

Abstract
Kinesin is a dimeric motor protein that can move along a microtubule for several microns without releasing (termed processive movement). The two motor domains of the dimer are thought to move in a coordinated, hand-over-hand manner. A region adjacent to kinesin's motor catalytic domain (the neck) contains a coiled coil that is sufficient for motor dimerization and has been proposed to play an essential role in processive movement. Recent models have suggested that the neck enables head-to-head communication by creating a stiff connection between the two motor domains, but also may unwind during the mechanochemical cycle to allow movement to new tubulin binding sites. To test these ideas, we mutated the neck coiled coil in a 560-amino acid (aa) dimeric kinesin construct fused to green fluorescent protein (GFP), and then assayed processivity using a fluorescence microscope that can visualize single kinesin–GFP molecules moving along a microtubule. Our results show that replacing the kinesin neck coiled coil with a 28-aa residue peptide sequence that forms a highly stable coiled coil does not greatly reduce the processivity of the motor. This result argues against models in which extensive unwinding of the coiled coil is essential for movement. Furthermore, we show that deleting the neck coiled coil decreases processivity 10-fold, but surprisingly does not abolish it. We also demonstrate that processivity is increased by threefold when the neck helix is elongated by seven residues. These results indicate that structural features of the neck coiled coil, although not essential for processivity, can tune the efficiency of single molecule motility.
Conventional kinesin is a motor protein that transports membranous organelles along microtubules in vivo (Bloom and Endow, 1995). Motility assays in vitro have shown that kinesin is capable of moving across hundreds of tubulin dimers (>1 μm) without detaching and diffusing away from the microtubule (Howard et al., 1989; Block et al., 1990; Vale et al., 1996). Such processive movement is an unusual feature for cytoskeletal motor proteins (Howard, 1997) and does not appear to occur in muscle myosin (Finer et al., 1994), ciliary dynein (Vale et al., 1992), or even other members of the kinesin superfamily (Case et al., 1997). In vivo, processivity may be an important adaptation that allows efficient transport of organelles with only a small number of kinesin motors.
The kinesin motor domain consists of a 320-amino acid (aa)1 core that contains the microtubule binding and ATP hydrolysis sites and whose three-dimensional structure has been solved by X-ray crystallography (Kull et al., 1996; Sablin et al., 1996). This core catalytic domain, also termed the motor “head”, is shared by all members of the kinesin superfamily. Adjacent to this core catalytic domain is the “neck” region, which is defined by sequence conservation among specific classes of kinesin motors and which is thought to act in concert with the catalytic domain to produce movement (Vale and Fletterick, 1997). In conventional kinesin, the neck consists of two parts: (a) an NH2-terminal ∼10-aa residue β sheet motif that is shared with many other plus end–directed motors and may be important for directional motion (Case et al., 1997; Henningsen and Schliwa, 1997); and (b) a subsequent ∼30-aa residue hydrophobic heptad repeat that gives rise to a coiled-coil structure (Huang et al., 1994; Morii et al., 1997; Tripet et al., 1997). The coiled-coil portion of the neck is highly conserved (∼65% aa identity) among conventional kinesins, suggesting a specialized function (Huang et al., 1994; Vale and Fletterick, 1997). A noteworthy conserved feature of this region is that it contains two unstable heptads in the middle of the coiled coil (Huang et al., 1994; Tripet et al., 1997). The neck coiled coil is terminated by a sequence containing glycine and proline residues. COOH-terminal to this potential hinge region, kinesin contains a long (∼400-aa) coiled-coil stalk domain followed by a globular tail domain that may help to dock the motor onto organelles.
Kinetic studies have suggested a model for kinesin processivity in which the motor remains in continuous contact with the microtubule as the result of an alternating interaction of the heads with the polymer (Hackney, 1994; Ma and Taylor, 1997). This idea has been supported by cryoelectron microscopy images that show dimeric kinesin bound to the microtubule by one head, whereas the other is detached and oriented perpendicular to the microtubule axis (Arnal et al., 1996; Hirose et al., 1996). Thus, binding of one head to the microtubule may inhibit binding of the partner head, at least during some intermediates of the mechanochemical cycle. However, experiments with monomeric kinesin also suggest that one head may help to release the other from its tubulin binding site at another stage of the cycle (Jiang and Hackney, 1997). By coordinating the two heads so that the binding of one head induces the release of the partner head, the motor could move hand-over-hand from one tubulin dimer to the next.
Truncations of kinesin have shown that monomeric motors are not processive, although these motors are still competent to produce motility when many proteins interact simultaneously with the microtubule (Berliner et al., 1995; Vale et al., 1996). However, if the truncation is less severe and yields a dimeric protein, then the motor remains processive. The protein sequence that enables these latter constructs to dimerize resides within the kinesin neck (Huang et al., 1994). Several recent models have proposed a mechanochemical cycle in which the neck also acts as a sophisticated communication link between the heads (Hackney, 1994; Hirose et al., 1996; Tripet et al., 1997). At the start of the proposed cycle, the neck joins the two heads tightly, so that only one head can interact strongly with the microtubule. A nucleotide-induced conformational change in the bound head is then transmitted to the neck, causing a segment of the coiled coil to unwind. This unwinding releases the second head so that it can now reach a new tubulin binding site through a diffusional search, creating an intermediate in which both heads are bound to the microtubule. A rezippering of the coiled coil at the end of the cycle then pulls the first head off the microtubule. This regenerates the motor state present at the beginning of the cycle with one head bound and the other detached, except that the motor has moved forward by one tubulin dimer. In support of such a model, the crystal structure of the kinesin dimer reveals that the neck coiled coil forms a tight connection between the two catalytic domains, suggesting that coiled coil unwinding could be necessary for simultaneous binding of both heads to the microtubule (Kozielski et al., 1997). Moreover, studies of synthetic kinesin neck peptides reveal an unstable region in the middle of the coiled coil segment that could facilitate partial unwinding of this structure (Tripet et al., 1997).
To test the role of the kinesin neck coiled coil in processivity, we have either deleted or made alterations in this region and then assayed the resultant proteins for processivity using a single molecule fluorescence motility assay. We show that stabilizing the neck coiled coil only reduced processivity by 45%. Thus, it is unlikely that the neck coiled coil needs to unwind substantially during motility. On the other hand, increasing the flexibility in the connection between the two heads by inserting a three-residue glycine linker at the beginning of the neck reduced processivity by 60%. A more drastic mutation of deleting virtually the entire neck coiled coil decreased processivity 10-fold but did not abolish it. Unexpectedly, duplicating the first heptad repeat of the coiled coil enhanced processivity by threefold. Collectively, these results suggest that neck coiled coil is not essential for processivity, but that features of this structure make single molecule motility more efficient.
Materials and Methods
Expression Constructs
A pET17b vector (Novagen, Inc., Madison, WI) containing the NH2-terminal 560 amino acids of human kinesin followed by a COOH-terminal histidine tag (Woehlke et al., 1997) was used as the starting point for mutagenesis of the kinesin neck region. To construct the desired mutations in the neck region, different strategies combining PCR, QuikChange mutagenesis (Stratagene, La Jolla, CA), or annealing followed by DNA synthesis with Sequenase (Amersham Corp., Arlington Heights, IL) were used. The neck region was then sequenced to confirm that only the correct changes were introduced, and then a Nco I–Hind III fragment containing the altered neck segment was subcloned back into the wild-type construct or into a 560-aa kinesin–green fluorescent protein (GFP) fusion (using the Ser65Thr variant of GFP) (Case et al., 1997; Pierce et al., 1997).
Protein Expression and Purification
At least four protein preparations of each neck mutant were made: two of the mutant kinesin alone and two of the kinesin–GFP fusion. For each preparation, Escherichia coli BL21 (DE3) was transformed with the expression plasmid construct and then a single colony was selected and grown in 0.5 ml Luria broth/50 μM ampicillin for 8 h at 37°C. 10 μl of this preculture was then inoculated into 2 liters of TPM media (20 g/liter tryptone, 15 g/liter yeast extract, 8 g/liter NaCl, 10 mM glucose, 2 g/liter KH2PO4, and 50 μg/ml ampicillin) and grown for 15 h at 25°C. The cells were induced with 0.1 mM isopropyl B-p-thiogalactopyranoside and then grown for an additional 8 h. Cells were spun at 2,000 g for 10 min and then the pellets were frozen in liquid nitrogen and stored at −80°C.
