

Abstract
Cytoplasmic dynein, a minus end–directed, microtubule-based motor protein, is thought to drive the movement of membranous organelles and chromosomes. It is a massive complex that consists of multiple polypeptides. Among these polypeptides, the cytoplasmic dynein heavy chain (cDHC) constitutes the major part of this complex. To elucidate the function of cytoplasmic dynein, we have produced mice lacking cDHC by gene targeting. cDHC−/− embryos were indistinguishable from cDHC+/−or cDHC+/+ littermates at the blastocyst stage. However, no cDHC−/− embryos were found at 8.5 d postcoitum. When cDHC−/− blastocysts were cultured in vitro, they showed interesting phenotypes. First, the Golgi complex became highly vesiculated and distributed throughout the cytoplasm. Second, endosomes and lysosomes were not concentrated near the nucleus but were distributed evenly throughout the cytoplasm. Interestingly, the Golgi “fragments” and lysosomes were still found to be attached to microtubules.
These results show that cDHC is essential for the formation and positioning of the Golgi complex. Moreover, cDHC is required for cell proliferation and proper distribution of endosomes and lysosomes. However, molecules other than cDHC might mediate attachment of the Golgi complex and endosomes/lysosomes to microtubules.
Microtubules are thought to play important roles in various cellular activities, including mitosis, determination of cellular shape, and the transport, organization, and distribution of organelles in higher eukaryotic cells. The involvement of microtubules in the transport, organization, and distribution of organelles requires the activity of motor proteins that interact with both microtubules and organelles (Hirokawa 1993, 1998; Holzbaur and Vallee, 1994; Schroer, 1994). Among these motor proteins, minus end–directed, microtubule-based motor proteins are thought to drive the centrosomal movement of membranous organelles (Hirokawa 1993, 1998; Holzbaur and Vallee, 1994; Schroer, 1994). Cytoplasmic dynein (Lye et al., 1987; Paschal et al., 1987), the most abundant minus end–directed motor protein, is a high–molecular mass complex primarily composed of multiple polypeptides, two heavy chains of ∼550,000 D, three to four 74,000-D intermediate chains, four light intermediate chains of ∼55,000 D, and light chains of 8,000–22,000 D. The heavy chain, containing the sites for ATP hydrolysis and microtubule binding, probably makes up the bulk of the head (Koonce et al., 1992; Mikami et al., 1993; Zhang et al., 1993), while the intermediate and light intermediate chains lie at the base of the molecule. Biochemical and immunocytochemical studies using anti–cytoplasmic dynein antibodies have shown that the motor is localized on late endosomes and lysosomes (Lin and Collins, 1992) and the TGN (Fath et al., 1994), suggesting its role in transporting these organelles in nonneuronal cells. In neurons it is believed to be involved in retrograde axonal transport (Schnapp and Reese 1989; Schroer et al., 1989; Hirokawa et al., 1990). Furthermore, colocalization of cytoplasmic dynein with kinetochores and mitotic spindles suggested its possible involvement in mitosis (Pfarr et al., 1990; Steuer et al., 1990).
