

Abstract
In budding yeast, accurate chromosome segregation requires that one and only one kinetochore assemble per chromosome. In this paper, we report the use of DNA–protein crosslinking and nondenaturing gel analysis to study the structure of CBF3, a four-protein complex that binds to the essential CDEIII region of Saccharomyces cerevisiae centromeres. We find that three subunits of CBF3 are in direct contact with CDEIII over a region of DNA that spans 80 bp. A highly asymmetric core complex containing p58CTF13 p64CEP3 and p110NDC10 in direct contact with DNA forms at the genetically defined center of CDEIII. This core complex spans ∼56 bp of CEN3. An extended complex comprising the core complex and additional DNA-bound p110NDC10 also forms. It spans ∼80 bp of DNA. CBF3 makes sequence-specific and -nonspecific contacts with DNA. Both contribute significantly to the energy of CBF3–DNA interaction. Moreover, important sequence-specific contacts are made with bases that are not conserved among yeast centromeres. These findings provide a foundation for understanding the organization of the CBF3–centromere complex, a structure that appears to initiate the formation of microtubule attachment sites at yeast kinetochores. These results also have implications for understanding centromere-binding proteins in higher cells.
Kinetochores are structures that assemble on centromeric DNA and attach chromosomes to the microtubules of the mitotic spindle. In most organisms, a single kinetochore is present on each chromosome. A chromosome lacking a correctly assembled kinetochore (an acentric chromosome) neither attaches correctly to microtubules nor segregates properly at mitosis. A chromosome with two kinetochores (a dicentric chromosome) is pulled toward both spindle poles and fragments. In either case, a cell will become aneuploid and may die. Because acentric and dicentric chromosomes are rapidly lost, the probability of incorrect kinetochore assembly cannot be higher than the observed frequency of chromosome nondisjunction, ∼10−5 per generation in budding yeast.
The stringent requirement for one and only one kinetochore per chromosome must reflect a tightly controlled assembly pathway. We are attempting to understand how kinetochores assemble on centromeric DNA in the budding yeast Saccharomyces cerevisiae. The comparison of centromeres from different yeast chromosomes reveals that they contain three DNA elements called CDEI, CDEII, and CDEIII (Clarke and Carbon, 1980; Fitzgerald-Hayes, et al., 1982; Hegemann and Fleig, 1993; see Fig. 1). CDEI is an 8-bp, well-conserved palindrome that binds to the dimeric helix–loop–helix domain protein Cbf1p (Baker and Masison, 1990; Cai and Davis, 1990). The deletion of CDEI DNA, or the gene that encodes Cbf1p, increases the rate of chromosome loss ∼10-fold, indicating that CDEI is important, but not essential for centromere function (Baker and Masison, 1990; Cai and Davis, 1990). CDEII is a 75–80-bp sequence that is 85–95% adenine–thymidine but has little sequence conservation and as yet, no known binding proteins. The deletion of CDEII abolishes centromere function, but point mutations within the element have little or no effect (Gaudet and Fitzgerald-Hayes, 1987).
Figure 1.

Structure of crosslinking probes. (note 1) “Filled-in” refers to the enzymatic synthesis of double-stranded DNA from a single-strand template using BrdU or BrdC, radionucleotide, and other deoxynucleotides. (note 2) Dark arrow indicates the region of DNA that was synthesized. (note 3) Numbers (−8, −14, −15, and −21) refer to the precise positions of BrdU/BrdC substitutions. Asterisks indicate additional specific sites substituted with BrdU or BrdC. (note 4) The box shows additional noncentromeric DNA that was added to promote oligo annealing.
CDEIII is an essential DNA element that binds to the multiprotein complex CBF3 (Lechner and Carbon, 1991). Point mutations in the absolutely conserved sequence at the center of CDEIII (5′-CCG-3′; the “CCG motif”) cause a 100–1,000-fold increase in chromosome loss in vivo and severely impair CBF3–DNA binding in vitro (McGrew et al., 1986; Ng and Carbon, 1987). Although superficially palindromic, two pieces of data demonstrate that CDEIII is inherently asymmetric. First, point mutations on the CDEII-proximal side of the CCG motif (referred to below as the left side) have a greater effect on centromere function in vivo and CBF3 binding in vitro than corresponding mutations on the CDEII-distal side (referred to as the right side; Hegemann et al., 1988; Jehn et al., 1991). Second, if the CDEIII region is inverted with respect to CDEI and CDEII in a manner that retains all of the bases present in a wild-type centromere, CBF3 binding is nearly as great as with wild-type CDEIII, but the CDEIII-inverted centromere is completely inactive in vivo (Murphy et al., 1991; Sorger et al., 1994).
CBF3 is a four-protein complex that appears to make both sequence-specific and -nonspecific contacts with centromeric DNA (Lechner and Carbon, 1991; Sorger et al., 1995). All four protein subunits of CBF3 are encoded by essential genes (Doheny et al., 1993; Goh and Kilmartin, 1993; Jiang et al., 1993; Lechner, 1994; Strunnikov et al., 1995; Connelly and Hieter, 1996). At semipermissive temperatures, temperature-sensitive mutations in either SKP1 (which encodes the CBF3p23 protein), CTF13 (which encodes the CBF3p58 protein), CEP3 (which encodes the CBF3p64 protein), or NDC10 (which encodes the CBF3p110 protein) elevates the rate of chromosome loss, demonstrating the importance of CBF3 for chromosome transmission. Of the four CBF3 subunits, only CBF3p64 has a recognizable DNA-binding motif (Lechner, 1994; Strunnikov et al., 1995). The amino terminus of p64 contains a GAL4-like C6 zinc cluster that functions like a zinc finger but has a different three-dimensional protein structure (Marmorstein et al., 1992). In extracts prepared from cells carrying a mutation in any one of the four CBF3 subunits, CDEIII-binding activity is abolished at nonpermissive temperatures (Sorger et al., 1995; Kaplan et al. 1997). Thus, all of the CBF3 subunits are required for DNA-binding activity. To understand why this is true, it is necessary to determine which proteins are in direct contact with DNA.
We have previously described the reconstitution of centromere–microtubule attachment in vitro and argued that CBF3 is necessary but not sufficient for this attachment (Sorger et al., 1994). Because CBF3 appears to bind to DNA throughout the vegetative cell cycle, we hypothesize that it forms a centromere-bound scaffold onto which other kinetochore proteins assemble (Sorger et al., 1995; Wilmen and Hegemann, 1996). This model implies that highly selective binding of CBF3 to centromeric DNA initiates kinetochore assembly and ensures that one and only one kinetochore forms per chromosome (Hyman and Sorger, 1995). Although the sequences of the CBF3 subunits are known, there is very little information about the architecture of the CBF3–DNA complex. As a result, elucidation of this architecture is an important first step in understanding how a microtubule attachment site forms. Moreover, we anticipate that there will be features of the three-dimensional organization of CBF3 that are common among the simple kinetochores of budding yeast and the much more complex centromeres of animal cells.
In this paper, we use nondenaturing gel analysis and DNA–protein crosslinking to probe the organization of the CBF3–DNA complex. To make the crosslinking of proteins to CEN3 DNA efficient, we inserted bromodeoxyuridine (BrdU)1 in place of T and bromodeoxycytidine (BrdC) in place of C. The exposure of BrdC- or BrdU-containing DNA to UV light generates a reactive free radical that is capable of forming a covalent bond with proteins that are within 2 to 4 Å of the brominated base. We will refer to these as proteins in “direct” contact with DNA. The bromines on both the top and bottom strands of BrdC- or BrdU-modified DNA project into the major groove. Because BrdC and BrdU can be introduced at single sites, the locations of CBF3 proteins along the major groove of binding site DNA can therefore be determined. From this information, a picture of the arrangement of CBF3 subunits with respect to each other can be inferred.
Using UV-activated crosslinking we find that three CBF3 subunits are in direct contact with bases in the major groove of CDEIII and that these contacts span >80 bp of DNA. There are remarkably few bases in CDEIII that are conserved among the 16 yeast centromeres, implying that sequence-specific binding involves contact with bases that are not part of the strict CDEIII consensus. Two related complexes assemble on CDEIII: a core complex and an extended complex. The core complex involves three DNA-bound CBF3 proteins and ∼56 bp of CEN3. The extended complex forms when p110 binds in an asymmetric manner to the core complex and to flanking CDEIII DNA to form a p110 multimer.
Materials and Methods
CBF3 Preparation
For whole-cell yeast extracts, cultures were grown to 2–5 × 107 cells/ml, washed by pelleting in breakage buffer (50 mM bis-tris-Propane, 100 mM β-glycerophosphate, 10 mM EDTA, 10 mM EGTA, 10% [vol/vol] glycerol), resuspended in a minimal amount of breakage buffer with protease inhibitors (1 mM PMSF, 10 μg/ml each of pepstatin, chymostatin, and leupeptin), frozen in liquid nitrogen, and ground in a chilled mortar and pestle as described (Sorger et al., 1995). Cell debris was removed by centrifugation at 15,000 g for 15 min. Typically, the total protein concentration was 25–50 mg/ml. Recombinant CBF3 was obtained from High Five insect cells (GIBCO BRL, Gaithersburg, MD) infected with baculoviruses expressing CBF3 subunits. Cells were coinfected with virus stocks to express each of the four CBF3 subunits. In some cases, however, subunits were expressed individually or in various combinations (see figure legends for details). The construction of baculovirus vectors and the production of recombinant CBF3 will be described elsewhere.
Probe Preparation
All centromere sequences were derived from S. cerevisiae CEN3. Probes are divided into crosslinking probes (X; see Fig. 1) and bandshift/competition probes (BC; see Fig. 2). The binding of CBF3 to both types of probes was assayed using nondenaturing polyacrylamide (bandshift) gels as described previously (Ng and Carbon, 1987; Sorger et al., 1995), but the primary purpose of the crosslinking probes was to analyze DNA–protein interactions. The primary purpose of the bandshift/competition probes was to examine the assembly of CBF3 on different DNA fragments.
Figure 2.

Structure of bandshift-competition probes. (note 5) See Sorger et al. (1995). (note 6) See Murphy et al. (1991).
