

Abstract
Cells of the macrophage lineage express a peculiar surface receptor for extracellular ATP, designated P2Z/P2X7 purinergic receptor, that induces pore formation and collapse of the plasma membrane potential. Although the function of the P2Z receptor is largely unknown, accumulating evidence implicates its role in cell signaling and immune reactions. Here, we investigated the effect of P2Z receptor ligation on the activation of NF-κB, a transcription factor controlling cytokine expression and apoptosis. Exposure of microglial cells to ATP but not other nucleotides resulted in potent NF-κB activation. This effect was specifically mediated by the P2Z receptor, because selective receptor antagonists prevented NF-κB activation. NF-κB activation required reactive oxygen intermediates and proteases of the caspase family, because it was abolished by antioxidants and specific protease inhibitors. The subunit composition of the ATP-induced NF- κB–DNA complex was rather unusual. Whereas exposure to LPS-induced prototypical NF-κB p50 homo- and p65 (RelA)/p50 heterodimers, ATP stimulation resulted in the sole appearance of a p65 homodimer. This is the first demonstration that a certain stimulus activates a particular NF-κB subunit. Because different NF-κB complexes exhibit distinct transcriptional and DNA-binding activities, ATP may control the expression of a subset of NF-κB target genes distinct from those activated by classical proinflammatory mediators.
There is increasing evidence that extracellular ATP serves as a mediator of cell-to-cell communication by triggering a variety of biological responses, particularly in the immune system (13, 14). Large amounts of ATP can be rapidly released from different sources, such as activated platelets, endothelial cells, nerve terminals, antigen-stimulated T cells, and other cell types following hypoxia, stress, and tissue damage. The biological activities of extracellular ATP are various and include mitogenic stimulation, excitatory transmitter function, gene expression, and induction of cell death. These effects are not the result of nonspecific membrane alterations, but rather, are mediated through the activation of specific surface molecules called purinergic receptors (13, 14). At least two subtypes of receptors for extracellular ATP are currently known: the G-protein–coupled P2Y receptors and the ATP-gated cation channels classified as P2X receptors (20). The purinergic receptor families have distinct agonist, inhibitor, and expression profiles and, in addition, require different ATP concentrations to trigger their biological responses.
Very recently the molecular structure of the P2Z receptor, also called P2X7, has been elucidated (36, 46). The P2Z receptor contains two transmembrane domains and a large extracellular loop, structural features that are characteristic of members of the P2X family. Unlike other P2X receptors, the P2Z receptor has an unusually long COOH-terminal domain that does not contain any known signaling motifs. P2Z receptor expression appears to be rather restricted to cells of the macrophage lineage, such as dendritic cells, mature macrophages, and microglial cells. Intriguingly, triggering of the P2Z receptor elicits two types of cellular responses (46). Like with all P2X receptors, ligation of ATP results in a transient membrane current through divalent cation channels. A second and unique response is the sustained membrane depolarization and increase in cytosolic-free calcium by the opening of a nonselective transmembrane pore which is permeable to hydrophilic molecules of up to 900 D. This increase in membrane permeability finally results in the induction of cell death. In many cases, ATP-induced cytotoxicity is mediated by classical alterations of apoptosis, including membrane blebbing, nuclear condensation, and DNA fragmentation (9, 18, 58). In addition, in lipopolysaccharide (LPS)1-primed macrophages and microglial cells, it has been observed that stimulation with ATP induces the rapid release of the proinflammatory cytokine interleukin (IL)-1β (19, 22). This event is presumably mediated by the activation of IL-1β-converting enzyme (ICE) which cleaves the IL-1β precursor to the active cytokine (49). ICE and related proteases, now classified as caspases, have been recently identified as the critical executioners of various forms of apoptosis (10, 27, 34, 43), which may explain that high levels of IL-1β secretion are often found in cells undergoing programmed cell death (25).
Although a sustained increase in cytosolic-free calcium occurs upon exposure of cells to extracellular ATP, the signaling events mediating P2Z receptor action are rather unknown. No studies have yet addressed transcriptional processes that might be involved in apoptosis or inflammatory activities of the P2Z receptor. An important regulator implicated in the control of apoptosis and expression of proinflammatory genes is the transcriptional activator NF-κB (1, 2, 4). The factor is ubiquitously found as an inactive complex in the cytoplasm bound to its inhibitory subunit IκB. In many cases, NF-κB is composed of a p50 and p65 (also called RelA) heterodimer but also other NF-κB subunits such as p52, c-Rel, and RelB may participate in dimer formation. Whereas p50 and p52 are solely required for DNA binding, p65, c-Rel, and RelB have, in addition, transactivating activity. It has been shown in some cases that combinatorial interactions between the different NF-κB subunits give rise to dimers with distinct DNA sequence and transcriptional specificity. A broad panel of different stimuli can activate NF-κB: for instance, LPS, viruses, inflammatory cytokines, UV light, and phorbol esters. Several lines of evidence indicated that reactive oxygen intermediates (ROIs), in particular hydrogen peroxide, serve as second messengers in the activation pathway of NF-κB. Treatment of cells with many structurally unrelated antioxidants or the overexpression of antioxidative enzymes, such as catalase, thioredoxin, or glutathione peroxidase, inhibit NF-κB activation (40–42). In some cell types, addition of hydrogen peroxide to the culture medium activated the transcription factor (42).
A key event in the activation of NF-κB is the inducible phosphorylation of the inhibitory subunit IκB-α on serines 32 and 36. This covalent modification by the recently identified IκB kinase induces the conjunction of IκB to ubiquitin, followed by a rapid degradation of the inhibitor by the proteasome (12, 37, 50). This reaction leaves the active form of NF-κB which is translocated to the nucleus where it binds to regulatory sequences of target genes. Among the numerous targets of NF-κB are those encoding inflammatory and chemotactic cytokines, cell adhesion molecules, major histocompatibility complex class I molecules, and cytokine receptors (1).
In the present study, we investigated the effects of extracellular ATP on NF-κB activation in microglial cells. Microglial cells are important cells of inflammatory processes within the central nervous system (21), and their expression of purinergic receptors has been previously analyzed in detail (17). We show that ATP but not other nucleotides lead to the potent and selective activation of NF-κB in microglial cells through a P2Z receptor-mediated pathway. The stimulatory effect of ATP required intracellular ROI formation and induction of ICE-like proteolytic activity. The subunit composition of the ATP-induced NF-κB–DNA complex was rather unique. In contrast to other classical proinflammatory inducers of NF-κB activation, such as LPS, which trigger formation of a p65/p50 NF-κB heterodimer, ATP-induced NF-κB activation resulted in the selective DNA binding of an unusual p65 homodimer. Our findings suggest that ATP and proinflammatory mediators may have distinct gene regulatory effects on NF-κB target genes which may be involved in cell death and inflammatory reactions within the nervous system.
