

Abstract
Recent evidence has suggested that subunits of the coatomer protein (COPI) complexes are functionally associated with endosomes in mammalian cells. We now provide genetic evidence that COPI plays a role in endocytosis in intact cells. The ldlF mutant CHO cell line bears a temperature-sensitive defect in the COPI subunit ε-COP. In addition to exhibiting conditional defects in the secretory pathway, we find that the cells are also defective at mediating endosome-associated functions. As found for cells microinjected with anti-COPI antibodies, ldlF cells at the restrictive temperature could not be infected by vesicular stomatitis (VSV) or Semliki Forest virus (SFV) that require delivery to acidic endosomes to penetrate into the cytosol. Although there was no temperature-sensitive defect in the internalization of receptor-bound transferrin (Tfn), Tfn recycling and accumulation of HRP were markedly inhibited at the restrictive temperature. Sorting of receptor-bound markers such as EGF to lysosomes was also reduced, although delivery of fluid-phase markers was only partially inhibited. In addition, lysosomes redistributed from their typical perinuclear location to the tips of the ldlF cells. Mutant phenotypes began to emerge within 2 h of temperature shift, the time required for the loss of detectable ε-COP, suggesting that the endocytic defects were not secondary to a block in the secretory pathway. Importantly, the mutant phenotypes were also corrected by transfection of wild-type ε-COP cDNA demonstrating that they directly or indirectly reflected the ε-COP defect. Taken together, the results suggest that ε-COP acts early in the endocytic pathway, most likely inhibiting the normal sorting and recycling functions of early endosomes.
Endocytosis is responsible for the internalization of extracellular fluid and macromolecules as well as plasma membrane proteins, receptors, and their bound ligands. Within early endosomes, this complex cargo must be sorted to ensure proper delivery to the appropriate intracellular destinations. Many receptors such as low density lipoprotein (LDL)1 receptor and some ligands such as transferrin (Tfn) are recycled back to the plasma membrane, whereas bulk fluid and ligands that dissociate from their receptors because of acidic endosomal pH are targeted to lysosomes (Gruenberg and Maxfield, 1995; Mellman, 1996). Other sorting events lead from endosomes back to the Golgi complex, specialized storage compartments, or in polarized cells, a plasma membrane domain other than the domain of entry (Mellman, 1996). The mechanisms responsible for sorting in early endosomes remain largely unknown, but seem likely to involve the formation of transport vesicles or tubules that selectively include or exclude individual components. As found for sorting or transport events in other organelles, endosomal sorting may directly or indirectly involve the assembly of cytosolic coatomer proteins (COP).
Several distinct coat complexes have been identified to be functionally important in molecular sorting and vesicle budding. These include clathrin and the clathrin adaptor proteins responsible for much of the transport of receptors from the plasma membrane and the trans-Golgi network to endosomes (Pearse and Robinson, 1990) and the COPI and COPII complexes, which are well known to play important roles in mediating transport through the secretory pathway. COPI was first identified as coating Golgi-derived transport vesicles (Orci et al., 1986, 1993) and shown to function in the transport between the ER and Golgi (Pepperkok et al., 1993). It is a multimeric complex termed coatomer consisting of α (160), β (110), β′ (102), γ (98), δ (61), ε (31), and ζ (20 kD) subunits (Waters et al., 1991).
The COPI coatomer is thought to associate with the cytoplasmic face of membranes under the regulatory control of small GTPases known as ADP ribosylation factors (ARFs; Takizawa and Malhotra, 1993). Inhibition of ARF nucleotide exchange by the fungal metabolite brefeldin A (BFA) prevents the binding of coatomer to membranes, and thus the normal sorting and transport functions of the ER and the Golgi complex (Donaldson et al., 1992; Helms and Rothman, 1992). In the secretory pathway, COPI may function in vesicle bud formation, selection of cargo for transport, or both (Schekman and Orci, 1996). A role in cargo selection was suggested by findings that COPI subunits can interact with KKXX or aromatic amino acid– containing motifs on the cytoplasmic domains of many membrane proteins (Letourneur et al., 1994; Fiedler et al., 1996; Lowe and Kreis, 1996; Tisdale et al., 1997).
Both clathrin and coatomer have been identified as candidates for endocytic sorting and vesicle budding, largely because of biochemical and immunocytochemical evidence that they can associate with endosomes in vitro (Whitney et al., 1995; Aniento et al., 1996; Stoorvogel et al., 1996; Takei et al., 1996). In the case of COPI, endosome-binding is specific and ARF dependent. Only a subcomplex of coatomer appears to bind, with γ- and δ-COP associating at lower than the normal 1:1 stoichiometry relative to the other subunits (Whitney et al., 1995; Aniento et al., 1996). Although the functional role for clathrin on endosomes has not yet been explored, several considerations have suggested that COPI may be associated with one or more endosomal activities. Initial hints came from observations that BFA caused a dramatic increase in long tubular elements of the endosomes reminiscent of those observed emanating from the Golgi complex in BFA-treated cells (Hunziker et al., 1991; Lippincott-Schwartz et al., 1991; Wood et al., 1991). Similarly, overexpression of inactive ARF mutants have been shown to alter endosome morphology or function (Zhang et al., 1994; D'Souza-Schorey et al., 1995; Peters et al., 1995). Although BFA did not greatly affect the uptake of receptor-bound or fluid-phase tracers, it did markedly alter endosomal sorting functions, altering recycling efficiency as well as the fidelity of basolateral to apical transcytosis in epithelial cells (Hunziker et al., 1991; Apodaca et al., 1993; Matter et al., 1993).
More direct evidence of a role for COPI in endosome function has come from experiments performed both in intact cells and in cell-free assays. Microinjection of antibodies to β-COP, previously shown to block transport between the ER and the Golgi, were found to block infection of vesicular stomatitis virus (VSV), an enveloped virus that must reach endosomes of a suitably acidic pH (<pH 6) to fuse with the endosome membrane and penetrate into the cytosol for replication (Whitney et al., 1995). A similar result was obtained for the low pH-dependent penetration of diptheria toxin from endosomes (Lemichez et al., 1997). In vitro, β-COP antibodies were found to inhibit the formation of endosome-derived structures thought to represent vesicles that mediate transport from early to late endosomes (Aniento et al., 1996).
In addition, a mutant CHO cell line (ldlF) found to have a temperature-sensitive defect in ε-COP was found not only to have a defect in the transport of newly synthesized LDL receptors, but also to perturb the behavior of LDL receptors that had already reached the plasma membrane (Hobbie et al., 1994; Guo et al., 1994, 1996). At the restrictive temperature, surface LDL receptors were not recycled properly but prematurely and ectopically degraded.
If COPI subunits were to play a role in endosome function, a minimal expectation would be that cells defective in ε-COP would exhibit one or more defects in the endocytic pathway. To this end, we have now examined endocytosis in ldlF cells in some detail. Our results clearly show that these cells exhibit significant alterations in the handling of endocytic tracers internalized as bulk fluid or receptor-bound ligands. Although the experiments do not pinpoint the precise step(s) at which COPI might act in the endocytic pathway, they do suggest that alterations in COPI activity directly affect the normal sorting functions of early endosomes in CHO cells.
Materials and Methods
Cell Culture
All cells were maintained in α-MEM containing 10% FCS. Wild-type (wt) and mutant CHO cells were maintained at 34°C and shifted to 40°C for experiments, whereas complementation ldlF[LDLF] CHO cells were maintained under selection at the nonpermissive temperature, 40°C, and then shifted to 34°C for experiments to parallel wt and ldlF cells at this temperature. Cells were plated 2 d before all experiments into multiwell plates for [125I]Tfn and HRP experiments or onto glass coverslips for immunofluorescence. Cells were used at 50–75% confluence.
Detection of ε-COP by Western Blot
After incubation at either 34° or 40° for 12 h, cells were washed extensively in PBS/900 μM Ca2+ per 500 μM Mg2+ and lysed in PBS containing 1% Triton X-100. Samples were then normalized for total cell protein using a modified biuret assay (BCA assay; Pierce Chemical Co., Rockford, IL), which was insensitive to the concentrations of Triton X-100 used. Samples were then subjected SDS-PAGE and then transferred to 0.2-μm nitrocellulose for Western blotting. A rabbit polyclonal anti–ε-COP (gift from F. Wieland, University of Heidelberg, Heidelberg, Germany) was used at 1:1,000 and detected using HRP-coupled goat anti–rabbit secondary diluted at 1:2,000, followed by enhanced chemiluminescence (Amersham Corp., Arlington Heights, IL).
Virus Infection of Cells
Endocytosis and infection of Semliki Forest virus (SFV) or tsO45 VSV was performed essentially as described (Kreis, 1986). Briefly, before infection, cells were preincubated at 34° or 40°C for ⩽10 h. After 1 h of preabsorption, cells were incubated with virus for 2.5 h. Cells were then fixed and extracted with methanol at −20°C for 4 min. Viral infection was detected by immunofluorescence using monoclonal antibody, P5D4, directed against VSV G protein (Kreis, 1986), or E2-1 (Kielian et al., 1990) directed against SFV E1/E2 spike glycoprotein. Delivery of SFV to an acidic compartment was monitored by immunofluorescence using the monoclonal antibody, E1-a1 (Kielian et al., 1990), directed against the acid-specific conformation of the E1 subunit of the SFV viral spike glycoprotein. For these experiments, endocytosis of SFV was carried out for 30 min in cells that were first preincubated at 34° or 40°C for 10 h.
