

Abstract
A novel clathrin adaptor-like complex, adaptor protein (AP)-3, has recently been described in yeast and in animals. To gain insight into the role of yeast AP-3, a genetic strategy was devised to isolate gene products that are required in the absence of the AP-3 μ chain encoded by APM3. One gene identified by this synthetic lethal screen was VPS45. The Vps pathway defines the route that several proteins, including carboxypeptidase Y, take from the late Golgi to the vacuole. However, vacuolar alkaline phosphatase (ALP) is transported via an alternate, intracellular route. This suggested that the apm3-Δ vps45 synthetic phenotype could be caused by a block in both the alternate and the Vps pathways. Here we demonstrate that loss of function of the AP-3 complex results in slowed processing and missorting of ALP. ALP is no longer localized to the vacuole membrane by immunofluorescence, but is found in small punctate structures throughout the cell. This pattern is distinct from the Golgi marker Kex2p, which is unaffected in AP-3 mutants. We also show that in the apm3-Δ mutant some ALP is delivered to the vacuole by diversion into the Vps pathway. Class E vps mutants accumulate an exaggerated prevacuolar compartment containing membrane proteins on their way to the vacuole or destined for recycling to the Golgi. Surprisingly, in AP-3 class E vps double mutants these proteins reappear on the vacuole. We suggest that some AP-3–dependent cargo proteins that regulate late steps in Golgi to vacuole transport are diverted into the Vps pathway allowing completion of transfer to the vacuole in the class E vps mutant.
The formation of vesicles for transport between membrane-bound organelles requires assembly of coat proteins that are recruited from the cytosol. These proteins direct the sequestration and concentration of cargo as well as invagination of the membrane. One of the best studied classes of coats involved in vesicle budding is comprised of clathrin and its adaptor proteins (APs)1, AP-1 and AP-2 (Schmid, 1997). In clathrin-mediated vesicle transport the AP complexes play the dual role of cargo selection and recruitment of clathrin to the membrane. These adaptors are heterotetramers containing two large chains (adaptins, α or γ and β), one medium chain (μ), and one small chain (σ). AP-1 (γ, β1, μ1, and σ1) functions in sorting at the TGN, whereas AP-2 (α, β2, μ2, and σ2) is involved in receptor capture at the PM during endocytosis.
Although there is a great deal of evidence supporting the involvement of adaptors in clathrin-mediated vesicle budding, recent studies in animal cells have led to the discovery of a novel adaptor-like complex, AP-3, that seems to function independently of clathrin (Newman et al., 1995; Simpson et al., 1996). AP-3 has identical subunit architecture to AP-1 and AP-2, with two adaptin-like subunits (δ and β3), a medium chain (μ3), and a small chain (σ3) (Simpson et al., 1996, 1997; Dell'Angelica et al., 1997a , b ). AP-3 antibodies label a perinuclear region, perhaps the TGN, and punctate structures extending to the cell periphery, which may be endosomal compartments (Simpson et al., 1996, 1997; Dell'Angelica et al., 1997a ). However, the mammalian AP-3 complex does not colocalize with clathrin or AP-1 and AP-2 adaptors in cells and it does not copurify with brain clathrin-coated vesicles (Newman et al., 1995; Simpson et al., 1996, 1997; Dell'Angelica et al., 1997b ). Clues to the function of AP-3 have come from the discovery that the garnet gene of Drosophila encodes a protein closely related to δ adaptin (Ooi et al., 1997; Simpson et al., 1997). Mutations in garnet cause decreased pigmentation of the eyes and other tissues and a reduced number of pigment granules, which may be lysosome-like organelles (Ooi et al., 1997; Simpson et al., 1997). Thus, AP-3 is proposed to function in clathrin-independent transport between the TGN, endosomes and/or lysosomes, although its exact sorting function is still not known.
Over the last several years, yeast homologues of the mammalian adaptor subunits have been identified, allowing for the examination of specific functions of these proteins in a genetically tractable organism. Genes encoding subunits sufficient for at least three complete AP complexes have been identified by sequence homology (Phan et al., 1994; Rad et al., 1995; Stepp et al., 1995) or by function (Panek et al., 1997). APL1-APL6 encode large chain/ adaptin-related subunits, APM1-APM4 encode μ-like chains, and APS1-APS3 are genes for σ-related proteins. Apl2p (β), Apl4p (γ), Apm1p (μ1), and Aps1p (σ1) are thought to be subunits of an AP-1–like complex that functions with clathrin at the late Golgi/TGN (Phan et al., 1994; Rad et al., 1995; Stepp et al., 1995; Payne, G., personal communication). Mutations in the yeast AP-1 genes enhance the growth and the α-factor processing defects of a temperature sensitive (ts) allele of the clathrin heavy chain gene (Phan et al., 1994; Rad et al., 1995; Stepp et al., 1995; Payne, G., personal communication). The latter phenotype is a hallmark of clathrin-deficient yeast, in which late Golgi/ TGN proteins, such as the α-factor processing enzymes Kex2p and dipeptidyl amino peptidase-A (DPAP)-A, are not retained in the late Golgi but escape to the cell surface (Seeger and Payne, 1992b ). To date, no yeast adaptor subunit has been shown to be important for endocytosis, although Apl3p, Apm4p, and Aps2p are most homologous to mammalian AP-2 α, μ2 and σ2, respectively.
Recently, a yeast adaptor related to AP-3 of animal cells was described (Panek et al., 1997). It is comprised of Apl5p, Apl6p, Apm3p, and Aps3p, which show preferential homology to mammalian δ, β3, μ3, and σ3, respectively. Mutations in each of these subunits were isolated by their ability to suppress the lethality resulting from loss of function of PM casein kinase 1 encoded by a gene pair, YCK1 and YCK2. Yck activity was found to be required for constitutive endocytosis of the a-factor receptor (Ste3p), and AP-3 subunit mutations partially rescued this internalization defect (Panek et al., 1997). However, the AP complex itself is not necessary for endocytosis, nor is it required for sorting of carboxypeptidase Y (CPY) or retention of late Golgi proteins. Furthermore, unlike disruption of the yeast AP-1 complex, loss of AP-3 function causes no synthetic phenotype in combination with chc1 mutations, suggesting it may function independently of clathrin. Although these data indicated that Apl5p, Apl6p, Apm3p, and Aps3p comprise an AP-3-like adaptor, its precise sorting role was still not known.
In this report we describe a genetic approach to determine the function of the yeast AP-3 complex. A colony sectoring screen was performed to identify genes that are essential in the absence of Apm3p, the yeast AP-3 μ chain. Such synthetic lethal screens can be used to identify functional homologues, genes whose proteins function in intersecting or parallel pathways, and genes whose proteins physically interact (Bender and Pringle, 1991). We have cloned the gene for the apm three synthetic lethal mutant, mts1-1, and found it encodes Vps45p, a protein involved in vacuolar protein sorting (Vps; Cowles et al., 1994; Piper et al., 1994). The Vps pathway is defined by >40 complementation groups whose proteins are required for the transport of a number of soluble and membrane-bound proteins, including CPY, protease A (PrA), and carboxypeptidase S (CPS) from the late Golgi/TGN to the vacuole (Stack et al., 1995; Cowles et al., 1997). This pathway is also essential for proper assembly of the vacuolar ATPase (Raymond et al., 1992). However, the type II vacuolar membrane protein alkaline phosphatase (ALP) follows an alternate intracellular pathway to the vacuole (Raymond et al., 1992; Nothwehr et al., 1995; Cowles et al., 1997; Piper et al., 1997). Few vps mutants prevent localization of ALP to the vacuolar membrane and its arrival at the vacuole is not dependent upon transport through the cell surface. The requirement for Apm3p in the absence of Vps45p suggested the possibility that at least one of these routes to the vacuole must be functional for survival and led us to examine ALP sorting in the AP-3 mutants. We show here that yeast AP-3 is essential for the transport of ALP via the alternative pathway to the vacuole.
Materials and Methods
Strains, Media, and Genetic Methods
Strains used in the study are listed in Table I. Rich yeast extract peptone dextrose (YEPD), rich yeast extract peptone glycerol (YEPG), synthetic minimal (SM), and synthetic dropout (C-nutrient) media were prepared as described previously (Stepp et al., 1995). Yeast mating, sporulation, and tetrad analyses were performed essentially as described in Guthrie and Fink (1991).
Table I.
Strains Used in This Study
| Strain no. | Genotype | Source | ||
|---|---|---|---|---|
| SL1463 | MATα leu2 ura3-52 trp1 his3-Δ200 | Panek et al. (1997) | ||
| SL1528 | MATa/MATα leu2/leu2 ura3-52/ura3-52 trp1/trp1 his3-Δ200/his3-Δ200 | This study | ||
| SL1650 | MATa/MATα apm3-Δ::HIS3/APM3 leu2/leu2 ura3-52/ura3-52 trp1/trp1 his3-Δ200/his3-Δ200 | This study | ||
| SL1652 | MATα apm3-Δ::HIS3 leu2 ura3-52 trp1 his3-Δ200 | This study | ||
| SL1653 | MATα apm3-Δ::HIS3 leu2 ura3-52 trp1 his3-Δ200 | This study | ||
| SL1904 | MATa apm3-Δ::HIS3 leu2 ura3-52 trp1 ade2-101 his3-Δ200 | This study | ||
| SL1922 | MATα apm3-Δ::HIS3 leu2 ura3-52 trp1 ade2-101 his3-Δ200 lys2 | This study | ||
| SL2163 | MATa apm3-Δ::HIS3 mts1-1 leu2 ura3-52 trp1 ade2-101 his3-Δ200 lys2 + pDS19 | This study | ||
| SL2284 | MATα mts1-1 leu2 ura3-52 trp1 his3-Δ200 ade2-101 lys2 | This study | ||
| SL2286 | MATa apm3-Δ::HIS3 mts2-1 leu-2 ura3-52 trp1 ade2-101 his3-Δ200 lys2 + pDS19 | This study | ||
| SL2385 | MATa apm3-Δ::HIS3 vpt21(vps21) leu2 ura3-52 trp1 his3-Δ200 lys2 | This study | ||
| SL2531 | MATα apm3-Δ::HIS3 vps1-Δ::LEU2 leu2 ura3-52 trp1 his3-Δ200 CAN1? | This study | ||
| SL2611 | MATα apm3-Δ::HIS3 pep12-Δ::TRP1 ura3-52 trp1 HIS3 | This study | ||
| SL2755 | MATα apm3-Δ::HIS3 vpl7-1(vps34) leu2 ura3-52 sde6 his4 HIS3? PEP4? | This study | ||
| SL2756 | MATa apm3-Δ:HIS3 vpl19-10(vps15) leu2 ura3-52 ade6 his4 HIS3? PEP4? | This study | ||
| SL2757 | MATa apm3-Δ::HIS3 vpl21-13(vps19) leu2 ura3-52 trp1 HIS3 PEP4? | This study | ||
| SL2758 | MATa apm3-Δ::HIS3 vpl22-7(vps39) leu2 ura3-52 ade6 HIS3 PEP4? | This study | ||
| SL2759 | MATα apm3-Δ::HIS3 vpl31-2(vps9) leu2 ura3-52 ade6 his4 HIS3? PEP4? | This study | ||
| SL2763 | MATα apm3-Δ::HIS3 sec1-1 leu2 ura3-52 | This study | ||
| SL2764 | MATa apm3-Δ::HIS3 sec4-8 ura3-52 trp1 HIS3 | This study | ||
| SL2765 | MATα apm3-Δ::HIS3 sec6-4 ura3-52 his3-Δ200 | This study | ||
| SL2766 | MATa apm3-Δ::HIS3 end3-1 leu2 ura3-52 trp1 his4 bar1-1 HIS3? | This study | ||
| SL2767 | MATa apm3-Δ::HIS3 end4-1 leu2 ura3-52 trp1 his4 bar1-1 HIS3? | This study | ||
| SL2820 | MATα apm3-Δ::HIS3 vpl25-6(vps33) leu2 ura3-52 trp1 ade6 HIS3? PEP4? | This study | ||
| SL2821 | MATα apm3-Δ::HIS3 pep3-Δ::LEU2 leu2 ura3-52 trp1 his1 HIS3? | This study | ||
| SL2822 | MATα apm3-Δ::HIS3 pep5-Δ::URA3 leu2 ura3-52 trp1 HIS3? | This study | ||
| SL2845 | MATα apm3-Δ::HIS3 vpl4-1(vps4) leu2 ura3-52 his4 pep4-3 HIS3? | This study | ||
| SL2846 | MATα apm3-Δ::HIS3 vpt4(vps26) leu2 ura3-52 trp1 his3 | This study | ||
| SL2850 | MATα apm3-Δ::HIS3 vps27-Δ::LEU2 leu2 ura3-52 trp1 his3 | This study | ||
| SL2864 | MATα apm3-Δ::HIS3 ypt7-Δ::URA3 leu2 ura3-52 trp1 HIS3? | This study | ||
| SL2951 | MATα apm3-Δ::HIS3 pep4::HIS3 prb1-Δ1.6R CAN1 leu2 ura3-52 trp1 HIS3 | This study | ||
| SL2955 | MATa apm3-Δ::HIS3 vps45-13 leu2 ura3-52 trp1 his3-Δ200 ade2 | This study | ||
| SL2956 | MATa apm3-Δ::HIS3 vps45-13 leu2 ura3-52 trp1 his3-Δ200 ade2 | This study | ||
| SL3034 | MATa apm3-Δ::HIS3 vat2-Δ::LEU2 leu2 ura3-52 his3 CAN1? | This study | ||
| SL3037 | MATa apm3-Δ::HIS3 vps8-Δ::URA3 leu2 ura3-52 trp1 his3 | This study | ||
| SL3089 | MATa apm3-Δ::HIS3 vps45-Δ leu2 ura3-52 his4 HIS3? | This study | ||
| SL3129 | MATa apm3-Δ::HIS3 mts1-1 leu2 ura3-52 trp1 his3-Δ200 | This study | ||
| SL3130 | MATa apm3-Δ::HIS3 vps35-Δ::HIS3 leu2 ura3-52 trp1 his3 SUC2? | This study | ||
| HPY20 | MATa ap15::HIS3 leu2 ura3-52 his3 | Panek et al. (1997) | ||
| LRB858 | MATa ap16::URA3 leu2 ura3-52 his3 | Panek et al. (1997) | ||
| LRB739 | MATa aps 3::LEU2 leu2 ura3-52 his3 | Panek et al. (1997) | ||
| SF838-1D | MATα vp14-1(vps4) leu2 ura3-52 his4 ade6 pep4-3 | T. Stevens | ||
| SNY17 | MATα pho8-Δ::LEU2 leu2 ura3-52 his3-Δ200 trp1-Δ901 lys2-801 | S. Nothwehr |
Synthetic growth phenotypes caused by apm3-Δ combined with other mutant loci were examined by crossing single mutant strains and isolating double mutant progeny from dissection of heterozygous diploids. The double mutants were then transformed with an APM3, CEN plasmid or no plasmid, and the growth of the isogenic pair at 25°, 30°, 34°, and 37°C was compared by streaking for singles colonies on the same YEPD plate.