Cells were resuspended in 40–80 ml buffer (50 mM NaPO4, pH 8, 20 mM imidazole, 250 mM NaCl, 1 mM MgCl2, 1 mg/ml Pefabloc (Boehringer Mannheim Biochemicals, Indianapolis, IN), 0.5 μg/ml leupeptin, 0.5 μg/ml aprotinin, 0.7 μg/ml pepstatin, 0.1 μg/ml chymostatin) per liter of culture and then disrupted in a French press at 0.8 megaPa (18,000 psi). Insoluble material was removed by centrifugation at 11,000 g for 30 min. 1 ml of Ni2+-NTA resin (QIAGEN, Inc., Santa Clarita, CA) was incubated with the supernatant on a roller at 4°C for 1 h before the resin was transferred to a column. The column was washed eight times with 12 ml of wash buffer (50 mM NaPO4, pH 6, 60 mM imidazole, 250 mM NaCl, 1 mM MgCl2, 25 μM ATP), or until the OD at 280 nm was below 0.05. 1 ml of elution buffer (wash buffer with 500 mM imidazole, pH 7) was then applied and the resin was allowed to equilibrate for 10 min. The protein was eluted with an additional 4 ml of elution buffer and then diluted fivefold with mono-Q column buffer (25 mM Pipes, pH 6.8, 1 mM MgCl2, 1 mM EGTA, 25 μM ATP). The protein was then applied to a mono-Q column (Pharmacia Biotech. Inc., Piscataway, NJ), which was washed with column buffer plus 100 mM NaCl or 200 mM NaCl for kinesin or kinesin–GFP proteins, respectively. The column was eluted with a gradient to 1 M NaCl and the kinesin was eluted at ∼0.35 M NaCl. Peak fractions were stored in liquid nitrogen after the addition of 10% sucrose. To quantitate protein concentrations, samples were run at varying dilutions on SDS-PAGE gels with a standard curve of BSA and were then stained with Coomassie dye. The gels were imaged with a charge-coupled device camera and then optical densities were calculated using the program NIH Image (National Institutes of Health, Bethesda, MD). Protein concentrations were between 0.2–1.4 mg/ml depending on the kinesin construct, and the purity of the full-length protein was ∼75 and 50% for the kinesin and kinesin–GFP constructs, respectively. Nearly all the contaminants were degradation products that contained the histidine-tagged COOH terminus of the protein and did not show detectable binding to microtubules.
ATPase Assays
Microtubule-stimulated ATPase rates were measured in a spectrophotometer using a coupled enzymatic assay (Catterall and Pederson, 1971) with details as described in Woehlke et al. (1997). In brief, microtubules were polymerized with 20 μM paclitaxel, 1 mM GTP, 4 mM MgCl2, and 10% DMSO, centrifuged through a 50% glycerol cushion, and then resuspended in low salt assay buffer (12 mM K-Pipes, pH 6.8, 2 mM MgCl2, 1 mM EGTA) with 20 μM paclitaxel. ATPase assays were performed in assay buffer with 12 mM NaCl, 1 mM ATP, and a coupled NADH oxidation system. The assays were performed with 65–250 nM kinesin and 10–15 different microtubule concentrations ranging from 0.02–30 μM, depending on the construct. The K m(MT) and k cat values were determined using a Kaleidagraph-based hyperbolic curve-fitting routine, and r values were between 0.97–0.99. At least two complete ATPase curves of varying microtubule concentrations were performed for each protein preparation.
Multiple Motor Motility Assays
Kinesin motility was assayed by using differential interference contrast microscopy to observe microtubules gliding across kinesin-coated coverslips. Kinesin (0.7–7.0 μM) was combined in BRB80 buffer (80 mM Pipes, pH 6.8, 2 mM MgCl2, 1 mM EGTA) with 0.1 mg/ml casein, paclitaxel-stabilized bovine brain microtubules (∼0.5 mM), an ATP regenerating system (40 μg/ml phosphokinase (Boehringer Mannheim Biochemicals), 150 μg/ml phosphoenol pyruvate, and 1 mM ATP), and an oxygen scavenging system (0.2 mg/ml glucose oxidase, 36 μg/ml catalase, 22 mM glucose, and 0.5% β-mercaptoethanol) (Harada et al., 1990). The mixture was then pipetted into a flow chamber consisting of a coverslip supported over a glass slide by two strips of double-stick tape. Microtubules at the coverslip surface were visualized using an Axioplan microscope (Carl Zeiss, Inc., Thornwood, NY) equipped with a 63×, 1.4 NA objective and a Newvicon camera (Hamamatsu Photonics, Hamamatsu City, Japan). Over the course of 5 min, images of several fields were recorded onto super VHS tape. The speeds of >30 microtubules per protein preparation were measured using a custom-written computer analysis program.
Single Motor Motility Assays
Individual kinesin–GFP fusion proteins were visualized moving along sea urchin sperm flagellar axonemes in a custom-built total internal reflection fluorescence microscope. The microscope (Pierce and Vale, 1997) and assay (Vale et al., 1996; Pierce et al., 1997) are described in detail elsewhere. In brief, 1–8 nM kinesin–GFP was diluted into the low-salt buffer described above for the ATPase assay, except that K-Pipes was used rather than Na-Pipes because it increased the number of kinesin molecules moving along axonemes. Additional components included 7.5 mg/ml BSA (to minimize kinesin–GFP adsorption to the surface), the oxygen scavenging system described above, 1 mM ATP and Cy-5 labeled sea urchin axonemes (Gibbons and Fronk, 1979; Vale et al., 1996). 5 μl of this solution was applied onto a fused silica slide and then sealed under a coverslip using rubber cement dissolved in heptane. Slides were illuminated with an argon laser (model 5490A, Ion Laser Technology, Salt Lake City, UT) (488 nm) at 10 mW, since this provided optimal single spot detection. Up to three fields were imaged for 5 min each, and then data was recorded onto super VHS tape after four-frame averaging with an Argus 20 image processor (Hamamatsu Photonics).
Analysis of Processivity
For analysis of single motor motility, a motor concentration was used in which movements on the axonemes were frequent but not overlapping. In addition, background fluorescence from motors diffusing in solution had to be sufficiently low so that fluorescent spots could be easily distinguished. Data was recorded from two different preparations of each construct.
For fluorescence intensity analyses, the intensity of all moving spots in a 1–3-μm area of an axoneme was measured; this was repeated until >50 spots had been measured for each construct. The fluorescence intensity of GFP diminishes slightly during illumination (Pierce et al., 1997). To minimize this contribution, intensities of spots were measured <1 s after contacting the axoneme. Data were taken from a single frame acquired using a four-frame rolling average. To determine the spot intensity, a 15 × 15 pixel area around a spot was selected and then the total intensity was measured using an Argus 20 image processor (Hamamatsu Photonics). The background intensity derived from an adjacent region was then subtracted from this value. As a control to determine the fluorescence intensity of the overall motor population, nonmoving spots that landed in an area on the slide adjacent to the axoneme during the same time period were analyzed in the same manner.
