

Abstract
Ligation of major histocompatability complex class I (MHC-I) molecules expressed on T cells leads to both growth arrest and apoptosis. The aim of the current study was to investigate the intracellular signal pathways that mediate these effects.
MHC-I ligation of human Jurkat T cells induced a morphologically distinct form of apoptosis within 6 h. A specific caspase inhibitor, which inhibited Fas-induced apoptosis, did not affect apoptosis induced by MHC-I ligation. Furthermore, MHC-I–induced apoptosis did not involve cleavage and activation of the poly(ADP- ribose) polymerase (PARP) endonuclease or degradation of genomic DNA into the typical fragmentation ladder, both prominent events of Fas-induced apoptosis. These results suggest that MHC-I ligation of Jurkat T cells induce apoptosis through a signal pathway distinct from the Fas molecule.
In our search for other signal pathways leading to apoptosis, we found that the regulatory 85-kD subunit of the phosphoinositide-3 kinase (PI-3) kinase was tyrosine phosphorylated after ligation of MHC-I and the PI-3 kinase inhibitor wortmannin selectively blocked MHC-I–, but not Fas-induced, apoptosis. As the c-Jun NH2-terminal kinase (JNK) can be activated by PI-3 kinase activity, and has been shown to be involved in apoptosis of lymphocytes, we examined JNK activation after MHC-I ligation. Strong JNK activity was observed after MHC-I ligation and the activity was completely blocked by wortmannin. Inhibition of JNK activity, by transfecting cells with a dominant-negative JNKK– MKK4 construct, led to a strong reduction of apoptosis after MHC-I ligation. These results suggest a critical engagement of PI-3 kinase–induced JNK activity in apoptosis induced by MHC-I ligation.
Apoptosis is an active form of cell death associated with certain characteristic morphological changes of the cell. These include cell shrinkage, condensation of chromatin, and usually, but not always, fragmentation of genomic DNA into specific oligonucleosomal fragments, also referred to as apoptotic DNA ladder (21). In addition, a morphologically distinct form of apoptosis has been described in germinal centers, thymocyte suspensions, and certain tumors with characteristic features of type B dark cells (7, 34). The condensed chromatin in these cells is not smoothly redistributed into the characteristic eye seen in the nucleus of classical apoptosis; the cytoplasm is darkened and the mitochondria and endoplasmatic reticulum tend to be swollen (7, 34).
The mammalian interleukin-1β–convertase enzyme (ICE)1 protease family (caspases) are known to be critically involved in Fas- and tumor necrosis factor α–induced apoptosis (12). All caspases share two features: (a) they are synthesized as proenzymes and they are activated by cleavage at specific aspartate residues, and (b) both have the same consensus sequence as their own protease activity (14). Thus, the caspases are presumed to be regulated within a hierarchy of auto- and trans-cleavage. In Fas- mediated apoptosis, a sequential activation of caspase 1 (ICE) and caspase 3 (CPP32) has been demonstrated (13). Recent evidence suggests that one subfamily of caspases (inhibitable by YVAD pseudo-substrate) works proximally in Fas-induced apoptosis, leading to release of cytochrome C and other factors from the mitochondria. The initially activated caspases, together with the released factors from the mitochondria, activate the effector caspases (inhibitable by DEVD pseudo-substrate) by a mechanism that is not well defined (22). One of the targets of the effector caspases is the nuclear enzyme poly(ADP-ribose) polymerase (PARP) involved in DNA damage sensing. PARP activity is thought to be critical for apoptosis induced through caspase 3 (5, 43). The PARP enzyme is a 116-kD protein that is cleaved into a 85-kD fragment upon activation (43).
Aurintricarboxylic acid (ATA), an inhibitor of Ca2+- dependent endonuclease activity (23, 29), has been shown to inhibit apoptosis that occurs without DNA fragmentation (25, 32). The mechanism by which ATA inhibits apoptosis is not fully understood. However, inhibition of endonuclease activity may not be the only function of ATA; rather, inhibition of topoisomerase II that induces chromatin condensation during apoptosis seems to be important (6). ATA has also been shown to inhibit the Ca2+-activated enzyme calpain, which may be involved in apoptosis (33).
A new member of the mitogen-activated protein kinase (MAPK) superfamily designated c-Jun NH2-terminal kinase (JNK), has recently been identified (16). A signal pathway functionally independent from extracellular signal-regulated kinase (ERK), which involves JNKK–MKK4, activates JNK (11, 49, 52). JNK is activated by dual phosphorylation of a Thr-Pro-Tyr motif during apoptosis induced by UV light, heat shock, and ligation of the Fas antigen (8, 17, 45, 50). Costimulation of T cells with T cell antigen receptor complex (TCR–CD3) and CD28 ligation, or CD40 ligation of B cells also results in activation of JNK (36, 41). Thus, JNK activity is involved in processes leading to both cell death and differentiation/activation. It has been demonstrated that apoptosis can be regulated through a balance between members of the MAPK superfamily; e.g., high JNK and low ERK activities may lead to apoptosis, whereas high JNK and ERK activities prevent apoptosis (17, 51).
Stimulation of T cells through the major histocompatability complex class I (MHC-I) molecule initiates a cascade of biochemical changes that can lead to either activation and growth or cell cycle arrest and apoptosis (1, 4, 31, 40, 44). One of the critical events that occurs in both cases is the activation of tyrosine kinases, resulting in tyrosine phosphorylation of a variety of proteins including phospholipase C-γ1 (PLC-γ1) and ZAP70 (38, 39). We have recently shown that tyrosine kinase activity appears to be critical for growth inhibition and apoptosis induced by ligation of MHC-I molecules (4, 38). The functional outcome of MHC-I ligation is tightly linked to regulation of peripheral T cell activity and tolerance induction (37, 42).
In the present study we have investigated the intracellular signal pathway leading to apoptosis after MHC-I ligation of T cells, in an attempt to find a cause-and-effect relationship between the biochemical and functional consequences of MHC-I ligation. We present evidence that MHC-I induces apoptosis through a distinct pathway involving phosphoinositide-3 (PI-3) kinase–induced JNK activity.