To generate crosslinking probes, 50 pmol of two complementary oligonucleotides of unequal length were annealed in 10 μl of 2 mM MgCl2, 50 mM NaCl, 20 mM Tris, pH 7.4, by heating at 90°C for 10 min and then slowly cooling to room temperature. Photocrosslinkable BrdU bases were introduced by filling in the single stranded region using the Klenow fragment of DNA polymerase I. 40-μl extension reactions containing 5 U Klenow fragment (NEB), 125 μM BrdU (5-bromo-2′-deoxyuridine-5′-triphosphate; Sigma Chemical Co., St. Louis, MO), 125 μM dCTP, 125 μM dGTP, and 32P-α-dATP were incubated for 15 min at 30°C and then 20 min at 37°C. The final concentration of dATP varied from 5 to 50 μM depending on the length of the region being incorporated and typically contained a mixture of 3,000 Ci/mmol 32P-α-dATP and cold dATP at a molar ratio of 1:10 to 1:20. In some probes, BrdC (5-bromo-2′-deoxycytidine-5′-triphosphate; Sigma Chemical Co.) was incorporated in place of BrdU. Probes were purified by electroelution from 2% agarose TBE gels. The concentration of the probe was calculated from its specific activity.
In some cases, crosslinkers were introduced at single positions during oligonucleotide synthesis. Unexpectedly, we were unable to get efficient enzymatic extension of oligos in which the brominated base was at the extreme 3′ end of the oligo. However, the addition of two or more native DNA bases at the 3′ end of oligo solved this problem (see Fig. 1).
Bandshift/competition probes were derived from constructs described previously (Sorger et al., 1995). For each probe, 10 PCR reactions were pooled and the DNA purified on agarose gels. Probe concentration was determined by comparing band intensities with a HaeIII-digested θX standard on ethidium bromide-stained agarose gels using a digital camera and analyzed using the IP LabGel program (Molecular Dynamics, Inc., Sunnyvale, CA). Purified DNA was labeled with 32P-γATP using polynucleotide kinase. Some DNA probes for bandshift experiments and controls were prepared by body labeling in the absence of BrdU/BrdC.
UV Crosslinking
Pilot experiments revealed a technical difficulty in CBF3–DNA crosslinking. Ideally, DNA probes containing crosslinkers are end labeled by kinasing. All probes then have the same specific activity, and the reactivities of individual bases can be reliably compared. Despite considerable effort, we were unable to find conditions in which end-labeled CDEIII probes could be used with CBF3. Heavy nuclease treatment was necessary to allow CBF3 complexes crosslinked to DNA to be resolved on SDS-containing gels. This is not unexpected for a large protein complex bound to a long piece of DNA. However, heavy nuclease treatment invariably removed the radiolabel from end-labeled probes. Fine results were obtained with body-labeled probes, presumably because the radiolabel was often close to the site of crosslinking and thus protected from nuclease digestion by the bound protein. It is necessary to bear in mind that the intensity with which a protein is radiolabeled by a body-labeled probe reflects not only the crosslinking efficiency but also the number of radioactive bases that remain linked to the protein after nuclease digestion as well as the specific activity of each probe and thus can vary from probe to probe. Thus, as negative controls, probes without BrdU or BrdC, but with equal specific activity, were compared in parallel with every BrdU- or BrdC-incorporated probe.
150 μl binding reactions with 200 fmol of 32P-labeled CEN3 probe DNA made with either BrdU, BrdC, or native bases, 5 μl of crude extract from CBF3-expressing insect cells, 8 μg of sheared salmon sperm DNA, 45 μg casein and 10 mM Hepes (pH 8.0), 6 mM MgCl2, 10% glycerol were adjusted with 1 M KCl to a final concentration of 150 mM. After 35 min incubation at 25°C, a 30-μl aliquot was removed and loaded onto a 4% nondenaturing polyacrylamide bandshift gel, as described (Sorger et al., 1995). The remaining 120 μl was immediately transferred to a 2.0-ml flat-bottom tube. Samples were exposed to 305 nm UV light at a distance of 3.5 mm for 20 min at 25°C.
After UV exposure, samples were adjusted to 0.01 M CaCl2 and incubated at 37°C for 20 min with 60 U of micrococcal nuclease (Worthington Biochemical Corp., Freehold, NJ) and 25 U of DNase I (Worthington Biochemical Corp.). Proteins were then precipitated with 0.20 vol of cold trichloroacetic acid and 4 mg/ml deoxycholate by incubation on ice for 15 min. The precipitate was recovered by centrifugation in a microfuge at 4°C for 10 min. Pellets were washed with acetone, resuspended in electrophoresis loading buffer containing 1% SDS, and denatured by boiling for 5 min. Electrophoresis was performed on discontinuous SDS–polyacrylamide gels. Gels were fixed in a 20% methanol/7% acetic acid solution, dried on Whatman paper, and analyzed using a PhosphorImager (Molecular Dynamics).
DNA-binding Assays
To measure DNA-binding affinity, 30-μl reactions containing 0.5 μl of crude extracts from CBF3-expressing insect cells, 40 fmol of DNA probe, 9 μg casein, 3 μg of sheared salmon sperm DNA, in 10 mM Hepes (pH 8.0), 6 mM MgCl2, 10% glycerol, and adjusted with 1 M KCl to a final concentration of 150 mM were analyzed as described above. Competition experiments were carried out by mixing unlabeled wild-type or mutant CDEIII DNA with labeled probe DNA at various ratios and then adding CBF3. Off-rate experiments were performed by preincubating CBF3 with radiolabeled probe for 40 min and then adding a 200-fold excess of unlabeled 89-bp CDEIII competitor DNA to the reaction mixture (probe BC1). At times between 5 min and 4 h after the addition of competitor, reactions were loaded onto bandshift gels, and CDEIII-binding activity was measured. Off-rate experiments suggested that protein–DNA dissociation continued throughout the time that the gel was electrophoresing, typically 90 min.
To measure the ratio of sequence-specific to nonspecific binding, sheared salmon sperm DNA was used as a competitor. The average length of the salmon sperm DNA was 500 bp. Random sequence DNA of this length contains ∼103 50-bp sequences, and we have converted macroscopic binding constants into microscopic binding constants by dividing the former by 103.
Results
How many CBF3 proteins are in contact with DNA? To determine this, we performed DNA–protein crosslinking with CDEIII DNA probes in which the photactivatable bases BrdU or BrdC had been introduced. Our strategy was to make a set of probes in which the region containing BrdU or BrdC was made progressively smaller. We could then narrow in on bases required for crosslinking and finally investigate individual crosslinks by inserting BrdU or BrdC at single positions. The 34 different DNA probes used in this study are divided into 20 crosslinking probes (numbers X1 to X20; Fig. 1) and 14 bandshift/competition probes (numbers BC1 to BC14; Fig. 2). We refer to the CDEI/ CDEII-proximal end of these probes as the left end and the CDEI/CDEII distal end as the right end. Bases on the top strand of CDEIII are labeled from 1 to 89 and complementary bases on the bottom strand of CDEIII are labeled −1 to −89 (Fig. 3).
Figure 3.

Structure of CEN3 sequences used in this study. (a) Boxes indicate CDEI and CDEIII sequences. (b) The boxed bases denote the conserved core of CDEIII as determined by Hegemann et al. (1988). A diamond marks the location of the essential CCG motif. Solid circles indicate bases conserved among all 16 S. cerevisiae centromeres. Open circles indicate bases conserved in 15 of 16 centromeres. The top strand of DNA is numbered from 1 to 89 and the corresponding bottom strand is numbered from −1 to −89. “Left” refers to bases that are CDEII proximal, and “right” refers to the bases that are centromere distal. (c) This schematic is used in subsequent figures to represent various crosslinking probes. The heavy arrow indicates the region of single-stranded DNA in which BrdC or BrdU, 32PαdATP and native bases were incorporated by enzymatic fill-in.
Three CBF3 Subunits Contact DNA
For simplicity, we analyzed CDEIII-CBF3 crosslinking one strand at a time. We began by examining crosslinking to a 56-bp fragment of CDEIII that corresponds to the CBF3-dependent DNaseI footprint (see below; Lechner and Carbon, 1991) and is the smallest fragment of DNA known to bind to CBF3 with high affinity (Fig. 1, probe X1). To generate sufficient CBF3 for biochemical experiments, CBF3 p23, p58, p64, and p110 were coexpressed in insect cells infected with recombinant baculoviruses, and nuclear extracts were prepared (we refer to this as rCBF3). A description of the production of recombinant CBF3 as well as a comparison of the DNA-binding specificities of native and recombinant complexes will be published elsewhere. In experiments in which the biological significance of various CBF3–DNA complexes was investigated, we have used yeast CBF3. In all cases however, yeast and recombinant CBF3 had fundamentally similar properties. To crosslink CBF3 to DNA, body-labeled probes were incubated with rCBF3 (or yeast extract) and then exposed to 305 nm UV light for 20 min. This produces a covalent bond between photoactivatable BrdU (BrdC) in the probe and any protein within ∼2 Å. Nuclease digestion with DNase I and micrococcal nuclease was used to remove any excess DNA and the proteins were run on an SDS-PAGE gel and analyzed by autoradiography. This allows identification of DNA-labeled proteins.
After crosslinking of rCBF3 to the 56-bp BrdU-containing probe, three prominent radiolabeled bands with apparent molecular weights of 58, 65, and 110 kD as well as several weaker bands were visible (Fig. 4, lane 1). Even on long exposures, there was no evidence of crosslinking to a protein of 23 kD (the mass of p23SKP1). A competition assay with wild-type CDEIII DNA or a CDEIII mutant that contains a 3-bp deletion in bases essential for centromere function (probe BC2; called 3bpΔ CDEIII in Sorger et al., 1995) established that the 58-, 65-, and 110-kD bands arise from sequence-specific crosslinking to CDEIII DNA (Fig. 4, lanes 2 and 3). Other bands visible on the gel probably represent crosslinking to proteins in the nuclear extract that bind to DNA nonspecifically. To show that the CDEIII-labeled proteins are indeed CBF3 subunits, p58 and p64 were fused to amino-terminal epitope tags and p110 was truncated to remove 122 carboxy-terminal residues. These modified proteins are functional because they can complement a chromosomal deletion of the appropriate CBF3 gene (data not shown). When rCBF3 preparations containing modified subunits were crosslinked to DNA, the predicted molecular weight changes were observed (Fig. 4, lanes 4–6). With a BrdC-containing probe, only p58 and p64 were labeled (Fig. 4, lane 7; below we discuss the diminution in CBF3–DNA binding caused by BrdC incorporation). We conclude that CBF3-p58, p64, and p110 can be crosslinked to the bottom strand of CDEIII DNA.
Figure 4.