To elucidate its function, several studies have been performed by various researchers. Depletion of cytoplasmic dynein blocked the in vitro movement of endosomes/ lysosomes on microtubules (Blocker et al., 1997) and prevented the centrosomal localization of exogenously applied Golgi-derived vesicles in semiintact cells (Corthesy-Theulaz et al., 1992). Injection of antibodies into cells induced the collapse of mitotic spindles (Vaisberg et al., 1993). Overexpression of the dynamitin subunit of the dynactin complex, which is believed to dissociate dyneins from their target organelles, leads to dispersion of the Golgi complex, endosomes, and lysosomes. The overexpression of dynamitin was also shown to block the ER to Golgi transport (Burkhardt et al., 1997; Presley et al., 1997). Recently, the molecular cloning of dynein genes by reverse transcriptase PCR using degenerate primers has resulted in the identification of several members of the cytoplasmic dynein family (Gibbons et al., 1994; Tanaka et al., 1995; Vaisberg et al., 1996). Using isoform-specific antibodies, immunolocalization of each isoform has revealed that DHC2 (DLP4 of Tanaka and DHC1B of Gibbons) is localized on the ER–Golgi intermediate compartment (Vaisberg, E.A., P.M. Grissom, S.R. Gill, T.A. Schroer, and J.R. McIntosh. 1996. Mol. Biol. Cell. 7:403a) and DHC3 (DLP2 of Tanaka and DHC7C of Gibbons) is localized on unidentified vesicular structures (Vaisberg et al., 1996). Injection of anti-DHC2 antibody leads to dispersion of the Golgi complex (Vaisberg et al., 1996), which is in contrast to the findings of a previous injection experiment using anti–cytoplasmic dynein heavy chain antibody (Vaisberg et al., 1993). Therefore, there is a possibility that previous immunolocalization and antibody injection studies may not have discriminated the unique function of one cytoplasmic dynein member from that of another. Generating mutants that specifically lack each dynein gene is the ideal way to determine the function of each cytoplasmic dynein heavy chain isoform. Mutational studies using yeast (Eshel et al., 1993; Li et al., 1993) have shown the involvement of cytoplasmic dynein in spindle orientation and anaphase chromosome segregation (Saunders et al., 1995). Fungal cytoplasmic dynein mutants exhibited abnormalities in nuclear positioning and movement (Plamann et al., 1994; Xiang et al., 1994), while Drosophila (Gepner et al., 1996) mutants were shown to be lethal, suggesting that cytoplasmic dynein is essential for proliferation. Although these studies have increased our knowledge regarding the functions of cytoplasmic dynein, its role in intracellular transport remains elusive because of the lack of appropriate mammalian mutants that lack the functional cytoplasmic dynein heavy chain (cDHC)1 gene.
To elucidate this, we generated mice lacking cytoplasmic dynein, the major species of numerous minus end– directed motor proteins, by targeted disruption of its heavy chain (cDHC [DHC1 of Vaisberg and DHC1A of Gibbons]), which is essential for the functioning of cytoplasmic dynein.
Materials and Methods
Generation of Mutant Mice
A 13-kb EcoRI fragment containing the cDHC gene was obtained from a 129/Sv mouse genomic library using a rat cDHC cDNA fragment, which contains the first coding exon (Zhang et al., 1993) and was subcloned into pBluescript. The sequence from the translation start ATG in the first coding exon was shown in Fig. 1 e. For construction of the targeting vector, the second half of the first coding exon (after the EagI site in Fig. 1 e) and a part of the following intron were deleted. The loxP sequence, splice- acceptor/splice-enhancer (Friedrich and Soriano, 1991; Watakabe et al., 1993) sequences, and the PGK-neo gene (McBurney et al., 1991) in the reverse transcriptional orientation to the cDHC gene were inserted into this deleted region. For negative selection, a diphtheria toxin A (Yagi et al., 1993) fragment cassette was introduced into the downstream polylinker site. The replacement-type cDHC construct (Fig. 1 a) was linearized with NotI before electroporation. The J1 line of ES cells used for these experiments were cultured essentially as described previously (Harada et al., 1994). ES cells carrying a disrupted cDHC gene were injected into C57BL/6 embryos at the blastocyst stage as described previously (Harada et al., 1994).
Figure 1.



