

Abstract
Glucose regulates the degradation of the key gluconeogenic enzyme, fructose-1,6-bisphosphatase (FBPase), in Saccharomyces cerevisiae. FBPase is targeted from the cytosol to a novel type of vesicle, and then to the vacuole for degradation when yeast cells are transferred from medium containing poor carbon sources to fresh glucose. To identify proteins involved in the FBPase degradation pathway, we cloned our first VID (vacuolar import and degradation) gene. The VID24 gene was identified by complementation of the FBPase degradation defect of the vid24-1 mutant. Vid24p is a novel protein of 41 kD and is synthesized in response to glucose. Vid24p is localized to the FBPase-containing vesicles as a peripheral membrane protein. In the absence of functional Vid24p, FBPase accumulates in the vesicles and fails to move to the vacuole, suggesting that Vid24p regulates FBPase targeting from the vesicles to the vacuole. FBPase sequestration into the vesicles is not affected in the vid24-1 mutant, indicating that Vid24p acts after FBPase sequestration into the vesicles has occurred. Vid24p is the first protein identified that marks the FBPase-containing vesicles and plays a critical role in delivering FBPase from the vesicles to the vacuole for degradation.
Protein degradation is an important process that is tightly regulated. In mammalian cells, serum starvation induces protein degradation by lysosomes (Dice, 1990; Hayes and Dice, 1996). Cytosolic proteins containing a pentapeptide sequence are targeted to the lysosome for degradation in a process mediated by a heat shock protein (Chiang and Dice, 1988; Chiang et al., 1989; Terlecky et al., 1992; Terlecky and Dice, 1993; Cuervo et al., 1994). The receptor protein for this selective proteolysis pathway has been identified recently to be LGP96 (Cuervo and Dice, 1996). Overexpression of the receptor protein increases the degradation of cytosolic proteins in lysosomes both in vivo and in vitro (Cuervo and Dice, 1996).
In Saccharomyces cerevisiae, the vacuole is functionally homologous to the lysosome and takes up proteins by several mechanisms. Most vacuole resident proteinases such as carboxypeptidase Y (CPY)1 enter the vacuole through the secretory pathway (Hasilik and Tanner, 1978; Hemmings et al., 1981; Rothman and Stevens, 1986; Banta et al., 1988; Jones, 1991). CPY is synthesized and processed sequentially in the ER and the Golgi. Sorting occurs in the late Golgi by the CPY receptor encoded by the PEP1/ VPS10 gene (Marcusson et al., 1994; Cooper and Stevens, 1996). CPY is delivered to the vacuole from the prevacuolar or endosomal compartment and the receptor protein recycles back to the Golgi (Marcusson et al., 1994; Cooper and Stevens, 1996). Other vacuolar proteins such as α-mannosidase or aminopeptidase I are imported from the cytosol to the vacuole, independent of the secretory pathway (Yoshihisa and Anraku, 1990; Klionsky et al., 1992; Harding et al., 1995, 1996; Scott et al., 1996). Plasma membrane proteins can be internalized by endocytosis and transported through early endosomes to late endosomes, from which they are directed to the vacuole for degradation (Davis et al., 1993; Raths et al., 1993; Kolling and Hollenberg, 1994; Schandel and Jennes, 1994; Lai et al., 1995; Riballo et al., 1995). Organelles such as peroxisomes or mitochondria can be engulfed by the vacuoles by autophagy (Takeshige et al., 1992; Tuttle and Dunn, 1995; Chiang et al., 1996).
The key gluconeogenic enzyme, fructose-1,6-bisphosphatase (FBPase), is induced when Saccharomyces cerevisiae cells are grown in medium containing poor carbon sources. When cells are transferred to medium containing fresh glucose, FBPase is rapidly inactivated (Gancedo, 1971). Using isogenic strains differing only at the PEP4 gene, we have demonstrated that FBPase is targeted from the cytosol to the vacuole for degradation when cells are transferred from poor carbon sources to fresh glucose (Chiang and Schekman, 1991). The PEP4 gene encodes proteinase A, whose activity is required for the maturation of proteinase B and proteinase C (Zubenko and Jones, 1981; Jones, 1991). As a result, the pep4 strain reduces the vacuolar proteolytic activity to 30% of the wild-type level (Zubenko and Jones, 1981; Jones, 1991; Chiang et al., 1996). The glucose-induced distribution of FBPase from the cytosol to the vacuole has been observed in the pep4 cell by cell fractionation techniques, immunofluorescence microscopy, and immunoelectron microscopy (Chiang and Schekman, 1991; Chiang et al., 1996). FBPase targeting into the vacuole always occurs, regardless of whether cells are transferred to glucose from acetate, ethanol, galactose, or oleate (Chiang and Schekman, 1994; Chiang et al., 1996).
To dissect the FBPase degradation pathway, we have taken a genetic approach. Several vid (vacuolar import and degradation) mutants that fail to degrade FBPase in response to glucose have been isolated (Hoffman and Chiang, 1996). Most vid mutants block FBPase in the cytosol. However, in the vid14-1, vid15-1, and vid16-1 mutants, FBPase is found in punctate structures in the cytoplasm. When cell extracts from one of these mutants are fractionated, a substantial amount of FBPase is found in the high speed pellet, suggesting that FBPase is associated with intracellular structures in these mutants (Hoffman and Chiang, 1996). This association is also observed in wild-type cells (Huang and Chiang, 1997).
The FBPase-containing vesicles have been purified from wild-type cells to near homogeneity using a combination of differential centrifugation, gel filtration, and equilibrium centrifugation in sucrose gradients (Huang and Chiang, 1997). The purified fractions contain 30–40-nm-diam vesicles and are essentially free of other organelles. Kinetic studies indicate that FBPase association with these vesicles is induced by glucose, occurs only transiently, and precedes the association with the vacuole. The FBPase-containing vesicles are distinct from mitochondria, peroxisomes, endosomes, vacuoles, ER, Golgi, or transport vesicles such as the coat protein (COPI or COPII)-containing vesicles as analyzed by protein markers and electron microscopy (Huang and Chiang, 1997).
The vesicles were predicted to contain proteins involved in FBPase targeting and sequestration into the vesicles, as well as proteins participating in carrying FBPase from the vesicles to the vacuole for degradation. To identify such factors, we cloned our first VID gene. The VID24 gene was identified by complementation of the degradation defect of the vid24-1 mutant. Vid24p is a novel 41-kD protein and is synthesized in response to glucose. A significant portion of the Vid24p is localized to the FBPase-containing vesicles as a peripheral protein. The deletion of Vid24p abolishes the degradation of FBPase, but does not cause significant change in growth, sporulation, germination, osmolarity sensitivity, or processing of CPY. In the absence of functional Vid24p, FBPase accumulates in the vesicles and fails to move to the vacuole. FBPase is sequestered inside the vesicles in the vid24-1 mutant, suggesting that Vid24p acts after FBPase sequestration into the vesicles has occurred. Vid24p is the first protein identified that is localized to the FBPase-containing vesicles and plays a critical role in delivering FBPase from the vesicles to the vacuole for degradation.