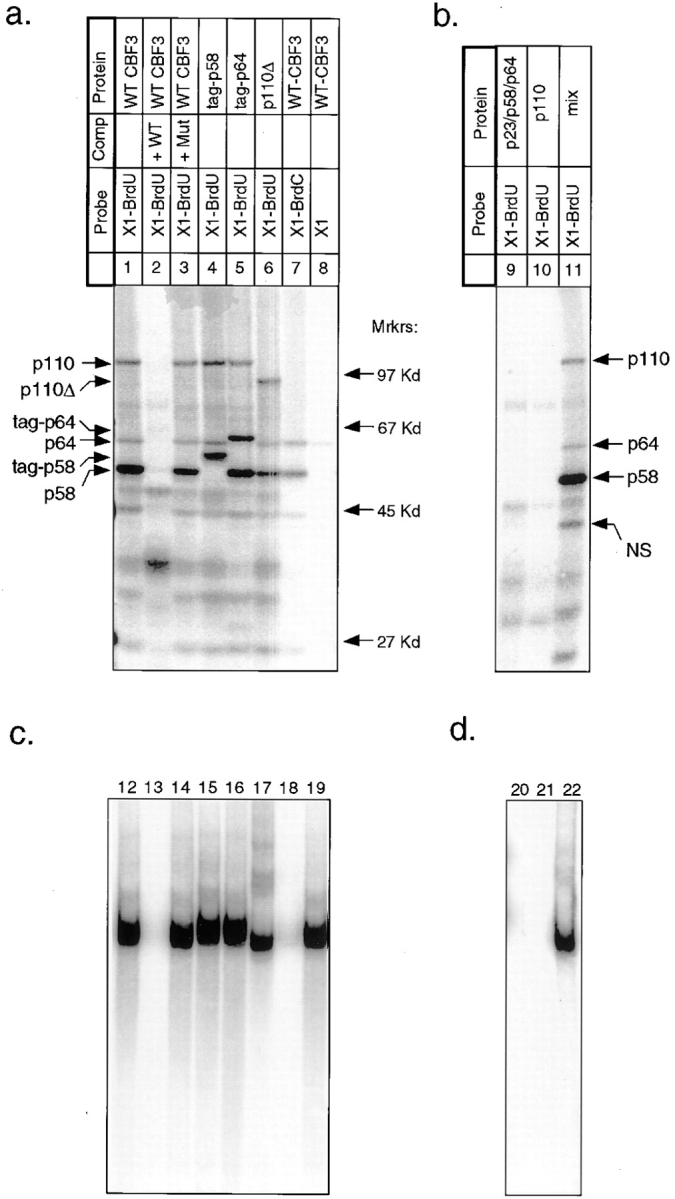
UV crosslinking of rCBF3 to the bottom strand of radiolabeled 56-bp CDEIII DNA. (a) rCBF3 proteins labeled by DNA crosslinking probes (as indicated) were run on an SDS-containing gel. The positions of molecular weight markers are shown. The types of crosslinking probe and CBF3 preparation used in each lane are indicated above the gel. The specificity of CBF3 binding was tested by including a 200-fold excess of unlabeled wild-type (WT; probe BC1) or mutant CDEIII competitor (Mut; probe BC2) DNA in the binding reaction. Some reactions contain rCBF3 with p58 (tag-p58) or p64 (tag-p64) proteins that are linked at their amino termini to a short epitope tag. p110Δ refers to rCBF3 containing p110 protein with a carboxy-terminal 122 amino acid deletion. For a complete description of probe X1 see Figs. 1 and 3. (b) Crosslinking reactions containing recombinant p64/p58/23 (lane 9), recombinant p110 protein (lane 10), or a mixture of these two extracts (lane 11) was analyzed as above. (c) One fifth of each crosslinking reaction from a was analyzed on bandshift gels (lanes 12–19). (d) One fifth of each crosslinking reaction from b was analyzed on bandshift gels (lanes 20–22).
To demonstrate the specificity of the crosslinking, we performed two control experiments. First, we established that CBF3 crosslinks to bromine-modified bases more efficiently than to native bases. When radiolabeled native DNA was used as a crosslinking probe, p64 was only weakly labeled, and p58 and p110 were not labeled, even after long exposures (Fig. 4, lane 8). As a second control, we showed that only assembled CBF3 complexes could be crosslinked to DNA. No crosslinking to BrdU-substituted CDEIII was observed in cell lysates containing rCBF3 subunits expressed individually or in two or three-subunit combinations (Fig. 4, lanes 9 and 10, and data not shown). The inability of individual CBF3 subunits to crosslink to DNA was not a trivial consequence of having inactive proteins because DNA binding by CBF3 (Fig. 4, lane 22) and crosslinking to BrdU-containing CDEIII DNA (Fig. 4, lane 11) were restored when extracts were mixed.
These data show that the crosslinking of CBF3 proteins to DNA involves sequence-specific binding to CDEIII, is stimulated by the incorporation of BrdU and requires functional CBF3 complexes. We conclude that CBF3-p58, p64, and p110 (but probably not p23) are in direct contact with bases in the major groove of CDEIII DNA.
Localizing p64 and p58 Crosslinks on the Bottom Strand of CDEIII
To localize CBF3–DNA crosslinks, we generated a set of probes in which BrdU or BrdC and 32PαdATP (the “filled-in” region; Fig. 1, probes X2–X4) were incorporated into progressively shorter regions of DNA. Crosslinking observed with these probes was compared to crosslinking observed with the probe X1 that contains a 39-bp filled-in region and labels p58, p64, and p110 (Fig. 5, lanes 1–3). When a probe with a filled-in region comprising 18 bases to the left of the CCG motif was used (Fig. 5, Probe X2), the labeling of p58 and p64 was as intense as with a probe X1 (Fig. 5, lanes 1–6). p110 labeling was observed only with BrdU, and was ∼50% as strong. A further reduction of the filled-in region to 11 bases (Fig. 5, Probe X3, lanes 7–9) eliminated p58 labeling, retained some p110 labeling, and left p64 labeling unaffected. A final reduction to seven filled-in bases (Fig. 5, Probe X4, lanes 10–12) eliminated all crosslinker-specific labeling. Thus, we infer that p58 crosslinks to the single T and single C present in the seven base interval that distinguishes probes X2 and X3 (i.e., −15T and −14C) and that p64 crosslinks to the T and C in the region that distinguishes probes X3 and X4 (i.e., −8C and −10T).
Figure 5.

UV crosslinking of rCBF3 to progressively shorter labeled regions of the bottom strand of the 56-bp CDEIII DNA. (a) Crosslinking reactions using differently labeled probes (as indicated) were analyzed on SDS-containing gels. Each set of three reactions was performed in parallel. The positions of the filled-in regions as well as the type of bromine-modified base (BrdU or BrdC) are indicated. The probe numbers and schematics are keyed to the full descriptions in Fig. 1. Lane 4 a is exposed only one fifth as long as lane 4 to allow better visualization of the crosslinked bands. (b) One fifth of each crosslinking reaction from a was separately analyzed on bandshift gels (lanes 13–24).
To establish directly the positions of individual crosslinks, two techniques were used. First, we generated a series of oligonucleotides in which single bromine substitutions were introduced during chemical synthesis. After UV irradiation, p64 was labeled by probes containing bromines at −8C and −21C, two- to threefold more strongly than by a native DNA control (Fig. 6, lanes 1–3 and 9–11). p58 was labeled by probes containing bromines at −14 C and −15 T but not by a native DNA control (Fig. 6, lanes 4–8). To overcome problems with low signal and incomplete digestion of crosslinked DNA (Fig. 6 a) we used mutant oligos that allowed crosslinker and radiolabel to be incorporated at successive bases (Fig. 1, X9 and X10). Although these mutated CDEIII sequences bound to CBF3 with 5- to 10-fold lower affinity than wild-type CDEIII (see below), competition analysis established that the binding was sequence specific (data not shown). When BrdC was present only at −14C, p58 was specifically labeled (Fig. 6 b, lanes 15–17, probe X9), and when BrdC was present only at −8C, p64 was specifically labeled (Fig. 6 b, lanes 18–20, probe X10). Taken together, these data demonstrate that on the bottom strand of CDEIII, p58 interacts with two successive bases at −14C and −15T, and that p64 interacts with two bases separated by slightly more than one turn of a B-form DNA helix, one at −21C and one at −8C.
Figure 6.
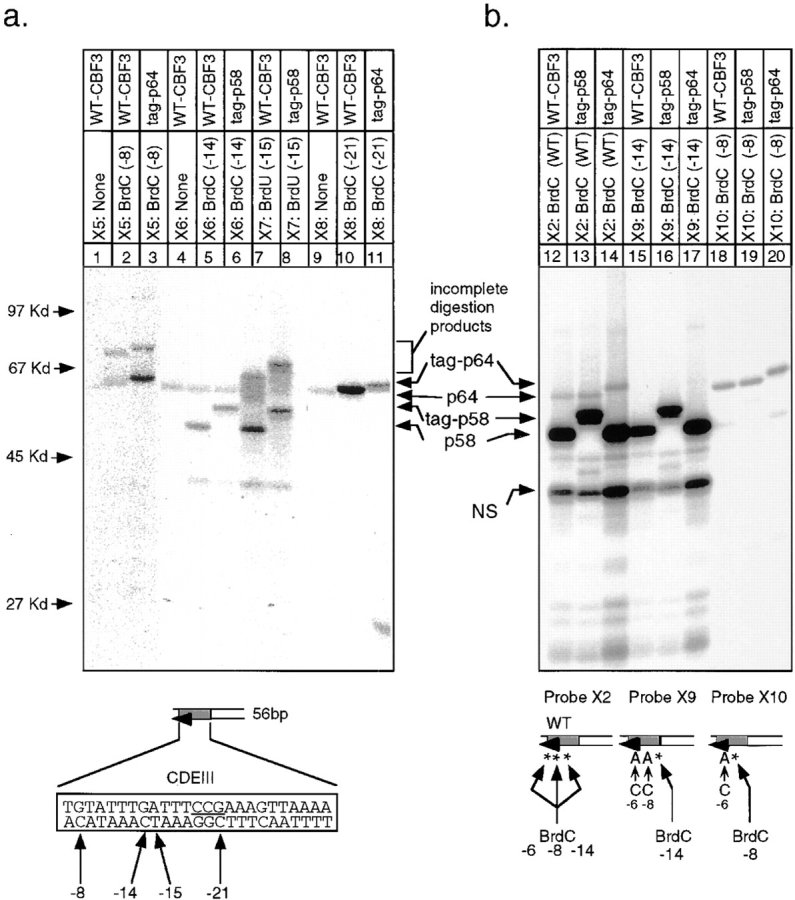
Localization of protein–DNA crosslinks on the bottom strand of 56-bp CDEIII DNA. (a) The site-specific crosslinkers, BrdC or BrdU, were incorporated at the positions indicated into the bottom strand of DNA. In each case “none” refers to the synthesis of probe in the absence of BrdC or BrdU. (b) Site-specific BrdC crosslinkers (indicated by asterisks) were incorporated using mutant top-strand oligos and appropriate bottom-strand oligos by enzymatic extension (see Fig. 1 for details about the probes). Crosslinking reactions using probes described in a and b were analyzed on SDS-containing gels as in Fig. 4. Faint labeling of p58 in lanes 18–20 are a result of non-BrdC–dependent binding as in Fig. 5, lane 12.
