

Abstract
Mice that harbor a targeted homozygous defect in the gene coding for the gap junctional protein connexin26 died in utero during the transient phase from early to midgestation. From day 10 post coitum onwards, development of homozygous embryos was retarded, which led to death around day 11 post coitum. Except for growth retardation, no gross morphological alterations were detected between homozygous connexin26-defective embryos and wild-type littermates.
At day 9 postcoitum, when chorioallantoic placenta started to function, connexin26 was weakly expressed in the yolk sac epithelium, between syncytiotrophoblasts I and II in the labyrinth region of the placenta, and in the skin of the embryo. At day 10 post coitum, expression of connexin26 in the placenta was much stronger than at the other locations. To analyze involvement of connexin26 in the placental transfer of nutrients, we have measured embryonic uptake of the nonmetabolizable glucose analogue 3-O-[14C]methylglucose, injected into the maternal tail vein. At day 10 post coitum, viable, homozygous connexin26-defective embryos accumulated only ∼40% of the radioactivity measured in wild-type and heterozygous littermates of the same size. We conclude that the uptake of glucose, and presumably other nutrients as well, from maternal blood into connexin26-deficient mouse embryos was severely impaired and apparently not sufficient to support the rapid organogenesis during midgestation. Our results suggest that connexin26 gap junction channels likely fulfill an essential role in the transfer of maternal nutrients and embryonic waste products between syncytiotrophoblast I and II in the labyrinth layer of the mouse placenta.
Gap junctions are cell-to-cell channels that mediate intercellular communication by exchanging small diffusible molecules like second messengers, metabolites, and ions of up to 1,000 D molecular mass. These channels can be formed after docking of two hemichannels (connexons) in the plasma membranes of adjacent cells. Each hemichannel is composed of six subunit proteins, the connexins (Cx),1 which are coded by a multigene family of at least 13 members in the murine genome (for reviews see Bruzzone et al., 1996; Goodenough et al., 1996; Kumar and Gilula, 1996). Connexin proteins are cell type specifically expressed, but many cells express more than one connexin. It has been shown that the permeability of connexin channels, which consist of one or two types of connexin proteins, is different with regard to ions and tracer molecules (Steinberg et al., 1994; Veenstra et al., 1994; Elfgang et al., 1995). The physiological relevance of these permeability differences is not clear.
Recently, targeted disruption of connexin genes has been used to study the consequences of lacking connexin proteins in mutant mice. Surprisingly, disruption of the connexin43 (Cx43) gene, which is expressed during early embryogenesis, revealed no severe developmental disturbances until birth, when the homozygous defective mice died with a morphological heart defect. This malformation led to obstruction of the right ventricular outflow tract to the lung and, thus, to death shortly after birth (Reaume et al., 1995). Homozygous connexin32 (Cx32)-deficient mice showed normal viability and fertility but exhibited significantly lower release of glucose from glycogen upon electrical stimulation of sympathetic liver nerves (Nelles et al., 1996).
In adult rats and mice, the Cx26 protein is coexpressed with Cx32 in hepatocytes (Nicholson et al., 1987), other exocrine glands (Meda et al., 1993), and, together with other connexin proteins, in kidney as well as skin. Cx26 was detected in rat brain after day 12 post coitum (dpc) and was found in ependyma, leptomeninges, and pinealocytes (Dermietzel et al., 1989). Recently Kelsell et al. (1997) have shown that a mutation of the Cx26 gene in humans can lead to a nonsyndromic sensorineural deafness. During pregnancy, Cx26 is induced in rodent uterine epithelium in response to embryo recognition at implantation (Winterhager et al., 1993). In addition, Cx26 has been shown to be abundantly expressed during the second half of gestation in the three-layered trophoblast of the rat placental labyrinth (Risek and Gilula, 1991; Reuss et al., 1996). These gap junctions have been suggested to serve as channels for diffusion of nutrients between maternal and fetal blood vessels (Metz et al., 1978). Takata et al. (1994) and Shin et al. (1996) immunolocalized Cx26 between syncytiotrophoblasts I and II, whereas the glucose transporter protein (GLUT1) was located in the plasma membrane on the maternal side of syncytiotrophoblast I and on the fetal side of syncytiotrophoblast II. These authors hypothesized that diffusion of glucose across both syncytial layers of the placental labyrinth into fetal blood is mediated by glucose transporter proteins and Cx26 gap junctional channels.
We inactivated the murine Cx26 channel by targeted disruption of the Cx26 gene in embryonic cells and found an essential role for placental function. The uptake of 3-O-[14C]methylglucose from maternal blood into the 10-dpc embryo was severely impaired compared with wild-type littermates. Thus, Cx26-containing gap junction channels serve for transplacental uptake of glucose and presumably other nutrients that appear to be necessary for growth and intrauterine survival of the mouse embryo during transition from early to midgestation.
Materials and Methods
Construction of Targeting Vector
The mouse Cx26 gene was isolated by screening a genomic library from BALB/c mice (Hennemann et al., 1992), using a Cx26 cDNA probe (Zhang and Nicholson, 1989). Positive clones were subcloned into pUC19 and pBluescript SK+ vectors (Stratagene, Heidelberg, Germany). Restriction mapping and DNA sequencing yielded the restriction map of the mouse Cx26 gene shown in Fig. 1 A. A SalI/BclI fragment (10.05 kb) upstream and a Sau3A fragment (1,254 bp) downstream of the Cx26 coding sequence gene were cloned into SalI/BamHI and BamHI sites, respectively, of the pBR322-based pTNe+ vector, which contained the herpes simplex virus thymidine kinase gene, the neomycin-resistance gene driven by the phosphoglycerate kinase (PGK) promoter, and two multiple cloning sites for two-step cloning of vectors for positive and negative selection. Between the upstream and downstream Cx26 fragment, the 1,065 bp that code for the amino acids 83–226 of Cx26 were missing.
Figure 1.

Targeted disruption of the Cx26 gene and transcriptional analysis. (A) Mouse Cx26 wild-type gene with noncoding exon 1 (Ex1) and exon 2 (Ex2) harboring the complete reading frame. (B) Targeting vector: a 1,065-bp fragment of Ex2 was replaced by the neo cassette driven by the PGK promoter. A herpes simplex virus TK cassette was inserted at the 5′ end. The construct was linearized by digestion with NotI, and sticky ends were filled with α-thio dNTP. (C) Mutated, recombinant Cx26 locus and informative restriction fragments, to be compared with the corresponding fragments, in A. The 12- and 7.3-kb fragments represent wild-type and targeted alleles, respectively. The localization of the 3′-external probe and the position of the primers used to genotype embryo yolk sac DNA are shown. PCR primers were selected to generate a 1,118-bp DNA product indicative of the wild-type allele or a 827-bp product due to the targeted Cx26 allele.
