

Abstract
In proliferating cells, DNA synthesis must be performed with extreme precision. We show that groups of replicons, labeled together as replicon clusters, form stable units of chromosome structure. HeLa cells were labeled with 5-bromodeoxyuridine (BrdU) at different times of S phase. At the onset of S phase, clusters of replicons were activated in each of ∼750 replication sites. The majority of these replication “foci” were shown to be individual replicon clusters that remained together, as stable cohorts, throughout the following 15 cell cycles. In individual cells, the same replication foci were labeled with BrdU and 5-iododeoxyuridine at the beginning of different cell cycles. In DNA fibers, 95% of replicons in replicon clusters that were labeled at the beginning of one S phase were also labeled at the beginning of the next. This shows that a subset of origins are activated both reliably and efficiently in different cycles.
The majority of replication forks activated at the onset of S phase terminated 45–60 min later. During this interval, secondary replicon clusters became active. However, while the activation of early replicons is synchronized at the onset of S phase, different secondary clusters were activated at different times. Nevertheless, replication foci pulse labeled during any short interval of S phase were stable for many cell cycles. We propose that the coordinated replication of related groups of replicons, that form stable replicon clusters, contributes to the efficient activation and propagation of S phase in mammalian cells.
The proliferation of eukaryotic cells requires that two key events, DNA synthesis and chromosome segregation, are highly regulated and performed with extreme fidelity. Over recent years, it has become clear how different cyclin-dependent kinases, together with associated cyclins, regulate cell cycle progression and serve to coordinate these vital events (Heichman and Roberts, 1994; Nurse, 1994). The activation of S phase is one aspect of this program that has been studied in detail. A critical part has been the isolation (Hsiao and Carbon, 1979; Stinchcomb et al., 1979) and characterization (for review see Fangman and Brewer, 1992; Marahrens and Stillman, 1996) of sequences from yeast (Saccharomyces cerevisiae) that are capable of sustaining the autonomous replication of extrachromosomal DNA. These autonomous replicating sequence (ARS) elements are pivotal in activating S phase (Fangman and Brewer, 1992; Marahrens and Stillman, 1996), providing binding sites for an origin recognition complex (Diffley and Cocker, 1992) that allows activation of replication at the required time of the cell cycle (Cocker et al., 1996).
In higher eukaryotes, genetically determined elements direct sites of initiation of DNA synthesis (for review see Coverley and Laskey, 1994; DePamphilis, 1996). In contrast to yeast, however, different origins do not appear to contain classical ARS-like sequence elements. In addition, sites where DNA synthesis initiates seem to be determined with lower precision, so that preferred sites of initiation appear to be surrounded by many potential secondary sites (for review see DePamphilis, 1996). To explain these differences, it has been proposed that structural criteria influence the efficiency of activation, in individual cells. However, the molecular mechanisms that might control this selection remain ill defined.
Once S phase begins, origins must be activated efficiently but only once in each cycle (for review see Coverley and Laskey, 1994). In yeast, origins fire at different times (Reynolds et al., 1989), even though they are thought to have the same origin recognition complexes present at the onset of S phase (Diffley and Cocker, 1992). The demonstration that the time of activation of different origins correlates with position (Reynolds et al., 1989) is supported by the fact that early replicating origins can become late replicating when transferred to new chromosomal sites (Fangman and Brewer, 1992). In mammalian cells, replication origins that are typically 150-kbp apart (Edenberg and Huberman, 1975; Hand, 1978) are activated at different times of an S phase lasting 8–10 h. Only 10–15% of replicons are active at any time; most active genes replicate early in S phase whereas inactive genes and repetitive elements replicate later (Hatton et al., 1988; O'Keefe et al., 1992). This program, and associated changes in the structure of active replication sites (Nakamura et al., 1986; van Dierendonck et al., 1989; Humbert and Usson, 1992), imply that an orderly series of events follow once S phase is activated. As the success of this S phase program is clearly fundamental to the completion of replication, it is perhaps surprising that mechanisms by which these events are propagated remain poorly characterized.
We describe experiments designed to establish how replicons activated at different times of S phase might be coordinated into an orderly S phase program. Synchronized human cells were pulse labeled with 5-bromodeoxyuridine (BrdU)1 and sites containing Br-labeled DNA were analyzed up to 14 d later. After the first mitosis, the random segregation of labeled and unlabeled chromosomes gives nuclei with progressively fewer labeled sites. Simplification of the labeled sites reveals that groups of labeled foci segregate together during mitosis and occupy discrete chromosome domains during interphase. Throughout this period, the density of Br-DNA–containing foci on the labeled mitotic chromosomes and their interphase counterparts does not change. As each foci contains many replicons, replicon clusters must be stable over many cell cycles. Cells synchronized at the beginning of S phase were labeled with BrdU and 5-iododeoxyuridine (IdU) in different cycles and the activation of replicons in replicon clusters assessed. To establish how replicon clusters are activated within S phase, sites of synthesis labeled for different periods of S phase were analyzed. 45–60 min from the onset of S phase, many forks of the first replicons activated terminate. This is followed by the activation of secondary replicons and appearance of new labeled sites, often adjacent to those labeled initially. As stable replicon clusters are seen when replication sites are pulse labeled during any short interval of S phase, these probably reflect some fundamental feature of chromosome structure.
Materials and Methods
Cell Culture and Synchronization
HeLa cells were grown in MEM modified for suspension culture and supplemented with 10% FCS. Cells were synchronized in mitosis using thymidine and nocodazole. Cells were first blocked in S phase (2.5 mM thymidine, 24 h), washed thoroughly, regrown for 4 h in fresh medium, and then >95% were arrested at mitosis using nocodazole (50 ng/ml, 8 h; Aldrich Chemical Co., Ltd., Gillingham, England). Cells were regrown in fresh medium, aphidicolin (5 μg/ml; Sigma Chemical Co., Poole, UK) was added after 7 h, and cells synchronized at the G1/S border were collected 2 h later (Jackson, 1995a ). In Fig. 6, synchrony was improved using a double thymidine block (2.5 mM thymidine, 24 h; medium, 12 h; 2.5 mM thymidine, 12 h) followed by nocodazole and aphidicolin, as above. This reduces the time cells are held in metaphase, decreasing the time taken to complete mitosis once nocodazole is removed. Note however, that whereas both approaches give synchronized S phase cells that reverse readily on removing aphidicolin and perform replication with normal characteristics, the timing used to maintain these criteria gives populations with only 30–40% S phase cells. In double synchrony experiments only ∼10% cells are double labeled.
Figure 6.

Early replicons are activated efficiently in different cell cycles. Cells synchronized at the beginning of S phase were grown for 20 min in medium containing BrdU, and then 4 d in medium, synchronized a second time, and grown for 20 min in medium containing IdU. Sites containing BrdU (A, D, and E) and IdU (B, F, and G) were indirectly immunolabeled. CLSM images showed foci containing Br-DNA (green) to colocalize with IdU-labeled (red) sites (C, H, and I; overlaps between A, D, or E, and B, F, or G show yellow in merge). Synchronized cells were also grown in medium supplemented with BrdU 5–20 min from the beginning of one S phase and IdU 10–30 min from the beginning of the next, after growing for 20 h in medium and synchronizing a second time (J). Spread DNA fibers were prepared and sites containing BrdU (J) and IdU (K) incorporation indirectly immunolabeled with FITC and Cy-3, respectively. Clusters of two to four replicons with at least one replicon labeled with both BrdU and IdU were collected; a typical example with three replicons (ori1–3) that are active in both cycles is shown (J–L). In 54 clusters with 126 replicons 120 were labeled with both analogues. Bars: (A–C) 2.5 μm; (D–I) 1 μm; (J–L) 5 μm.
Labeling Sites of DNA or RNA Synthesis
Synchronized cells were washed with fresh medium to remove aphidicolin. Cells were regrown in fresh medium (5 × 106 cells/ml) and BrdU added (100 μM, 20 min or 10 μM, 3–10 h).
After labeling, cells were washed once and regrown in medium containing thymidine (100 μM, 30 min; then 10 μM, 24 h). Cultures containing BrdU or cells with Br-DNA were protected from light.