Plasmids
APM3 subclones were generated from pJW4, a SNF5 clone from M. Carlson (Columbia University, New York, NY). (APM3 lies upstream of SNF5 on chromosome II.) pDS13 contains the APM3 2.3-kb Pvu2 fragment from pJW4 cloned into the Sma1 site of pBluescript SK+ (APM3 is oriented in the Sac1 to Kpn1 direction of the polylinker). pDS14 is identical to pDS13 except the insert is in the opposite orientation. pDS19 contains the APM3 2.3-kb BamH1–Pst1 fragment from pDS13 cloned into pASZ11, an ADE2, CEN vector (Stotz and Linder, 1990). pDS20 (APM3, TRP1, CEN) contains the 2.3-kb BamH1–Pst1 fragment from pDS13 cloned into pRS314 (Sikorski and Hieter, 1989). pDS24 (APM3, URA3, CEN) contains the 2.3-kb BamH1–Sal1 fragment from pDS14 cloned into pRS316 (Sikorski and Hieter, 1989). pDS27, containing VPS45, was cloned from the YCp50 library of Rose et al. (1987). To generate the VPS45, URA3 integrating vector (pDS31), the 2.1-kb Sma1–Sph1 fragment from pDS27 was subcloned into YIp352 (Hill et al., 1986). pDS31 was linearized with Nhe1 to target integration at the VPS45 locus. pRCP56, carrying the vps45-13 ts for function allele of VPS45 was described in Piper et al. (1994). pEND4, CEN was from H. Riezman (University of Basel, Basel, Switzerland). In YCpKX22, the KEX2 gene is under the control of the TDH3 glyceraldehyde-3-phosphatase dehydrogenase (GAPDH) promoter in YCp50. This results in an ∼25-fold overproduction of Kex2p (Wilcox et al., 1992).
Gene Disruption of APM3
The apm3-Δ::HIS3 disruption was generated by integration of a PCR fragment (Baudin et al., 1993) using the following oligonucleotides: 5′ primer: 5′-GTGCAACAGCTCCCTCCTTCAAGCACCTGTGGACCCGCGTTCGTACTCTTGGCCTCCTCTAG-3′, and 3′ primer: 5′-GCCTTACTTGAAAATTGCCTGTCTGGGTCTTATACTTTGCACCCTCGTTCAGAATGACACG-3′. This results in the replacement of the APM3 sequence coding for amino acids 36–468 (Apm3p is 483 amino acids long) by the HIS3 gene. PCR reactions (100 μl) contained 1.4 ng HIS3 template DNA (pUC-HIS3; Rothstein, 1991), 100 pmol each primer, 20 mM Tris-HCl, pH 8.3, 25 mM KCl, 0.05% Tween 20, 1.5 mM MgCl2, 100 μg/ml gelatin, and 200 μM dNTPs. Amplification was performed as follows: 5 min at 95°C; 2 cycles of 30 s at 94°C, 30 s at 45°C, 2 min at 74°C; 30 cycles of 30 s at 94°C, 30 s at 50°C, 2 min at 74°C; 7 min at 72°C. The ∼950-bp product was purified and transformed into diploid SL1528 to generate the apm3-Δ/APM3 heterozygote, SL1650. As expected for apm3-Δ (Panek et al., 1997), tetrad analysis of this diploid yields four viable spores (two His+ and two His−) that have wild-type growth at all temperatures. The disruption was confirmed by DNA blot analysis (data not shown).
Synthetic Lethal Screen and Cloning of MTS1
Unless otherwise noted, growth was performed at 30°C. The synthetic lethal screen was a modification of the ADE2/ade2 sectoring method originally described in Bender and Pringle (1991). Strains SL1904 (MAT a ade2 apm3-Δ::HIS3) and SL1922 (MATα ade2 apm3-Δ::HIS3) each carrying the ADE2, APM3, CEN plasmid, pDS19, were mutagenized with methane sulfonic acid ethyl ester (EMS) to ∼50% killing and then plated on YEPD. The parental strains (Ade2+) formed white colonies that readily sector to red (Ade2−), since they do not require the APM3-plasmid for survival. A screen of ∼13,000 colonies yielded 204 candidates that had lost the ability to sector (Sect−) and could be dependent on plasmid-borne APM3 for survival. These were restreaked for singles on YEPD twice and rescored for sectoring each time. Since ade2 pet strains remain white and would give false positives in this screen, Sect− candidates were also tested for mitochondrial function by growth on glycerol medium (YEPG) and by a 2,3,5-triphenyltetrazolium chloride overlay assay (Ogur et al., 1957). The remaining 79 candidates were then tested for dependence of the Sect− phenotype upon the plasmid-borne APM3, as opposed to other plasmid sequences, by a plasmid shuffle test. Each candidate was transformed with pDS24 (APM3, URA3, CEN) and then tested for the ability to now lose the APM3, ADE2 plasmid. Only 10 candidates were able to sector to red upon transformation with the second APM3 plasmid. These candidates were backcrossed to an apm3-Δ strain of the opposite mating type (SL1904 or SL1922) to test for dominance of the sectoring phenotype. All backcrossed diploids were Sect+, indicating that all of the mutations were recessive. These diploids were then dissected and tetrads were scored for 2:2 segregation of the Sect− phenotype. Eight candidates were discarded because tetrads from backcrosses showed nonstandard segregation, or there was complete loss of the Sect− phenotype. The remaining two candidates showed 2:2 segregation of the Sect− phenotype and were designated mts1-1 and mts2-1.
MTS1 was cloned by complementation of the ts growth phenotype of mts1-1 cells. The original mts1-1 isolate was backcrossed three times to SL1904, resulting in SL2163 (ade2 apm3-Δ::HIS3 mts1-1 + pDS19). This strain was transformed with a YCp50 yeast genomic library (Rose et al., 1987) and spread plated onto C-uracil (C-Ura) plates. Colonies were allowed to grow at 25°C for 24 h and were then incubated at 37°C for 2 d. One colony (designated A1), which grew much faster than the others, was restreaked for single colonies onto C-Ura and incubated at 25° or 37°C. The A1 transformant was also tested for restoration of the sectoring phenotype on YEPD at 30°C. Both ts and loss of sectoring were rescued by this plasmid (redesignated pDS27). pDS27 also complemented the mts1-1 mutation after retransformation of the parental strain (SL2163) with the isolated plasmid that had been shuttled through bacteria. Both ends of the pDS27 insert were sequenced using primers that recognize the 5′ and 3′ sides of the BamH1 library cloning site in YCp50 as described previously (Stepp et al., 1995). The sequence was compared to the Saccharomyces Genome Database (SGD).
Antibodies
ALP, CPY, and affinity purified Kex2p rabbit antisera were gifts from S. Nothwehr (University of Missouri, Columbia, MO), E.W. Jones (Carnegie Mellon University, Pittsburgh, PA) and R. Fuller (University of Michigan, Ann Arbor, MI), respectively. Mouse monoclonal antibody 13D11 (anti-Vat2p) was the gift of P. Kane (Syracuse University, Syracuse, NY). ALP antibodies were affinity purified against the maltose binding protein–ALP (MBP–ALP) fusion protein construct described in Nothwehr et al. (1996) using standard methods (Harlow and Lane, 1988).
Preparation of Whole Cell Extracts and Subcellular Fractions for Immunoblotting
Whole cell extracts were generated by glass bead homogenization in 4% SDS and prepared for immunoblotting as described previously (Nelson and Lemmon, 1993). Subcellular fractionation was based on the procedure described in Horazdovsky and Emr (1993). Strains were grown to mid-log phase and 109 cells were harvested by centrifugation. Cell pellets were resuspended and incubated in 6 ml spheroplast buffer (1.2 M sorbitol, 50 mM KPO4, 40 mM β-mercaptoethanol, pH 7.5) plus 100 μg oxalyticase (Enzogenetics, Corvallis, OR) for 30 min at 30°C. Spheroplasts were pelleted and then resupended with 1.5 ml lysis buffer (0.2 M sorbitol, 50 mM Tris-HCl, 1 mM EDTA, pH 7.5) plus a protease inhibitor cocktail (PIC; Stepp et al., 1995). Lysis was achieved by dounce homogenization (10 strokes), and unlysed cells and debris were removed by centrifugation at 450 g for 3 min, and then washed with lysis buffer plus PIC, and recentrifuged. The two supernatants were combined and centrifuged at 13,000 g for 15 min, generating S13K and P13K. The S13K was further centrifuged at 100,000 g for 60 min resulting in S100K and P100K. Pellets were resuspended to equal volume and samples of each fraction were taken. Equivalent volumes were separated by SDS-PAGE (7.5% acrylamide) and transferred to nitrocellulose. Blots were probed with affinity purified anti-ALP (1: 500), followed by goat anti–rabbit HRP (Zymed Labs, South San Francisco, CA) (1:5,000) as the secondary antibody. Immunoblots were developed by incubation with a mixture of equal parts solution A (2.5 mM luminol, 400 μM p-coumaric acid, 100 mM Tris, pH 8.5) and solution B (5.4 mM H2O2, 100 mM Tris, pH 8.5) for 1 min, and then exposure to X-ray film.