Materials and Methods
Antibodies (Abs) and Reagents
Purified anti–human β2 microglobulin (anti–β2m) Ab from rabbit serum (A072), purified Ig from rabbit serum (X903), and UCHT-1 (anti–CD3, mAb, IgG1; M835) were from Dako Corp. (Roskilde, Denmark). Anti– human Fas, mAb, IgM (05-201) was from Upstate Biotechnology Inc., (Lake Placid, NY). Anti–human PARP, mAb, IgG1 65191A was from Pharmingen (San Diego, CA). Anti–PI-3 kinase Ab from rabbit serum (06-195) was from Upstate Biotechnology Inc. Anti–PI-3 kinase Ab from rabbit serum (P13030) was from Transduction Laboratories (Lexington, KY). Anti–JNK1, mAb, IgG1 (15701A), which only recognize the activated form of JNK1, was from Pharmingen. Anti–active ERK Ab from rabbit serum (V6671) was from Promega Corp. (Madison, WI). Peroxidase-conjugated anti–mouse Ig from rabbit serum (P260) and peroxidase-conjugated anti–rabbit Ig from swine serum (Z196) were from Dako Corp. Anti-phosphotyrosine, mAb, IgG2b (05-321) was from Upstate Biotechnology Inc. Antibodies used for cell stimulation were dialyzed against PBS before use. Biotin-conjugated antibody was prepared by reacting the antibody with biotinsuccinimide (B-2643; Sigma Chemical Co., St. Louis, MO), as described in reference 28. Peroxidase-conjugated protein A (P40045) was from Transduction Laboratories. Avidin (A9275; Sigma Chemical Co.), was used to cross-link biotin-conjugated antibodies. Natrium-orthovanadate (Na3VO4; S6508) was from Sigma Chemical Co. Protein A–Sepharose CL-4B was from Pharmacia (Uppsala, Sweden). Ripa buffer (10 Mm Tris-HCl buffer, pH 7.5, 1% NP-40, 0.25% deoxycholate wt/vol, 2 mM EDTA, 10 mM orthovanadate). Protease inhibitor cocktail (2697498) was from Boehringer Mannheim (Mannheim, Germany). Ac-Y-V-A-D-chloromethylketone ICE inhibitor (N-1330) was from Bachem Bioscience (Heidelberg, Germany). Proteinase K (P2308) and ribonuclease A (R5503) were from Sigma Chemical Co. Wortmannin (ST-415) was from Biomol (Hørsholm, Denmark). PD98059 (513000) was from Calbiochem-Novabiochem (La Jolla, CA).
Cells
Jurkat cells J76.25 were provided by C. Geisler (University of Copenhagen, Copenhagen, Denmark). Jurkat cells JE6-1 were obtained from the American Type Culture Collection (Rockville, MD). Cells were grown in RPMI 1640 with 5% FCS, fresh l-glutamine, and antibiotics. All cells continuously tested mycoplasma free.
Cell Stimulation
Cells were preincubated with saturating amounts of biotinylated anti–β2m Ab or biotinylated control rabbit Ig (1 μl/106 cells/ml) for 10 min at room temperature and then cross-linked with avidin (20 μg/106 cells/ml) or reacted with UCHT-1 Ab (1 μl/106 cells/ml) or anti–Fas Ab (1 μl/106 cells/ml) at 37°C for various times.
Apoptosis Analysis
106 cells were stimulated as described above. After 30 min of stimulation at 37°C, the cells were resuspended in RPMI 1640 supplemented with 10% heat-inactivated FCS (106 cells/ml), and then cultured for 6 h at 37°C. At the end of the culture period the cells were pelleted, washed once in 2 ml 0.03% saponin (S7900; Sigma Chemical Co.) in PBS, and reacted with 1 ml 0.4 μg/ml 7-aminoactinomycin d (7-AAD) (A9400; Sigma Chemical Co.) in 0.03% saponin for 25 min at room temperature in the dark. The samples were analyzed immediately by flow cytometry in a FACScan® (Becton and Dickinson Co., Mountain View, CA), using a logarithmic fluorescence scale.
Electron Microscopy
5 × 106 cells were pelleted at 1,000 g in FCS. Cell pellets were fixed for 18 h in 2% glutaraldehyde–PBS and postfixed in 1% osmium tetraoxid–PBS, pH 7.4. Samples were dehydrated in ethanol and propylene oxide and then embedded in epon. Ultrathin sections were examined in an electron microscope (model JEM 100CX; JEOL USA Inc., Peabody, MA) at 4,800×.
Wortmannin Treatment
Cells were incubated with 500 ηM wortmannin in PBS or RPMI 1640 with 5% FCS, fresh l-glutamine, and antibiotics for 1–18 h at 37°C before stimulation. Cells were subjected to Western blotting or apoptosis analysis as described elsewhere.
DNA Fragmentation Assay
1.5 × 106 cells were stimulated as described above. Cells were washed once in PBS and the cell pellet was resuspended in 0.5 ml lysis buffer (10 mM EDTA, 50 mM Tris, pH 8, 0.5% sarcosyl) with 10 μl proteinase K (20 mg/ml) and 10 μl RNase A (10 mg/ml). The lysate was incubated for 1 h at 50°C and 12 h at 37°C. DNA was extracted with phenol/chloroform/isoamyl alcohol (25:24:1), precipitated with 2-propanol, and washed with 70% ethanol according to standard procedures. The DNA was subsequently electrophoresed in a 2% agarose gel and stained with ethidium bromide.
Western Blot
4 × 106 cells were stimulated as described above. The cells were pelleted and lysed in 100 μl hot SDS sample buffer containing 2% 2-ME and 10 mM Na3VO4. The lysate was boiled for 5 min, centrifuged at 178,000 g for 20 min in an airfuge (Beckman and Dickinson Co.), and then 70 μl of the supernatant was electrophoresed on SDS–polyacrylamide gels and blotted onto a Hybond-enhanced chemiluminescence (ECL) nitrocellulose membrane (RPN 2020D; Amersham Corp., Arlington Heights, IL). The immunoblot was incubated with antibody (normally 1:1,000 diluted in 3% milkpowder/PBS) for 2 h, followed by incubation with 1:1,000 peroxidase-conjugated rabbit anti–mouse Ig, swine anti–rabbit, or protein A for 1 h, and were then washed and developed by ECL (RPN 2106; Amersham Corp.) using the manufacturer's instruction.
Immunoprecipitation
3 × 107 cells were stimulated as described above and then pelleted and lysed in 1 ml Ripa buffer and precleared several times with protein A–Sepharose in a 50% wt/vol slurry. Proteins were immunoprecipitated with saturating amounts of Ab and 30 μl protein A–Sepharose in a 50% wt/vol slurry. Immunoprecipitated proteins were washed in Ripa buffer, subjected to SDS– polyacrylamide electrophoresis, and immunoblotted with antibodies as described above.
Cell Transfection
3 × 106/ml cells were transiently transfected with 10 μg of pcDNA3.1 vector alone or containing a dominant-negative JNKK–MKK4 insert (Ala substituted at Ser-257 and Thr-261), provided by R. Davis (University of Massachusetts Medical Center, Howard Hughes Medical Institute [HHMI], Worcester, MA; 11, 49), using 10 μl LipofectAmine (Life Technologies, Inc., Gaithersburg, MD) according to the manufacturer's instructions. Cells were grown for 48 h and subsequently assayed for apoptosis and JNKK–MKK4 expression.