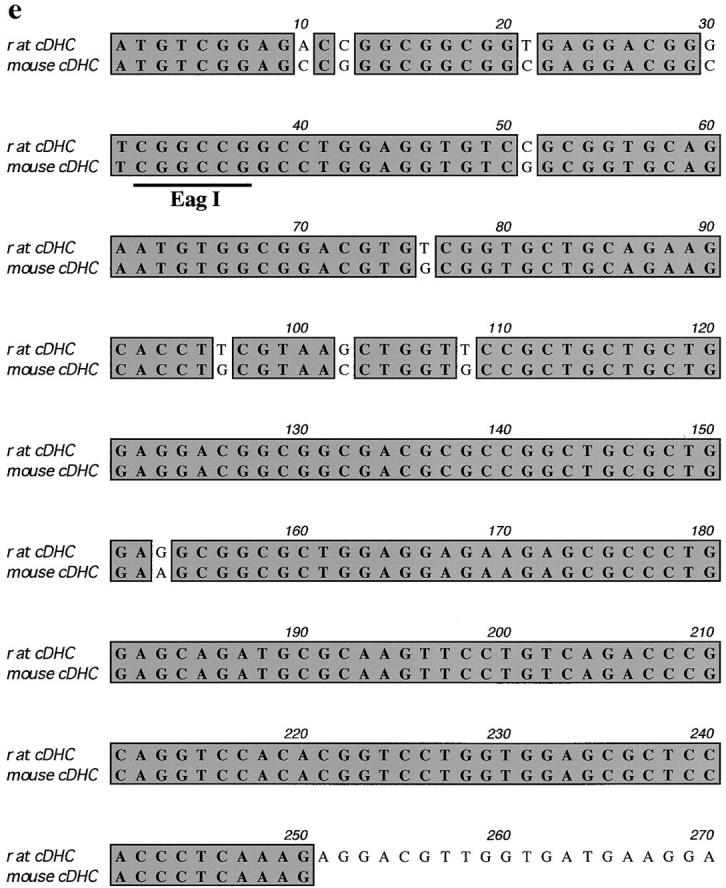
Targeted inactivation of the cDHC gene in ES cells and mice. (a) The cDHC gene locus and targeting vector. (Top) cDHC wild-type allele. (Middle) Replacement-type targeting vector. (Bottom) Targeted allele after homologous recombination. (b) Southern blot analysis of representative genomic DNA of 12.5-dpc embryos from cDHC heterozygote intercrosses. Embryonic DNA was digested with SacI, and Southern blot analysis was performed using the 3′-flanking probe. WT, wild-type band; HR, homologous recombinant band. Genotypes: +/+, wild-type;+/−, heterozygote; −/−, homozygote. (c) PCR analysis of genomic DNA of 3.5-dpc embryos from a cDHC heterozygote intercross. DNA samples were subjected to PCR using the primer pairs b/c (wild-type allele) or b/d (mutated allele). (d) Immunoblotting of cDHC (left) and DHC2 (right). (Left) Crude cell extracts from: lane 1, NRK; lane 2, Cos; lane 3, wild-type ES; lane 4, heterozygous mutant ES; lane 5, wild-type ES (half amount); lane 6, wild-type ES; lane 7, heterozygous mutant ES cells were loaded on the gel and blotted with anti-cDHC sera. Arrowheads represent the positions of molecular mass markers (200, 116, 97, and 66 kD, from top to bottom, respectively). (Right) Crude cell extracts from: lane 1, NRK; lane 2, Cos; lane 3, wild-type ES; lane 4, heterozygous mutant ES; lane 5, wild-type ES (half amount) cells. Crude extracts were loaded and blotted with anti-DHC2 sera. (e) The sequence of the open reading frame of the mouse cDHC first coding exon and the cDNA sequence from rat cDHC. The EagI site used for constructing the targeting vector was underlined.
Immunoblotting
Immunoblotting was performed largely as described previously (Harada et al., 1994). Wild-type (cDHC+/+) and heterozygous mutant (cDHC+/−) ES cells were cultured as described. Cos, NRK, and NIH 3T3 cells were grown in DME (GIBCO BRL, Gaithersburg, MD) supplemented with 10% FCS. All types of cells were grown to confluency, washed twice with PBS, collected by scraping into ice-cold PBS, pelleted down by centrifugation, and suspended in 25 mM TrisCl, pH 6.8, 2% SDS. Cell suspension was sonicated and then boiled to lower the viscosity. Crude extracts were made by centrifuging the homogenates at 20,000 g for 15 min at 4°C. Protein concentrations were determined by BCA Protein Assay Reagent (Pierce Chemical Co., Rockford, IL). Equal amounts of crude extracts were separated with polyacrylamide gel. Proteins were electrophoretically transferred to nylon filters (Millipore Corp., Bedford, MA). Nylon filters with transferred proteins were blocked with 2% skim milk in TBS, incubated in anti-cDHC (DHC1) antisera or anti-DHC2 antisera (Vaisberg et al., 1996) (gifts from E.A.Vaisberg and J.R. McIntosh) for up to 6 h at 37°C, rinsed in TBS containing 0.05% Tween 20, and incubated for 2 h with 125I-labeled protein A. Binding was detected by autoradiography using an imaging analyzer (model BAS-2000; Fuji-Film, Tokyo, Japan).