To label sites of transcription, G1 phase HeLa cells (4 h after removing nocodazole) encapsulated in agarose microbeads (2.5 × 106/ml; Jackson and Cook, 1985) were incubated for 5 min in medium supplemented with 2.5 mM bromouridine.
Chromosome and DNA Spreads
Chromosomes and nuclei were spread onto glass slides following a standard procedure. Cell cultures (5 × 105 cells/ml) were incubated in medium containing colcemid (50 ng/ml, 30 min; 37°C; Sigma Chemical Co., Ltd.), washed once in HBSS, once in PBS, swollen (0.075 M KCl, 15 min; 37°C), fixed with methanol/acetic acid (3:1), dropped (three drops; 2.5 × 106 cells/ml) onto washed slides, and then air dried.
To prepare extended DNA fibers (Parra and Windle, 1993), 2 μl cells resuspended in PBS (106 cells/ml) were spotted onto cleaned glass slides and lysed with 5 μl of 0.5% SDS in 200 mM Tris-HCl, pH 7.4, 50 mM EDTA (10 min, 20°C). Slides were tilted (15° to horizontal), allowing a stream of DNA to run slowly down the slide, air dried, and then fixed in methanol/acetic acid (3:1). For most purposes, cells containing halogenated DNA were diluted 30- and 100-fold with untreated HeLa cells, before spreading. This simplifies the spreads, allows isolated labeled DNA fibers to be found with relative ease, and makes possible the identification of replicons from a single labeled cell (see Fig. 4).
Figure 4.

Replication foci contain multiple replicons. Cells synchronized at the beginning of S phase were grown for 5 min in medium, and then 10 (A), 20 (B), 40 (C), 60 (D), or 120 min (E–G) in medium containing BrdU. Spread DNA fibers were prepared and sites of BrdU incorporation indirectly immunolabeled with Cy-3 (A–G). Each panel (A–G) shows a single- labeled DNA fiber with three origins of replication (ori1–3, small arrows from left to right). In A–E, replication is assumed to have initiated at the center of a short, unlabeled region flanked by symmetrical growing forks. Replicons activated within S phase (E and F) are uniformly labeled, their origins are assumed to lie centrally. In E, three panels show two early origins (ori1, and 2) apparently adjacent to one that fired much later (ori3); for orientation, ori1 is indicated in both top and center panels. The rate of replication fork movement was established (H). For the times shown, 50 replicons (A–C) were measured and the average lengths (± SD) of 100 forks determined. The separation of 250 adjacent origins activated at the onset of S phase (I) and number of replicons activated in 100 clusters (J) are shown. In J, open bars show the distribution of clusters and closed bars the distribution of DNA. Bar, 5 μm.
To estimate the extension of DNA fibers, spreads were prepared from HeLa cells infected with adenovirus serotype 2 and grown in medium supplemented with 100 μM BrdU 15–20 h after infection (Pombo et al., 1994). Abundant Br-labeled DNA molecules measured 13.9 ± 1.3 μm (mean ± SD; n = 50). As the viral genome is 36 kbp, the extension of these DNA fibers is 2.59 ± 0.24 kbp/μm.
Cell Extractions
In Fig. 7, cells were washed once in PBS, once in PB (10 mM Na2HPO4, 1 mM MgCl2, 100 mM K-acetate, 30 mM KCl, 1 mM Na2ATP, 1 mM DTT, 0.2 mM PMSF, and 5 units/ml ribonuclease inhibitor (Amersham Intl., Little Chalfont, UK) adjusted to pH 7.4 (Jackson, 1995a ), lysed in PB with 0.25% Triton X-100 (10 min, 0°C), washed twice in PB, and then extracted with 2 M NaCl (10 min, 0°C).
Figure 7.

Early replication foci and transcription sites have common structural features. Synchronized cells encapsulated in agarose microbeads were grown in medium containing BrdU for 20 min either at the beginning of S phase (A) or 3 h later (B). Samples were extracted with 2 M NaCl and Br-DNA indirectly immunolabeled with FITC. G1 phase cells were grown in medium supplemented with BrU for 5 min and transcription sites labeled in the same way (C). Hypertonic extraction removes histones and spreads ∼60% DNA into a “halo” that surrounds the remnant nuclear matrix (D; DAPI staining of C). Bar, 2 μm.
Immunolabeling
To label sites of incorporation of BrdU in cells fixed onto glass, sites were stabilized by treatment with 4% paraformaldehyde (10 min, 20°C), washed, and then incubated with 2.5 M HCl (1 h, 20°C). Slides were rinsed three times in 0.1 M NaBorate, twice in PBS, twice in PBS containing 1% BSA and 0.1% Tween 20 (PBS+), incubated (1 h, 20°C) with a mouse anti-BrdU IgG (1 μg/ml; Boehringer Mannheim, Sussex, UK) in PBS+, washed four times in PBS+, incubated (1 h, 20°C) with a donkey anti– mouse IgG conjugated with either FITC or Cy-3 (1:500 dilution in PBS+; Jackson Laboratories, Bar Harbor, ME), and then washed three times in PBS+. Finally, slides were incubated with 20 μM TOTO-3 in PBS+ (20 min, 20°C; Molecular Probes, Inc., Eugene, OR), rinsed twice in PBS, and then mounted in Vectashield (Vector Labs, Inc., Burlingame, CA). DNA fibers were denatured directly (2.5 M HCl, 1 h; 20°C) and immunostained with mouse anti-BrdU IgG and donkey anti–mouse IgG conjugated with Cy-3, as described above (see also Tomilin et al., 1993).
Cells or DNA fibers containing both BrdU and IdU were labeled following the approach described by Manders et al. (1992). DNA was denatured as described above, samples were washed in borate and PBS+, and then incubated (30 min, 20°C) with 5% normal donkey serum in PBS+, then (1 h, 20°C) with a rat anti-BrdU IgG (20 μg/ml; Harlan Sera-lab Ltd., Leicestershire, England), and (1 h, 20°C) donkey anti–rat F(ab′)2 conjugated with biotin (Jackson Laboratories). Samples were washed and blocked with normal donkey serum, fixed with 4% paraformaldehyde in PBS (10 min, 20°C), and then washed in PBS+. Slides were incubated (1 h, 20°C) with a mixture of mouse anti-BrdU IgG (1 μg/ml; Boehringer Mannheim) and goat anti-biotin IgG (1 μg/ml; Sigma Chemical Co.) washed, incubated (1 h, 20°C) with donkey anti–goat IgG conjugated with FITC, and donkey anti–mouse IgG conjugated with Cy-3 (both 1:500 dilution in PBS+; Jackson Laboratories), washed, and then mounted. This approach relies on the very low affinity of the rat anti-BrdU antibody for IdU. As the mouse anti-Br antibody binds both BrdU and IdU, a high concentration of the rat antibody is used to saturate Br-containing sites. The conditions used here were optimized to saturate Br-containing sites with the lowest possible background and loss of access to I-containing sites. Nevertheless, some reduction in the intensity of I-containing sites was always seen when these were also Br labeled (see Fig. 6 B). Fixation after adding anti–rat-biotin F(ab′)2 stabilizes the first complex and reduces cross-talk.
Cells extracted after encapsulation in agarose and fixed with 4% paraformaldehyde (10 min, 0°C) were washed three times in PBS, and DNA was denatured by incubating in 2.5 M HCl (1 h, 0°C). Samples were neutralized and immunolabeled as described above, incubated with 4′,6′-diamidino-2-phenylindole (DAPI; 0.05 μg/ml, 5 min; Boehringer Mannheim), washed twice in PBS, and then mounted in Vectashield.
Fluorescence Microscopy
Conventional photographs were taken using an Axiophot microscope (Carl Zeiss Inc., Thornwood, NY) fitted with standard filter sets (sets 02, 09, and 15; Carl Zeiss Inc.), ×100 oil-immersion lens (NA 1.3), and Optovar (×1, ×1.25, or ×1.6) using Tmax 400 black and white film.