Materials and Methods
Yeast Strains and Media
Yeast strains used in this study were HLY404 (MATα trp1 ade2 his3-Δ200 ura3-52 leu2,3-112), vid24-1 (MATa trp1 ade2 his3-Δ200 ura3-52 leu2,3-112 vid24-1), vid15-1 (MATa trp1 ade2 his3-Δ200 ura3-52 leu2,3-112 vid15-1), MCY1 (MATa trp1 his3-Δ200 ura3-52 leu2,3-112 lys2-801), and MCY2 (MATa trp1 his3-Δ200 ura3-52 leu2,3-112 lys2-801 Δ vid24::TRP1). YPD contained 1% yeast extract (Difco Laboratories Inc., Detroit, MI), 2% peptone (Difco Laboratories Inc.), and 2% glucose (Fisher Scientific Co., Pittsburgh, PA). YPKG consisted of 1% yeast extract, 2% peptone, 1% potassium acetate (Fisher Scientific Co.), and 0.5% glucose. YPA sporulation medium is composed of 1% yeast extract, 2% peptone, and 1% potassium acetate. Synthetic dextrose (SD) (-N) medium contained 6.7 g/liter yeast nitrogen base (YNB) without amino acids (Difco Laboratories Inc.) supplemented with 2% glucose. SD plates contained 6.7 g/liter of YNB, 2% glucose, 2% Bacto agar (Difco Laboratories Inc.), and 2 μg/ml of adenine, uracil, l-tryptophan, and l-histidine, and also 3 μg/ml of l-leucine and l-lysine (Guthrie and Fink, 1991). Lysine, tryptophan, or uracil drop-out plates were prepared as described above except that l-lysine, l-tryptophan, or uracil was omitted.
Pulse Chase of FBPase
Pulse-chase experiments were performed as described by Hoffman and Chiang (1996). Cells were precultured overnight, labeled in 10 ml of yeast nitrogen base without ammonium sulfate and amino acids, and then supplemented with necessary nutrients and 300 μCi of [35S]methionine and cysteine for 24 h. Cells (2 ml) were transferred to YPD medium containing 2% glucose for 0, 45, 90, 180, or 300 min, harvested, and then resuspended in 100 μl of buffer containing 50 mM Hepes-NaOH, pH 7.2, 100 μg/ml PMSF, 5 mM MgSO4, 40 mM (NH4)2SO4, 10 mM NaN3 and 0.1 mM EDTA. Cell extracts were obtained by adding an equal volume of glass beads and then agitating vigorously. Cell lysates were immunoprecipitated with anti-FBPase antibodies followed by protein A–Sepharose (Pharmacia Biotechnology inc., Piscataway, NJ). Proteins were separated by SDS-PAGE, exposed in phosphorimager cassettes, and then quantitated with a phosphorimager (Molecular Dynamics, Inc., Sunnyvale, CA). The kinetics of FBPase degradation was determined using the amount of FBPase at t = 0 min as 100%. The half life of FBPase degradation is defined as the time required for 50% of the FBPase to be degraded after glucose readdition.
Cloning of VID24
Our results suggested that VID24 was closely linked to LYS2 on chromosome II (see text). The genomic sequence covering a region of 36 kb that surrounds LYS2 was obtained from American Type Culture Collection (Rockville, MD) cosmid clone No. 70930. This region contains 13 open reading frames, including five known and eight unknown genes. It was fragmented with either BamHI or XbaI and subcloned into a yeast centromeric vector to produce pMC1-pMC5. They were introduced into the vid24-1 strain and tested for complementation by Western blotting. pMC5, which tested positive, was further digested to yield pMC5-1 and pMC5-2, which were tested for complementation of the FBPase degradation defect. To construct the deletion strain, the TRP1 sequence was integrated into BclI sites on the VID24 gene to produce MCY2 (MATa trp1 his3-Δ200 ura3-52 leu2,3-112 lys2-801 Δ vid24::TRP1). The transformants were selected on the tryptophan drop-out plates and the site of integration was confirmed by PCR. The DNA from the original vid24-1 mutant was sequenced from both strands using the Biopolymer Facility at Harvard Medical School (Cambridge, MA). Computer searches for domains, motifs or homologies with Vid24p were performed using the GCG Wisconsin package to search the databases from Genbank, EMBL, and SwissProt.
Hemagglutinin-tagged Vid24p
Double-stranded DNA containing the hemagglutinin (HA) epitope coding sequence was produced by mixing the synthetic oligonucleotide GATCCCAGGTGGTTACCCATATGTTCCAGATTACGCTGGTAA with the oligonucleotide GATCTTACCAGCGTAATCTGGAACATCGTATGGGTAACCACTGG. The DNA was ligated to the BclI site of the VID24 gene (pMC5-2). The resulting construct contained the HA coding sequence fused in frame with the VID24 gene on the NH2 terminus. The entire VID24 coding sequence was preserved except that the first methionine residue was replaced with lysine. The construct was digested with BspEI restriction enzyme and transformed into the vid24 null allele (MCY2) by integration into the BspEI site located in the promoter region. The construct was also transformed into the pep4 and vid15-1 cells. The transformants were selected on the SD minus uracil plates and the integration was confirmed by PCR reactions.
Immunofluorescence Microscopy
Immunofluorescence microscopy was performed as described by the supplier (Boehringer Mannheim Corp., Indianapolis, IN) with the following modifications. The cells were fixed and stained with 4 μg/ml of 12CA5 monoclonal antibodies for 30 min at room temperature and a 5 μg/ml dilution of FITC-conjugated, goat anti–mouse antibodies (Sigma Chemical Co., St. Louis, MO) for 1 h at room temperature. The staining of HA-Vid24p was visualized using an Axiophot microscope (Carl Zeiss Inc., Thornwood, NY) and then analyzed with Photoshop software (Adobe Systems Inc., Mountain View, CA).
Cell Fractionation on S-1000 Columns and Sucrose Density Gradients
The wild-type, pep4, and vid15-1 cells expressing the HA-Vid24p were cultured in 1 liter of YPKG for 2 d. Cells (OD600 = 7,000–10,000) were transferred to YPD medium for the indicated times and temperatures. Cells were harvested, converted to spheroplasts, and then homogenized as described by Hoffman and Chiang (1996). Cell lysates were centrifuged at 15,000 g for 20 min and the medium speed supernatant was further centrifuged at 100,000 g for 2 h to obtain the high speed pellet that was resuspended in 1 ml of TEA buffer (10 mM triethanolamine, pH 7.5, 100 μg/ml PMSF, 10 mM NaF, 10 mM NaN3, 1 mM EDTA, and 0.8 M sorbitol) and loaded onto S-1000 columns (Pharmacia Biotechnology Inc.). Fractions 25–30, which contained FBPase, were collected from the S-1000 columns and centrifuged at 100,000 g for 4 h. The pellets were resuspended in 0.5 ml of TEA buffer, loaded onto the top of 10 ml of 20–50% (wt/wt) sucrose gradients and centrifuged to equilibrium at 100,000 g for 20 h at 4°C. Fractions were collected from the top (0.8 ml), precipitated with 10% TCA, washed twice in cold acetone, and then solubilized in SDS sample buffer. Proteins were separated using 10% SDS-PAGE and transferred to nitrocellulose membranes. FBPase was detected by Western blotting with anti-FBPase antibodies, and HA-Vid24p was detected by monoclonal antibodies against the HA tag.