Cell Labeling and Immunoprecipitation
For CPY processing, a modification of the method of Raymond et al. (1992) was used. Cells were grown to log phase (1 to 2 × 107 cells/ml) in SM plus requirements for nutritional markers. Cells (5 × 106) were harvested, washed, and resuspended with 500 μl fresh growth medium containing 50 mM KPO4, pH 5.7, and 1 mg/ml BSA. Pulse labeling was initiated by addition of 35S-EasyTag (NEN Life Science Products, Boston, MA) to 200 μCi/ml for 10 min at 30°C. Samples were chased by addition of 100× CHASE (0.3 M NH3SO4, 1% methionine, 1% cysteine) and further incubated for 30 min. Cells (I, intracellular) were harvested by centrifugation and the medium (E, extracellular) was transferred to another tube containing 20 μl of PIC (see above). The E samples were diluted with 415 μl water and 100 μl 10× immunoprecipitation (IP) buffer (0.9 M Tris-HCl, pH 8.0, 1% SDS, 1% Triton X-100, 20 mM EDTA), heated at 100°C for 5 min, and then stored on ice. The cell pellets (I) were resuspended in 100 μl 2% SDS, transferred to a tube containing 0.15 g glass beads, vortexed on high for 3 min, and heated at 100°C for 5 min. Glass beads and insoluble material were sedimented in a microcentrifuge for 1 min and the soluble cell extract was transferred to a new tube. The glass beads were washed once with 100 μl TBS (50 mM Tris-HCl, 150 mM NaCl, pH 8.0), and the extract supernatants were combined. The I samples were then diluted with 80 μl 10× IP buffer and 720 μl of water. I and E samples were preabsorbed with preswollen protein A–Sepharose (Stepp et al., 1995). The supernatants from this treatment were then incubated with 3 μl of anti-CPY antiserum for 1 h. Immune complexes were collected with protein A–Sepharose and then beads were washed twice with 1× IP buffer. Immunoprecipitates were eluted in 40 μl 2× SDS-PAGE sample buffer and separated on an SDS 10% polyacrylamide gel. Fixation, enhancement, and autoradiography were performed as described previously (Stepp et al., 1995).
Pulse-chase analysis of ALP processing was examined using methods described in Nothwehr et al. (1995) with the following modifications. Cells were labeled with 300 μCi/ml 35S-EasyTag (NEN Life Science Products), and chased for the indicated times as described above. Cell lysis was performed in the presence of PIC and preabsorbed with protein A–Sepharose as described for CPY above. For IP, samples were incubated with 1 or 2 μl of crude ALP antiserum for 2 h. Fixation, enhancement, and autoradiography were performed as described above for CPY. Quantification of ALP was performed by densitometry on autoradiograms using a Sci Scan 5000 (United States Biochemical Corp., Cleveland, OH), or by quantification of pixel density using a phosphorimager (Molecular Dynamics Inc., Sunnyvale, CA).
Immunofluorescence Microscopy
Preparation of cells and staining with antibodies was performed essentially as described in Roberts et al. (1991). Primary antibodies were used at a dilution of 1:50 (affinity purified anti-ALP, affinity purified anti-Kex2p, and anti–Vat2p 13D11 monoclonal). Secondary antibodies were FITC-conjugated goat anti–rabbit Ig (1:100, Kirkegaard and Perry Laboratories, Gaithersburg, MD) and Cy3-conjugated goat anti–mouse Ig (1: 1,000, gift of D. Loyaza and S. Michaelis, Johns Hopkins University, Baltimore, MD). All antibody incubations were for 1–2 h. Immunofluorescence visualization was performed on an epifluorescence microscope (Axioplan; Carl Zeiss, Inc., Thornwood, NY), and images were captured on a camera (CH250 CCD; Photometrics Ltd., Tucson, AZ) using CCD ImageCapture (Yale Univerity, New Haven, CT). Images were imported into Adobe Photoshop 3.0, labeled in Canvas 3.5 (Deneba Software, Miami, FL), and printed on a Tetronix 450 dye sublimation printer.
Results
Synthetic Lethal Screen for Isolation of Mutants That Require Yeast AP-3 Function
Strains deleted for any of the subunits of the yeast AP-3 complex grow normally and show no defects in secretion or sorting pathways previously found to be affected in chc1 mutants (Fig. 1; Panek et al., 1997; data not shown). These pathways include trafficking of soluble vacuolar hydrolases (Seeger and Payne, 1992a ), retention of late-Golgi resident proteins (Seeger and Payne, 1992b ), and endocytosis (Tan et al., 1993). Therefore, we undertook a genetic approach to determine where the AP-3–like complex functions within the cell. A colony-sectoring synthetic lethal screen (Bender and Pringle, 1991) was used to identify mutations in other genes that require the presence of Apm3p for survival. In this strategy, parental strains were of ade2 apm3-Δ::HIS3 genotype, carrying the wild-type (wt) ADE2 and APM3 genes on a plasmid (pDS19). These strains form white colonies because of the presence of the plasmid-borne ADE2, but can readily sector red (Ade2−) on nonselective medium, because the APM3 gene is not essential. A pair of strains of opposite mating type were mutagenized with EMS and then colonies scored for the requirement of the APM3 plasmid as measured by inability to sector red (Sect−). From a total of ∼13,000 colonies screened, two Sect− mutants were isolated and designated mts1-1 and mts2-1.
Figure 1.

Precursor CPY is secreted from mts1-1 cells. Cells were pulse labeled with [35S]methionine and [35S]cysteine for 10 min and then chased for 30 min with excess cold amino acids. CPY was then immunoprecipitated from internal (I) and external (E) fractions. Strains indicated are: wt, SL1652 + pDS24 (pAPM3, CEN); apm3-Δ, SL1652; mts1-1, SL2284; mts2-1, SL2286. p2CPY indicates precursor CPY; mCPY indicates mature CPY.
Initial characterization of the mts mutants indicated that mts1-1, but not mts2-1, is ts for growth at elevated and low temperatures (34°, 37° and 16°C; data not shown). In addition, sorting assays demonstrated that mts1-1 has a vps defect (Fig. 1). Normally, CPY is made as a precursor whose terminal glycosylation in the Golgi yields 69-kD p2CPY. Vacuolar delivery results in PEP4-dependent cleavage of CPY to the active, mature 61-kD form (mCPY). In vps mutants, p2CPY is secreted from the cell (Stack et al., 1995). The pulse-chase analysis in Fig. 1 illustrates the accumulation of p2CPY in the extracellular fraction of the mts1-1, but not the mts2-1 mutant strain. As we described previously (Panek et al., 1997), normal CPY processing and sorting were observed in an apm3-Δ strain. The CPY sorting defect associated with mts1-1 suggested that it may be allelic to an already identified VPS gene.
MTS1 Is VPS45
The MTS1 gene was cloned by complementation of the mts1-1 ts growth phenotype at 37°C by transformation with a yeast genomic DNA library. One complementing plasmid was obtained and the sequence was determined from each end of the genomic DNA insert. The resulting sequences were compared to the SGD and found to correspond to a region on the left arm of chromosome VII. The only complete open reading frame within the insert was the class D VPS gene VPS45, which encodes a member of the Sec1 protein family (Cowles et al., 1994; Piper et al., 1994). Most class D VPS gene products are believed to be involved in the fusion of postGolgi vesicles with an endosome-like organelle referred to as the prevacuolar compartment (PVC; Fig. 2; Raymond et al., 1992; Cowles et al., 1994; Piper et al., 1994; Becherer et al., 1996; Burd et al., 1997; Webb et al., 1997). Mutants in this vps class accumulate intracellular vesicles, have a large central vacuole, and exhibit defects in vacuole inheritance and acidification. Upon further analysis we also found that mts1-1 mutant cells have vacuolar acidification and inheritance defects (data not shown). Confirmation that mts1-1 is allelic to VPS45 was obtained by showing that the temperature sensitivity of mts1-1 cells was not complemented upon crossing to a vps45-4 strain and by integrative transformation and segregation analysis (data not shown).
Figure 2.

Proposed sorting steps blocked by vps and other trafficking mutants analyzed in Table II. The sorting steps shown and the placement and grouping of mutants are based on a large number of studies (Raymond et al., 1992; Stack et al., 1995, and citations therein). In the Vps pathway cargo molecules are sorted at the late Golgi/TGN into transport vesicles that fuse with a PVC/ endosomal compartment. This latter step is blocked in many of the original class D vps mutants, such as vps45. Exit from the PVC for transport to the vacuole and for recycling back to the Golgi is thought to require the function of the class E mutants. Class C vps mutants block the final step of transport to the vacuole. Some of the sorting mutants analyzed in Table II are indicated with a question mark or are not shown in the figure (vps8, vps39, pep8, and dnm1). In these cases the placement in the sorting pathways is more ambiguous. Mutants showing synthetic growth phenotypes with apm3-Δ are indicated with an asterisk.
apm3-Δ Causes Synthetic Growth Phenotypes with Nearly All Class D vps Mutants
Since apm3-Δ showed synthetic lethality with the class D mutant vps45 in the sectoring screen, we investigated whether other vps mutations cause synthetic growth phenotypes when combined with apm3-Δ (Fig. 2; Table II). The vps mutants have been classified into six groups (A–F) based primarily on vacuolar morphology and localization of vacuolar proteins (Raymond et al., 1992). In general, these classes represent distinct intermediate steps along the transport pathway that CPY and other vacuolar hydrolases take from the Golgi to the vacuole (for review see Stack et al., 1995). Growth was examined at various temperatures in isogenic pairs of apm3-Δ vps double mutants with and without a complementing APM3 plasmid (Table II). We observed synthetic growth defects when apm3-Δ was combined with all class D vps mutants, with the exception of vps34. However, it has been proposed that Vps34p and Vps15p may function at a step in the vps pathway distinct from other class D mutants (Fig. 2) (Raymond et al., 1992; Munn and Riezman, 1994; Stack et al., 1995). In addition, we saw no synthetic interaction of apm3-Δ with vps8-Δ, which has recently been reclassified as a potential class D vps mutant (Horazdovsky et al., 1996).
Table II.
Genetic Interactions of apm3-Δ with Vesicle Transport Mutants*
| vps class | Genotype | vps allele | Growth at 25° or 30°C | Growth at 34° or 37°C | ||||
|---|---|---|---|---|---|---|---|---|
| vps mutants | ||||||||
| A/D | vps8 APM3 | Δ | ++ | ++ | ||||
| vps8 apm3-Δ | Δ | ++ | ++ | |||||
| A | vps35 APM3 | Δ | ++ | ++ | ||||
| vps35 apm3-Δ | Δ | ++ | ++ | |||||
| B | vam6 (vps39) APM3 | vpl18-9 | ++ | ++ | ||||
| vam6 (vps39) apm3-Δ | vpl18-9 | ++ | ++ | |||||
| B | ypt7 APM3 | Δ | ++ | ++ | ||||
| ypt7 apm3-Δ | Δ | ++ | ++ | |||||
| C | pep5 (vps11, end1) APM3 | Δ | ++ | ± | ||||
| pep5 (vps11, end1) apm3-Δ | Δ | ++ | ± | |||||
| C | pep3 (vps18) APM3 | Δ | ++ | + | ||||
| pep3 (vps18) apm3-Δ | Δ | ++ | + | |||||
| C | vps33 (pep14, slp1) APM3 | vp125-6 | ++ | + | ||||
| vps33 (pep14, slp1) apm3-Δ | vpl25-6 | ++ | ± | |||||
| D | vps3 (pep6) APM3 | vpl3-1 | ++ | ++ | ||||
| vps3 (pep6) apm3-Δ | vpl3-1 | ++ | − | |||||
| D | pep12 (vps6) APM3 | Δ | ++ | ++ | ||||
| pep12 (vps6) apm3-Δ | Δ | ++ | − | |||||
| D | vps9 APM3 | vpl31-2 | ++ | ++ | ||||
| vps9 apm3-Δ | vpl31-2 | ++ | + | |||||
| D | vps15 APM3 | vpl19-10 | ++ | ++ | ||||
| vps15 apm3-Δ | vpl19-10 | ++ | ± | |||||
| D | pep7 (vps19) APM3 | Δ | ++ | ++ | ||||
| pep7 (vps19) apm3-Δ | Δ | ++ | ± | |||||
| D | vps21 APM3 | vpt21 | ++ | ++ | ||||
| vps21 apm3-Δ | vpt21 | ++ | − | |||||
| D | vps34 (pep15, end12) APM3 | vpl7-1 | ++ | + | ||||
| vps34 (pep15, end12) apm3-Δ | vpl7-1 | ++ | + | |||||
| D | vps45 APM3 | Δ | ++ | ++ | ||||
| vps45 apm3-Δ | Δ | ++ | − | |||||
| D | vps45 APM3 | mts1-1 | ++ | ± | ||||
| vps45 apm3-Δ | mts1-1 | ± | − | |||||
| D | vps45 APM3 | vps45-13 | ++ | ++ | ||||
| vps45 apm3-Δ | vps45-13 | ++ | ++ | |||||
| E | vps4 (end13) APM3 | vpl4-1 | ++ | ++ | ||||
| vps4 (end13) apm3-Δ | vpl4-1 | ++ | ++ | |||||
| E | vps27 APM3 | Δ | ++ | ++ | ||||
| vps27 apm3-Δ | Δ | ++ | ++ | |||||
| F | vps1 APM3 | Δ | ++ | + | ||||
| vps1 apm3-Δ | Δ | + | ± | |||||
| F | pep8 (vps26) APM3 | vpt4 | ++ | ++ | ||||
| pep8 (vps26) apm3-Δ | vpt4 | ++ | ++ | |||||
| Genotype | Mutant allele | Growth at 25° or 30°C | Growth at 34° or 37°C | |||||
| Other mutants tested | ||||||||
| sec1 APM3 | sec1-1 | ++ | ± | |||||
| sec1 apm3-Δ | sec1-1 | ++ | ± | |||||
| sec4 APM3 | sec4-8 | ++ | − | |||||
| sec4 apm3-Δ | sec4-8 | ++ | − | |||||
| sec6 APM3 | sec6-4 | ++ | ± | |||||
| sec6 apm3-Δ | sec6-4 | ++ | ± | |||||
| dnm1 APM3 | Δ | ++ | ++ | |||||
| dnm1 apm3-Δ | Δ | ++ | ++ | |||||
| end3 APM3 | end3-1 | ++ | ++ | |||||
| end3 apm3-Δ | end3-1 | ++ | − | |||||
| end4 APM3 | end4-1 | ++ | ± | |||||
| end4 apm3-Δ | end4-1 | ++ | − | |||||
| vac8 APM3 | Δ | ++ | ++ | |||||
| vac8 apm3-Δ | Δ | ++ | ++ | |||||
| vma2 (vat2) APM3 | Δ | ++ | ++ | |||||
| vma2 (vat2) apm3-Δ | Δ | ++ | ++ |
Isogenic pairs of strains were generated by transforming the indicated double mutants with an APM3 plasmid. For simplicity, apm3-Δ strains complemented with pAPM3 are shown as APM3 genotype. In the crosses of apm3-Δ to vac8-Δ and dnm1-Δ isogenic pairs were not made, but all spores from crosses (±apm3-Δ) grew normally at all temperatures tested. Growth of strains was scored at 25°, 30°, 34°, and 37°C. The score shown is for the temperature under which the observed differences ±APM3 was greatest, or if no difference was seen, at the highest temperature that allowed growth. For example, for some strains the strongest synthetic interaction was observed at 34°C, but at 37° both strains were completely inviable, since the nonpermissive temperature for the vesicle transport mutant had been reached. Scores are designated: ++, wt growth; +, poor growth; ±, very poor growth; −, dead/no growth.