Localizing p64 and p110 Crosslinks on the Top Strand of CDEIII
To investigate crosslinking to the top strand of CDEIII, we again used a set of probes with progressively shorter labeled regions. When BrdU was incorporated in place of T between bases 2 and 56 on the top strand (probe X11), p64 and p110 were efficiently labeled in a sequence-specific manner, but only very weak labeling of p58 was observed (Fig. 7 a, lanes 1–3, and data not shown). By using probes with progressively shorter filled-in regions (Fig. 1, probes X12–X18), we were able to localize a p64 crosslink to +7T (Fig. 7 a, compare lanes 5 and 6). This site lies immediately across the major groove from the bottom-strand crosslink at −8C, strengthening the argument that this region of CDEIII is in close association with p64.
Figure 7.
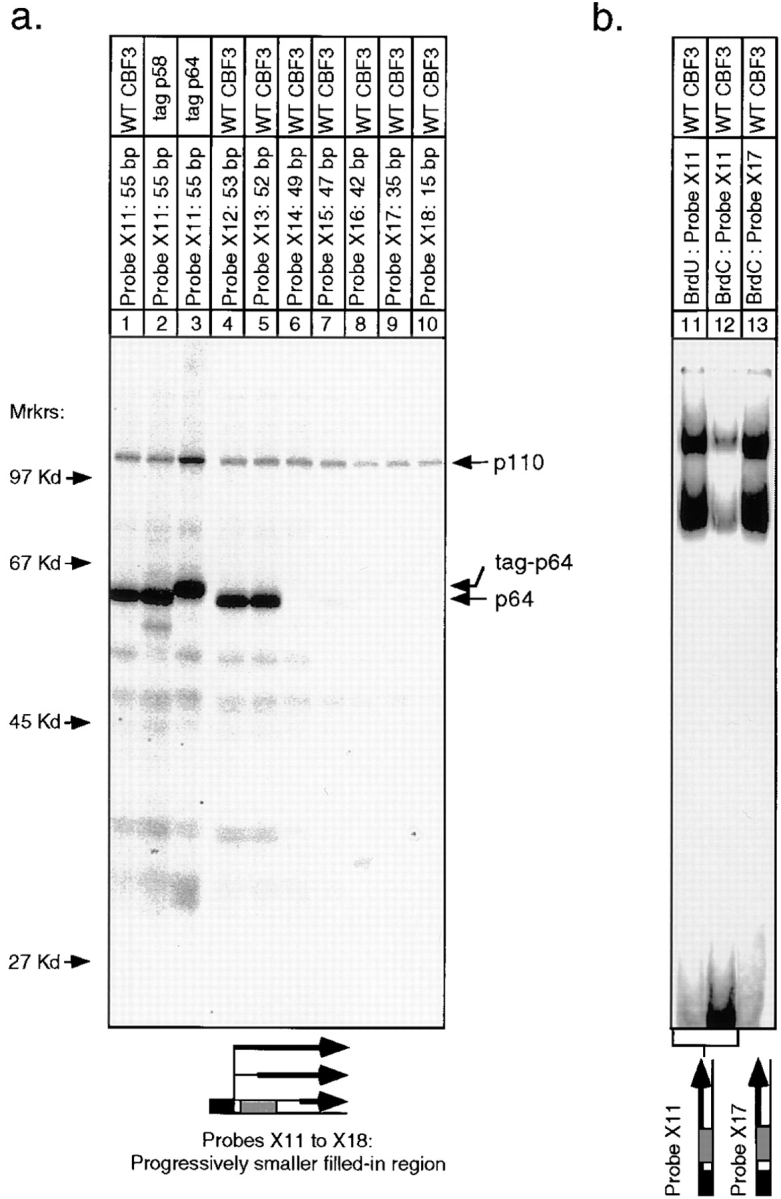
UV crosslinking of rCBF3 to progressively shorter labeled regions of the top strand of the 56-bp CDEIII DNA. (a) BrdU was incorporated into each crosslinking probe as in Fig. 5. Reactions were carried out in the presence of either wild-type or tagged CBF3 proteins and analyzed on an SDS-containing gel. (b) Probes with either BrdU or BrdC incorporated into the entire top strand (+2 to +56; lanes 11 and 12) or a probe with BrdC incorporated only to the right of the CCG (+22 to 56; lane 13) were mixed with rCBF3 and analyzed on bandshift gels.
When BrdU was incorporated as a crosslinker, p110 was labeled in a sequence-specific manner by most of the top- and bottom-strand probes that were tested. In no case did we observe p110 crosslinking to BrdC-containing probes, implying that p110 does not bind to the major groove of CDEIII within 3 Å of a cytosine. As described in the Discussion, p110 appears to be multimeric. Attempts to localize the crosslinking of at least one p110 monomer to a single base or a small set of bases were not successful (Figs. 5–7), suggesting that p110 contacts an extended region of the major groove. However, we cannot exclude the possibility that there are a few localized regions of p110–DNA contact that we have not been able to identify because of technical limitations in the crosslinking methodology. We can conclude, however, that p110 can be crosslinked to the middle of CDEIII and also to the left end on the bottom strand and to the right end on the top strand (Figs. 5, lanes 2–12; and 7, lanes 9 and 10). Thus, the contacts that p110 makes with CDEIII are not restricted to a single stretch of DNA.
BrdC-dependent Binding Interference
In several cases, we found that the incorporation of BrdC into CDEIII DNA interfered with CBF3–DNA binding. With the bottom-strand probe X1 and the top-strand probe X11, CBF3 binding affinity was 10-fold weaker than with native DNA (Figs. 5, lanes 13 and 15; and 7, lanes 11 and 12). On both DNA strands, interference with CBF3– DNA binding occurred primarily when BrdC was incorporated into the CCG motif (e.g., Fig. 7, compare lanes 12 and 13). The bromine in BrdC lies in the C-5 position and replaces a hydrogen in the native nucleotide that projects into the major groove. BrdC incorporation substantially occludes the major groove of DNA and, independently from its use as a crosslinker, BrdC-dependent interference can been used to identify critical regions of protein–DNA contact (de Vries et al., 1987). In the case of CBF3-CDEIII interaction, we conclude that the presence of BrdC at −21C, +19C, or +20C in the CCG motif interferes significantly with DNA binding. This presumably reflects a direct interaction of CBF3 with these bases and is consistent with the important role that the CCG motif plays in centromere function in vivo (Hegemann et al., 1988).
CBF3-crosslinked Bases Are Important for DNA Binding
Of the bases in CDEIII that can be crosslinked to CBF3, only those in the CCG motif at positions 19–21 have previously been shown to be important for CBF3–DNA binding in vitro (Ng and Carbon, 1987; Sorger et al., 1995). To investigate the importance of other sites of CBF3–DNA contact, we performed a competition analysis with a mutant probe (Comp. BC3) in which both −8C (which crosslinks to p64) and −14C (which crosslinks to p58) were changed to T. CBF3 bound to this sequence 50-fold more weakly than to wild-type DNA (Fig. 8 a, compare BC4 and BC3). We also examined sequences to the right of 38T that crosslink to p110 but that lie outside of the CDEIII region as conventionally defined (Cottarel et al., 1989). A variant 56-bp CEN3 fragment was constructed in which the order of bases 38 to 56 was scrambled but the base composition retained (Fig. 8 a, Comp. BC5). The binding of CBF3 to this probe was ∼50-fold weaker than to wild-type DNA (Fig. 8, Comp. BC4) but stronger than to the mutant 3bpΔ CDEIII (Fig. 8, Comp. BC2). The removal of bases between +38 and +56 to generate a 38-bp-long CDEIII fragment eliminated even residual binding to BC4 (Fig. 8 b, lanes 1–3). Thus, CDEIII bases identified by crosslinking as being in close proximity with CBF3 are also required for sequence-specific CBF3–DNA binding.
Figure 8.

Analysis of centromeres containing point mutations in sequences that crosslink to CBF3 subunits. (a) rCBF3 and radiolabeled probe BC4 were mixed with a 4- to 200-fold excess of unlabeled centromeric DNA competitors (as indicated) and analyzed on bandshift gel. Values are expressed as a percentage of core complex CDEIII binding observed in the absence of competitor. Error bars represent the range of values obtained in duplicate experiments. (b) rCBF3 was mixed with radiolabeled 56-bp wild-type DNA (Probe BC4; lane 1), linker-modified 56-bp DNA (Probe BC5; lane 2), or 38-bp probe DNA (Probe BC6; lane 3), and CDEIII binding was analyzed on bandshift gels.
Multiple p110-dependent CBF3–DNA Complexes
In chromosomes, centromeric DNA is imbedded in flanking DNA. We therefore investigated the effect of lengthening the DNA probe on CBF3–DNA complex formation. Previously, we and others had noted that when centromeric probes longer than 56 bp are used for bandshift analysis, more than one CBF3–protein complex can form (Lechner and Carbon, 1991; Jiang and Carbon, 1993; Sorger et al., 1995). We started with a wild-type 89-bp probe (probe BC1) containing the 56 bp of DNA in probe BC4 as well as 33 bp of CEN DNA lying to its right. This encompasses the region of CDEIII that is partially protected in the DNase I footprint (Lechner and Carbon, 1991). Gel analysis with this probe revealed upper and lower bands. Both corresponded to sequence-specific DNA–protein complexes (Fig. 9, lanes 2–4). The lower band electrophoresed to the same position as the band corresponding to the CBF3 complex formed with the 56-bp probe (Fig. 9, lane 1). To define more precisely the amount of DNA required to produce the upper band, we generated an 89-bp probe (probe BC7) containing 56 bp of CEN DNA and 33 bp of linker DNA. The linker contained four restriction sites, allowing the probe to be cleaved into 80-, 72-, 64-, and 56-bp DNA fragments (probes BC8 to BC11). The 89-bp linker-modified probe gave rise to upper and lower bands in essentially the same ratios as wild-type CDEIII DNA (Fig. 9, compare lanes 2 and 5). The amount of upper band remained high with an 80-bp probe (Fig. 9, lane 7), fell 2-fold with a 72-bp probe (lane 8), and was reduced at least 20-fold with a 64-bp probe (lane 9). From these findings, we conclude that two discrete CBF3–DNA complexes can form on centromeric DNA and that formation of the more slowly migrating complex requires DNA between positions 57 and 80.