In Fig. 3, images were captured using a charge-coupled device (CCD; C4742; 1000 × 1018 pixel; −40°C Peltier-cooled; Hamamatsu Photonics, Hamamatsu City, Japan), run under Image 1.41 (National Institutes of Health, Bethesda, MD). The camera settings were adjusted so that all images were collected using the full range of intensities, without saturation. Raw images were noise reduced using Image 1.41, incorporated into Photoshop (Adobe Systems, Mountain View, CA) and the peaks of intensities of discrete replication foci determined. The intensity of background auto-fluorescence (see Fig. 3, A–C), a consequence of acid treatment, was measured and subtracted; in Fig. 3, we deliberately show this uniform, low level, background to indicate the area occupied by each nucleus. The intensity profiles shown in Fig. 3 D were compared using the Kolmogorov-Smirnov test.
Figure 3.

The fluorescence intensities of replication foci. Cells synchronized at the beginning of S phase were grown for 20 min in medium containing BrdU, and then 20 h or 5 d in medium. Sites of BrdU incorporation were indirectly immunolabeled with FITC and images recorded using a CCD camera. A shows a typical cell in the first G1 phase after labeling and (B and C) two cells that indicate the range of labeling 5 d later. Raw images were collected, noise removed, and the intensities of individual foci were determined (D). 621 sites (D, open bars) chosen from random areas of six cells like A, and 430 sites (D, closed bars) all foci from seven cells like B and C were measured. Bar, 2.5 μm.
In Fig. 6, images were collected on a confocal laser scanning microscope (MRC 1000; Bio-Rad Laboratories, Hercules, CA) equipped with an argon/krypton laser (running under Comos 7.0a software; Bio-Rad Laboratories) and coupled to a Nikon Diaphot 200 inverted microscope. Samples were analyzed with a ×60 PlanApo objective (NA 1.4), using a pinhole aperture of 0.7 mm, and Kalman averaging of eight images. After contrast stretching, the images were incorporated into Photoshop, converted into TIFF format, and printed on a Phaser 440 (Tektronix, Inc., Beaverton, OR) dye sublimation printer.
Results
Sites of Replication in Human Cells
The DNA precursor analogue BrdU provides a facile means of labeling DNA in eukaryotic cells (Dolbeare, 1995, 1996). S phase cells grown for 20 min in medium supplemented with BrdU incorporate the analogue into active sites of DNA synthesis (Nakamura et al., 1986). Samples immunostained immediately show (Fig. 1 A) the distribution of active replication sites or foci. HeLa cells synchronized at the S phase border using aphidicolin (Jackson, 1995a ) incorporated BrdU into 749 ± 154 (n = 50) replication foci.
Figure 1.

Sites of DNA synthesis in synchronized HeLa cells. Cells synchronized at the beginning of S phase were grown for 20 min in medium containing BrdU. Sites of incorporation were indirectly immunolabeled with FITC immediately (A and E), and 10 (B and F), 15 (C and G), and 20 h (D and H) later. All the labeled sites of typical cells (A–D), and details from different cells (E–H) are shown. Arrowheads (C) mark the ends of a TOTO- 3–stained chromosome that labeled poorly. Based on size, it is probably the predominantly late-replicating, inactive X chromosome. Double arrowheads in F highlight daughter replication foci seen in G2 phase of the cell cycle; typical low (small arrowheads) and high (large arrowheads) intensity sites are indicated. Bars: (A–D) 2 μm; (E–F) 1 μm.
Replication Foci Persist Throughout the Cell Cycle
Individual replication sites are only active for about 1 h (Nakamura et al., 1986) before adjacent sites are activated. However, labeling with BrdU in vivo allows the distribution of replicons labeled during any interval of S phase to be visualized in any subsequent cell cycle phase (Sparvoli et al., 1994). DNA pulse labeled at the onset of S phase was shown to persist as labeled foci independently of DNA synthesis (Fig. 1, B–D). In G2 phase, these cells had 1275 ± 205 foci (n = 25). This 1.7-fold increase in the number of labeled foci follows the appearance of apparent sister foci (Fig. 1 F) that presumably arise from the separation of the duplicated DNA soon after synthesis (Selig et al., 1992; Kitsberg et al., 1993). Sister foci first appeared 1 h into S phase (not shown) and remained paired until mitosis (Fig. 1, C and G). As expected, predominantly single foci were seen in the following G1 phase (Fig. 1, D and H).
Replication Foci Are Stable Over Many Cell Generations
Similarities in the appearance of active replication foci (Fig. 1 A) and foci containing BrdU in the following G1 phase (Fig. 1 D; Sparvoli et al., 1994) suggest that different replicons of individual foci (see below) remain associated throughout the cell cycle, surviving structural changes that accompany mitosis. To confirm the stability of this association, cells synchronized at the beginning of S phase, were grown for 20 min in medium containing BrdU, and up to 14 d in medium. During this time, random segregation of labeled and unlabeled chromosomes will give rise to cells with progressively fewer Br-labeled chromosomes. If different replicons of individual foci remain associated throughout this period, the average number and intensity of foci labeled in each chromosome in mitosis and equivalent chromosome domains, during interphase (we call these interphase chromosome domains), should not change. If, on the other hand, replicons of one cluster were to reassort with those of adjacent clusters, the appearance of mitotic chromosomes or interphase chromosome domains with more labeled sites of reduced intensity would be expected.
5 d after incorporating BrdU, all labeled nuclei contained some unlabeled areas and some areas with labeled foci similar to those seen soon after incorporating BrdU (Fig. 2 A). At this time, labeled nuclei had Br-containing foci that appeared clustered in three to eight groups. 5 d later, 89% labeled cells retained a single group of labeled foci (Fig. 2, B–D) and after a further 4 d all labeled cells had a single group of labeled foci (not shown).
Figure 2.


Replicon clusters are stable over many cell generations. Cells synchronized at the beginning of S phase were grown for 20 min in medium containing BrdU and then 5 or 10 d in medium. Sites of BrdU incorporation were indirectly immunolabeled with FITC 5 (A) or 10 d (B–E) later. A shows a single cell with many clusters of labeled foci and (B–D) isolated clusters of labeled foci, the only labeled sites within three different cells. Foci are scattered along metaphase chromosomes in the same preparations (E); double exposure shows that foci are restricted to only one chromatid. The foci of 200 discrete clusters immunolabeled 5–10 d after incorporating BrdU in cells at the beginning of S phase (F), or unsynchronized cells (G) were counted and their distribution (open bars) compared with foci of 100 metaphase chromosomes (closed bars). Bars: (A) 2 μm; (B–E) 1 μm.
Samples were immunolabeled 5–10 d after incorporating BrdU at the beginning of S phase, and labeled foci in discrete groups of nuclear foci and individual mitotic chromosomes were counted. The number of labeled foci (Fig. 2 F) in the interphase groups (12.1 ± 4.8; n = 200) and mitotic chromosomes (10.4 ± 4.3; n = 100) was similar. As the HeLa cells used have 62 ± 8 (n = 50) chromosomes this confirms that a typical cell will have ∼750 Br-containing foci at the time of labeling (Fig. 1). Note that the distribution (Fig. 2 F) of foci reflects the length of different human chromosomes (Morton, 1991); with the obvious exception of the poorly labeled inactive X chromosome (Fig. 1 C), chromosomes have similar densities (foci/Mbp DNA) of replication foci at the onset of S phase. Unsynchronized cells showed a broader labeling profile (Fig. 2 G), their nuclei and chromosomes had 10.7 ± 6.0 (n = 200) and 10.3 ± 5.2 (n = 100) labeled sites, respectively. These experiments demonstrate that BrdUMP is incorporated into sites that appear stable for at least 15 cell generations. After many cycles of cell division, nuclei contain discrete groups or clusters of labeled foci that reflect the grouping seen on chromosomes, during mitosis.
Organization of Replicon Domain Clusters
Different replication forks elongate at similar rates after cell synchronization (Jackson, 1995a ). It is likely, therefore, that sites with the range of intensities seen above (e.g., Fig. 1) will contain different numbers of replicons. To confirm this, the intensities of individual foci were measured after capturing images with a CCD camera (Fig. 3). The range of intensities seen soon after BrdUMP was incorporated and in later generations indicates that most sites contain multiple replicons and that replicons labeled together as one cluster remained together over many generations; note that the sensitivity of the detection system used ensures that any fragmentation of labeled replicon clusters would be seen clearly. To confirm this point, we applied the Kolmogorov-Smirnov test to the intensity profiles of the two populations (Fig. 3 D). The probability that these two profiles arise from populations of foci with the same median value is >95%. Small differences in the shapes of the intensity profiles (Fig. 3 D) are consistent with cell cycle–dependent variations in chromatin compaction and inconsistent with fragmentation of the foci and appearance of new labeled sites. We conclude that groups of replicons labeled together at the onset of S phase remain clustered together over many cell generations.