Significant variation in the severity of the growth defects was seen when apm3-Δ was combined with the class D mutants. This may be because of the fact that vps null alleles result in stronger genetic interactions with apm3-Δ than vps alleles that result in partial loss of function (e.g., compare apm3-Δ vps45-Δ and apm3-Δ vps45-13ts). However, in other cases, it appears that the differences in the strength of the growth phenotype reflect differences in the functions of the Vps proteins. For example, apm3-Δ pep12-Δ and apm3-Δ vps45-Δ have much more severe growth phenotypes than apm3-Δ pep7-Δ. Interestingly, in crosses we found apm3-Δ mts1-1 spore segregants were viable, although they showed a very severe synthetic growth phenotype. Similar results were obtained when mutants in each of the other components of the yeast AP-3 complex were crossed with mts1-1 (data not shown).
Among the other vps mutants, a synthetic growth defect was observed when apm3-Δ was combined with a deletion of VPS1, which codes for the dynamin homologue involved in vesicle budding from the late Golgi for transport to the vacuole (Vater et al., 1992; Nothwehr et al., 1995). Note that the other class F mutant (pep8/vps26) showed no synthetic growth phenotype when combined with apm3-Δ. We also found a weak genetic interaction between apm3-Δ and vps33, but no other class C vps mutant tested (Table II). Class C mutants have little or no visible vacuoles and affect a late transport step from the prevacuole/endosome to the vacuole (Raymond et al., 1992; Darsow et al., 1997). Interestingly, Vps33p is another member of the Sec1p/Vps45p family involved in vesicle fusion (Banta et al., 1990; Wada et al., 1990), suggesting possible overlap in the function of Vps45p and Vps33p. However, there does not seem to be a general effect of apm3-Δ on mutants for all Sec1p family members, since no such interaction was observed between apm3-Δ and sec1-1 (Table II).
We also tested for genetic interactions between apm3-Δ and several other mutations affecting the late secretory and endocytic pathways (Table II; Fig. 2). No synthetic growth phenotypes were observed when apm3-Δ was combined with the late secretory mutants sec1-1, sec4-8, or sec6-4 (Novick et al., 1981), indicating that the simple accumulation of vesicles is not sufficient for the synthetic growth defects described above. No effect of apm3-Δ was observed in a dnm1-Δ mutant, which lacks a dynamin homologue involved in late endocytic transport (Gammie et al., 1995). The apm3-Δ mutant did lower the permissive growth temperature of end3-1 and end4-1 mutants, which are defective in the internalization step of endocytosis (Raths et al., 1993). This contrasts with our earlier findings showing that AP-3 mutants, in fact, suppress the growth and a-factor receptor internalization defects of strains mutant for yeast PM casein kinase 1 (Panek et al., 1997). Therefore, loss of AP-3 function does not suppress the growth phenotype of all endocytic mutants.
Since class D vps mutants have multiple phenotypes, additional crosses were made in an effort to gain information about which defect(s) was responsible for the genetic interaction with apm3-Δ (Table II). Mutations in several of the VAC genes (e.g., VAC8) result in vacuole inheritance defects, yet sorting of CPY is not affected (Wang et al., 1996). However, the growth of the apm3-Δ vac8-Δ double mutant was normal. Class D vps mutants also are defective for acidification of the vacuole (Raymond et al., 1992). VMA2/VAT2 encodes the 60-kD peripheral membrane regulatory subunit of the vacuolar ATPase (Yamashiro et al., 1990), but, again, apm3-Δ did not alter the growth of vat2-Δ cells. These data indicate that interruption of the transport function in class D vps mutants is most likely responsible for the synthetic growth phenotypes observed when combined with apm3-Δ.
Transport and Processing of ALP Are Defective in apm3-Δ Cells
The genetic interactions described above suggested that in the absence of the vesicular transport step performed by the class D Vps proteins the function of Apm3p, and hence the AP-3 complex, becomes crucial. We considered the possibility that essential endosomal or vacuolar processes require the function of at least one route to the vacuole, and that the AP-3 mutants blocked one of these pathways. The endocytic pathway to the vacuole was not a likely candidate because we have previously shown that AP-3–deficient cells have normal internalization and degradation of Ste3p (Panek et al., 1997). Therefore, we tested whether the second internal pathway from the late Golgi to the vacuole, parallel to the Vps pathway, was affected in the absence of the AP-3 complex. This pathway has been defined by the vacuolar membrane protein, ALP, whose localization on the vacuole is independent of many of the VPS gene products, including the class D proteins (Raymond et al., 1992; Cowles et al., 1997; Piper et al., 1997). First, we examined the processing of ALP in apm3-Δ strains by pulse-chase/immunoprecipitation analysis (Fig. 3 A). In wt cells precursor ALP (pALP) was transported to the vacuole and underwent PEP4-dependent proteolytic maturation with a half-time of ∼6 min. In contrast, in the apm3-Δ mutant ALP processing was severely delayed, extending the half-life of pALP to ∼45 min (average of 5 experiments). Interestingly, by 120 min the major species in apm3-Δ cells was a smaller, abberantly processed form (*ALP), which migrated at ∼66 kD. Vacuolar proteases were required for processing of ALP in apm3-Δ cells, as only pALP accumulated in an apm3-Δ pep4 prb1 strain (Fig. 3 B). These data suggested that some ALP was delivered to the vacuole in apm3-Δ cells, albeit with slowed kinetics. Alternatively, this ALP processing may have occurred in a novel compartment containing PEP4-dependent hydrolytic activity.
Figure 3.

ALP processing is defective in AP-3 mutant cells. (A) Pulse-chase immunoprecipitation of ALP from isogenic wt (SL1652 + pDS24 [pAPM3, CEN]) and apm3-Δ (SL1652) cells. Cells were labeled with [35S]methionine and [35S]cysteine for 10 min, chased for indicated times (min), and subjected to IP with ALP antiserum. (B) Steady state levels of ALP forms in cells of the indicated genotypes (wt, SL1463; pho8-Δ, SNY17; aps3-Δ, LRB739; apm3-Δ, SL1652; apl5-Δ, HPY20; apl6-Δ, LRB858). Cells were lysed and prepared for immunoblotting with affinity-purified ALP antibodies. pALP, precursor ALP; mALP, mature ALP; *ALP, soluble lumenal ALP are indicated.
The ALP processing defect in apm3-Δ cells was also observed at steady state as shown by immunoblotting of whole cell extracts (Fig. 3 B). In wt cells ALP exists predominantly as mature ALP (mALP) with a small amount of *ALP at steady state. However, in apm3-Δ cells, pALP was also a major species. Cells carrying a deletion in any of the four AP-3 subunit-encoding genes (apl5-Δ, apl6-Δ, apm3-Δ, and aps3-Δ) gave the identical distribution of ALP (Fig. 3 B). ALP processing was normal in strains carrying a mutation in each of the other yeast AP μ chain-encoding genes (apm1-Δ, apm2-Δ, and apm4-Δ) or combinations of these, and none of these other mutants enhanced the ALP phenotype when combined with apm3-Δ (data not shown).
Since ALP was delayed in transport to the vacuole in apm3-Δ cells, we investigated the location of ALP by subcellular fractionation (Fig. 4). As expected, in wt cells the vast majority of mALP pelleted at 13,000 g, which sediments vacuoles as well as PM, ER, and other large membrane organelles (Horazdovsky and Emr, 1993). However, in apm3-Δ cells the three forms of ALP fractionated independently. The distribution of mALP was similar to that in wt cells, predominantly in the P13K fraction. In contrast, pALP was found in the high speed pellet (P100K) where Golgi, endosomes, and other small organelles and vesicles sediment (Vida et al., 1993), suggesting accumulation in a novel compartment. *ALP remained soluble even after the high speed spin (S100K). Because of its solubility and size, it is likely that *ALP is a product of a cleavage(s) at the lumenal juxtamembrane region of the protein, resulting in a soluble form that is released during lysis and fractionation from a compartment(s) that is osmotically fragile. The cleavage of ALP at the juxtamembrane region would remove as many as half of the sulfur-containing amino acids from the protein. This could explain the apparent loss of total ALP signal during the course of the pulse-chase analysis (Fig. 3 A).
Figure 4.

Fractionation of ALP forms in AP-3 mutant cells. Cells from an isogenic pair of strains (wt, SL1652 + pDS24 [pAPM3, CEN]; apm3-Δ, SL1652) were subjected to subcellular fractionation, followed by immunoblot analysis with anti-ALP antibodies. ALP forms are as indicated in Fig. 3. S13K, 13,000 g supernatant; S100K, 100,000 g supernatant; P100K, 100,000 g pellet.
ALP Is Mislocalized in apm3-Δ cells
The delayed processing and subcellular fractionation of ALP suggested that ALP is missorted and may localize to a novel compartment. Therefore, we used immunofluorescence microscopy to localize ALP in apm3-Δ cells (Fig. 5). As expected, in wt cells affinity-purified ALP antibodies stained the vacuole membrane, colocalizing with the vacuolar ATPase. The vacuolar staining pattern for the ATPase was normal in the apm3-Δ mutant, consistent with our findings that the Vps pathway is intact in AP-3 mutants. In contrast, in the apm3-Δ mutant ALP staining was detected primarily in a large number of small punctate structures throughout the cell. Infrequently, faint rim staining of the vacuole was seen, which was often more visible in pep4 strains (see Fig. 8 C). Nevertheless, even in pep4 strains, the punctate pattern was predominant.
Figure 5.