Results
MHC-I Ligation of Jurkat T Cells Induces Apoptosis
Ligation of MHC-I molecules expressed on Jurkat T cells induces cell death. Fig. 1 shows the ultrastructural changes in Jurkat cells exposed to 6 h of MHC-I ligation and, for comparison, anti–Fas antibody. MHC-I–induced apoptosis shows striking similarities with a morphologically distinct form of apoptosis with characteristic features of type B dark cells (34). This involves ultrastructural changes in both the cytoplasm and nucleus, whereas Fas-induced ultrastructural changes are primarily confined to the nucleus. The early signs of MHC-I–induced apoptosis shown in Fig. 1 (B and C) include darkening of the cytoplasm, aggregation of ribosomes, and condensation and shrinkage of the nucleus with dilatation of the perinuclear cisterna. In severely damaged cells (Fig. 1, D and E), the nucleus disintegrates and the condensed chromatin is surrounded by a leaky nuclear envelope. Occasionally, typical apoptotic bodies are observed Fig. 1 (F). Fig. 1, (G–I) shows a typical Fas-induced apoptosis that includes apoptotic bodies composed of homogenous broken chromatin surrounded by an intact nuclear envelope in a relatively well-preserved cytoplasm.
Figure 1.
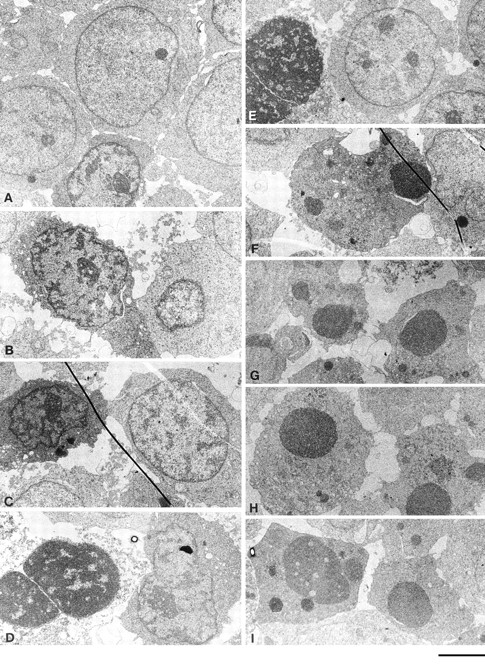
(A) Electron micrographs of control Jurkat cells 6 h after exposure to biotinylated control antibody plus avidin. Notice the normal ultrastructure of the cell. (B–F) Varying stages of decay in Jurkat cells 6 h after exposure to biotinylated anti–MHC-I antibody plus avidin. Notice the simultaneous disintegration of the cytoplasm and the nucleus. (B and C) Darkening of cytoplasm, shrinkage of the nucleus, and dispersed DNA condensation of heterochromatin. (D and E) Disintegration of the cytoplasm and homogenization of the chromatin surrounded by nuclear envelope. (F) Apoptotic-like bodies of condensed homogenous chromatin in a relatively well-preserved cytoplasm. (G–I) Typical apoptotic bodies in Jurkat cells after a 6-h exposure to anti-Fas antibody. The nuclear fragments consist of homogenized chromatin encircled by an intact nuclear envelope surrounded by a relatively well-preserved cytoplasm. Bar, 2 μm.
MHC-I–induced Apoptosis Is Not Blocked by Inhibitors of ICE Enzymes
Using the fluorescent dye 7-AAD that binds to DNA, it is possible to measure apoptosis by flow cytometry (46). A selective decrease in the DNA stainability is seen in apoptotic cells, because of DNA condensation and/or cleavage (10). Fig. 2 (a) shows examples of 7-AAD staining of Jurkat cells after control, MHC-I, and Fas ligation. The apoptosis data is presented as percent subG1 staining, where background staining is subtracted. Fig. 2 (b) shows that severe apoptosis is observed after 6 h of ligation of either MHC-I or Fas. Ligation of the T cell antigen receptor (TCR– CD3) complex induced only minor apoptosis within 6 h. Interestingly, simultaneous ligation of the MHC-I and TCR– CD3 complex inhibited apoptosis (see Discussion). Supernatants from cells ligated with MHC-I antibody for 6 h did not induce apoptosis in untreated cells (supernatants were purified with protein A to remove stimulating antibodies). Thus, the apoptosis signal is not induced by secreted molecules. No apoptosis was observed with avidin cross-linking of biotinylated control antibody, or without secondary avidin cross-linking of biotinylated anti–MHC-I antibody. Another novel method of measuring apoptosis through fluorescein-labeled annexin V staining of phosphatidylserine expression on the outer cell membrane of early apoptotic cells (46) detected apoptosis in Jurkat cells after 6 h of MHC-I or Fas ligation, respectively (data not shown). Fig. 2 (c) shows that a specific YVAD pseudo-substrate ICE inhibitor could block apoptosis induced by Fas ligation. In contrast, the ICE inhibitor could not inhibit apoptosis induced after 6 h of MHC-I ligation, suggesting that the MHC-I molecule induced apoptosis through a signal pathway distinct from the signal mediated by the Fas molecule.
Figure 2.