The observed run lengths and speeds of 150–250 moving fluorescent kinesin–GFP spots were measured using a custom computer analysis program. Only movements on long axonemes were measured (generally 8–18 μm, although for the less processive mutants, axonemes as short as 5 μm were sometimes analyzed) to minimize the chance that a kinesin–GFP would release from the microtubule simply by running off the end. To measure run lengths, small segments of the axonemes were first viewed to locate moving spots and then observed frame-by-frame to ensure that the exact starting and ending points of each run were determined. Only kinesin–GFP spots that were well-separated from other fluorescent spots could be clearly distinguished from the background, and moved smoothly and continuously for at least 0.5 s, were chosen for analysis. By visually tracking the center of the diffraction-limited spots, movements as short as 0.1 μm could be detected. However, since the error in measuring the length of these runs is high, only runs >0.2 μm were used in velocity calculations. The efficiency of detecting extremely short runs was low because the dwell time of the motor on the microtubule was very brief. To locate short runs more efficiently for the least processive proteins, GLY3 and DEL, the axonemes were divided into very short segments for visual inspection (∼1-μm versus ∼3-μm segments for the other constructs). In addition, because these two constructs moved more slowly than wild type, their dwell times on the microtubule were increased, raising the likelihood that very short movements could be detected. Therefore, for these mutants, a more complete set of data at the shorter run lengths could be gathered.
Histograms of observed run length were plotted for each construct and fit to an exponential curve using the program Origin (MicroCal Inc., Northampton, MA). Although all runs detected were plotted, only data from runs longer than 0.25 μm were used for the curve fitting because of the inefficient detection of runs below this limit. Runs as short as 0.2 and 0.15 μm were used for fitting GLY3 and DEL, respectively, because of the more efficient data collection described above. The average run length values generated are referred to as the observed run lengths. However, the rate constant for photobleaching (k bleach), as well as the motor release rate constant (k rel), contributes to the observed rate of disappearance of kinesin–GFP spots (k obs) according to the equation: k obs = k rel + k bleach. k obs is the reciprocal of the dwell time of the motor on the microtubule, where the dwell time = observed run length/motor speed. k bleach was determined by counting the disappearance of kinesin–GFP spots adsorbed to the surface at 2.5, 5, 10, and 20 mW. In each case, the disappearance showed an exponential decay with time (Vale, et al., 1996), and the rate constant varied linearly with the laser power (Pierce and Vale, 1997). At the 10-mW laser power used in this study, the photobleaching rate was 0.10 s−1 for a kinesin–GFP dimer (Pierce and Vale, 1997). From k obs and k bleach, the actual run length of the motor in the absence of GFP photobleaching (k rel) could be calculated from the above equations. This correction for photobleaching was confirmed experimentally by measuring run lengths of the DUP mutant at 4- and 10-mW laser illumination as described in the text.
Results
Design of Neck Coiled-Coil Mutants
To design mutations in the neck, we used information derived from circular dichroism studies of kinesin neck peptides as well as functional studies of kinesin proteins truncated within this region. Structural studies by Morii et al. (1997) and Tripet et al. (1997) revealed that peptides containing the heptad repeat sequence from the neck will form a coiled coil, and suggested that the COOH-terminal boundary of this coiled coil lies at Gly371. The NH2-terminal boundary was less clearly defined, but was unlikely to begin before Cys330 (aa numbers quoted in this paper correspond to residues in the human kinesin sequence). Functional studies also showed that kinesin truncated at residue 332 displays a low microtubule gliding velocity (Stewart et al., 1993; Vale et al., 1996) and an abnormal basal ATP rate and K m for microtubule-stimulated ATPase activity (Huang and Hackney, 1993; Jiang et al., 1997). Kinesin with an additional 10 residues displays more normal motility and ATPase properties. Therefore, to insure robust motor activity, mutations were made after Trp340 for all constructs in this study. Very recently, the crystal structure of dimeric rat kinesin has been solved and reveals a coiled coil between residues 337–370 (Kozielski et al., 1997). Thus, the region chosen for mutagenesis in this study encompasses virtually the entire neck coiled coil. All mutations described below were made in the context of a human kinesin protein that remains dimeric irrespective of the neck structure due to the presence of the first 110 amino acids of the coiled coil in the kinesin stalk domain (Fig. 1, K560).
Figure 1.

Mutant neck constructs. (a) WT K560: The wild-type kinesin construct used as the basis for mutagenesis contains the first 560 amino acids of the human kinesin gene followed by a histidine tag. *, residues at the hydrophobic interface of the coiled coil; (*), destabilizing residues in this interface. Domains of this construct are (I) core catalytic domain, (II) neck β sheet region, (III) neck coiled-coil region, (IV) hinge region, (V) coiled-coil stalk. The boundaries of the core catalytic domain are defined by conservation throughout the kinesin superfamily. The boundary of the neck is defined by strong class-specific conservation among conventional kinesins (Vale and Fletterick, 1997). (b) DEL: aa 341–370 of the neck coiled coil were deleted. (c) GLY3: three glycines were inserted between K342 and K343. (d) DUP: Residues T336–K342, one complete turn of the α-helix, were duplicated. (e) YEN→ ILI: Three destabilizing residues at the “a” and “d” position of the coiled coil were changed to stabilizing hydrophobic residues (Y344I, E347L, and N351I). (f) STABLE COIL: Four heptad repeats (aa 343–370), were replaced by a highly stable model coiled coil consisting of four repeats of the sequence EIEALKA. (g) WT K560–GFP: The above neck mutations (b–f) were also inserted into K560 with GFP, the Ser65Thr mutant (Heim et al., 1995), fused to its COOH terminus.
To test whether the neck coiled coil is essential for processivity in the context of a dimer, the residues 341–370 were deleted from K560 (Fig. 1, DEL). In this mutant protein, the initial β strand region of the neck is connected to a nonconserved region preceding the stalk. This nonconserved region contains several glycines and prolines and may serve as a hinge that could account for the flexible behavior of kinesin observed in motility assays (Hunt and Howard, 1993; Huang et al., 1994).
To perturb any tight connection between the two heads that could serve to communicate tension or positional information, a flexible linker of three glycines was inserted at residue 342 (GLY3). 6 aa from the heptad repeat remain NH2-terminal to the glycine linker, but this sequence alone should be insufficient to form a stable coiled coil (Su et al., 1994; Tripet et al., 1997). As a control for aa insertions at residue 342, the construct DUP was created in which the previous 7 aa (T336–K342) were duplicated. This construct would be expected to extend the neck coiled coil by one heptad repeat.
Finally, two constructs were made to test the model that the neck coiled coil needs to unwind during the mechanochemical cycle. In the construct STABLE COIL, residues 343–370 were replaced by four copies of the heptad repeat sequence EIEALKA. Thermodynamic studies of this 28-aa peptide have shown that it forms an extremely stable coiled coil with a ΔG for dissociation that is >20 kcal/mol (Su et al., 1994; Tripet et al., 1997). Since the energy derived from ATP hydrolysis is ∼12 kcal/mol, it is unlikely that this sequence would readily unwind during motility. A second and less drastic construct (YEN→ ILI) was made by changing three nonideal amino acids at the hydrophobic interface of the coiled coil to more stabilizing residues (Y344I, E347L, N351I). These three substitutions have been shown to stabilize dimers of a neck peptide (aa 344–383), increasing the dissociation energy from 8.5 to 11.2 kcal/mol (Tripet et al., 1997).
ATPase and Multiple Motor Motility Assays
All mutant proteins were first assayed for ATPase and in vitro motility activities to determine whether mutations in the neck coiled coil affect basic enzymatic and motor functions (Table I). The microtubule-stimulated ATP turnover rates for four of the five of the neck constructs were similar to that of the wild-type motor (k cat of 15–19 ATP/s per head for mutants versus 22 ATP/s per head for wild type). On the other hand, STABLE COIL displayed a slightly higher k cat (31 ATP/s per head). In addition, most of the mutants exhibited an apparent K m for microtubule stimulation of the ATPase activity (K mMT) that was similar to that of the wild-type construct. An exception, however, was DUP, whose K mMT was significantly lower (0.3 μM tubulin for DUP versus 0.96 μM tubulin for wild type). All of the neck mutants were also able to move microtubules across a microscope slide surface under conditions where multiple motors were contacting each microtubule (Table I). The speeds of movement ranged from 44–72% of wild-type K560. The above assays confirm that none of the neck coiled-coil mutations severely damaged either motile or enzymatic activity.
Table I.