Electroporation and Selection of Embryonic Stem Cells
E14 embryonic stem (ES) cells (Hooper et al., 1987) derived from the 129/ Ola mouse strain were cultured as monolayer on mitomycin C–treated mouse embryonic feeder fibroblast in DME. After linearizing by NotI digestion, the targeting vector (25 μg) was treated with Klenow enzyme in the presence of α-thio dNTP and electroporated into trypsinized and suspended ES cells (2 × 107) in 0.5 ml medium of a Hepes-based electroporation buffer, using a gene pulser (Bio-Rad, Munich, Germany). Cells were plated and allowed to recover for 24 h before grown in DME, containing 250 μg/ml G418 and 2 μM gancyclovir. After 10 d of selection, double-resistant ES clones were picked and transferred to a 24-well plate with feeder cells. 2 d later, the cells were split: one half was frozen, whereas the other half was plated into a 12-well plate for growth and preparation of DNA.
Analysis of Targeted ES Cell Clones
Cells, grown in a 12-well plate, were washed with PBS and lysed in 0.5 ml lysis buffer containing 50 μM Tris-HCl, pH 7.5, 100 mM NaCl, 10 mM EDTA, 0.5% SDS, and 50 μg proteinase K. The lysate was incubated overnight at 37°C and transferred into an Eppendorf tube. An equal volume of isopropanol was added, and the DNA was precipitated by centrifugation, washed with 70% ethanol, dried, and resuspended in water. 15 μg of DNA was digested overnight at 37°C in 50 μl of enzyme reaction mixture (1× restriction buffer, 50 μg/ml RNaseA, and 15 U of EcoRI). Electrophoresis and Southern blot hybridization of digested DNA under high stringency were performed according to standard conditions (Sambrook et al., 1989). A 32P-labeled 1.5-kb SalI/SacI-A fragment was used as 3′ external probe to identify targeted and wild-type alleles by the corresponding EcoRI fragments (Fig. 1 C).
Generation of Mice Carrying the Disrupted Cx26 Allele
Two different targeted ES clones, 4IVB5 and 4IVC5, were used to generate chimeric mice by injection of C57/Bl6 blastocysts with 10–20 ES cells. The blastocysts were implanted into pseudopregnant U.S. Naval Medical Research Institute out-bred mouse strain foster mothers. Progeny mice were mated with C57/BL6 mice. Offspring with agouti coat color were tested for the presence of the targeted Cx26 allele by Southern blot analysis. Heterozygous mice were interbred, and resulting animals were genotyped.
DNA Isolation and PCR Genotyping of Embryos
In timed pregnancies, conception was assumed to have occurred around midnight before the morning when the vaginal plug was detected. The light phase in the mouse room was from 4 a.m. to 4 p.m. At 9.5, 10.5, and 11.5 dpc, embryos were dissected from maternal uterus for further analysis. The visceral yolk sac was collected and subjected to genotype determination by PCR. Yolk sac tissue was lysed overnight at 55°C in 500 μl lysis buffer containing 50 mM Tris HCl, pH 7.5, 100 mM NaCl, 10 mM EDTA, 0.5% SDS, and 50 μg proteinase K. DNA was precipitated by centrifugation after adding 500 μl of isopropanol. After washing with 70% ethanol, drying, and resuspending in 10 μl water, DNA samples were used for PCR as follows: 1 μl of sample DNA was mixed with 24 μl PCR solution, containing 1× reaction buffer, 2.5 mM MgCl2, 0.4 mM each of the four dNTPs, and 25 pmol of each primer. The mixture was overlaid with 50 μl of mineral oil. PCR reactions were carried out for 33 cycles of denaturation at 94°C for 2 min, annealing at 64°C for 1.5 min, and extension at 72°C for 2 min.
PCR primers were selected to generate a product specific for either the wild-type or the mutant Cx26 alleles. For the wild-type allele, a 1,118-bp PCR product was generated by primers: 5′-AGCCCAGCACGCCAGTGATGAATACAATA-3′ (P2, located in Cx26 exon 2) and complementary to 5′-CCTACGGGAGACATGAAAAGAAACGGAAG-3′ (P1), a region of Cx26 exon 2, which was deleted in the mutant allele (Fig. 1 A). An 827-bp PCR product was generated by primers indicative of the mutant Cx26 allele: P2 (same primer as used for detection of the wild-type allele) and 5′-GGATCGGCCATTGAACAAGATGGATTGCAC-3′, which annealed to the PGKneo cassette (Fig. 1 C). In wild-type mice, a PCR product was found only with primers to the wild-type Cx26 allele. Both PCR products were detected in heterozygous mice, and only the PCR product, generated by primers to the mutant allele, was found in homozygous Cx26-deficient mice.
Reverse Transcriptase PCR
Individual embryos and placentas were collected at 9.5 and 10.5 dpc of gestation. Total RNA was isolated from lysates using the RNAeasy-Kit (Qiagen, Hilden, Germany). Each RNA preparation was tested for the presence of contaminating genomic DNA by PCR using primers that flanked an intron in the β-actin gene (De Sousa et al., 1993).
RNA preparations were reverse-transcribed using AMV reverse transcriptase (RT) (Promega, Heidelberg, Germany) with 600 pg oligo(dT)20 as primer (Promega). The RT buffer consisted of 1× reaction buffer (Promega), 4.5 mM MgCl2, 0.6 ml RNasin (Promega), and 0.4 mM of each dNTP (GIBCO BRL, Eggenstein, Germany). Aliquots of the reaction solution (30 μl total volume) were incubated for 1 h at 42°C. The cDNA preparations were stored at −20°C or used immediately.
To avoid amplification of genomic DNA, intron-spanning primers were used for detection of Cx26 mRNA. Aliquots of cDNA were amplified using primer pairs specific for exon 1 (5′-GCTTCAGACCTGCTCCTTAC-3′) and exon 2 (5′-GGTCTTTTGGACTTTCCTGAGCA-3′) of the Cx26 gene. 25 μl of the reaction mixture in 1× Taq-reaction buffer (Eurogentec, Seraing, Belgium) contained 2 mM MgCl2, 0.4 mM dNTPs, 50 pmol of each primer, and 2.5 U Taq-DNA-polymerase. PCR was carried out for 33 cycles of 94°C for 1.5 min, 56°C for 1 min, and 72°C for 1 min. PCR products were separated by electrophoresis through 1.5% agarose gel and visualized after staining with ethidium bromide.
Immunohistochemistry
Isolated embryos as well as placentas were freshly frozen on dry ice or liquid nitrogen. Cryostat sections (4–7 μm) were fixed in ice-cold absolute ethanol for 5–10 min. Immunoincubation was performed as previously reported (Winterhager et al., 1991), using different affinity-purified primary antibodies: rabbit anti-Cx26 (Traub et al., 1989), rabbit antibodies, directed to a COOH-terminal polypeptide of Cx31 (Butterweck et al., 1994a ), rabbit anti-Cx32 (Traub et al., 1989), rabbit anti-Cx40 and anti-Cx43 (Traub et al., 1994), and anti-Cx45 (Butterweck et al., 1994b). Furthermore, rabbit antibodies to a COOH-terminal peptide of E-cadherin were applied (Vestweber and Kemler, 1984). Appropriate fluorescein-isothiocyanate–conjugated secondary antibodies were used for staining (Sigma, Deisenhofen, Germany).