Steady-state Distribution of Tfn Receptors
Cells were first incubated at 34° and 40°C for ⩽12 h. To rid the cells and medium of unlabeled Tfn, a brief incubation in serum-free medium (30 min, α-MEM + 2 mg/ml BSA) was performed. The cells were then labeled for 60 min at 34° or 40°C with 2.5 μg/ml [125I]Tfn and were then transferred to ice to prevent any further endocytic traffic. Iodination of Tfn was performed as previously described (Podbilewicz and Mellman, 1990). External [125I]Tfn was removed by extensive washing with PBS2+ at 4°C. Surface-bound [125I]Tfn was determined by cycling several times between acid (α-MEM + 2 mg/ml BSA, pH 2.6) and neutral (PBS/900 μM Ca2+ per 500 μM Mg2+ per 2 mg/ml BSA, pH 7.4) washes at 4°C to dissociate the ligand from its receptor. The combined washes were assayed to determine total amount of ligand from the cell surface. After removal of cell surface [125I]Tfn, the internal fraction was assayed by lysing cells in 1% Triton X-100 and γ counting. Values are represented as percent total of bound [125I]Tfn. Total [125I]Tfn was calculated as the sum of cell surface and internal [125I]Tfn.
Internalization of [125I]Tfn
After incubation at 34° or 40°C, 2.5 μg/ml [125I]Tfn was internalized for 0–8 min. The cells were then washed extensively and surface-bound Tfn was removed as described above. The cells were then lysed in 1% Triton X-100 and total cell protein was determined by a modified biuret assay (BCA assay; Pierce Chemical Co.) that was insensitive to the concentrations of Triton X-100 used.
[125I]Tfn Recycling
After incubation at 34° or 40°C for ⩽12 h, the cells were labeled to steady state as described above. Cells were then washed extensively with ice-cold PBS/900 μM Ca2+ per 500 μM Mg2+. Recycling was then initiated by warming the cells simultaneously to 34°C with α-MEM + 2 mg/ml BSA in the presence of 100-fold excess unlabeled Tfn. At each time point the medium was removed for γ counting and replaced with fresh medium. At the end of the recycling assay, cell-associated [125I]Tfn was obtained by lysing in 1% Triton X-100 and scraping the wells with a rubber policeman. Total [125I]Tfn was calculated as the sum of all recycled and cell-associated [125I]Tfn. Essentially the same kinetics were obtained when cells were labeled to steady state and surface-bound Tfn was acid stripped as described above before initiation of recycling (data not shown).
Specificity of [125I]Tfn
After incubation at 34° and 40°C, all cell lines were labeled to steady state as described above in the absence or presence of 100-fold excess unlabeled Tfn. Under all conditions, noncompetable uptake of [125I]Tfn represented >2% of the total specific signal.
Fluid-phase Endocytosis
Cells were labeled at either 34° or 40°C with 2.5 mg/ml HRP (Type VI; Sigma Chemical Co., St. Louis, MO) in α-MEM + 2 mg/ml BSA for various times ⩽120 min after incubation at the permissive and nonpermissive temperatures for ⩽12 h. The cells were then transferred to ice and washed three times with α-MEM + 5% BSA, three times with α-MEM + 2 mg/ml BSA, and three times with PBS/900 μM Ca2+ per 500 μM Mg2+. The cells were then lysed in 1% Triton X-100 and spun at 1,000 rpm for 10 min in an Eppendorf microcentrifuge (model 1514C; Brinkman Instruments, Westbury, NJ). The supernatants were assayed for HRP activity as previously described (van der Sluijs et al., 1991) and normalized to total cell protein (BCA assay; Pierce Chemical Co.).
Density Gradient Fractionation
Cells were labeled either with [125I]Tfn or by uptake of HRP as described above. For each gradient, three confluent 10-cm culture dishes were scraped into 0.6 ml of cracking buffer (78 mM KCl, 4 mM MgCl2, 8.4 mM CaCl2, 10 mM EGTA, 50 mM Hepes/KOH, pH 7.0) plus 250 mM sucrose and passed four times through a ball-bearing cell homogenizer using an 0.2498′′ ball bearing. The homogenate was centrifuged at 1,000 g for 5 min to produce a postnuclear supernatant (PNS) that was loaded on top of a 12-ml preformed linear 5 to 20% gradient (optiprep; Nycomed, Oslo, Norway) made with cracking buffer. The gradients were developed at 100,000 g for 20 h and then decanted mechanically. Fractions were tested for β-hexosaminidase activity using 4-methyl-umbelliferyl-N-acetyl-β-d-glucosaminidine (Sigma Chemical Co.) as a fluorescent substrate. Activity was assayed with an excitation wavelength of 365 nm and an emission wavelength of 450 nm.
Immunofluorescence
Cells grown on coverslips were first incubated at the permissive or nonpermissive temperatures for up to 12 h. The cells were then fixed in 2.5% paraformaldehyde and subsequently processed for immunofluorescence. All steps were performed using PBS/2% BSA per 0.03% saponin. Fixed cells were permeabilized for 30 min, and then incubated in primary antibodies for 1 h (for lysosomes: a mouse monoclonal antibody against lysosomal glycoprotein (lgp)-B, diluted 1:10 [Harter and Mellman, 1992]). After incubation in the primary antibodies, the cells were washed for 30 min and incubated in secondary antibodies (Texas red or FITC goat anti– mouse or goat anti–rabbit; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). The cells were then washed for 30 min, rinsed briefly in PBS and then water, and mounted in Moviol (Calbiochem-Novabiochem Corp., La Jolla, CA) containing 2.5% 1,4-diazabicyclo(2.2.2)octane (Sigma Chemical Co.).
Labeling of Lysosomes by Lucifer Yellow, EGF, and lgp
For Lucifer yellow (LY), cells were first incubated for 6 h at either 34° or 40°C and then labeled with 2 mg/ml LY (Molecular Probes Inc., Eugene, OR) for 90 min and washed for 90 min at either 34° or 40°C. For EGF, cells were preincubated for ⩽12 h at either 34° or 40°C, and fed 100 μg/ml of biotinylated EGF complexed to Texas red-conjugated avidin (Molecular Probes Inc.) for 30 min at either 34° or 40°C. The EGF or LY labeled cells were then rinsed briefly in PBS/900 μM Ca2+ per 500 μM Mg2, fixed in 2.5% paraformaldehyde, and counterstained for late endosomes and lysosomes as described above. For internalization of the anti–lgp-B antibody, cells were first preincubated for 6 h at 34° or 40°C and then incubated for 60 min at either temperature in antibody-containing culture supernatant before fixation in 2.5% paraformaldehyde. The cells were then permeabilized and the internalized primary antibody was detected using FITC goat anti–mouse secondary antibodies as described above. The lgp-β antibody concentration was sufficiently low to prevent detectable uptake of antibody by fluid-phase endocytosis.
Confocal Fluorescence Microscopy
Fluorescently labeled cells were examined using a modified confocal microscope (model MRC600; Bio-Rad Laboratories, Hercules, CA) attached to a microscope (model 451888 Axiovert; Carl Zeiss, Inc., Thornwood, NY) using separate filters for each fluorochrome viewed; FITC: ex = 488 nm, em = 515 LP; Texas red: ex = 568 nm, em = 585 LP. Control slides (single-labeled with each fluorophore) were examined to ensure that no cross-talk or bleed-through was occurring for the given confocal conditions. Images were imported into Adobe Photoshop and processed.
FITC–Tfn Labeling of Endosomes
Cells grown on coverslips were first incubated at the permissive or nonpermissive temperatures and then serum-starved for 30 min at either 34° or 40°C in α-MEM + 2 mg/ml BSA to deplete the cells and medium of unlabeled Tfn. The cells were then incubated for 90 min in 50 μg/ml FITC– Tfn (human holo form; Molecular Probes Inc.) in α-MEM + 2 mg/ml BSA per 20 mM Hepes, pH 7.4, at either 34° or 40°C. The cells were then rinsed briefly with PBS/900 μM Ca2+ per 500 μM Mg2+, fixed in 2.5% paraformaldehyde, rinsed briefly in PBS and then water, and mounted in Moviol (Calbiochem- Novabiochem Corp.) containing 2.5% 1,4-diazabicyclo (2.2.2)octane (Sigma Chemical Co.).
Intracellular pH Measurement
Intracellular pH was determined essentially as described (Kaplan and Boron, 1994). Briefly, cells were grown on rectangular coverslips designed for use in a 2-ml cuvette. After an 8-h incubation at either 34° or 40°C, cells were loaded for 15 min with 1 μM BCECF-AM (Molecular Probes Inc.) diluted in loading buffer (125 mM NaCl, 5 mM KCl, 1 mM CaCl2·2H2O, 1.2 mM MgSO4·7H2O, 2 mM NaH2PO4·H2O, 32 mM Hepes, 10.5 mM glucose, 18.8 mM NaOH). Removal of uncleaved dye was achieved by washing in dye-free loading buffer for 15 min. A standard curve was then generated for each cell line at either 34° or 40°C by incubating cells in 1 μM nigericin (Sigma Chemical Co.) in calibration buffer containing 105 mM KCl, 1 mM CaCl2·2H2O, 1.2 mM MgSO4·7H2O, 2 mM H3PO4, 32 mM Hepes, 10.5 mM glucose, 40 mM HCl, 45.8 mM N-methyl-d-glucamine (Sigma Chemical Co.) at pH 8.0, 7.5, 7.0, 6.5, 6.0, and 5.5.
Endosomal pH Measurement
Endosomal pH was measured using a modification of the method of Yamashiro and Maxfield (Yamashiro and Maxfield, 1987). Cells were labeled with FITC–Tfn as described above. Coverslips were then transferred to a chamber to permit confocal imaging of live cells. The coverslips were then rinsed in loading buffer (see Intracellular pH Measurement) and images were acquired using the BHS filter set (ex = 488 nm, em = 515 LP) and the same confocal settings for all cell lines. The pH of the endosomes was then artificially raised to pH 7.4 using 1 μM nigericin (Sigma Chemical Co.) in calibration buffer described in intracellular pH measurement, and a second set of images were acquired using the same confocal settings. The percent increase in fluorescence intensity was calculated using NIH image. A greater percent increase in fluorescence intensity represents a lower endosomal pH.