Deconstructing the labeled sites of isolated cells using sarkosyl or SDS demonstrates that individual foci contain multiple replicons (not shown). To elaborate this point, extended DNA fibers prepared by spreading cells lysed in SDS (Fig. 4), confirmed that runs of adjacent replicons were labeled together. The distribution of replicons in different clusters (Fig. 4 J) was similar to the intensity profile of different replication foci (Fig. 3 D).
Propagating S Phase
DNA fiber spreads prepared from cells labeled for different periods at the beginning of S phase (Fig. 4) provide a detailed picture of the distribution and growth of replicons active at this time. Synchronized HeLa cells were resuspended in fresh medium, and BrdU was added 5 min later. After various times (Fig. 4), cells were lysed in SDS, DNA spreads were prepared, and Br-containing fibers were immunolabeled. This labeling strategy gives labeled replicons with short unlabeled patches containing the origin flanked by BrdUMP-containing growing forks. For example, when pulse labeling was performed for 10 min 97.5% (n = 200) of labeled replicons contained two labeled patches separated by an unlabeled region of similar length (Fig. 4 A).
As the majority of replicons activated at the onset of S phase are grouped in clusters (Fig. 4 J), details of their separation and the rate of replication fork movement can be used to estimate how long synthesis continues before opposing forks meet. As an important control, we used Br-labeled adenovirus DNA (see Materials and Methods) to establish the extension of DNA under the spreading and labeling condition used. With a DNA extension factor of 2.59 kbp/μm (see Materials and Methods), adjacent replicons, active at the onset of S phase, were 144 ± 66-kbp (n = 250) apart (Fig. 4). For the first 45 min of S phase, replication forks grew at an average rate of 1.7 ± 0.3 kbp/min (n = 300; maximum is 2.3 kbp/min; Fig. 4 H). During this period, 36% of forks met an oncoming fork and so completed synthesis. At this stage, replication of the majority of outward growing forks continued normally; only 5% of outward growing forks appeared to have terminated or stalled (see for example right fork of ori2 in Fig. 4 E, bottom panel). After 60 min of S phase, 87% of internal forks terminated and many new sites of synthesis had appeared. At this time, replication was propagated in three ways: (a) most outward growing forks from individual clusters continued to grow for ⩽2 h, the longest time analyzed (Fig. 4 E, top panel), independently of the activity of internal (sister) forks in the same cluster; (b) secondary replicons were activated adjacent to the primary replicons (Fig. 4 E, bottom panel); (c) secondary replicon clusters were activated elsewhere (Fig. 4, F and G). In the latter case, it is notable that replicons within secondary clusters were activated together while different secondary clusters from individual cells were activated at different times (Fig. 4, F and G).
Replication Foci Form Stable Structures Throughout S Phase
Experiments on unsynchronized cells (Fig. 2 G) indicated that replicon clusters labeled during any short interval of S phase might form labeled foci that are stable over many cell generations. To address this point, synchronized cells were grown in medium supplemented with BrdU either for times longer than required to complete synthesis of replicon clusters activated at the onset of S phase or for short periods within S phase. Cells labeled continuously for the first third or entire S phase had many more labeled sites than seen after pulse labeling, consistent with the progressive activation of discrete replicon clusters at different times of S phase. Cells that incorporated BrdU for 3 h, at the beginning of S phase, had 26.7 ± 7.7 (n = 100; range 12–43) foci/interphase chromosome domain, when analyzed 5 (Fig. 5 A) and 10 (Fig. 5, B–D) d later. Under these conditions, the labeling on mitotic chromosomes appears banded; because of their high density, individual foci appear to coalesce with their neighbors. Even during interphase, the replicon clusters labeled over this period of S phase often appeared fused or clustered in strings. When cells were labeled with BrdU for the entire S phase (Fig. 5, F–J) the clusters of labeled foci (Fig. 2) that remained 10 d later, appeared to occupy distinct nuclear domains (Fig. 5, G–I). These labeled interphase domains clearly represent individual chromosome “territories” as they give rise to chromosomes, with single-labeled chromatids, in mitosis (Fig. 5 J).
Figure 5.
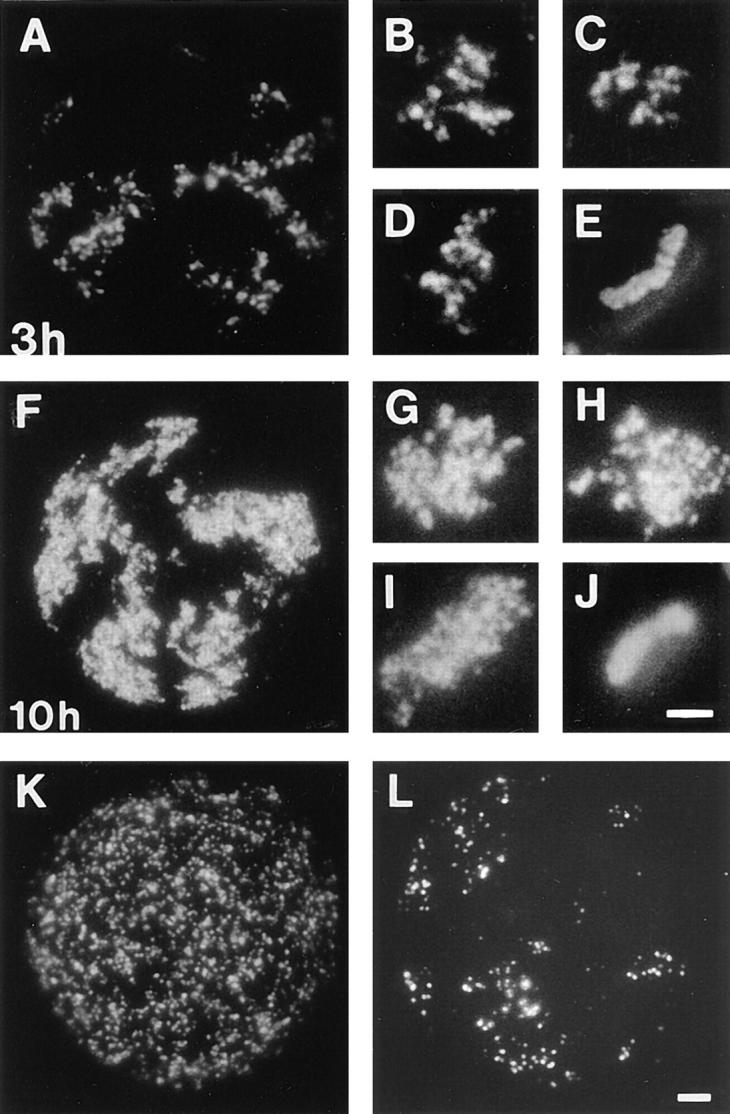
Replication during different periods of S phase. Cells synchronized at the beginning of S phase were grown for 3 (A–E) or 10 h (F–J) in medium containing BrdU, and then 5 (A and F) or 10 d (B–E, and G–J) in medium. Sites of BrdU incorporation were indirectly immunolabeled with FITC 5 (A and F) or 10 d (B–E, and G–J) later. A and F show single cells with many clusters of labeled foci and (B–D, and G–I) isolated clusters of labeled foci, the only labeled sites within three different cells. Metaphase chromosomes in the same preparations appeared banded (E) or fully labeled (J); double exposures show that labeling is restricted to only one chromatid. Synchronized cells were also grown in medium for 3 h and then medium was supplemented with BrdU for 20 min. Sites of incorporation were indirectly immunolabeled with FITC immediately (K) or 5 d later (L). Bars: (A, F, K, and L) 2 μm; (B–E, and G–J) 1 μm.
Cells labeled 3 h into S phase showed patterns of labeling (Fig. 5 K) similar to those seen at the onset of S phase; though the size and intensity of labeled foci is characteristic of those labeled at the onset of S phase, their distribution is less uniform. This aggregation of foci, that is obvious at the time of labeling (Fig. 5 K), was not apparent when cells labeled in mid–S phase were analyzed 5 d later (Fig. 5 L). These experiments suggest that replication foci, containing groups of replicons that are duplicated together, are a general feature of chromatin organization.