Proteinase K Treatment
To test the sensitivity of FBPase and Vid24p to proteinase K, the vesicles were isolated from the vid15-1 strain and treated with or without proteinase K (1 mg/ml) in the absence or presence of Triton X-100 (2%) for 0, 10, and 20 min. The reactions were terminated by adding 10% TCA and the samples were processed as described above. To test whether FBPase was sequestered inside the vesicles, both pep4 and vid24-1 mutant were shifted to glucose for 0, 30, and 60 min. Total cell extracts (OD600 = 20) were treated with proteinase K in the absence or presence of 2% Triton X-100 for the indicated times. The reactions were terminated by adding 10% TCA, and the samples were processed as described above.
Extraction of Vid24p
The vesicles were isolated and resuspended directly in 1 ml of TE buffer alone (10 mM Tris, 1 mM EDTA, pH 8.0); 0.1 M Na2CO3, pH 11.5; 5 M urea or 2% Triton X-100 for 30 min on ice. Samples were centrifuged at 100,000 g for 1 h using an RP 555 rotor (Sorvall, Newtown, CT). The supernatant was precipitated with 10% TCA, processed, and then solubilized in 20 μl of SDS sample buffer. The pellet was resuspended in 1 ml of deionized water, precipitated with TCA, processed, and then resuspended in 20 μl of SDS sample buffer. Samples from the supernatant and pellet fractions from each treatment were loaded on SDS–polyacrylamide gels, transferred to nitrocellulose membranes, and then blotted with anti-HA antibodies.
Differential Centrifugation
The pep4 and vid24-1 were cultured in 1 liter of YPKG for 2 d. Cells (OD600 = 7,000–10,000) were divided and aliquots with OD600 = 3,500– 5,000 were transferred to YPD medium for the indicated times and temperatures. Cells were harvested, converted to spheroplasts, and homogenized as described above. Cell lysates were first centrifuged at 500 g for 10 min, and centrifuged at 15,000 g for 20 min, and then the medium speed supernatant (T) was further centrifuged at 100,000 g for 2 h to obtain the high speed pellet (P) and the high speed supernatant (S). Proteins from the T, S, and P fractions were diluted 5,000-fold, separated by electrophoresis using 10% SDS–polyacrylamide gels, and then transferred to nitrocellulose membranes. The distributions of FBPase or glucose-6-phosphate dehydrogenase (G-6-P dehydrogenase) were detected by Western blotting with a 1:1,000 dilution of affinity-purified anti-FBPase antibodies or a 1:10,000 dilution of G-6-P dehydrogenase antibodies (Sigma Chemical Co.) for 1 h, followed by a 1:10,000 dilution of HRP-conjugated anti– rabbit antibodies for 1 h (Amersham Corp., Arlington Heights, IL), and then detected by enhanced chemiluminescence reactions.
Miscellaneous Assays and Methods
Tetrad dissecting, colony-blotting procedure, and regrowth of cells on fresh glucose were performed as described by Hoffman and Chiang (1996). Survival rates during nitrogen starvation were measured according to Finley et al. (1987). The numbers of survival cells at each day were counted using t = 0 d as 100%. Sporulation efficiency was conducted as described by Betz and Weiser (1976). Cells were pre-grown in medium containing 1% potassium acetate and 0.67% yeast nitrogen base (without amino acids). Cells were transferred to YPA sporulation medium containing 1% potassium acetate, 2% peptone, and 1% yeast extract for 4 d. CPY sorting was performed as described by Robinson et al. (1988). Cells were spheroplasted, pulsed for 20 min, and chased for 30 min. Cells were then separated into intracellular and extracellular fractions. CPY in these fractions was precipitated with CPY serum and quantitated with a phosphorimager. The growth of cells on high salt plates was performed as described by Latterich and Watson (1991).
Results
vid24-1 Shows a Strong Defect in FBPase Degradation
We have isolated and characterized vid1-vid20 mutants that are defective in the glucose-induced degradation of FBPase (Hoffman and Chiang, 1996). Using the same colony-blotting scheme, we obtained the vid24-1 mutant that showed a strong defect in FBPase degradation. The vid24-1 mutant was back-crossed to wild-type cells four times. Analysis of tetrads derived from vid24-1/wild-type diploid cells demonstrated that the FBPase degradation defect segregated 2:2, suggesting that a single gene lesion was responsible for the FBPase degradation defect. The half-life of FBPase degradation was determined by pulse-chase experiments (Fig. 1). In wild-type cells, FBPase was degraded in response to glucose with a t 1/2 = 30 min. The vid24-1 mutant had a strong defect in the glucose-induced degradation of FBPase with a t 1/2 > 300 min. This defect was seen whether the cells were transferred to glucose at 22° or 30°C.
Figure 1.

Pulse-chase analysis of FBPase degradation in wild-type and vid24-1 cells. Wild-type and vid24-1 cells were pulse labeled in poor carbon sources and chased in glucose for 0, 45, 90, 180, or 300 min at 22°C as described in Materials and Methods. Cell lysates were immunoprecipitated with FBPase antibodies, separated by SDS-PAGE, and the FBPase levels quantitated using a phosphorimager.
Cloning of the VID24 Gene
We observed a tight linkage between the FBPase degradation defect and the lysine auxotrophy in the vid24-1 mutant. Of the 26 tetrads that we have dissected, we obtained 22 parental ditypes, 4 tetratypes, and 0 nonparental ditypes. Based on this, we calculated the distance between LYS2 and VID24 to be 7–8 map units. This corresponded to 15–20 kb, assuming 1 map unit equals 2.5 kb. The clone containing 36-kb genomic sequences flanking LYS2 was obtained from American Type Culture Collection. This region has been completely sequenced by the yeast genome project and contained 13 open reading frames. To determine which open reading frame contained the authentic VID24 gene, five genomic fragments were generated by restriction digestion and subcloned into a yeast centromeric vector. They were introduced into the vid24-1 cell and complementation of the FBPase degradation defect was examined. Fig. 2 A shows that the plasmids pMC1 and pMC5 complemented the defect, whereas pMC2, pMC3, and pMC4 did not. pMC5, which was the smallest fragment that complemented the degradation defect of vid24-1, had two open reading frames. They were further subcloned to yield pMC5-1 and pMC5-2. We found that pMC5-2 complemented the degradation defect, but pMC5-1 did not. pMC5-2 has an open reading frame previously called YBR105C by the yeast genome project and encodes a 41-kD protein of unknown function.
Figure 2.

Cloning of VID24. (A) A 36-kb DNA sequence flanking LYS2 was obtained from the American Type Culture Collection, digested by either XbaI (X) or BamHI (B), and then subcloned into a yeast centromeric vector to generate pMC1-pMC5. These plasmids were introduced into the vid24-1 mutant and complementation of FBPase degradation defect was examined by Western blotting (right-hand column). pMC5 complemented the degradation defect and was subcloned to yield pMC5-1 and pMC5-2. pMC5-2, but not pMC5-1, complemented the defect. (B) The VID24 gene was deleted by replacing the entire gene with TRP1. The deleted gene was introduced into wild-type cells and the transformants were selected on the tryptophan drop-out plates. The deletion was confirmed by PCR reactions. (C) The degradation of FBPase in wild-type, vid24-1, and the Δvid24:: TRP1 strains after glucose shift for 0–180 min.