Motility and Enzymatic Characterization of Neck Mutant Constructs
| Kinesin construct | Microtubule gliding speed* | ATPase‡ | ||||
|---|---|---|---|---|---|---|
| k cat | K m (MT) | |||||
| μm/s | ATP/s per head | μM tubulin | ||||
| Wild type | 0.32 ± 0.08 | 22 ± 3 | 0.97 ± 0.26 | |||
| DEL | 0.14 ± 0.02 | 15 ± 3 | 0.87 ± 0.12 | |||
| GLY3 | 0.23 ± 0.04 | 17 ± 3 | 1.36 ± 0.28 | |||
| DUP | 0.21 ± 0.04 | 16 ± 6 | 0.30 ± 0.07 | |||
| YEN→ ILI | 0.14 ± 0.04 | 19 ± 3 | 0.85 ± 0.22 | |||
| STABLE COIL | 0.17 ± 0.04 | 31 ± 5 | 0.96 ± 0.59 | |||
Microtubules gliding on kinesin-coated surfaces and microtubule-stimulated ATPase assays were performed as described in Materials and Methods. Values are as follows:
Mean ± SD of velocities of >60 microtubules were measured from assays with at least two independent protein preparations.
Mean ± SD of at least four independent assays were derived from two different protein preparations. k cat and K m (MT) values were obtained by the best fit to a hyperbolic curve of 10–15 turnover rates at varying microtubule concentrations.
Single Molecule Motility Assays
The behavior of single molecules of the neck coiled-coil mutant proteins was tested in a motility assay using a total internal reflection microscope. This low background fluorescence microscope is capable of visualizing individual fluorescent molecules (Funatsu et al., 1995), and has been used to observe individual kinesin proteins moving along axonemes (a nine + two array of microtubules) from sea urchin sperm flagella (Vale et al., 1996). As the fluorescent tag for assaying mutants in this study, we fused the Ser65Thr mutant of GFP (Heim et al., 1995) to the COOH terminus of the mutant K560 constructs (Pierce et al., 1997). The kinesin–GFP fusion proteins were first tested in the multiple motor microtubule gliding assay to ensure that the constructs remained active when fused to GFP. All mutant–GFP proteins moved microtubules at speeds between 48–115% of wild-type kinesin–GFP (Table II).
Table II.
Motility of GFP–Kinesin Constructs
| K560–GFP construct | Velocity* | Run length | P off/step‖ | Movement frequency ¶ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| movements per min·μm axoneme·nM kinesin | ||||||||||||
| Many motors | Single motors | Observed‡ | Corrected§ | |||||||||
| μm/s | μm | % | ||||||||||
| Wild type | 0.40 ± 0.05 | 0.31 ± 0.07 | 0.93 ± 0.07 | 1.33 | 0.60 | 0.05 | ||||||
| DEL | 0.19 ± 0.07 | 0.17 ± 0.07 | 0.13 ± 0.01 | 0.14 | 5.6 | 0.05 | ||||||
| GLY3 | 0.28 ± 0.06 | 0.16 ± 0.09 | 0.38 ± 0.02 | 0.50 | 1.6 | 0.04 | ||||||
| DUP | 0.46 ± 0.1 | 0.31 ± 0.07 | 1.81 ± 0.17 | 4.35 | 0.18 | 0.07 | ||||||
| 3.00 ± 0.43** | 4.89** | 0.16** | ||||||||||
| YEN→ ILI | 0.28 ± 0.05 | 0.32 ± 0.08 | 0.83 ± 0.06 | 1.12 | 0.71 | 0.06 | ||||||
| STABLE COIL | 0.28 ± 0.04 | 0.29 ± 0.09 | 0.58 ± 0.04 | 0.73 | 1.1 | 0.37 | ||||||
Microtubules gliding on kinesin–GFP coated surfaces and single molecule assays for kinesin–GFP using a total internal reflection microscope were performed as described in the Materials and Methods. Determination of the values were performed as described below:
Means and standard deviations were derived from measurements from two independent protein preparations. For microtubule gliding assays, >30 microtubule measurements were made per protein preparation. For single motor assays, >70 measurements were made per preparation and velocity data was derived only from those molecules that moved >0.2 μm.
The data from Fig. 3 was fit to an exponential curve using the equation y = A
* e(−×/λ), where λ = the average run length. Errors listed are the 95% confidence limits. Assays were performed using a laser power of 10 mW except where indicated by the double asterisk (
) for the DUP construct, in which case 4 mW laser power was used.
To account for the photobleaching of GFP, the rate constant for release of kinesin from the microbutule (k rel) was calculated according to the equation k obs = k rel + k bleach, where k obs = single motor velocity/observed run length, and k bleach is the rate constant for bleaching of GFP–kinesin under a given laser power (0.1 s−1 for 10 mW and 0.04 s−1 for 4 mW (Pierce and Vale, 1997)). The corrected run length = velocity/k rel (Refer to Materials and Methods).
The probability that a kinesin will release from the microtubule rather than completing its next step was calculated using the equation P off/step = 1 − e(−s/λ), where s is 8 nm (the kinesin step size [Svoboda et al., 1993]) and λ is the average run length.
For calculating the protein activity level, the total number of movements was divided by the total length of axonemes, time of observation, and the kinesin concentration. Since the percentage of runs that are <0.2 μm depends on the processivity of the mutant being measured, and the efficiency of detecting these runs is low, the total number of movements was derived from integrating the exponential curve fit to the data in Fig. 3. The values listed are derived from the combined data from two preparations of the same mutant protein; when quantitated independently, assay to assay variability could be as great as two- to threefold.
To observe single motor motility, the kinesin–GFP proteins were diluted to nanomolar concentrations in a buffer containing 1 mM ATP and Cy5-labeled fluorescent axonemes. By total internal reflection microscopy, fluorescent spots corresponding to individual wild-type kinesin– GFP appeared along the length of an axoneme and moved continuously in a given direction before releasing (Pierce et al., 1997). In contrast, kinesin–GFP in solution contributed to a faint blurred fluorescent background due to its rapid Brownian motion. Surprisingly, for all the mutant kinesin– GFP proteins, fluorescent spots were observed moving unidirectionally along axonemes. The velocities of the moving spots for the DUP, YEN→ ILI, and STABLE COIL constructs were similar to wild type (Table II). In contrast, DEL and GLY3 moved at approximately half the speed of the wild-type protein. These single motor speeds largely mirror those from multimotor assays, and are probably the more reliable measure of relative motor velocity because only active motors contribute to the data collected.
It was important to establish that the single spot motility observed for kinesin mutants was produced by kinesin dimers and not by motor aggregates. Using similar assay conditions, previous experiments showed that fluorescent spots of K560–GFP were twice as bright as monomeric GFP, indicating that K560–GFP is dimeric under the conditions of the assay (Pierce et al., 1997). Here, we measured the single spot fluorescence intensities of neck coiled coil mutants fused to GFP and found that they all have similar average intensities to K560–GFP (within 30%), indicating that these proteins are dimeric under our assay conditions as well.
It was still possible, however, that a small population of aggregated protein could account for the moving fluorescent spots observed in assays with the mutant proteins. To rule out this possibility, we compared the fluorescence intensities of moving spots to that of spots that were adhered to the slide surface. For all constructs, the intensity histograms for moving spots were similar to the glass-adsorbed population (Fig. 2). However, for several, most notably GLY3 and YEN→ ILI, there is a shoulder of brighter spots in the moving kinesin population that is not present in the nonmoving histograms. Nonetheless, even in the most extreme case of GLY3, it is apparent that the majority of the moving spots are of the correct intensity. Thus, protein multimers are likely to constitute only a small percentage of the moving spots observed in these assays.
Figure 2.