Clustered Replicons Are Activated Efficiently
We next attempted to establish if stable replicon clusters could contribute to the efficient activation of S phase. To determine if the same replication foci were activated in different cell cycles, synchronized cultures were pulse labeled with BrdU at the onset of S phase, grown, synchronized a second time, pulse labeled with IdU, and then analyzed in G1 phase of the following cycle. Immunostaining sites containing BrdU (Fig. 6, A, D, and E) or IdU (Fig. 6, B, F, and G) showed that the same replication foci were active at equivalent times in different cell cycles (Fig. 6, C, H, and I). The confocal sections shown highlight regions of double-labeled cells with Br-labeled foci clustered in two to four groups, and many more I-labeled foci (groups of I-labeled foci are not evident even though a typical optical section contains ∼25% of the nuclear volume and a corresponding fraction of I-labeled sites). In the images shown, sites containing DNA substituted with both halogens show a reduced labeling by the second antibody (see Materials and Methods); control experiments showed that this reflects competition for binding sites. At high magnification (Fig. 6, D–I), it is clear that almost all Br-labeled foci are also I-labeled; though exact colocalization of the two labels is rarely seen.
To improve the resolution of this analysis, DNA fibers were prepared from cells labeled with both BrdU and IdU. Synchronized cells were labeled with BrdU, grown through mitosis, synchronized a second time, labeled with IdU, and then DNA fibers were immunostained to reveal Br- and I-containing replicons (Fig. 6, J–L). Cells synchronized so that >90% are in S phase are known to develop a variety of replication defects (Jackson 1995a ); hence, it is not possible to label authentic sites of initiation in two consecutive cell cycles with high efficiency. To circumvent this problem, we use a synchronization protocol (see Materials and Methods) that avoids known artifacts. Then, to accommodate a high proportion of non–S phase cells, we restrict our analysis to replicons in replicon clusters containing both BrdUMP and IdUMP. We cannot analyze single replicons, as it is impossible to establish whether individual replicons, labeled with a single analogue, arise from cells in G1 phase during one round of labeling or from S phase cells that activate replication inefficiently.
DNA fiber spreads were prepared and isolated fibers containing replicon clusters with two to four active replicons were identified (see Materials and Methods). 54 clusters with 126 labeled replicons were analyzed. Of these, 120 (95%) were active in both cycles. In the example shown (Fig. 6, J–L), all three replicons were active in both cycles. This suggests that under the conditions used, replicons in replicon clusters activated at the onset of S phase initiate DNA synthesis with an efficiency of ∼97% in each cell cycle.
Nuclear Organization Influences the Activation of Replication Origins at the Onset of S Phase
As origins of replication are activated efficiently at the beginning of S phase, in different cell cycles, the activation process must distinguish replicons that are used at this time from those activated later. As transcriptionally active genes are known to be replicated early in S phase (Hatton et al., 1988), we wanted to establish if transcription sites and replicon clusters, active at different times of S phase, had any structural features in common. Replicons were pulse labeled with BrdU for different periods of S phase and extracted with buffer containing 2 M NaCl, 5 d later (Fig. 7). Cells labeled at the onset of S phase contained foci similar in appearance to those of unextracted cells (Fig. 2); though foci often appeared fragmented or more diffuse, once extracted, they always maintained a tight association with the nuclear matrix (Fig. 7 A). In contrast, when cells were labeled 3 h after the onset of S phase, hypertonic extraction dispersed labeled foci into the DNA halo (Fig. 7 B). This experiment shows that replicons from clusters labeled in early– and mid–S phase respond differently to hypertonic extraction. Note that whereas all replicons associate with the nuclear matrix as replication occurs (data not shown; Pardoll et al., 1980), this analysis is performed on unsynchronized cells, so that only a small fraction will have previously labeled foci that are undergoing replication at the time of extraction. The structure of replicon clusters active at the onset of S phase mimics the appearance of labeled sites of transcription, in cells extracted in the same way (Fig. 7, C and D).
Discussion
Duplicating any eukaryotic genome is a complex task that must be performed with high fidelity. Simple eukaryotic models, notably yeast, have facilitated a detailed description of ARS elements (Marahrens and Stillman, 1996) that define sites where DNA synthesis initiates (Fangman and Brewer, 1992) and shown how these direct a single initiation event at the appropriate time of each cell cycle (Diffley and Cocker, 1992; Cocker et al., 1996). In higher eukaryotes, with much more complex genomes, the elusive nature of replication origins (Coverley and Laskey, 1994) has prompted the suggestion that an efficient S phase will rely on “the interplay between specific DNA sequences, chromosome structure and nuclear organization” (DePamphilis, 1996). Here we describe a series of experiments designed to assess how such an interplay might be achieved.
Chromosome Structure and Nuclear Function
Whereas the characteristic appearance of mitotic chromosomes confirms that they are highly organized (Holmquist, 1992), the molecular features responsible remain ill-defined (Earnshaw, 1988; Manuelidis and Chen, 1990; Saitoh and Laemmli, 1994; Cook, 1995). However, different aspects of chromosome structure are known to correlate with nuclear function. For example, when cells are pulse labeled with BrdU, gene-rich regions, corresponding to chromosomal R bands in mitosis, are labeled at the beginning of S phase and must complete synthesis before late synthesis in regions corresponding to mitotic G bands can proceed (for review see Drouin et al., 1994). This distinction implies that elements of the mitotic organization must persist throughout interphase when each chromosome occupies a discrete “territory” (Cremer et al., 1993) that provides a reasonably stable environment for chromosome structure (Abney et al., 1997).
Units of Eukaryotic Replication
In mammalian cells, the synchronous activation of groups of contiguous replicons allows the separation of adjacent replicons to be measured (Huberman and Riggs, 1968). Detailed analyses of different mammalian cells suggests that most relicons are 50–300 kbp (for review see Edenberg and Huberman, 1975; Hand, 1978). This range was calculated from the length of labeled DNA fibers assuming that B form DNA in solution spreads so that 3-kbp DNA measures 1 μm. Indeed, whereas DNA fibers spread as described here can stretch to as much as twice their expected length (Parra and Windle, 1993) the average values for different sequences visualized by fluorescence in situ hybridization (FISH) are usually within 10% of that expected for DNA in solution. For example, the average extension of an integrated Epstein-Barr virus genome (Lestou et al., 1996) and various Arabidopsis sequences of 4–63 kbp (Fransz et al., 1996) were 2.99 kbp/μm and 3.27 kbp/μm, respectively. In our experiments, spread DNA was shown to stretch to 2.59 kbp/μm; we used Br-substituted adenovirus DNA so that immunolabeling could be performed under our standard conditions.
In our analysis, 92% of adjacent origins activated at the onset of S phase in HeLa cells were 50–300-kbp apart (Fig. 4). The average and extreme separations measured were 144, 21, and 406 kbp, respectively. Under these conditions, the anatomy of sites labeled at the onset of S phase confirms that most replicons have growing forks that emanate from a single point (Fig. 4) and that aberrant initiation events arise rarely (Jackson, 1995a ).
Visualizing Sites of DNA Synthesis Labeled In Vivo
It is a routine matter to label sites of replication using BrdU, a DNA precursor analogue that can be detected after incorporation into nascent DNA (for review see Dolbeare, 1995, 1996). The appearance of the labeled replication sites has been described in detail (Nakamura et al., 1986; Humbert and Usson, 1992; Tomilin et al., 1995), and is known to reflect the natural distribution of replication proteins (Kill et al., 1991; Krude, 1995), ancillary proteins (Leonhardt et al., 1992; Krude, 1995), and particular DNA sequences that replicate at known times of S phase (O'Keefe et al., 1992). Once labeled, replication foci also present a useful tool for the analysis of genome organization (for review see Drouin et al., 1994; Dolbeare, 1996). For example, human lymphocytes labeled with BrdU for 15 min give rise to mitotic chromosomes that appear labeled at discrete sites (e.g., Vogel et al., 1989). Indeed, as labeled replication foci appear to survive mitosis and persist throughout the subsequent cell cycle, this approach provides an opportunity to analyze how certain aspects of nuclear structure and function might be linked (Sparvoli et al., 1994; first observed by Meng, C., and R. Berezney. 1991. J. Cell Biol. 115:9a; as discussed in Jackson, 1995b and Berezney et al., 1995a ).