Morphology
Embryos and placentas were fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer overnight, postfixed in 2% osmium tetroxide for 4 h, and, after dehydration, embedded in araldite. For freeze fracturing, tissue specimen of placentas were fixed in 2.5% glutaraldehyde and processed according to routine methods (Winterhager and Kühnel, 1982). Replicas were obtained in a Balzers device BAF 400 D. Semithin sections as well as cryostat sections were viewed in a microscope (model Axiophot; Carl Zeiss, Oberkochen, Germany) equipped with epifluorescence. Replicas were examined in an electron microscope (model 902A; Carl Zeiss).
Transplacental Uptake of 3-O-[14C]methylglucose
Heterozygous defective Cx26 mice were interbred. At 10 dpc of pregnancy, radiolabeled 3-O-[14C]methylglucose (30 μCi, 56 mCi/mmol) (Amersham International, Buckinghamshire, UK) in 150 μl of isotonic NaCl solution was injected into the tail vein of female pregnant mice from Cx26 (+/−) intercrosses. After equilibration of the 3-O-[14C]methylglucose for 20 min in the maternal blood circulation, the animals were killed, and the uterus was prepared and collected in PBS. The myometrium was carefully dissected, and the embryos were separated from their extra-embryonic membranes and placentas. After inspection of heart beat and morphology of the embryo under the stereo microscope, embryos and placentas were transferred separately into 500 μl Protosol (New England Nuclear, Boston, MA). The time required for preparation of the embryos from the uterus to the Protosol solution was 2–4 min. Embryos with no heart beat, reduced size, or altered morphology were discarded. The yolk sac tissue of each preparation was washed three times in PBS and transferred to lysis buffer for isolation of DNA.
For lysis of the embryo and placental tissue, the samples were incubated overnight at 55°C. After addition of 4.5 ml scintillation solution (Rotiszint, Karlsruhe, Germany), the 14C radioactivity of each sample was measured in a liquid-scintillation counter (model LS-1801; Beckman Instruments, Duesseldorf, Germany). The maximum uptake of radioactivity into an embryo of one litter was set to 100%, and all uptake measurements within the same litter were evaluated relative to the maximum value. All data are represented as mean ± SD.
Results
Generation of Heterozygous Defective Mice by Targeted Disruption of the Cx26 Gene
Like all other known murine connexin genes, the total coding sequence of the mouse Cx26 gene is contained in exon 2. It is preceded by an intron of ∼4 kb and a nontranslated exon 1 (Fig. 1 A). The targeting vector for disruption of the mouse Cx26 gene contained a 10.05-kb upstream fragment and a 1,254-bp genomic downstream fragment of flanking DNA (Fig. 1 B). The vector was designed to eliminate a 1,065-bp fragment within exon 2, corresponding to amino acids 83–226 of the Cx26 protein. Instead, a 1.1-kb fragment containing the neomycin-resistance gene under control of the PGK promoter was inserted into the coding region of the Cx26 gene. Furthermore, the PGK herpes thymidine kinase gene was linked to the upstream Cx26 flanking DNA in the vector.
The targeting vector was transfected into E14 ES cells, and transfectants were subjected to positive and negative selection in the presence of G418 and gancyclovir, respectively. DNA from surviving colonies was isolated, and a total of 100 samples were analyzed by Southern blot hybridization to identify clones in which one of the Cx26 alleles was disrupted (Fig. 1 C). Homologous recombination was identified by an EcoRI fragment of the predicted size (7.3 kb), using a 3′ external hybridization probe (Fig. 2). Positive clones were further analyzed by Southern blot hybridization using as internal hybridization probe the 1-kb EcoRI/XbaI DNA fragment, located shortly upstream of exon 1 (cf. Fig. 1), and the 673-bp PstI/BamHI DNA fragment of the pPGKneo plasmid to characterize the targeted gene disruption (data not shown).
Figure 2.

Southern blot hybridization using EcoRI-digested DNA from ES cell clones and the 3′-external probe shown in Fig. 1 C. Lanes 1 and 2, heterozygous recombinant ES cell clones 4IVB5 and 4IVC5, identified by the 12-kb wild-type and the 7.3-kb targeted fragments. Lanes 3–7, wild-type ES cell clones.
ES cells from two homologously recombined clones were independently injected into C57/Bl6 blastocysts, which were then transferred into uteri of pseudopregnant NMRI female foster mice. Both ES cell clones gave rise to germ line chimeras. Heterozygous mice exhibited normal phenotype and were fertile.
Absence of Wild-Type Cx26 Alleles Resulted in Embryonic Lethality Around Day 11 pc
To investigate the in vivo effect of homozygous Cx26 deletion, heterozygous mice were intercrossed. Among ∼250 animals tested 2–3 wk after birth, no homozygous Cx26-deficient mouse was found. In addition, genotyping of all litters demonstrated that heterozygous and wild-type animals appeared at the expected ratio.
To determine the exact time of intrauterine death, pregnant females from Cx26 (+/−) intercrosses were killed, and the embryos were examined at different time points of gestation between 9.5 to 12.5 dpc. Homozygous Cx26- defective embryos, which were identified by PCR using DNA from yolk sac tissue (Fig. 3), developed normally up to day 10 pc. The same results were obtained with homozygous Cx26-defective embryos derived from two independent ES cell clones.
Figure 3.

PCR analysis of DNA from embryonic yolk sac tissues of all embryos from one litter at day 10 pc. The presence of the wild-type (WT) allele was indicated by the 1,118-bp PCR product generated by the primers P1 and P2 (Fig. 1 A). The targeted allele was indicated by the 827-bp PCR product using primers P3 and P2 (Fig. 1 C). Lane 5, wild-type embryo; lanes 1, 2, 4, and 7, heterozygous Cx26-deficient embryos; lanes 3, 6, and 8, homozygous Cx26-deficient embryos.
At 10.5 dpc, Cx26 (−/−) embryos were significantly smaller than the wild-type or heterozygous littermates (Fig. 4). No obvious malformation could be detected in semithin sections of embryos at day 10.5 pc (data not shown). Around day 11 pc, however, the homozygous defective embryos died, as diagnosed by the absence of heart beat. Subsequently, embryos became necrotic and were reabsorbed.
Figure 4.

External appearance of two embryos of the same litter around 10.5 dpc of pregnancy. At this time, the homozygous, Cx26 defective embryo (left) was significantly smaller than the wild-type littermate (right). Bar, 1.5 mm.