Materials and Methods
Cell Culture and Reagents
The mouse microglial cell lines N9 and N13 have been described (38) and were kindly provided by Dr. Ricciardi-Castagnoli (University of Milan, Milan, Italy). Cells were grown in RPMI-1640 medium supplemented with 10% FCS and 2 mM glutamine, and were routinely passaged by trypsinization. ATP, ATPγS, and other nucleotides were purchased from Boehringer Mannheim (Mannheim, Germany). 2′,3′-(4-benzoyl)-benzoyl-ATP, apyrase, polymyxin B, pyrrolidine dithiocarbamate (PDTC), and LPS were obtained from Sigma Chemical Co. (Deisenhofen, Germany). The ICE tetrapeptide inhibitor YVAD-CHO was purchased from Bachem (Heidelberg, Germany), and proteasome inhibitor lactacystin from Calbiochem Corp. (Bad Soden, Germany). Periodate-oxidized ATP was a kind gift of Dr. Hanau and Dr. Di Virgilio (University of Ferrara, Ferrara, Italy) and synthesized as described (33). Anti–mouse IL-1β was purchased from Endogen (Woburn, MA), rabbit anti–ICE antibodies from Santa Cruz (Heidelberg, Germany), and recombinant IL-1 receptor antagonist (IL-1RA) from R&D Systems Inc. (Wiesbaden, Germany). A rabbit anti– mouse tumor necrosis factor (TNF) antiserum was kindly provided by Dr. C. Libert (University of Ghent, Ghent, Belgium).
Cell Extracts
Cells were plated at 106/well in six-well plates and allowed to adhere overnight. Unless otherwise indicated, cells were pretreated for 30 min with the inhibitors, followed by the addition of 3 mM ATP for an additional 3 h. Total cell extracts were prepared after cell stimulation and used for electrophoretic mobility shift assays (EMSAs) and Western blot analysis. Cell extracts were prepared by resuspending PBS-washed cell pellets in a high-salt buffer containing 20 mM Hepes, pH 7.9, 350 mM NaCl, 20% glycerol, 1% NP-40, 1 mM MgCl2, 0.5 mM EDTA, 0.1 mM EGTA, 0.5 mM DTT, 2 mM PMSF, and 2 μg/ml aprotinin. Extracts were incubated on ice for 10 min and then cleared by centrifugation.
EMSAs
EMSAs were carried out essentially as described (24). Equal amounts of the extracts (∼10-μg crude protein; determined by the Bio-Rad Laboratories assay kit; Munich, Germany) were incubated with the 32P-labeled NF-κB– or AP-1–specific oligonucleotides. Binding reactions were performed in a 20-μl vol containing 4 μl of extract, 4 μl 5× binding buffer (20 mM Hepes, pH 7.5, 50 mM KCl, 1 mM DTT, 2.5 mM MgCl2, 20% Ficoll), 2 μg poly(dI-dC) as nonspecific competitor DNA, 2 μg BSA, and 20,000– 40,000 cpm (Cerenkov) of the labeled oligonucleotide. After 15 min binding reaction at room temperature, samples were loaded on a 4% non- denaturing polyacrylamide gel and run in 0.5 × TBE buffer (89 mM Tris, 89 mM boric acid, 1 mM EDTA), pH 8.3. Gels were dried under a gel dryer and exposed to an x-ray film. The oligonucleotides with a high-affinity NF-κB binding and AP-1 DNA-binding motif (Promega, Heidelberg, Germany) were labeled using γ[32P]ATP (3,000 Ci/mmol; Amersham Buchler GmbH, Braunschweig, Germany) and T4 polynucleotide kinase (Boehringer Mannheim) followed by P-10 gel filtration (Bio-Rad Laboratories) to remove nonincorporated radioactivity. To characterize the NF-κB–DNA complex, supershift analyses were performed using bacterially expressed IκB-α (5) and antibodies directed against NF-κB subunits (Santa Cruz).
Transfections and Luciferase Assays
To measure the transactivating activity of NF-κB, N9 cells were transfected with a luciferase reporter gene construct controlled by six κB-binding sites (24). Briefly, cells were washed in TBS, resuspended at 108 cells/ 0.4 ml TBS and transfected with 15 μg of expression plasmids using a Bio-Rad electroporator (1,015 μF, 165 V). After electroporation, cells were seeded in a 6-well plate at 106 cells/well. 24 h after transfection, cells were stimulated with the indicated agents for 4 h. Cells were then harvested for luciferase assays, lysed in 25 mM glycylglycine, pH 7.8, 1% Triton X-100, 15 mM MgSO4, 4 mM EGTA, 1 mM DTT, and centrifuged at 13,000 g at 4°C for 5 min. 50 μl of the supernatant were assayed in 100 μl assay buffer (25 mM glycylglycine, 15 mM MgSO4, 4 mM EGTA, 15 mM KPi, pH 7.8, 1 mM DTT, 1 mM ATP) using a ML2200 luminometer (Dynatech Corp., Denkendorf, Germany). After injection of 100 μl luciferin (0.3 mg/ml) light emission was recorded as relative light units.
Measurement of Cell Viability
Cell viability was determined by measuring the release of lactate dehydrogenase (LDH) using a commercial cytotoxicity detection kit (Boehringer Mannheim). Briefly, cells were incubated for the indicated times in the presence of 3 mM ATP, and the fraction of LDH activity released from dead cells was measured spectrophotometrically in the supernatants. To obtain total LDH activity, cells were lysed with 1% Triton X-100. The percentage of LDH release represents the fraction of LDH activity found in the supernatants with respect to the overall enzyme activity.
Western Blotting
Proteolytic cleavage and activation of ICE was detected by Western blotting. After treatment with 3 mM ATP for the indicated times, cell lysates were prepared. Cellular proteins from 2.5 × 106 cells were loaded in each lane, separated on a 15% SDS-PAGE under reducing conditions, and transferred to polyvinylidene fluoride membranes (Amersham Buchler GmbH). The membranes were blocked for 1 h with 5% nonfat dry milk powder in PBS and incubated for 1 h with an ICE-specific rabbit antiserum. Membranes were washed three times with TBS/0.05% Tween-20 and incubated with peroxidase-conjugated affinity-purified rabbit anti–mouse IgG for 1 h. After extensive washing, the reaction was developed by enhanced chemoluminescent staining using enhanced chemoluminescent reagents (Amersham Buchler GmbH).
Results
Extracellular ATP Induces NF-κB Activation in Microglial Cells
The effect of extracellular ATP on the activation of NF-κB was investigated in the mouse microglial cell lines N9 and N13. The cells have been previously characterized in detail and found to express functional P2Y and P2Z purinergic receptors (17). Cells were treated for 3 h with 3 mM ATP and, subsequently, total cell extracts were prepared and analyzed for DNA-binding activity to a 32P-labeled κB-specific oligonucleotide. As a positive control, cells were stimulated with LPS, a potent NF-κB inducer in microglial cells (5). As shown in Fig. 1 A, in both N9 and N13 cells, treatment with ATP caused the appearance of a novel NF-κB protein–DNA complex that comigrated with the upper LPS-inducible complex. A faster migrating, nonspecific DNA complex was not significantly affected by the treatments and provided an internal control for the amount and integrity of the cell extracts.
Figure 1.