Here we confirm and extend these earlier reports using the organization of replication sites in HeLa cells as a model system. At the onset of S phase, a typical HeLa cell has ∼750 sites of DNA synthesis (Fig. 1). This is many more than the ∼126 sites identified in rat fibroblasts, using indirect immunofluorescence of early–S phase cells labeled with BrdU (Nakamura et al., 1986). However, in later studies, technical improvements increased the number of sites in mouse 3T3 cells to ∼250 (Fox et al., 1991), and then ∼600 (Berezney et al., 1995b ), in line with the number seen here.
After 2 h or more from the start of S phase, the first replicons activated have mostly completed synthesis and the majority give rise to discrete daughter foci (Fig. 1). Though daughter chromatids (Sumner, 1991) are not resolved until late in prophase, daughter replicons are known to be resolved soon after replication is complete (Selig et al., 1992; Kitsberg et al., 1993). Foci clearly persist into mitosis (Sparvoli et al., 1994), where the expected daughter foci (Baumgartner et al., 1991) can be seen on adjacent chromatid arms (Fig. 1). When nuclei reform after mitosis, the daughter cells contain foci similar in appearance to those seen in the previous S phase (Fig. 1; Sparvoli et al., 1994).
Replication Foci Are Replicon Clusters
Simple calculations show that most replication foci must contain many replicons (Nakamura et al., 1986). Throughout S phase, an average human chromosome of 145 Mbp has ∼12 active sites; if each were a single replicon with forks growing at ∼2 kbp/min S phase would take ∼50 h. As S phase is ∼10 h in human cells an average site must contain approximately five replicons.
We have shown that 84% replicons are activated in clusters (Fig. 4) and demonstrated that the number of replicons in different clusters (Fig. 4 J) is similar to the distribution of intensities in different labeled foci (Fig. 3 D). This suggests that most replication foci are individual replicon clusters; overabundant single replicons could be because of breaks during spreading, but are also consistent with a minority (<10%) of foci having single replicons and unrelated replicon clusters labeled together.
Replicon Clusters Are Stable Units of Chromosome Structure
Once BrdU is incorporated into DNA, successive cycles of growth, in the absence of the analogue, lead to a progressive simplification of labeled sites through random chromosome segregation (Fig. 2). After 10 cell generations, most cells contain a single group of BrdU-containing foci; these correspond with individual chromosome domains during interphase, and give rise to single-labeled chromatids, at mitosis. As replication foci also contain several replicons, these units must be stable over many cell cycles. If these structures became unstable, during the 15 generations analyzed here, we would expect the mixing of replicons from adjacent labeled and unlabeled clusters to give rise to new labeled sites. The possiblity that replicons from different pulse-labeled clusters might reassort seems unlikely, as most are adjacent to unlabeled clusters. Whether or not the same DNA sequences contribute to different replication foci, in all cells of a population, can only be established by FISH.
It is clearly important to understand the molecular features that give rise to stable replicon clusters. Preliminary experiments show that replicons activated at the onset of S phase, unlike those activated later, are tightly associated with the nuclear matrix throughout interphase (Fig. 7). This dramatic distinction is probably related to the established replication of active genes early in S phase (Hatton et al., 1988) and the stability of replicon clusters active at this time. We can only speculate on mechanisms responsible for stable replicon clusters seen in mid– and late–S phase, but imagine that these must reflect some fundamental feature of chromosome structure. We prefer this explanation to alternatives where the spatial organization of different replicon clusters is maintained “passively” as a result of other structural constraints; this type of mechanism could apply over short periods but would be unlikely to account for the long periods of stability described here.
As replication foci of unsynchronized cells behave similarly, stable replicon clusters must be a general feature of chromosome structure. Indeed, labeling for the first third or entire S phase or pulse labeling for short intervals within S phase (Fig. 5) suggests that replicons labeled together in different clusters, at any time of S phase, persist as stable units over many cell generations. Extended labeling also gives a clear impression of the appearance of chromosome territories during interphase (Fig. 5); single interphase chromosome domains clearly occupy a restricted nuclear volume (Fig. 5, H and I). Furthermore, individual territories appear to be separated by interchromatin channels, with chromatin from one territory rarely invading the space occupied by its neighbors (Fig. 5 F). This approach allows interphase chromosome domains to be seen using conditions that are much less destructive than those used to visualize chromosome territories by FISH (Schardin et al., 1985; Manuelidis, 1985).
In our preparations, a typical interphase chromosome domain is ∼4 μm (Fig. 5), and typical replication foci contain five replicons with ∼0.8-Mbp DNA, approximately one-tenth this size (Fig. 1). It follows, therefore, that stable replication foci with ∼1-Mbp DNA will impose significant constraints on the organization of DNA within chromosome domains in situ. Indeed, it may seem remarkable that two sequences separated by 1-Mbp DNA should be ∼1.5-μm apart in nuclei (van den Engh et al., 1992; Yokota et al., 1995) when the same sequences folded as a 10-nm chromatin fiber might be separated by as much as ∼55 μm (this extent of separation is clearly not possible in a typical human nucleus). It seems likely that replicon clusters contribute to the local organization of interphase chromosome domains, with small groups of adjacent clusters forming individual chromosome bands, during mitosis.
Efficient Activation of Replication within Replicon Clusters
In higher eukaryotes the pathway of replicon activation remains poorly defined. Though it is clear that genetically determined elements play a role, difficulties in isolating chromosomal ARS-like elements on extrachromosomal episomes, and the apparent promiscuity of DNA fragments that are >20 kbp, imply that simple sequence elements are not sufficient to drive S phase in mammalian cells (for review see Coverley and Laskey, 1994). As a result, it has been argued that chromosome structure and/or nuclear organization might play important roles during the S phase of higher eukaryotic cells (Coverley and Laskey, 1994; DePamphilis, 1996).
In S. cerevisiae, different ARS elements are activated with different efficiency—some are used each S phase in almost all cells, and others used rarely (Fangman and Brewer, 1992). In mammalian cells, this efficiency is difficult to assess as procedures that yield the desired level of cell synchrony alter the natural patterns of origin activation (see Jackson, 1995a for discussion). To overcome this problem, we have used a cell synchrony with normal S phase activation and analyzed only clustered replicons; if single replicons are labeled in only one cycle it is impossible to assess whether the lack of labeling is due to inefficient origin activation (in S phase cells), or a failure to reach S phase. We have shown that replicons activated at the beginning of S phase, in a particular cell, are activated with high efficiency in the same cell in the following S phase. Clearly this implies that a precise mechanism determines sites of synthesis and rules out the possibility that early origins are selected at random from a large pool of origins that are activated randomly, at any time during S phase.
Approaches using double-labeled DNA fibers should also allow detailed analyses of sites where replication begins. For example, labeled fibers (like those in Fig. 6) will allow us to establish if replication initiates at the same sites in two successive S phases (Jackson, D.A., and A. Pombo, manuscript in preparation) and, in conjunction with FISH, should allow the position of initiation to be established relative to any known sequence.
Nuclear Organization and the Propagation of S Phase
Once S phase begins, active replication forks grow at a constant rate of ∼2 kbp/min. No additional forks are activated during the next 30 min. 30–60 min after the onset of S phase, many elongating forks meet oncoming ones and terminate. At the same time, new sites are activated (Fig. 4). As synthesis progresses, secondary sites are activated adjacent to the primary ones (Manders et al., 1992, 1996; Berezney et al., 1995b ), and in clusters that are activated at different times, but that contain replicons activated together (Fig. 4, F and G).
This clearly distinguishes the activation of primary and secondary replicons and suggests that a cell cycle–dependent “switch” that activates synthesis at the beginning of S phase does not initiate synthesis of replicons activated later. As the activation of origins that fire within S phase is dependent on the completion of replicons activated earlier (Jackson, 1995a ; Fig. 4), it follows that events occurring during any one phase of synthesis are coupled to the activation of the next phase. As very similar numbers of synthetic sites are active at any time during the first 3 h of S phase, it is possible that once activated, each replication factory (Hozák et al., 1993) recruits and replicates consecutive replicon clusters in turn. If the propagation of S phase is programmed by the arrangement of replicon clusters, these units would perform a vital function and might reflect some fundamental feature of chromosome structure in mammalian cells.