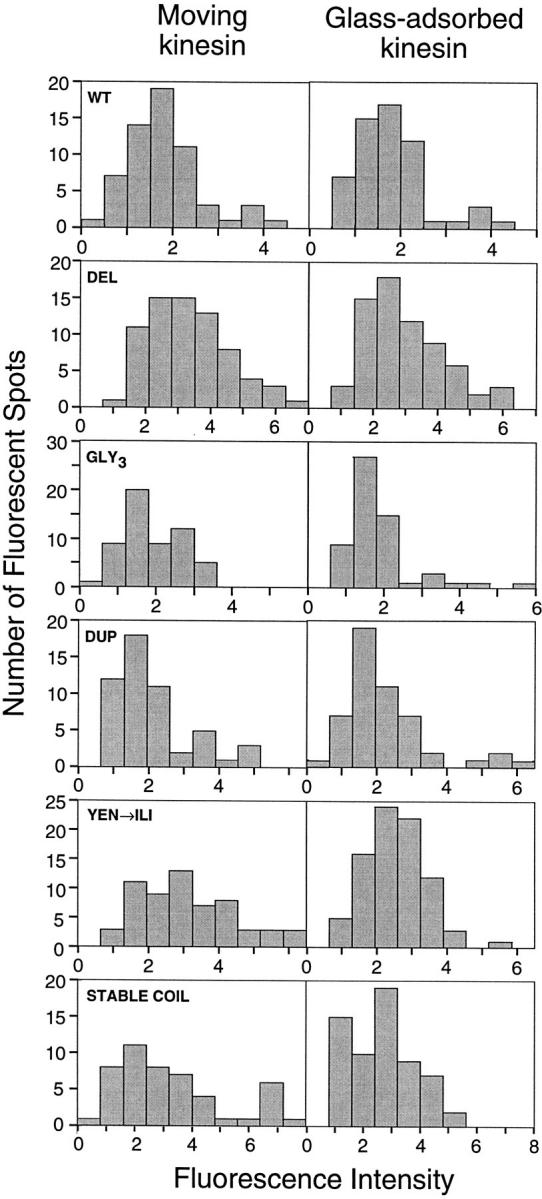
Fluorescent intensity of individual kinesin molecules. The histograms show the fluorescent intensity of kinesin molecules either moving along axonemes or nonspecifically adsorbed onto the slide surface nearby (refer to Materials and Methods for details). Tick marks represent one arbitrary fluorescent unit; fluorescent intensities cannot be directly compared between preparations because of small variations in laser alignment during different assays.
To further confirm that a minor population of aggregated motors does not account for the motility seen, the activity level (movements/μm MT per min per nM kinesin) was determined for each protein preparation (Table II). All mutant proteins displayed similar activity to the wild-type protein, and one mutant, STABLE COIL, was even more active. These data further argue against a rare protein aggregate as the source of moving spots for the mutant proteins.
Single Molecule Run Lengths
To examine whether the neck coiled-coil mutations changed the extent of processivity, the distance that a fluorescent spot moved from the time that it appeared on the axoneme to the time when it disappeared was measured. As found previously (Block et al., 1990; Vale et al., 1996), these distributions could be fit to an exponential curve (Fig. 3), indicating that kinesin has a constant probability of releasing and diffusing away from the microtubule each time it takes a step. The observed disappearance of kinesin from axonemes, however, is the sum of two independent exponential processes: (a) the release of kinesin from the axoneme, and (b) the photobleaching of the GFP. The true rate of release of the motor can therefore be calculated from the equation: k rel = k obs − k bleach. The photobleaching rate for GFP was previously determined to be 0.10 s−1 at 10 mW (Pierce and Vale, 1997; refer to Materials and Methods). The observed run lengths multiplied by k obs/k rel define the actual motor run lengths, which are shown in Table II and quoted throughout the text. For wild-type kinesin, the corrected run length is 1.3 μm, which is similar to previous reported values (Block et al., 1990; Vale et al., 1996). Assuming a kinesin step size of 8 nm (the distance between tubulin dimers) (Svoboda et al., 1993), the wild-type protein takes 166 steps on average before detaching and has a release probability per step (P off/ step) of 0.6%.
Figure 3.

Run lengths of single, fluorescently labeled kinesin molecules. Run lengths of 150–270 individual GFP–kinesin molecules moving on axonemes were measured from two independent preparations of each construct. Histograms of the data were plotted using bin widths derived from the formula 2.6σn(−1/3) (Scott, 1979), where σ is the standard deviation of the data and n is the number of data points collected. Exponential curves were fit to the data using only runs >0.25 μm (or 0.2 and 0.15 μm for GLY3 and DEL, respectively), as described in Materials and Methods. Run-length values are shown in Table II.
The two constructs that increase the stability of the neck coiled coil, YEN→ ILI and STABLE COIL, exhibited run lengths that were similar to those of wild-type kinesin (Table II). The run length of YEN→ ILI was 1.1 μm, or 84% of the wild-type distance. Even in the 28-aa replacement of the coiled coil (STABLE COIL), the run lengths were 55% of the wild-type protein. These results suggest that it is unlikely that the neck coiled coil needs to unwind significantly for the motor to move processively.
Two constructs, DEL and GLY3, had significantly shorter run lengths than wild type, moving only 0.14 and 0.50 μm, respectively. These two GFP constructs were also the only ones that moved at significantly slower speeds than wild type in the single motor assays (0.16 and 0.17 μm/s versus 0.31 μm/s for wild type). Nevertheless, these mutants have a high probability of stepping from one tubulin subunit to the next without detaching. For DEL, P off/ step is 5.6%, indicating that this motor has a ∼95% chance of remaining attached after completing a step. Therefore, although the tight coupling of the two heads by the neck coiled coil may make processivity more efficient, the neck coiled coil is not essential for processive motion.
Interestingly, the construct DUP moved ∼3.5-fold farther than wild type, a phenomenon shown by two independent preparations of the protein. The fluorescence intensity data in Fig. 2 indicates that the lengthened runs are not simply due to a large population of multimeric protein. However, because DUP has the longest dwell time on the microtubule, the photobleaching rate contributes substantially to the determination of corrected run length (the corrected run length is 2.5-fold greater than the observed run length). To confirm that the calculated run length of this motor is correct, we measured DUP movements at two laser powers and found that the observed run length at 4 mW laser power was significantly greater than at 10 mW (3.0 versus 1.8 μm). The corrected run length remained approximately the same (4.9 versus 4.4 μm), confirming both the validity of the photobleaching correction procedure and the long-run length of the DUP construct.
Discussion
Recent models for processivity have suggested that the two heads of the motor alternate in their interaction with the microtubule and that the kinesin neck coiled-coil is involved in coordinating this process (Hackney, 1994; Hirose et al., 1996; Kozielski et al., 1997; Tripet et al., 1997). These models are supported by experiments showing that kinesin dimers containing the coiled coil are processive, whereas monomers that lack the coiled coil are not (Berliner et al., 1995; Hackney, 1995; Vale et al., 1996). However, in these previous truncation studies, specific roles that the neck coiled coil might have in processivity, beyond simply dimerizing two motor domains, could not be assessed. In this study, we have altered the neck coiled coil in the context of a stable kinesin dimer to evaluate how specific structural features of the neck contribute to motor processivity. The resultant proteins were assayed using a motility assay that involves direct visualization of single, fluorescently labeled kinesin molecules (Vale et al., 1996; Pierce et al., 1997). Surprisingly, we show that deletion of virtually the entire neck coiled coil (aa 341–370) does not completely abolish processivity, since the resultant protein could still take 18 steps on average. Therefore, we conclude that the neck coiled coil is not essential to processivity, provided that the motor is dimerized by downstream sequences. However, we also demonstrate that mutations that alter structural features in the neck affect single motor run length. This provides information on the mechanism by which kinesin steps along the microtubule lattice, as discussed below.