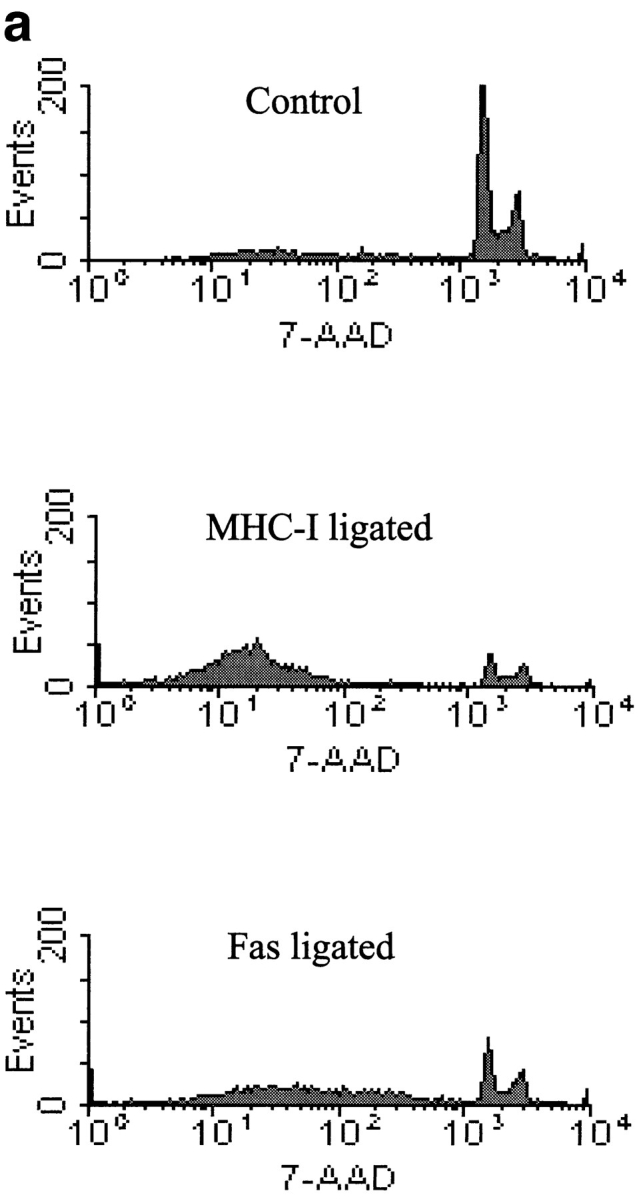


Measurement of apoptosis (cells < 2n DNA). 106 cells were stimulated for 6 h and apoptosis was measured using 7-AAD as described in Materials and Methods. (a) 7-AAD flow cytometric profile of Jurkat cells after control, MHC-I and Fas ligation. (b) Apoptosis measurement after different stimuli. “Post” supernatant (dark grey bar) is supernatant from anti–MHC-I antibody and avidin stimulated cells; the supernatants were purified with protein A before stimulation. (c) Cells were incubated with or without 100-μM peptide ICE inhibitor for 1 h before stimulation. The data show results of at least three independent experiments.
MHC-I–induced Apoptosis Does Not Involve Activation of the PARP Enzyme and DNA Ladder Formation
Next, we examined the activation of the PARP endonuclease which is activated by caspase 3 (5). Despite the clear evidence for apoptosis after 6 h of MHC-I ligation, Western blot analysis of whole cell extracts showed that the PARP enzyme was not cleaved into the 85-kD active form (Fig. 3 a, lane 4). In contrast and as expected, PARP was cleaved into the active form after Fas ligation (Fig. 3 a, lane 5).
Figure 3.



(a) Anti-PARP Western blot of whole Jurkat cell lysate. 4 × 106 cells were stimulated for 6 h as described in Materials and Methods. (b) DNA ladder analysis. 106 cells were stimulated for 6 h and then DNA was extracted and electrophoresed on a 2% agarose gel as described in Materials and Methods. (c) ATA inhibition of apoptosis. 106 cells were incubated with or without 300 μM ATA for 1 h before stimulation. Cells were stimulated for 6 h and apoptosis was measured using 7-AAD as described in Materials and Methods. All data show results of at least three independent experiments.
As activation of the PARP endonuclease could be involved in the DNA ladder formation of apoptotic cells (53), we examined DNA fragmentation after MHC-I ligation. Fig. 3 (b) shows that 6 h of MHC-I ligation did not induce a detectable DNA ladder, in contrast to Fas ligation (compare Fig. 3 b, lanes 4 and 5).
The endonuclease inhibitor ATA has been shown to block apoptosis that occurs without formation of DNA ladders (32). Similarly, preincubation of Jurkat cells with 300 μM ATA inhibited apoptosis induced by MHC-I ligation, but not the apoptosis induced after Fas ligation (Fig. 3 c). These results show that the MHC-I molecule activates an apoptotic signal pathway distinctly different from the Fas molecule.
Apoptosis Induced by MHC-I Ligation Involves PI-3 Kinase Activity
PI-3 kinase activity has been shown to be involved in growth arrest and apoptosis especially in lymphocytes (3, 19). Therefore, we investigated the involvement of PI-3 kinase activity in MHC-I–induced apoptosis. Through treatment of cells with the specific PI-3 kinase inhibitor wortmannin, MHC-I–induced apoptosis, in contrast to Fas, was significantly inhibited by preincubating Jurkat cells with 500 ηM wortmannin before stimulation (Fig. 4 a).
Figure 4.


(a) Wortmannin inhibition of apoptosis. 106 cells were incubated with or without 500 ηM wortmannin for 2 h before stimulation. Cells were stimulated for 6 h, with 500 ηM wortmannin present, and apoptosis was measured using 7-AAD as described in Materials and Methods. (b) Immunoprecipitates of the P85 subunit of the PI-3 kinase obtained from lysates of Jurkat cells. 3 × 107 cells were stimulated for the indicated time, lysed, and subjected to immunoprecipitation as described in Materials and Methods (top). Blots were probed with anti-phosphotyrosine antibody, stripped, and reprobed with anti–PI-3 kinase antibody (bottom). All data show results of at least three independent experiments.
As activation of the PI-3 kinase is normally (9, 15, 19), but not always (48), associated with tyrosine phosphorylation of the 85-kD regulatory PI-3 kinase subunit, we investigated whether MHC-I ligation mediated tyrosine phosphorylation of this subunit. Fig. 4 (b) shows a phosphotyrosine Western blot of the 85-kD PI-3 kinase subunit immunopurified from cell lysates. A marked increase in the tyrosine phosphorylation status of the 85-kD subunit was observed within 2–10 min of MHC-I ligation (Fig. 4 b, lanes 4–6). Fas ligation did not induce phosphorylation of the PI-3 kinase (Fig. 4 b, lane 7). Collectively, these data indicate that MHC-I–induced PI-3 kinase activity is involved in apoptosis.
MHC-I–induced PI-3 Kinase Activity Activates the JNK1 Enzyme
To look for downstream effector enzymes of the PI-3 kinase, we investigated the activation of the newly described JNK enzyme, which is involved in a number of stimuli leading to apoptosis (45, 51). Furthermore, JNK can be activated by the PI-3 kinase (3, 19). Using a newly developed antibody that only recognizes the activated form of JNK1, we observed that MHC-I ligation within 5 min resulted in activation of both the 46- and 55-kD isoforms of the JNK1 enzyme (Fig. 5 a). Afterwards, the activity declined but was still detectable 120 min after MHC-I ligation (data not shown). As shown previously, Fas ligation also resulted in activation of the JNK1 enzyme (Fig. 5 a, lane 8; 50). Fig. 5 b shows that MHC-I–induced JNK1 activity was totally inhibited by preincubating the cells with 500 ηM wortmannin. These results show that the MHC-I molecule activates the JNK1 enzyme by a signal pathway involving PI-3 kinase activity.
Figure 5.