At 9.5 dpc, Cx26 Is Expressed in the Labyrinth Layer of the Placenta, Yolk Sac Epithelium, and Embryonic Skin
To search for the reasons of early death of Cx26 (−/−) embryos, the spatial and temporal expression pattern of Cx26 was analyzed in the wild-type embryo, and in extra-embryonic tissue at 9.5–11.5 dpc by RT-PCR and immunofluorescence. After 9.5 dpc, Cx26 mRNA was detected in the embryo (Fig. 5). At this time, Cx26 protein was found only in embryonic skin (cf. Fig. 6 A), although all other embryonic tissues were analyzed on serial sections. At 10.5 dpc, the most prominent extra-embryonic immunohistochemical staining of the Cx26 protein was found within the labyrinth layer of the developing placenta (Fig. 6 B) in those parts that are functioning in maternal–fetal exchange. Furthermore, Cx26 transcripts had also been detected in the placenta at 9.5 and 10.5 dpc (cf. Fig. 5).
Figure 5.

Results of RT-PCR analysis, using intron-spanning primers and Cx26 mRNA from wild-type embryos and placentas at 9.5 and 10 dpc. Lane 1, size marker; lane 2, adult liver (control); lane 3, adult heart (control). RT-PCR products due to Cx26 mRNA were found in embryos on day 9.5 pc (lane 4) and at higher amounts in embryos on 10.5 pc (lane 6). A strong signal for Cx26 was demonstrated in placentas of day 9.5 (lane 5) and 10.5 pc (lane 7). Primers to β actin cDNA were used as amplification control.
Figure 6.
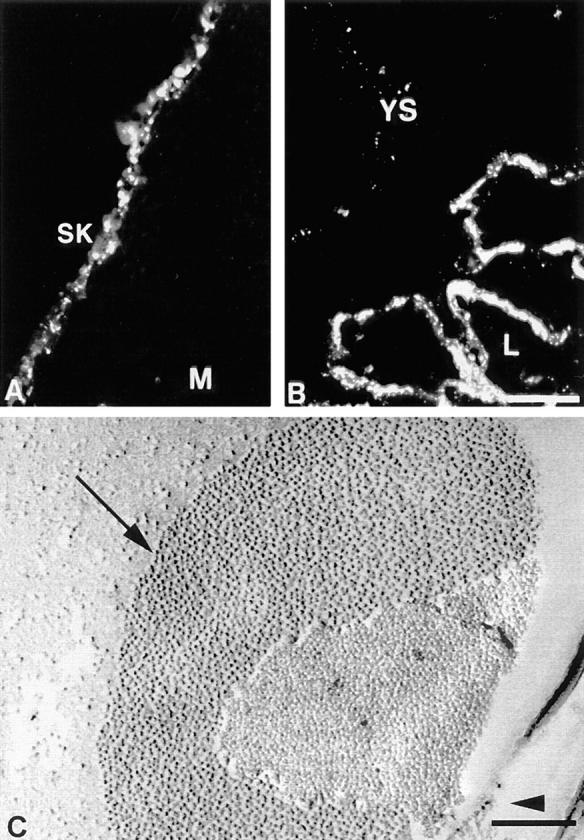
Expression of Cx26 protein in embryonic skin (A) and placenta (B) of wild-type embryos on day 10.5 pc. (A) Punctate immunoreaction was detected in developing skin (SK), M, mesenchyme. (B) Intensive staining of Cx26 protein in the labyrinthin (L) part of the placenta was observed. In addition, the yolk sac (YS) epithelium showed some staining at lateral cell borders. (C) Freeze-fractured placental labyrinth of a wild-type embryo on 9.5 dpc. Abundant gap junctional plaques (arrow) were found within the labyrinth layer. The arrowhead indicates direction of shadowing. Bars: (A and B) 57 μm; (C) 0.2 μm.
Immunolabeling revealed already at 9 dpc the typical punctate staining of gap junctional plaques at the lateral membrane of the visceral yolk sac epithelium, a derivative of extra-embryonic endoderm (shown for 10.5 dpc in Fig. 6 B). At the same time, strong immunoreactivity of the Cx26 protein was restricted to the three-layered trophoblast of the developing placental labyrinth. On day 9.5 pc, large gap junctional plaques were seen between trophoblast cells in the labyrinth by electron microscopy of freeze-fractured specimens (Fig. 6 C).
As expected, no immunohistochemical Cx26 staining could be detected in Cx26-deficient mice, in contrast to strong signals in the labyrinthine trophoblast of wild-type placenta (cf. Fig. 7 A with B). Cadherins appear to have a synergic effect on gap junction mediated cell–cell communication (Jongen et al., 1991; Meyer et al., 1992; Wang and Rose, 1997). E-cadherin, normally coexpressed in the labyrinth with Cx26, was still observed and served as marker of the labyrinthine region in the placenta of Cx26 mutant mice (Fig. 7 C).
Figure 7.
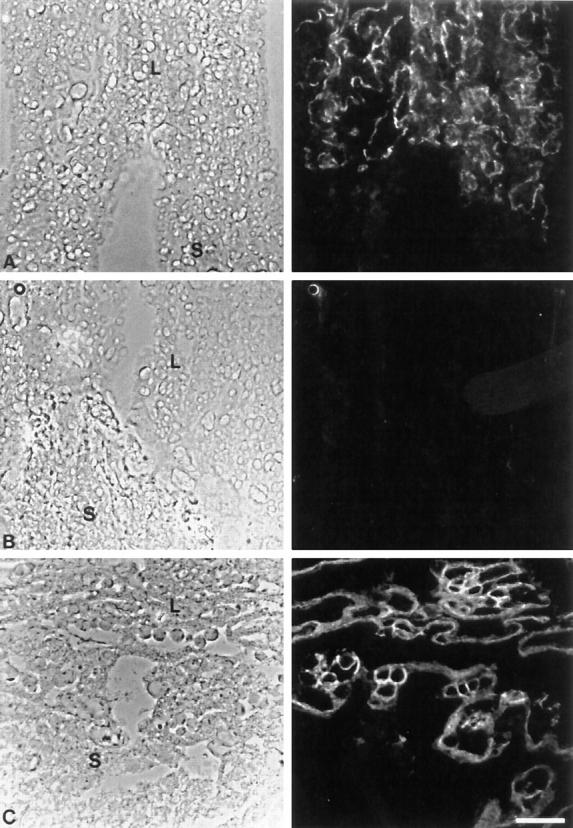
Placenta on day 10.5 pc of wild-type and Cx26-deficient placentas, as seen on phase contrast (left) and immunofluorescence micrographs (right). (A) Phase contrast micrograph of cryostat section showing the transition of the labyrinth to the spongiotrophoblast in wild-type placenta. Cx26 immunoreactivity was only seen in the trophoblast layers of the labyrinth. (B) Cryostat section of the same region as in A. No immunostaining of Cx26 antigen was observed in the labyrinth of Cx26 (−/−) placenta. (C) Region between labyrinth and spongiotrophoblast of Cx26 (−/−) placenta. E-cadherin was detected by immunostaining, similar as in wild-type placentas where it was coexpressed with Cx26. L, labyrinth; S, spongiotrophoblast. Bar, 100 μm.