Results
ldlF Cells Lack ε-COP
The ldlF CHO cell mutant was identified in a screen intended to isolate cell lines with temperature-sensitive defects in LDL receptor activity. The cells were isolated because of their inability to transport newly synthesized LDL receptors to the plasma membrane and to maintain previously synthesized LDL receptors at the surface (Hobbie et al., 1994). cDNA transfection experiments and cloning of the mutant ldlF allele identified the site of the mutation as being a point mutation in the coding region of the gene encoding ε-COP (Guo et al., 1994, 1996). To confirm that the cells used for these experiments were deficient in ε-COP, Western blot analysis was performed on wt, mutant (ldlF cells), and mutant cells transfected with a cDNA expressing wt ε-COP (ldlF[LDLF] cells) after incubation at either the permissive (34°C) or nonpermissive temperature (40°C) for 12 h. As shown in Fig. 1, ldlF cells have more than fivefold less ε-COP than wt or ldlF[LDLF] cells even at 34°C and after incubation at 40°C for 12 h, ε-COP was undetectable in the mutants, but barely affected in cells expressing wt ε-COP. The time course of the loss of ε-COP from ldlF cells indicated that at 40°C, the t1/2 of ε-COP degradation was ∼1–2 h (Guo et al., 1996).
Figure 1.

ldlF cells lack ε-COP. After incubation of wt and mutant cells at either 34° or 40°C for 12 h, cell lysates were normalized for total cell protein and then subjected to SDS-PAGE and immunoblotting. At 34°C, ldlF cells have much less ε-COP than wt cells, and after incubation at 40°C for 12 h, ε-COP is undetectable in the mutant cells. The defect was complemented by transfection with wt ε-COP in ldlF[LDLF] cells.
Virus Infection Is Inhibited in ldlF Cells
In previous work, we found that microinjection of antibodies to β-COP, another COPI subunit that binds to endosomes, blocked infection by VSV (Whitney et al., 1995). Microinjection of VSV nucleocapsids, however, circumvented this block. Since the normal infectious pathway of enveloped viruses such as VSV involves delivery to endosomes of sufficiently acidic pH to trigger fusion of the viral membrane with the endosomal membrane, we reasoned that the anti–β-COP antibody affected the delivery of the virus to endosomes capable of hosting viral penetration or inhibited some feature (perhaps acidification) of endosomes themselves. A minimal expectation, therefore, was that ldlF cells would be similarly unable to host virus infection. For these experiments, we used both VSV and a second enveloped virus, SFV. Whereas VSV has a fairly broad pH threshold for fusion (pH 5.3–6.3; Matlin et al., 1982), the SFV E1/E2 spike glycoproteins exhibit a pH-induced conformational change leading to fusion at pH < 6.2 (White et al., 1980). Thus, SFV may penetrate from an earlier or less acidic site on the endocytic pathway than VSV (Schmid et al., 1989).
Cells were incubated at 34° or 40°C for 10 h and then exposed to wt SFV or the tsO45 mutant of VSV and fixed, and then infection was monitored by immunofluorescence using antibodies to SFV E1/E2 or VSV G protein. As shown in Fig. 2, SFV was incapable of infecting the mutant cells at the restrictive temperature. However, clear synthesis of E1/E2 protein was observed in ldlF cells incubated at 34°C or in wt or complemented ldlF[LDLF] cells at either temperature. Similar results were obtained using tsO45 VSV (data not shown). These experiments were quantified by counting several hundred cells stained by immunofluorescence for the SFV E1/E2 or VSV G spike glycoproteins (Whitney et al., 1995). Although at the permissive temperature, >90% of the ldlF cells were infected by VSV or SFV; after 12 h at 40°C the infection frequency was reduced to 5 and 10%, respectively.
Figure 2.

Virus infection is inhibited in ldlF cells. After incubation for 10 h at 34° or 40°C, cells were exposed to SFV for 2.5 h to allow for viral endocytosis as described in Materials and Methods. Infection was monitored by immunofluorescence detection of newly synthesized SFV–E2 spike glycoprotein. SFV infection occurred normally in ldlF cells preincubated at 34°C and wt and ldlF[LDLF] cells preincubated at either temperature. wt and ldlF[LDLF] are representative of results obtained at either temperature. No viral infection was observed in ldlF cells that were preincubated at 40°C. Similar results were obtained for tsO45 mutant of VSV.
The failure of VSV and SFV to infect CHO cells in the absence of functional ε-COP suggested that virus particles were not delivered to endosomal compartments of sufficiently acidic pH to trigger fusion, or that the endosomes were rendered defective for some other secondary reason. To distinguish between these possibilities, we monitored the delivery of SFV to acidic compartments using a monoclonal antibody (E1a-1) specific for the acid conformation of the E1 spike glycoprotein (Kielian et al., 1990). After 30 min of uptake, E1a-1 reactive virus (i.e., virus particles that had reached pH <6.1) was detected in wt cells and ldlF[LDLF] cells preincubated at either 34° and 40°C and in ldlF cells preincubated at 34°C (Fig. 3). The acid conformation was not detected after SFV internalization by ldlF cells incubated at the restrictive temperature. Thus, in the absence of ε-COP, the virus was unable to reach a compartment of sufficiently low pH to trigger fusion. This seemed unlikely to reflect a global acidification defect in ldlF cells, since they accumulated the pH-sensitive probe acridine orange in a fashion similar to wt cells (see Endosomal pH Assessment). Additionally, ldlF cells were capable of viral infection when virus was microinjected directly into the cytosol. Thus, the inhibition of viral infection may reflect a block in intracellular transport.
Figure 3.

SFV does not reach an acidic compartment in ldlF cells. After SFV uptake for 30 min, delivery of SFV to an acidic compartment was monitored by immunofluorescence detection of the acid-specific (pH < 6.2) conformation of SFV–E1 protien. Cells were preincubated at 34° or 40°C for 10 h. The acid conformation was detected in ldlF cells preincubated at 34°C, wt, and ldlF[LDLF] cells, but not ldlF cells preincubated at 40°C. wt and ldlF[LDLF] are representative of results obtained at either temperature.
ldlF Cells Have a Partial Defect in Tfn Endocytosis
To study transport along the endocytic pathway in ldlF cells, suitable probes were required. Since LDL receptor was prematurely degraded in mutant cells at the restrictive temperature (Hobbie et al., 1994), we analyzed the internalization and recycling of receptor-bound Tfn, another well-characterized endocytic marker. Extracellular Tfn binds to its receptor (TfnR) at the cell surface, and is then internalized via clathrin coated pits. The Tfn–TfnR complex is then delivered to early endosomes where it resides only briefly to discharge bound iron before being sorted into recycling vesicles that carry the Tfn–TfnR complex back to the plasma membrane (Dautry-Varsat et al., 1983; Hopkins and Trowbridge, 1983; Yamashiro et al., 1984). The recycling pathway actually seems to bifurcate, with the bulk of Tfn returning to the surface rapidly via peripheral recycling vesicles and a fraction (∼25%) residing in a distinct perinuclear recycling compartment (PNRC) before recycling with somewhat slower kinetics (Sheff, D., E. Davo, and I. Mellman, manuscript in preparation). Tfn does not normally reach late endosomes or lysosomes during its route through the cell (Schmid et al., 1988). Unlike LDL receptor, we found that TfnR was not prematurely degraded in ldlF cells incubated at the restrictive temperature (data not shown). Thus, Tfn was a useful probe to study the endocytic pathway in the mutant cells.
We first asked whether ldlF cells exhibited any major overall defects in Tfn endocytosis. At steady state in wt CHO cells, ∼20% of TfnRs are found on the cell surface, while 80% are found intracellularly (van der Sluijs et al., 1992). Even after 8 h at the restrictive temperature, ldlF cells exhibited the characteristic 80:20% intracellular/surface distribution of TfnR (Fig. 4 A).
Figure 4.



ldlF cells have a partial defect in transferrin endocytosis. (A) Loss of ε-COP does not affect the distribution of Tfn receptors. After incubation of wt and mutant cells at 34° and 40°C for 8 h, intracellular and cell surface receptors were saturated with [125I]Tfn by labeling for 60 min at 34° or 40°C. Surface and internal Tfn were determined as described in Materials and Methods. Values are represented as percent total of bound [125I]Tfn. In all conditions, cells exhibited a TfnR distribution with ∼20% on the cell surface, and 80% intracellular. n = 9. (B) Tfn internalization is inhibited in ldlF cells. After incubation of wt and mutant cells at 40°C for 8 h, cells were incubated with [125I]Tfn at 40°C for various amounts of time to allow for internalization of receptor-bound Tfn. Internalized Tfn was assayed after removal of cell surface Tfn (% surface-bound Tfn was similar for wt and ldlF cells) and normalized for total cell protein as described in Materials and Methods. Internalization of Tfn was inhibited in ldlF cells lacking ε-COP. n = 6. (C) Tfn internalization defect is complemented in ldlF[LDLF] cells and is partially temperature sensitive. After incubation at 34° or 40°C for 8 h, cells were allowed to internalize [125I]Tfn for 5 min and internalized Tfn was assayed as in B. wt cells internalized similar amounts of [125I]Tfn at either temperature, but ldlF cells internalized somewhat less [125I]Tfn at 34°C and even less at 40°C as compared to wt cells. The defect was complemented by addition of wt ε-COP in ldlF[LDLF] cells. n = 6.