Blastocyst Culture and Immunofluorescence
Blastocysts were collected by flushing the oviducts of female mice at 3.5 d postcoitum (dpc) and cultured individually for 3 d on gelatinized coverslips in embryonic stem (ES) cell medium. Cultured blastocysts were fixed in 2% paraformaldehyde, PBS for 10 min at room temperature (RT). Some samples were fixed in 2% paraformaldehyde and 0.2% glutaraldehyde for 10 min at RT and blocked in 1 mg/ml NaBH4 for 30 min at 4°C. They were permeabilized with cold methanol for 15 s, washed in PBS twice, blocked in 1% BSA/PBS for 5 min at RT, and then incubated with the first antibodies in the blocking solution for 1 h at 37°C. Subsequently, samples were extensively washed in PBS, blocked in 1% BSA/PBS for 5 min at RT, and incubated with the second antibodies for 1 h at 37°C. As for anti-Arp1 and Glued antibodies, samples were fixed in 2% paraformaldehyde, washed in PBS twice and in 20 mM glycine/PBS once, permeabilized in 0.1% saponin in 20 mM glycine/PBS for 20 min at RT, and incubated with the first antibody in 0.1% saponin in 20 mM glycine/PBS for 1 h at 37°C. The following washing, blocking, and incubation processes were the same as described above.
For the first antibodies, the following antibodies or antisera were used: anti-cDHC antisera (Vaisberg et al., 1996) (a gift from E.A.Vaisberg and J.R. McIntosh); anti-GM130 antisera (Nakamura et al., 1995) (a gift from N. Nakamura and G. Warren); CTR433 (Jasmin et al., 1989) (a gift from M. Bornens); anti-Arp1 monoclonal antibody (45A) (Schafer et al., 1994) and anti-p150Glued monoclonal antibody (150B) (Blocker et al., 1997) (gifts from T. Schroer); anti-PDI antibody (StressGen Biotechnologies Corp., Victoria, British Columbia); anti-LAMP2 antibody ABL-93 (Chen et al., 1986); DM1A (Sigma Chemical Co., St. Louis, MO); and YL1/2 (BIOSYS S.A., Compiegne, France) for tubulin. The samples were observed either using a confocal laser scanning microscope (model MRC-1000; Bio-Rad Laboratories, Hercules, CA) or using an AXIOPHOT microscope (Carl Zeiss, Inc., Thornwood, NY). Images were transferred to a Macintosh computer for editing and were printed with a Fujix Pictrography Digital Printer.
Conventional Electron Microscopy
Blastocysts grown on gelatinized coverslips in ES media for 3 d were washed once with PBS and fixed in 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M cacodylate buffer for 2 h. Sections were processed as described previously (Harada et al., 1990) and viewed under an electron microscope (model 2000EX; JEOL, Inc., Tokyo, Japan) at 100 kV.