We also searched in Cx26 wild-type mice for expression of additional connexin proteins in mouse placenta and yolk sac epithelium using affinity-purified antibodies to Cx31, Cx32, Cx37, Cx40, Cx43, and Cx45. In addition to Cx26, only Cx32 and Cx43 immunosignals were detected at lateral membranes of the yolk sac epithelium. In the placenta, Cx43 and to a lesser extent Cx31 immunoreactivity was noticed in the spongiotrophoblast, but no further connexin protein could be detected in the labyrinth region (data not shown).
The Morphology of Cx26 (−/−) Placentas Did Not Show Dramatic Alterations
Semithin sections of Cx26 (−/−) placenta, compared with wild-type mouse placenta, revealed the same morphological organization (cf. Fig. 8 A with C). Both placentas exhibited the same trophoblastic layers of labyrinthine trophoblast, spongiotrophoblast, and trophoblast giant cells without obvious differences in cell morphology. There were some indications for retarded placental development at 10.5 dpc. Ingrowth of embryonic mesenchyme from the chorionic plate seemed to be more extended in wild-type than in Cx26 (−/−) placentas. As a consequence, the placental labyrinth of wild-type animals contained more embryonic blood vessels and appeared more enlarged (cf. Fig. 8 B with D).
Figure 8.
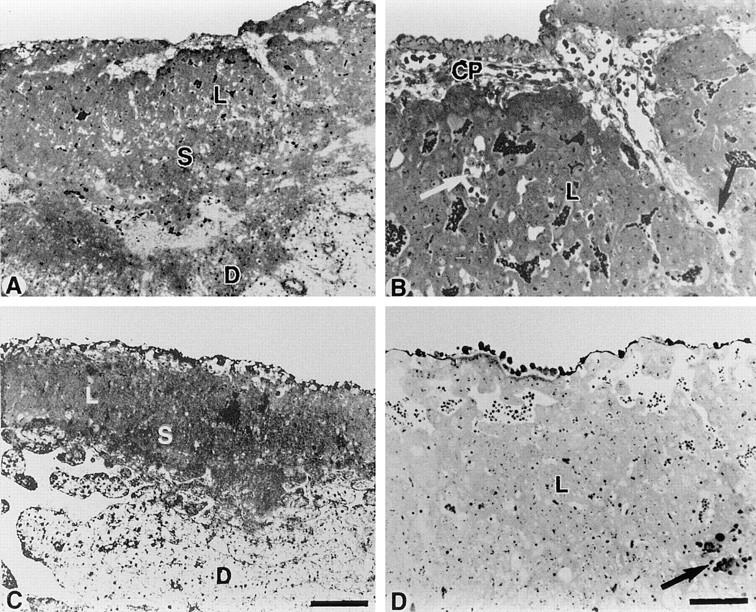
Morphology of placentas of homozygous Cx26 mutant and wild-type embryos on 10.5 dpc. (A) Overview of wild-type placenta showing the normal organization with chorionic plate, labyrinth (L), and spongiotrophoblast (S). (B) Higher magnification revealed chorionic plate (CP), ingrowth of fetal stroma, and several fetal blood vessels (arrows) within the labyrinth (L). (C) Overview of a Cx26 (−/−) placenta revealing the same organization of labyrinth (L), and spongiotrophoblast (S) and decidua (D), as shown in A, but less extended than in wild-type placenta. (D) Higher magnification showed a more compact labyrinth trophoblast with only few fetal vessels (arrow). L, labyrinth. Bars: (A and C) 400 μm; (B and D) 100 μm.
Transplacental Uptake of 3-O-[14C]methylglucose Was Decreased in Cx26 (−/−) Embryos
To investigate alterations in maternal–fetal transfer of glucose in Cx26 mutant mice (for scheme see Fig. 9 A), we measured the uptake of the nonmetabolizable glucose analogue 3-O-[14C]methylglucose at day 10 pc into all embryos of several litters from Cx26 (+/−) intercrosses. As mentioned above, great care was taken to analyze only embryos of the same size, as determined by visual comparison. The radioactive compound was injected into the tail vein of pregnant mice, and radioactivity was measured in embryos, removed 20 min after injection. The viability of all embryos was confirmed by the presence of heartbeat. Heterozygous and wild-type embryos showed no significant differences in the uptake of the glucose analogue. Homozygous Cx26-defective embryos revealed, however, a 60% decrease (P < 0.01) in uptake of 3-O-[14C]methylglucose, compared with wild-type and heterozygous embryos of the same litter (Fig. 9 B).
Figure 9.

(A) Schematic drawing of the trophoblast layers of the labyrinth in the murine placenta. The localization of gap junctional Cx26 channels and glucose transporter GLUT1 proteins as well as the likely route of glucose transfer from maternal to fetal blood vessels are illustrated. Glucose enters via GLUT1 the cytoplasm of the syncytiotrophoblast I, diffuses mainly via gap junction channels into syncytiotrophoblast layer II, and is released via GLUT1 at the basal side of the trophoblast membrane into fetal blood circulation through fenestrated endothelial cells (adapted from Takata et al., 1994). M, maternal blood; Cyt, cytotrophoblast; Syn I, syncytiotrophoblastic layer I; GJ, Cx26-containing gap junctions; Syn II, syncytiotrophoblastic layer II; BL, basal lamina; E, endothelial cell; F, fetal blood circulation. (B) Uptake of 3-O-[14C]methylglucose into whole embryos at day 10 pc from maternal blood across the placenta. Significantly decreased accumulation (P < 0.01) of radioactivity was measured in homozygous Cx26-defective (−/−) embryos compared with heterozygous (+/−) and wild-type embryos (+/+). n designates the number of embryos investigated.
Discussion
This study demonstrates that homozygous mutant mice lacking functional Cx26 protein died in utero during the transient stage between early and midgestation. This lethality is likely due to functional disturbances of the chorioallantoic placenta lacking gap junctional Cx26 channels in the labyrinthine part. The mutant embryos were smaller, presumably because of starvation, but exhibited no obvious malformations. Thus, the early embryonic death was probably not caused by embryonic malformations but by dysfunction of the placenta. This notion was supported by the finding that, at this critical time of development, Cx26 was exclusively expressed in wild-type embryonic skin. Extra-embryonically, Cx26 was detected, among other connexins (Cx32 and Cx43), in wild-type yolk sac epithelium and abundantly in the labyrinth part of the chorioallantoic placenta after 9.5 dpc, where no other connexins were found. At this stage of development, the function of the yolk sac to mediate nutrition and waste disposal of the embryo is more and more taken over by the chorioallantoic placenta.