We next measured the relative rates of Tfn endocytosis. Control or ldlF cells were incubated for 8 h at 40°C and then incubated with [125I]Tfn for 0–30 min at 40°C for 0–10 min. As shown in Fig. 4 B, the amount of Tfn internalized at early time points in ldlF cells incubated at the restrictive temperature was ∼60% of that of wt CHO cells. However, after incubations of >1 h, the total amount of Tfn accumulated by the mutants was not significantly different from wt cells (data not shown; Fig. 4 A). The defect in the initial rate of Tfn uptake was not completely temperature sensitive, however. Even at 34°C, Tfn endocytosis by ldlF cells was partially reduced relative to wt cells (Fig. 4 C). Thus, the inability of enveloped viruses to infect ldlF cells at 40°C was unlikely to reflect a defect in virus uptake.
The partial Tfn endocytosis defect might reflect the presence of a temperature-sensitive mutation in ε-COP or an as yet unknown second site mutation(s) harbored by ldlF cells. To distinguish these possibilities, we attempted to complement the defect by using an ldlF cell line, ldlF[LDLF], that was stably transfected with wt ε-COP cDNA (Guo et al., 1994); these cells were found to express ε-COP protein at levels comparable to wt cells (Fig. 1). Indeed, even after 8 h at 40°C, Tfn internalization was restored to wild-type levels in ldlF[LDLF] cells (Fig. 4 C).
ldlF Cells Exhibit a Temperature-sensitive Defect in Tfn Recycling
The observation that ldlF cells did not exhibit an alteration in equilibrium distribution of TfnR suggested the existence of an inhibition of Tfn recycling to balance the slowed rate of endocytosis. To measure recycling rates, cells were first incubated at 34° or 40°C for ⩽12 h and then labeled to steady state with [125I]Tfn at either temperature. After washing at 4°C, recycling of internalized Tfn was initiated by warming the cells back to 34° or 40°C in the presence of excess, unlabeled Tfn to ensure quantitative dissociation of recycled [125I]Tfn. The amount of [125I]Tfn recycled back to the medium was determined at various times. As shown in Fig. 5 A, the recycling curves were very similar for wt CHO cells incubated at either 34° or 40°C with an overall t 1/2 of ∼3.5 min. At 34°C, ldlF cells exhibited an inhibition in recycling with an overall t 1/2 of ∼4.5 min, and at 40°C, this inhibition was exacerbated with an overall t 1/2 of ∼11.5 min.
Figure 5.


Transferrin recycling is defective in ldlF cells. (A) Loss of ε-COP inhibits Tfn recycling. After incubation at 34° and 40°C for ⩽12 h, cells were labeled to steady state with [125I]Tfn and then transferred to ice and washed extensively. Recycling was then initiated by warming the cells to 34° or 40°C and the medium was assayed at various time points. After 60 min, the remaining cell-associated [125I]Tfn was determined by lysing the cells in 1% Triton X-100. Total [125I]Tfn is the sum of cell associated and recycled Tfn at each time point. Values from each time point were normalized to % total Tfn. There is a slight slowing in the rate of Tfn recycling in ldlF cells compared to wt and ldlF[LDLF] cells at 34°C. There is a significant inhibition of recycling at 40°C in the ε-COP–deficient ldlF cells and the defect is complemented in ldlF[LDLF] cells at 40°C. n = 15 for wt and ldlF cells and n = 6 for ldlF[LDLF] cells. (B) The recycling defect occurs within 1 h of shift to 40°C. wt and ldlF cells were preincubated at 40°C for 0–8 h before measuring the amount of [125I]Tfn recycled at the 15 min time point in A. Recycling decreased within 1 h of shifting to the nonpermissive temperature, and the inhibition was qualitatively half-maximal by 2 h. n = 3.
Importantly, expression of wt ε-COP in ldlF cells almost completely complemented the temperature-sensitive defect in Tfn recycling (Fig. 5 A, ldlF[LDLF] cells) with an overall t 1/2 of recycling of ∼3.7 min at 34°C and ∼3.4 min at 40°C. These results demonstrate that the temperature-sensitive recycling defect was a direct or indirect consequence of the loss of ε-COP, but not because of a second site mutation in ldlF cells.
The fact that ε-COP, as well as most other coatomer subunits, bind to endosomes suggests that the loss of ε-COP function may directly affect the recycling activity of one or more populations of early endosomes. If ε-COP was required for endosome function, the kinetics of the onset of the recycling defect should mirror the kinetics of loss of ε-COP upon shifting to the restrictive temperature. To determine the time of onset of the recycling defect, ldlF cells were incubated at 40°C for 0–9 h before loading with [125I]Tfn. Recycling was assayed by measuring the amount of [125I]Tfn released into the medium during a 15-min chase (the time at which the difference between wt and mutant cells was maximal; see Fig. 5 [A]). As shown in Fig. 5 A, recycling activity began to decrease 1 h after shifting to the restrictive temperature and the defect was qualitatively half-maximal by 2 h. These data agree well with the kinetics of ε-COP loss upon shifting ldlF cells to 40°C and with the onset of the transport defect in the secretory pathway, both of which appear with t 1/2 of ∼1–2 h (Hobbie et al., 1994). The simultaneous loss of recycling activity and ε-COP is consistent with the possibility that ε-COP plays a direct role in the endocytic pathway.
Endosomal pH Assessment
Since defects in endocytosis and recycling are sometimes observed after treatment with drugs that alter endosomal pH, we next asked if the early endosomes accessed by Tfn in ldlF cells exhibited an altered internal pH. These determinations were made by quantitative fluorescence microscopy of single cells using a modification of previously published methods (Yamishiro, 1987). After incubation at 34° or 40°C for 8 h, wild-type, ldlF, or ldlF[LDLF] cells were loaded with FITC–Tfn for 60 min, washed, and then viewed by confocal microscopy to establish baseline values. The percent increase in FITC fluorescence intensity was then monitored after nigericin addition and subsequent equilibration of endosomal pH to the pH of the medium (pH 7.4). The greater the percentage increase, the lower the starting endosomal pH. As shown in Fig. 6, within the limits of this technique, we were unable to detect any significant differences in the relative pH values of Tfn-containing endosomes in wt or mutant cells at the permissive or nonpermissive temperatures. Given the nature of these measurements, we did not attempt to assign specific pH values. Since wt and ldlF cells were able to deliver FITC–Tfn to early and/or recycling endosomes of comparable internal pH values, these results strongly suggest that the inability of SFV and VSV to infect ldlF cells at 40°C does not reflect a failure of endosomal acidification, but rather a failure of delivery of virus particles to acidic endosomes.
Figure 6.

Endosomal pH is similar in wt, ldlF, and ldlF[LDLF] cells. After incubation of wt and mutant cells at 34° and 40°C for 8 h, cells were loaded with FITC–Tfn. FITC fluorescence was quantitated both before and after equilibration to pH 7.4 with nigericin. The recorded change in fluoresence intensity is proportional to the difference between the pH of the FITC–Tfn-containing endosomes and that of the equilibration buffer (pH 7.4). Within the limits of these experiments, there was no significant difference observed between ldlf mutant cells and either the wt or ldlf[LDLF] mutant cells. n = 8 for wt cells at 40°C, n = 9 for ldlF cells at 40°C, and n = 6 for all others.
Loss of ε-COP Inhibits Accumulation of Fluid-phase Endocytic Tracers
We next measured the capacity of ldlF cells to accumulate the extracellular tracer, HRP by fluid-phase endocytosis. HRP is known to be internalized and delivered to lysosomes where it concentrates, because of its relative resistance to degradation. After incubation at either 34° or 40°C for 8.5 h, cells were incubated with HRP for 90 min. As shown in Fig. 7, wt cells accumulated similar amounts of HRP at either temperature. However, at the restrictive temperature, mutant cells exhibited a marked defect in HRP accumulation. Uptake of HRP at early time points (0–10 min) was also inhibited in ldlF cells (data not shown). The defect was restored to wt levels by complementation with wt ε-COP in ldlF[LDLF] cells, indicating that the phenotype was because of the defective ε-COP allele in ldlF cells (Fig. 7).
Figure 7.

Loss of ε-COP inhibits accumulation of fluid-phase markers. Cells were incubated for 8.5 h at either 34° or 40°C before labeling with the fluid-phase marker, HRP, for 90 min at either temperature. After extensive washing, cells were lysed and assayed for HRP activity and total cell protein. Loss of ε-COP in ldlF cells at 40°C resulted in an inhibition of HRP accumulation. The defect was restored in ldlF[LDLF] cells which were complemented with wt ε-COP. n = 12 for wt and ldlF cells and n = 6 for ldlF[LDLF] cells.
Endosomes and Lysosomes Are Redistributed in ldlF Cells
Our analysis of endocytosis in ldlF cells revealed that loss of ε-COP in ldlF cells resulted in inhibition of the initial rate of internalization of solutes and ligands, as well as a decrease in the ability of the cells to recycle ligands back to the cell surface or accumulate solutes in lysosomes. Together, these phenotypes suggested that the mutation disrupted transport or sorting in early endosomes. Since the Golgi complex exhibits an altered morphology in the mutant cells (Guo et al., 1994), we next asked if there was any alteration in the distribution or organization of endocytic organelles.
We examined the distribution of late endosomes and lysosomes by staining wild-type and mutant cells with an antibody directed against a major lysosomal membrane protein lgp-B (lgp 110, lamp-2; Harter and Mellman, 1992). As shown in Fig. 8 A, wt cells at 34° and 40°C exhibited the characteristic distribution of late endosomes and lysosomes throughout the cytoplasm with a more concentrated accumulation near the nucleus (large arrows). ldlF cells, on the other hand, appeared to exhibit somewhat less perinuclear clustering at 34°C. Strikingly, after incubation at 40°C for 5–12 h, the lysosomes in the mutant cells redistributed to the cell periphery, being concentrated at the very tips of the cells (small arrows). This is in contrast to the pattern observed with FITC–Tfn (Fig. 8 B) that labels the recycling pathway. Thus, the lgp-B–containing structures in the cell periphery represent redistributed lysosomes rather than mistargeting of lgp-B to peripheral early endosomes. Complementation with wt ε-COP in ldlF[LDLF] cells restored the lysosomal distribution to the juxtanuclear region (Fig. 8 A, large arrows), indicating that the phenotype was indeed because of the loss of ε-COP. Occasionally, mutant cells were observed to display both centrally (large arrow, bottom right) and peripherally clustered lysosomes (small arrows, bottom right).