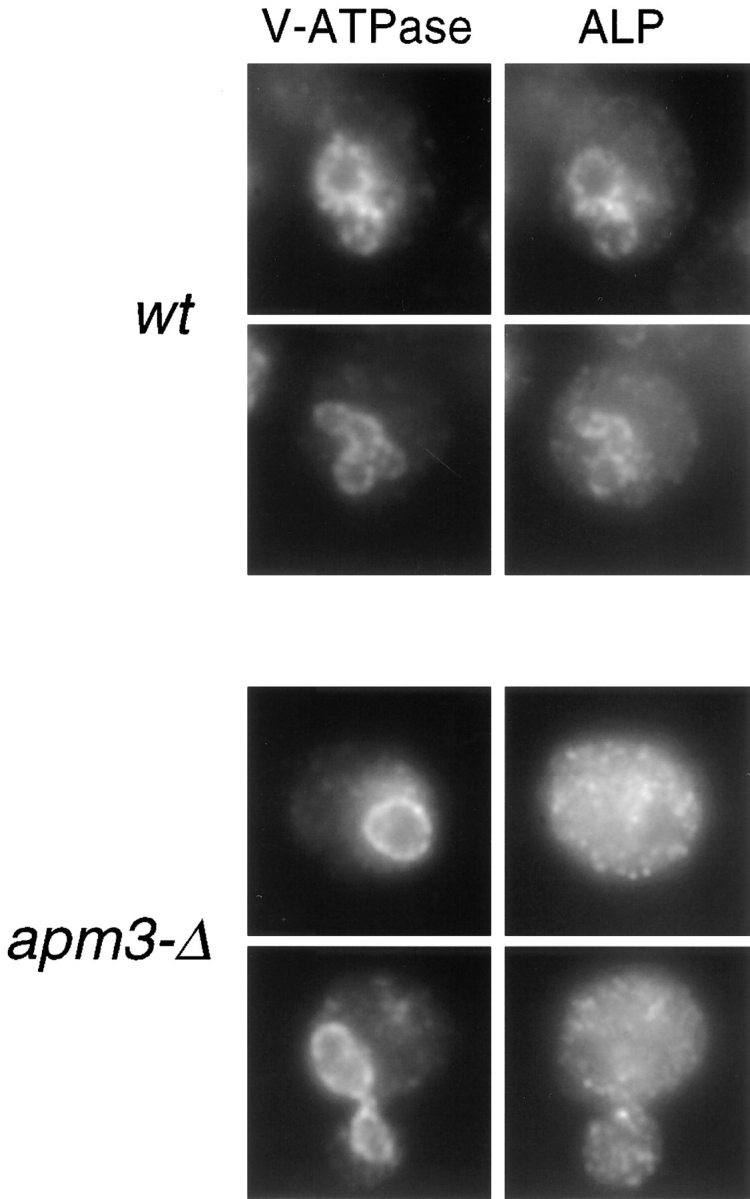
Immunofluorescence localization of ALP in AP-3 mutant cells. wt (SL1653 + pDS24 [pAPM3, CEN]) and apm3-Δ (SL1653) cells were prepared for immunofluorescence. The V-ATPase was detected with monoclonal antibody 13D11 and goat anti–mouse Cy3. ALP was visualized with affinity-purified ALP antibodies and goat anti–rabbit FITC secondary.
Figure 8.
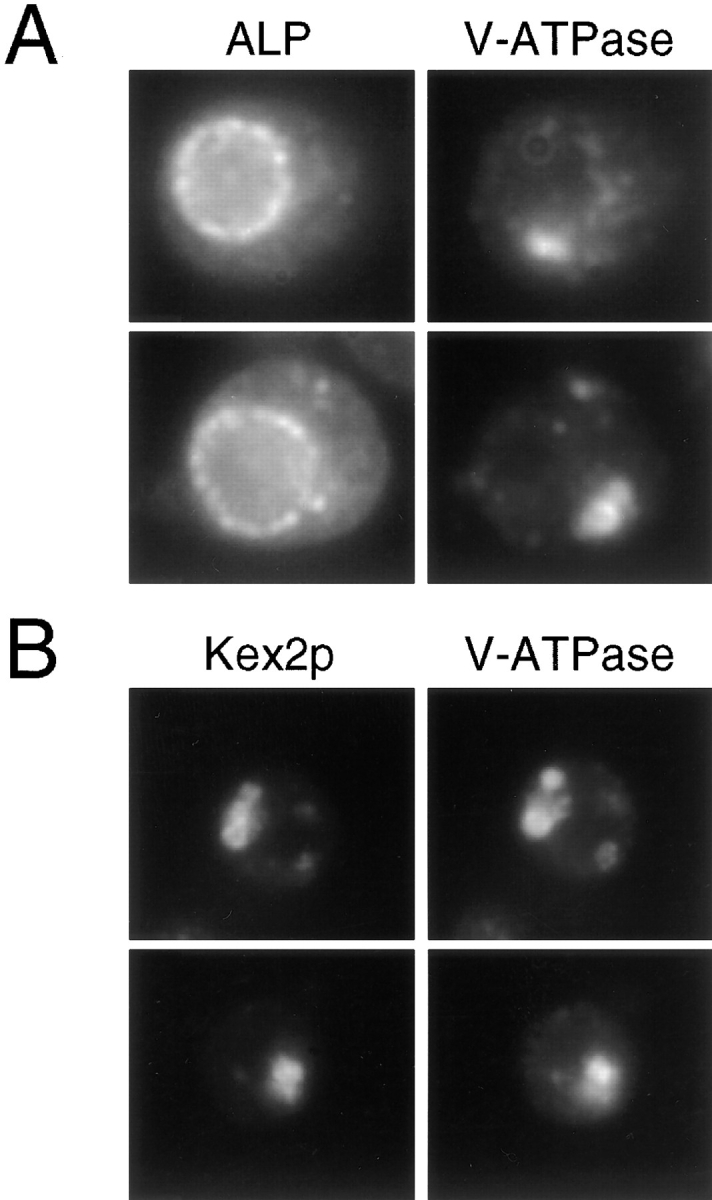
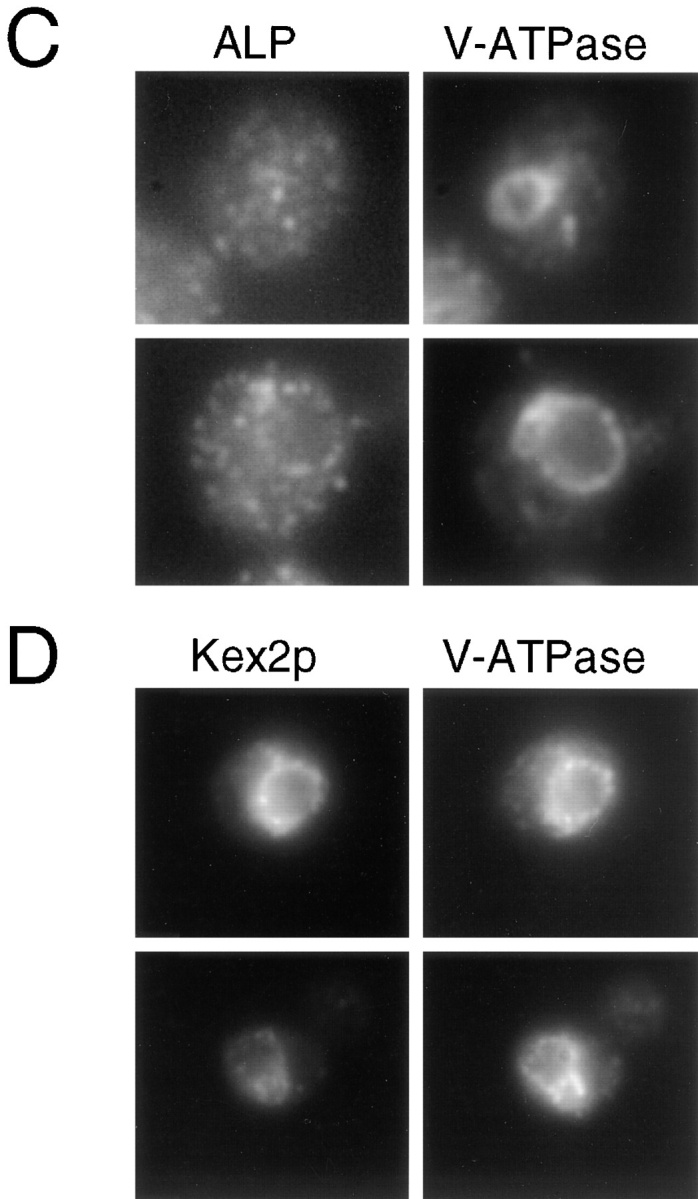
Loss of AP-3 function results in disappearance of the class E PVC. ALP, V-ATPase, and Kex2p were localized by immunofluorescence as described in Figs. 5 and 6. (A) Localization of ALP and the V-ATPase in the class E prevacuole in vps4 strain, SF838-1D. (B) vps4 (SF838-1D + YCpKX22) cells stained for Kex2p and the V-ATPase. (C) Localization of ALP and the V-ATPase in the vps4 apm3-Δ strain, SL2845. (D) Colocalization of Kex2p and the V-ATPase to the vacuole in vps4 apm3-Δ cells (SL2845 + YCpKX22).
Previous immunofluorescence studies have shown that yeast Golgi or late Golgi proteins display a distinctive staining pattern with several spots localized throughout the cytoplasm (Redding et al., 1991; Graham et al., 1994). Therefore, we examined whether the ALP staining in AP-3 deficient cells was consistent with such a Golgi pattern by localizing Kex2p, a late Golgi/TGN marker. As shown in Fig. 6, the Kex2p localization was identical in wt and apm3-Δ cells, each showing the characteristic Golgi pattern of ∼10 large spots in each cell. This was quite distinct from the overall smaller size and larger number of spots observed for ALP staining in the apm3-Δ mutant (Fig. 5). We conclude that the overall structure of the Golgi and vacuole appear normal in cells lacking AP-3 function, but substantial ALP accumulates in a distinct compartment.
Figure 6.

Golgi staining is normal in AP-3 mutant cells. wt (SL1463 + YCpKX22) and apm3-Δ (SL1652 + YCpKX22) cells were stained with affinity-purified Kex2p antibodies followed by goat anti–rabbit FITC secondary. The plasmid YCpKX22 was used for overexpression of Kex2p to enable intracellular visualization as described previously (Wilcox et al., 1992).
Since immunofluorescence analysis indicated that ALP accumulates in small membrane structures in apm3-Δ cells, we examined isogenic wt and mutant cells by thin section transmission EM using two different fixation/staining methods (Kaiser and Schekman, 1990; Govindan et al., 1995). Upon examination of >50 cells of each genotype by each method, we found that wt and apm3-Δ cells had the same types and similar numbers of membrane structures (data not shown). Surprisingly, we observed no accumulation of small vesicles in the apm3-Δ cells. This suggests that ALP may accumulate in a normally occurring, preexisting membrane compartment(s) in AP-3–deficient yeast.
Partial Rerouting of ALP through the Vps Pathway in apm3-Δ Mutants
The pulse-chase and immunoblot analyses of AP-3 mutants (Fig. 3) indicated that some ALP arrives at the vacuole and is processed to the mature form, albeit slowly. If the normal ALP pathway is blocked, it could arrive at the vacuole by rerouting through the cell surface followed by endocytosis, as it does in a vps1 mutant (Nothwehr et al., 1995). Alternatively it could be delivered to the vacuole by the Vps pathway as it does when the sorting information in the ALP cytoplasmic tail is removed (Cowles et al., 1997; Piper et al., 1997). To investigate whether ALP traffics through the cell surface in AP-3 mutants, we compared ALP processing in an apm3-Δ end4-1 strain with its isogenic apm3-Δ END4 counterpart. If rerouting of ALP to the vacuole requires endocytosis, we would expect the end4 mutation to further slow or block ALP processing in the apm3-Δ mutant. However, the kinetics and degree of processing of ALP at 37°C were identical in the apm3-Δ strain with or without End4p function (Fig. 7 A). Combining apm3-Δ with end3-1 or sec1-1 also did not further delay the processing of ALP (data not shown), demonstrating that ALP does not transit the PM in AP-3–deficient cells.
Figure 7.

ALP reaches the vacuole via the Vps Pathway in AP-3 mutant cells. Isogenic pairs of strains were shifted to 37°C for 30 min, pulse labeled for 10 min, chased for the indicated times (min), and then subjected to IP with ALP antiserum. (A) apm3-Δ (SL2767 + pEND4, CEN) and apm3-Δ end4-1 (SL2767). (B) apm3-Δ (SL1904) and apm3-Δ vps45-13 (SL2956).