Both types of placentas, the yolk sac and chorioallantoic, have their own essential roles in development (for review see Garbis-Berkvens and Peters, 1987). The yolk sac is of endodermal origin and surrounds the embryo as well as an extra-embryonic part in a double sac. In early stages of development, the mouse embryo is totally dependent on the yolk sac for supply of nutrients. During this so-called histiotrophic phase, the epithelial cells of the parietal yolk sac absorb proteins as well as glucose from the yolk sac cavity and hydrolyze the proteins (Beck et al., 1967; Record et al., 1982; Miki and Kugler, 1984). After formation of vitelline blood vessels, the transcytotic pathway via the yolk sac epithelium to the embryonic blood vessels starts to function (Beck et al., 1967; Poelmann and Mentink, 1982a ,b; Freeman and Loyd, 1983). Although Cx26 is missing in the yolk sac epithelium, the Cx26 (−/−) mouse embryo seems to develop normally during this early phase, and the yolk sac placenta does not show obvious changes in morphology. Because of the localization of Cx26 at lateral membrane borders between visceral yolk sac epithelial cells, the channels do not seem to be involved in the transcellular transport of nutrients. Dysfunction of the yolk sac placenta due to the lack of Cx26 is probably compensated by the other connexins expressed. If Cx26 deficiency had resulted in a defective function of the yolk sac placenta, embryonic defects would have been expected to become obvious already during early gestation. Thus, we think it unlikely that Cx26 channels are involved in transport function of the yolk sac epithelium. Around 8.5 dpc, the chorioallantoic placenta starts to develop by fusion of the allantois with the extra-embryonic ectoderm and ectoplacental cone (Müntener and Hsu, 1977), forming the chorionic plate and stimulating the differentiation of trophoblast cells into the placental labyrinth. It has been considered that the labyrinthine trophoblast is a derivative of the extra-embryonic ectoderm, which fits with observations by Reuss et al. (1996), who detected E-cadherin and weak expression of Cx26, both markers of the labyrinth, in the extra-embryonic ectoderm at this stage of development. Concomitant with establishment of the labyrinth, the materno–fetal exchange is initiated, resulting in rapid growth and progressing organogenesis of the embryo.
In contrast to human placenta that contains one giant syncytiotrophoblast, the murine placenta consists of syncytiotrophoblast layers I and II, which are interconnected through gap junctions to facilitate the transport of nutrients, such as glucose (Metz et al., 1978; Takata, 1994). These morphological differences explain why mutations in the human Cx26 gene do not appear to affect placental efficiency (Kelsell et al., 1997). In the present study and by investigations in the rat, it has been shown that gap junctions of the labyrinth contain Cx26 protein, which is expressed shortly after chorioallantoic fusion (Reuss et al., 1996). Takata et al. (1994) and Shin et al. (1996) reported that the glucose transporter protein GLUT1 was localized in the rat placenta in the apical membrane of syncytiotrophoblast layer I and the basal membrane of syncytiotrophoblast layer II, close to the fetal blood stream. Cx26 immunoreactivity was found in membranes between syncytiotrophoblast layers I and II. These authors suggested that uptake and release of glucose occurred via GLUT1 and intercellular transport via Cx26 channels (cf. Fig. 9 A). This hypothesis is supported by our experimental data. We found 60% decrease in the uptake of 3-O-[14C]methylglucose on 10 dpc, when the functional chorioallantoic placenta was established. The remaining 40% of embryonic glucose uptake are likely to occur mainly through the yolk sac epithelium, since GLUT1 has been found in rat yolk sac placenta very early in development (Trocino et al., 1994). Some transfer of glucose between syncytiotrophoblasts I and II might also be mediated via the extracellular space by GLUT1 proteins that were expressed at low level in the same plasma membrane where Cx26 was detected (cf. Fig. 9 A; Shin et al., 1996). Recently, in addition to GLUT1, localization of high-affinity glucose transporter GLUT3 in the labyrinth region has been reported (Shin et al., 1997). This could also contribute to the alternative transplacental transfer route of glucose. In addition, exchange of many other small molecules, such as essential amino acids, is likely to be mediated by Cx26-containing gap junction channels in murine placenta. In Cx26 (−/−) placentas, defective uptake of maternal nutrients or defective removal of embryonic waste products may contribute to early embryonic death.
The lack of Cx26 channels did not seem to directly affect labyrinth formation, although there were some indications of a retardation in placental development. We found that fetal mesenchymal ingrowth, concomitant with fetal vessels, seemed to be less extensive. Like in the embryo, retardation in the establishment of the placenta could be caused by malnutrition of compact trophoblast cell aggregates at this developmental stage, when the lack of Cx26 channels restricted supplementation with nutrients from the maternal blood sinus. This phenomenon may have enhanced malnutrition leading to embryonic death. In contrast to Cx26-deficient mice, disruption of the gene coding for hepatocyte growth factor/scatter factor led to a severely impaired placenta with a size reduction in labyrinthine trophoblast and to embryos that died in utero around 13 dpc (Uehara et al., 1995; Schmidt et al., 1995). Copp (1995) has reviewed targeted gene defects that affect routes of nutritional interaction of the embryo with the mother, including formation of the chorioallantoic placenta.
The embryonic lethality of Cx26 (−/−) mice precluded studies of functional importance of Cx26 gap junction channels in the adult animal. As mentioned, Cx26 is coexpressed in adult rats and mice with Cx32 in exocrine glands. This coexpression has been most intensively studied in liver. Recently, it has been shown (Nelles et al., 1996) that Cx32-defective mice were viable and fertile but exhibited decreased release of glucose from glycogen upon stimulation of hepatic sympathetic nerves. Furthermore, Cx32 (−/−) mice developed spontaneous and chemically induced liver tumors (Temme et al., 1997). Cultured Cx32 (−/−) hepatocytes showed still ∼40% dye transfer, compared with wild-type hepatocytes, due to functional Cx26 gap junctional channels. Thus, a decreased level of functional Cx26 channels in Cx32 (−/−) mice appears to be consistent with normal development and function of liver as well as other exocrine glands but cannot propagate among hepatocytes sufficient inositol 1,4,5 trisphosphate to stimulate wild-type level of glucose mobilization and does not protect against hepatocarcinogenesis. Therefore, Cx26 and Cx32 gap junctional channels are likely to have different biological functions. In the placenta, probably mainly glucose, small molecular weight nutrients and ions are transferred through Cx26-containing gap junctions between syncytiotrophoblast layers I and II from maternal to fetal blood. This is different from the function of gap junctions between hepatocytes, where efficient intercellular communication by second messenger molecules, such as inositol 1,4,5 trisphosphate, may require Cx32-containing gap junctional channels to be expressed. We are only beginning to gain insight into the reasons for functional diversity of connexin channels. In the future it should be possible to study the biological function of Cx26 channels in adult mice by cell type–specific deletion of the Cx26 gene or by functional replacement (“knock-in”) with other connexin genes.
Acknowledgments
We thank Ulrike Ebertshäuser (Institut für Genetik, Universität Bonn) for technical assistance during the initial phase of this work.