Figure 8.

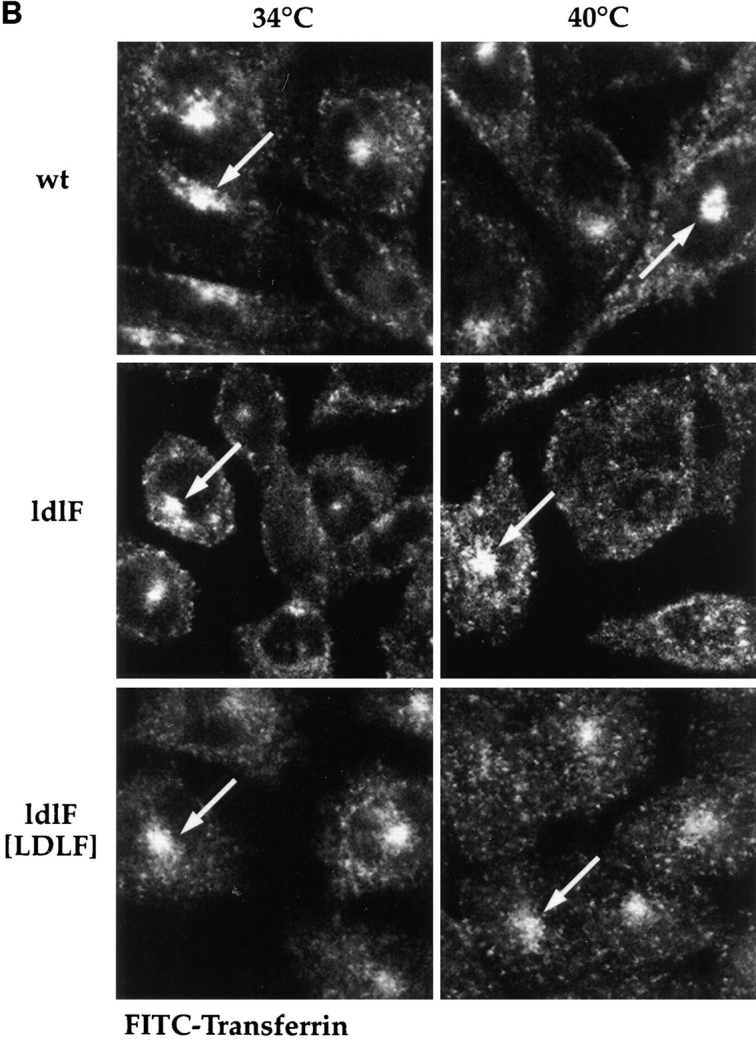
Lysosome, but not endosome, distribution is altered in ldlF cells. Cells were incubated at either 34° or 40°C to compare the distribution of internalized FITC–Tfn and the lysosomal glycoprotein lgp-B. (A) Distribution of lysosomes. ldlf and wt cells were fixed and immunostained for lgp-B. Wt cells exhibited the characteristic clustering of lysosomes in the perinuclear region (large arrowheads), whereas the lysosomes in the mutant cells seem to be more randomly dispersed at the permissive temperature. Upon incubation at the nonpermissive temperature for ⩽12 h, the lysosomes of the mutant cells seem to redistribute to the cell periphery (small arrowheads), extending to the very tips of the cells. Occasionally, mutant cells were observed to display both centrally (large arrowheads, bottom right) and peripherally (small arrowheads, bottom right) clustered lysosomes. Addition of wt ε-COP in the ldlF[LDLF] cells restored this phenotype (large arrowheads). Similar results were obtained when cells were incubated at 40°C for only 5 h. wt and ldlF[LDLF] panels are representative of results obtained at either temperature. (B) Distribution of FITC–Tfn-containing early endosomes. Wt, ldlf, and ldlf[LDLF] cells were incubated at the permissive or nonpermissive temperature for 12 h and then allowed to internalize FITC– Tfn for 1 h. Most (>80%) wild-type and 30–60% of ldlf CHO cells exhibited the characteristic clustering of recycling endosomes at the pericentriolar region (arrows) at both temperatures. ldlF[LDLF] cells were similar to wt cells. Arrows indicate the position of the PNRC. There was no redistribution of Tfn-containing endosomes to the cell periphery.
It remained possible that the lysosomal marker enzymes have shifted from the lysosomes to an endosomal compartment. To address this possibility, we separated endosomes from lysosomes on an optiprep density gradient. Subcellular fractionation of ldlf and wt cells at both the permissive and nonpermissive temperatures revealed no difference in the distribution of the lysosomal marker β-hexosaminidase, suggesting that redistribution of this lysosomal marker was not occurring (data not shown).
The redistribution of lgp-B–positive structures to the periphery of ldlF cells at 40°C was not because of a reorganization of the microtubule network in these cells, since by immunofluorescence, there were no apparent differences in methanol-fixed microtubules in wt or ldlF cells grown at the permissive or restrictive temperatures (data not shown). Microtubule disruption has been shown to disperse endosomes (McGraw et al., 1993; Daro et al., 1996), late endosomes, and lysosomes (Gruenberg et al., 1989) from the perinuclear region of CHO and other cells to a more random distribution.
CHO cells exhibit a characteristic perinuclear accumulation of early endosomes containing recycling markers (Dunn and Maxfield, 1992; Hopkins and Trowbridge, 1983). These structures are thought to represent a distinct endosome population based on their unique rab protein composition (Daro et al., 1996; Ullrich et al., 1996), ability to accumulate recycling markers such as FITC–Tfn, and ability to be physically separated from early endosomes by centrifugation (Sheff, D., E. Daro. and I. Mellman, manuscript in preparation). On average, fewer (∼60%) ldlF cells demonstrated the characteristic distribution of Tfn-containing endosomes as compared to wt cells (∼80%; data not shown). However, this difference was observed at both 34° and 40°C, making it unlikely that the altered distribution was responsible for or reflected any of the temperature-sensitive endocytic defects. Although the distribution of Tfn was slightly altered in ldlF cells, Tfn-containing structures did not redistribute to the periphery or tips of the cells (Fig. 8 B).
Acidification of the cytosol to pH < 6.9 has also been shown to redistribute endosomes and lysosomes to the cell periphery in a fashion remarkably similar to that exhibited by ldlF cells at 40°C (Heuser, 1989; Parton et al., 1991). We therefore measured the intracellular pH of wt, ldlF, and ldlF[LDLF] cells after incubation at 34° and 40°C for 8 h, and found that the pH of the mutant cells was ∼pH 7.2 whereas that of wt and ldlF[LDLF] was ∼pH 7.4 at either temperature (Table I). Thus, it was unlikely that cytosol acidification was responsible for the organelle redistribution observed at 40°C. In any event, the fact that the redistribution was not observed in ldlF[LDLF] cells strongly suggested that the effect was at least indirectly the consequence of an inactivation of ε-COP activity.
Table I.
Cytosolic pH Is Slightly Acidified in ldlF Cells
| pH/pKa | 34°C | 40°C | ||
|---|---|---|---|---|
| wt | 7.44/7.21 | 7.43/7.15 | ||
| ldlF | 7.18/7.30 | 7.19/7.35 | ||
| ldlF[LDLF] | 7.30/7.32 | 7.41/7.24 |
Acidification of cytosolic pH can cause the redistribution of endocytic structures. We therefore measured cytosolic pH using the fluorescent probe BCECF in each of the cell types after incubation for 8 h at either the permissive (34°C) or nonpermissive (40°C) temperatures. For each condition, a standard curve was generated to calculate both the pKa of the dye and the intracellular pH. The mutant cells were only slightly acidified. Values are from one experiment wherein a standard curve was prepared for each condition. Similar results were obtained for wt cells and ldlF cells at 40°C in several other experiments.
Delivery of Markers to Lysosomes Is Inhibited in ldlF Cells
Given the apparent defects in endosomal sorting combined with the dramatic redistribution of endocytic organelles, we next asked if internalized fluid-phase and receptor-bound tracers could reach their appropriate destinations using fluorescence microscopy.
To determine if any of the fluid-phase marker could reach the peripheral late endosomes or lysosomes, wt, ldlF, or ldlF[LDLF], cells were incubated for 6 h at 34° or 40°C and then labeled with LY for 90 min, washed, and then chased for 90 min at 34° or 40°C in the absence of LY. The cells were then fixed and stained for lysosomes using the lgp-B antibody. wt and ldlF[LDLF] cells exhibited the characteristic perinuclear clustering of lysosomes together with a high degree of colocalization with internalized LY, indicating that this internalized marker reached late endosomes/lysosomes at either temperature in these cells. At 34°C, there was also a high degree of colocalization between the internalized LY and late endosomes/lysosomes in the mutant ldlF cells, although the perinuclear clustering was less pronounced. Upon incubation of ldlF cells at 40°C (Fig. 9), the lgp-B–positive late endosomes/lysosomes (red) redistributed to the tips of the cells (arrowheads), but these structures were still accessible to internalized LY (green; visualized using a higher excitation intensity than wt or ldlF[LDLF] cells because of the decreased amount of LY accumulated under these conditions). Interestingly, lgp-B–positive structures that remained more towards the center of the cell were relatively devoid of LY. Thus, at the restrictive temperature, ldlF cells remained capable of delivering LY to at least a subpopulation of lgp-positive structures. These peripheral lgp/LY-containing structures may correspond to late endosomes, or may simply represent a very low number of both late endosomes and lysosomes that receive a minimal amount of LY but become visually apparent because of their concentration in a small area of the cell. In any event, it was clear that at least a portion of the internalized LY could reach late endosomes/lysosomes.