Genotyping of Blastocysts
Both intact and cultured blastocysts were genotyped by hemi-nested PCR method. Cultured blastocysts were lysed in 50–100 μl lysis buffer (1% SDS, 10 mM TrisCl, pH 8.0, 25 mM EDTA, 75 mM NaCl) containing proteinase K (100 μg/ml) for 10–20 min at RT and stored at −20°C until use. For samples for electron microscopy, the inner cell masses (ICM) of fixed cultured blastocysts were removed with a finely drawn glass micropipette on an inverted microscope (model Diaphot; Nikon Corp., Tokyo, Japan), washed with PBS once, lysed in the lysis buffer, and stored at −20°C until use. The lysates were thawed, overlaid with mineral oil, heat-denatured at 95°C for 35 min, and then subjected to ethanol precipitation. Ethanol precipitates were dissolved in 10 μl distilled water and were used as PCR templates. Intact blastocysts were lysed in 10 μl of autoclaved distilled water and frozen immediately at −20°C, and the lysates were directly used as PCR templates. The first round PCR was carried out in a reaction buffer containing 10 mM Tris-Cl, pH 8.3, 50 mM KCl, 1.5 mM MgCl2, 0.001% (wt/vol) gelatin, 200 μM each of dATP, dCTP, dGTP, and dTTP, 1 μM each of PCR primers, and 1 U of Taq DNA polymerase (Perkin-Elmer Corp., Norwalk, CT) using three primers (Fig. 1 a): the forward primer “a” (5′-CCTGGGTGTTCAAGACAAGCTGGTTC-3′), located upstream of the insertion site; the reverse primer “c” (5′-GTTATGGGGGCTGAGGTCTTGTCGT-3′), located near the 3′ end of the first coding exon; and the reverse primer “d” (5′-TAAGGGCCAGCTCATTCCTCCACT-3′), located within the polyA signal of the PGK-neo gene. PCR schedules were employed for 25 cycles: 94°C for 30 s, 63°C for 30 s, and 72°C for 90 s. For reamplification, 2 μl of the first round PCR mix was removed and added to 48 μl of a fresh PCR mix containing nested primers (forward primer “b” [5′-ACGCCCCTCACTGACCGTTGCTATT-3′] [located between a and the insertion site] and c or d) and amplified for an additional 30 cycles: 94°C for 30 s, 63°C for 30 s, and 72°C for 40 s.
Quantification of Lysosome Distribution
Cultured blastocysts were incubated for 8 h at 37°C in ES cell media containing 1 mg/ml FITC-dextran (Sigma Chemical Co.), washed with PBS, and fixed in 2% paraformaldehyde, PBS for 10 min. The samples were observed using a confocal laser scanning microscope (model MRC-100; Bio-Rad Laboratories) in the photon counting mode to ensure that the fluorescein intensity reflects the amount of FITC-dextran uptake, and the data were quantified using National Institutes of Health (NIH) Image software.
Taxol Treatment of Cultured Blastocysts
Taxol was obtained from the National Cancer Institute (Bethesda, MD) and was dissolved in dimethyl sulfoxide at a concentration of 3.5 mM and stored at −20°C. Blastocysts cultured for 3 d were incubated with ES media containing either 5 or 20 μM taxol for 24 h. Similar results were obtained under both conditions.
Results
Targeting of the Cytoplasmic Dynein Heavy Chain Gene