To determine whether YBR105C was the authentic VID24 gene, we performed targeted disruption to essentially delete the entire YBR105C coding sequence (Fig. 2 B). Fig. 2 C shows that the deletion of YBR105C caused the wild-type cell to become defective in FBPase degradation. The half life of FBPase degradation was >300 min in the deletion strain, similar to that found in the original vid24-1 mutant. Tetrad analysis indicated that the FBPase degradation defect always cosegregated with TRP1 (data not shown). Since all spores were viable, VID24 appeared to be nonessential for viability.
The vid24-1 gene was sequenced from both strands. A point mutation from A to T was found in position 811 of the coding sequence, which resulted in early translation termination (ArgAGA → TermTGA) at amino acid residue 271. Since the pMC5-2 that contained YBR105C complemented the vid24-1 defect, and the deletion of YBR105C rendered wild-type cells defective in FBPase degradation, and because the original vid24-1 mutant had a point mutation in YBR105C, we concluded that YBR105C was the authentic VID24 gene.
VID24 Encodes a Novel Protein of 41 kD
Fig. 3 shows that the nucleic acid sequence of VID24 encodes a protein of 362 amino acid resides. The calculated molecular weight of Vid24p is 41 kD, and the calculated isoelectric point is 6.5. There are no hydrophobic stretches of sufficient length to serve as a signal sequence or transmembrane domain. Computer database searches failed to identify functional domains or motifs in the Vid24p protein sequence, except for some potential glycosylation and phosphorylation sites. Vid24p shares 56% sequence similarity or 32% sequence identity with another protein of unknown function encoded by YGR066C. The function of YGR066C is probably not related to the degradation of FBPase, since the deletion of YGR066C had no effect on FBPase degradation (data not shown).
Figure 3.
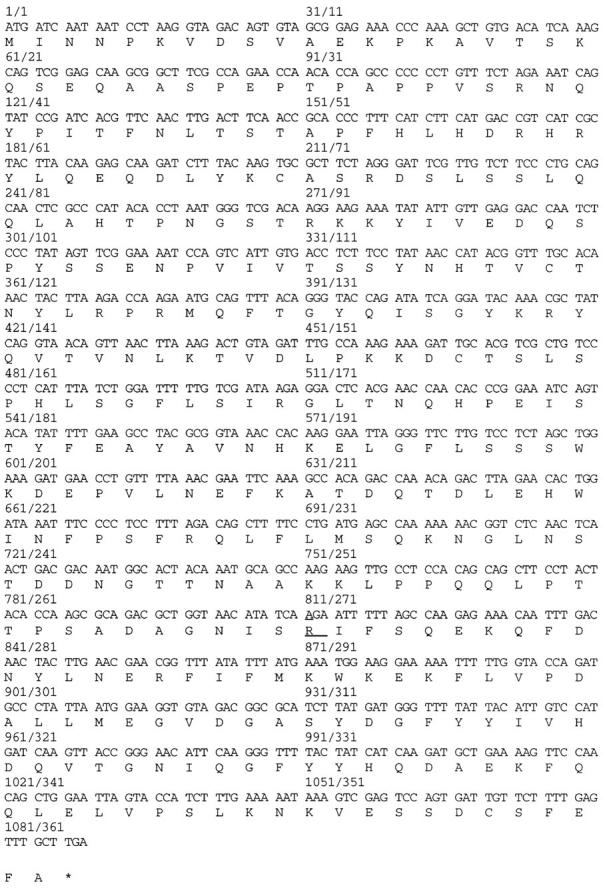
Analysis of the VID24 gene and Vid24p. The nucleic acid sequence and corresponding amino acid sequence were obtained directly from the yeast genome database. (These sequence data are available from Genbank/EMBL/DDBJ under accession number U86750.) Both strands of the DNA of the original vid24-1 mutant were sequenced. The mutation was found to be a point mutation at position 811 of the coding sequence.
Vid24p Is Synthesized in Response to Glucose
If Vid24p participated in the glucose-induced degradation of FBPase, the expression of Vid24p might be regulated by glucose. To test this, the HA coding sequence was fused in frame with the VID24 gene. The construct was integrated to replace the endogenous VID24 gene and the expression of the HA-Vid24p was under its own promoter control. Cells were grown in poor carbon sources and shifted to glucose for various periods of time. Fig. 4 shows that in the untransformed control cell FBPase was degraded at a normal rate and the anti-HA antibodies failed to detect any signal (Fig. 4, lanes 1–5). In the wild-type cell expressing the HA-Vid24p, anti-HA antibodies detected a 42-kD protein that appeared after glucose shift for 20 min (Fig. 4, lanes 7–10). This protein was undetectable at t = 0 min (Fig. 4, lane 6). The degradation of FBPase was normal (Fig. 4, lanes 6–10), suggesting that HA tagging on the NH2 terminus preserved the function of Vid24p. The synthesis of HA-Vid24p was blocked when cycloheximide was added with glucose (Fig. 4, lanes 11 and 12). The degradation of FBPase was also prevented by cycloheximide (Fig. 4, lanes 11 and 12).
Figure 4.

Vid24p is synthesized in response to glucose. Wild-type cells expressing the HA-Vid24p were cultured and shifted to glucose for 0, 20, 40, 60, and 100 min at 22°C. The induction of HA-Vid24p and the degradation of FBPase in response to glucose were examined by Western blotting with anti-HA and anti-FBPase antibodies. Lanes 1–5, untransformed wild-type cells were shifted to glucose for 0–100 min. Lanes 6–10, wild-type cells expressing the HA-Vid24p were shifted to glucose for 0–100 min in the absence of cycloheximide. Lanes 11 and 12, wild-type cells expressing the HA-Vid24p were shifted to glucose for 0 and 100 min in the presence of 10 μg/ml of cycloheximide.
Vid24p Is a Peripheral Protein Localized to the FBPase-containing Vesicles
FBPase is targeted from the cytosol to intermediate vesicles and then to the vacuole for degradation. The FBPase-associated vesicles were expected to contain proteins that functioned in targeting and sequestration of FBPase into the vesicles and also proteins involved in delivering FBPase from the vesicles to the vacuole for degradation. If Vid24p played a role in the vesicle trafficking pathway, Vid24p itself might be localized to the FBPase-containing vesicles. We studied the localization of Vid24p in the wild-type cells that were shifted to glucose for 0, 15, 30, and 60 min at 22°C. The cells were visualized by Nomarski images (Fig. 5, a–d), and localization of HA-Vid24p was examined by immunofluorescence microscopy using anti-HA antibodies, followed by FITC-conjugated, goat anti–mouse antibodies. At t = 0 min, there was no detectable fluorescence (Fig. 5 e). After a glucose shift of 15 min, the fluorescence remained low (Fig. 5 f). At t = 30 or 60 min, the fluorescence increased. Most of the staining appeared in punctate structures within cells (Fig. 5, g and h), suggesting that a substantial amount of HA-Vid24p was associated with intracellular organelles. A similar staining of Vid24p was seen in the vid15-1 mutant that accumulates FBPase in the vesicles (data not shown). The structures were most likely to be the FBPase-containing vesicles or aggregates of the vesicles.
Figure 5.