Figure 9.

Peripheral lysosomes are accessible to fluid-phase markers in ldlF cells. To determine if the peripheral lysosomes in Fig. 7 were accessible to a fluid-phase tracer, cells were first incubated for 6 h at 40°C and then labeled with LY for 90 min and washed for 90 min at 40°C. The cells were then fixed and stained for lysosomes using the lgp-B antibody shown in Fig. 7. In ldlF cells, the lysosomes (red) are found at the tips of the cells (arrowheads), but these structures are still accessible to internalized LY (green) as indicated by the yellow color in the merge panel.
We next examined the ability of the receptor-bound ligand EGF to be delivered to lysosomes. Cells were permitted to internalize Texas red–labeled biotinylated EGF for 30 min at 34° or 40°C, fixed, and then stained using the anti–lgp-B antibody. In wt cells at either temperature, EGF (red) was found to colocalize extensively with lgp-positive late endosomes and lysosomes (green) in the perinuclear region yielding yellow double-positive structures (Fig. 10, top two rows). In ldlF cells, however, colocalization was already limited at 34°C and even further reduced at 40°C (Fig. 10, lower two rows). EGF was strikingly absent from the lgp-positive structures at the cell tips which stained green (arrowheads). Thus, the delivery of EGF to late endosomes and lysosomes was inhibited far more strongly than the delivery of LY. This difference may in part reflect the fact that unlike fluid-phase tracers, EGF must dissociate from its receptor after reaching acidic endosomes to reach lysosomes efficiently.
Figure 10.
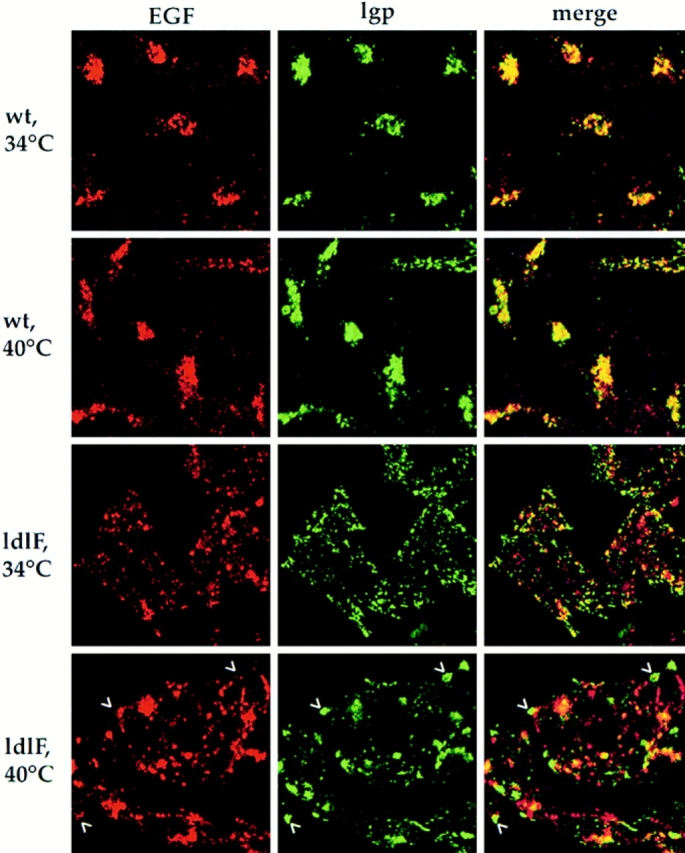
EGF does not reach lysosomes in ldlF cells. To follow the intracellular destination of a lysosomally targeted ligand, Texas red–conjugated EGF was internalized for 30 min after the cells were first preincubated for ⩽12 h at 34° or 40°C. The cells were then fixed and counter-stained for lysosomes using the same lgp-B antibody shown in Figs. 7 and 8. In wt cells at either temperature, the internalized EGF (red) reached lgp-positive structures (green). Overlap is yellow. In ldlF cells, there is very little overlap between the red and green channels at either temperature, indicating that very little of the internalized EGF reached lgp-positive structures. In ldlF cells at 40°C, the lysosomes (green) are found at the tips of the cells (arrowheads), but EGF (red) is not found in these structures.
An additional assay further demonstrated that the lysosomal pathway was, in general, very inefficient in ldlF cells at 40°C. A small fraction of lgp-B made in CHO cells reaches late endosomes and lysosomes after insertion into the plasma membrane (Harter and Mellman, 1992). By binding anti–lgp-B IgG to cell surface lgps, and incubating to permit uptake of the IgG–lgp-B complex, we were able to monitor delivery to late endosomes and lysosomes in cells that were preincubated at 34° or 40°C (data not shown). Delivery of internalized anti-lgp antibody to characteristic perinuclear lysosomal structures was found in wt and ldlF- [LDLF] cells at either temperature. However, in ldlF cells that were preincubated at 40°C, the antibody failed to reach the peripheral lysosomal structures found in the tips of cells. Thus, mutant cells even failed to deliver a resident lysosomal membrane protein to its intrinsic destination at 40°C.
Discussion
Our results demonstrate that multiple aspects of the endocytic pathway are disrupted in CHO cells expressing a temperature-sensitive point mutation in the COPI complex subunit ε-COP. Although it is well established that COPI coatomer functions in early stages of the secretory pathway, our results provide genetic evidence of a role for at least one COPI subunit in endocytic transport. These data do not define the precise transport step or steps under the control of COPI in endosomes. Nor do they prove that COPI is directly required for endosome function, since it remains formally possible that COPI is required for a transport step that delivers a rapid turn over or recycling component from the Golgi complex to endosomes. However, given recent biochemical data showing that COPI components physically associate with endosomes and appear to inhibit certain endosome-related activities, the availability of the ldlF mutants provided a critical initial test for the concept that COPI indeed is required for endosome function: the failure of ldlF cells to exhibit any defect in the endocytic pathway would have dismissed a role for COPI in endosomes. Importantly, the fact that these defects could be complemented by transfection of a wt copy of the ε-COP gene proved that the mutant phenotype was directly or indirectly because of the loss of ε-COP.
Our analysis of endocytosis in ldlF cells revealed that temperature-dependent loss of ε-COP resulted in a complex but interconnected series of temperature-sensitive and -insensitive phenotypes, each appearing to inhibit some aspect of early endosome function. The first of these was a dramatic inhibition of virus infection at 40°C. In the case of SFV, the use of a conformation-specific antibody to the E1/E2 spike glycoprotein revealed that at the restrictive temperature, the virus never reached an endosome compartment of suitably low pH to acquire the acid conformation required for fusion of the viral envelope with the endosome membrane. Yet, there appeared to be no global acidification defect, since acridine orange was accumulated normally by ldlF cells at all temperatures. Moreover, quantitative fluorescence analysis of the pH within FITC–Tfn-containing endosomes indicated again the absence of obvious differences between ldlF and wt cells at either 34° or 40°C. Although early endosomes have generally been assumed to be the site of penetration of viruses such as SFV (which fuses at pH < 6.1), our previous results noted the presence of an early endocytic compartment that does not have sufficiently low pH to support even SFV entry (Schmid et al., 1989). Conceivably, in ldlF cells, incoming virus cannot be efficiently transferred from this compartment (which may reflect early endosomes or primary endocytic vesicles such as coated vesicles) to a compartment of suitably low pH to host fusion. In any event, resolving the actual site of virus entry will require some further exploration.
Also consistent with a defect in early endosome function was the observation that ldlF cells exhibit a marked temperature-sensitive inhibition of Tfn recycling. It is becoming clear that the pathway of Tfn recycling may be somewhat more complex than originally conceived, involving the existence of two parallel pathways: the faster, predominant one corresponding to a short circuit pathway that returns receptors back to the cell surface directly from early endosomes, and a second, slower pathway, which involves passage through (and transient residence within) the perinuclear population of recycling endosomes (Mellman, I., and D. Sheff, manuscript in preparation). The data are consistent with an inhibition of the short circuit pathway in ldlF cells lacking ε-COP. Conceivably, a greater fraction of Tfn in ldlF cells at the restrictive temperature passes through the perinuclear recycling endosome population (Fig. 11).
Figure 11.

Potential sites for COP-I affected steps in endosomal transport. Arrows indicate pathways of endosomal traffic. Double lines indicate pathways that may be blocked when COP-I is inactivated. Recycling of receptors may occur through both a rapid return pathway from the early endosomes and a slower pathway passing through the PNRC. COP-I may be involved in budding and/or sorting events in the early endosome required for normal trafficking to late endosomes and through the rapid return pathway.
Finally, we observed a defect in transport from early to late endocytic compartments such as late endosomes and lysosomes. It is well established that the lysosomal accumulation of extracellular macromolecules internalized as fluid-phase tracers or receptor-bound ligands depends on the ability of early endosomes to sort recycling receptors from the fluid content of the endosome lumen (Mellman, 1996). To some extent, this is a nonspecific, geometric phenomenon, but signal-mediated targeting events must also play a role particularly in polarized cells. Thus, the inability of HRP and EGF to efficiently reach lysosomes is consistent with an inhibition of early endosomes in ldlF cells to properly sort membrane from content. The fact that anti–lgp-B antibodies failed to reach the peripheral lysosomal structures in ldlF cells at 40°C suggests that the cells may also be defective at targeting even resident membrane components to late endocytic compartments. This would be consistent with previous suggestions that late endosome-like structures cannot be generated from early endosomes in vitro in the absence of β-COP (Aniento et al., 1996).
