10.1073/pnas.77.6.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JM. Viral and cellular membrane fusion proteins. Annu Rev Physiol. 1990;52:675–697. doi: 10.1146/annurev.ph.52.030190.003331. [DOI] [PubMed] [Google Scholar]
- White JM. Membrane fusion: the influenza paradigm. Cold Spring Harbor Symp Quant Biol. 1995;60:581–588. doi: 10.1101/sqb.1995.060.01.062. [DOI] [PubMed] [Google Scholar]
- White JM, Wilson IA. Anti-peptide antibodies detect steps in a protein conformational change: low-pH activation of the influenza virus hemagglutinin. J Cell Biol. 1987;105:2887–2896. doi: 10.1083/jcb.105.6.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigler M, Silverstein S, Lee L-S, Pellicer A, Cheng Y-C, Axel R. Transfer of purified herpes virus thymidine kinase gene to cultured mouse cells. Cell. 1977;11:223–232. doi: 10.1016/0092-8674(77)90333-6. [DOI] [PubMed] [Google Scholar]
- Wolfsberg TG, White JM. ADAMs in fertilization and development. Dev Biol. 1996;180:389–401. doi: 10.1006/dbio.1996.0313. [DOI] [PubMed] [Google Scholar]
- Yagami-Hiromasa T, Sato T, Kurisaki T, Kamijo K, Nabeshima Y, Fujisawa-Sehara A. A metalloprotease-disintegrin participating in myoblast fusion. Nature. 1995;377:652–656. doi: 10.1038/377652a0. [DOI] [PubMed] [Google Scholar]
- Young JA, Bates P, Varmus HE. Isolation of a chicken gene that confers susceptibility to infection by subgroup A avian leukosis and sarcoma viruses. J Virol. 1993;67:1811–1816. doi: 10.1128/jvi.67.4.1811-1816.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingler K, Young JAT. Residue Trp-48 of Tva is critical for viral entry but not for high-affinity binding to the SU glycoprotein of subgroup A avian leukosis and sarcoma viruses. J Virol. 1996;70:7510–7516. doi: 10.1128/jvi.70.11.7510-7516.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingler K, Belanger C, Peters R, Agard E, Young JA. Identification and characterization of the viral interaction determinant of the subgroup A avian leukosis virus receptor. J Virol. 1995;69:4261–4266. doi: 10.1128/jvi.69.7.4261-4266.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]