Figure 9.
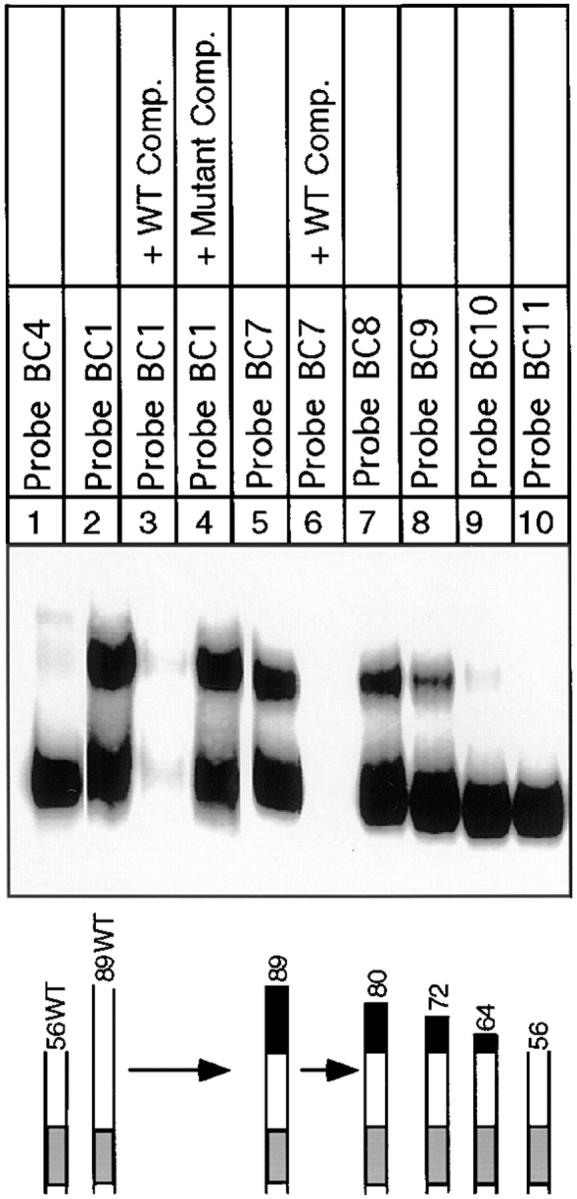
Analysis of extended complex formation on progressively shorter CDEIII probes. rCBF3 and the indicated probes were analyzed by bandshift gel. Specificity was demonstrated by adding excess unlabeled wild-type (lanes 3 and 6) or mutant (lane 4) CDEIII competitor DNA.
To investigate the difference in protein composition between upper and lower bands, we altered CBF3 subunit stoichiometry by expressing p110, p64, or p58/p23 separately and then adding them to rCBF3 (see Fig. 10, legend). In general, the total amount of DNA-bound CBF3 increased when subunits were added, but only in the case of p110 addition did we observe a substantial increase in the ratio of the upper to lower bands (Fig. 10 a, lanes 1–6). We interpret this to mean that the upper band corresponds to a CBF3 complex with more p110 than the lower band. Even when large amounts of p110 were added, the upper band was seen only with the 89- and not with the 56-bp probe (Fig. 11 a, lanes 1–4). To demonstrate that the additional p110 in the upper band is DNA bound, we crosslinked rCBF3 to a probe in which BrdU had been incorporated only in the 33 bases that lie to the right of 56C (Fig. 11, Probe X19). This probe specifically labeled only p110, proving that p110 indeed touches DNA in the 56C to 89G interval (Fig. 11 c, lane 11). Because the more slowly migrating complex involves a longer piece of DNA and more p110 molecules, we will refer to it as the extended complex. The complex that forms on 56-bp DNA will be referred to as the core complex. The formation of the extended complex on the 89-bp probe does not appear to radically alter the architecture of the core CBF3 complex, because the patterns of bottom strand crosslinks with the 56- and 89-bp probes were nearly indistinguishable (compare Figs. 11 b, lanes 5–7, with 2, lanes 1, 7, and 8). Thus, we conclude that the extended CBF3 complex forms when additional p110 binds to the core complex, without substantially altering its organization.
Figure 10.
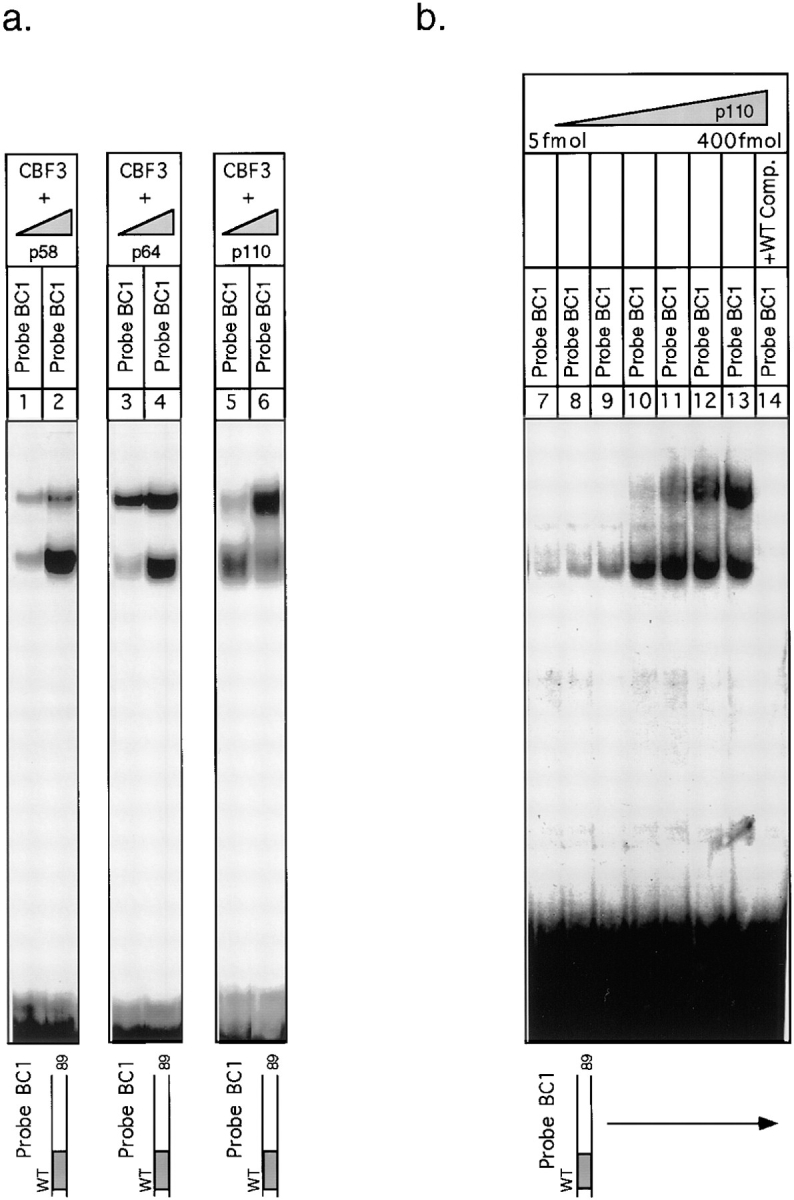
Effect of varying the stoichiometry of CBF3 subunits on CBF3–DNA complex formation. (a) Extract containing recombinant p110 and p64 (lanes 1 and 2), p110, p58, and p23 (lanes 3 and 4) or p64, p58, and p23 (lanes 5 and 6) were mixed with 10 to 20 fmol or with 60 to 120 fmol of p58/p23, p64, or p110 and complexes analyzed on bandshift gels. (b) Extract from ndc10-42 cells (80 μg) was mixed with 5, 10, 20, 50, 100, 200, or 400 fmoles of recombinant p110 (lanes 7–14). Specificity was demonstrated by adding excess wild-type CDEIII competitor DNA (lane 14).
Figure 11.

Analysis of p110 binding to 89-bp CDEIII probe. (a) rCBF3 on its own or mixed with excess p110 (as indicated) was bound to a 56-bp (lanes 1 and 2) or 89-bp (lanes 3 and 4) probe and analyzed on a bandshift gel. (b) 89-bp probes containing either BrdU, BrdC, or no crosslinkers in the bottom strand were reacted with rCBF3 and analyzed on an SDS-containing gel. (c) 89-bp probes with BrdU incorporated in the entire top strand or only in the region between +56 and +89 bp were added to extracts containing rCBF3 as indicated and crosslinking analyzed on an SDS-containing gel.
Properties of the Extended CBF3–DNA Complex
To characterize the extended complex, we investigated its concentration dependence, DNA-binding affinity, and stability. To determine the concentration of p110 required for extended complex formation, we prepared extracts from yeast cells carrying a temperature-sensitive mutation in the p110 subunit (ndc10-42) and then added increasing amounts of recombinant p110. ndc10-42 extracts lack CDEIII-binding activity (Sorger et al., 1995), but this defect is complemented by the addition of recombinant p110 (Fig. 10 b, lanes 7–14). The concentration of p110 at which the extended CBF3 complex formed (1.7 nM) was only fivefold higher than the concentration at which the core complex formed (0.4 nM). Similar results were obtained when using rCBF3 components. In extracts from most wild-type yeast strains, we have observed that p110 is present in substantial excess over the other CBF3 subunit and that roughly equimolar core and extended complexes form on 89-bp CDEIII probe (Sorger et al., 1995). It therefore seems likely that the concentration of p110 in cells is sufficient to drive extended complex formation.
Next, we estimated the DNA-binding affinities of the core and extended complexes by performing a series of bandshift reactions with increasing amounts of the 89-bp probe (probe BC1). This data is plotted in Fig. 12 a as a saturation curve. Half-maximal formation of the extended complex occurred at 8 × 10−10 M and of the core complex at 2 × 10−10 M (these are not true binding constants because the reactions were not at equilibrium). We also measured the half-lives of the core and extended complexes by incubating yeast CBF3 with an 89-bp probe (probe BC1) for 40 min, adding a large excess of unlabeled CDEIII, and then measuring the amounts of the two complexes as a function of time. Both the core and extended complexes were relatively stable, with half-lives of ∼2 h (Fig. 12 b). Taken together, these data argue that the extended complex has a high affinity for DNA and that it is kinetically as stable as the core complex.
