

Abstract
Both phosphoinositides and small GTP-binding proteins of the Rho family have been postulated to regulate actin assembly in cells. We have reconstituted actin assembly in response to these signals in Xenopus extracts and examined the relationship of these pathways. We have found that GTPγS stimulates actin assembly in the presence of endogenous membrane vesicles in low speed extracts. These membrane vesicles are required, but can be replaced by lipid vesicles prepared from purified phospholipids containing phosphoinositides. Vesicles containing phosphatidylinositol (4,5) bisphosphate or phosphatidylinositol (3,4,5) trisphosphate can induce actin assembly even in the absence of GTPγS. RhoGDI, a guanine-nucleotide dissociation inhibitor for the Rho family, inhibits phosphoinositide-induced actin assembly, suggesting the involvement of the Rho family small G proteins. Using various dominant mutants of these G proteins, we demonstrate the requirement of Cdc42 for phosphoinositide-induced actin assembly. Our results suggest that phosphoinositides may act to facilitate GTP exchange on Cdc42, as well as to anchor Cdc42 and actin nucleation activities. Hence, both phosphoinositides and Cdc42 are required to induce actin assembly in this cell-free system.
The actin cytoskeleton is involved in most aspects of cellular morphogenesis where it is dynamically regulated by both extra- and intracellular signals (Stossel, 1993; Fishkind and Wang, 1995; Mitchison and Cramer, 1996). In almost all cases, this regulation is posttranslational and the locus of this posttranslational control is not on actin, but rather on actin binding proteins that control actin dynamics in the cell. There are ∼50 actin binding proteins identified so far that directly interact with actin and modulate its assembly properties (Kreis and Vale, 1993). They can be grouped into six functional families: monomer sequestering, filament severing, capping, nucleating, cross-linking, and bundling. The presence of such a large number of regulatory proteins makes it very difficult to study the physiological mechanisms controlling actin functions. Although it is relatively easy to identify the mechanism by which individual proteins act from in vitro studies, it is difficult to reveal the network of interactions that occur in vivo. It is even more challenging to connect the actin response to extracellular signals, which undoubtedly involve a different assemblage of signaling factors.
One very promising hint of in vivo control that has come from in vitro studies is the role of phosphoinositides, particularly phosphatidylinositol (4,5) bisphosphate (PI(4,5)P2),1 as potential signaling intermediates (Janmey, 1994). PI(4,5)P2 interacts with several actin binding proteins. For example, PI(4,5)P2 binds profilin and dissociates actin from profilin/ actin complex (Lassing and Lindberg, 1985); it also inhibits gelsolin functions, including severing, nucleation, and actin monomer binding (Janmey and Stossel, 1989). Studies carried out in intact cells have also implicated phosphoinositides in signaling from activated cell surface receptors to intracellular actin cytoskeletal responses, making them attractive places to look for the linkage. The level of phosphoinositide synthesis, for example, has been correlated with the amount of actin assembled in basophilic cells (Apgar, 1995) and platelets (Hartwig et al., 1995). Phosphoinositide 3-kinase (PI 3-kinase), a lipid kinase that phosphorylates the D-3 position of phosphoinositides, is known to be required for PDGF-induced membrane ruffling and chemotaxis in fibroblasts (Kundra et al., 1994; Wennstrom et al., 1994; Wymann and Arcaro, 1994; Nobes et al., 1995). Though lipid binding has been studied extensively in individual actin binding proteins, it is not clear which phosphoinositides are able to modulate actin assembly in cells. Only recently has the specificity been studied in permeabilized platelets (Hartwig et al., 1995, 1996).
Another source of linkage between signal transduction pathways and actin assembly are the small GTP-binding proteins (G proteins) of the Rho family (Machesky and Hall, 1996; Ridley, 1996; Zigmond, 1996). This family, including Rac, Rho, and Cdc42, are Ras-like small GTPases that act as molecular switches cycling from GTP-bound active forms to GDP-bound inactive forms (Bourne et al., 1991; Hall, 1992). The GTP/GDP cycle is mainly controlled by GTPase-activating proteins, guanine-nucleotide exchange factors (GEFs), and guanine-nucleotide dissociation inhibitors (GDIs) (Boguski and McCormick, 1993). When injected into fibroblasts in a constitutively active form, each member of the Rho family elicits distinct morphological changes that involve remodeling of the actin cytoskeleton (Ridley and Hall, 1992; Ridley et al., 1992; Nobes and Hall, 1995). In yeast cells, Cdc42 is required for polarized cell growth and bud formation, two actin-dependent processes (Chant, 1994). Despite the central role postulated for them, it is not clear how these small G proteins are activated or how they regulate actin assembly. Surprisingly, several target-binding domain mutants of these G proteins do not interfere with their ability to induce actin assembly (Lamarche et al., 1996; Tapon and Hall, 1997). G proteins are usually activated by GEFs, but nothing is known about where this occurs. All GEFs for the Rho family are soluble proteins (Cerione and Zheng, 1996), but the function that G proteins execute is membrane associated. It is not clear whether G proteins are activated in the cytosol before binding to the membrane or whether they are associated with the membrane first, and then activated there.
Although both phosphoinositides and small G proteins seem to be involved in regulating the actin cytoskeleton, it is not clear whether these two signaling pathways are related to each other. Several studies have suggested a role of G proteins in lipid kinase activation. Phosphoinositide kinases are associated with activated G proteins in both cell lysates and tissue homogenates (Tolias et al., 1995). A phosphatidylinositol 4-phosphate 5-kinase is associated with Rho in Swiss 3T3 cells (Ren et al., 1996). Activation of PI 3-kinase requires Rho in platelets (Zhang et al., 1993). However, additional evidence suggests that lipid kinases may be involved in controlling G proteins. In fibroblast cells, PI 3-kinase is required for PDGF-stimulated Rac activation (Hawkins et al., 1995) and constitutively active PI 3-kinase is able to activate Rac/Rho (Reif et al., 1996). In addition, activation of small G proteins appears to correlate with the increase in phosphoinositide synthesis. The level of PI(4,5)P2 is increased in response to the activation of Rho in Swiss 3T3 cells (Chong et al., 1994) and Rac in platelets (Hartwig et al., 1995). These mixed results make it difficult to draw conclusive lines among the two signaling molecules and the actin cytoskeleton.
Xenopus egg extracts have proven to be a powerful cell-free system for reconstituting heterogeneous and complex reactions including nuclear assembly and disassembly (Newport and Spann, 1987), chromosome condensation (Ohsumi et al., 1993), spindle assembly (Sawin et al., 1992), DNA replication (Hutchison et al., 1989), and the control of cell cycle (Murray and Kirschner, 1989). Recently, the actin-based motility of Listeria has also been reconstituted in egg extracts (Theriot et al., 1994; Marchand et al., 1995), suggesting that the extracts contain all the necessary components in somatic cells that are required to modulate actin assembly. However, egg extracts have not been used to reconstitute the interplay of membrane-based actin assembly with soluble components. It should be possible to reconstitute membrane-dependent cytoskeletal responses by including endogenous membranes or providing exogenous membranes to the extracts. We report on the success of that approach here and its use in unraveling aspects of the physiological control of actin polymerization.
Materials and Methods
Preparation of Extracts
Low speed mitotic extracts from unfertilized eggs were prepared from Xenopus eggs in a buffer (XB) containing 10 mM Hepes, pH 7.7, 100 mM KCl, 2 mM MgCl2, 0.1 mM CaCl2, 5 mM EGTA, 50 mM sucrose, 1 mM DTT, and protease inhibitors as described (Murray, 1991), except that cytochalasin was omitted in all steps. Extracts were either used immediately or supplemented with 200 mM sucrose and frozen in liquid nitrogen.
High speed supernatants were prepared from low speed extracts according to King et al. (1995). Briefly, low speed extracts were diluted 10-fold in XB and then centrifuged at 400,000 g for 1 h. The clear supernatant was carefully removed and reconcentrated to its original volume in Centriprep-3 or -10 concentrators (Amicon Corp., Danvers, MA), giving a final concentration of 40–50 mg/ml. The membrane pellet was resuspended in XB to the same original volume and broken up in a Dounce homogenizer. All extract fractions were supplemented with energy regenerating mix containing 1 mM ATP, 1.25 mM MgCl2, 7.5 mM creatine phosphate, and 0.1 mM EGTA, and stored at −80°C. Actin concentration is ∼2 mg/ ml in low speed extracts and 1 mg/ml in high speed supernatants as estimated by Western blot.