This work was supported by grants of the Dr. Mildred Scheel Stiftung, the Deutsche Forschungsgemeinschaft through SFB 284, project C1, and the Fonds der Chemischen Industrie to K. Willecke, as well as SFB 354 to E. Winterhager.
Abbreviations used in this paper
- Cx
connexin
- dpc
days post coitum
- ES
embryonic stem
- PGK
phosphoglycerate kinase
- RT
reverse transcriptase
Footnotes
H.-D. Gabriel and D. Jung contributed equally to this work.
Address all correspondence to Dr. Klaus Willecke, Institut für Genetik, Abt. Molekulargenetik, Römerstr. 164, 53117 Bonn. Tel.: (0228) 734210. Fax: (0228) 734263. E-mail: genetik@uni-bonn.de
References
- Beck F, Lloyd JB, Griffiths A. A histochemical and biochemical study of some aspects of placental function in the rat using maternal injection of horseradish peroxidase. J Anat. 1967;101:461–478. [PMC free article] [PubMed] [Google Scholar]
- Bruzzone R, White TW, Goodenough DA. The cellular internet: on-line with connexins. BioEssays. 1996;18(9):709–718. doi: 10.1002/bies.950180906. [DOI] [PubMed] [Google Scholar]
- Butterweck A, Elfgang C, Willecke K, Traub O. Differential expression of the gap junction proteins connexin45, -43, -40, -31, and -26 in mouse skin. Eur J Cell Biol. 1994a;65:152–163. [PubMed] [Google Scholar]
- Butterweck A, Gergs U, Elfgang C, Willecke K, Traub O. Immunochemical characterization of the gap junction protein connexin45 in mouse kidney and transfected human HeLa cells. J Membr Biol. 1994b;141:247–256. doi: 10.1007/BF00235134. [DOI] [PubMed] [Google Scholar]
- Copp AJ. Death before birth, clues from gene knockouts and mutations. Trends Genet. 1995;11:87–93. doi: 10.1016/S0168-9525(00)89008-3. [DOI] [PubMed] [Google Scholar]
- De Sousa PA, Valdimarsson G, Nicholson BJ, Kidder GM. Connexin trafficking and the control of gap junction assembly in mouse preimplantation embryos. Development (Camb) 1993;117:1355–1367. doi: 10.1242/dev.117.4.1355. [DOI] [PubMed] [Google Scholar]
- Dermietzel R, Traub O, Hwang TK, Beyer E, Bennett MVL, Spray DC, Willecke K. Differential expression of three gap junction proteins in developing and mature brain tissues. Proc Natl Acad Sci USA. 1989;86:10148–10152. doi: 10.1073/pnas.86.24.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfgang C, Eckert R, Lichtenberg-Frate H, Butterweck A, Traub O, Klein RA, Hülser DF, Willecke K. Specific permeability and selective formation of gap junction channels in connexin-transfected HeLa cells. J Cell Biol. 1995;129:805–817. doi: 10.1083/jcb.129.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman SJ, Lloyd JB. Evidence that protein ingested by the rat visceral yolk sac yields amino acids for synthesis of embryonic protein. J Embryol Exp Morph. 1983;73:307–315. [PubMed] [Google Scholar]
- Garbis-Berkvens, J.M., and P.W.J. Peters. 1987. Comparative morphology and physiology of embryonic and fetal membranes. In Pharmacokinetics in Teratogenesis. Vol. 1. H. Nau and W.J. Scott, Jr., editors. CRC Press, Boca Raton, FL. 14–44.
- Goodenough DA, Goliger JA, Paul DL. Connexins, connexons, and intercellular communication. Annu Rev Biochem. 1996;65:475–502. doi: 10.1146/annurev.bi.65.070196.002355. [DOI] [PubMed] [Google Scholar]
- Hennemann H, Kozjek G, Dahl E, Nicholson B, Willecke K. Molecular cloning of mouse connexins26 and -32: similar genomic organization but distinct promoter sequences of two gap junction genes. Eur J Cell Biol. 1992;58:81–89. [PubMed] [Google Scholar]
- Hooper M, Hardy K, Handyside A, Hunter S, Monk M. HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature. 1987;326:292–295. doi: 10.1038/326292a0. [DOI] [PubMed] [Google Scholar]
- Jongen WMF, Fitzgerald DJ, Asamoto M, Piccoli C, Slaga TJ, Gros D, Takeichi M, Yamasaki H. Regulation of connexin-43–mediated gap junctional intercellular communication by Ca2+in mouse epidermal cells is controlled by E-cadherin. J Cell Biol. 1991;114:545–555. doi: 10.1083/jcb.114.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsell, D.P., J. Dunlop, H.P. Stevens, N.J. Lench, J.N. Liang, G. Parry, R.F. Mueller, and I.M. Leigh. 1997. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature, 387:80–83. [DOI] [PubMed]
- Kumar NM, Gilula NB. The gap junction communication channel. Cell. 1996;84:381–388. doi: 10.1016/s0092-8674(00)81282-9. [DOI] [PubMed] [Google Scholar]
- Meda P, Pepper MS, Traub O, Willecke K, Gros D, Beyer E, Nicholson B, Paul D, Orci L. Differential expression of gap junction connexins in endocrine and exocrine glands. Endocrinology. 1993;133:2371–2378. doi: 10.1210/endo.133.5.8404689. [DOI] [PubMed] [Google Scholar]
- Metz J, Aoki A, Forssmann WG. Studies on the ultrastructure and permeability of the hemotrichorial placenta I. Intercellular junctions of layer I and tracer administration into the maternal compartment. Cell Tissue Res. 1978;192:391–407. doi: 10.1007/BF00212321. [DOI] [PubMed] [Google Scholar]
- Meyer RA, Laird DW, Revel JP, Johnson RG. Inhibition of gap junction and adherens junction assembly by connexin and A-CAM antibodies. J Cell Biol. 1992;119:179–189. doi: 10.1083/jcb.119.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki A, Kugler P. Comparative enzyme histochemical study on the visceral yolk sac endoderm in the rat in vivo and in vitro. Histochemistry. 1984;81:409–415. doi: 10.1007/BF00514337. [DOI] [PubMed] [Google Scholar]
- Müntener M, Hsu Y-C. Development of trophoblast and placenta of the mouse: a reinvestigation with regard to the in vitro culture of mouse trophoblast and placenta. Acta Anat. 1977;98:241–252. [PubMed] [Google Scholar]
- Nelles E, Bützler C, Jung D, Temme A, Gabriel HD, Dahl U, Traub O, Stumpel F, Jungermann K, Zielasek J, et al. Defective propagation of signals generated by sympathetic nerve stimulation in the liver of connexin32-deficient mice. Proc Natl Acad Sci USA. 1996;93:9565–9570. doi: 10.1073/pnas.93.18.9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson B, Dermietzel R, Teplow D, Traub O, Willecke K, Revel J-P. Two homologous protein components of hepatic gap junctions. Nature. 1987;329:732–734. doi: 10.1038/329732a0. [DOI] [PubMed] [Google Scholar]