Figure 12.

Analysis of core and extended CBF3 complexes. (a) Data for the saturation curve was obtained by adding an 89-bp wild-type probe (probe BC1) at increasing concentrations to a fixed amount of semi-pure yeast CBF3 and then determining the amounts of core and extended complexes on bandshift gels. (b) The off-rate for yeast CBF3 DNA complexes was measured by adding 200-fold excess of unlabeled BC1 competitor to CBF3 prebound to a radiolabeled 89 bp wild-type probe (BC1). At time points between 5 and 270 min, reactions were loaded on bandshift gels for analysis. Data points and best-fit exponential lines are shown. The lines extrapolate back to 100% binding at −90 min. This was the running time of the gel, suggesting that CBF3 was equilibrating between labeled and unlabeled CDEIII DNA throughout this period. (c) rCBF3 and radiolabeled probe BC4 were mixed with a 4- to 200-fold excess of unlabeled centromeric DNA competitors (as indicated) and analyzed on bandshift gels (see Fig. 6). (d) 89-bp radiolabeled CEN3 DNA (probe BC1) was mixed with unlabeled 89-bp wild-type CDEIII DNA (solid lines) or sheared salmon sperm testes DNA (broken lines) at a range of concentrations and rCBF3 was then added. Values are expressed as a percentage of CDEIII binding observed in the absence of competitor. Upper (more slowly migrating) and lower (more rapidly migrating) complexes are indicated by right-side-up and inverted triangles, respectively.
Among S. cerevisiae centromeres, there is no apparent conservation of the sequences that lie between bases 56T and 89G. To determine whether p110 makes sequence-specific contact with this region of CDEIII, we generated a linker-modified probe with 56 bp of wild-type CDEIII DNA and 33 bases of scrambled DNA to the right that had the same base composition as wild-type CEN3 but was unrelated in sequence (this probe is identical to probe BC7 described above). Competition analysis showed that the 56-bp wild-type DNA (Comp. BC4), 89-bp wild-type (Comp. BC1), and 89-bp linker–modified probes (Comp. BC7) had similar affinities for CBF3 (Fig. 12 c). From this, we conclude that the contacts p110 makes with bases 56–89 as part of the extended CBF3 complex are nonsequence specific.
If p110 contacts bases 56–89 nonspecifically, can p110 nevertheless assemble selectively on CDEIII DNA in the context of the large amount of noncentromeric DNA found in a chromosome? To answer this, we compared the sequence-specific and -nonspecific binding affinities of core and extended CBF3 complexes. This was done by binding CBF3 to an 89-bp wild-type probe (probe BC1) in the presence of either specific competitor DNA (unlabeled BC1 CDEIII DNA) or nonspecific competitor DNA (bulk DNA from salmon sperm testes). The amounts of core and extended CBF3 complexes were then determined on bandshift gels for a range of specific and nonspecific competitor DNA concentrations. The relative affinities of CBF3 for specific and nonspecific DNA could then be approximated by comparing the concentrations of competitor at which complex formation was reduced to 50% of its original value. We found that a 50% reduction in the amount of labeled core and extended complexes occurred at an identical concentration of specific CDEIII DNA, ∼3 × 10−9 M. With salmon sperm DNA as a competitor, a 50% reduction in the amount of core CBF3 complex occurred at 2 × 10−6 M and a 50% reduction in the amount of extended complex occurred at 5 × 10−7 M (Fig. 12 d). From this we conclude that the macroscopic ratio of affinities for the binding of the core CBF3 complex to CDEIII DNA and to bulk DNA is ∼1,000-fold. Because the preparation of bulk DNA we used had an average length of 500 bp, it contained ∼1,000 random 50-bp sites per molecule. Thus, the molar ratio of specific to nonspecific binding sites at half-maximal CBF3 binding was ∼1 × 106 (this is the ratio of microscopic affinities). The microscopic ratio of binding affinities for formation of the extended complex was ∼fivefold lower. Because p110 does not appear to make sequence-specific contact with bases 56 to 89, to form an extended complex, it must recognize bases 56 to 89 in the context of the immediately adjacent and highly sequence-specific core complex.
Asymmetric Binding of CBF3 to DNA
The yeast centromere is strikingly asymmetric. Bases on the left side of CDEIII are more important than the corresponding bases on the right side of CDEIII (Hegemann et al., 1988; Jehn et al., 1991; Sorger et al., 1995). We can now understand this asymmetry, at least in part, because both p58 and p64 make sequence-specific contacts on the left side but not on the right side of CDEIII. To determine if asymmetry is also observed in the formation of the extended complex, we asked whether p110 could bind to DNA on both the left and right sides of the core. A set of three 89-bp probes was constructed, each containing the 56 bases bound by the CBF3 core complex but differing by having either wild-type CDEIII or linker DNA to the right or wild-type CDEII DNA to the left. Whereas both core and extended complexes formed on probe containing wild-type bases (probe BC1) or linker DNA (probe BC7) to the right, only the core complex formed efficiently on probe having extra DNA to the left (probe BC12), even under conditions of p110 excess (Fig. 13 a, lanes 1–6; the upper band formed on BC12 is at least 20-fold weaker than the upper band formed on BC1). We conclude that the extended CBF3 complex can form by the addition of p110 molecules to the right of the core complex (on the CDEII-distal side) but not to the left.
Figure 13.
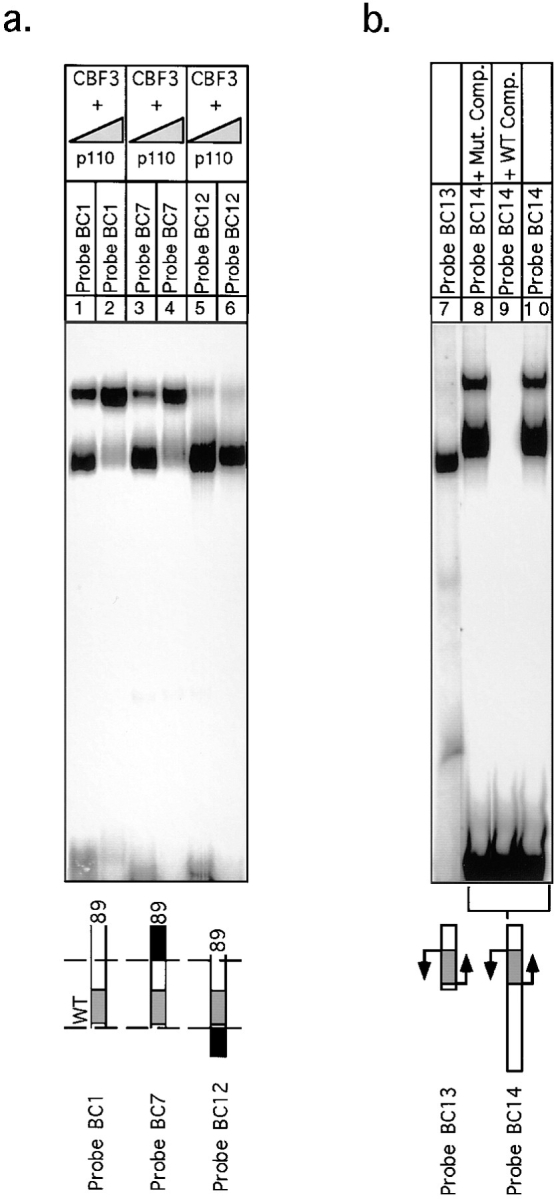
Formation of the extended CBF3 complex on probes containing wild-type and inverted CDEII sequences. (a) rCBF3 on its own or mixed with excess p110 (as indicated) was bound to an 89-bp wild-type probe (lanes 1 and 2), an 89-bp probe containing a linker to the right of base +56 (lanes 3 and 4), or an 89-bp probe containing 33 bp of CDEII to the left of base +1 (lanes 5 and 6) and analyzed on a bandshift gel. (b) rCBF3 was mixed with a radiolabeled 56-bp inverted-CDEIII DNA probe (lane 7) or radiolabeled 106-bp inverted-CDEIII DNA probe (lanes 8–10) and analyzed on a bandshift gel. Specificity was demonstrated by the addition of wild-type or mutant CDEIII DNA competitors, as indicated.
The implication of this finding is that the asymmetry of CDEIII is a property of the core sequences and is not imposed by flanking CDEII and centromere-distal DNA sequences. If this is correct, one prediction is that we can switch the left–right asymmetry in p110 binding by inverting the orientation of the central bases of CDEIII. A centromere variant containing an inverted CDEIII region had previously been shown to bind to CBF3 but to be nonfunctional in vivo (Murphy et al., 1991; Sorger et al., 1995). Using CDEIII-inverted DNA we generated two probes: one that contained only the flipped-core sequence (probe BC13) and one that contained the flipped core and 33 bases of CDEII sequence to its left (probe BC14). Bandshift analysis revealed that with CDEIII-inverted DNA, the extended complex formed with an orientation specificity opposite to the specificity observed with wild-type CDEIII DNA (Fig. 13 b, lanes 7–10); p110 could now bind to the DNA that lies to the left of the core and form an extended complex with reverse orientation. We therefore conclude that p110 indeed interacts with the CBF3 core complex in an asymmetric fashion and that this asymmetry is imposed by the core itself and not by the flanking DNA.
Discussion
The crosslinking experiments described here had three immediate goals: to establish which CBF3 proteins are in contact with DNA; to investigate the basis of sequence-specific binding by CBF3; and to determine the basis for the asymmetry of CDEIII–CBF3 interaction. We have found that in the context of an assembled CBF3 complex, the CBF3 subunits p58CTF13, p64CEP3, and p110NDC10 are in close contact with bases in the major groove of DNA but that p23SKP1 does not appear to touch DNA. Mutations in any one of the four CBF3 subunits abolish the CDEIII-binding activity of CBF3. In the cases of p58, p64, and p110 this probably reflects a direct contribution to the affinity of the CBF3–DNA interaction. In the case of p23, we have found that this reflects its role in the phosphorylation-dependent activation of p58 and that CBF3–DNA complexes containing activated p58 can form in the absence of p23 (Kaplan et al., 1997).