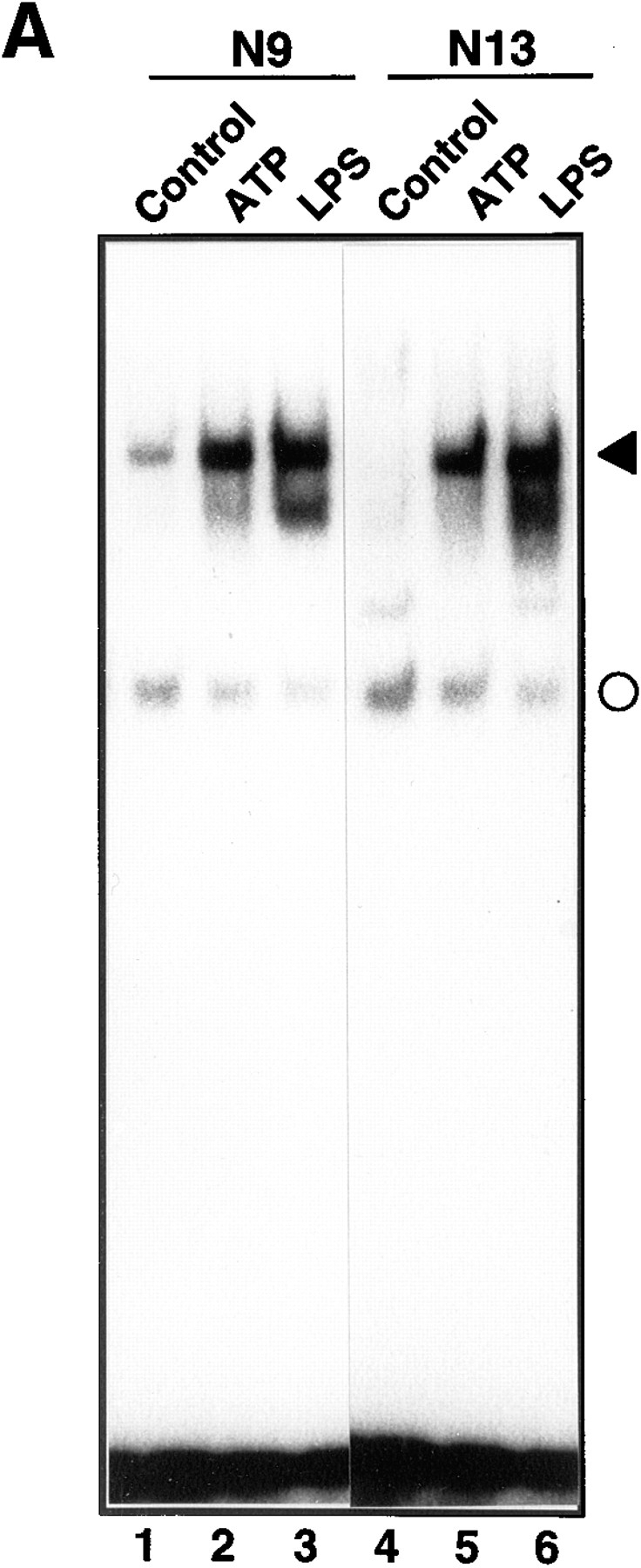

Induction of NF-κB activation by ATP. (A) The effect of ATP on NF-κB activation in N9 and N13 microglial cells. N9 (lanes 1–3) or N13 cells (lanes 4–6) were either left untreated or treated with 3 mM ATP or 100 ng/ml LPS. After 3 h total cell extracts were prepared and analyzed by EMSA using a 32P-labeled oligonucleotide containing a high affinity κB-binding motif. The NF-κB–DNA complex is indicated by a filled arrowhead. A faster migrating non-specific complex is marked by a circle. (B) Dose dependency of NF-κB activation in response to ATP or LPS. N9 cells were treated for 3 h with the indicated concentrations of ATP or LPS and analyzed for NF-κB activation. (C) The effect of ATP and LPS on the DNA-binding activity of AP-1. The same cellular extracts and protein amounts as in Fig. 1 A were analyzed with an AP-1 specific oligonucleotide in an EMSA.
The dose-dependence of ATP-induced NF-κB activation was studied by stimulating N9 cells for 3 h with different concentrations of ATP. As shown in Fig. 1 B, maximal NF-κB activation was obtained with 3 mM ATP, whereas a higher concentration (5 mM) was less effective. In contrast to ATP, NF-κB activation in response to LPS was already strongly induced with 10 ng/ml LPS (Fig. 1 B). We further investigated the effect of ATP on the DNA-binding activity of AP-1, a transcription factor implicated in various proliferation and differentiation processes. In the same cell extracts used in Fig. 1 A, ATP did not increase but considerably inhibited the basal DNA-binding activity of AP-1 (Fig. 1 C). These data indicate that ATP specifically activated NF-κB and show, in line with previous reports (40), that activation of NF-κB and AP-1 are regulated in opposite ways.
Time Course and NF-κB Subunit Composition in Response to ATP
In the next series of experiments, the time course of NF-κB activation was studied by stimulating cells with 3 mM ATP or 100 ng/ml LPS for different time periods. The kinetics by which ATP-induced NF-κB activation was rather delayed and significant DNA-binding activity was not observed before 3 h after exposure to ATP (Fig. 2 A). In contrast, LPS induced NF-κB activation very rapidly and strong NF-κB activity was obtained after 15 min of cell stimulation. To investigate whether the presence of ATP was required for the whole incubation of 3 h, a pulse-chase experiment was performed. Cells were treated for different time points with ATP and cultured subsequently in the absence of ATP for a total incubation time of 3 h. Fig. 2 B demonstrates that stimulation of cells with ATP for less than 60 min was sufficient to induce NF-κB activation.
Figure 2.

Time dependence of NF-κB activation in response to ATP and LPS. (A) Kinetics of NF-κB activation: total extracts of cells treated for the indicated periods of time with 3 mM ATP or 100 ng/ml LPS were prepared and analyzed by EMSA. Only a section of the autoradiogram is shown. The filled arrowhead indicates the position of the inducible NF-κB–DNA complex; the open circle denotes a non-specific complex. (B) Pulse-chase experiment: N9 cells were stimulated for different times (0.5–3 h) with 3 mM ATP and further incubated for the indicated time periods in culture medium without ATP. After a total incubation time of 3 h, cells were harvested and analyzed for NF-κB DNA-binding activity.
In the previous experiments, it was observed that after exposure of cells to ATP, a single NF-κB–DNA complex was induced, whereas LPS consistently triggered the formation of two inducible protein–DNA complexes. Because NF-κB complexes may constitute a variety of different homo- and heterodimers, we analyzed the subunit composition of the LPS- and ATP-induced DNA complexes by supershift assays. Rabbit sera against the NF-κB subunits p50 and p65, c-Rel and RelB, were added to an extract of cells stimulated with either LPS or ATP (Fig. 3). In extracts from LPS-treated cells, the lower NF-κB-specific–DNA complex was retarded completely by anti-p50, whereas the appearance of the upper DNA complex was reduced (Fig. 3 A, lane 3). Formation of the upper DNA complex was also strongly reduced by anti-p65 (Fig. 3 A, lane 4), whereas anti–c-Rel or anti-RelB had no effect. This indicates that in LPS-stimulated cells the slowly migrating NF-κB complex consisted of a classical p65/p50 heterodimer and the lower complex of a p50 homodimer. In contrast, in extracts from ATP-stimulated cells, neither anti-p50 nor antibodies against c-Rel or RelB reacted with the inducible protein–DNA complex. Only anti-p65 was able to abolish the formation of the NF-κB–DNA complex (Fig. 3 B, lane 4), indicating that it predominantly consisted of a p65 homodimer. In addition, recombinant IκB-α, which removes NF-κB from bound DNA (57), inhibited formation of the various protein–DNA complexes (Fig. 3, lanes 7), thus confirming their NF-κB specificity. Together, the distinct immunoreactivities indicated that ATP and LPS trigger the activation of different NF-κB subunit combinations.