To test whether residual ALP processing in AP-3 mutants occurred by rerouting through the Vps pathway, we combined apm3-Δ with the vps45-13 mutation, which causes a temperature-dependent vps phenotype (Piper et al., 1994). It was important to use an allele that is ts for function, since a constitutive block of the Vps pathway, as would occur with vps45-Δ, would deplete the vacuole of hydrolytic activity and prevent pALP processing with or without apm3-Δ. An apm3-Δ vps45-13 mutant and an isogenic apm3-Δ VPS45 strain were grown at 25°C and then shifted to 37°C for pulse-chase labeling and IP (Fig. 7 B). In this case pALP was significantly stabilized by the presence of the vps45-13 mutation. In other control experiments (data not shown) we found that at 25°C, the processing of ALP in the apm3-Δ vps45-13 mutant was identical to that of apm3-Δ alone. Also, the isogenic APM3 vps45-ts strain gave wt processing of ALP at 37°C. Moreover, the CPY processing/sorting defect of vps45-13 was dependent upon the shift to 37°C, but independent of APM3 (data not shown). This indicates that vacuolar hydrolytic activity was present in the cell at the time of the shift to the nonpermissive temperature even though the vps45-ts allele caused a temperature-dependent block in the Vps pathway. Therefore, pALP was rerouted through the Vps pathway in the AP-3–deficient strain and was unable to continue to the vacuole in the apm3-Δ vps45-13 strain at 37°C. Interestingly, there was still a significant accumulation of *ALP in the apm3-Δ vps45-13 mutant at 37°C, although it appeared with slower kinetics than in the apm3-Δ VPS45 strain (Fig. 7 B). The compartment where this processing occurs is still not clear.
Loss of AP-3 function Allows Membrane Cargo Trapped in the Vps Class E Prevacuole to Be Delivered to the Vacuole
The above studies suggested that some ALP was delivered to the vacuole via the Vps pathway and not via endocytosis in apm3-Δ cells. We thought it should also be possible to confirm these findings by immunofluorescence localization of ALP in appropriate double mutants. Consistent with the pulse-chase analysis, ALP did not accumulate at the cell surface in apm3-Δ end4-1 cells shifted to 37°C, but showed the identical staining pattern to that of apm3-Δ END4 cells (data not shown). This supports our conclusion that ALP takes an intracellular pathway(s) in AP-3 mutants.
To examine traffic through the Vps pathway, we took advantage of a class E vps mutant, vps4, which accumulates an exaggerated endosomal/prevacuolar compartment (class E compartment/PVC; Raymond et al., 1992; Babst et al., 1997; Finken-Eigen et al., 1997). In class E mutants late Golgi proteins (e.g., DPAP-A), the vacuolar ATPase, endocytosed cell surface receptors (e.g., Ste2p), and the CPY sorting receptor (Vps10p) become trapped in the class E PVC and appear as one or two large bright dots adjacent to the vacuole by immunofluorescence (Raymond et al., 1992; Piper et al., 1995; Babst et al., 1997; Bryant and Stevens, 1997; Finken-Eigen et al., 1997). In contrast, ALP localization to the vacuolar membrane is unaffected in class E vps mutants (Raymond et al., 1992; Piper et al., 1997). Therefore, we reasoned that if ALP is partially rerouted through the Vps pathway in the absence of AP-3 function, we might observe accumulation of ALP in the class E PVC in an apm3-Δ vps4 double mutant. In agreement with previous studies with class E mutants, the V-ATPase and the late Golgi marker, Kex2p, localized to one or two large perivacuolar spots, whereas ALP resided in the vacuole membrane in vps4 cells expressing Apm3p (Fig. 8, A and B). However, in the vps4 apm3-Δ mutant, ALP did not accumulate in a perivacuolar region, but showed a similar dispersed punctate staining throughout the cell (Fig. 8 C) as seen with the apm3-Δ mutation alone. More to our surprise, when vps4 apm3-Δ cells were stained to localize the V-ATPase, the class E compartment had disappeared and the ATPase subunit was found on the vacuole membrane (Fig. 8 C). Kex2p also appeared on the vacuolar membrane colocalizing with the V-ATPase in vps4 apm3-Δ cells (Fig. 8 D). These results indicate that the apm3-Δ mutation did not actually suppress the class E vps phenotype, since Kex2p (and presumably other late Golgi proteins) did not recycle to the Golgi, but rather was transported to the vacuole. In addition, p2CPY was still secreted from the cell in the double mutant (data not shown). Similar results were obtained in vps27-Δ apm3-Δ cells (data not shown), indicating that this was not specific to the vps4 class E mutant. These findings suggest that the loss of Apm3p allowed the class E prevacuole to fuse directly with the vacuole membrane. Alternatively, prevacuolar structures might have acquired the ability to fuse with themselves and/or mature into a larger vacuole-like organelle (see Discussion). However, substantial ALP was still trapped in small vesicular organelles, distinctive to the AP-3 mutants.
Discussion
Since the identification of AP-3 in animal cells, much effort has been directed at determining the function of this novel adaptor-related complex. This complex does not appear to colocalize or copurify with clathrin and, therefore, may function independently of clathrin coats (Newman et al., 1995; Simpson et al., 1996; Dell'Angelica et al., 1997b ; Simpson et al., 1997). Cytological studies have suggested that the mammalian AP-3 complex is involved in transport from the TGN or functions in an endosomal compartment (Simpson et al., 1996; Dell'Angelica et al., 1997a ; Simpson et al., 1997). Further information on the function of AP-3 came from the finding that the garnet gene of Drosophila encodes a protein closely related to the δ subunit of AP-3 (Ooi et al., 1997; Simpson et al., 1997). Mutations in garnet lead to reduced pigmentation of the eyes and other tissues. Pigment cells are present, but these have few or no pigment granules, which are thought to be lysosome-like organelles. Therefore, it was suggested that AP-3 might be involved in sorting to or biogenesis of lysosomal organelles. Our studies showing a requirement of AP-3 for processing and sorting of ALP support this idea, and provide direct functional evidence that the AP-3 complex is required for sorting to the vacuole, a lysosome-like compartment. Interestingly, the pathway that AP-3 mutants block is not the well studied Vps pathway, but the alternate route. This is consistent with the hypothesis that the garnet mutations affect formation of a specialized lysosome-like organelle, which might have different trafficking requirements for biogenesis than normal lysosomes.
Where the yeast AP-3 complex functions in the alternative pathway from the late Golgi to the vacuole still needs to be defined. Presumably, the AP-3 complex serves as a coat protein; binding to cytoplasmic tails, sequestering cargo proteins, and directing vesicle formation. It is possible that the AP-3 complex binds to the cytoplasmic face of the late Golgi/TGN, directing selective cargo (e.g., ALP) away from the secretory and Vps pathways. The pALP that accumulated in the apm3-Δ mutant sedimented at 100,000 g where Golgi, endosomes, and small vesicles are found. However, immunofluorescence staining indicated that ALP localized to structures distinct from the late Golgi protein Kex2p, which showed a normal localization pattern. Also, ER and Golgi modifications of ALP occurred with normal kinetics in AP-3 mutants (Fig. 3 A), suggesting that both early and late compartments of the Golgi are unaffected in the absence of the adaptor. Nonetheless, the AP-3 complex could function at the late Golgi/ TGN and ALP becomes trapped in a subcompartment budding from this organelle.
We also found that substantial ALP was diverted to the Vps pathway in the AP-3 mutants, but its processing was very slow. Therefore, the ALP-staining structures may represent a normal subcompartment along the Vps pathway and this was visualized in the apm3 mutant because ALP's transit through this compartment was delayed. Independent studies have indeed shown that when the sorting information in the cytoplasmic tail of ALP is mutated, it traffics through the Vps pathway, resulting in a kinetic delay in the mutant protein's arrival at the vacuole (Piper et al., 1997). However, the mutant ALP did not appear to localize with the punctate staining pattern we observed in the AP-3 mutant.
This raises an alternative possibility: ALP is able to enter its normal trafficking pathway but becomes trapped in an intermediate compartment in the adaptor mutant. In this case, AP-3 might not be involved directly in cargo selection at the late Golgi, but could function at some later compartment, such as another endosomal/prevacuole intermediate. Over time, sorting receptors or other membrane components of the sorting machinery might not be recycled to the Golgi, resulting in ALP slowly leaking into the Vps pathway. The punctate staining pattern suggests that the ALP might be trapped in vesicles that cannot fuse with their target membrane, such as an endosomal compartment or the vacuole itself, although by EM there was no dramatic accumulation of vesicles like that seen with class D vps mutants. If such a fusion defect existed, it might be caused by reduced recycling of some component(s) (e.g., a vesicle-soluble-N-ethylmaleimide-sensitive factor attachment protein receptor [v-SNARE]) to the Golgi that would need to be included in transport vesicles for fusion at a later step.
An endosomal function for AP-3 is also consistent with the findings that AP-3 mutants partially suppress the growth and Ste3p internalization defects caused by yeast PM casein kinase 1 deficiency (Panek et al., 1997). If AP-3 functions at an endosomal compartment where the endocytic and Golgi to vacuole pathways overlap, loss of function of the complex might affect both the ALP pathway and a recycling pathway to the cell surface, resulting in the suppression of the kinase endocytic defect. The genetic interactions of apm3-Δ with end3-1 and end4-1 also suggest a possible functional overlap of an endocytic route and the ALP sorting pathway. Alternative possibilities are that AP-3 has more than one sorting function in the cell or that some of the proteins that normally traverse the ALP pathway are diverted to secretory vesicles and traffic to the cell surface in AP-3 mutants, where they might somehow suppress the yck endocytic defect. Identification of the kinase substrates and localization of the AP-3 complex within the cell should help clarify the connections of the AP-3 complex with both Golgi to vacuole transport and endocytosis.
Although it is not clear at what step the AP-3 complex functions in sorting ALP to the vacuole, existing mutations seem to define at least three distinct steps along this pathway (Fig. 9). The late acting Vps proteins, including Vps33p, a Sec1p homologue (Banta et al., 1990; Wada et al., 1990), Vam3p, a syntaxin-related target (t)-SNARE of the vacuole membrane (Darsow et al., 1997; Nichols et al., 1997; Wada et al., 1997), and Ypt7p, a Rab homologue (Wichmann et al., 1992; Haas et al., 1995; Wada et al., 1996), are thought to function in fusion of endosome- derived membranes or vesicles directly with the vacuolar membrane. These gene products appear to be required for the final stages of transport in both the Vps and ALP pathways. Vps41p/Vam2p (Cowles et al., 1997; Nakamura et al., 1997) and Vps39p/Vam6p, which were recently shown to physically interact (Nakamura et al., 1997), may also act at a distinct, possibly later, step than AP-3. Null mutants of these genes show a block in both CPY sorting and ALP processing and also accumulate fragmented vacuoles containing the V-ATPase and ALP (Raymond et al., 1992; Cowles et al., 1997; Nakamura et al., 1997). VPS41/VAM2 and VPS39/VAM6 were also identified in a screen for cvt mutants, which are defective in the cytoplasm to vacuole transport pathway (Harding et al., 1995). However, recent studies using a vps41-ts allele suggested that the primary defect resulting from loss of Vps41p function is in the ALP pathway, since only after prolonged periods at the nonpermissive temperature did the vps sorting defect surface (Cowles et al., 1997). These phenotypes are quite distinct from those found in AP-3 null mutants which exhibit no obvious vps phenotypes, localize the V-ATPase to the vacuole normally and show no extensive elaboration of any abnormal membrane organelles. AP-3 is also not required for the Cvt pathway (Huang, K., and S. Lemmon, unpublished observations). Therefore, the AP-3 complex most likely acts at a different step than the Vps41p(Vam2p)– Vps39p(Vam6p) complex.
Figure 9.

Models for the Vps-dependent and alternative ALP pathways to the vacuole. (A) Proteins such as the Vps10p CPY sorting receptor, Kex2p and the vacuolar ATPase membrane protein, Vph1p, exit the late Golgi in transport vesicles, which then fuse with the prevacuole. This fusion is dependent upon the class D Vps proteins, such as Vps45p. In a step dependent upon the class E Vps proteins (e.g., Vps4p), late Golgi proteins and Vps10p are recycled to the Golgi from the PVC, whereas resident vacuolar proteins, such as Vph1p and soluble vacuolar hydrolases continue onto the vacuole in a step dependent on late acting Vps proteins (e.g., Vps33p and Vam3p). As shown in this study, ALP transport through the alternative route to the vacuole is dependent upon AP-3 function. It is not clear how many intermediate steps are required for transport of ALP to the vacuole and at what step AP-3 actually functions. Shown here is the possibility that AP-3 is involved in cargo selection at the late Golgi and that Vps41p/Vam2p acts at second, later step in the ALP pathway. Final delivery of ALP to the vacuole is also dependent on the Vps33p and Vam3p. The vacuolar t-SNARE, Vam3p, may also transit the alternative ALP pathway to the vacuole. (B) Much of the existing information on transport of cargo molecules to the vacuole is consistent with a model in which the Vps prevacuole identified in class E mutants is an early endosomal compartment that intersects with endocytic traffic. In this case, convergence of the alternative ALP and Vps pathways would occur at a late endosome. The final delivery of constituents from both the Vps and ALP pathways would still be dependent upon the class C and other late acting Vps components. The possibility that AP-3 is involved in recycling from an endosomal compartment back to the Golgi is also indicated in this model (see Discussion).