How might the loss of ε-COP affect recycling, sorting, and transport to lysosomes? It seems unlikely that COPI coats are responsible for each of these events individually. However, given that early endosomal sorting activities must be completed within 1–2 min after internalization (Mellman, 1996), any loss of function affecting any one early endosome function would be expected to have rapid consequences for the activities of compartments both upstream and downstream. A similar situation may prevail in the early secretory pathway, where inhibition of COPI function might block ER to Golgi transport as a rapid consequence of an arrest of retrograde transport back from the cis-Golgi. It should be pointed out, however, that the precise site(s) of action of COPI coats in the secretory pathway remains controversial, with evidence suggesting its involvement not just in retrograde Golgi to ER transport, but also anterograde transport through the Golgi stack itself (Schekman and Mellman, 1997). In this context, it is useful to conceptualize the endocytic pathway as being somewhat similar to the secretory pathway in the organization of transport steps (Fig. 11). The rapid bidirectional traffic of membrane between the ER and the Golgi complex is thought to involve a rapid turn over of an intermediate compartment that may occupy a position analogous to that occupied by early endosomes between the plasma membrane and late endosomes and/or the perinuclear recycling endosomes. As such, any perturbation of early endosome function due to the action of COPI can be seen as causing both proximal and distal effects.
An important unknown remains whether COPI acts directly or indirectly to affect endosome function. Upon shifting to 40°C, the onset kinetics of the endocytosis and recycling defects are very similar to the onset of the secretory defect as well as to the loss of immunodetectable ε-COP. This is consistent with the idea, suggested by the physical association of COPI components with endosomes, that COPI plays a direct role in endosome function. However, it remains possible the effect is indirect, perhaps blocking the delivery of a Golgi-derived component. Such an indirect effect is unlikely to involve the delivery of a newly synthesized component since inhibition of protein synthesis is without any inhibition of endocytosis in most mammalian cell types (Steinman et al., 1983).
Thus far, the participation of COPI in the formation of small diameter coated vesicles, analogous to those believed to function during intercisternal Golgi transport, has not been unambiguously detected in endosomes, although small Tfn-containing vesicles associated with COPI have been detected by immunocytochemistry (Whitney et al., 1995). Entire COPI-coated regions on larger endosomal structures have also been observed. Conceivably, COPI coats act to selectively include cargo as is thought to occur in the secretory pathway, where KKXX motif proteins are thought to be sequestered in COPI-coated vesicles for retrograde transport from the cis-Golgi back to the ER. The KKXX motif appears to interact with the α, β, and ε-COP subunits (Cosson and Letourneur, 1994; Fiedler et al., 1996; Lowe and Kreis, 1996), and indeed there is evidence that KKXX-containing proteins such as the KDEL receptor can act to retrieve ligands internalized from the plasma membrane (Miesenbock and Rothman, 1995). It is also of interest that COPI complexes can interact with a second, more degenerate sequence motif containing an aromatic amino acid (Fiedler et al., 1996). Conceivably, this second motif, which is not restricted to ER proteins, is the motif decoded by endosomal COPI.
It has been suggested that the regulated assembly of COPI coats may act to control vesicular transport in ways that do not always involve the formation of discrete COPI-coated vesicles (Klausner et al., 1992). According to this view, long membrane tubules reminiscent of the tubular extensions characteristic of early endosomes may form when coats are not allowed to bind. In the Golgi complex and in endosomes this is most obvious upon the inhibition of COPI binding by the drug brefeldin A (Hunziker et al., 1991; Lippincott-Schwartz et al., 1991; Wood et al., 1991). At 40°C, ldlF cells do not exhibit a brefeldin-like phenotype, but this is probably because of the fact that the ε-COP– free COPI complex can still bind to membranes, perhaps inhibiting tubule formation in a dominant negative fashion (Gomez, M., and T. Kreis, unpublished data). Were COPI complexes to regulate the formation of endosomal tubules, an ldlF-like phenotype might be easily explained.
Acknowledgments
We would like to thank V. Malhotra (University of California, San Diego, CA), A. Whitney (University of Geneva, Geneva, Switzerland), and the members of the Mellman–Helenius group for many valuable discussions, and J. Gruenberg (University of Geneva) for sharing results with us before publication. M. Veseree (Yale University) provided much appreciated advice and reagents for intracellular pH measurement. Supplies, equipment, and additional advice for intracellular pH measurement and live cell imaging were kindly provided by N. Andrews (Yale University).
Abbreviations used in this paper
- ARF
ADP ribosylation factor
- BFA
brefeldin A
- COP
coatomer proteins
- LDL
low density lipoprotein
- lgp
lysosomal glycoprotein
- PNRC
perinuclear recycling compartment
- SFV
Semliki Forest virus
- Tfn
transferrin
- TfnR
transferrin receptor
- VSV
vesicular stomatitis virus
- LY
Lucifer yellow
Footnotes
This work was made possible by the generosity and support of M. Krieger (MIT, Cambridge, MA). Funding for D. Sheff provided by the Patrick and Catherine Weldon Donaghue Medical Research Foundation.
E. Daro's present address is Immunex Corporation, 51 University St., Seattle, WA 98110.
Address all correspondence to Ira Mellman, Department of Cell Biology, Yale University School of Medicine, 333 Cedar St., P.O. Box 208002, New Haven, CT 06520-8002. Tel.: (203) 785-5058. Fax: (203) 785-7226. E-mail: ira.mellman@yale.edu
References
- Aniento F, Gu F, Parton RG, Gruenberg J. An endosomal β COP is involved in the pH-dependent formation of transport vesicles destined for late endosomes. J Cell Biol. 1996;133:29–41. doi: 10.1083/jcb.133.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apodaca G, Aroeti B, Tang K, Mostov KE. Brefeldin-A inhibits the delivery of the polymeric immunoglobulin receptor to the basolateral surface of MDCK cells. J Biol Chem. 1993;268:20380–20385. [PubMed] [Google Scholar]
- Cosson P, Letourneur F. Coatomer interaction with di-lysine endoplasmic reticulum retention motifs. Science. 1994;263:1629–1632. doi: 10.1126/science.8128252. [DOI] [PubMed] [Google Scholar]
- Daro E, van der Sluijs P, Galli T, Mellman I. Rab4 and cellubrevin define different early endosome populations on the pathway of transferrin receptor recycling. Proc Natl Acad Sci USA. 1996;93:9559–9564. doi: 10.1073/pnas.93.18.9559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dautry-Varsat A, Ciechanover AA, Lodish HF. pH and the recycling of transferrin during receptor mediated endocytosis. Proc Natl Acad Sci USA. 1983;80:2258–2262. doi: 10.1073/pnas.80.8.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG, Finazzi D, Klausner RD. Brefeldin A inhibits Golgi membrane catalyzed exchange of guanine nucleotide bound to ARF. Nature. 1992;360:350–352. doi: 10.1038/360350a0. [DOI] [PubMed] [Google Scholar]
- D'Souza-Schorey C, Li G, Colombo MI, Stahl PD. A regulatory role for Arf6 in receptor mediated endocytosis. Science. 1995;267:1175–1178. doi: 10.1126/science.7855600. [DOI] [PubMed] [Google Scholar]
- Dunn KW, Maxfield FR. Delivery of ligands from sorting endosomes to late endosomes occurs by maturation of sorting endosomes. J Cell Biol. 1992;117:301–310. doi: 10.1083/jcb.117.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler K, Veit M, Stamnes MA, Rothman JE. Bimodal interaction of coatomer with the p24 family of putative cargo receptors. Science. 1996;273:1396–1399. doi: 10.1126/science.273.5280.1396. [DOI] [PubMed] [Google Scholar]
- Gruenberg J, Griffiths G, Howell K. Characterization of the early endosome and putative endocytic carrier vesicles in vivo and with an assay of vesicle fusion in vitro. J Cell Biol. 1989;108:1301–1317. doi: 10.1083/jcb.108.4.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenberg J, Maxfield FR. Membrane transport in the endocytic pathway. Curr Opin Cell Biol. 1995;7:552–563. doi: 10.1016/0955-0674(95)80013-1. [DOI] [PubMed] [Google Scholar]
- Guo Q, Vasile E, Krieger M. Disruptions in Golgi structure and membrane traffic in a conditional lethal mammalian cell mutant are corrected by ε-COP. J Cell Biol. 1994;125:1213–1224. doi: 10.1083/jcb.125.6.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Penman M, Trigatti BL, Krieger M. A single point mutation in ε-COP results in temperature-sensitive, lethal defects in membrane transport in a Chinese hamster ovary cell mutant. J Biol Chem. 1996;271:11191–11196. doi: 10.1074/jbc.271.19.11191. [DOI] [PubMed] [Google Scholar]
- Harter C, Mellman I. Transport of the lysosomal membrane glycoprotein lgp120 (lgp-A) to lysosomes does not require appearance on the plasma membrane. J Cell Biol. 1992;117:311–325. doi: 10.1083/jcb.117.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms JB, Rothman JE. Inhibition by brefeldin A of a Golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nature. 1992;360:352–355. doi: 10.1038/360352a0. [DOI] [PubMed] [Google Scholar]