Preparation of Lipid Vesicles
All phospholipids were obtained from Sigma Chemical Co. (St. Louis, MO) or Avanti Polar Lipids (Alabaster, AL), except PI(3,4)P2 and PI(3,4,5)P3, which were gifts from Dr. Ching-Shih Chen (University of Kentucky, Lexington, KY). Lipids were dissolved in chloroform and stored at −80°C. Lipid vesicles were prepared as follows. Equal amounts of phosphatidylcholine (PC) and phosphatidylinositol (PI) were mixed with or without each phosphoinositide in chloroform and dried in air. The dried lipid mixture was resuspended in a lipid buffer (20 mM Hepes, pH 7.7, 1 mM EDTA) to a final concentration of 1 mM. The mixture was then sonicated in a water bath with a W-385 ultrasonic processor (Heat Systems Inc., Farmingdale, NY) at its maximum power (475 Watts) for 5 min.
Video Microscopy and Image Analysis
Extract samples were viewed with a 63×/1.4 NA plan-apochromatic objective on a Zeiss Axiovert 135 microscope (Carl Zeiss, Inc., Thornwood, NY). A multi-band pass dichroic and emission filter set (Chroma Technology Co., Brattleboro, VT) was used for fluorescence observation and a dual filter wheel (Ludl, Hawthorne, NY) was used to choose appropriate excitation and neutral density filters. All images were collected with a cooled charge coupled device (CCD; Photometrics Inc., Tucson, AZ) and stored digitally on a hard disk. Device control, image acquisition, and data analysis were carried out using Metamorph 2.0 (Universal Imaging Corp., West Chester, PA) software running on an IBM-compatible computer. To look for the relative position of lipid vesicles, the nitrobenzoxadiazole image was taken immediately after the rhodamine image with a time delay estimated at ∼1 s. The two images were then pseudocolored and overlaid.
Visual Assay for Actin Polymerization
All reactions were carried out at room temperature. For experiments using low speed extracts, 1 μl rhodamine actin (0.2–0.4 mg/ml) was mixed with 6-μl extracts (either concentrated or diluted twofold with XB). Rhodamine actin was prepared according to Symons and Mitchison (1991) using purified rabbit skeletal muscle actin and tetramethylrhodamine-iodoacetamide rhodamine (Molecular Probes, Inc., Eugene, OR). After the addition of either GTPγS or vanadate (1 μl) to the extract to a final concentration of 1 mM, 5 μl of the reaction mixture was squashed between two coverslips. The coverslips were then mounted on the microscope stage for imaging as described above. For experiments using phosphoinositides, 3 μl high speed supernatants were diluted with 3 μl XB and supplemented with 1 μl rhodamine actin. 1 μl lipid vesicles (1 mM) were then added.
Pyrene Actin Assay
Pyrene actin was labeled according to an established protocol (Kouyama and Mihashi, 1981) using rabbit skeletal muscle actin and N-(1-pyrene)iodoacetamide (Molecular Probes, Inc.). Due to the high background fluorescence in the extracts, high speed supernatants were diluted three- to fourfold in XB to a final concentration of ∼10 mg/ml. 3–5 min after 100 μl diluted high speed supernatants were mixed with pyrene actin in a quartz cuvette, 5-μl lipid vesicles (1 mM) were added. Pyrene fluorescence was measured at 407 nm with excitation at 365 nm in a Aminco-Bowman Series 2 Luminescence Spectrometer (SLM-Aminco Inc., Rochester, NY) with 4-nm slit width. Kinetics of the reaction was analyzed and plotted in Origin (Microcal Software, Inc., Northampton, MA).
Purification of Recombinant Proteins
Small G proteins were expressed in COS cells as glutathione-S-transferase (GST) fusion proteins. COS cells were transfected using the calcium phosphate method and allowed at least 60 h to express fusion proteins. Protein purification followed (Heyworth et al., 1993) with some modifications. Briefly, cells were broken by sonication in a buffer with 10 mM HEPES, pH 7.3, 100 mM KCl, 3 mM NaCl, 3.5 mM MgCl2, 1 mM EGTA, 1 mM DTT, and protease inhibitors. Nuclei and cell debris were then removed by centrifugation at 10,000 g·min. The supernatant was then centrifuged at 200,000 g·h to pellet the membrane. Except for Cdc42V12C189S, the membrane for all other G proteins was solubilized in an extraction buffer (25 mM Tris-HCl, pH 7.7, 50 mM NaCl, 1 mM EDTA, 5 mM MgCl2, 1 mM DTT, 1% cholate, and protease inhibitors). After a clarifying spin at 200,000 g·h, the supernatant was then incubated with glutathione-Sepharose beads (Pharmacia Biotech, Piscataway, NJ) at 4°C overnight. After wash, the beads were then eluted with an elution buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 5 mM MgCl2, 2.5 mM CaCl2, 5 mM DTT, 0.1% cholate, and 10 mM reduced glutathione). Eluted proteins were cleaved with thrombin and analyzed by SDS-PAGE. Cdc42V12C189S was precipitated using glutathione beads directly from the supernatant after centrifugation at 200,000 g·h. RhoGDI was expressed in Escherichia coli as a GST fusion protein and was purified using glutathione beads.
To activate them with GTPγS, processed G proteins were incubated with 20 mM EDTA and 40 μM GTPγS for 15 min at 30°C. GTPγS was then locked in place by adding 20 mM extra MgCl2.
Results
GTPγS and Sodium Orthovanadate Each Induce Localized Actin Polymerization and Vesicle Motility in Low Speed Egg Extracts
As agonists in several signaling systems, GTPγS and sodium orthovanadate were added to low speed extracts (4,000 g·h) of Xenopus eggs in an attempt to induce actin assembly. To visualize actin, we used a rhodamine actin-based visual assay, which had been used to study Listeria-induced actin assembly in extracts (Theriot et al., 1994; Marchand et al., 1995). In this assay, a trace amount of rhodamine actin was added to mark the behavior of endogenous actin. Despite the presence of a large number of heterogeneous endogenous vesicles in low speed extracts, the fluorescence of rhodamine actin was uniform across the field, signifying no apparent assembly (Fig. 1 A). Localized foci of actin assembly could be induced by adding 1 mM GTPγS or sodium orthovanadate to the extracts. Approximately 5 min after the addition of either compound, bright fluorescent foci appeared, some in the shape of comet tails (Fig. 1). Actin assembly occurred only around a subset of the phase-dense vesicles, which were propelled through the cytoplasm along gently curved trajectories (Fig. 1 E). The average velocity was 10.0 μm/min (SEM = 1.3; measured from the vanadate experiments), similar to that observed with Listeria (Theriot et al., 1994; Marchand et al., 1995). Cytochalasin D (20 μM) completely inhibited the formation of foci and comet tails, indicating that motility and assembly reflected the accumulation of polymerized actin (data not shown).
Figure 1.