Immunolocalization of Vid24p. Wild-type cells expressing the HA-Vid24p were shifted to glucose at 22°C for 0, 15, 30, and 60 min. Cells were fixed and immunofluorescence microscopy was performed as described. Left, Nomarski images of cells (a–d). Right, FITC staining of HA-Vid24p using 12CA5 monoclonal antibodies (e–h). The same immunofluorescence results were obtained using the vid15-1 mutant (data not shown).
We investigated whether HA-Vid24p was indeed localized to the FBPase-containing vesicles. Colocalization studies using immunofluorescence microscopy were not suitable for this purpose, because a high level of FBPase remained in the cytosol and the diffuse cytosolic staining could mask the staining of FBPase in the vesicles. For this reason, cell fractionation experiments were undertaken using the vid15-1 mutant that accumulates FBPase in the vesicles (Hoffman and Chiang, 1996). Cells were shifted to glucose for 0, 30, and 60 min at 22°C, and the vesicles were fractionated by differential centrifugation, S-1000 columns, and sucrose gradients as described earlier (Huang and Chiang, 1997). Each fraction from the sucrose gradients was divided into two portions and blotted with anti-HA and anti-FBPase antibodies separately. Fig. 6 shows that the amount of FBPase was low at t = 0 min (Fig. 6 a). The amount of FBPase increased in fractions 9–11 at t = 30 min (Fig. 6 b) and also at t = 60 min (Fig. 6 c). Fractions 9–11 were isolated from the vid15-1 mutant and examined by electron microscopy. A homogeneous population of vesicles was seen. These vesicles were indistinguishable from the ones isolated from wild-type cells (Huang and Chiang, 1997).
Figure 6.

Vid24p is localized to the FBPase-containing vesicles. The vid15-1 mutant was shifted to glucose for 0, 30, and 60 min at 22°C. Cells were fractionated by differential centrifugation, gel filtration on S-1000 column, and sucrose equilibrium density gradient as described (Huang and Chiang, 1997). Fractions were collected from the top of the sucrose gradients. Fractions were divided into two portions and blotted with FBPase and HA-Vid24p antibodies separately. The distributions of FBPase on the sucrose gradients at t = 0 min (a), 30 min (b), and 60 min (c), and the distributions of HA-Vid24p on the sucrose gradients at t = 0 min (d), 30 min (e), and 60 min (f) were detected by enhanced chemiluminescence reactions.
Colocalization of HA-Vid24p with FBPase in the vesicles was observed after the vid15-1 mutant was shifted to glucose for 30 min (Fig. 6 e) and also 60 min (Fig. 6 f). At t = 0 min, HA-Vid24p was low (Fig. 6 d). These results suggest that Vid24p is localized to the FBPase-containing vesicles.
We studied how Vid24p was associated with the vesicles by proteinase K treatment and carbonate extraction. The vesicles were isolated and treated with proteinase K for 0, 10, and 20 min in the absence or presence of Triton X-100. Fig. 7 A shows that FBPase was stable in the absence of both proteinase K and Triton X-100 (Fig. 7 A, a). FBPase remained resistant to proteinase K in the absence of Triton X-100 (Fig. 7 A, b); it was digested by proteinase K in the presence of Triton X-100 (Fig. 7 A, c). Vid24p was also stable in the absence of both proteinase K and Triton X-100 (Fig. 7 A, d). When proteinase K was added, Vid24p was digested by proteinase K whether Triton X-100 was present or not (Fig. 7 A, e and f).
Figure 7.


Vid24p is a peripheral protein. (A) The vesicles were isolated from the vid15-1 mutant and incubated with 1 mg/ml of proteinase K for 0, 10, and 20 min in the absence or presence of 2% Triton X-100. The amounts of FBPase and Vid24p without proteinase K and without Triton X-100 (a and d), with proteinase K and without Triton X-100 (b and e), with proteinase K and with Triton X-100 (c and f). (B) The vesicles were resuspended in buffer alone (lanes 1 and 2); 0.1 M Na2CO3, pH 11.5 (lanes 3 and 4); 5 M urea (lanes 5 and 6) or 2% Triton X-100 (lanes 7 and 8) and incubated for 30 min on ice. The samples were centrifuged to separate the supernatants (S) and pellets (P). The distribution of HA-Vid24p was examined by Western blotting with anti-HA antibodies.
As an additional test, the FBPase-associated vesicles were resuspended in buffer alone, 0.1 M Na2CO3, 5 M urea, or 2% Triton X-100. After incubation for 30 min on ice, samples were separated into supernatants and pellets, and the distributions of HA-Vid24p in these fractions were determined by Western blotting. The control experiment shows that most of the HA-Vid24p was detected in the pellet when incubated in buffer alone (Fig. 7 B, lanes 1 and 2). When 0.1 M Na2CO3, pH 11.5, was added to remove peripheral membrane proteins, HA-Vid24p was extracted to the supernatant fraction (Fig. 7 B, lanes 3 and 4). Similarly, the addition of 5 M urea caused HA-Vid24p to distribute to the supernatant (Fig. 7 B, lanes 5 and 6). When 2% Triton X-100 was added to solubilize the membranes, HA-Vid24p was found in the supernatant (Fig. 7 B, lanes 7 and 8). These results suggest that Vid24p is localized to the FBPase-containing vesicles as a peripheral protein.
FBPase Accumulates in the Vesicles in the vid24-1 Mutant
We studied which step of the FBPase targeting pathway is affected in the vid24-1 or the vid24 null mutant. Since indistinguishable results were obtained with either the vid24-1 mutant or the vid24 null mutant, the sets of results obtained with the vid24-1 mutant were shown from Fig. 8 to Fig. 11 to avoid redundancy. If Vid24p played a role in FBPase targeting and sequestration into the vesicles, FBPase might accumulate in the cytosol in the vid24-1 mutant. If Vid24p participated in the delivery of FBPase from the vesicles to the vacuole, FBPase might accumulate in the vesicles in the vid24-1 mutant. To distinguish between these possibilities, differential centrifugation experiments were performed. The vid24-1 mutant was transferred to glucose for 60 min at 22°C, homogenized, and then centrifuged to obtain the high speed supernatant and high speed pellet. The distributions of FBPase and a control cytosolic protein, G-6-P dehydrogenase, were compared in the vid24-1 mutant and the pep4 strain.
Figure 8.

FBPase is recovered in the high speed pellet in the vid24-1 mutant. pep4 and vid24-1 cells were grown in YPKG and transferred to YPD containing 2% glucose for 60 min at 22°C. Cell lysates were separated into medium speed supernatant (T), high speed supernatant (S) and high speed pellet (P) as described in Materials and Methods. The distribution of FBPase in pep4 (a and b) and vid24-1 cells (c and d) at t = 0 and t = 60 min was detected by Western blotting. The distribution of G-6-P dehydrogenase in the pep4 (e and f) and vid24-1 cells (g and h) at t = 0 min and t = 60 min was detected by Western blotting with G-6-P dehydrogenase antibodies.
Figure 11.

The vid24-1 mutant shows normal growth, survival during nitrogen starvation, sporulation efficiency, and processes CPY to the mature form. (a) Regrowth of wild type and vid24-1 in glucose for 9 h. (b) The percentage of cells surviving after nitrogen starvation for 3 d in wild-type, pep4, and vid24-1 cells. (c) Sporulation efficiency in homozygous diploids of wild-type, pep4, and vid24-1 cells. (d) Pulse chase and immunoprecipitation of CPY in the intracellular (I) or extracellular (E) fractions in wild-type, pep4, and vid24-1 cells.