To disrupt the endogenous cDHC gene, a replacement-type targeting vector was constructed (Fig. 1 a). Genomic PCR and Southern blot analysis revealed a targeting frequency of 4 in ∼600 G418R clones. To assess the levels of cDHC and DHC2 in wild-type (cDHC+/+) and heterozygous mutant (cDHC+/−) ES cells, we performed immunoblotting of crude extracts prepared from wild-type ES cells, heterozygous mutant ES cells, and other cell lines (NRK and Cos cells) using antibodies specific for cDHC and DHC2 (Vaisberg et al., 1996). In wild-type ES cells, cDHC is expressed at similar or slightly higher levels compared with other cell lines (NRK and Cos cells) (Fig. 1 d, left, lanes 1–3). On the contrary, the amount of DHC2 in wild-type ES cells is much lower than that in Cos cells, which express high amounts of DHC2, and at similar levels to that in NRK cells, which express the protein much lower than Cos cells (Fig. 1 d, right, lanes 1–3) (see also Vaisberg et al., 1996). There was no apparent compensatory increase in DHC2 in cDHC+/− ES cells (Fig. 1 d, right, lanes 3 and 4). In cDHC+/− ES cells, the amount of cDHC was reduced to approximately one half that of cDHC+/+ ES cells (Fig. 1 d, left, lanes 4 and 5), and proteins of aberrant sizes were not detected with this antibody (Fig. 1 d, left, lanes 6 and 7), suggesting that the allele generated by this gene targeting experiment is likely to be null. Chimeric male mice generated from injections with three different cell lines transmitted the ES cell genome through the germline. Approximately 50% of these agouti pups were found to be heterozygous for the mutant cDHC allele when Southern blot analysis was carried out (data not shown). These mice were indistinguishable from their wild-type littermates and displayed no discernible abnormalities. To determine the phenotype of homozygous mutant mice, we interbred heterozygous animals and genotyped litters, but we could not identify cDHC−/− animals either at birth or after weaning. When DNA isolated from fetuses at different gestation times from 8.5 to 12.5 dpc (Fig. 1 b) was analyzed by genomic Southern blot or by PCR, we could not identify any cDHC−/− among morphologically normal fetuses. Instead, we found much smaller decidua that contained completely resorbed embryos. This observation indicated that cDHC−/− embryos seemed to develop until the blastocyst stage and were able to hatch and be implanted, but they died before 8.5 dpc. Therefore, we genotyped blastocysts after heterozygote interbreedings. As shown in Fig. 1 c, cDHC−/− blastocysts were identified, and they appeared to be indistinguishable from control (cDHC+/− or cDHC+/+) blastocysts at the light microscopic level (Fig. 2, a and b). To check for the presence of cDHC mRNA, we performed reverse transcriptase PCR using total RNA from homozygote blastocyst cultures and control blastocyst cultures as templates. Contrary to our expectations, we found PCR products from both homozygotes and controls. Hence, we conclude that detectable amounts of mRNA of maternal origin still remain even at this developmental stage.
Figure 2.
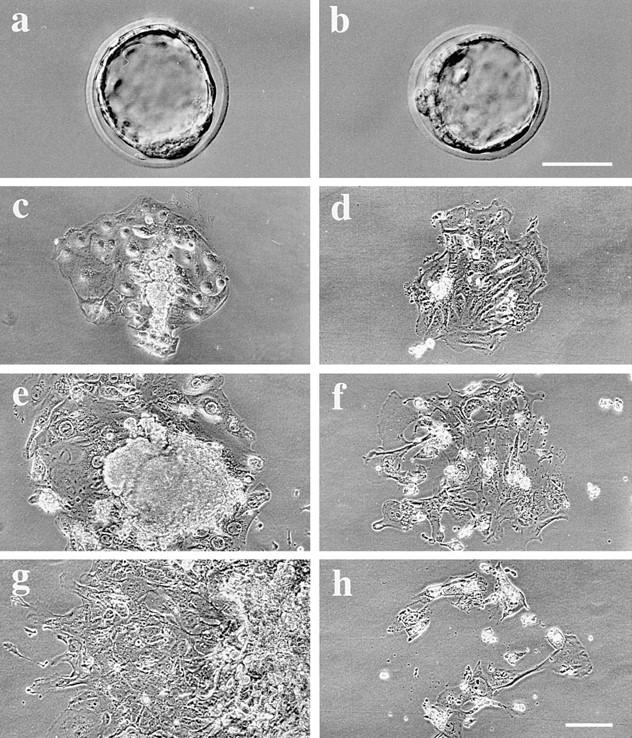
Outgrowths of control and cDHC−/− blastocysts in vitro. A control blastocyst (a) and a cDHC−/− blastocyst (b) appear to be morphologically normal. Representative example of control (c, e, and g) and cDHC−/− (d, f, and h) blastocysts cultured for 3 d (c and d), 6 d (e and f), and 9 d (g and h) in vitro, respectively. Bars, 50 μm.