- Heuser J. Changes in lysosome shape and distribution correlated with changes in cytoplasmic pH. J Cell Biol. 1989;108:855–864. doi: 10.1083/jcb.108.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbie L, Fisher AS, Lee S, Flint A, Krieger M. Isolation of three classes of conditional lethal Chinese hamster ovary cell mutants with temperature-dependent defects in low density lipoprotein receptor stability and intracellular membrane transport. J Biol Chem. 1994;269:20958–20970. [PubMed] [Google Scholar]
- Hopkins CR, Trowbridge IS. Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. J Cell Biol. 1983;97:508–521. doi: 10.1083/jcb.97.2.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunziker W, Whitney JA, Mellman I. Selective inhibition of transcytosis in MDCK cells by Brefeldin A. Cell. 1991;67:617–627. doi: 10.1016/0092-8674(91)90535-7. [DOI] [PubMed] [Google Scholar]
- Kaplan DL, Boron WF. Long-term expression of c-H-ras stimulates Na-H and Na(+)-dependent Cl-HCO3 exchange in NIH-3T3 fibroblasts. J Biol Chem. 1994;269:4116–4124. [PubMed] [Google Scholar]
- Kielian M, Jungerwirth S, Sayad KU, deCandido S. Biosynthesis, maturation, and acid activation of the semliki forest virus fusion protein. J Virol. 1990;64:4614–4624. doi: 10.1128/jvi.64.10.4614-4624.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausner RD, Donaldson J, Lippincott-Schwartz J. Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis TE. Microinjected antibodies against the cytoplasmic domain of vesicular stomatitis virus glycoprotein block its transport to the cell surface. EMBO (Eur Mol Biol Organ) J. 1986;5:931–941. doi: 10.1002/j.1460-2075.1986.tb04306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemichez E, Bomsel M, Devilliers G, van der Spek J, Murphy JR, Lukianov EV, Olsnes S, Boquet P. Membrane translocation of diphtheria toxin fragment A exploits early to late endosome trafficking machinery. Mol Microbiol. 1997;23:445–457. doi: 10.1111/j.1365-2958.1997.tb02669.x. [DOI] [PubMed] [Google Scholar]
- Letourneur F, Gaynor EC, Hennecke S, Demoliiere C, Duden R, Emr SD, Riezman H, Cosson P. Coatomer is essential for retrieval of dilysine tagged proteins to the endoplasmic reticulum. Cell. 1994;74:1199–1208. doi: 10.1016/0092-8674(94)90011-6. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Yuan L, Tipper C, Amherdt M, Orci L, Klausner RD. Brefeldin A's effect on endosomes, lysosomes, and the TGN suggest a general mechanism for regulating organelle structure and membrane traffic. Cell. 1991;67:601–616. doi: 10.1016/0092-8674(91)90534-6. [DOI] [PubMed] [Google Scholar]
- Lowe M, Kreis TE. In vivo assembly of coatomer, the COP-I coat precursor. J Biol Chem. 1996;271:30725–30730. doi: 10.1074/jbc.271.48.30725. [DOI] [PubMed] [Google Scholar]
- Matlin KS, Reggio H, Helenius A, Simons K. Pathway of vesicular stomatitis virus entry leading to infection. J Mol Biol. 1982;156:609–631. doi: 10.1016/0022-2836(82)90269-8. [DOI] [PubMed] [Google Scholar]
- Matter K, Whitney A, Yamamoto EM, Mellman I. Common signals control low density lipoprotein receptor sorting in endosomes and the Golgi complex of MDCK cells. Cell. 1993;74:1053–1063. doi: 10.1016/0092-8674(93)90727-8. [DOI] [PubMed] [Google Scholar]
- McGraw TE, Dunn KW, Maxfield FR. Isolation of a temperature-sensitive variant Chinese hamster ovary cell line with a morphologically altered endocytic recycling compartment. J Cell Physiol. 1993;155:579–594. doi: 10.1002/jcp.1041550316. [DOI] [PubMed] [Google Scholar]
- Mellman I. Endocytosis and molecular sorting. Annu Rev Cell Dev Biol. 1996;12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- Miesenbock G, Rothman JE. The capacity to retrieve escaped ER proteins extends to the trans-most cisterna of the Golgi stack. J Cell Biol. 1995;129:309–319. doi: 10.1083/jcb.129.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L, Glick BS, Rothman JE. A new type of coated vesicular carrier that appears not to contain clathrin: its possible role in protein transport within the Golgi stack. Cell. 1986;46:171–184. doi: 10.1016/0092-8674(86)90734-8. [DOI] [PubMed] [Google Scholar]
- Orci L, Palmer DJ, Amherdt M, Rothman JE. Coated vesicle assembly in the Golgi requires only coatomer and ARF proteins from the cytosol. Nature. 1993;364:732–734. doi: 10.1038/364732a0. [DOI] [PubMed] [Google Scholar]
- Parton RG, Dotti CG, Bacallao R, Kurtz I, Simons K, Prydz K. pH-induced microtubule-dependent redistribution of late endosomes in neuronal and epithelial cells. J Cell Biol. 1991;113:261–274. doi: 10.1083/jcb.113.2.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse BMF, Robinson MS. Clathrin, adaptors, and sorting. Annu Rev Cell Biol. 1990;6:151–171. doi: 10.1146/annurev.cb.06.110190.001055. [DOI] [PubMed] [Google Scholar]
- Pepperkok R, Scheel J, Horstmann H, Hauri HP, Griffiths G, Kreis TE. β-COP is essential for biosynthetic membrane transport from the endoplasmic reticulum to the Golgi complex in vivo. Cell. 1993;74:71–81. doi: 10.1016/0092-8674(93)90295-2. [DOI] [PubMed] [Google Scholar]
- Peters PJ, Hsu VW, Ooi CW, Finazzi D, Teal SB, Oorschot V, Donaldson JG, Klausner RD. Overexpression of wild-type and mutant ARF1 and ARF6: distinct perturbations of nonoverlapping membrane compartments. J Cell Biol. 1995;128:1003–1017. doi: 10.1083/jcb.128.6.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podbilewicz B, Mellman I. ATP and cytosol requirements for transferrin recycling in intact and disrupted MDCK cells. EMBO (Eur Mol Biol Organ) J. 1990;9:3477–3487. doi: 10.1002/j.1460-2075.1990.tb07556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schekman R, Orci L. Coat proteins and vesicle budding. Science. 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- Schekman R, Mellman I. Does COPI go both ways? . Cell. 1997;90:197–200. doi: 10.1016/s0092-8674(00)80326-8. [DOI] [PubMed] [Google Scholar]
- Schmid SL, Fuchs R, Male P, Mellman I. Two distinct subpopulations of endosomes involved in membrane recycling and transport to lysosomes. Cell. 1988;52:73–83. doi: 10.1016/0092-8674(88)90532-6. [DOI] [PubMed] [Google Scholar]
- Schmid S, Fuchs R, Kielian M, Helenius A, Mellman I. Acidification of endosome subpopulations in wild type chinese hamster ovary cells and temperature sensitive acidification mutants. J Cell Biol. 1989;10:1291–1300. doi: 10.1083/jcb.108.4.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM, Mellman I, Muller WA, Cohn ZA. Endocytosis and the recycling of plasma membrane. J Cell Biol. 1983;96:1–27. doi: 10.1083/jcb.96.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoorvogel W, Oorschot V, Geuze HJ. A novel class of clathrin-coated vesicles budding from endosomes. J Cell Biol. 1996;132:21–33. doi: 10.1083/jcb.132.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei K, Mundigl O, Daniell L, De Camilli P. The synaptic vesicle cycle: a single vesicle budding step involving clathrin and dynamin. J Cell Biol. 1996;133:1237–1250. doi: 10.1083/jcb.133.6.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa PA, Malhotra V. Coatomers and SNAREs in promoting membrane traffic. Cell. 1993;75:593–596. doi: 10.1016/0092-8674(93)90477-8. [DOI] [PubMed] [Google Scholar]
- Tisdale EJ, Plutner H, Matteson J, Balch WE. p53/58 binds COPI and is required for selective transport through the early secretory pathway. J Cell Biol. 1997;137:581–593. doi: 10.1083/jcb.137.3.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich O, Reinsch S, Urbe S, Zerial M, Parton RG. Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol. 1996;135:913–924. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sluijs P, Hull M, Zahraoui A, Tavitian A, Goud B, Mellman I. The small GTP binding protein rab4 is associated with early endosomes. Proc Natl Acad Sci USA. 1991;88:6313–6317. doi: 10.1073/pnas.88.14.6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sluijs P, Hull M, Webster P, Goud B, Mellman I. The small GTP binding protein rab4 controls an early sorting event on the endocytic pathway. Cell. 1992;70:729–740. doi: 10.1016/0092-8674(92)90307-x. [DOI] [PubMed] [Google Scholar]
- Waters MG, Serafini T, Rothman JE. Coatomer: a cytosolic protein complex containing subunits of non-clathrin-coated Golgi transport vesicles. Nature. 1991;349:248–251. doi: 10.1038/349248a0. [DOI] [PubMed] [Google Scholar]
- White J, Kartenbeck J, Helenius A. Fusion of Semliki forest virus with the plasma membrane can be induced by low pH. J Cell Biol. 1980;87:264–272. doi: 10.1083/jcb.87.1.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney JA, Gomez M, Sheff D, Kreis TE, Mellman I. Cytoplasmic coat proteins involved in endosome function. Cell. 1995;83:703–713. doi: 10.1016/0092-8674(95)90183-3. [DOI] [PubMed] [Google Scholar]
- Wood SA, Park JE, Brown WJ. Brefeldin A causes a microtubule mediated fusion of the Trans Golgi Network and early endosomes. Cell. 1991;67:591–600. doi: 10.1016/0092-8674(91)90533-5. [DOI] [PubMed] [Google Scholar]
- Yamashiro DJ, Tycko B, Fluss SR, Maxfield FR. Segregation of transferrin to a mildly acidic (pH 6.5) para-Golgi compartment in the recycling pathway. Cell. 1984;37:789–800. doi: 10.1016/0092-8674(84)90414-8. [DOI] [PubMed] [Google Scholar]
- Yamashiro D, Maxfield FR. Kinetics of endosome acidification in mutant and wild type chinese hamster ovary cells. J Cell Biol. 1987;105:2713–2721. doi: 10.1083/jcb.105.6.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Rosenwald AG, Willingham MC, Skuntz S, Clark C, Kahn RA. Expression of a dominant allele of human ARF1 inhibits membrane traffic in vivo. J Cell Biol. 1994;124:289–300. doi: 10.1083/jcb.124.3.289. [DOI] [PMC free article] [PubMed] [Google Scholar]