Organization of the CBF3–DNA Core Complex
Based on our analysis, we can draw the following model for the architecture of the CBF3–DNA complex. Most of the CBF3–DNA contacts occur within the 56 bases of CDEIII that correspond (at least approximately) to the smallest functional CBF3-binding site. Within this binding site, p64 appears to touch two regions of centromeric DNA. One is the CCG motif at bp 19 to 21 and one is at 7T and −8C (and possibly also −10T). The CCG motif is present in all 16 yeast centromeres, and single point mutations in the motif increase chromosome loss rates 100– 1,000-fold (McGrew et al., 1986). Thus, CBF3-CDEIII binding in vitro is sensitive to changes in major groove atoms at bases that are essential for centromere function in vivo; this argues strongly that the mode of CBF3 binding observed in vitro is fundamentally similar to the binding that occurs in vivo.
The amino terminus of p64 contains a C6 DNA-binding motif whose structure was first determined in the yeast transcriptional activator Gal4p (Marmorstein et al., 1992). CBF3–DNA binding is substantially impaired by mutations in p64 residues that, by analogy to residues in Gal4p, should touch DNA (Lechner, 1994). This argues that p64 probably contains a C6 cluster similar in fold to the cluster found in Gal4p. C6–Zn clusters are found in a large number of dimeric transcriptional activators and are folded into a compact domain that fits into the major groove of DNA (Marmorstein et al., 1992; Bohm et al., 1997). The binding sites for dimeric C6 cluster proteins contain two CCG sequences, and one of the two DNA-binding domains of a Gal4p-type dimer lies in the major groove of one CCG half-site, and the second binding domain lies in the major groove of the other CCG half-site (Reece and Ptashne, 1993). Specificity in DNA binding arises because different members of the C6 cluster family recognize CCG half-sites with different orientations and spacing (Zhang and Guarente, 1994). We have recently obtained data on the hydrodynamic properties of p64 showing that it is a dimer (Grancell, A., and P.K. Sorger, unpublished results; Lechner, 1994). These findings suggest that one C6–Zn cluster of a p64 dimer binds to the major groove of CDEIII at 20C and a second Zn cluster binds to the major groove near 7T (Fig. 14). A striking feature of this arrangement is that only one of the p64–Zn clusters appears to make contact with a CCG half-site. Perhaps this is why p64 is unable to bind to CDEIII DNA on its own, whereas proteins such as Gal4p bind to their operators with high affinity. It will be interesting to determine whether the difference between p64 and Gal4p lies in the DNA-binding sites or whether some structural property of the p64 protein makes it inherently unable to bind to DNA unless other CBF3 subunits are present.
Figure 14.

A model for the organization of the CBF3–DNA complex. (a) The positions of BrdU- and BrdC-mediated crosslinks (solid arrows) and BrdC-dependent interference of the CBF3–DNA interaction (open arrows) are shown in the diagram. Sequences with which CBF3 makes sequence-specific and -nonspecific contacts are indicated. (b) CBF3 subunits have been positioned at their sites of DNA crosslinking and the core and extended CBF3 complexes indicated with brackets. See Results for details.
Neither p58 nor p110 have recognizable DNA-binding motifs, but both contact DNA. p58 appears to bind to CDEIII on the opposite side of the DNA helix from p64 near −14C and −15T. The close physical proximity of p64 and p58 suggests that these proteins could touch each other, and there is evidence that a p58–p64 complex can form in the absence of p110 and DNA (Stemmann and Lechner, 1996). Preliminary hydrodynamic data suggests that p58 is a monomer, although its oligimerization state has yet to be determined reliably (Grancell, A., and P.K. Sorger, unpublished observations). p110 can be crosslinked to BrdU (but not BrdC) bases throughout the length of CDEIII, suggesting that p110 makes multiple contacts with DNA. p110 appears to be a dimer in solution (Grancell, A., and P.K. Sorger, unpublished observations), and for simplicity we speculate that a single dimer of p110 is bound to the CDEIII DNA between bases 1 and 56.
The positions of CBF3–DNA crosslinks and of BrdC-dependent interference are summarized in Fig. 14 along with a speculative model for the organization of the CBF3– DNA complex. It postulates that a p64 dimer binds via C6 cluster domains to two regions of the major grove ∼1 1/4 helical turns apart. p58 binds to the major groove between these sites and on the opposite side of DNA. p110 dimers bind along the top and bottom strands in an as yet unknown manner. Missing from this model is any sense of the geometry of CDEIII DNA; it is possible, for example, that it bends upon CBF3 binding. It is also possible that there are contacts between CBF3 proteins and other regions of centromeric DNA, or their associated proteins, lying to the left and right of the 89-bp sequence we have examined here (Hegemann et al., 1988).
Formation of a p110-dependent Extended CBF3–DNA Complex
The structure of the CBF3 complex that forms on CDEIII in vitro depends on the length of the DNA and on the concentration of p110. When CBF3 is bound to CDEIII DNA that is 56 bp long, only the core DNA–protein complex described above is formed. However, when the CDEIII DNA is 80 bp or longer, two CBF3–DNA complexes with different electrophoretic mobilities are formed (Fig. 9; Lechner and Carbon, 1991; Sorger et al., 1994). The faster-migrating or core complex is similar in composition to the complex that forms on the 56-bp DNA. The more slowly migrating complex assembles when additional p110 binds to the 33 bases of DNA that lie to the right of core complex. The interaction of p110 with CDEIII bases 56 to 89 is sequence nonspecific, implying that p110 can bind selectively to these bases only because it recognizes them in the context of the immediately adjacent CBF3 core complex.
We cannot yet test directly the importance of the extended CBF3 complex for kinetochore function in cells. However, several properties of the extended complex argue that it does indeed form on centromeric DNA in vivo. First, its apparent affinity for DNA is as high as that of the CBF3–DNA core complex. Second, it is as stable as the core complex with an off rate of ∼2 h. Third, it forms at a concentration of p110 that is only three- to fourfold higher than the concentration at which the core complex forms and p110 concentrations in yeast extracts are indeed great enough to drive extended complex formation. We propose that the functional CBF3–DNA complex in vivo is similar to the extended complex that forms in vitro and that it contains two DNA-bound dimers of p110, 80 bp of centromeric DNA, and has an overall mass of 680 kD.
Assembling One and Only One Kinetochore Per Chromosome
In organisms such as S. cerevisiae, one of the most important properties of the kinetochore is that it assembles at one and only one site on each chromosome. This requires that one 125-bp centromeric sequence be recognized in the context of a chromosome with an average length of 800 kb. We believe that this selectivity is achieved by four mechanisms: the sequence-specific binding of CBF3 to CDEIII; the juxtaposition of multiple DNA elements so that a single element (such as CDEIII) is not active on its own and must be flanked by a second element (CDEII; Gaudet and Fitzgerald-Hayes, 1987); tight control over the amounts and activities of CBF3 subunits (Kaplan et al., 1997); and the operation of an error-sensing checkpoint that detects those assembly errors that do occur (Murray, 1992). In this paper we are concerned only with the first of these mechanisms.
There are two ways in which sequence selectivity can be achieved. One possibility, exemplified by the HO endonuclease, is a short, highly conserved binding site. 17 bases of the 24 bp in the HO-binding site are conserved between MATa and MATα (Nickoloff et al., 1986). A match to a 17-bp consensus occurs at random once every 2 × 1010 bases, easily accounting for the selectivity of the endonuclease (although other mechanisms are also involved in site specificity in a cell). A second possibility is that the binding site is long and selectivity is achieved by summing over many DNA–protein contacts, only some of which are completely base selective. The interaction of CBF3 with CDEIII DNA appears to be an example of the latter type of interaction.
We do not yet understand what aspects of CDEIII are important for sequence-specific binding. The highly conserved bases clearly play a role, but there are only seven of them and they would be found in random-sequence DNA every 104 bases or so. This is insufficient to explain the 106 sequence-selectivity observed for CBF3-CDEIII interaction in vitro. A second possibility is that the geometry or deformability of a sequence is important for CBF3 binding. This might not manifest itself as a simple conservation of sequence. A final possibility is that there are multiple degenerate elements within CDEIII that can substitute for each other (p110-binding sites for example). In this case, it might be quite difficult to detect a consensus sequence. It should be possible however, to develop a mutagenic strategy to test each of these possibilities.
Implications for Complex Kinetochores
A fundamental and unanswered question in centromere biology is the relationship between the simple centromeres found in budding yeast and the complex centromeres found in fission yeast and animal cells. S. cerevisiae centromeres are 125 bp long, whereas S. pombe centromeres are up to 100 kb in length and animal cell centromeres span many megabases. It is widely assumed that simple centromeres contain conventional sequence-specific DNA-binding proteins, whereas complex centromeres involve large regions of chromatin that have few, if any, sequence-specific contacts within them. Consistent with this view, existing models for the S. cerevisiae kinetochore propose a simple DNA–protein connection in which the Zn cluster domains of p64 are primarily responsible for attaching CBF3 to the conserved bases of CDEIII (Pluta et al., 1995). The data presented in this paper suggests that there may be important similarities in the modes of DNA binding between yeast CBF3 and the DNA-binding components of complex kinetochores. Outside of the 25-bp CDEIII core, it is not possible to discern any conservation of DNA sequence in the 80 bp that appear to comprise the functional CBF3 binding site. Similarly, it has been difficult to detect any highly conserved motifs in the sequences of the complex centromeres found in S. pombe (Fishel et al., 1988; Clarke, 1990; Takahashi et al., 1992).
Future Prospects
Our model poses a number of interesting questions about the CBF3–DNA complex. Does CDEIII contain only a single p64 half-site, and would p64 therefore bind on its own to a DNA sequence that contains two appropriately spaced half sites? If so, what does this imply about the assembly of CBF3–DNA complexes? What is the basis of sequence-specific binding by CBF3, and why does an interaction that must be highly specific appear to involve so few conserved bases? Finally, is centromeric DNA bent, and does this bring elements at CDEI and CDEII into contact with CDEIII-bound CBF3?
We anticipate that yeast kinetochores will be embedded in a specialized region of chromatin (Bloom and Carbon, 1982). It is therefore intriguing that the core complex of CBF3 seems to form a nucleating structure on which a sequence-nonspecific p110 multimer can assemble. Perhaps this p110 multimer can be much larger in cells than the complex we have observed in vitro. One speculative idea is that p110 polymers can direct the formation of a centromeric chromatin domain much as Sir3 polymers lead to the assembly of a silent region of chromatin (Hecht et al., 1996). To address these issues, the association of CBF3 with other centromere elements must be studied, and the structure of the CBF3–DNA complex in the context of an intact kinetochore in a living cell needs to be examined.