Figure 3.

Subunit composition of the induced NF-κB–DNA complexes after treatment with LPS (A) or ATP (B). Total cell extracts of non-stimulated (lanes 1) or stimulated (lanes 2) N9 cells were prepared and either left untreated (lanes 1 and 2) or incubated with specific antibodies against the NF-κB subunits p50 (lanes 3), p65 (lanes 4), c-Rel (lanes 5) and RelB (lanes 6). Lanes 7 denote an extract of ATP-treated cells incubated with recombinant IκB-α that releases NF-κB–specific complexes from bound DNA. Only sections of the autoradiograms are shown.
ATP-induced NF-κB Activation Involves a P2Z Receptor-mediated Pathway
We further investigated the specificity of the ATP effect and the involved subtype of purinergic receptors in experiments using pharmacological ATP analogues. Strong NF-κB activation was exclusively triggered upon incubation with ATP but not other nucleotides such as ADP and CTP (Fig. 4 A). UTP, which activates the P2Y receptor, had also no effects, whereas GTP only weakly induced NF-κB activation.
Figure 4.

Specificity of ATP-induced NF-κB activation. (A) The effect of different nucleotides on NF-κB activation. N9 cells were treated with 3 mM ATP, ADP, UTP, CTP, or GTP. After 3 h, cell extracts were prepared and analyzed by EMSA. (B) Induction of NF-κB activation by ATP or its pharmacological analogues ATPγS (3 mM) and benzoylbenzoyl-ATP (BzATP, 1 mM). (C) Inhibition of ATP-induced NF-κB activation by oxidized ATP (oATP). Cells were either left untreated or stimulated with ATP following a 2 h pretreatment with the indicated concentrations of oxidized ATP. (D) Lack of ATP-induced NF-κB activation in the P2Z receptor-deficient N9 derivative clones N9R14 and N9R17. Parental N9 cells and the ATP-resistant N9R14 and N9R17 were either left untreated or incubated for 3 h in the presence of 3 mM ATP or LPS (100 ng/ml).
ATPγS, a nonhydrolyzable ATP derivative, also activated NF-κB (Fig. 4 B), demonstrating that degradation of ATP was not required for this process. In addition, benzoylbenzoyl ATP, which is a more potent agonist of the P2Z receptor, was able to activate NF-κB even at concentrations of 1 mM (Fig. 4 B).
P2Z receptor effects have been reported to be specifically antagonized by oxidized ATP which covalently binds to and inhibits the P2Z receptor (33). Indeed, preincubation of N9 cells with oxidized ATP completely prevented ATP-induced NF-κB activation (Fig. 4 C), indicating that the effects of ATP were mediated by the P2Z but not by other subtypes of purinergic receptors. This assumption was supported in experiments with N9 derivative clones which lack the P2Z receptor but still express P2Y purinergic receptors (17, 18). As shown in Fig. 4 D, NF-κB DNA binding was not inducible by ATP in the clones N9R14 and N9R17, whereas LPS was still able to activate the transcription factor.
ATP-induced NF-κB Activation Requires ROI Production and Proteasome Activity
Because microglial cells are highly sensitive to LPS, we further evaluated the effect of the LPS inhibitor polymyxin B. Preincubation of cells with polymyxin B did not interfere with ATP-induced NF-κB, suggesting that the observed effects were specific and not mediated by a contamination with LPS (Fig. 5 A).
Figure 5.

The effect of the LPS inhibitor polymyxin B (A), the antioxidant PDTC (B), and the proteasome inhibitor lactacystin (C) on NF-κB activation in response to ATP. N9 cells were pretreated for 30 min with polymyxin B (PMB, 10 μg/ml; A) or 50 μM PDTC (B), or for 90 min with the indicated concentrations of lactacystin (LC; C), followed by the activation with 3 mM ATP for 3 h. Total cell extracts were then prepared and analyzed by EMSA for NF-κB DNA-binding activity. Only sections of the autoradiograms are shown.
Antioxidants such as dithiocarbamates have been described as potent inhibitors of NF-κB activation (40, 42). We therefore assessed whether PDTC, which acts as a radical scavenger, was capable of blocking ATP-induced NF-κB–DNA activation. As shown in Fig. 5 B, PDTC led to a strong inhibition of NF-κB DNA binding, indicating that reactive oxygen intermediates were involved in the mechanism of ATP-induced NF-κB activation.
A key event in most pathways leading to NF-κB activation is the proteolytic degradation of its inhibitor IκB at the proteasome (50). We investigated the involvement of the proteasome in ATP-induced NF-κB activation using lactacystin, a microbial metabolite that was recently found to be a potent and highly specific inhibitor of the 20S proteasome (16). For unknown reasons, incubation of N9 cells with 2.5 μM lactacystin alone activated the transcription factor (Fig. 5 C). Pretreatment of cells with lactacystin at the same concentrations, however, strongly suppressed ATP-induced NF-κB DNA-binding. These data indicate that proteasome activity was required for ATP to induce NF-κB activation.
ATP-induced NF-κB Activation Involves Induction of ICE Proteolytic Activity
Stimulation of LPS-primed microglial cells with ATP has been reported to result in the rapid release of IL-1β (17). This event is presumably mediated by the activation of ICE or related proteases that process the IL-1β precursor to the active cytokine (49). ICE-like proteases, now called caspases, are also centrally involved in the execution of apoptosis and thus may be implicated in ATP-induced cytotoxicity. We therefore investigated the relationship between the induction of ICE proteolytic activity and NF-κB activation. Preincubation of N9 cells with the tetrapeptide YVAD-CHO, a specific inhibitor of ICE and related caspases (32), suppressed ATP-induced NF-κB activation in a dose- dependent manner (Fig. 6 A). LPS-induced NF-κB activation was also slightly reduced (data not shown). We further observed that DEVD-CHO, another inhibitor of caspases, suppressed ATP-induced NF-κB activation, whereas tosyl-Arg-methlylester, an inhibitor of trypsin-like proteases, was ineffective (data not shown).
Figure 6.