In the pep4 strain, most of the FBPase was detected in the high speed supernatant and very little FBPase was found in the high speed pellet at t = 0 min. (Fig. 8 a). After a glucose shift of 60 min, ∼20–30% of the FBPase was detected in the pellet (Fig. 8 b). These results are consistent with FBPase being localized to the cytosol at t = 0 min and then distributed to intracellular organelles after a 60-min glucose shift in the pep4 cell. In the vid24-1 mutant, most of the FBPase was present in the supernatant at t = 0 min (Fig. 8 c). After a 60-min glucose shift, ∼20–30% of the FBPase was recovered in the pellet (Fig. 8 d). The control cytosolic protein, G-6-P dehydrogenase, was distributed mostly in the supernatants both in the pep4 strain (Fig. 8, e and f) and the vid24-1 mutant (Fig. 8, g and h). Furthermore, its distribution was not affected by glucose readdition in either strain. Similar amounts of FBPase were recovered in the high speed pellet when the pep4 or vid24-1 cells were shifted to glucose at t = 30 min.
We followed the kinetics of FBPase distribution in the pep4 and vid24-1 strains that were shifted to glucose for 0, 30, 60, and 90 min at 22°C. The high speed pellets were fractionated on S-1000 columns and then by sucrose density equilibrium gradients. Fractions were collected from the sucrose gradients and FBPase distribution was detected by Western blotting with FBPase antibodies. In the pep4 strain, the amount of FBPase was minimal at t = 0 min. (Fig. 9 a). At t = 30 min, the amount of FBPase in fractions 9–11 containing the vesicles increased. A small amount of FBPase was detected in the lighter fractions containing the vacuole (Fig. 9 b). At t = 60 min, FBPase was detected in the vacuole and also in the vesicles (Fig. 9 c). At t = 90 min, FBPase distribution in the vesicles disappeared and a small amount of FBPase was recovered in the vacuole (Fig. 9 d).
Figure 9.

FBPase accumulates in the vesicles in the vid24-1 mutant. pep4 and vid24-1 cells were transferred to glucose for 0, 30, 60, or 90 min at 22°C. The high speed pellets were fractionated on S-1000 columns first and then on equilibrium sucrose density gradients using the protocol described by Huang and Chiang (1997). Fractions were collected from the top. FBPase distribution on the sucrose gradient was detected by Western blotting after a shift of the pep4 cell (a–d) and the vid24-1 mutant (e–h) to glucose for 0, 30, 60, and 90 min.
In the vid24-1 mutant, the amount of FBPase was low on the sucrose gradients at t = 0 min (Fig. 9 e). At t = 30 min and t = 60 min, the amounts of FBPase in the vesicles increased (Fig. 9, f and g). At t = 90 min, FBPase in the vesicles persisted in the mutant and failed to move to the vacuole (Fig. 9 h), suggesting that the delivery of FBPase from the vesicles to the vacuole is impaired in the vid24-1 mutant.
To determine whether the sequestration of FBPase into the vesicles occurred at a similar rate, the pep4 and vid24-1 cells were shifted to glucose for 0, 30, and 60 min at 22°C. Total cell extracts were obtained and treated with or without proteinase K in the absence or presence of Triton X-100. At t = 0 min, most of the FBPase was sensitive to proteinase K in the pep4 and the vid24-1 mutant (Fig. 10, a and d), suggesting that FBPase was in the cytosol in these strains. After a shift of cells to glucose for 30 min, ∼30% of the FBPase became resistant to proteinase K in the pep4 cell (Fig. 10 b) and also the vid24-1 mutant (Fig. 10 e). Similar results were obtained after shifting of both strains to glucose for 60 min (Fig. 10, c and f), suggesting that the sequestration of FBPase occurred at a similar rate in the pep4 and the vid24-1 strains.
Figure 10.

FBPase is sequestered inside the vesicles in the vid24-1 mutant. Both pep4 and vid24-1 cells were shifted to glucose for 0, 30, and 60 min. Total cell extracts were treated with 0, 1, and 2 mg/ml of proteinase K for 20 min in the pep4 cell (a–c) or in the vid24-1 cell (d–f). Samples were processed and transferred to nitrocellulose membranes. FBPase was detected by Western blotting.
Other Functions Proceed Normally in the vid24-1 Mutant
To determine whether Vid24p was involved in other cellular processes, we studied glucose response, sporulation efficiency, response to nitrogen starvation, processing of vacuolar protein CPY, and osmolarity regulation in the vid24-1 mutant. Glucose response was examined by starving cells in poor carbon sources and then diluting them to resume growth in medium containing fresh glucose. The vid24-1 mutant grew as well as wild-type cells with a similar doubling time (Fig. 11 a), indicating that the growth response to glucose was normal in the vid24-1 mutant.
Nitrogen starvation induces accumulation of autophagic bodies in the vacuole (Takeshige et al., 1992; Tsukada and Ohsumi, 1993). apg mutants defective in this process lose viability when cultured in medium lacking nitrogen source (Tsukada and Ohsumi, 1993). We measured the number of viable cells after nitrogen starvation and found that the vid24-1 mutant survived well under nitrogen starvation (Fig. 11 b). It has been reported that homozygous diploids of pep4 strain (Zubenko and Jones, 1981), or apg strains defective in the nitrogen-induced autophagy (Tsukada and Ohsumi, 1993) could not sporulate. We tested sporulation frequency in homozygous diploids of vid24-1 and found that the spore formation was as efficient as the wild-type cell (Fig. 11 c). Therefore, vid24-1 did not affect the autophagy stimulated by nitrogen starvation. The pep4 strain that reduces the vacuolar proteolytic activity (Zubenko and Jones, 1981) lost viability during nitrogen starvation (Fig. 11 b). In addition, the sporulation efficiency was severely impaired in the homozygous pep4 strain (Fig. 11 c).
We determined the kinetics of processing and sorting of CPY in wild-type, pep4, and vid24-1 cells using the protocol described by Robinson et al. (1988). The cells were pulsed for 20 min and then chased for 30 min. The CPY in the extracellular or intracellular fractions were immunoprecipitated with anti-CPY antibodies, separated by SDS-PAGE, and then quantitated by a phosphorimager. Both the wild-type and the vid24-1 strains processed CPY to the mature form (Fig. 11 d). Essentially all the CPY (>95%) was recovered intracellularly in the vid24-1 cell (Fig. 11 d). The pep4 strain contained the p2 form of CPY intracellularly, suggesting that p2-CPY was not processed but was targeted correctly to the vacuole. Finally, the vid24-1 mutant grew well on the high salt plates that selected for mutants defective in osmolarity regulation by the vacuole (Latterich and Watson, 1991), suggesting that regulation of osmolarity by the vacuole was not significantly altered in the vid24-1 mutant (data not shown).