The Neck Coiled Coil Does Not Need to Dissociate during Processive Motility
A notable feature of the kinesin neck is the placement of destabilizing residues with low hydrophobicity (Y344, E347, N351) at the interface of the coiled coil (Kozielski et al., 1997; Morii et al., 1997; Tripet et al., 1997). The high conservation of this unstable region has led to the suggestion that the NH2-terminal three-fifths of the coiled coil may unwind during motility (Huang et al., 1994; Tripet et al., 1997). According to such a model, stabilizing this region of the neck should decrease processivity and might even interfere with movement entirely. However, we find that the constructs YEN→ ILI and STABLE COIL move 0.8 and 0.6 times as far as the wild-type protein, respectively, demonstrating that increased stability in the neck coiled coil does not prevent processive motility. The free energy required to dissociate the entire coiled coil structure of the STABLE COIL heptad sequence is >20 kcal/ mol (Tripet et al., 1997), which exceeds the energy from ATP hydrolysis. The energy barrier for a partial unwinding of STABLE COIL is expected to be significant as well, since the unfolding of coiled-coil structures is highly cooperative (Su et al., 1994). In addition, because of the free energy differences between the altered neck sequences, unwinding would be expected to be slower in the mutant proteins compared to the wild-type motor. However, single motor motility of YEN→ ILI and STABLE COIL occurs at wild-type speeds, suggesting that unwinding of the coiled coil is not a rate-limiting step in cycle.
The results with STABLE COIL raise interesting questions as to how the motor steps along the microtubule. For the motor to be processive, a transient intermediate in which both heads are bound to the microtubule is expected to exist (Hackney, 1994; Hirose et al., 1996; Tripet et al., 1997). However, the crystal structure of the kinesin dimer reveals that the length of the linker between the two heads is insufficient to allow both heads to bind simultaneously to adjacent tubulin dimers 8-nm apart on the protofilament (Kozielski et al., 1997; Fig. 4 A). Thus, to extend the connection between the heads, one of the structured regions in the crystal structure must dissociate or unfold. In our study, we have not obtained evidence for coiled-coil dissociation. However, we have not replaced the first heptad of the neck coiled coil (aa 337–342) in the STABLE COIL construct, and it is possible that this region could unwind during motility. Nevertheless, even if this region adopted an extended conformation, the added linker would be insufficient to enable simultaneous binding of both heads.
Figure 4.
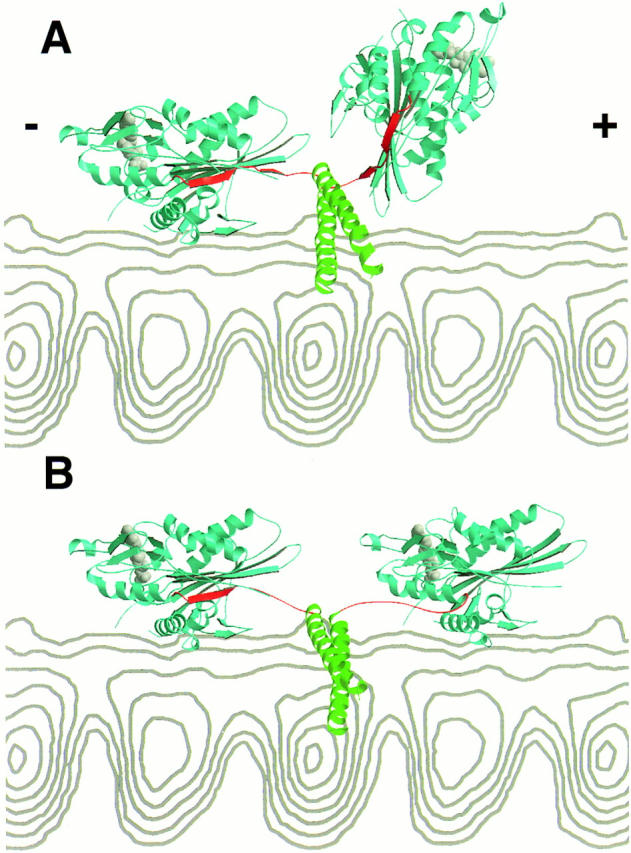
A structural model for how the kinesin dimer might span the eight nanometers between adjacent α/β tubulin binding sites. In this crystal structure of the rat kinesin dimer (Kozielski et al., 1997), the catalytic core domain is colored blue, the nucleotide is colored gray, the β strand region of the neck (β9 and β10; rat aa 321–336) is colored red, and the neck coiled coil (rat aa 337–370) is colored green (note: the rat kinesin aa numbers differ by −2 aa compared to human kinesin in this region). A side view of a microtubule protofilament from cryoelectron microscopy reconstructions (Hoenger et al., 1995) is shown in gray. The microtubule plus end (the direction of travel for kinesin) is at the right. In A, the unaltered crystal structure of the rat kinesin dimer is shown with one head docked onto the microtubule. The approximate orientation of the bound head was defined by having the half of the molecule containing the nucleotide pointing towards the minus end, the “arrowhead tip” pointing towards the plus end (Hoenger and Milligan, 1997; Sosa et al., 1997), and the main microtubule binding loop (L12) in contact with tubulin surface (Sosa et al., 1997; Woehlke et al., 1997). As noted by Kozielski et al. (1997), the neck coiled coil runs perpendicular to the long axis of the protofilament and is located near but not sterically clashing with the microtubule surface. In the crystal structure, the distance between the two heads is insufficient to enable the second head to dock onto the microtubule. It is important to mention that the structure shown here may not exactly correspond to one that occurs normally in the motility cycle, since the geometry of the heads could be partially determined by crystal contacts and since microtubule or nucleotide binding may change the solution conformation. In B, the β strands between aa 327–336 were separated from the catalytic core in the leading head using the program O (T.A. Jones and M. Kjeldgaard), obeying restraints of bond distances and geometries. This generates a sufficiently long linker to enable the leading head to dock to the adjacent tubulin binding site in the identical orientation to the lagging head. Only modest adjustments need to be made to the neck β strands of the lagging head, since they are already extended and pointing towards the microtubule plus end. In this model, the neck coiled coil does not unwind. The nucleotide (ADP) from the crystal structure is shown in both heads in these panels, although it is more likely that the two heads are in different nucleotide states during the motility cycle.
Since our results do not support extensive unwinding of the neck coiled coil, we favor a model in which the bonds that hold the β sheet region of the neck (β9 and β10; residues 323–334) to the catalytic domain are broken during the hydrolysis cycle, enabling these residues to adopt an extended conformation (Fig. 4 B). Consistent with this hypothesis, the neck adopts a well-ordered β sheet structure in one crystal form (Kozielski et al., 1997), but is disordered in another (Kull et al., 1996). This difference, which may reflect different crystallization conditions, suggests that β9 and β10 may be able to adopt different conformations in the mechanochemical cycle. As an alternate to the model shown, however, it is also possible that the necessary linkage is generated by dissociating the first coiled-coil heptad in combination with more modest changes in the neck β strand region.
Based upon docking of atomic structures to cryo-EM images of motor–microtubule complexes (Hoenger and Milligan, 1997; Sosa et al., 1997), the neck β strands are thought to point towards the microtubule plus end. Therefore, the dissociation of the neck β strands from the leading head is expected to be particularly important for enabling concurrent head binding, since the β strands in the lagging head are already extended towards the next binding site (Fig. 4 B). An interesting consequence of this structural model is that the reformation of the β9 and β10 strands in the leading head during subsequent events in the enzymatic cycle could generate a “power stroke” that is directed toward the plus end of the microtubule. The asymmetry in our structural model is consistent with the known enzymatic asymmetry in the two heads (Hackney, 1994; Ma and Taylor, 1997). Kinetic studies also indicate that changes in the nucleotide on the tightly bound head (Fig. 4, left head) are communicated to the partner head to enable its tight binding to the microtubule (Ma and Taylor, 1997). The structural model shown does not indicate how this could be achieved, although the linker between the heads or head–head interactions could be involved in the communication.
Since extensive unwinding of the neck coiled coil is not essential for processive movement, why are the destabilizing residues at the interface (Y344, E347, and N351) so well conserved? One possibility is that they play a role in the regulation of the kinesin motor. The tail of the full-length kinesin protein has been shown to fold onto the head and inhibit activity (Hackney et al., 1993), and sequences in the neck coiled-coil may be important for this interaction (Hackney, D.D., and T.G. Huang, 1993, Mol. Biol. Cell, 4 [Suppl.]:49a). It is possible that partial dissociation of the kinesin neck coiled coil could serve some function in the regulation of motor activity by the tail domain.