(a) Anti–active JNK-1 Western blot of whole Jurkat cell lysate. 5 × 106 cells were stimulated for the indicated time as described in Materials and Methods. The strong band at 56 kD after TCR–CD3 ligation is an artefact from the stimulatory antibody. (b) Wortmannin inhibition of JNK-1: 5 × 106 cells were incubated 1 h with 500 ηM wortmannin (lanes 1–4) or without (lanes 5–7) before stimulation for the indicated time as described in Materials and Methods. All data show results of at least three independent experiments.
MHC-I–induced Apoptosis Is Inhibited by Transfection of a Dominant-Negative JNKK–MKK4 Construct
To investigate the direct involvement of JNK in MHC- I–induced apoptosis, Jurkat cells were transiently transfected with a dominant-negative JNKK–MKK4 construct. JNKK–MKK4 is a kinase upstream from JNK, which is essential for its activation of JNK (11, 49). Fig. 6 shows that transfection of the dominant-negative JNKK–MKK4 construct resulted in an ∼50% reduction in apoptosis induced after MHC-I ligation, suggesting an essential role for JNK activity in MHC-I–induced apoptosis. Besides activating JNK, JNKK–MKK4 is an upstream activator of the p38 MAP kinase (11). Thus, the inhibiting effect of the dominant-negative JNKK–MKK4 construct could also be because of inhibition of p38 activity. However, p38 activity does not seem to be involved in MHC-I–induced apoptosis based on the following experiments: (a) Neither an antibody specifically recognizing activated p38, nor specific immunoprecipitation with anti–p38 antibodies and subsequent detection with an anti-phosphotyrosine antibody, detected activated/phosphorylated p38 after MHC-I ligation (data not shown); and (b) SB203580, a newly developed, highly specific inhibitor of p38 (54), did not affect MHC-I–induced apoptosis at all (data not shown). In conclusion, our results point towards a critical role of JNK activity in MHC-I–induced apoptosis.
Figure 6.

Apoptosis measurement after transfection of dominant-negative JNKK–MKK4 construct. Cells were transfected with pcDNA3.1 vector containing a dominant-negative JNKK– MKK4 construct (Ala substituted at Ser-257 and Thr-261), or with the empty vector as described in Materials and Methods. Subsequent apoptosis was measured as described in Fig. 2.
TCR–CD3-induced ERK Activity Rescues MHC-I–induced Apoptosis
As activation of ERK enzymes belonging to the MAPK family has been shown to inhibit apoptosis induced through JNK activity (17, 51), we examined ERK1 and ERK2 activity in Jurkat cells. Fig. 7 (a) shows a Western blot against the active forms of the 42-kD ERK1 and the 44-kD ERK2 after MHC-I ligation (Fig. 7 a, lane 4), Fas ligation (Fig. 7 a, lane 5), and TCR–CD3 ligation (Fig. 7 a, lane 6). Both MHC-I and Fas ligation induce minor activation of ERK2 and no activation of ERK1, whereas TCR–CD3 ligation of Jurkat cells induced strong activation of both ERK1 and ERK2.
Figure 7.


(a) Anti–active ERK Western blot of whole Jurkat cell lysate. 5 × 106 cells were stimulated for the indicated time as described in Materials and Methods. (b) PD98059 inhibition of the TCR–CD3 rescue signal on MHC-I–induced apoptosis. 106 cells were incubated with or without 100 μM PD98059 for 1 h before stimulation. Cells were stimulated for 6 h and apoptosis was measured using 7-AAD as described in Materials and Methods. All data show results of at least three independent experiments.
Our data in Fig. 2 (a), showing that TCR–CD3 coligation inhibits MHC-I–induced apoptosis supports the idea that ERK activity can inhibit JNK-induced apoptosis. To examine this in greater detail, we used the Mek inhibitor PD98059 that inhibits ERK activity (30, 47) and data not shown. As shown in Fig. 7 b, the Mek inhibitor did not influence MHC-I–induced apoptosis of Jurkat cells, but the TCR–CD3-induced rescue signal on MHC-I–induced apoptosis was inhibited by treating the cells with PD98059 before stimulation. These results strongly suggest that TCR– CD3-induced ERK activity inhibits MHC-I–induced apoptosis.
Discussion
Results in this paper describe the intracellular signal pathway leading to apoptosis after ligation of MHC-I molecules expressed on T cells. We present strong evidence that MHC-I–induced apoptosis uses a signal pathway that involves PI-3 kinase–induced JNK activity.
MHC-I–induced apoptosis is characterized by distinct morphological changes resembling type B dark cells initially observed in thymocyte suspensions, germinal centers, and tumors (7, 34). In particular, dispersed condensed heterochromatin encircled by a leaky nuclear envelope and surrounded by a dilatated perinuclear space in a disintegrated dark cytoplasm is characteristic of MHC-I–induced apoptosis. Although purely speculative, this MHC-I–induced way of chromatin condensation may reflect the lack of DNA ladder formation after MHC-I ligation (see below).
Until a few years ago, DNA fragmentation of the genomic DNA was considered to be a prerequisite, and even essential, for apoptosis. Recent studies have, however, revealed that DNA fragmentation is a consequence rather than a prerequisite for apoptosis (26). Especially in lymphocytes, it has been demonstrated that apoptosis induced in alloreactive T cells (32) or germinal center B cells (27) can occur without DNA fragmentation. Our results show that apoptosis after MHC-I ligation also occurs without the characteristic DNA ladder fragmentation observed in agarose gels. Similarly, we did not observe involvement of ICE enzymes and activation of the PARP endonuclease after MHC-I–induced apoptosis. PARP activation normally correlates with DNA ladder formation (53), and is thus supposed to be involved in DNA fragmentation, although this has not been formally proved.
We observed that ATA, a Ca2+-dependent endonuclease inhibitor, inhibits MHC-I–, but not Fas-induced apoptosis. This is in agreement with our earlier results showing that MHC-I ligation, in contrast to Fas (2), induces an immediate and sustained rise in the intracellular-free Ca2+ concentration (39). It is currently unclear how ATA inhibits apoptosis. However, several studies have demonstrated that ATA not only inhibits endonucleases, but also calpain and topoisomerase II activities (6, 33). The latter enzyme may be involved in certain forms of DNA condensation (6).
The ability of wortmannin to selectively inhibit apoptosis after MHC-I ligation strongly suggests that PI-3 kinase activity is involved in this process. In conjunction with this we observed that the regulatory 85-kD subunit of the PI-3 kinase was tyrosine phosphorylated within 2 min after MHC-I ligation. Phosphorylation of this subunit is often associated with the PI-3 kinase activity (48). The wortmannin pretreatment of Jurkat cells does not completely inhibit apoptosis after MHC-I ligation; thus, it is likely that other signal pathways are also involved in this process. However, wortmannin is known to be very unstable and its PI-3 kinase suppressive effect may weaken during culture (18). Thus, taken together, our data suggest that PI-3 kinase activity is involved in a substantial part of apoptosis induced by MHC-I ligation. Recently, Kitanaka et al. have demonstrated that CD38-mediated growth suppression of immature B cells involves PI-3 kinase activity which could be inhibited by wortmannin (19). In addition, Beckwith et al. have demonstrated that anti–Ig-mediated growth inhibition of B cells requires a wortmannin-sensitive PI-3 kinase activity (3).