- Poelmann RE, Mentink MMT. The maternal-embryonic barrier in the early postimplantation mouse embryo: a morphological and functional study. Scanning Electron Microsc. 1982a;3:1237–1247. [PubMed] [Google Scholar]
- Poelmann RE, Mentink MMT. Parietal yolk sac in early gestation mouse embryos: structure and function. Bibl Anat. 1982b;22:123–127. [PubMed] [Google Scholar]
- Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, Juneja SC, Kidder GM, Rossant J. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267:1831–1834. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- Record IR, Dreosti IE, Manuel SJ. Inhibition of rat yolk sac pinocytosis by cadmium and its reversal by zinc. J Nutr. 1982;112:1994–1998. doi: 10.1093/jn/112.10.1994. [DOI] [PubMed] [Google Scholar]
- Reuss B, Hellmann P, Dahl E, Traub O, Butterweck A, Grümmer R, Winterhager E. Connexins and E-cadherin are differentially expressed during trophoblast invasion and placenta differentiation in the rat. Dev Dyn. 1996;205:172–182. doi: 10.1002/(SICI)1097-0177(199602)205:2<172::AID-AJA8>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Risek B, Gilula NB. Spatiotemporal expression of three gap junction gene products involved in fetomaternal communication during rat pregnancy. Development (Camb) 1991;113:165–181. doi: 10.1242/dev.113.1.165. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Schmidt C, Bladt F, Goedecke S, Brinkmann V, Zschiesche W, Sharpe M, Gherardi E, Birchmeier C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature. 1995;373:699–702. doi: 10.1038/373699a0. [DOI] [PubMed] [Google Scholar]
- Shin B-C, Suzuki T, Matsuzaki T, Tanaka S, Kuraoka A, Shibata Y, Takata K. Immunolocalization of GLUT1 and connexin26 in the rat placenta. Cell Tissue Res. 1996;285:83–89. doi: 10.1007/s004410050623. [DOI] [PubMed] [Google Scholar]
- Shin B-C, Fujikura K, Suzuki T, Tanaka S, Takata K. Glucose transporter GLUT3 in the rat placental barrier: a possible machinery for the transplacental transfer of glucose. Endocrinology. 1997;138:3997–4004. doi: 10.1210/endo.138.9.5369. [DOI] [PubMed] [Google Scholar]
- Steinberg TH, Civitelli R, Geist ST, Robertson AJ, Hick E, Veenstra RD, Wang HZ, Warlow PM, Westphale EM, Laing JG, Beyer EC. Connexin43 and connexin45 form gap junctions with different molecular permeabilities in osteoblastic cells. EMBO (Eur Mol Biol Organ) J. 1994;13:744–750. doi: 10.1002/j.1460-2075.1994.tb06316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata K. Structural basis of glucose transport in the placental barrier: role of GLUT1 and the gap junction. Endocrine J. 1994;41(Suppl.):S3–S8. [Google Scholar]
- Takata K, Kasahara T, Kasahara M, Ezaki O, Hirano H. Immunolocalization of glucose transporter GLUT1 in the rat placental barrier: possible role of GLUT1 and the gap junction in the transport of glucose across the placental barrier. Cell Tissue Res. 1994;276:411–418. doi: 10.1007/BF00343939. [DOI] [PubMed] [Google Scholar]
- Temme A, Buchmann A, Gabriel H-D, Nelles E, Schwarz M, Willecke K. High incidence of spontaneous and chemically induced liver tumors in mice deficient for comexin 32. Curr Biol. 1997;7:713–716. doi: 10.1016/s0960-9822(06)00302-2. [DOI] [PubMed] [Google Scholar]
- Traub O, Look J, Dermietzel R, Brümmer F, Hülser D, Willecke K. Comparative characterization of the 21-kD and 26-kD gap junction proteins in murine liver and cultured hepatocytes. J Cell Biol. 1989;108:1039–1051. doi: 10.1083/jcb.108.3.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub O, Eckert R, Lichtenberg-Frate H, Elfgang C, Bastide B, Scheidtmann KH, Hülser DF, Willecke K. Immunochemical and electrophysiological characterization of murine connexin40 and -43 in mouse tissues and transfected human cells. Eur J Cell Biol. 1994;64:101–112. [PubMed] [Google Scholar]
- Trocino RA, Akazawa S, Takino H, Takao Y, Matsumoto K, Maeda Y, Okuno S, Nagataki S. Cellular-tissue localization and regulation of the GLUT-1 protein in both the embryo and the visceral yolk sac from normal and experimental diabetic rats during the early postimplantation period. Endocrinology. 1994;134:869–878. doi: 10.1210/endo.134.2.8299581. [DOI] [PubMed] [Google Scholar]
- Uehara Y, Minowa O, Mori C, Shiota K, Kuno J, Noda T, Kitamura N. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature. 1995;373:702–705. doi: 10.1038/373702a0. [DOI] [PubMed] [Google Scholar]
- Veenstra RD, Wang HZ, Beyer EC, Ramanan SV, Brink PR. Connexin37 forms high conductance gap junction channels with subconductance state activity and selective dye and ionic permeabilities. Biophys J. 1994;66:1915–1928. doi: 10.1016/S0006-3495(94)80985-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestweber D, Kemler R. Rabbit antiserum against a purified surface glycoprotein decompacts mouse primplantation embryos and reacts with specific adult tissues. Exp Cell Biol. 1984;152:169–178. doi: 10.1016/0014-4827(84)90241-6. [DOI] [PubMed] [Google Scholar]
- Wang Y, Rose B. An inhibition of gap-junctional communication by cadherins. J Cell Sci. 1997;110:301–309. doi: 10.1242/jcs.110.3.301. [DOI] [PubMed] [Google Scholar]
- Winterhager E, Kühnel W. Alterations in intercellular junction of the uterine epithelium during the preimplantation phase in the rabbit. Cell Tissue Res. 1982;224:517–526. doi: 10.1007/BF00213749. [DOI] [PubMed] [Google Scholar]
- Winterhager E, Stutenkemper R, Traub O, Beyer E, Willecke K. Expression of different connexin genes in rat uterus during decidualization and at term. Eur J Cell Biol. 1991;55:133–142. [PubMed] [Google Scholar]
- Winterhager E, Grümmer R, Jahn E, Willecke K, Traub O. Spatial and temporal expression of connexin26 and connexin43 in rat endometrium during trophoblast invasion. Dev Biol. 1993;157:399–409. doi: 10.1006/dbio.1993.1144. [DOI] [PubMed] [Google Scholar]
- Zhang JT, Nicholson BJ. Sequence and tissue distribution of a second protein of hepatic gap junctions, Cx26, as deduced from its cDNA. J Cell Biol. 1989;109:3391–3401. doi: 10.1083/jcb.109.6.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]