Growth Abnormalities of cDHC− /− Cells
To assess possible defects in cDHC−/− cells, we cultured blastocysts and examined them by light microscopy. On the third day of culture, both mutant and control blastocysts had completed adhesion onto the surface of coverslips and produced trophoblast giant cell outgrowths (Fig. 2, c and d). However, the ICM appeared smaller in size in most of the mutant blastocysts compared with control blastocysts. After longer periods of blastocyst culture, we found cDHC−/− cells incapable of growing in vitro (Fig. 2, f and h). After 9 d in culture (6 d after attachment onto the substrate surface) (Fig. 2, g and h), all of the cDHC−/− blastocysts had failed to grow, and most of the trophoblast cells had died while control cells were proliferating vigorously (Fig. 2 g). To examine if there was any mitotic phenotype, we took photographs of four additional cultured cDHC−/− blastocysts every day. However, we were not able to find any mitotic figures nor an increase in cell number.
The Golgi Complex in cDHC− /− Cells Were Highly Fragmented and Distributed throughout the Cytoplasm
When we examined the distribution of the Golgi complex, significant changes were found in cDHC−/− cells compared with control cells. In cDHC−/− cells, the Golgi complex stained either by antisera against GM130 (cis-Golgi marker) or by CTR433 monoclonal antibody (medial-Golgi marker) appeared to be highly vesiculated and distributed throughout the cytoplasm (Fig. 3 b), which was in marked contrast to the case for the control cells, in which it appeared as large flattened cisternae located around the nucleus (Fig. 3 a). To further investigate the morphological changes in cDHC−/− cells, we examined control and cDHC−/− cells by electron microscopy. The rough endoplasmic reticulum, mitochondria, and the nuclei of cDHC−/− cells were indistinguishable from those of control (cDHC+/− or cDHC+/+) cells (Fig. 3, c and d). However, a gross change in the Golgi complex was observed in cDHC−/− cells (Fig. 3 d). First, the Golgi complex (arrows) was not located near the nucleus as observed in the control cells but distributed throughout the cytoplasm as observed by confocal microscopy. Second, the Golgi complex was much smaller in size, but it consisted of stacks of several small flattened cisternae (Fig. 3 e) similar to the Golgi complex observed in control cells.
Figure 3.

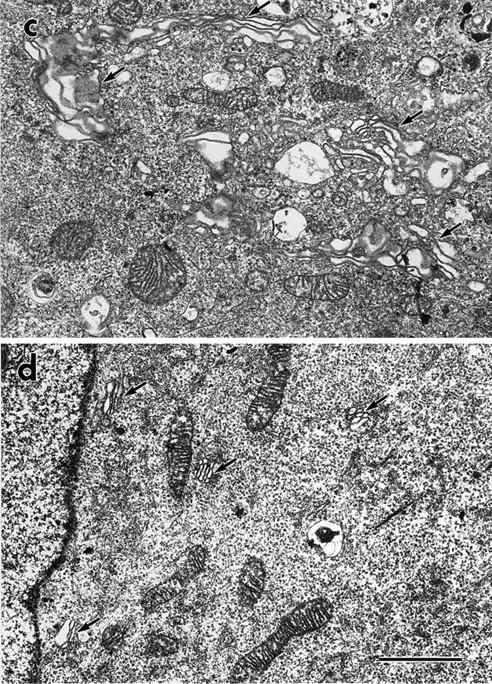

The Golgi complex was fragmented and dispersed throughout the cytoplasm of cDHC−/− cells. Control (a) and cDHC−/− (b) cells were stained with anti-GM130 antisera. Electron micrographs compare representative areas of the trophoectodermal cells of (c) control and (d and e) cDHC−/− cells. Arrows indicate the Golgi complex. (e) At higher magnification, the Golgi fragments (arrows) clearly consisted of stacks of several small flattened cisternae and associated with microtubules (arrowheads) in cDHC−/− cells. Bars: (a and b) 10 μm; (c and d) 2 μm; (e) 500 nm.