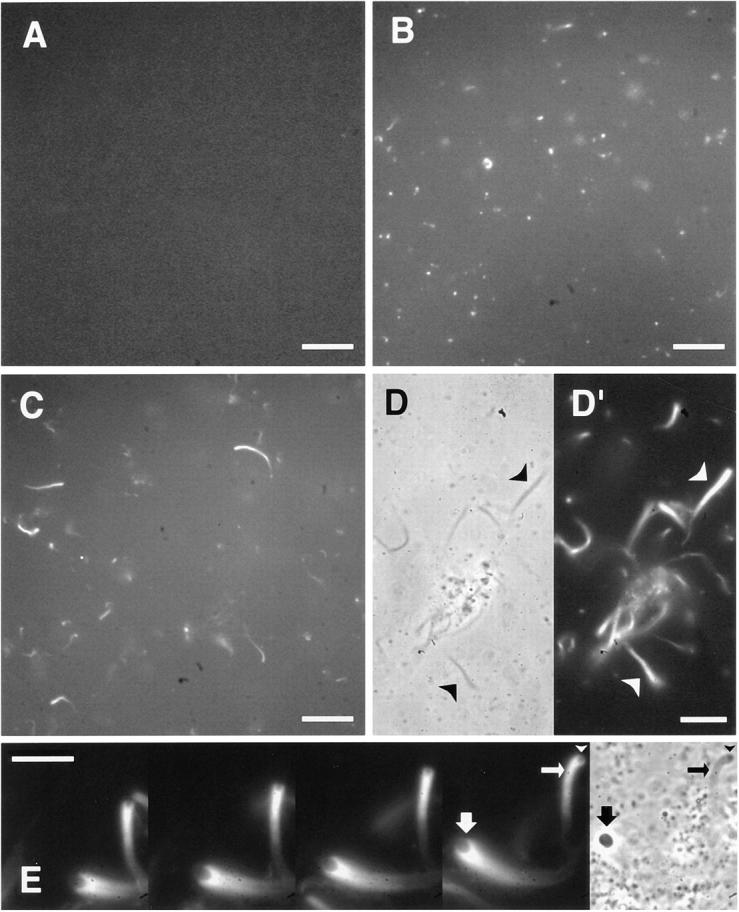
Localized actin assembly induced by GTPγS or sodium orthovanadate in low speed extracts as viewed with rhodamine actin. (A) Control extracts; (B) extracts with 1 mM GTPγS; (C–E) extracts with 1 mM sodium orthovanadate. The first four sequential frames 30 s apart in E show the motility of actin comet tails and associated vesicles. Corresponding phase images are shown in D and the last frame in E. Actin tails appear as phase-dense structures (D, arrowheads). Note in E the presence of a phase-dense vesicle (arrowheads) in front of the dark actin tail (small arrows) that has moved. Rhodamine fluorescence clearly delineates the contour of an associated vesicle (large arrows). Bars: (A–C) 20 μm; (D and E) 10 μm.
Comet tails appeared as phase-dense structures under the phase contrast microscope. These comet tails often had distinct vesicles at one end. When the tails were large (up to 4 μm in width), rhodamine fluorescence clearly delineated the contour of associated vesicles (Fig. 1 E). While many phase-dense vesicles showed no association with actin, the majority of comet tails were associated with visible vesicles of variable sizes. Only when the tails were small was it difficult to find any vesicles at their ends. After the phase-dense vesicles were removed from extracts by sedimentation at 400,000 g·h, the remaining high speed supernatants did not induce actin assembly in the presence of GTPγS or vanadate. When the vesicle fraction was added back, actin foci and comet tails reformed (data not shown). The intimate physical association of these phase dense vesicles with foci and comet tails suggests that actin assembly is nucleated from the vesicles.
Lipid Vesicles Containing Purified Phosphoinositides Can Induce Localized Actin Polymerization in Vesicle-free Extracts
Membrane vesicles seem indispensable for actin polymerization in this system since GTPγS is unable to induce actin assembly in high speed supernatants lacking endogenous vesicles. Though it is possible that these membrane vesicles simply provide a hydrophobic docking surface for activated G proteins, some specific constituents of the membrane, such as modified phospholipids or membrane-associated proteins, might be required for the induction of actin assembly. To examine the role of specific phospholipids in regulating actin assembly, we prepared synthetic lipid vesicles from purified phospholipids and tested their ability to induce actin assembly in vesicle-free supernatants.
Lipid vesicles of diameter estimated to be <200 nm (Janmey and Stossel, 1989) were constructed from PC and PI. When added to high speed supernatants containing labeled actin, they did not induce localized actin assembly (Fig. 2 A). However, inclusion of 33% phosphatidylinositol (3,4,5) trisphosphate (PI(3,4,5)P3) in PC/PI vesicles stimulated the formation of actin foci and comet tails (Fig. 2 B). In a typical field of 10,000 μm2, there were ∼41.2 ± 10 foci and 4.0 ± 0.5 comet tails with an average length of 4.3 ± 0.2 μm. These comet tails moved at a rate of 5.4 μm/ min (SEM = 1.1) (Fig. 2 G). PI(3,4,5)P3-containing lipid vesicles themselves could be visualized by including in them nitrobenzoxadiazole (NBD)-tagged PC (4%). The vast majority of rhodamine signals were associated with the tagged vesicles (data not shown). Strikingly, each comet tail had only one NBD-vesicle attached to its end (Fig. 2 H). All assembly could be blocked completely by cytochalasin D.
Figure 2.



Induction of actin assembly in high speed supernatants by lipid vesicles containing different phosphoinositides. (A–F) Rhodamine images show the difference of actin assembly. High speed supernatants were mixed with PC/PI vesicles containing no phosphoinositides (A), 33% PI(3,4,5)P3 (B), 33% PI(4,5)P2 (C), 33% PI(3,4)P2 (D), 33% PI(4)P (E), or 33% PI(3)P (F). (G) Two moving comet tails induced by PI(3,4,5)P3 are shown in sequential frames 30 s apart. (H) Four examples of color-overlaid images. Note that NBD-vesicles (green) containing PI(3,4,5)P3 are associated with rhodamine-actin comet tails (red). Bars: (A–G) 10 μm; (H) 5 μm.
The induction of actin foci was dependent on the type of phosphoinositide used. Vesicles containing PI(4,5)P2 produced both foci (19.7 ± 4.4) and comet tails (1.5 ± 0.6) (Fig. 2 C) as did vesicles containing PI(3,4,5)P3. However, vesicles containing phosphatidylinositol (3,4) bisphosphate or phosphatidylinositol (3) phosphate (PI(3)P) produced fewer foci (6.5 ± 0.8 or 9.5 ± 0.7 per 10,000 μm2 field) and no comet tails (Fig. 2, D and F). Vesicles containing phosphatidylinositol (4) phosphate (PI(4)P) induced foci (35.9 ± 7.5 per field) but fewer tails (1.2 ± 0.6 per field) (Fig. 2 E).
Quantitative Pyrene Actin Assays for Assembly Induced by Phosphoinositides
Although the visual assay gave a reproducible qualitative picture of the effectiveness of different phosphoinositides, it was not readily quantifiable. Vesicles varied in size, as did the amount of actin associated with them and there was no simple measure of the volume in the field to be used for comparison. To give a more accurate measurement of the polymerized actin, we used a well-established pyrene actin assay (Cooper et al., 1983). Pyrene actin has increased fluorescence when it is incorporated into filamentous actin (F-actin). Therefore, an increase in fluorescence intensity is a direct measure of actin polymerization and the kinetics can be easily monitored in a spectrofluorometer.
Like purified actin, pyrene actin assembles spontaneously into filaments in high salt buffers, with a lag phase followed by a nearly linear increase (Cooper et al., 1983). After the addition of 1 μM pyrene actin to high speed supernatants containing physiological salts, there was no appreciable increase in fluorescence for at least 1 h (data not shown). The magnitude of pyrene signal in the supernatant did not differ much from that in a low salt buffer, indicating that most of the actin in the extract remained unpolymerized. Under the condition used in the experiments, nearly 90% of the basal fluorescence signal came from the extracts diluted to 10 mg/ml, and 10% was due to pyrene actin (1 μM final concentration).
Kinetics of Actin Assembly Induced by Phosphoinositides.
When vesicles containing only PC/PI were added to high speed supernatants, the pyrene signal again remained unchanged after an immediate 5–10% increase due to the light scattering of the lipids (Fig. 3 A). However, the addition of vesicles containing only 4% PI(3,4,5)P3 or PI(4,5)P2 to the supernatant caused a rapid increase in fluorescence (Fig. 3 A) during the first 3–5 min. Within 5–10 min, the fluorescence was at 10× the original signal. At the peak, we estimated that ∼10% of total actin in the extract had been incorporated into F-actin, after normalizing to the fluorescence intensity from extracts treated with 2.5 μM phalloidin. The kinetics and the magnitude of the response were similar for both phosphoinositides. The increase in pyrene signal could be completely inhibited by 5 μM cytochalasin D (Fig. 3 C).
Figure 3.