In our attempt to further characterize the pathway responsible for the residual ALP processing in AP-3 mutant cells we obtained an astonishing result. In the context of a vps class E mutant, loss of AP-3 function resulted in reappearance of the V-ATPase on the vacuolar membrane. Strictly speaking, this was not suppression of the class E phenotype(s) since Kex2p did not return to the Golgi, but was also transported to the vacuole. In addition, p2CPY was secreted from the cell (data not shown), indicating these cells retained their vps phenotype. Interestingly, ALP localization was still predominantly in small vesicular structures throughout the cytosol. One model to explain these observations is that a regulatory protein(s) normally sorted into the ALP–AP-3–dependent pathway becomes diverted through the Vps pathway in the AP-3 mutant. This might now enable fusion of the aberrant class E compartment with the vacuole or permit prevacuole–prevacuole fusion, which could mature into a large vacuolar-like organelle. A candidate for such a protein is Vam3p, which has recently been suggested to traffic through the ALP– AP-3 pathway (Piper et al., 1997). In a normal cell it would be highly desirable for a t-SNARE, such as Vam3p, to bypass at least some compartments of the Vps pathway to prevent premature interaction with its v-SNARE partner, perhaps the newly identified Nyv1p (Nichols et al., 1997). This sort of quality control system would be essential for maintenance of an endosomal/prevacuole compartment, from which proteins need to recycle to the Golgi and through which endocytic traffic must pass.
This model could also explain why the genetic interactions we observed were largely restricted to the combination of apm3-Δ with class D vps mutants, which are blocked before fusion with the prevacuole. Mutants which can form a functional prevacuole or acidifying compartment like the vacuole (such as class E, B, and A vps mutants; Raymond et al., 1992) would be able to survive quite well even in the absence of AP-3 function. There is also evidence that some class D mutants have endocytic defects (pep7; Webb et al., 1997, and vps45; our unpublished observations), suggesting that some components involved in vesicle fusion with the prevacuole may be shared by the endocytic and Golgi to vacuole pathways. In the absence of AP-3 and certain class D genes all three pathways to the vacuole would be blocked.
The model in Fig. 9 shows the requirement of the AP-3 complex for transport of ALP from the late Golgi to the vacuole. At the present time, it is not clear how many intermediate steps there are along the ALP–AP-3 pathway. It is possible that there is a single vesicle intermediate for transport of ALP from the late Golgi directly to the vacuole, or that ALP traffics through a Vps-independent endosomal compartment. Although the model shown suggests AP-3 acts at the level of the late Golgi, as discussed above, it could act at such an endosomal intermediate (e.g., Fig. 9 B). A more speculative model is considered in Fig. 9 B in which Golgi-derived vesicles that follow the Vps pathway initially fuse with an early endosome, and that the ALP- (and Vam3p-) containing vesicles fuse with the late endosome. The Vps pathway would then converge with the ALP pathway at the late endosome/prevacuole. This idea allows for maintenance of an early endosome recycling compartment and is consistent with the observation that Vam3p, Vps33p, Ypt7p, and presumably other late acting Vps proteins are required for a late stage of transport to the vacuole of both ALP and proteins of the Vps pathway. It has been proposed that the class E compartment is an abnormal late endosome/prevacuolar compartment, since it appears as one or two bright spots next to the vacuole (Raymond et al., 1992; Piper et al., 1995; Rieder et al., 1996; Babst et al., 1997), and by EM it appears as stacks of curved membrane cisternae (Rieder et al., 1996; Babst et al., 1997). However, recent studies indicate that the tubular structures of the aberrant class E organelle are more reminiscent of early endosomes observed in yeast (Prescianotto-Baschong and Riezman, 1998). Moreover, morphological analysis of an end13 mutant (end13 is an allele of VPS4 [Munn and Riezman, 1994]) indicates endocytosis is retarded at an early, postinternalization endocytic step (Riezman, H., personal communication). As more gene products involved in the alternative ALP pathway to the vacuole are identified, it should be possible to resolve these two models.
Acknowledgments
This work was supported by a grant from the National Science Foundation (NSF MCB-9305287). J.D. Stepp was supported by National Institutes of Health predoctoral training grant (AG-00105) and K.M. Huang was supported by an individual National Research Service Award (NIH GM17370). S.K. Lemmon is the recipient of a National Science Foundation Career Advancement Award (MCB-9707583).
Abbreviations used in this paper
- *ALP
aberrantly processed ALP
- ALP
alkaline phosphatase
- AP
adaptor protein
- CPY
carboxypeptidase Y
- IP
immunoprecipitation
- mALP
mature ALP
- mts
apm three synthetic lethal mutant
- pALP
precursor ALP
- PIC
protease inhibitor cocktail
- PVC
prevacuolar compartment
- SNARE
soluble N-ethymaleimide-sensitive factor attachment protein receptor
- ts
temperature sensitive
- Vps
vacuolar protein sorting
- wt
wild type
Note Added in Proof
Another report on the role of yeast AP-3 in the sorting of ALP via the alternate pathway to the vacuole has been published recently (Cowles, C.R., G. Odorizzi, G.S. Payne, and S.D. Emr. 1997. The AP-3 adaptor complex is essential for cargo-selective transport to the yeast vacuole. Cell. 91:109–118). In addition, this work provided further evidence that Vam3p also traffics through the alternate pathway.
Footnotes
Address all correspondence to Sandra K. Lemmon, Department of Molecular Biology and Microbiology, Case Western Reserve University, School of Medicine, 10900 Euclid Avenue, Cleveland, OH 44106-4960. Tel.: (216) 368-6279. Fax: (216) 368-3055. E-mail: skl@po.cwru.edu
We are particularly indebted to S. Nothwehr (University of Missouri) for his generous supply of anti-alkaline phosphatase antibodies. M. Carlson (Columbia University), S. Emr (University of California, San Diego, CA), R. Fuller (University of Michigan), E. Jones (Carnegie Mellon University), P. Kane (SUNY Health Science Center at Syracuse, Syracuse, NY), D. Loyaza (Johns Hopkins University, Baltimore, MD), S. Michaelis (Johns Hopkins University), R. Piper (University of Iowa, Iowa City, IA), H. Riezman (University of Basel), L. Robinson (Louisiana State University, Shreveport, LA), T. Stevens (University of Oregon, Eugene, OR), and L. Weisman (University of Iowa) also generously provided strains, plasmids, and/or antibodies. S. Nothwehr, R. Piper, L. Conibear (University of Oregon), P. Kane, L. Robinson, and L. Weisman provided many helpful suggestions. We thank H. Riezman and G. Payne (University of California, Los Angeles, CA) for communicating results before publication. We thank G. Matera (all from case Western Reserve University) and members of his lab for providing frequent use of their Zeiss Axioplan, J. Polak and R.-Z. Wang for their assistance with EM and T. Nilsen for use of his phosphorimager. We also acknowledge E. Noss and M. Otsuka for their help in generation of some of the strains and plasmids used in these studies. Finally, we thank M. Snider, H. Riezman, and members of the Lemmon and Riezman labs for stimulating discussions and critical reading of this manuscript.
References
- Babst M, Sato TK, Banta LM, Emr SD. Endosomal transport function in yeast requires a novel AAA-type ATPase, Vps4p. EMBO (Eur Mol Biol Organ) J. 1997;16:1820–1831. doi: 10.1093/emboj/16.8.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banta LM, Vida TA, Herman PK, Emr SD. Characterization of Vps33p, a protein required for vacuolar protein sorting and vacuole biogenesis. Mol Cell Biol. 1990;10:4638–4649. doi: 10.1128/mcb.10.9.4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin A, Ozierkalogeropoulos O, Denouel A, Lacroute F, Cullin C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. . Nuc Acids Res. 1993;21:3329–3330. doi: 10.1093/nar/21.14.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becherer KA, Rieder SE, Emr SD, Jones EW. Novel syntaxin homologue, Pep12p, required for the sorting of lumenal hydrolases to the lysosome-like vacuole in yeast. Mol Biol Cell. 1996;7:579–594. doi: 10.1091/mbc.7.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Pringle JR. Use of a screen for synthetic lethal and multicopy suppressed mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. . Mol Cell Biol. 1991;11:1295–1305. doi: 10.1128/mcb.11.3.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant NJ, Stevens TH. Two separate signals act independently to localize a yeast late Golgi membrane protein through a combination of retrieval and retention. J Cell Biol. 1997;136:287–297. doi: 10.1083/jcb.136.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd CG, Peterson M, Cowles CR, Emr SD. A novel Sec18p/ NSF-dependent complex is required for Golgi-to-endosome transport in yeast. Mol Biol Cell. 1997;8:1089–1104. doi: 10.1091/mbc.8.6.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowles CR, Emr SD, Horazdovsky BF. Mutations in the VPS45 gene, a SEC1homologue, result in vacuolar protein sorting defects and accumulation of membrane vesicles. J Cell Sci. 1994;107:3449–3459. doi: 10.1242/jcs.107.12.3449. [DOI] [PubMed] [Google Scholar]
- Cowles CR, Snyder WB, Burd CG, Emr SD. Novel Golgi to vacuole delivery pathway in yeast: identification of a sorting determinant and required transport component. EMBO (Eur Mol Biol Organ) J. 1997;16:2769–2782. doi: 10.1093/emboj/16.10.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darsow T, Rieder SE, Emr SD. A multispecifity syntaxin homologue, Vam3p, essential for autophagic and biosynthetic protein transport to the vacuole. J Cell Biol. 1997;138:517–529. doi: 10.1083/jcb.138.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Angelica EC, Ohno H, Ooi CE, Rabinovich E, Roche KW, Bonifacino JS. AP-3: an adaptor-like protein complex with ubiquitous expression. EMBO (Eur Mol Biol Organ) J. 1997a;16:917–928. doi: 10.1093/emboj/16.5.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Angelica EC, Ooi CE, Bonifacino JS. Beta 3A-adaptin, a subunit of the adaptor-like complex AP-3. J Biol Chem. 1997b;272:15078–15084. doi: 10.1074/jbc.272.24.15078. [DOI] [PubMed] [Google Scholar]
- Finken-Eigen M, Rohricht RA, Kohrer K. The VPS4gene is involved in protein transport out of a yeast pre-vacuolar endosome-like compartment. Curr Genet. 1997;31:469–480. doi: 10.1007/s002940050232. [DOI] [PubMed] [Google Scholar]
- Gammie AE, Kurihara LJ, Vallee RB, Rose MD. DNM1, a dynamin-related gene, participates in endosomal trafficking in yeast. J Cell Biol. 1995;130:553–566. doi: 10.1083/jcb.130.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindan B, Bowser R, Novick P. The role of MYO2, a yeast class V myosin, in vesicular transport. J Cell Biol. 1995;128:1055–1068. doi: 10.1083/jcb.128.6.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TR, Seeger M, Payne GS, Mackay VL, Emr SD. Clathrin-dependent localization of alpha 1,3 mannosyltransferase to the Golgi complex of Saccharomyces cerevisiae. . J Cell Biol. 1994;127:667–678. doi: 10.1083/jcb.127.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink GR eds. Guide to yeast genetics and molecular biology. Methods Enzymol. 1991;194:1–933. [PubMed] [Google Scholar]
- Haas A, Scheglmann D, Lazar T, Gallwitz D, Wickner W. The GTPase Ypt7p of Saccharomyces cerevisiaeis required on both partner vacuoles for the homotypic fusion step of vacuole inheritance. EMBO (Eur Mol Biol Organ) J. 1995;14:5258–5270. doi: 10.1002/j.1460-2075.1995.tb00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding TM, Morano KA, Scott SV, Klionsky DJ. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow, E., and D. Lane. 1988. Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, NY. 726 pp.