Acknowledgments
We would like to thank Tony Hyman, Steve Bell, Tania Baker, and Dan Lee for helpful advice and gifts of reagents, and Tania Baker, Steve Bell, Bob Sauer, Caroline Shamu, and members of the Sorger lab for help with this manuscript.
Abbreviations used in this paper
- BrdC
bromodeoxycytidine
- BrdU
bromodeoxyuridine
Footnotes
The work in this paper was supported by the National Institutes of Health Grant GM51464, NATO Grant CRG960763 and the Lucille P. Markey Charitable Trust. K.B. Kaplan is supported by the Cancer Research Fund of the Damon Runyon Walter Winchell Foundation Fellowship, DRG-1321.
Address all correspondence to Peter K. Sorger, Department of Biology, 68-371, MIT, 77 Massachusetts Avenue, Cambridge, MA 02139. Tel.: (617) 252-1648. Fax: (617) 253-8550. E-mail: psorger@mit.edu
References
- Baker RE, Masison DC. Isolation of the gene encoding the Saccharomyces cerevisiaecentromere binding protein CP1. Mol Cell Biol. 1990;10:2458–2467. doi: 10.1128/mcb.10.6.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom K, Carbon J. Yeast centromere DNA is in a unique and highly ordered structure in chromosomes and small circular minichromosomes. Cell. 1982;29:305–317. doi: 10.1016/0092-8674(82)90147-7. [DOI] [PubMed] [Google Scholar]
- Bohm S, Frishman D, Mewes H. Variations of the CH2zinc finger motif in the yeast genome and classification of the yeast zinc finger proteins. Nucleic Acids Res. 1997;25:2464–2469. doi: 10.1093/nar/25.12.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai M, Davis RW. Yeast centromere binding protein CBF1, of the helix-loop-helix family, is required for chromosome stability and methionine prototrophy. Cell. 1990;61:437–446. doi: 10.1016/0092-8674(90)90525-j. [DOI] [PubMed] [Google Scholar]
- Clarke L. Centromeres of budding and fission yeast. TIG (Trends Genet) 1990;6:150–154. doi: 10.1016/0168-9525(90)90149-z. [DOI] [PubMed] [Google Scholar]
- Clarke L, Carbon J. Isolation of a yeast centromere and construction of functional small circular chromosomes. Nature. 1980;287:504–509. doi: 10.1038/287504a0. [DOI] [PubMed] [Google Scholar]
- Connelly C, Hieter P. Budding yeast SKP1 encodes an evolutionarily conserved kinetochore protein required for cell cycle progression. Cell. 1996;86:275–285. doi: 10.1016/S0092-8674(00)80099-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottarel G, Shero JH, Hieter P, Hegemann JH. A 125-bp CEN6 DNA fragment is sufficient for complete meiotic and mitotic centromere functions in Saccharomyces cerevisiae. . Mol Cell Biol. 1989;9:3342–3349. doi: 10.1128/mcb.9.8.3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries E, Bloemers SM, van der Vliet PC. Incorporation of 5-bromodeoxycytidine in the adenovirus 2 replication origin interferes with nuclear factor 1 binding. Nucleic Acids Res. 1987;15:7223–7234. doi: 10.1093/nar/15.18.7223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doheny KF, Sorger PK, Hyman AA, Tugendreich S, Spencer F, Hieter P. Identification of essential components of the S. cerevisiaekinetochore. Cell. 1993;73:761–774. doi: 10.1016/0092-8674(93)90255-O. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishel B, Amstutz H, Baum M, Carbon J, Clarke L. Structural organization and functional analysis of centromeric DNA in the fission yeast Schizosaccharomyces pombe. . Mol Cell Biol. 1988;8:754–763. doi: 10.1128/mcb.8.2.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald-Hayes M, Clarke L, Carbon J. Nucleotide sequence comparisons and functional analysis of yeast centromere DNAs. Cell. 1982;29:235–244. doi: 10.1016/0092-8674(82)90108-8. [DOI] [PubMed] [Google Scholar]
- Gaudet A, Fitzgerald-Hayes M. Alterations in the Adenine-plus thymidine-rich region of CEN3 affect centromere function in Saccharomyces cerevisiae. . Mol Cell Biol. 1987;7:68–75. doi: 10.1128/mcb.7.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh P-Y, Kilmartin JV. NDC10: a gene involved in chromosome segregation in Saccharomyces cerevisiae. . J Cell Biol. 1993;121:503–512. doi: 10.1083/jcb.121.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht A, Strahl-Bolsinger S, Grunstein M. Spreading of transcriptional repressor SIR3 from telomeric heterochromatin. Nature. 1996;383:92–96. doi: 10.1038/383092a0. [DOI] [PubMed] [Google Scholar]
- Hegemann JH, Fleig UN. The centromere of budding yeast. Bioessays. 1993;15:451–460. doi: 10.1002/bies.950150704. [DOI] [PubMed] [Google Scholar]
- Hegemann JH, Shero JH, Cottarel G, Philippsen P, Hieter P. Mutational analysis of centromere DNA from chromosome VI of Saccharomyces cerevisiae. . Mol Cell Biol. 1988;8:2523–2535. doi: 10.1128/mcb.8.6.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA, Sorger PK. Structure and function of kinetochores in budding yeast. Annu Rev Cell Biol. 1995;11:471–495. doi: 10.1146/annurev.cb.11.110195.002351. [DOI] [PubMed] [Google Scholar]
- Jehn B, Niedenthal R, Hegemann JH. In vivo analysis of the Saccharomyces cerevisiaecentromere CDEIII sequence: requirements for mitotic chromosome segregation. Mol Cell Biol. 1991;11:5212–5221. doi: 10.1128/mcb.11.10.5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, W., and J. Carbon. 1993. Molecular analysis of the budding yeast centromere/kinetochore. Cold Spring Harbor Symp. Quant. Biol. LVIII:669–676. [DOI] [PubMed]
- Jiang W, Lechner J, Carbon J. Isolation and characterization of a gene (CBF2) specifying a protein component of the budding yeast centromere. J Cell Biol. 1993;121:513–519. doi: 10.1083/jcb.121.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan KB, Hyman AA, Sorger PK. Regulating the yeast kinetochore by ubiquitin-dependent degradation and Skp1p mediated phosphorylation. Cell. 1997;91:491–500. doi: 10.1016/s0092-8674(00)80435-3. [DOI] [PubMed] [Google Scholar]
- Lechner J. A zinc finger protein, essential for chromosome segregation, constitutes a putative DNA binding subunit of the Saccharomyces cerevisiaekinetochore complex Cbf3. EMBO (Eur Mol Biol Organ) J. 1994;13:5203–5211. doi: 10.1002/j.1460-2075.1994.tb06851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner J, Carbon J. A 240kD multisubunit protein complex, CBF3, is a major component of the budding yeast centromere. Cell. 1991;64:717–726. doi: 10.1016/0092-8674(91)90501-o. [DOI] [PubMed] [Google Scholar]
- Marmorstein R, Carey M, Ptashne M, Harrison SC. DNA recognition by GAL4: structure of a protein-DNA complex. Nature. 1992;356:408–414. doi: 10.1038/356408a0. [DOI] [PubMed] [Google Scholar]
- McGrew J, Diehl B, Fitzgerald-Hayes M. Single base-pair mutations in centromere element III cause aberrant chromosome segregation in Saccharomyces cerevisiae. . Mol Cell Biol. 1986;6:530–538. doi: 10.1128/mcb.6.2.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MR, Fowlkes DM, Fitzgerald-Hayes M. Analysis of centromere function in Saccharomyces cerevisiaeusing synthetic centromere mutants. Chromosoma. 1991;101:189–197. doi: 10.1007/BF00355368. [DOI] [PubMed] [Google Scholar]
- Murray AW. Creative blocks: cell cycle checkpoints and feedback controls. Nature. 1992;359:599–604. doi: 10.1038/359599a0. [DOI] [PubMed] [Google Scholar]
- Ng R, Carbon J. Mutational and in vitro protein-binding studies on centromere DNA from Saccharomyces cerevisiae. . Mol Cell Biol. 1987;7:4522–4534. doi: 10.1128/mcb.7.12.4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickoloff JA, Chen EY, Heffron F. A 24-base-pair DNA sequence from the MATlocus stimulates intergenic recombination in yeast. Proc Natl Acad Sci USA. 1986;83:7831–7835. doi: 10.1073/pnas.83.20.7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluta AF, Mackay AM, Ainsztein AM, Goldberg IG, Earnshaw WC. The centromere: hub of chromosomal activities. Science. 1995;270:1591–1594. doi: 10.1126/science.270.5242.1591. [DOI] [PubMed] [Google Scholar]
- Reece RJ, Ptashne M. Determinants of binding-site specificity among yeast C6zinc cluster proteins. Science. 1993;261:909–911. doi: 10.1126/science.8346441. [DOI] [PubMed] [Google Scholar]
- Sorger PK, Severin FF, Hyman AA. Factors required for the binding of reassembled yeast kinetochores to microtubules in vitro. J Cell Biol. 1994;127:995–1008. doi: 10.1083/jcb.127.4.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorger PK, Doheny KF, Hieter P, Kopski KM, Huffaker TC, Hyman AA. Two genes required for the binding of an essential S. cerevisiaekinetochore complex to DNA. Proc Natl Acad Sci USA. 1995;92:12026–12030. doi: 10.1073/pnas.92.26.12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmann O, Lechner J. The Saccharomyces cerevisiaekinetochore contains a cyclin-CDK complexing homologue, as identified by in vitro reconstitution. EMBO (Eur Mol Biol Organ) J. 1996;15:3611–3620. [PMC free article] [PubMed] [Google Scholar]
- Strunnikov AV, Kingsbury J, Koshland D. CEP3 encodes a centromere protein of Saccharomyces cerevisiae. . J Cell Biol. 1995;128:749–760. doi: 10.1083/jcb.128.5.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Murakami S, Chikashige Y, Funabiki H, Niwa O, Yanagida M. A low copy number central sequence with strict symmetry and unusual chromatin structure in fission yeast centromere. Mol Biol Cell. 1992;1992:819–835. doi: 10.1091/mbc.3.7.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmen A, Hegemann JH. The chromatin of the Saccharomyces cerevisiaecentromere shows cell-type specific changes. Chromosoma. 1996;104:489–503. doi: 10.1007/BF00352113. [DOI] [PubMed] [Google Scholar]
- Zhang L, Guarente L. The yeast activator HAP1—a GAL4 family member—binds DNA in a directly repeated orientation. Genes Dev. 1994;8:2110–2119. doi: 10.1101/gad.8.17.2110. [DOI] [PubMed] [Google Scholar]