Characterization of phosphoinositide-induced actin assembly in high speed supernatants using pyrene actin. (A) Time course of change in pyrene fluorescence in high speed supernatants treated with PC/PI alone (dotted line), or PC/PI vesicles containing 4% PI(3,4,5)P3 (solid line), or 4% PI(4,5)P2 (dashed line). The lipid concentration was 50 μM. The arrow indicates the time when the lipid was added. (B) Comparison of the inductive activity among different phosphoinositides in PC/PI vesicles (4% phosphoinositides in 50 μM total lipids) as measured by the initial rate of actin assembly and the maximum F-actin at the peak. All activities are normalized to that of PI(4,5)P2. (C) Inhibition of PI(4,5)P2-induced actin assembly in high speed supernatants by cytochalasin D or a peptide (QRLFQVKGRR) derived from gelsolin. Each reaction included 50-μM lipid vesicles containing 4% PI(4,5)P2. Inhibition of PI(3,4,5)P3-induced activity is similar and is not shown here. (D) Dose response of actin assembly to phosphoinositides. (a) F-actin induced by 50-μM PC/PI vesicles containing different percentages of PI(3,4,5)P3 (solid line) or PI(4,5)P2 (dashed line). (b) F-actin induced by vesicles containing 4% PI(4,5)P2 in high speed supernatants at various total lipid concentrations (expressed as total PI(4,5)P2). The data fit into a linear curve (dashed line). The initial rate has very similar dose– response profiles (not shown). All activities are normalized to that of phosphoinositides at 4% in 50 μM total lipids. All data in B–D are expressed as mean ± SEM calculated from at least two sets of normalized data.
The role of these specific phosphoinositides to induce actin assembly was confirmed by incubation of extracts with a lipid-binding peptide (QRLFQVKGRR) derived from gelsolin. This peptide has been shown to bind phosphoinositides and can compete for phosphoinositide-binding with gelsolin (Janmey et al., 1992). The peptide completely inhibited actin assembly induced by PI(4,5)P2 at 20 μM with a half inhibitory concentration of 5–10 μM (Fig. 3 C), consistent with its estimated micromolar affinity with PI(4,5)P2 micelles (Janmey et al., 1992).
Specificity of Phosphoinositides in Actin Assembly.
Vesicles containing three other phosphoinositides, 4% PI(3,4,)P2, (an isomer of PI(4,5)P2), 4% PI(4)P, or 4% PI(3)P induced actin assembly at very slow rates and to a much lower extent when compared with PI(4,5)P2 (Fig. 3 B). The rate of actin assembly for PI(3,4)P2 was only 5% that for PI(4,5)P2; the total amount of F-actin after 5 min was 29% that of PI(4,5)P2. These results are consistent with those from the visual assay.
Dose Response of Actin Assembly to Phosphoinositides.
To ascertain whether the effective levels of phosphoinositides in vesicles were physiologically meaningful, we varied the phosphoinositide concentration in the vesicles and compared the initial rate of actin polymerization and the total amount of F-actin at the peak. Fig. 3 D, a, shows the dose response to PI(3,4,5)P3 and PI(4,5)P2 at a total lipid concentration of 50 μM. Vesicles containing only 1% of PI(3,4,5)P3 or 1% PI(4,5)P2 gave measurable stimulation of assembly. The extent of assembly increased with the percentage of phosphoinositides in lipid vesicles to a maximum at 12% for PI(3,4,5)P3 and 4% for PI(4,5)P2. Above 4%, the activity induced by PI(4,5)P2 plateaued. Interestingly, a higher concentration of PI(3,4,5)P3 (33%) reduced the activity to about one third of that at 12%.
These results suggested that there might be a limiting component in the extract, depleted above 12% PI(3,4,5)P3 or 4% PI(4,5)P2 at 50 mM total lipids. We therefore varied the concentration of total lipids, holding the phosphoinositide composition at 4%. F-actin increased linearly with increasing total lipids, from 3.8% of the total actin in the extract at 20 μM total lipids to 15.8% at 100 μM lipids (Fig. 3 D, b). The initial rate also increased linearly (data not shown). This argues against a shortage of cytoplasmic factors and suggests that the plateau of effects at 12% or more PI(4,5)P2 or PI(3,4,5)P3 reflects limits on each vesicle.
GTPγS Broadens the Specificity of Phosphoinositides for Actin Assembly
The effect of GTPγS in stimulating actin assembly off endogenous vesicles suggested that it might be able to overcome unfavorable lipid or protein composition. We therefore retested the lipid specificity for actin assembly in the presence of GTPγS. Although PI(3,4)P2-containing vesicles did not induce actin assembly in vesicle-free supernatants, incubation with 100 μM GTPγS before the addition of PI(3,4)P2 stimulated these vesicles to induce actin assembly. In this case, there was a noticeable lag of ∼100 s between the addition of lipids and the beginning of increase in fluorescence (Fig. 4). Actin assembly was sensitive to the GTPγS concentration. Concentrations below 1 μM GTPγS had no stimulatory effect, but at 10 μM GTPγS, PI(3,4)P2 was as effective as PI(4,5)P2 (Fig. 4 B). In control experiments, GTPγS (100 μM) alone did not stimulate actin assembly (data not shown).
Figure 4.

Effects of GTPγS on phosphoinositide-induced actin assembly in high speed supernatants. (A) Time course of actin assembly induced by lipid vesicles containing 25% PI(3,4)P2 or PI(4,5)P2 followed by pyrene fluorescence. High speed supernatants were treated with (solid line) or without (dashed line) 0.1 mM GTPγS for 5 min, and then stimulated with lipid vesicles (50 μM). (B) Comparison of the effect of GTPγS on different lipid vesicles. Experiments were done as in A and lipid vesicles contained 25% of each phosphoinositide. All activities were normalized to that of PI(4,5)P2 in the absence of GTPγS. (C) Effects of GTPγS dose on phosphoinositide-induced actin assembly. Again, experiments were done as in A.
The stimulation by GTPγS was not limited to PI(3,4)P2-containing vesicles (Fig. 4 B). Both the rate and amount of F-actin formation increased several fold in the presence of GTPγS for vesicles containing PI(4)P or PI(3)P. For vesicles containing PI(4,5)P2 or PI(3,4,5)P3, the rate was almost doubled by 10 μM GTPγS, and the effect was dependent on the dose of GTPγS (Fig. 4 C). The effect of GTPγS was specific for phosphoinositides, since there was very little stimulatory effect of GTPγS on PC/PI vesicles (Fig. 4 B). These results suggest that some but not all of the lipid specificity can be suppressed by GTPγS.
It is worth noting that GTPγS, at concentrations >100 μM, also induced a delayed fluorescence increase followed by a drop to almost the basal line (Fig. 4 A, right). This was due to the aggregation of the added lipid vesicles because we noticed a visible lipid aggregate in the reaction mixture and the loss of NBD-PC signal after the spike appeared. We speculated that an actin-based contraction and/or gelation system was activated by a G protein at high GTPγS doses.
Involvement of the Rho Family Small G Proteins in Phosphoinositide-induced Actin Assembly
GTPγS is an agonist with a broad spectrum and can activate any GTP-binding proteins in the extracts. To determine whether the Rho family small G proteins were required for GTPγS-induced actin assembly, we tested whether the activity was sensitive to RhoGDI, a protein that binds preferentially to GDP-bound proteins and prevents nucleotide exchange (Boguski and McCormick, 1993; Takai et al., 1993).
To test whether RhoGDI could inhibit actin assembly from the endogenous vesicles stimulated by GTPγS, recombinant RhoGDI was purified from E. coli and incubated in high speed supernatants supplemented with endogenous vesicles before the addition of GTPγS. Actin assembly was completely abolished in the presence of RhoGDI at concentrations between 0.5 and 2.5 μM (Fig. 5 A). Since RhoGDI interacts with all three members of the family (Takai et al., 1993), we can only conclude that some or all G proteins of the Rho family are involved in GTPγS-induced actin assembly in egg extracts.
Figure 5.


Inhibition of actin assembly by RhoGDI. (A) Inhibition of vesicle-dependent actin assembly in low speed extracts as viewed with rhodamine actin. Endogenous vesicles were stimulated with 100 μM GTPγS in extracts that were preincubated without (a) or with (b) 2.5 μM recombinant RhoGDI for 5 min. (B) Inhibition of phosphoinositide-induced actin assembly. Actin assembly induced by vesicles containing 25% PI(3,4)P2 (a and c) or 25% PI(4,5)P2 (b and d) in the absence (solid line) or presence (dashed line) of 2 μM RhoGDI. In a and c, high speed supernatants were preincubated with 10 μM GTPγS and RhoGDI for 5 min. In b and d, GTPγS was omitted. Lipid vesicles were then added to 50 μM final concentration. a and b are time courses of actin assembly monitored with pyrene actin. c and d show the dose effect of RhoGDI.
We further tested whether RhoGDI could block GTPγS- stimulated actin assembly by a variety of phosphoinositides in high speed supernatants. RhoGDI (2 μM) abolished the ability of GTPγS to stimulate PI(3,4)P2-induced actin assembly (Fig. 5 B, a). The dose for half maximum inhibition by RhoGDI was between 80 and 400 nM (Fig. 5 B, c).