- Hill JE, Meyers AM, Koerner TJ, Tzagoloff A. Yeast/E. colishuttle vectors with multiple unique restriction sites. Yeast. 1986;2:163–167. doi: 10.1002/yea.320020304. [DOI] [PubMed] [Google Scholar]
- Horazdovsky BF, Emr SD. The VPS16gene product associates with a sedimentable protein complex and is essential for vacuolar protein sorting in yeast. J Biol Chem. 1993;268:4953–4962. [PubMed] [Google Scholar]
- Horazdovsky BF, Cowles CR, Mustol P, Holmes M, Emr SD. A novel RING finger protein, Vps8p, functionally interacts with the small GTPase, Vps21p, to facilitate soluble vacuolar protein localization. J Biol Chem. 1996;271:33607–33615. doi: 10.1074/jbc.271.52.33607. [DOI] [PubMed] [Google Scholar]
- Kaiser C, Schekman R. Distinct sets of SECgenes govern transport vesicle formation and fusion early in the secretory pathway. Cell. 1990;61:723–733. doi: 10.1016/0092-8674(90)90483-u. [DOI] [PubMed] [Google Scholar]
- Munn AL, Riezman H. Endocytosis is required for the growth of vacuolar H+-ATPase-defective yeast: identification of six new ENDgenes. J Cell Biol. 1994;127:373–386. doi: 10.1083/jcb.127.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Hirata A, Ohsumi Y, Wada Y. Vam2/Vps41p and Vam6/Vps39p are components of a protein complex on the vacuolar membranes and involved in the vacuolar assembly in the yeast Saccharomyces cerevisiae. . J Biol Chem. 1997;272:11344–11349. doi: 10.1074/jbc.272.17.11344. [DOI] [PubMed] [Google Scholar]
- Nelson KK, Lemmon SK. Suppressors of clathrin deficiency: overexpression of ubiquitin rescues lethal strains of clathrin-deficient Saccharomyces cerevisiae. . Mol Cell Biol. 1993;13:521–532. doi: 10.1128/mcb.13.1.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman LS, Mckeever MO, Okano HJ, Darnell RB. β-NAP, a cerebellar degeneration antigen, is a neuron-specific vesicle coat protein. Cell. 1995;82:773–783. doi: 10.1016/0092-8674(95)90474-3. [DOI] [PubMed] [Google Scholar]
- Nichols BJ, Ungermann C, Pelham HRB, Wickner WT, Haas A. Homotypic vacuolar fusion mediated by t- and v-SNAREs. Nature. 1997;387:199–202. doi: 10.1038/387199a0. [DOI] [PubMed] [Google Scholar]
- Nothwehr SF, Conibear E, Stevens TH. Golgi and vacuolar membrane proteins reach the vacuole in vps1mutant yeast cells via the plasma membrane. J Cell Biol. 1995;129:35–46. doi: 10.1083/jcb.129.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothwehr SF, Bryant NJ, Stevens TH. The newly identified yeast GRDgenes are required for retention of late-Golgi membrane proteins. Mol Cell Biol. 1996;16:2700–2707. doi: 10.1128/mcb.16.6.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P, Ferro S, Schekman R. Order of events in the yeast secretory pathway. Cell. 1981;25:461–469. doi: 10.1016/0092-8674(81)90064-7. [DOI] [PubMed] [Google Scholar]
- Ogur M, St. John R, Nagai S. Tetrazolium overlay technique for population studies of respiration deficiency in yeast. Science. 1957;125:928–929. doi: 10.1126/science.125.3254.928. [DOI] [PubMed] [Google Scholar]
- Ooi CE, Moreira JE, Dell'Angelica EC, Poy G, Wassarman DA, Bonifacino JS. Altered expression of a novel adaptin leads to defective pigment granule biogenesis in the Drosophila eye color mutant garnet. . EMBO (Eur Mol Biol Organ) J. 1997;16:4508–4518. doi: 10.1093/emboj/16.15.4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panek HR, Stepp JD, Engle HM, Marks KM, Tan P, Lemmon SK, Robinson LC. Suppressors of YCK-encoded yeast casein kinase 1 define four subunits of a novel clathrin AP-like complex. EMBO (Eur Mol Biol Organ) J. 1997;16:4194–4204. doi: 10.1093/emboj/16.14.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan HL, Finlay JA, Chu DS, Tan P, Kirchhausen T, Payne GS. The Saccharomyces cerevisiae APS1gene encodes a homologue of the small subunit of the mammalian clathrin AP-1 complex: evidence for functional interaction with clathrin at the Golgi complex. EMBO (Eur Mol Biol Organ) J. 1994;13:1706–1717. doi: 10.1002/j.1460-2075.1994.tb06435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper RC, Whitters EA, Stevens TH. Yeast Vps45p is a Sec1p-like protein required for the consumption of vacuole-targeted, post-Golgi transport vesicles. Eur J Cell Biol. 1994;65:305–318. [PubMed] [Google Scholar]
- Piper RC, Cooper AA, Yang H, Stevens TH. VPS27 controls vacuolar and endocytic traffic through a prevacuolar compartment in Saccharomyces cerevisiae. . J Cell Biol. 1995;131:603–617. doi: 10.1083/jcb.131.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper RC, Bryant NJ, Stevens TH. The membrane protein alkaline phosphatase is delivered to the vacuole by a route that is distinct from the VPS-dependent pathway. J Cell Biol. 1998;138:531–545. doi: 10.1083/jcb.138.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescianotto-Baschong, C., and H. Riezman. 1998. Morphology of the yeast endocytic pathway. Mol. Biol. Cell. In press. [DOI] [PMC free article] [PubMed]
- Rad MR, Phan HL, Kirchrath L, Tan PK, Kirchhausen T, Hollenberg CP, Payne GS. Saccharomyces cerevisiaeApl2p, a homologue of the mammalian clathrin AP β subunit, plays a role in clathrin-dependent Golgi functions. J Cell Sci. 1995;108:1605–1615. doi: 10.1242/jcs.108.4.1605. [DOI] [PubMed] [Google Scholar]
- Raths S, Rohrer J, Crausaz F, Riezman H. end3 and end4: two mutants defective in receptor-mediated and fluid-phase endocytosis in Saccharomyces cerevisiae. . J Cell Biol. 1993;120:55–65. doi: 10.1083/jcb.120.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CK, Howald-Stevenson I, Vater CA, Stevens TH. Morphological classification of the yeast vacuolar protein sorting mutants: evidence for a prevacuolar compartment in class E vpsmutants. Mol Biol Cell. 1992;3:1389–1402. doi: 10.1091/mbc.3.12.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redding K, Holcomb C, Fuller RS. Immunolocalization of Kex2 protease identifies a putative late Golgi compartment in the yeast Saccharomyces cerevisiae. . J Cell Biol. 1991;113:527–538. doi: 10.1083/jcb.113.3.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder SE, Banta LM, Kohrer K, Mccaffery JM, Emr SD. Multilamellar endosome-like compartment accumulates in the yeast vps28vacuolar protein sorting mutant. Mol Biol Cell. 1996;7:985–999. doi: 10.1091/mbc.7.6.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CJ, Raymond CK, Yamashiro CT, Stevens TH. Methods for studying the yeast vacuole. Methods Enzymol. 1991;194:644–661. doi: 10.1016/0076-6879(91)94047-g. [DOI] [PubMed] [Google Scholar]
- Rose MD, Novick P, Thomas JH, Botstein D, Fink GR. A Saccharomyces cerevisiaegenomic plasmid bank based on a centromere-containing shuttle vector. Gene. 1987;60:237–243. doi: 10.1016/0378-1119(87)90232-0. [DOI] [PubMed] [Google Scholar]
- Rothstein R. Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol. 1991;194:281–301. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- Schmid SL. Clathrin-coated vesicle formation and protein sorting: an integrated process. Annu Rev Biochem. 1997;66:511–548. doi: 10.1146/annurev.biochem.66.1.511. [DOI] [PubMed] [Google Scholar]
- Seeger M, Payne GS. A role for clathrin in the sorting of vacuolar proteins in the Golgi complex of yeast. EMBO (Eur Mol Biol Organ) J. 1992a;11:2811–2818. doi: 10.1002/j.1460-2075.1992.tb05348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger M, Payne GS. Selective and immediate effects of clathrin heavy chain mutations on Golgi membrane protein retention in Saccharomyces cerevisiae. . J Cell Biol. 1992b;118:531–540. doi: 10.1083/jcb.118.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson F, Bright NA, West MA, Newman LS, Darnell RB, Robinson MS. A novel adaptor-related protein complex. J Cell Biol. 1996;133:749–760. doi: 10.1083/jcb.133.4.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson F, Peden AA, Christopoulou L, Robinson MS. Characterization of the adaptor-related protein complex, AP-3. J Cell Biol. 1997;137:835–845. doi: 10.1083/jcb.137.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack JH, Horazdovsky B, Emr SD. Receptor-mediated protein sorting to the vacuole in yeast: roles for a protein kinase, a lipid kinase and GTP-binding proteins. Annu Rev Cell Dev Biol. 1995;11:1–33. doi: 10.1146/annurev.cb.11.110195.000245. [DOI] [PubMed] [Google Scholar]
- Stepp JD, Pellicena-Palle A, Hamilton S, Kirchhausen T, Lemmon SK. A late Golgi sorting function for Saccharomyces cerevisiaeApm1p, but not for Apm2p, a second yeast clathrin AP medium chain-related protein. Mol Biol Cell. 1995;6:41–58. doi: 10.1091/mbc.6.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stotz A, Linder P. The ADE2 gene from Saccharomyces cerevisiae: sequence and new vectors. Gene. 1990;95:91–98. doi: 10.1016/0378-1119(90)90418-q. [DOI] [PubMed] [Google Scholar]
- Tan PK, Davis NG, Sprague GF, Payne GS. Clathrin facilitates the internalization of seven transmembrane segment receptors for mating pheromones in yeast. J Cell Biol. 1993;123:1707–1716. doi: 10.1083/jcb.123.6.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vater CA, Raymond CK, Ekena K, Howald-Stevenson I, Stevens TH. The VPS1 protein, a homologue of dynamin required for vacuolar protein sorting in Saccharomyces cerevisiae, is a GTPase with two functionally separable domains. J Cell Biol. 1992;119:773–786. doi: 10.1083/jcb.119.4.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vida TA, Huyer G, Emr SD. Yeast vacuolar proenzymes are sorted in the late Golgi complex and transported to the vacuole via a prevacuolar endosome-like compartment. J Cell Biol. 1993;121:1245–1256. doi: 10.1083/jcb.121.6.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y, Kitamoto K, Kanabe T, Tanaka K, Anraku Y. The SLP1 gene of Saccharomyces cerevisiaeis essential for vacuolar morphogenesis and function. Mol Cell Biol. 1990;10:2214–2223. doi: 10.1128/mcb.10.5.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y, Ohsumi Y, Kawai E, Ohsumi M. Mutational analysis of Vam4/Ypt7p function in the vacuolar biogenesis and morphogenesis in the yeast, Saccharomyces cerevisiae. . Protoplasma. 1996;191:126–135. [Google Scholar]
- Wada Y, Nakamura N, Ohsumi Y, Hirata A. Vam3p, a new member of syntaxin related protein, is required for vacuolar assembly in the yeast Saccharomyces cerevisiae. . J Cell Sci. 1997;110:1299–1306. doi: 10.1242/jcs.110.11.1299. [DOI] [PubMed] [Google Scholar]
- Wang YX, Zhao HR, Harding TM, Demesquita DSG, Woldringh CL, Klionsky DJ, Munn AL, Weisman LS. Multiple classes of yeast mutants are defective in vacuole partitioning yet target vacuole proteins correctly. Mol Biol Cell. 1996;7:1375–1389. doi: 10.1091/mbc.7.9.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb GC, Zhang ZJ, Garlow SJ, Wesp A, Riezman H, Jones EW. Pep7p provides a novel protein that functions in vesicle-mediated transport between the yeast Golgi and endosomes. Mol Biol Cell. 1997;8:871–895. doi: 10.1091/mbc.8.5.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann H, Hengst L, Gallwitz D. Endocytosis in yeast—evidence for the involvement of a small GTP-binding protein (Ypt7p) Cell. 1992;71:1131–1142. doi: 10.1016/s0092-8674(05)80062-5. [DOI] [PubMed] [Google Scholar]
- Wilcox CA, Redding K, Wright R, Fuller RS. Mutation of a tyrosine localization signal in the cytosolic tail of yeast Kex2 protease disrupts Golgi retention and results in default transport to the vacuole. Mol Biol Cell. 1992;3:1353–1371. doi: 10.1091/mbc.3.12.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro C, Kane PM, Wolczyk DF, Preson RA, Stevens TH. Role of vacuolar acidification in protein sorting and zymogen activation: a genetic analysis of the yeast vacuolar proton-translocating ATPase. Mol Cell Biol. 1990;10:3737–3749. doi: 10.1128/mcb.10.7.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]