The Neck Coiled Coil Enhances Processivity
Although the neck coiled coil is not essential to single molecule motility, mutant proteins with the coiled coil deleted or containing a three glycine insertion move only ∼10 and ∼40% as far as wild-type, respectively. In addition, these DEL and GLY3 constructs moved at half the wild-type speed in single molecule assays. Thus, the native neck coiled coil does make processive motility more efficient.
The presence of the coiled coil in the neck might enhance single motor motility by at least three mechanisms. First, the coiled coil could help to align the two polypeptides of the dimer so that regions involved in positioning the two heads (Kozielski et al., 1997) could more readily interact. The positioning of the heads relative to one another is thought to cause the motor domains to undergo alternate cycles of microtubule-catalyzed ATP hydrolysis (Hackney, 1994; Hirose et al., 1996; Amos and Hirose, 1997). Second, processivity may require tension to be transmitted through a stiff connection between the motor domains to release the posterior head from the microtubule (Jiang and Hackney, 1997). The slower single motor speeds of the DEL and GLY3 mutants might be explained by inefficient release of the posterior head from the microtubule, resulting in futile cycles of ATP hydrolysis without forward progress along the microtubule. Finally, a cluster of basic residues on the outer surface of the coiled coil could potentially interact with the negatively charged tubulin subunit (Kozielski et al., 1997; Tucker and Goldstein, 1997), which would help to maintain contact between the motor and the microtubule, thereby helping processivity.
Surprisingly, we also discovered that duplicating the first heptad repeat of the neck (T336–K342) results in a striking “gain-of-function” phenotype, increasing processivity more than threefold. Thus, the length of the neck helix may play a role in the efficiency of processive movement. Interestingly, the neck helix of conventional kinesin motors is 10 residues longer than the predicted neck helices of other NH2-terminal motors in the kinesin superfamily (Vale and Fletterick, 1997). At the present time, it is difficult to explain why duplicating seven amino acids in the neck improves processivity. For myosin, a long α helix in the neck is thought to act as a lever arm to swing the motor between binding sites (Block, 1996), and changes in the length of this helix correlate with changes in the velocity of the motor (Uyeda et al., 1996). However, as shown in this and previous studies (Inoue et al., 1997), deletion of the kinesin neck coiled coil does not significantly diminish the velocity of movement, which argues that the kinesin neck helix does not act as a lever arm essential for movement, as is generally envisioned for myosin (Vale, 1996). It remains possible, however, that the neck helix increases processivity by increasing the chance that the partner head will reach and bind to the next tubulin dimer. Alternatively, if the seven duplicated aa do not form part of the coiled coil structure, they might enhance processivity by increasing the length of the linker between the two heads. However, addition of three glycine residues in the same location produces the opposite effect. Thus, if this idea is correct, then the specific structure of the linker element must be important for eliciting this effect.
Location of Processivity Elements in Kinesin
Our results, in conjunction with previous studies (Berliner et al., 1995; Vale et al., 1996), indicate that processivity requires a kinesin dimer and is optimized when the heads are connected by the neck coiled coil. However, this work also eliminates the neck coiled coil as a primary determinant for single motor motility, which implies that the catalytic domain and/or the neck β sheet region contain important elements for processivity. This conclusion is consistent with the results of Case et al. (1997), who found that the neck and stalk of kinesin are not sufficient to confer processivity on the catalytic domain of NCD, a kinesin superfamily member. Further mutagenesis studies in conjunction with single molecule assays can be used to better define the regions of kinesin that are essential to processive motion.
Acknowledgments
We wish to thank N. Hom-Booher and C. Hart, who provided valuable assistance with cloning, and J. Kull, who prepared Fig. 4. We also extend thanks to E. Mandelkow for discussions providing structure coordinates for figure preparation, R. Cooke for helpful discussion, and R. Case and G. Woehlke (all from University of California, San Francisco, CA, except Woehlke [University of Munich, Munich, Germany]) for their comments on the manuscript. L. Romberg received a Chancellor's graduate student fellowship from the University of California, San Francisco, and D. Pierce received postdoctoral support from the Jane Coffin Childs Foundation.
Abbreviations used in this paper
- aa
amino acid
- GFP
green fluorescent protein
Note Added in Proof
We have extended the STABLE COIL replacement to include the first heptad of the coiled coil (kinesin residues 337–342 replaced by residues IEALKA in the STABLE COIL construct) and find that the expressed motor shows good processive motility.
Footnotes
Address all correspondence to Ron Vale, Department of Cellular and Molecular Pharmacology, University of California at San Francisco, 513 Parnassus Avenue, San Francisco, CA 94143. Tel.: (415) 476-6380. Fax: (415) 476-5233. E-mail: vale@phy.ucsf.edu
References
- Amos LA, Hirose K. The structure of microtubule-motor complexes. Curr Opin Cell Biol. 1997;9:4–11. doi: 10.1016/s0955-0674(97)80145-7. [DOI] [PubMed] [Google Scholar]
- Arnal I, Metoz F, DeBonis S, Wade RH. Three-dimensional structure of functional motor proteins on microtubules. Curr Biol. 1996;6:1265–1270. doi: 10.1016/s0960-9822(02)70712-4. [DOI] [PubMed] [Google Scholar]
- Berliner E, Young EC, Anderson K, Mahtani H, Gelles J. Failure of a single-headed kinesin to track parallel to microtubule protofilaments. Nature. 1995;373:718–721. doi: 10.1038/373718a0. [DOI] [PubMed] [Google Scholar]
- Block SM. Fifty ways to love your lever: myosin motors. Cell. 1996;87:151–157. doi: 10.1016/s0092-8674(00)81332-x. [DOI] [PubMed] [Google Scholar]
- Block SM, Goldstein LS, Schnapp BJ. Bead movement by single kinesin molecules with optical tweezers. Nature. 1990;348:348–352. doi: 10.1038/348348a0. [DOI] [PubMed] [Google Scholar]
- Bloom G, Endow S. Motor proteins 1: kinesin. Protein Profile. 1995;2:1112–1138. [PubMed] [Google Scholar]
- Case RB, Pierce DW, Hom-Booher N, Hart CL, Vale RD. The directional preference of kinesin motors is specified by an element outside of the motor catalytic domain. Cell. 1997;90:959–966. doi: 10.1016/s0092-8674(00)80360-8. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Pederson PL. Adenosine triphosphatase from rat liver mitochondria. J Biol Chem. 1971;246:4987–4994. [PubMed] [Google Scholar]
- Finer JT, Simmons RM, Spudich JA. Single myosin molecule mechanics: piconewton forces and nanometer steps. Nature. 1994;368:113–119. doi: 10.1038/368113a0. [DOI] [PubMed] [Google Scholar]
- Funatsu T, Harada Y, Tokunaga M, Saito K, Yanagida Y. Imaging of single fluorescent molecules and individual ATP turnovers by single myosin molecules in aqueous solution. Nature. 1995;374:555–559. doi: 10.1038/374555a0. [DOI] [PubMed] [Google Scholar]
- Gibbons IR, Fronk E. A latent adenosine triphosphatase form of dynein 1 from sea urchin sperm flagella. J Biol Chem. 1979;254:187–196. [PubMed] [Google Scholar]