Involvement of ICE proteases in ATP-induced NF-κB activation. (A) The effect of the ICE tetrapeptide inhibitor YVAD-CHO (YVAD) on NF-κB activation. Cells were pretreated for 30 min with the indicated concentrations of YVAD and then stimulated with 3 mM ATP for 3 h. (B) Proteolytic processing of ICE by ATP treatment. N9 cells were incubated for the indicated times with 3 mM ATP. Total cell extracts were then prepared and analyzed by SDS-PAGE and immunoblotting. Proteolytic cleavage of the ICE precursor to the active protease was monitored using an anti-ICE specific antiserum that detects pro-ICE and the p20 proteolytic cleavage product. (C) Effect of ATP on cell viability. N9 cells were treated with 3 mM ATP. After the indicated time points, LDH released from dead cells was measured in the supernatants and calculated as the percentage of total cellular LDH activity from an untreated sample.
Caspases are synthesized as proenzymes that are cleaved by an activation step to yield an active tetrameric complex. Thus, the 45-kD precursor of ICE is converted to an active protease composed of two heterodimeric 10 and 20-kD subunits (35, 36). To investigate ICE activation, we monitored the processing of the proenzyme by Western blot analysis. N9 cells were treated with ATP, and after different times cellular lysates were fractionated by SDS-PAGE. As shown in Fig. 6 B, ATP treatment resulted in the appearance of the proteolytically cleaved p20 subunit within 3 h of incubation. The processing of ICE coincided with the time course of NF-κB activation as shown in Fig. 2. Activation of both ICE and NF-κB, however, preceded the onset of ATP-induced cell death, as no significant LDH release into the culture supernatants occurred within the first 3 h after ATP stimulation (Fig. 6 C).
Secretion of Soluble Proteins Is Not Required for ATP-induced NF-κB Activation
Because ATP-induced NF-κB activation, in contrast to many proinflammatory stimuli, was rather delayed, it was analyzed whether this effect was mediated by a para- or autocrine loop involving ATP-induced IL-1β secretion. Incubation of N9 cells with even high concentrations of a neutralizing IL-1RA (Fig. 7 A) or anti–mouse IL-1β (data not shown) was ineffective in preventing NF-κB activation after ATP treatment. Because TNF is a potent NF-κB stimulus that can be released by microglial cells, we further analyzed the involvement of TNF in the ATP effects. As shown in Fig. 7 B, also neutralizing anti-TNF did not interfere with NF-κB DNA-binding activity. Furthermore, we found that IL-1β itself was unable to induce NF-κB activation in N9 cells (data not shown), consistent with a previous report showing that NF-κB is activated in primary microglial cultures following treatment with LPS but not IL-1β, TNF, or IL-6 (5). In addition, inhibition of de novo protein synthesis by cycloheximide did not prevent ATP-induced NF-κB activation (data not shown). Instead, there was a superinduction of NF-κB by ATP following pretreatment with cycloheximide, and as previously reported (11), inhibition of protein synthesis itself activated the transcription factor. To further exclude the possibility that NF-κB was indirectly mediated through ATP-induced soluble factors, we performed transfer experiments with supernatants from ATP-stimulated cells (Fig. 7 C). To this end, cells were treated with ATP, and after different times, culture supernatants were transferred to N9 cells that were subsequently analyzed for NF-κB activation. As shown in Fig. 7 C, supernatants from ATP-treated cells were unable to either induce or to alter the kinetics of NF-κB activation which was elicited not earlier than within 3 h. As expected, virtually no NF-κB activation was observed when supernatants from ATP-treated cells were preincubated with the ATP-degrading enzyme apyrase, before the transfer to cells (data not shown). We therefore consider it unlikely that NF-κB activation is a secondary response to ATP treatment, and that either IL-1β, TNF, or other ATP-induced soluble factors are required to induce NF-κB DNA binding.
Figure 7.

Lack of involvement of IL-1β, TNF-α, or soluble factors in ATP-induced NF-κB activation. (A) Effect of IL-1RA. N9 cells were either left untreated or incubated with 3 mM ATP for 3 h in the presence of the indicated concentrations of IL-1RA. (B) Neutralizing anti-TNF-α does not interfere with ATP-induced NF-κB activation. N9 cells were either left untreated or incubated with 3 mM ATP for 3 h in the presence of the indicated concentrations of rabbit anti–mouse TNF antibodies. (C) Secretion of soluble proteins is not required for NF-κB activation. In lanes 1–6, cells were incubated for the indicated times with 3 mM ATP. In lanes 10–13, cells were treated for 0.5–3 h with ATP. Supernatants (SN) of these cultures were then transferred to untreated N9 cells which were analyzed for NF-κB activation after the indicated incubation periods. In lane 9, N9 cells were incubated for 4 h with a culture supernatant from control cells.
ATP Induces NF-κB Controlled Gene Expression
To investigate whether exposure of cells to ATP is able to induce functional gene expression, reporter gene assays were performed. A luciferase construct controlled by six κB-binding sites was transiently transfected into N9 cells. Fig. 8 shows that increasing concentrations of ATP induced the expression of the NF-κB–controlled reporter gene. The dose response roughly corresponded to the activation of NF-κB DNA-binding activity. Preincubation of N9 cells with the antagonist oxidized ATP abolished activation of gene expression, indicating that the ATP effect was specifically mediated by a P2Z receptor-dependent pathway. In conclusion, the data demonstrate that the ATP-induced NF-κB complex is transcriptionally active and able to induce expression of NF-κB target genes.
Figure 8.

Effect of ATP on NF-κB target gene expression. N9 cells were transfected by electroporation with an NF-κB-controlled luciferase construct. 24 h after transfection, cells were either left untreated (Control) or stimulated with the indicated amounts of ATP. In some samples, cells were pretreated with 600 μM oATP before ATP stimulation. Cells were harvested after an additional 4 h and assayed for luciferase activity. Mean values ±SD of NF-κB activity given as relative light units from triplicate experiments are shown.
Discussion
Cells of the macrophage lineage express a receptor for extracellular ATP, which has been originally termed P2Z receptor and, after its recent cloning, has been also classified as P2X7 receptor (46). The most characteristic cellular response upon binding of ATP to the P2Z receptor is a fast collapse of the membrane potential resulting from ATP-induced opening of a large transmembrane pore. The physiological function of the P2Z receptor is rather unknown but there is increasing evidence for its implication in immune and inflammatory reactions (13). Thus, it has been shown that P2Z receptor action mediates rapid cell death, secretion of IL-1β, and formation of multinucleated giant cells (13, 15, 18).
In the present study, we investigated the effects of ATP on the activation of NF-κB, an important transcriptional activator involved in proinflammatory cytokine synthesis and apoptosis. As a cellular system, we employed microglial cells which have been previously characterized in detail for purinergic receptor expression (17). These cells are a good model since they are able to release large amounts of proinflammatory cytokines and may be stimulated by ATP from adjacent neurons (8, 21, 59). We demonstrate that NF-κB is potently activated by ATP but not by other nucleotides. NF-κB activation was selectively triggered through the P2Z receptor pathway, because: (a) oxidized ATP, a P2Z receptor antagonist, abrogated NF-κB activation in response to ATP, and (b) ATP failed to induce NF-κB activation in N9 clones that lack P2Z receptors but still express P2Y receptors. We further show that ATP- induced NF-κB activation in microglial cells exhibits some unusual features. In contrast to traditional proinflammatory stimuli, ATP-induced NF-κB activation was rather delayed and seemed to require induction of ICE-proteolytic activity. In addition, ATP selectively induced the formation of a rather unusual p65 NF-κB homodimer, indicating that different NF-κB target genes are regulated by ATP and proinflammatory stimuli.