Surprisingly, RhoGDI inhibited not only the enhancement of PI(4,5)P2-dependent activity by GTPγS, but the basal activity as well. Fig. 5 B, b shows that F-actin-inducing activity of PI(4,5)P2 in the absence of GTPγS was blocked by RhoGDI (2 μM). The inhibition was dependent on the concentration of RhoGDI, with similar effective doses for the GTPγS-stimulated assembly of PI(3,4)P2 vesicles (Fig. 5 B, d). This result suggests that the Rho family small G proteins may play a role downstream of all phosphoinositides in regulating actin assembly.
Special Role of Cdc42
Cdc42 Is Required for PI(4,5)P2-induced Actin Assembly.
To characterize the small G proteins that were required for PI(4,5)P2-induced activity, we first tested the dominant active forms of small G proteins of the Rho family and asked whether they could rescue RhoGDI inhibition. Dominant active Cdc42V12 was purified from COS cell membranes and added directly to the extracts without loading with GTPγS. Fig. 6 A shows that Cdc42V12 restored F-actin, inducing activity of PI(4,5)P2 in a dose- dependent manner. In the absence of GTPγS, Cdc42V12 itself did not induce any actin assembly in the extracts. We further tested whether Cdc42 was necessary using Cdc42N17, a dominant negative form. As shown in Fig. 6 B, 0.17 μM Cdc42N17 blocked PI(4,5)P2-induced actin assembly. In contrast, the dominant negative form RacN17 was not able to do so, even when present at a much higher concentration (1.67 μM). These results suggest that Cdc42 may be both necessary and sufficient for PI(4,5)P2-induced actin polymerization. Interestingly, Cdc42N17 only partially inhibited PI(3,4)P2- and PI(4,5)P2-induced actin assembly in the presence of GTPγS (Fig. 6 C). This suggests that GTPγS may activate other small G proteins in this system.
Figure 6.

Involvement of Cdc42 in PI(4,5)P2-induced actin assembly in high speed supernatants. (A) Cdc42V12 rescues the inhibition of PI(4,5)P2-induced actin assembly by RhoGDI. Cdc42V12 without GTPγS was added to high speed supernatants along with RhoGDI (0.4 μM). After a 5-min incubation, vesicles containing 25% PI(4,5)P2 were added to a final concentration of 60 μM. (B) Inhibition of PI(4,5)P2-induced actin assembly in high speed supernatants by the dominant negative form of Cdc42, but not that of Rac. High speed supernatants were preincubated with either Cdc42N17 or RacN17 for 5 min, and then stimulated with 60 μM PI(4,5)P2-containing vesicles (12%). (C) Partial inhibition of GTPγS-stimulated actin assembly by Cdc42N17 in high speed supernatants. Extracts were incubated with or without 0.34 μM Cdc42N17 for 5 min before the addition of 10 μM GTPγS and 60 μM phosphoinositide-containing vesicles (12% for PI(4,5)P2 and 25% for PI(3,4)P2). In all experiments, actin assembly was monitored using pyrene actin and the activities were normalized to the controls.
Under Special Conditions, Activated Cdc42 Can Induce Actin Assembly in Vesicle-free Extracts.
To test whether Cdc42 alone could induce actin assembly, both dominant active Cdc42V12 and RacV12 were added to the extracts. Neither had an effect on actin assembly (data not shown). We reasoned that there might not be sufficient exchange activities present in the extracts to activate the proteins with GTP. We then loaded Cdc42V12 and RacV12 with GTPγS in the presence of EDTA before adding them to high speed supernatants. As shown in Fig. 7 A, GTPγS-Cdc42 induced a significant amount of F-actin in a lipid- independent manner, with an assembly rate more than half of that induced by PI(4,5)P2. In contrast, GTPγS-RacV12 did not generate any noticeable amount of F-actin. The free GTPγS carried over from the loading reaction was diluted to <4 μM and did not have any effect on its own (Fig. 7 A, Control). In the visual assay using rhodamine actin, vesicle-free extracts supplemented with GTPγS-Cdc42V12 contained many rhodamine foci as well as tail structures similar to those observed in the low speed extract (Fig. 7 B). These structures were completely absent in GTPγS-RacV12-treated extracts. Actin assembly stimulated by activated Cdc42V12 requires prenylated proteins. Cdc42V12C189S, a CAAX box mutant devoid of prenylation, was unable to stimulate actin assembly even when charged with GTPγS (Fig. 7 A).
Figure 7.


Actin assembly induced by GTPγS-charged Cdc42 in high speed supernatants. (A) Time course of actin assembly in high speed supernatants incubated with 0.17 μM Cdc42V12-GTPγS (solid line), 0.83 μM RacV12-GTPγS (dashed line), buffer (dotted line) or 0.17 μM Cdc42V12C189S-GTPγS (dotted and dashed line). G proteins were preloaded with 40 μM GTPγS, and then added to high speed supernatants containing pyrene actin. (B) As in A, but viewed with rhodamine actin.
Discussion
Molecular and pharmacological studies in intact cells have suggested many signaling pathways that regulate the actin cytoskeleton under physiological conditions (Machesky and Hall, 1996; Ridley, 1996; Zigmond, 1996). Recently, permeabilized cells have also been used to study actin assembly (Hartwig et al., 1995; Tardif et al., 1995; Barkalow et al., 1996). Due to the intimate association of actin assembly with the plasma membrane (Symons and Mitchison, 1991), it is difficult to use conventional biochemical approaches, which have been developed to study the cytoskeleton in soluble systems, to study membrane-dependent regulation of the actin cytoskeleton.
In this report, we have demonstrated the use of Xenopus egg extracts to reconstitute membrane-dependent actin assembly and to study both actin assembly and aspects of signal transduction. Endogenous membranes can be stimulated to nucleate actin assembly in the presence of GTPγS or sodium orthovanadate. We have been able to define some of the important functional membrane components and distinguish them from the soluble factors contributing to actin assembly. We also demonstrate a key role for Cdc42 in mediating phosphoinositide-stimulated actin assembly in this cell-free system. Finally, as described below, we suggest a regulatory pathway involving phosphoinositides in both the catalysis of guanine-nucleotide exchange (directly or indirectly) and the localization of activated small G proteins.
We have shown that PI(4,5)P2 and PI(3,4,5)P3 are able to induce actin assembly in high speed supernatants. Since other phosphoinositide isoforms and phospholipids are ineffective, it cannot be simply charge that is important for PI(4,5)P2 and PI(3,4,5)P3 in regulating actin assembly. The specificity of phosphoinositides is likely to be universally applicable because high speed extracts contain a physiological mixture of well balanced actin binding proteins under which actin polymerization is globally inhibited but locally activatable. For this reason, we propose that PI(4,5)P2 and PI(3,4,5)P3 are important membrane signals for actin assembly in vivo. At present we are unable to determine whether the apparently equal effectiveness of PI(3,4,5)P3 and PI(4,5)P2 in inducing actin assembly is due to the fact that one may be converted to another in the egg extract. Wortmannin, a potent PI 3-kinase inhibitor (Arcaro and Wymann, 1993), does not interfere with PI(4,5)P2- induced actin assembly and we have not seen phosphate incorporation into PI(4,5)P2 in the extracts (data not shown). These data suggest that PI(4,5)P2 itself is sufficient. However, the presence of a wortmannin-insensitive PI 3-kinase could not be ruled out, and the conversion of a small amount of PI(4,5)P2 into PI(3,4,5)P3 would be hard to detect. On the other hand, PI(3,4,5)P3 could be converted back to PI(4,5)P2 if a 3-phosphatase were present. It is also possible that both phosphoinositides are equally potent in the extract.