Conclusion
We have shown that the HeLa cell genome is organized into a series of stable replicon clusters. At the onset of S phase, the extent of replication at different active sites suggests that the replicons within most replicon clusters are labeled together, at a single nuclear site. The replicon clusters activated at this time initiate synthesis both reliably and efficiently, in consecutive cell cycles. At different times of S phase, replicons labeled together in different replication sites appear to remain together, forming stable units of chromosome structures over many cell generations. As S phase proceeds, different replication sites are active for ∼1 h before new sites are activated. The spatial arrangement of replicon clusters, active at different times of S phase, suggests that their organization might contribute to an orderly program that ensures the success of replication in higher eukaryotes.
Acknowledgments
We thank P. Cook, E. Manders, and S. Kearsey for their help and encouragement. We also thank the Cancer Research Campaign and Program PRAXIS XXI (Portugal) for support.
Abbreviations used in this paper
- BrdU
5-bromodeoxyuridine
- IdU
5-iododeoxyuridine
Footnotes
Address all correspondence to D.A. Jackson, CRC Nuclear Structure and Function Research Group, Sir William Dunn School of Pathology, University of Oxford, South Parks Road, Oxford, OX1 3RE United Kingdom. Tel.: 44-1865-275527. Fax: 44-1865-275501. E-mail: Dean.Jackson@Path.OX.AC.UK
References
- Abney JR, Cutler B, Fillbach ML, Axelrod D, Scalettar BA. Chromatin dynamics in interphase nuclei and its implications for nuclear structure. J Cell Biol. 1997;137:1459–1468. doi: 10.1083/jcb.137.7.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner M, Dutrillaux B, Lemieux N, Paulin D, Viegas-Pequignot E. Genes occupy a fixed and symmetrical position on sister chromatids. Cell. 1991;64:761–767. doi: 10.1016/0092-8674(91)90505-s. [DOI] [PubMed] [Google Scholar]
- Berezney R, Mortillaro MJ, Ma H, Wei X, Samarabandu J. The nuclear matrix: a structural milieu for genomic function. Int Rev Cytol. 1995a;162:1–65. doi: 10.1016/s0074-7696(08)61228-0. [DOI] [PubMed] [Google Scholar]
- Berezney R, Ma H, Meng C, Samarabandu J, Cheng PC. Connecting genomic architecture and DNA replication in three dimensions. Zool Studies. 1995b;1(Suppl.):29–32. [Google Scholar]
- Cocker JH, Piatti S, Santocanale C, Nasmyth K, Diffley JFX. An essential role for the Cdc6 protein in forming the pre-replicative complexes of budding yeast. Nature. 1996;379:180–182. doi: 10.1038/379180a0. [DOI] [PubMed] [Google Scholar]
- Cook PR. A chromomeric model for nuclear and chromosome structure. J Cell Sci. 1995;108:2927–2935. doi: 10.1242/jcs.108.9.2927. [DOI] [PubMed] [Google Scholar]
- Coverley D, Laskey RA. Regulation of eukaryotic DNA replication. Annu Rev Biochem. 1994;63:745–776. doi: 10.1146/annurev.bi.63.070194.003525. [DOI] [PubMed] [Google Scholar]
- Cremer T, Kurz A, Zirbel R, Dietzel S, Rinke B, Schrock E, Speicher MR, Mathieu U, Jauch A, Emmerich P, et al. Role of chromosome territories in the functional compartmentalization of the cell nucleus. Cold Spring Harbor Symp Quant Biol. 1993;58:777–792. doi: 10.1101/sqb.1993.058.01.085. [DOI] [PubMed] [Google Scholar]
- DePamphilis, M.L. 1996. Replication origins in metazoan chromosomes. In Eukaryotic DNA Replication. J.J. Blow, editor. IRL Press, Oxford. 96–123.
- Diffley JFX, Cocker JH. Protein-DNA interactions at a yeast replication origin. Nature. 1992;357:169–172. doi: 10.1038/357169a0. [DOI] [PubMed] [Google Scholar]
- Dolbeare F. Bromodeoxyuridine: a diagnostic tool in biology and medicine, Part I. Historical perspectives, histochemical methods and cell kinetics. Histochem J. 1995;27:339–369. [PubMed] [Google Scholar]
- Dolbeare F. Bromodeoxyuridine: a diagnostic tool in biology and medicine, Part III. Proliferation in normal, injured and diseased tissue, growth factors, differentiation, DNA replication sites and in situhybridization. Histochem J. 1996;28:531–575. doi: 10.1007/BF02331377. [DOI] [PubMed] [Google Scholar]
- Drouin R, Holmquist GP, Richer C-L. High-resolution replication bands compared with morphologic G- and R-bands. Adv Hum Genet. 1994;22:47–115. doi: 10.1007/978-1-4757-9062-7_2. [DOI] [PubMed] [Google Scholar]
- Earnshaw WC. Mitotic chromosome structure. Bioessays. 1988;9:147–150. doi: 10.1002/bies.950090502. [DOI] [PubMed] [Google Scholar]
- Edenberg HJ, Huberman JA. Eukaryotic chromosome replication. Annu Rev Genet. 1975;9:245–284. doi: 10.1146/annurev.ge.09.120175.001333. [DOI] [PubMed] [Google Scholar]
- Fangman WL, Brewer BJ. A question of time: replication origins of eukaryotic chromosomes. Cell. 1992;71:363–366. doi: 10.1016/0092-8674(92)90505-7. [DOI] [PubMed] [Google Scholar]
- Fox MH, Arndt-Jovin DJ, Jovin TM, Baumann PH, Robert-Nicaud M. Spatial and temporal distribution of DNA replication sites localized by immunofluorescence and confocal microscopy. J Cell Sci. 1991;99:247–255. doi: 10.1242/jcs.99.2.247. [DOI] [PubMed] [Google Scholar]
- Fransz PF, Alonso-Blanco C, Liharska TB, Peeters AJM, Zabel P, de Jong JH. High-resolution physical mapping in Arabidopsis thaliana and tomato by fluorescence in situhybridization to extended DNA fibers. Plant J. 1996;9:421–430. doi: 10.1046/j.1365-313x.1996.09030421.x. [DOI] [PubMed] [Google Scholar]
- Hand R. Eukaryotic DNA: organization of the genome for replication. Cell. 1978;15:317–325. doi: 10.1016/0092-8674(78)90001-6. [DOI] [PubMed] [Google Scholar]
- Hatton KS, Dhar V, Brown EH, Iqbal MA, Stuart S, Didamo VT, Schildkraut CL. Replication program of active and inactive multigene families in mammalian cells. Mol Cell Biol. 1988;8:2149–2158. doi: 10.1128/mcb.8.5.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heichman KA, Roberts JM. Rules to replicate by. Cell. 1994;79:557–562. doi: 10.1016/0092-8674(94)90541-x. [DOI] [PubMed] [Google Scholar]
- Holmquist GP. Chromosome bands, their chromatin flavors, and their functional features. Am J Hum Genet. 1992;51:17–37. [PMC free article] [PubMed] [Google Scholar]
- Hozák P, Hassan AB, Jackson DA, Cook PR. Visualization of replication factories attached to a nucleoskeleton. Cell. 1993;73:361–373. doi: 10.1016/0092-8674(93)90235-i. [DOI] [PubMed] [Google Scholar]
- Hsiao C-L, Carbon J. High-frequency transformation of yeast by plasmids containing the yeast ARG4 gene. Proc Natl Acad Sci USA. 1979;76:3829–3833. doi: 10.1073/pnas.76.8.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberman JA, Riggs AD. On the mechanism of DNA replication in mammalian chromosomes. J Mol Biol. 1968;32:327–341. doi: 10.1016/0022-2836(68)90013-2. [DOI] [PubMed] [Google Scholar]
- Humbert C, Usson Y. Eukaryotic DNA replication is a topologically ordered process. Cytometry. 1992;13:603–614. doi: 10.1002/cyto.990130608. [DOI] [PubMed] [Google Scholar]
- Jackson DA. S phase progression in synchronized human cells. Exp Cell Res. 1995a;220:62–70. doi: 10.1006/excr.1995.1292. [DOI] [PubMed] [Google Scholar]