The PI-3 kinase can activate several downstream effector molecules (48). One of the most intriguing is the JNK enzyme that phosphorylates the NH2-terminal domain of the c-Jun transcription factor. JNK activity is required for stress-induced apoptosis (17, 45), and is also activated by Fas ligation (21, 50). Furthermore, costimulatory JNK activity is needed for agonistic stimulation of T cells (41). Hence, JNK is hypothesized to be an integral kinase that is involved in both lymphocyte activation and apoptosis, depending on other intracellular stimuli. Recent studies have led to the hypothesis that the balance between ERK and JNK activation is crucial for the fate of the cell upon the activation of stimuli. Xia et al. have demonstrated that high JNK activity leads to apoptosis, whereas high ERK and JNK activity together prevent apoptosis and promote cell survival (51). Concordantly, Johnson et al. have shown that apoptosis by UV-induced JNK activity can be inhibited by ERK activators (17).
Our present observations show that JNK1 is activated after MHC-I ligation. Since wortmannin inhibits JNK1 activity induced by MHC-I ligation, the most likely and straightforward explanation is that the MHC-I molecule activates JNK1 through PI-3 kinase activity. Moreover, since wortmannin can inhibit MHC-I–induced apoptosis, it is tempting to speculate that JNK activity is the molecular basis for this kind of cell death. By using a dominant-negative construct of the kinase upstream from JNK (JNKK–MKK4), we provide direct evidence that JNKK–MKK4 activity is involved in MHC-I–induced apoptosis. The JNKK–MKK4 enzyme is an upstream kinase activating both JNK and p38 (11). Hence, p38 activity could theoretically be responsible for MHC-I–induced apotosis. However, our results do not support this hypothesis, as we do not observe p38 activity after MHC-I ligation. Also, a specific inhibitor of p38 activity was unable to inhibit MHC-I–induced apoptosis.
In conclusion, our results strongly suggest that JNK activity is essential for MHC-I–induced apoptosis. This model is consistent with our observation that TCR–CD3 ligation, which induces strong ERK activity, totally inhibits MHC-I–induced apoptosis. Furthermore, specific blockage of the TCR–CD3-induced ERK activity with the Mek inhibitor PD98059 prevents the effect of TCR–CD3 on MHC- I–induced apoptosis.
Our previously published results have demonstrated a critical role for tyrosine kinase activity in MHC-I–mediated signaling leading to PLC-γ1 phosphorylation and increased intracellular Ca2+ concentration (39). Recently, we have described that the tyrosine kinase inhibitor herbimycin A also inhibits the subsequent growth arrest and induction of apoptosis after MHC-I ligation (38). Based on the data in the present report, a likely model for MHC- I–induced apoptosis could be that the initial tyrosine kinase activity leads to phosphorylation and activation of the PI-3 kinase, which then activates the JNK enzyme.
The MHC-I molecule is expressed on both naive and activated T cells (20), as opposed to the Fas antigen that is only expressed on activated T cells (24). We have observed MHC-I–induced JNK activity and apoptosis in activated as well as naive T cells from purified peripheral blood (data not shown), suggesting a role for MHC-I signaling in regulation of naive T cells. Furthermore, we have recently shown that physiological concentrations of immobilized MHC-I antibody inhibit interleukin 2–induced growth of normal peripheral T cells (4). Sambhara et al. and Röpke et al. have also shown that cytotoxic T lymphocyte (CTL) reactivity can be anergized by antibodies against the MHC-I molecules, in particular against the α3 domain (35, 37). Negative signals via MHC-I molecules were also demonstrated by Takahashi et al., who recently showed that exposure of free antigenic peptide to CTL's almost completely inhibited subsequent CTL activity toward target cells presensitized with that peptide (42). The authors showed that the minimal requirement for such inhibition is simultaneous occupancy of the TCR–CD3 and MHC-I molecule on the same CTL, emphasizing the role of MHC-I signaling in this kind of self-veto mechanism. Together, these results imply that ligation of MHC-I molecules on T cells is generally involved in their regulation. The results in the present paper suggest that MHC-I molecules regulate cell survival through a distinct mechanism involving PI-3 kinase–induced JNK activity.
Acknowledgments
We thank R. Davis (University of Massachusetts Medical Center, HHMI) for providing the dominant-negative JNKK–MKK4 construct, B. Mikkelsen and T. Stokkendahl for excellent technical assistance, and K. Pedersen for providing the electron micrographs.
This work was supported by the Danish Medical Research Council, the Novo Nordic Foundation, Chairman Leo Nielsen og Wife Karen Magrethe Nielsens Grant for Medical Research, M. Kristjan Kjær and Wife Magrethe Kjær, born la Cour-Holmen's grant, Chairman Jacob Madsen and Wife Olga Madsens Grant, the Beckett Foundation, Kong Christian den Tiendes Foundation, Einar Willumsens Grant, the Leo Research Foundation, and the Lundbeck Foundation.