The levels of PI(3,4,5)P3 and PI(4,5)P2 needed to generate actin assembly in egg extracts are surprisingly low. As little as 1% will induce polymerization. The overall level of PI(4,5)P2 and PI(3,4,5)P3 in cells is ∼0.4% and <0.01%, respectively (Auger et al., 1989). Since it is likely that their amount is not uniformly distributed in the cell, our results suggest that local concentrations of PI(3,4,5)P3 and PI(4,5)P2 could be high enough to drive actin assembly in cells. Actin assembly induced by these phosphoinositides is saturable at relatively low levels of PI(3,4,5)P3 and PI(4,5)P2 and this is not due to saturation of downstream factors. Saturation has also been observed for the inhibition of gelsolin (Janmey and Stossel, 1989) and the activation of β-adrenergic receptor kinase (Pitcher et al., 1995). This is possibly due to the limited surface area of the vesicle. Alternatively, phosphoinositides in a PC/PI bilayer might form microdomains or boundaries within the bilayer, affecting their interaction with downstream proteins. The extracts also contain excess downstream factors because actin polymerization increases linearly with the concentration of lipid vesicles up to 100 μM. If the lipid/protein ratio used in our experiments was close to that in cells, we would conclude that Xenopus eggs and perhaps somatic cells would never exhaust their downstream machinery for nucleating actin polymerization.
Small G proteins of the Rho family are also able to induce actin assembly in Xenopus extracts. GTPγS can stimulate both endogenous membranes and purified lipid vesicles to assemble actin. RhoGDI blocks not only the GTPγS- induced actin assembly, but also phosphoinositide-induced activity. All these results can be explained by the participation of the Rho family G proteins. Cdc42V12 rescues RhoGDI inhibition (Fig. 6 A). Dominant negative Cdc42N17, but not RacN17, inhibits phosphoinositide-induced actin assembly (Fig. 6 B). These results suggest that Cdc42 is involved in actin assembly induced by PI(3,4,5)P3 and PI(4,5)P2. Cdc42 also appears to be the target for GTPγS, since Cdc42N17 inhibits GTPγS-stimulated phosphoinositide-dependent actin assembly (Fig. 6). However, the inhibition is partial, suggesting that other members of the Rho family also mediate GTPγS-induced assembly. More strikingly, exogenous Cdc42, when charged with GTPγS, is sufficient to induce actin assembly.
Actin polymerization induced by GTPγS and Cdc42 has been observed in cell lysates from polymorphonuclear leukocytes and Dictyostelium (Zigmond et al., 1997). Bundles and meshworks of F-actin are observed using phalloidin in the low speed lysates from these cells. In our extracts when rhodamine actin is used, the morphology of F-actin induced by GTPγS is similar but has additional features, possibly due to different extract concentrations and assay methods. F-actin does not only assemble into localized foci around endogenous vesicles, but also incorporates into moving comet tails. The comet tails and vesicle movements bear striking similarity to Listeria motility (Theriot et al., 1994; Marchand et al., 1995), endosome rocketing induced by lanthanides in macrophages (Heuser, J.E., and J.H. Morisaki. 1992. Mol. Biol. Cell. 3:172a) and “inductopodia” observed in Aplysia growth cones (Forscher et al., 1992). Conceivably, the mechanism for these actin-based motilities might be very similar.
The effects of phosphoinositides and Rho-family small G proteins on actin assembly in Xenopus egg extracts appear to be dependent on each other. First, phosphoinositide-containing membranes are required for localized actin assembly: (a) GTPγS induces actin assembly only in low speed extracts, but not in extracts depleted of endogenous membranes, presumably containing phosphoinositides; (b) exogenous lipid vesicles containing phosphoinositides induce actin assembly in these vesicle-depleted extracts; (c) only in the presence of exogenous phosphoinositide-containing vesicles does GTPγS induce actin assembly. Second, phosphoinositide-induced assembly requires small G proteins: (a) RhoGDI inhibits GTPγS-induced actin assembly; (b) RhoGDI inhibits phosphoinositide- induced assembly; (c) dominant negative Cdc42 blocks phosphoinositide-induced assembly.
The requirement of phosphoinositides for GTPγS-stimulated actin assembly can be explained by two scenarios: (a) they stimulate an GEF activity to activate G proteins, or (b) they promote translocation of G proteins to the membrane and serve as docking surfaces for activated G proteins to nucleate actin assembly. It has been shown recently that GEF activities are required for GTPγS-stimulated actin assembly in high speed supernatants from polymorphonuclear leukocytes, and anionic phospholipids allow GTPγS to induce actin assembly (Zigmond et al., 1997). However, the latter scenario would explain our results better because we have observed different lipid specificity depending on the nucleotides used (see below). Small G proteins contain a CAAX box at their COOH terminals and are modified by prenylation. Although they may interact with other proteins, prenyl groups are known to be important for protein-membrane association. In the cytosol, the attached prenyl group is embedded in RhoGDI, which keeps G proteins in GDP forms (Boguski and McCormick, 1993). Phosphoinositides have recently been shown to dissociate RhoGDI from the inactive Rac-GDI complex (Chuang et al., 1993). Therefore, the requirement of phosphoinositides in our system is consistent with the model that phosphoinositides translocate G protein to membranes. Endogenous small G proteins are likely to be brought to the membrane by phosphoinositides and activated by nonhydrolyzable GTPγS through basal nucleotide exchange. Actin assembly then occurs from membrane-associated GTPγS-activated G proteins. Further studies will be necessary to investigate the mechanism of this protein translocation and determine whether other cytosolic factors are required.
The requirement of phosphoinositides for actin assembly has another interesting aspect. We have observed very different lipid specificities depending on the nucleotide used. In the presence of endogenous GTP (but in the absence of GTPγS), only extremely specific phosphoinositides, PI(4,5)P2 and PI(3,4,5)P3, induce actin assembly (Fig. 3 B). In contrast, the specificity is broadened when GTPγS is added and PI(3,4)P2, PI(4)P, and PI(3)P are now able to induce actin assembly (Fig. 4 B). A simple explanation for the difference in specificity is that only PI(3,4,5)P3 and PI(4,5)P2, but not other phosphoinositides, stimulate a GEF activity, a physiological means to activate G proteins. In the presence of GTPγS, the GEF activity is no longer required for G protein activation. Lipid specificity is relaxed because phosphoinositides are only needed for the G protein-membrane interaction (Fig. 8). All known GEFs for the Rho family contain the pleckstrin homology (PH) domain (Cerione and Zheng, 1996), a 120 amino acid–long sequence thought to be important for interaction with phosphoinositides (Shaw, 1996). Three related proteins, cytohesin-1, ARNO, and Grp1, which all have an amino-terminal PH domain and a sequence highly similar to an exchange factor have recently been identified (Chardin et al., 1996; Kolanus et al., 1996; Klarlund et al., 1997). In addition, some PH domains have been shown to preferentially bind to PI(4,5)P2 and PI(3,4,5)P3 compared with PI(3,4)P2 or other phosphoinositides (Rameh et al., 1997). These data suggest that PI(3,4,5)P3 and PI(4,5)P2 may recruit a GEF to the membrane and activate G proteins in the vicinity. Alternatively, PI(4,5)P2 may itself activate G proteins since in vitro studies have indicated that PI(4,5)P2 can function as an exchange factor (Zheng et al., 1996).
Figure 8.

A schematic diagram of Cdc42 activation by phosphoinositides. GDP-Cdc42 is complexed with RhoGDI in the cytosol. Phosphoinositides of broad specificity dissociate RhoGDI from the complex and bring Cdc42 to the membrane through the prenyl group. Cdc42 is then activated in the presence of specific phosphoinositides (i.e., PI(4,5)P2 or PI(3,4,5)P3), which either recruits a GEF or directly acts as a GEF. Alternatively, GTPγS can activate Cdc42 at this step (not shown here). In both cases, activated Cdc42 leads to actin assembly at the cell membrane.
Our data suggest that actin assembly through small G proteins is dependent on phosphoinositides at two steps: association with membranes and activation by GEF activities. However, the dependence on phosphoinositides seems to be bypassed when exogenous GTPγS-activated Cdc42 is added to the extracts, as long as it contains an intact CAAX motif for prenylation. In this nonphysiological situation, Cdc42 is activated by GTPγS instead of GEFs, and actin assembly is associated with Cdc42 aggregates instead of lipid membranes. We believe that the aggregation of Cdc42 is essential for actin polymerization, since soluble forms of Cdc42 (Cdc42V12C189S) are completely ineffectual in stimulating actin assembly in the absence of phosphoinositides (Fig. 7 A). However, since prenylation at the CAAX box may have other functions, we can not rule out that soluble Cdc42 proteins are also able to stimulate actin assembly. The establishment of quantitative assays for actin assembly in response to Cdc42 and specific phosphoinositides offers us an opportunity to identify and purify factors regulating actin assembly at the cell membrane.