- Jackson DA. Nuclear organization: uniting replication foci, chromatin domains and chromosome structure. Bioessays. 1995b;17:587–591. doi: 10.1002/bies.950170704. [DOI] [PubMed] [Google Scholar]
- Jackson DA, Cook PR. A general method for preparing chromatin containing intact DNA. EMBO (Eur Mol Biol Organ) J. 1985;4:913–918. doi: 10.1002/j.1460-2075.1985.tb03718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kill IR, Bridger JM, Campbell KHS, Maldonado-Codina G, Hutchison CJ. The timing of the formation and usage of replicase clusters in S phase nuclei of human diploid fibroblasts. J Cell Sci. 1991;100:869–876. doi: 10.1242/jcs.100.4.869. [DOI] [PubMed] [Google Scholar]
- Kitsberg D, Selig S, Keshet I, Cedar H. Replication structure of the human β-globin gene domain. Nature. 1993;366:506–507. doi: 10.1038/366588a0. [DOI] [PubMed] [Google Scholar]
- Krude T. Chromatin assembly factor 1 (CAF-1) colocalizes with replication foci in HeLa cell nuclei. Exp Cell Res. 1995;220:304–311. doi: 10.1006/excr.1995.1320. [DOI] [PubMed] [Google Scholar]
- Leonhardt H, Page AW, Weier H-U, Bestor TH. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell. 1992;71:865–873. doi: 10.1016/0092-8674(92)90561-p. [DOI] [PubMed] [Google Scholar]
- Lestou VS, Strehl S, Lion T, Gadner H, Ambros PF. High-resolution FISH of the entire integrated Epstein-Barr virus genome on extended human DNA. Cytogenet Cell Genet. 1996;74:211–217. doi: 10.1159/000134416. [DOI] [PubMed] [Google Scholar]
- Manders EMM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S phase, analyzed by double labelling of DNA and confocal microscopy. J Cell Sci. 1992;103:857–862. doi: 10.1242/jcs.103.3.857. [DOI] [PubMed] [Google Scholar]
- Manders EMM, Stap J, Strackee J, van Driel R, Aten JA. Dynamic behaviour of DNA replication domains. Exp Cell Res. 1996;226:328–335. doi: 10.1006/excr.1996.0233. [DOI] [PubMed] [Google Scholar]
- Manuelidis L. Individual interphase chromosome domains revealed by in situ hybridization. Hum Genet. 1985;71:288–293. doi: 10.1007/BF00388453. [DOI] [PubMed] [Google Scholar]
- Manuelidis L, Chen TL. A unified model of eukaryotic chromosomes. Cytometry. 1990;11:8–25. doi: 10.1002/cyto.990110104. [DOI] [PubMed] [Google Scholar]
- Marahrens, Y., and B. Stillman. 1996. The initiation of DNA replication in the yeast Saccharomyces cerevisiae. In Eukaryotic DNA Replication. J.J. Blow, editor. IRL Press, Oxford. 66–95.
- Morton NE. Parameters of the human genome. Proc Natl Acad Sci USA. 1991;88:7474–7476. doi: 10.1073/pnas.88.17.7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H, Morita T, Sato C. Structural organisation of replicon domains during DNA synthetic phase in the mammalian nucleus. Exp Cell Res. 1986;165:291–297. doi: 10.1016/0014-4827(86)90583-5. [DOI] [PubMed] [Google Scholar]
- Nurse P. Ordering S phase and M phase in the cell cycle. Cell. 1994;79:547–550. doi: 10.1016/0092-8674(94)90539-8. [DOI] [PubMed] [Google Scholar]
- O'Keefe RT, Henderson SC, Spector DL. Dynamic organization of DNA replication in mammalian cell nuclei: Spatially and temporally defined replication of chromosome-specific α-satellite DNA sequences. J Cell Biol. 1992;116:1095–1110. doi: 10.1083/jcb.116.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll DM, Vogelstein B, Coffey DS. A fixed site of DNA replication in eukaryotic cells. Cell. 1980;19:527–536. doi: 10.1016/0092-8674(80)90527-9. [DOI] [PubMed] [Google Scholar]
- Parra I, Windle B. High resolution visual mapping of stretched DNA by fluorescent hybridization. Nat Genet. 1993;5:17–21. doi: 10.1038/ng0993-17. [DOI] [PubMed] [Google Scholar]
- Pombo A, Ferreira J, Bridge E, Carmo-Fonseca M. Adenovirus replication and transcription sites are spatially separated in the nucleus of infected cells. EMBO (Eur Mol Biol Organ) J. 1994;13:5075–5085. doi: 10.1002/j.1460-2075.1994.tb06837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AE, McCarroll RM, Newlon CS, Fangman WL. Time of replication of ARS elements along yeast chromosome III. Mol Cell Biol. 1989;9:4488–4494. doi: 10.1128/mcb.9.10.4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh N, Laemmli UK. Metaphase chromosome structure: bands arise from a differential folding path of the highly AT-rich scaffold. Cell. 1994;76:609–622. doi: 10.1016/0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- Schardin M, Cremer T, Hager HD, Lang M. Specific staining of human chromosomes in Chinese hamster x man hybrid cell lines demonstrates interphase chromosome territories. Hum Genet. 1985;71:281–287. doi: 10.1007/BF00388452. [DOI] [PubMed] [Google Scholar]
- Selig S, Okumura K, Ward DC, Cedar H. Delineation of DNA replication time zones by fluorescence in situhybridization. EMBO (Eur Mol Biol Organ) J. 1992;11:1217–1225. doi: 10.1002/j.1460-2075.1992.tb05162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparvoli E, Levi M, Rossi E. Replicon clusters may form structurally stable complexes of chromatin and chromosomes. J Cell Sci. 1994;107:3097–3103. doi: 10.1242/jcs.107.11.3097. [DOI] [PubMed] [Google Scholar]
- Stinchcomb DT, Struhl K, Davis RW. Isolation and characterisation of a yeast chromosomal replicator. Nature. 1979;282:39–43. doi: 10.1038/282039a0. [DOI] [PubMed] [Google Scholar]
- Sumner AT. Scanning electron microscopy of mammalian chromosomes from prophase to telophase. Chromosoma (Berlin) 1991;100:410–418. doi: 10.1007/BF00337519. [DOI] [PubMed] [Google Scholar]
- Tomilin N, Rosanov Y, Zenin V, Bozhkov V, Vig B. A new and rapid method for visualising DNA replication in spread DNA by immunofluorescence detection of incorporated 5-iododeoxyuridine. Biochem Biophys Res Commun. 1993;190:257–262. doi: 10.1006/bbrc.1993.1039. [DOI] [PubMed] [Google Scholar]
- Tomilin N, Solovjeva L, Krutilina R, Chamberland C, Hancock R, Vig B. Visualization of elementary DNA replication units in human nuclei corresponding in size to DNA loop domains. Chromosome Res. 1995;3:32–40. doi: 10.1007/BF00711159. [DOI] [PubMed] [Google Scholar]
- van den Engh G, Sachs R, Trask BJ. Estimating genomic distance from DNA sequence location in cell nuclei by a random walk model. Science. 1992;257:1410–1412. doi: 10.1126/science.1388286. [DOI] [PubMed] [Google Scholar]
- van Dierendonck JH, Keyzer R, van de Velde CJH, Cornelisse CJ. Subdivision of S-phase by analysis of nuclear 5-bromo-deoxyuridine staining patterns. Cytometry. 1989;10:143–150. doi: 10.1002/cyto.990100205. [DOI] [PubMed] [Google Scholar]
- Vogel W, Autenrieth M, Mehnert K. Analysis of chromosome replication by a BrdU antibody technique. Chromosoma (Berlin) 1989;98:335–341. doi: 10.1007/BF00292386. [DOI] [PubMed] [Google Scholar]
- Yokota H, van den Engh G, Hearst JE, Sachs RK, Trask BJ. Evidence for the organization of chromatin in megabase pair-sized loops arranged along a random walk path in the human G0/G1 interphase nucleus. J Cell Biol. 1995;130:1239–1249. doi: 10.1083/jcb.130.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]