Abbreviations used in this paper
- 7-AAD
7-aminoactinomycin d
- Ab
antibody
- ATA
aurintricarboxylic acid
- CTL
cytotoxic T lymphocyte
- ERK
extracellular signal-regulated kinase
- ICE
interleukin-1β–convertase enzyme
- JNK
c-Jun NH2-terminal kinase
- MAPK
mitogen-activated protein kinase
- MHC-I
major histocompatibility complex class I
- PARP
poly(ADP-ribose) polymerase
- PI-3
phosphoinositide-3 kinase
- TCR–CD3
T cell antigen receptor complex
Footnotes
Address all correspondence to Søren Skov, Laboratory of Experimental Immunology, Department of Medical Anatomy, The Panum Institute, University of Copenhagen, Blegdamsvej 3, Building 18.3, 2200 Copenhagen N, Denmark. Fax: 45.35.32.72.69. E-mail: s.skov@mai.ku.dk
References
- 1.Amirayan N, Furrie E, Deleuil F, Mellor A, Leserman L, Machy P. Influence of MHC class I molecules on T cell proliferation induced by CD3 or Thy-1 stimulation. Immunology. 1995;86:71–78. [PMC free article] [PubMed] [Google Scholar]
- 2.Apasov S, Redegeld F, Sitkovsky M. Cell-mediated cytotoxicity: contact and secreted factors. Curr Opin Immunol. 1993;5:404–410. doi: 10.1016/0952-7915(93)90060-6. [DOI] [PubMed] [Google Scholar]
- 3.Beckwith M, Fenton RG, Katona IM, Longo DL. Phosphatidylinositol-3-kinase activity is required for the anti–ig-mediated growth inhibition of a human B-lymphoma cell line. Blood. 1996;87:202–210. [PubMed] [Google Scholar]
- 4.Bregenholt S, Röpke M, Skov S, Claesson MH. Ligation of MHC class I molecules on peripheral blood T lymphocytes induces new phenotypes and functions. JImmunol. 1996;157:993–999. [PubMed] [Google Scholar]
- 5.Casciola-Rosen L, Nicholson DW, Chong T, Rowan KR, Thornberry NA, Miller DK, Rosen A. Apopain/CPP32 cleaves proteins that are essential for cellular repair: a fundamental principle of apoptotic death. J Exp Med. 1996;183:1957–1964. doi: 10.1084/jem.183.5.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Catchpoole DR, Stewart BW. Inhibition of topoisomerase II by aurintricarboxylic acid: implications for mechanisms of apoptosis. Anticancer Res. 1994;14:853–856. [PubMed] [Google Scholar]
- 7.Claesson MH, Jorgensen O, Olsson L. Comparative light and electron microscopical studies of decaying thymic lymphoid cells. APMIS (Acta Pathol Microbio Scand) 1972;80:821–826. doi: 10.1111/j.1699-0463.1972.tb00353.x. [DOI] [PubMed] [Google Scholar]
- 8.Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind S, Spiegel S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature. 1996;381:800–803. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- 9.Dadi H, Ke S, Roifman CM. Activation of phosphatidylinositol-3 kinase by ligation of the interleukin-7 receptor is dependent on protein tyrosine kinase activity. Blood. 1994;84:1579–1586. [PubMed] [Google Scholar]
- 10.Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P, Traganos F. Features of apoptotic cells measured by flow cytometry. Cytometry. 1992;13:795–808. doi: 10.1002/cyto.990130802. [DOI] [PubMed] [Google Scholar]
- 11.Derijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ, Davis RJ. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- 12.Enari M, Hug H, Nagata S. Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature. 1995;375:78–81. doi: 10.1038/375078a0. [DOI] [PubMed] [Google Scholar]
- 13.Enari M, Talanian RV, Wong WW, Nagata S. Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature. 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- 14.Fraser A, Evan G. A license to kill. Cell. 1996;85:781–784. doi: 10.1016/s0092-8674(00)81005-3. [DOI] [PubMed] [Google Scholar]
- 15.Gold MR, Duronio V, Saxena SP, Schrader JW, Aebersold R. Multiple cytokines activate phosphatidylinositol 3-kinase in hemopoietic cells. Association of the enzyme with various tyrosine-phosphorylated proteins. J Biol Chem. 1994;269:5403–5412. [PubMed] [Google Scholar]
- 16.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 17.Johnson NL, Gardner AM, Diener KM, Lange-Carter CA, Gleavy J, Jarpe MB, Minden A, Karin M, Zon LI, Johnson GL. Signal transduction pathways regulated by mitogen-activated/extracellular response kinase kinase kinase induce cell death. J Biol Chem. 1996;271:3229–3237. doi: 10.1074/jbc.271.6.3229. [DOI] [PubMed] [Google Scholar]
- 18.Kimura K, Hattori S, Kabuyama Y, Shizawa Y, Takayanagi J, Nakamura S, Toki S, Matsuda Y, Onodera K, Fukui Y. Neurite outgrowth of PC12 cells is suppressed by wortmannin, a specific inhibitor of phosphatidylinositol 3-kinase. J Biol Chem. 1994;269:18961–18967. [PubMed] [Google Scholar]
- 19.Kitanaka A, Ito C, Nishigaki H, Campana D. CD38-mediated growth suppression of B cell progenitors requires activation of phosphatidylinositol 3-kinase and involves its association with the protein product of the c-cbl proto-oncogene. Blood. 1996;88:590–598. [PubMed] [Google Scholar]
- 20.Klein, J. 1986. Natural History of the Major Histocompatibility Complex. John Wiley and Sons, New York. 152 pp.
- 21.Latinis KM, Koretzky GA. Fas ligation induces apoptosis and Jun kinase activation independently of CD45 and Lck in human T cells. Blood. 1996;87:871–875. [PubMed] [Google Scholar]
- 22.Martins LM, Earnshaw WC. Apoptosis: alive and kicking in 1997. Trends Cell Biol. 1997;7:111–114. doi: 10.1016/S0962-8924(96)10053-2. [DOI] [PubMed] [Google Scholar]
- 23.McConkey DJ, Chow SC, Orrenius S, Jondal M. NK cell- induced cytotoxicity is dependent on a Ca2+increase in the target. FASEB (Fed Am Soc Exp Biol) J. 1990;4:2661–2664. doi: 10.1096/fasebj.4.9.2347464. [DOI] [PubMed] [Google Scholar]
- 24.Miyawaki T, Uehara T, Nibu R, Tsuji T, Yachie A, Yonehara S, Taniguchi N. Differential expression of apoptosis-related Fas antigen on lymphocyte subpopulations in human peripheral blood. J Immunol. 1992;149:3753–3758. [PubMed] [Google Scholar]
- 25.Montpied P, Weller M, Paul SM. N-methyl-d-aspartate receptor agonists decrease protooncogene bcl-2 mRNA expression in cultured rat cerebellar granule neurons. Biochem Biophys Res Commun. 1993;195:623–629. doi: 10.1006/bbrc.1993.2091. [DOI] [PubMed] [Google Scholar]
- 26.Nagataki S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 27.Nakamura M, Yagi H, Kayaba S, Ishii T, Gotoh T, Ohtsu S, Itoh T. Death of germinal center B cells without DNA fragmentation. Eur J Immunol. 1996;26:1211–1216. doi: 10.1002/eji.1830260604. [DOI] [PubMed] [Google Scholar]