Acknowledgments
We thank Dr. Ching-Shih Chen of University of Kentucky for synthesizing PI(3,4)P2 and PI(3,4,5)P3, Dr. Rolands Vegners of the Latvian Institute of Organic Synthesis (Riga, Latvia) for providing the gelsolin peptide, Drs. Julie Theriot and Rong Li for protocols. We also thank Margaret Chou and Kimberly Tolias for plasmid DNAs. We are grateful to Drs. James Sabry, Tim Mitchison, Rong Li, John Hartwig, Jack Taunton, Chris Carpenter, and members of the Kirschner lab for helpful discussion and critical reading of the manuscript. We especially thank Sally Zigmond for discussing her results on the role of Cdc42 in actin assembly before publication.
This work was supported by grants from the National Institutes of Health (GM26875 to M.W. Kirschner, P50-HL 56993 and R01-36622 to L.C. Cantley, and AR38910 to P.A. Janmey).
Abbreviations used in this paper
- F-actin
filamentous actin
- G proteins
GTP-binding proteins, GDI, guanine-nucleotide dissociation inhibitor
- GEF
guanine-nucleotide exchange factor
- NBD
nitrobenzoxadiazole
- PC
phosphatidylcholine
- PI
phosphatidyinositol
- PI(4,5)P2
PI (4,5) bisphosphate
- PI(3,4)P2
PI (3,4) bisphosphate
- PI(3,4,5)P3
PI (3,4,5) trisphosphate
Footnotes
Address all correspondence to Marc Kirschner, Department of Cell Biology, Harvard Medical School, 240 Longwood Avenue, Boston, MA 02115. Tel.: 617-432-2250; Fax: 617-432-0420. E-mail: marc@hms.harvard.edu
References
- Apgar JR. Activation of protein kinase C in rat basophilic leukemia cells stimulates increased production of phosphatidylinositol 4-phosphate and phosphatidylinositol 4,5-bisphosphate: correlation with actin polymerization. Mol Biol Cell. 1995;6:97–108. doi: 10.1091/mbc.6.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcaro A, Wymann MP. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: the role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem J. 1993;296:297–301. doi: 10.1042/bj2960297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger KR, Serunian LA, Soltoff SP, Libby P, Cantley LC. PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell. 1989;57:167–175. doi: 10.1016/0092-8674(89)90182-7. [DOI] [PubMed] [Google Scholar]
- Barkalow K, Witke W, Kwiatkowski DJ, Hartwig JH. Coordinated regulation of platelet actin filament barbed ends by gelsolin and capping protein. J Cell Biol. 1996;134:389–399. doi: 10.1083/jcb.134.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguski MS, McCormick F. Proteins regulating Ras and its relatives. Nature. 1993;366:643–654. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: conserved structure and molecular mechanism. Nature. 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- Cerione RA, Zheng Y. The Dbl family of oncogenes. Curr Opin Cell Biol. 1996;8:216–222. doi: 10.1016/s0955-0674(96)80068-8. [DOI] [PubMed] [Google Scholar]
- Chant J. Cell polarity in yeast. Trends Genet. 1994;10:328–333. doi: 10.1016/0168-9525(94)90036-1. [DOI] [PubMed] [Google Scholar]
- Chardin P, Paris S, Antonny B, Robineau S, Beraud DS, Jackson CL, Chabre M. A human exchange factor for ARF contains Sec7- and pleckstrin-homology domains. Nature. 1996;384:481–484. doi: 10.1038/384481a0. [DOI] [PubMed] [Google Scholar]
- Chong LD, Traynor KA, Bokoch GM, Schwartz MA. The small GTP-binding protein Rho regulates a phosphatidylinositol 4-phosphate 5-kinase in mammalian cells. Cell. 1994;79:507–513. doi: 10.1016/0092-8674(94)90259-3. [DOI] [PubMed] [Google Scholar]
- Chuang TH, Bohl BP, Bokoch GM. Biologically active lipids are regulators of Rac.GDI complexation. J Biol Chem. 1993;268:26206–26211. [PubMed] [Google Scholar]
- Cooper JA, Walker SB, Pollard TD. Pyrene actin: documentation of the validity of a sensitive assay for actin polymerization. J Muscle Res Cell Motil. 1983;4:253–262. doi: 10.1007/BF00712034. [DOI] [PubMed] [Google Scholar]
- Fishkind DJ, Wang YL. New horizons for cytokinesis. Curr Opin Cell Biol. 1995;7:23–31. doi: 10.1016/0955-0674(95)80041-7. [DOI] [PubMed] [Google Scholar]
- Forscher P, Lin CH, Thompson C. Novel form of growth cone motility involving site-directed actin filament assembly. Nature. 1992;357:515–518. doi: 10.1038/357515a0. [DOI] [PubMed] [Google Scholar]
- Hall A. Ras-related GTPases and the cytoskeleton. Mol Biol Cell. 1992;3:475–479. doi: 10.1091/mbc.3.5.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig JH, Bokoch GM, Carpenter CL, Janmey PA, Taylor LA, Toker A, Stossel TP. Thrombin receptor ligation and activated Rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell. 1995;82:643–653. doi: 10.1016/0092-8674(95)90036-5. [DOI] [PubMed] [Google Scholar]
- Hartwig JH, Kung S, Kovacsovics T, Janmey PA, Cantley LC, Stossel TP, Toker A. D3 phosphoinositides and outside-in integrin signaling by glycoprotein IIb-IIIa mediate platelet actin assembly and filopodial extension induced by phorbol 12-myristate 13-acetate. J Biol Chem. 1996;271:32986–32993. doi: 10.1074/jbc.271.51.32986. [DOI] [PubMed] [Google Scholar]
- Hawkins PT, Eguinoa A, Qiu R-G, Stokoe D, Cooke FT, Walters R, Wennstrom S, Claesson-Welsh L, Evans T, Symons M, Stephens L. PDGF stimulates an increase in GTP-Rac via activation of phosphoinositide 3-kinase. Curr Biol. 1995;5:393–403. doi: 10.1016/s0960-9822(95)00080-7. [DOI] [PubMed] [Google Scholar]
- Heyworth PG, Knaus UG, Xu X, Uhlinger DJ, Conroy L, Bokoch GM, Curnutte JT. Requirement for posttranslational processing of Rac GTP-binding proteins for activation of human neutrophil NADPH oxidase. Mol Biol Cell. 1993;4:261–269. doi: 10.1091/mbc.4.3.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison CJ, Brill D, Cox R, Gilbert J, Kill I, Ford CC. DNA replication and cell cycle control in Xenopus egg extracts. J Cell Sci Suppl. 1989;12:197–212. doi: 10.1242/jcs.1989.supplement_12.17. [DOI] [PubMed] [Google Scholar]
- Janmey PA. Phosphoinositides and calcium as regulators of cellular actin assembly and disassembly. Annu Rev Physiol. 1994;56:169–191. doi: 10.1146/annurev.ph.56.030194.001125. [DOI] [PubMed] [Google Scholar]
- Janmey PA, Lamb J, Allen PG, Matsudaira PT. Phosphoinositide-binding peptides derived from the sequences of gelsolin and villin. J Biol Chem. 1992;267:11818–11823. [PubMed] [Google Scholar]
- Janmey PA, Stossel TP. Gelsolin-polyphosphoinositide interaction. Full expression of gelsolin-inhibiting function by polyphosphoinositides in vesicular form and inactivation by dilution, aggregation, or masking of the inositol head group. J Biol Chem. 1989;264:4825–4831. [PubMed] [Google Scholar]
- King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 1995;81:279–288. doi: 10.1016/0092-8674(95)90338-0. [DOI] [PubMed] [Google Scholar]
- Klarlund JK, Guilherme A, Holik JJ, Virbasius JV, Chawla A, Czech MP. Signaling by phosphoinositide-3,4,5-trisphosphate through proteins containing pleckstrin and sec7 homology domains. Science. 1997;275:1927–1930. doi: 10.1126/science.275.5308.1927. [DOI] [PubMed] [Google Scholar]
- Kolanus W, Nagel W, Schiller B, Zeitlmann L, Godar S, Stockinger H, Seed B. Alpha L beta 2 integrin/LFA-1 binding to ICAM-1 induced by cytohesin-1, a cytoplasmic regulatory molecule. Cell. 1996;86:233–242. doi: 10.1016/s0092-8674(00)80095-1. [DOI] [PubMed] [Google Scholar]
- Kouyama T, Mihashi K. Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labelled F-actin. Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur J Biochem. 1981;114:33–38. [PubMed] [Google Scholar]
- Kreis, T., and R. Vale. 1993. Actin and associated proteins. Oxford University Press Inc., New York. 3–100.