The time course of ATP-induced NF-κB activation was rather slow and induction of DNA binding was not observed before 3 h after cell stimulation. This is in contrast to most other inflammatory stimuli that generally induce NF-κB activation within minutes. Similar to ATP, a delayed NF-κB activation has been observed in other conditions inducing cell death, such as growth factor withdrawal, virus infection, or treatment of cells with chemotherapeutic drugs (7, 11, 24, 30). Due to the delayed time course, we investigated whether NF-κB activation was mediated by an auto- or paracrine secretion of ATP-triggered NF-κB inducing factors. Neither neutralizing antibodies against TNF nor IL-1β interfered with NF-κB activation. Incubation of cells with IL-1RA, which captures secreted IL-1β, was also ineffective to inhibit the ATP response. Moreover, as IL-1β is unable to activate NF-κB in N9 cells (data not shown), IL-1β is presumably not involved in ATP-induced NF-κB activation. Furthermore, because supernatant transfer experiments did not alter the kinetics of NF-κB activation, we consider it rather unlikely that the effects of ATP are a secondary response.
It has been proposed that activation of NF-κB is regulated by the intracellular production of ROIs that may be used as second messengers molecules integrating the diverse variety of NF-κB inducing agents in the final pathway (1, 45). Thus, it was shown that NF-κB activation in response to diverse stimuli such as LPS, cytokines, and phorbolesters, was commonly inhibited by antioxidants. Consistent with this idea, NF-κB activation in response to ATP also seemed to require ROI formation, since it was completely abolished by PDTC. For most stimuli, the signal transduction pathways leading to ROI formation and subsequent NF-κB activation are unknown. In the case of TNF, a very potent activator of NF-κB, it has been suggested that the respiratory chain of mitochondria is the site of ROI formation (44). Triggering of the CD28 receptor also results in ROI formation and NF-κB activation (31). Inhibitors of 5-lipoxygenase block NF-κB activation by CD28, suggesting that this enzyme generates ROIs in this pathway. In addition, it has been shown that NF-κB is activated by a variety of agents which perturb the function of the endoplasmic reticulum (35). In this case, calcium released from the endoplasmic reticulum was found to be responsible for ROI production and NF-κB activation. Since incubation of cells with ATP induces a rapid increase in intracellular free calcium (13), it may be presumed that oxidative stress and subsequent NF-κB activation are mediated by perturbation of calcium homeostasis.
An interesting finding was that inhibitors of caspases suppressed ATP-induced NF-κB activation. As these proteases have been identified as critical executioners of various apoptotic pathways, NF-κB activation may be related to the ATP-mediated induction of the apoptotic program. Indeed, ICE activation, as measured by the proteolytic processing of its proenzyme, was detected in ATP-treated N9 cells with a kinetic similar to NF-κB activation. We also observed a characteristic cleavage of caspase-specific substrates such as the DNA repair enzyme PARP, which undergoes proteolysis in a number of apoptotic conditions (data not shown). Both the activation of ICE and NF-κB, however, preceded the onset of ATP-induced cell death. An involvement of caspases has been recently also proposed for the activation of NF-κB/v-Rel in chicken spleen cells which was prevented by CrmA, a viral inhibitor of some proteases of the caspase family (54).
ATP-mediated NF-κB activation was further found to require proteasome activity, because lactacystin, a specific inhibitor of the 20S proteasome, suppressed induction of NF-κB DNA binding. Interestingly, we observed that not only NF-κB activation but also the proteolytic processing of ICE was affected by lactacystin (data not shown). Inhibition of ICE activation by proteasome inhibitors has been recently noted in cell death of sympathetic neurons following NGF withdrawal (39). Because caspases must themselves be cleaved to yield active proteases, it is possible that the proteasome may act directly or indirectly on pro-ICE.
Besides inflammatory processes, there is increasing evidence that NF-κB is involved in the control of apoptosis, although it is far from clear whether, depending on the cell type, NF-κB plays a pro-apoptotic or an anti-apoptotic role (3). Depletion of NF-κB in fibroblasts resulted in the enhanced susceptibility towards TNF or chemotherapeutic drug-induced cytotoxicity, most likely by preventing the inducible expression of anti-apoptotic genes (6, 51, 53). In contrast, glutamate-induced toxicity in neurons has been found to be accompanied by NF-κB induction (23). In this case, NF-κB seemed to cause cell death, as blocking its activation with aspirin and salicylate protected cells from glutamate-induced apoptosis.
Up to now, five different subunits including p65 (RelA), c-Rel, RelB, p50, and p52 have been identified that may constitute different NF-κB–DNA binding complexes. Recent studies using targeted disruptions provided evidence that the NF-κB proteins play distinct biological roles (2, 4). It has also been shown that combinatorial interactions between different NF-κB subunits give rise to dimers with distinguishable DNA-binding specificity and transcriptional activity (29). Besides the classical p65/p50 and p65/ c-Rel heterodimeric complexes that exhibit strong transactivation activity, in many cell types a p50 homodimer can be found. Because of the absence of a transactivation domain, p50 homodimers are transcriptionally inactive and function as repressors of gene expression (26). In our experiments, we investigated the subunit composition of the inducible NF-κB complexes using specific antibodies against different NF-κB proteins. Exposure of microglial cells to LPS resulted in the appearance of two complexes consisting of a p65/p50 and a p50 dimer. Surprisingly, ATP induced a single complex that only reacted with anti-p65 and was not supershifted by antibodies against p50 or other NF-κB proteins, indicating that a p65 homodimer constituted the major ATP-inducible complex. To our knowledge, this is the first report demonstrating that a selective stimulus triggers the formation of a particular NF-κB subunit combination within the same cells. In contrast to the p65/p50 complexes, p65 homodimers bind preferentially to an alternative κB motif which has been recognized in the promoter regions of the tissue factor, IL-8, MAdCAM-1 and ICAM-1 genes (28, 47). Therefore, ATP presumably triggers expression of NF-κB target genes different from those induced by proinflammatory pathways that mainly use classical NF-κB complexes.
Besides different DNA-binding subunits, also several inhibitors have been identified including IκB-α, -β, and -ε as the major NF-κB inhibitory proteins (48, 55). Similar to IκB-α, IκB-β and -ε undergo signal-induced degradation dependent on phosphorylation of two regulatory serine residues. Evidence emerges that different IκB proteins target distinct NF-κB subunits. Very recently, IκB-ε has been found as a major IκB protein expressed in macrophages that predominantly associates with p65 and c-Rel (55). Future studies therefore have to address which IκB protein is targeted by the P2Z receptor pathway.
Acknowledgments
We thank U. Gern for expert technical assistance. We are also grateful to Dr. P. Ricciardi-Castagnoli for providing microglial cells, Dr. C. Libert for valuable reagents, and Dr. F. Di Virgilio for helpful discussions.