Discussion
Several lines of evidence indicate that FBPase is targeted from the cytosol to a novel type of vesicles before uptake by the vacuole. The vid14-1, vid15-1, and vid16-1 mutants accumulate FBPase in punctate structures in the cytoplasm as detected by indirect immunofluorescence microscopy (Hoffman and Chiang, 1996). In the vid15-1 mutant, a substantial amount of FBPase sediments in the high speed pellet, suggesting that FBPase associates with intracellular structures in these mutants. The FBPase-containing fractions have been purified to near homogeneity from wild-type cells and the vid15-1 mutant. The purified vesicles are distinct from the ER, Golgi, vacuole, endosomes, peroxisomes, mitochondria, or transport vesicles such as the COPI- or COPII-containing vesicles (Huang and Chiang, 1997).
The vid24-1 mutant also accumulates FBPase in the vesicles. When these vesicles were isolated from the vid24-1 mutants and examined under electron microscopy, they were indistinguishable from the ones isolated from the wild-type strain. Furthermore, these vesicles equilibrate at a density of 1.20–1.22 g/ml, a value similar to that of the vesicles purified from the wild-type strain or the vid15-1 mutant. These results suggest that the wild-type, the vid15-1, and vid24-1 strains use the same type of vesicles as intermediates in the FBPase degradation pathway.
The vid24-1 mutant exhibits a specific defect in the FBPase degradation pathway. When Vid24p is mutated, deleted, or not synthesized, the degradation of FBPase is prevented. Other cellular functions such as regrowth in glucose, processing and sorting of the vacuolar protein CPY, osmotic regulation, sporulation efficiency, and viability during nitrogen starvation are not significantly altered in the vid24-1 mutant. FBPase targeting and sequestration into the vesicles occur at normal rates, suggesting that Vid24p acts after FBPase sequestration into the vesicles has occurred. Since FBPase accumulates in the vesicles and fails to move to the vacuole in the vid24-1 mutant, a functional Vid24p is required to deliver FBPase from the vesicles to the vacuole for degradation. Vid24p is most likely to be part of the protein machinery that regulates the trafficking of the vesicles in the FBPase degradation pathway.
The proteins that reside in the FBPase-containing vesicles are largely unidentified. The vesicles are expected to contain proteins that function in the sequestration of FBPase into the vesicles and also proteins mediating the delivery of FBPase from the vesicles to the vacuole. Consistent with this prediction, Vid24p is localized to the FBPase-containing vesicles. Vid24p is a peripheral protein, as it can be removed from the membranes by extraction with sodium carbonate and urea. In addition, Vid24p is sensitive to proteinase K digestion.
We have reported previously that the glucose-induced degradation of FBPase is inhibited by cycloheximide (Chiang and Schekman, 1991), suggesting that FBPase degradation requires the synthesis of new proteins. Our present studies indicate that Vid24p is undetectable at t = 0 min and is induced in response to glucose. Furthermore, the synthesis of Vid24p is blocked by the addition of cycloheximide. Vid24p induction by glucose is consistent with the idea that Vid24p plays a role in the FBPase targeting pathway, which is specially activated by glucose. Therefore, Vid24p is not only the first protein shown to be localized to the FBPase-containing vesicles, but also the first cycloheximide-sensitive factor identified whose de novo synthesis is important for FBPase degradation to occur.
Based on these results, we propose a model for the FBPase degradation pathway (Fig. 12). When cells are transferred from poor carbon sources to glucose, the signal transduction pathways activate several important events. One of the glucose-stimulated events is the targeting and sequestration of FBPase into the vesicles. The vesicles then carry FBPase to the vacuole for degradation in a process that is dependent on Vid24p. Another glucose-induced process is the synthesis and binding of Vid24p to the vesicles. The downstream events after Vid24p binding to the vesicles have not been established. We envision several possibilities that FBPase can be delivered from the vesicles to the vacuole for degradation. The vesicles may fuse directly with the vacuole. Alternatively, the vesicles may fuse with each other or with different types of vesicles before fusion with the vacuole. It is also possible that the vesicles aggregate to form large structures before fusion with the vacuole. Major questions remain as to whether the vesicles are newly formed or whether they are derived from existing structures. We suggest that Vid24p is part of the protein machinery that mediates recognition, fusion or docking of the FBPase-containing vesicles with the downstream compartment. Using a targeting event that remains to be determined, FBPase is correctly delivered to the final destination—the vacuole—for degradation.
Figure 12.

Pathway of FBPase degradation. Glucose induces FBPase targeting and sequestration into the vesicles. FBPase is then delivered from the vesicles to the vacuole for degradation. Glucose also induces the synthesis and binding of Vid24p to the vesicles. Vid24p is required to deliver FBPase from the vesicles to the vacuole for degradation. In the absence of functional Vid24p, FBPase accumulates in the vesicles. The downstream events after Vid24p binding to the vesicles have not been established. The vesicles may fuse directly with the vacuole. Alternatively, the vesicles may fuse with each other or with different types of vesicles and then with the vacuole. It is also possible that the vesicles aggregate before fusion with the vacuole.
Acknowledgments
We thank Dr. H.-L. Shieh for insightful discussions, and Dr. K.J. Barrett for editing this manuscript.
This work was supported by a grant (No. BE-212 A) from the American Cancer Society and a grant (No. RO1 GM49267) from National Institutes of Health to H.-L. Chiang.
Abbreviations used in this paper
- COP
coat protein
- CPY
carboxypeptidase Y
- ECL
enhanced chemiluminescence
- FBPase
fructose-1,6-bisphosphatase
- G-6-P
glucose-6-phosphate
- HA
hemagglutinin
- VID
vacuolar import and degradation
References
- Banta LM, Robinson JS, Klinosky DJ, Emr S. Organelle assembly in yeast: Characterization of yeast mutants defective in vacuolar biogenesis and protein sorting. J Cell Biol. 1988;107:1369–1383. doi: 10.1083/jcb.107.4.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz H, Weiser U. Protein degradation and proteinases during yeast sporulation; enzymes and cytochrome patterns. Eur J Biochem. 1976;62:65–76. doi: 10.1111/j.1432-1033.1976.tb10098.x. [DOI] [PubMed] [Google Scholar]
- Chiang H-L, Dice JF. Peptide sequences that target proteins for enhanced degradation during serum withdrawal. J Biol Chem. 1988;263:6797–6805. [PubMed] [Google Scholar]
- Chiang H-L, Schekman R. Regulated import and degradation of a cytosolic protein in the yeast vacuole. Nature. 1991;350:313–318. doi: 10.1038/350313a0. [DOI] [PubMed] [Google Scholar]
- Chiang H-L, Schekman R. Site of catabolite inactivation. Nature. 1994;369:284. doi: 10.1038/369283a0. [DOI] [PubMed] [Google Scholar]