- Kundra V, Escobedo JA, Kazlauskas A, Kim HK, Rhee SG, Williams LT, Zetter BR. Regulation of chemotaxis by the platelet-derived growth factor receptor-beta. Nature. 1994;367:474–476. doi: 10.1038/367474a0. [DOI] [PubMed] [Google Scholar]
- Lamarche N, Tapon N, Stowers L, Burbelo PD, Aspenstrom P, Bridges T, Chant J, Hall A. Rac and Cdc42 induce actin polymerization and G1 cell cycle progression independently of p65PAK and the JNK/SAPK MAP kinase cascade. Cell. 1996;87:19–29. doi: 10.1016/s0092-8674(00)81371-9. [DOI] [PubMed] [Google Scholar]
- Lassing I, Lindberg U. Specific interaction between phosphatidylinositol 4,5-bisphosphate and profilactin. Nature. 1985;314:472–474. doi: 10.1038/314472a0. [DOI] [PubMed] [Google Scholar]
- Machesky LM, Hall A. Rho: a connection between membrane receptor signalling and the cytoskeleton. Trends Cell Biol. 1996;6:304–310. doi: 10.1016/0962-8924(96)10026-x. [DOI] [PubMed] [Google Scholar]
- Marchand JB, Moreau P, Paoletti A, Cossart P, Carlier MF, Pantaloni D. Actin-based movement of Listeria monocytogenes: actin assembly results from the local maintenance of uncapped filament barbed ends at the bacterium surface. J Cell Biol. 1995;130:331–343. doi: 10.1083/jcb.130.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- Murray AW. Cell cycle extracts. Methods Cell Biol. 1991;36:581–605. [PubMed] [Google Scholar]
- Murray AW, Kirschner MW. Cyclin synthesis drives the early embryonic cell cycle. Nature. 1989;339:275–280. doi: 10.1038/339275a0. [DOI] [PubMed] [Google Scholar]
- Newport J, Spann T. Disassembly of the nucleus in mitotic extracts: membrane vesicularization, lamin disassembly, and chromosome condensation are independent processes. Cell. 1987;48:219–230. doi: 10.1016/0092-8674(87)90425-9. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hawkins P, Stephens L, Hall A. Activation of the small GTP-binding proteins rho and rac by growth factor receptors. J Cell Sci. 1995;108:225–233. doi: 10.1242/jcs.108.1.225. [DOI] [PubMed] [Google Scholar]
- Ohsumi K, Katagiri C, Kishimoto T. Chromosome condensation in Xenopus mitotic extracts without histone H1. Science. 1993;262:2033–2035. doi: 10.1126/science.8266099. [DOI] [PubMed] [Google Scholar]
- Pitcher JA, Touhara K, Payne ES, Lefkowitz RJ. Pleckstrin homology domain-mediated membrane association and activation of the beta-adrenergic receptor kinase requires coordinate interaction with G beta gamma subunits and lipid. J Biol Chem. 1995;270:11707–11710. doi: 10.1074/jbc.270.20.11707. [DOI] [PubMed] [Google Scholar]
- Rameh LE, Arvidsson A-K, Carraway KL, III, Couvillon AD, Rathbun G, Crampton A, VanRenterghem B, Czech MP, Ravichandran KS, Burakoff SJ, et al. A comparative analysis of the phosphoinositide binding specificity of pleckstrin homology domains. J Biol Chem. 1997;372:22059–22066. doi: 10.1074/jbc.272.35.22059. [DOI] [PubMed] [Google Scholar]
- Reif K, Nobes CD, Thomas G, Hall A, Cantrell DA. Phosphatidylinositol 3-kinase signals activate a selective subset of rac/rho-dependent effector pathways. Curr Biol. 1996;6:1445–1455. doi: 10.1016/s0960-9822(96)00749-x. [DOI] [PubMed] [Google Scholar]
- Ren XD, Bokoch GM, Traynor KA, Jenkins GH, Anderson RA, Schwartz MA. Physical association of the small GTPase Rho with a 68-kDa phosphatidylinositol 4-phosphate 5-kinase in Swiss 3T3 cells. Mol Biol Cell. 1996;7:435–442. doi: 10.1091/mbc.7.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ. Rho: theme and variations. Curr Biol. 1996;6:1256–1264. doi: 10.1016/s0960-9822(02)70711-2. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Sawin KE, LeGuellec K, Philippe M, Mitchison TJ. Mitotic spindle organization by a plus-end-directed microtubule motor. Nature. 1992;359:540–543. doi: 10.1038/359540a0. [DOI] [PubMed] [Google Scholar]
- Shaw G. The pleckstrin homology domain: an intriguing multifunctional protein module. Bioessays. 1996;18:35–46. doi: 10.1002/bies.950180109. [DOI] [PubMed] [Google Scholar]
- Stossel TP. On the crawling of animal cells. Science. 1993;260:1086–1094. doi: 10.1126/science.8493552. [DOI] [PubMed] [Google Scholar]
- Symons MH, Mitchison TJ. Control of actin polymerization in live and permeabilized fibroblasts. J Cell Biol. 1991;114:503–513. doi: 10.1083/jcb.114.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai Y, Kaibuchi K, Kikuchi A, Sasaki T, Shirataki H. Regulators of small GTPases. Ciba Found Symp. 1993;176:128–138. doi: 10.1002/9780470514450.ch9. [DOI] [PubMed] [Google Scholar]
- Tapon N, Hall A. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr Opin Cell Biol. 1997;9:86–92. doi: 10.1016/s0955-0674(97)80156-1. [DOI] [PubMed] [Google Scholar]
- Tardif M, Huang S, Redmond T, Safer D, Pring M, Zigmond SH. Actin polymerization induced by GTPγS in permeabilized neutrophils is induced and maintained by free barbed ends. J Biol Chem. 1995;270:28075–28083. doi: 10.1074/jbc.270.47.28075. [DOI] [PubMed] [Google Scholar]
- Theriot JA, Rosenblatt J, Portnoy DA, Goldschmidt-Clermont PJ, Mitchison TJ. Involvement of profilin in the actin-based motility of L. monocytogenes in cells and in cell-free extracts. Cell. 1994;76:505–517. doi: 10.1016/0092-8674(94)90114-7. [DOI] [PubMed] [Google Scholar]
- Tolias KF, Cantley LC, Carpenter CL. Rho family gtpases bind to phosphoinositide kinases. J Biol Chem. 1995;270:17656–17659. doi: 10.1074/jbc.270.30.17656. [DOI] [PubMed] [Google Scholar]
- Wennstrom S, Hawkins P, Cooke F, Hara K, Yonezawa K, Kasuga M, Jackson T, Claesson-Welsh L, Stephens L. Activation of phosphoinositide 3-kinase is required for PDGF-stimulated membrane ruffling. Curr Biol. 1994;4:385–393. doi: 10.1016/s0960-9822(00)00087-7. [DOI] [PubMed] [Google Scholar]
- Wymann M, Arcaro A. Platelet-derived growth factor-induced phosphatidylinositol 3-kinase activation mediates actin rearrangements in fibroblasts. Biochem J. 1994;3:517–520. doi: 10.1042/bj2980517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, King WG, Dillon S, Hall A, Feig L, Rittenhouse SE. Activation of platelet phosphatidylinositide 3-kinase requires the small GTP-binding protein Rho. J Biol Chem. 1993;268:22251–22254. [PubMed] [Google Scholar]
- Zheng Y, Glaven JA, Wu WJ, Cerione RA. Phosphatidylinositol 4,5-bisphosphate provides an alternative to guanine nucleotide exchange factors by stimulating the dissociation of GDP from Cdc42Hs. J Biol Chem. 1996;271:23815–23819. doi: 10.1074/jbc.271.39.23815. [DOI] [PubMed] [Google Scholar]
- Zigmond SH. Signal transduction and actin filament organization. Curr Opin Cell Biol. 1996;8:66–73. doi: 10.1016/s0955-0674(96)80050-0. [DOI] [PubMed] [Google Scholar]
- Zigmond SH, Joyce M, Borleis J, Bokoch GM, Devrotes PN. Regulation of actin polymerization in cell-free systems by GTPγS and Cdc42. J Cell Biol. 1997;138:363–374. doi: 10.1083/jcb.138.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]