Abbreviations used in this paper
- EMSA
electrophoretic mobility shift assay
- ICE
IL-1β-converting enzyme
- IL
interleukin
- LDH
lactate dehydrogenase
- LPS
lipopolysaccharide
- PDTC
pyrrolidine dithiocarbamate
- RA
receptor antagonist
- ROI
reactive oxygen intermediate
- TNF
tumor necrosis factor
Footnotes
D.F. acknowledges a fellowship from FEBS and the Vigoni (DAAD/ CRUI) programme. This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 364/A7, Schu 1180/1-1).
Address all correspondence to Klaus Schulze-Osthoff, Medical Clinics, Department of Internal Medicine I, Otfried-Müller-Str. 10, D-72076 Tübingen, Germany. Tel.: 49 7071 29 84113. Fax: 49 7071 29 5865.
References
- 1.Baeuerle PA, Henkel T. Function and activiation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 2.Baeuerle PA, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 3.Baichwal VR, Baeuerle PA. Activate NF-κB or die? . Curr Biol. 1997;7:R94–R96. doi: 10.1016/s0960-9822(06)00046-7. [DOI] [PubMed] [Google Scholar]
- 4.Baldwin AS. The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;242:540–546. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 5.Bauer MKA, Lieb K, Schulze-Osthoff K, Bauer J, Gebicke-Haerter PJ, Fiebich BI. Expression and regulation of cyclooxygenase-2 in rat microglia. Eur J Biochem. 1997;243:726–731. doi: 10.1111/j.1432-1033.1997.00726.x. [DOI] [PubMed] [Google Scholar]
- 6.Beg AA, Baltimore D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 7.Boland MP, Foster SJ, O'Neill LA. Daunorubicin activates NF-κB and induces κB-dependent gene expression in HL-60 promyelocytic and Jurkat T lymphoma cells. J Biol Chem. 1997;272:12952–12960. doi: 10.1074/jbc.272.20.12952. [DOI] [PubMed] [Google Scholar]
- 8.Burnstock G, Wood JN. Purinergic receptors: their role in nociception and primary afferent neurotransmission. Curr Opin Cell Biol. 1996;6:526–532. doi: 10.1016/s0959-4388(96)80060-2. [DOI] [PubMed] [Google Scholar]
- 9.Chvatchko Y, Valera S, Aubry J-P, Renno T, Buell G, Bonnefoy J-Y. The involvement of ATP-gated ion channel, P2X1, in thymocyte apoptosis. Immunity. 1996;5:275–283. doi: 10.1016/s1074-7613(00)80322-2. [DOI] [PubMed] [Google Scholar]
- 10.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das W, White CW. Activation of NF-κB by antineoplastic agents. Role of protein kinase C. J Biol Chem. 1997;272:14914–14920. doi: 10.1074/jbc.272.23.14914. [DOI] [PubMed] [Google Scholar]
- 12.DiDonato JA, Hayakawa M, Rothwarf D, Zandi E, Karin M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 13.Di Virgilio F. The purinergic P2Z receptor: an intriguing role in immunity, inflammation and cell death. Immunol Today. 1995;16:524–528. doi: 10.1016/0167-5699(95)80045-X. [DOI] [PubMed] [Google Scholar]
- 14.Dubyak GR, El-Moatassim C. Signal transduction via P2-purinergic receptors for extracellular ATP and other nucleotides. Am J Physiol. 1993;265:C577–C606. doi: 10.1152/ajpcell.1993.265.3.C577. [DOI] [PubMed] [Google Scholar]
- 15.Falzoni S, Munerati M, Ferrari D, Spisani S, Moretti S, Di Virgilio F. The purinergic P2Zreceptor of human macrophage cells: characterization and possible physiological role. J Clin Invest. 1995;95:1207–1216. doi: 10.1172/JCI117770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activity and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 17.Ferrari D, Villalba M, Chiozzi P, Falzoni S, Ricciardi-Castagnoli P, Di Virgilio F. Mouse microglial cells express a plasma membrane pore gated by extracellular ATP. J Immunol. 1996;156:1531–1539. [PubMed] [Google Scholar]
- 18.Ferrari D, Chiozzi P, Falzoni S, Dal M, Susino, Collo G, Buell G, Di Virgilio F. ATP-mediated cytotoxicity in microglial cells. J Neuropharmacol. 1997;36:1295–1301. doi: 10.1016/s0028-3908(97)00137-8. [DOI] [PubMed] [Google Scholar]
- 19.Ferrari D, Chiozzi P, Falzoni S, Hanau S, Di Virgilio F. Purinergic modulation of interleukin-1β release from microglial cells stimulated with bacterial endotoxin. J Exp Med. 1997;185:579–582. doi: 10.1084/jem.185.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, Williams M. Nomenclature and classification of purinoreceptors. Pharmacol Rev. 1994;46:143–156. [PMC free article] [PubMed] [Google Scholar]
- 21.Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immune effector cells of the brain. Brain Res Rev. 1996;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- 22.Griffith RJ, Stam EJ, Downs JT, Otterness IG. ATP induces the release of IL-1 from LPS-primed cells in vivo. J Immunol. 1995;154:2821–2828. [PubMed] [Google Scholar]
- 23.Grilli M, Pizzi M, Memo M, Spano PF. Neuroprotection by aspirin and sodium salicylate through blockade of NF-κB activation. Science. 1996;274:1383–1385. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- 24.Grimm S, Bauer M, Baeuerle PA, Schulze-Osthoff K. Bcl-2 attenuates the activity of NF-κB which is induced upon apoptosis. J Cell Biol. 1996;134:13–23. doi: 10.1083/jcb.134.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hogquist KA, Nett MA, Unanue ER, Chaplin DD. Interleukin-1 is processed and released during apoptosis. Proc Natl Acad Sci USA. 1991;88:8485–8489. doi: 10.1073/pnas.88.19.8485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang S-M, Tran A-C, Grilli M, Lenardo M. NF-κB subunit regulation in nontransformed CD4+T lymphocytes. Science. 1992;256:1452–1456. doi: 10.1126/science.1604322. [DOI] [PubMed] [Google Scholar]
- 27.Kumar S, Harvey NL. Role of multiple cellular proteases in the execution of programmed cell death. FEBS Lett. 1995;375:169–173. doi: 10.1016/0014-5793(95)01186-i. [DOI] [PubMed] [Google Scholar]
- 28.Kunsch C, Rosen CA. NF-κB subunit-specific regulation of the interleukin-8 promoter. Mol Cell Biol. 1993;13:6137–6146. doi: 10.1128/mcb.13.10.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kunsch CA, Ruben SM, Rosen CA. Selection of optimal κB/ Rel DNA-binding motifs: interaction of both subunits of NF-κB with DNA is required for transcriptional activation. Mol Cell Biol. 1992;12:4412–4421. doi: 10.1128/mcb.12.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin K-I, Lee S-H, Narayanan R, Baraban JM, Hardwick JM, Ratan RR. Thiol agents and Bcl-2 identify an alphavirus-induced apoptotic pathway that requires activation of the transcription factor NF-kappa B. J Cell Biol. 1995;131:1149–1161. doi: 10.1083/jcb.131.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Los M, Schenk H, Hexel K, Baeuerle PA, Dröge W, Schulze-Osthoff K. IL-2 gene expression and NF-κB activation through CD28 requires reactive oxygen production by 5-lipoxygenase. EMBO J. 1995;14:3731–3740. doi: 10.1002/j.1460-2075.1995.tb00043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Los M, Van deCraen M, Penning LC, Schenk H, Westendorp M, Baeuerle PA, Dröge W, Krammer PH, Fiers W, Schulze-Osthoff K. Requirement of an ICE/Ced-3 protease for Fas/APO-1-mediated apoptosis. Nature. 1995;375:81–83. doi: 10.1038/375081a0. [DOI] [PubMed] [Google Scholar]