- 28.Odum N, Martin PJ, Schieven GL, Norris NA, Grosmaire LS, Hansen JA, Ledbetter JA. Signal transduction by HLA-DR is mediated by tyrosine kinase(s) and regulated by CD45 in activated T cells. Hum Immunol. 1991;32:85–94. doi: 10.1016/0198-8859(91)90104-h. [DOI] [PubMed] [Google Scholar]
- 29.Okamoto M, Matsumoto M, Ohtsuki T, Taguchi A, Mikoshiba K, Yanagihara T, Kamada T. Internucleosomal DNA cleavage involved in ischemia-induced neuronal death. Biochem Biophys Res Commun. 1993;196:1356–1362. doi: 10.1006/bbrc.1993.2402. [DOI] [PubMed] [Google Scholar]
- 30.Pang L, Sawada T, Decker SJ, Saltiel AR. Inhibition of MAP kinase kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- 31.Pettersen RD, Hestdal K, Lie SO, Gaudernack G. Role of the TCR binding region of the HLA class I alpha 2 domain in regulation of cell adhesion and proliferation. J Immunol. 1996;156:1415–1424. [PubMed] [Google Scholar]
- 32.Pohl T, Pechhold K, Oberg HH, Wilbert OM, Kabelitz D. Antigen-induced death of alloreactive human T-lymphocytes occurs in the absence of low molecular weight DNA fragmentation. Cell Immunol. 1995;166:187–195. doi: 10.1006/cimm.1995.9979. [DOI] [PubMed] [Google Scholar]
- 33.Posner A, Raser KJ, Hajimohammadreza I, Yuen PW, Wang KK. Aurintricarboxylic acid is an inhibitor of mu- and m-calpain. Biochem Mol Biol Int. 1995;36:291–299. [PubMed] [Google Scholar]
- 34.Pulendran B, van Driel R, Nossal GJV. Immunological tolerance in germinal centres. Immunol Today. 1997;18:27–32. doi: 10.1016/s0167-5699(97)80011-4. [DOI] [PubMed] [Google Scholar]
- 35.Ropke M, Ropke C, Claesson MH. T cell activation. VI. Inhibitory and stimulatory effects of anti–major histocompatibility complex class I antibodies in allogeneic mixed lymphocyte culture. Immunol. 1993;79:263–269. [PMC free article] [PubMed] [Google Scholar]
- 36.Sakata N, Patel HR, Terada N, Aruffo A, Johnson GL, Gelfand EW. Selective activation of c-Jun kinase mitogen-activated protein kinase by CD40 on human B cells. J Biol Chem. 1995;270:30823–30828. doi: 10.1074/jbc.270.51.30823. [DOI] [PubMed] [Google Scholar]
- 37.Sambhara SR, Miller RG. Programmed cell death of T cells signaled by the T cell receptor and the alpha 3 domain of class I MHC. Science. 1991;252:1424–1427. doi: 10.1126/science.1828618. [DOI] [PubMed] [Google Scholar]
- 38.Skov S, Odum N, Claesson MH. MHC-class I signaling in T cells leads to tyrosine kinase activity and PLC gamma1 phosphorylation. J Immunol. 1995;154:1167–1176. [PubMed] [Google Scholar]
- 39.Skov S, Bregenholt S, Claesson MH. MHC class I ligation of human T cells activates the ZAP70 and the P56lck tyrosine kinases, leads to an alternative phenotype of the TCR/CD3 zeta-chain, and induces apoptosis. J Immunol. 1997;158:3189–3196. [PubMed] [Google Scholar]
- 40.Smith DM, Bluestone JA, Jeyarajah DR, Newberg MH, Engelhard VH, Thistlethwaite JR, Jr, Woodle ES. Inhibition of T cell activation by a monoclonal antibody reactive against the alpha 3 domain of human MHC class I molecules. J Immunol. 1994;153:1054–1067. [PubMed] [Google Scholar]
- 41.Su B, Jacinto E, Hibi M, Kallunki T, Karin M, Ben-Neriah Y. JNK is involved in signal integration during costimulation of T lymphocytes. Cell. 1994;77:727–736. doi: 10.1016/0092-8674(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 42.Takahashi H, Nakagawa Y, Leggatt GR, Ishida Y, Saito T, Yokomuro K, Berzofsky JA. Inactivation of human immunodeficiency virus (HIV)-1 envelope-specific CD8-positive cytotoxic T lymphocytes by free antigenic peptide: a self-veto mechanism? . J Exp Med. 1996;183:879–889. doi: 10.1084/jem.183.3.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32 beta, a mammalian homologue of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 44.Tscherning T, Claesson MH. Signal transduction via MHC class-I molecules in T cells. Scand J Immunol. 1994;39:117–120. doi: 10.1111/j.1365-3083.1994.tb03349.x. [DOI] [PubMed] [Google Scholar]
- 45.Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, et al. Requirement for ceramide-initiated SAPK/JNK signaling in stress-induced apoptosis. Nature. 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 46.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labeled Annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 47.Virdee K, Tolkovsky AM. Inhibition of p42 and p44 mitogen-activated protein kinase activity by PD98059 does not suppress nerve growth factor-induced survival of sympathetic neurones. J Neurochem. 1996;67:1801–1805. doi: 10.1046/j.1471-4159.1996.67051801.x. [DOI] [PubMed] [Google Scholar]
- 48.Ward GW, June CH, Olive D. PI 3-kinase: a pivotal pathway in T cell activation. Immunol Today. 1996;17:187–197. doi: 10.1016/0167-5699(96)80618-9. [DOI] [PubMed] [Google Scholar]
- 49.Whitmarsh AJ, Shore P, Sharrocks AD, Davis RJ. Integration of MAP kinase signal transduction pathways at the serum response element. Science. 1995;269:403–407. doi: 10.1126/science.7618106. [DOI] [PubMed] [Google Scholar]
- 50.Wilson DJ, Fortner KA, Lynch DH, Mattingly RR, Macara IG, Posada JA, Budd RC. JNK, but not MAPK, activation is associated with Fas-mediated apoptosis in human T cells. Eur J Immunol. 1996;26:989–994. doi: 10.1002/eji.1830260505. [DOI] [PubMed] [Google Scholar]
- 51.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 52.Yang D, Tournier C, Wysk M, Lu HT, Xu J, Davis RJ, Flavell RA. Targeted disruption of the MKK4 gene causes embryonic death, inhibition of c-Jun NH2-terminal kinase activation, and defects in AP-1 transcriptional activity. Proc Natl Acad Sci USA. 1997;94:3004–3009. doi: 10.1073/pnas.94.7.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoon YS, Kim JW, Kang KW, Kim YS, Choi KH, Joe CO. Poly(ADP-ribosyl)ation of histone H1 correlates with internucleosomal DNA fragmentation during apoptosis. J Biol Chem. 1996;271:9129–9134. doi: 10.1074/jbc.271.15.9129. [DOI] [PubMed] [Google Scholar]
- 54.Young PR, McLaughlin MM, Kumar S, Kassis S, Doyle ML, McNulty D, Gallagher TF, Fisher S, McDonnell PC, Carr SA, et al. Pyridinyl imidazole inhibitors of p38 mitogen-activated protein kinase bind in the ATP site. J Biol Chem. 1997;272:12116–12121. doi: 10.1074/jbc.272.18.12116. [DOI] [PubMed] [Google Scholar]