- Chiang H-L, Terlecky SR, Plant CP, Dice JF. A role for a 70 kD heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989;246:382–385. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- Chiang H-L, Schekman R, Hamamoto S. Selective uptake of cytosolic, peroxisomal and plasma membrane proteins by the yeast vacuole. J Biol Chem. 1996;271:9934–9941. doi: 10.1074/jbc.271.17.9934. [DOI] [PubMed] [Google Scholar]
- Cooper AA, Stevens TH. Vps10p cycles between the late-Golgi and prevacuolar compartments in its function as the sorting receptor for multiple yeast vacuolar hydrolases. J Cell Biol. 1996;133:529–541. doi: 10.1083/jcb.133.3.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Terlecky SR, Dice JF, Knecht E. Selective binding and uptake of RNase A and glyceraldehyde-3-phosphate dehydrogenase by isolated rat liver lysosome. J Biol Chem. 1994;269:26374–26380. [PubMed] [Google Scholar]
- Davis NG, Horecka JL, Sparague GF. Cis- and trans-acting functions required for endocytosis of the yeast pheromone receptor. J Cell Biol. 1993;122:53–65. doi: 10.1083/jcb.122.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends in Biochem. 1990;15:305–309. doi: 10.1016/0968-0004(90)90019-8. [DOI] [PubMed] [Google Scholar]
- Finley D, Ozakaynak E, Varshavsky A. The yeast polyubiquitin gene is essential for resistance to high temperatures, starvation and other stresses. Cell. 1987;48:1035–1046. doi: 10.1016/0092-8674(87)90711-2. [DOI] [PubMed] [Google Scholar]
- Gancedo C. Inactivation of fructose-1,6-bisphosphatase by glucose in yeast. J Bacteriol. 1971;107:401–405. doi: 10.1128/jb.107.2.401-405.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink G. Guide to yeast genetics and molecular biology. Methods Enzymol. 1991;194:21–37. [PubMed] [Google Scholar]
- Harding TM, Morano KA, Scott SV, Klionsky DJ. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding TM, Hefner-Gravink A, Thumn M, Klionsky DJ. Genetic and phenotypic overlap between autophagy and the cytoplasm to vacuole protein targeting pathway. J Biol Chem. 1996;30:17621–17624. doi: 10.1074/jbc.271.30.17621. [DOI] [PubMed] [Google Scholar]
- Hasilik A, Tanner W. Biosynthesis of the vacuolar yeast glycoprotein. Carboxypeptidase Y conversion of precursor into the enzyme. Eur J Biochem. 1978;85:599–608. doi: 10.1111/j.1432-1033.1978.tb12275.x. [DOI] [PubMed] [Google Scholar]
- Hayes S, Dice JF. Roles of molecular chaperones in protein degradation. J Cell Biol. 1996;132:255–258. doi: 10.1083/jcb.132.3.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmings B, Zubenko G, Hasilik A, Jones EW. Mutants defective in processing of an enzyme located in the lysosome-like vacuole of Saccharomyces cerevisiae. . Proc Natl Acad Sci USA. 1981;78:435–439. doi: 10.1073/pnas.78.1.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman M, Chiang H-L. Isolation of degradation-deficient mutants defective in the targeting of fructose-1,6-bisphosphatase into the vacuole for degradation in Saccharomyces cerevisiae. . Genetics. 1996;143:1555–1566. doi: 10.1093/genetics/143.4.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P-H, Chiang H-L. Identification of novel vesicles in the cytosol to vacuole protein degradation pathway. J Cell Biol. 1997;136:803–810. doi: 10.1083/jcb.136.4.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EW. Three proteolytic systems in the yeast Saccharomyces cerevisiae. . J Biol Chem. 1991;266:7963–7966. [PubMed] [Google Scholar]
- Klionsky DJ, Cueva R, Yaver DS. Aminopeptidase I of Saccharomyces cerevisiaeis localized to the vacuole independent of the secretory pathway. J Cell Biol. 1992;119:287–299. doi: 10.1083/jcb.119.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolling R, Hollenberg C. The ABC transporter Ste6 accumulates in the plasma membrane in a ubiquitinated form in endocytosis mutants. EMBO (Eur Mol Biol Organ) J. 1994;13:3261–3271. doi: 10.1002/j.1460-2075.1994.tb06627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai K, Bolognese C, Swift S, McGraw P. Regulation of inositol transport in Saccharomyces cerevisiaeinvolves inositol-induced changes in permease stability and endocytic degradation in the vacuole. J Biol Chem. 1995;270:2525–2534. doi: 10.1074/jbc.270.6.2525. [DOI] [PubMed] [Google Scholar]
- Latterich M, Watson MD. Isolation and characterization of osmosensitive vacuolar mutants of Saccharomyces cerevisiae. . Mol Microbiol. 1991;5:2417–2426. doi: 10.1111/j.1365-2958.1991.tb02087.x. [DOI] [PubMed] [Google Scholar]
- Marcusson EG, Horadovsky BF, Cereghino JL, Gharakhanian E, Emr S. The sorting receptor for yeast vacuolar carboxypeptidase Y is encoded by the VPS10gene. Cell. 1994;77:579–586. doi: 10.1016/0092-8674(94)90219-4. [DOI] [PubMed] [Google Scholar]
- Raths S, Rohrer J, Crausaz F, Riezman H. end3 and end 4: Two mutants defective in receptor-mediated and fluid phase endocytosis in S. cerevisiae. . J Cell Biol. 1993;120:55–65. doi: 10.1083/jcb.120.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riballo E, Herwerjer M, Wolf DH, Lagunas R. Catabolite inactivation of the yeast maltose transporter occurs in the vacuole after internalization by endocytosis. J Bacteriol. 1995;177:5622–5627. doi: 10.1128/jb.177.19.5622-5627.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JS, Klionsky DJ, Banta LM, Emr SD. Protein sorting in Saccharomyces cerevisiae: isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol Cell Biol. 1988;8:4936–4948. doi: 10.1128/mcb.8.11.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman J, Stevens T. Protein sorting in yeast: mutants defective in vacuolar biogenesis mislocalize vacuolar proteins into the late secretory pathway. Cell. 1986;47:1041–1051. doi: 10.1016/0092-8674(86)90819-6. [DOI] [PubMed] [Google Scholar]
- Schandel K, Jennes D. Direct evidence for ligand-induced internalization of the yeast α-factor pheromone receptor. Mol Cell Biol. 1994;14:7245–7255. doi: 10.1128/mcb.14.11.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott SV, Hefner-Gravink A, Morano KA, Noda T, Ohsumi Y, Klionsky D. Cytoplasm to vacuole targeting and autophagy employ the same machinery to deliver proteins to the yeast vacuole. Proc Natl Acad Sci USA. 1996;93:12304–12308. doi: 10.1073/pnas.93.22.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol. 1992;119:301–311. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terlecky SR, Chiang H-L, Oslon TS, Dice JF. Protein and peptide binding and stimulation of in vitro lysosomal proteolysis by 73 kD heat shock cognate protein. J Biol Chem. 1992;267:9202–9209. [PubMed] [Google Scholar]
- Terlecky SR, Dice JF. Polypeptide import and degradation by isolated lysosomes. J Biol Chem. 1993;268:23490–23495. [PubMed] [Google Scholar]
- Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. . FEBS (Fed Eur Biochem Soc) Lett. 1993;333:169–174. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- Tuttle DL, Dunn WA. Divergent modes of autophagy in the methylotrophic yeast Pichia pastoris. . J Cell Sci. 1995;108:25–35. doi: 10.1242/jcs.108.1.25. [DOI] [PubMed] [Google Scholar]
- Yoshihisa T, Anraku Y. A novel pathway of import of α-mannosidase, a marker enzyme for vacuolar membrane, in Saccharomyces cerevisiae. . J Biol Chem. 1990;265:22418–22425. [PubMed] [Google Scholar]
- Zubenko GS, Jones EW. Protein degradation, meiosis and sporulation in proteinase-deficient mutants of Saccharomyces cerevisiae. . Genetics. 1981;97:45–64. doi: 10.1093/genetics/97.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]