- 33.Murgia M, Hanau S, Pizzo P, Rippa M, Di Virgilio F. Oxidized ATP. An irreversible inhibitor of the macrophage purinergic P2Z receptor. J Biol Chem. 1993;268:8199–8203. [PubMed] [Google Scholar]
- 34.Nicholson DN, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 35.Pahl HL, Baeuerle PA. Activation of NF-κB by ER stress requires both Ca2+and reactive oxygen intermediates. FEBS Lett. 1996;392:129–136. doi: 10.1016/0014-5793(96)00800-9. [DOI] [PubMed] [Google Scholar]
- 36.Rassendren F, Buell GN, Virginio C, Collo G, North A, Surprenant A. The permeabilizing ATP receptor, P2X7. Cloning and expression of a human cDNA. J Biol Chem. 1997;272:5482–5486. doi: 10.1074/jbc.272.9.5482. [DOI] [PubMed] [Google Scholar]
- 37.Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IκB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 38.Righi M, Mori L, De Libero G, Sironi M, Biondi A, Mantovani A, Denis-Donini S, Ricciardi-Castagnoli P. Monokine production by microglial cells. Eur J Immunol. 1989;19:1443–1448. doi: 10.1002/eji.1830190815. [DOI] [PubMed] [Google Scholar]
- 39.Sadoul R, Fernandez P-A, Quiquerez A-L, Martinou I, Maki M, Schröter M, Becherer JD, Irmler M, Tschopp J, Martinou J-C. Involvement of the proteasome in the programmed cell death of NGF-deprived sympathetic neurons. EMBO J. 1996;15:3845–3852. [PMC free article] [PubMed] [Google Scholar]
- 40.Schenk H, Klein M, Erdbrügger W, Dröge W, Schulze-Osthoff K. Distinct effects of thioredoxin and other antioxidants on the activation of NF-κB and AP-1. Proc Natl Acad Sci USA. 1994;91:1672–1676. doi: 10.1073/pnas.91.5.1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidt KN, Amstad P, Cerutti P, Baeuerle PA. The roles of hydrogen peroxide and superoxide as messengers in the activation of transcription factor NF-κB. Chem Biol. 1995;2:13–22. doi: 10.1016/1074-5521(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 42.Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intemediates as apparently widely used messengers in the activation of the NF-κB transcription factor and HIV-1. EMBO J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schulze-Osthoff K, Bauer M, Vogt M, Los M. Role of ICE- related and other proteases in Fas-mediated apoptosis. Cell Death Diff. 1996;3:177–184. [PubMed] [Google Scholar]
- 44.Schulze-Osthoff K, Beyaert R, Vandevoorde V, Haegeman G, Fiers W. Depletion of the mitochondrial electron transport abrogates the cytotoxic and gene-inductive effects of TNF. EMBO J. 1993;12:3095–3104. doi: 10.1002/j.1460-2075.1993.tb05978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schulze-Osthoff K, Los M, Baeuerle PA. Redox regulation of transcription factor NF-κB and AP-1 in lymphocytes. Biochem Pharmacol. 1995;50:735–741. doi: 10.1016/0006-2952(95)02011-z. [DOI] [PubMed] [Google Scholar]
- 46.Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7) Science. 1996;272:735–738. doi: 10.1126/science.272.5262.735. [DOI] [PubMed] [Google Scholar]
- 47.Takeuchi M, Baichwal VR. Induction of the gene encoding mucosal vascular addressin cell adhesion molecule 1 by tumor necrosis factor α is mediated by NF-κB proteins. Proc Natl Acad Sci USA. 1995;92:3561–3565. doi: 10.1073/pnas.92.8.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thompson JE, Phillips RJ, Erdjumant-Bromage H, Tempst P, Ghosh S. IκB-β regulates the persistent response in a biphasic activation of NF-κB. Cell. 1995;80:573–582. doi: 10.1016/0092-8674(95)90511-1. [DOI] [PubMed] [Google Scholar]
- 49.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J. A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 50.Traenckner EB-M, Wilk S, Baeuerle PA. A proteasome inhibitor prevents activation of NF-κB and stabilizes a newly phosphorylated form of IκB-α that is still bound to NF-κB. EMBO J. 1994;13:5433–5441. doi: 10.1002/j.1460-2075.1994.tb06878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Antwerp DJ, Martin SJ, Kafri T, Green D, Verma IM. Suppression of TNF-α-induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 52.Walker NPC, Talanian RV, Brady KD, Dang LC, Bump NJ, Ferenz CR, Franklin S, Ghayur T, Hackett CR, Hamill LD, et al. Crystal structure of the cysteine protease interleukin-1β-converting enzyme: a (p20/p10)2heterodimer. Cell. 1994;78:343–352. doi: 10.1016/0092-8674(94)90303-4. [DOI] [PubMed] [Google Scholar]
- 53.Wang C-Y, Mayo MW, Jr AS, Baldwin TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 54.White DW, Gilmore TD. Bcl-2 and CrmA have different effects on transformation, apoptosis, and the stability of IκB-α in chicken spleen cells transformed by temperature-sensitive v-Rel oncoproteins. Oncogene. 1996;13:891–899. [PubMed] [Google Scholar]
- 55.Whiteside ST, Epinat J-C, Rice NR, Israel A. IκBε, a novel member of the IκB family, controls RelA and c-Rel NFκB binding activity. EMBO J. 1997;16:1413–1426. doi: 10.1093/emboj/16.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilson KP, Black R, Thomson JA, Kim EE, Griffith JP, Navia MA, Murcko MA, Chambers SP, Aldape RA, Raybuck SA, Livingston DJ. Structure and mechanism of interleukin-1β converting enzyme. Nature. 1994;370:270–275. doi: 10.1038/370270a0. [DOI] [PubMed] [Google Scholar]
- 57.Zabel U, Baeuerle PA. Purified human IκB can rapidly dissociate the complex of the NF-κB transcription factor with its cognate DNA. Cell. 1990;61:255–265. doi: 10.1016/0092-8674(90)90806-p. [DOI] [PubMed] [Google Scholar]
- 58.Zanovello P, Bronte V, Rosato A, Pizzo P, Di Virgilio F. Responses of mouse lymphocytes to extracellular ATP. II. Extracellular ATP causes cell type-dependent lysis and DNA fragmentation. J Immunol. 1990;145:1545–1550. [PubMed] [Google Scholar]
- 59.Zimmermann H. Signaling via ATP in the nervous system. Trends Neurosci. 1994;17:420–426. doi: 10.1016/0166-2236(94)90016-7. [DOI] [PubMed] [Google Scholar]