- Hackney DD. Evidence for alternating head catalysis by kinesin during microtubule-stimulated ATP hydrolysis. Proc Natl Acad Sci USA. 1994;91:6865–6869. doi: 10.1073/pnas.91.15.6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackney DD. Highly processive microtubule-stimulated ATP hydrolysis by dimeric kinesin head domains. Nature. 1995;377:448–450. doi: 10.1038/377448a0. [DOI] [PubMed] [Google Scholar]
- Harada Y, Sakurada K, Aoki T, Thomas DD, Yanagida T. Mechanochemical coupling in actomyosin energy transduction studied by in vitromovement assay. J Mol Biol. 1990;216:49–68. doi: 10.1016/S0022-2836(05)80060-9. [DOI] [PubMed] [Google Scholar]
- Heim R, Cubitt AB, Tsien RY. Improved green fluorescence. Nature. 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- Henningsen U, Schliwa M. Reversal in the direction of movement of a molecular motor. Nature. 1997;389:93–95. doi: 10.1038/38022. [DOI] [PubMed] [Google Scholar]
- Hirose K, Lockhart A, Cross RA, Amos LA. Three-dimensional cryoelectron microscopy of dimeric kinesin and ncd motor domains on microtubules. Proc Natl Acad Sci USA. 1996;93:9539–9544. doi: 10.1073/pnas.93.18.9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose K, Amos WB, Lockhart A, Cross RA, Amos LA. Three- dimensional cryoelectron microscopy of 16-protofilament microtubules: structure, polarity, and interaction with motor proteins. J Struct Biol. 1997;118:140–148. doi: 10.1006/jsbi.1997.3840. [DOI] [PubMed] [Google Scholar]
- Hoenger A, Milligan RA. Motor domains of kinesin and ncd interact with microtubule protofilaments with the same binding geometry. J Mol Biol. 1997;265:553–564. doi: 10.1006/jmbi.1996.0757. [DOI] [PubMed] [Google Scholar]
- Hoenger A, Sablin EP, Vale RD, Fletterick RJ, Milligan RA. Three-dimensional structure of a tubulin-motor-protein complex. Nature. 1995;376:271–274. doi: 10.1038/376271a0. [DOI] [PubMed] [Google Scholar]
- Howard J. Molecular motors: structural adaptations to cellular functions. Nature. 1997;389:561–567. doi: 10.1038/39247. [DOI] [PubMed] [Google Scholar]
- Howard J, Hudspeth AJ, Vale RD. Movement of microtubules by single kinesin molecules. Nature. 1989;342:154–158. doi: 10.1038/342154a0. [DOI] [PubMed] [Google Scholar]
- Huang TG, Hackney DD. Drosophila kinesin minimal motor domain expressed in Escherichia coli: purification and kinetic characterization. J Biol Chem. 1993;269:16493–16501. [PubMed] [Google Scholar]
- Huang TG, Suhan J, Hackney DD. Drosophila kinesin motor domain extending to amino acid position 392 is dimeric when expressed in Escherichia coli. . J Biol Chem. 1994;269:16502–16507. [PubMed] [Google Scholar]
- Hunt AJ, Howard J. Kinesin swivels to permit microtubule movement in any direction. Proc Natl Acad Sci USA. 1993;90:11653–11657. doi: 10.1073/pnas.90.24.11653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Toyoshima YY, Iwane AH, Morimoto S, Higuchi H, Yanagida T. Movements of truncated kinesin fragments with a short or an artificial flexible neck. Proc Natl Acad Sci USA. 1997;94:7275–7280. doi: 10.1073/pnas.94.14.7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Hackney DD. Monomeric kinesin head domains hydrolyze multiple ATP molecules before release from a microtubule. J Biol Chem. 1997;272:5616–5621. doi: 10.1074/jbc.272.9.5616. [DOI] [PubMed] [Google Scholar]
- Jiang W, Stock MF, Li X, Hackney DD. Influence of the kinesin neck domain on dimerization and ATPase kinetics. J Biol Chem. 1997;272:7626–7632. doi: 10.1074/jbc.272.12.7626. [DOI] [PubMed] [Google Scholar]
- Kozielski F, Sack S, Marx A, Thormahlen M, Schonbrunn E, Biou V, Thompson A, Mandelkow E-M, Mandelkow E. The crystal structure of dimeric kinesin and implications for microtubule-dependent motility. Cell. 1997;91:985–994. doi: 10.1016/s0092-8674(00)80489-4. [DOI] [PubMed] [Google Scholar]
- Kull FJ, Sablin EP, Lau R, Fletterick RJ, Vale R D. Crystal structure of the kinesin motor domain reveals a structural similarity to myosin. Nature. 1996;380:550–555. doi: 10.1038/380550a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y-Z, Taylor EW. Interacting head mechanism of microtubule-kinesin ATPase. J Biol Chem. 1997;272:724–730. doi: 10.1074/jbc.272.2.724. [DOI] [PubMed] [Google Scholar]
- Morii H, Takenawa T, Arisaka F, Shimizu T. Identification of kinesin neck region as a stable α-helical coiled-coil and its thermodynamic characterization. Biochemistry. 1997;36:1933–1942. doi: 10.1021/bi962392l. [DOI] [PubMed] [Google Scholar]
- Pierce DW, Hom-Booher N, Vale RD. Imaging individual green fluorescent proteins. Nature. 1997;388:338. doi: 10.1038/41009. [DOI] [PubMed] [Google Scholar]
- Pierce, D.W., and R.D. Vale. 1998. Visualization of single GFP molecules and applications to assaying single protein dynamics. Meth. Cell Biol. In press. [DOI] [PubMed]
- Sablin EP, Kull FJ, Cooke R, Vale RD, Fletterick RJ. Crystal structure of the motor domain of the kinesin-related motor ncd. Nature. 1996;380:555–559. doi: 10.1038/380555a0. [DOI] [PubMed] [Google Scholar]
- Scott DW. On optimal and data-based histograms. Biometrika. 1979;66:605–610. [Google Scholar]
- Sosa H, Dias DP, Hoenger A, Whittaker M, Wilson-Kubalek E, Sablin E, Fletterick RJ, Vale RD, Milligan RA. A model for the microtubule-Ncd motor protein complex obtained by cryo-electron microscopy and image analysis. Cell. 1997;90:217–224. doi: 10.1016/s0092-8674(00)80330-x. [DOI] [PubMed] [Google Scholar]
- Stewart RJ, Thaler JP, Goldstein LS. Direction of microtubule movement is an intrinsic property of the motor domains of kinesin heavy chain and Drosophilancd protein. Proc Natl Acad Sci USA. 1993;90:5209–5213. doi: 10.1073/pnas.90.11.5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su JY, Hodges RS, Kay CM. Effect of chain length on the formation and stability of synthetic alpha-helical coiled coils. Biochemistry. 1994;33:15501–15510. doi: 10.1021/bi00255a032. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Schmidt CF, Schnapp BJ, Block SM. Direct observation of kinesin stepping by optical trapping interferometry. Nature. 1993;365:721–727. doi: 10.1038/365721a0. [DOI] [PubMed] [Google Scholar]
- Tripet B, Vale RD, Hodges RS. Demonstration of coiled-coil interactions within the kinesin neck region using synthetic peptides: implications for motor activity. J Biol Chem. 1997;272:8946–8956. doi: 10.1074/jbc.272.14.8946. [DOI] [PubMed] [Google Scholar]
- Tucker C, Goldstein LSB. Probing the kinesin-microtubule interaction. J Biol Chem. 1997;272:9481–9488. doi: 10.1074/jbc.272.14.9481. [DOI] [PubMed] [Google Scholar]
- Uyeda TQP, Abramson PD, Spudich JA. The neck region of the myosin motor domain acts as a lever arm to generate movement. Proc Natl Acad Sci USA. 1996;93:4459–4464. doi: 10.1073/pnas.93.9.4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD. Switches, latches, and amplifiers: common themes of molecular motors and G proteins. J Cell Biol. 1996;135:291–302. doi: 10.1083/jcb.135.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Fletterick RJ. The design plan of kinesin motors. Annu Rev Cell Dev Biol. 1997;12:745–777. doi: 10.1146/annurev.cellbio.13.1.745. [DOI] [PubMed] [Google Scholar]
- Vale RD, Malik F, Brown D. Directional instability of microtubule transport in the presence of kinesin and dynein, two opposite polarity motor proteins. J Cell Biol. 1992;119:1589–1596. doi: 10.1083/jcb.119.6.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Funatsu T, Pierce DW, Romberg L, Harada Y, Yanagida T. Direct observation of single kinesin molecules moving along microtubules. Nature. 1996;380:451–453. doi: 10.1038/380451a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woehlke G, Ruby AK, Hart CL, Ly B, Hom-Booher N, Vale RD. Microtubule interaction site of the kinesin motor. Cell. 1997;90:207–216. doi: 10.1016/s0092-8674(00)80329-3. [DOI] [PubMed] [Google Scholar]