The Distributions of Endosomes/Lysosomes and the Dynactin Components Were Also Altered in cDHC− /− Cells
When endosomes and lysosomes were labeled by uptake of extracellular FITC-labeled dextran, they were found to be distributed around the nuclei in control cells (Fig. 4, a and c). However, in cDHC−/− cells, they were rather uniformly distributed or often remained close to the cell periphery (Fig. 4, b and d, arrows). To quantify the distribution of these organelles, the intensity of FITC-dextran was measured by confocal microscopy. The ratio of the FITC intensity in the inner one third against the intensity in the outer two thirds of the cytoplasm (inner/outer) was found to be significantly lower in the cDHC−/− cells (0.81 ± 0.31 [mean ± SD]; n = 11) than in the control cells (2.96 ± 1.48; n = 11) (P < 0.001; Student's t test). We obtained similar results when lysosomes were labeled by anti-LAMP2 antibody (Fig. 4, e and f). Moreover FITC-dextran uptake was reduced in cDHC−/− cells. When we quantified the intensity of endocytosed FITC-dextran by cDHC−/− cells and control cells cultured on the same coverslip, there was a statistically significant difference in intensity between cDHC−/− cells (12.7 ± 6.84 arbitrary units; n = 10) and control cells (20.0 ± 6.55 arbitrary units; n = 21) (P < 0.01; Student's t test). In contrast, the distributions of the endoplasmic reticulum network (Fig. 4, g and h) and mitochondria (data not shown) in cDHC−/− cells were not significantly different from those in the control cells. The dynactin complex, a protein complex known to bind cytoplasmic dyneins, is considered to serve as a linker between membranous organelles and cytoplasmic dyneins (Schroer, 1994). To examine their distribution in the absence of cDHC, we used monoclonal antibodies against Arp1(45A) and p150Glued (150B) to stain cDHC−/− cells and control cells (Fig. 4, i–l). While both dynactin components are localized to centrosomally dominant punctate structures in control cells as described previously (Gill et al., 1991) (Fig. 4, i and k), they are localized to punctate structures distributed throughout the cytoplasm in cDHC−/− cells, although some centrosomal staining still remained (Fig. 4, j and l). To assess the amount of cytoplasmic dynein heavy chain, immunostaining using antibodies that specifically react with cDHC was performed (Vaisberg et al., 1996). Though control cultured blastocysts showed prominent staining as described in a previous paper (Vaisberg et al., 1996), marked reduction in the level of staining was observed in cDHC−/− cultured blastocysts (Fig. 4, m and n).
Figure 4.


Localization of various organelles in control and cDHC−/− blastocysts cultured in vitro. (a–f) Localization of endosomes/lysosomes (labeled by internalized FITC-dextran) and lysosomes (labeled by anti-LAMP-2 antibody). Control (a, c, e, g, i, k, and m) and cDHC−/− (b, d, f, h, j, l, and n) cells were labeled by uptake of extracellular FITC-dextran (a and b) or by anti-LAMP-2 antibody (e and f). They were also stained by anti-PDI antibody (g and h), anti-Arp1 antibody (i and j), anti-p150Glued antibody (k and l), or anti-cDHC antibody (m and n). Cells stained by 4′,6-diamidino-2-phenylindole (DAPI) are shown in c and d. Bars: (a–f and i–n) 50 μm; (g and h) 20 μm.
Association of the Golgi Fragments and Endosomes/Lysosomes with Microtubules
Next we examined the spatial relationship between the Golgi fragments, lysosomes, and microtubules by double or triple immunofluorescence analysis of cDHC−/− cells (Fig. 5, a–f). The Golgi “fragments” were located quite close to microtubules (Fig. 5, a and b), which was clearly shown in the cytoplasmic region below the nucleus, where a relatively small number of microtubules were observed (Fig. 5 c). To demonstrate the association of the Golgi fragments with microtubules more clearly, we performed two additional experiments. First, cDHC−/− cells were incubated with the microtubule-stabilizing agent taxol. Taxol is known to have the unique ability to promote the formation of discrete microtubule bundles in cells. In the presence of taxol, microtubule bundles were formed in cDHC−/− cells (Fig. 6 a) as well as in cDHC+/− and cDHC+/+ cells (data not shown). The Golgi fragments (arrows) in cDHC−/− cells were found to be associated with these bundles (Fig. 6 a). In the cellular region where microtubule bundles were not found, the density of microtubules was sometimes so low that each microtubule filament could easily be distinguished. In this region the Golgi fragments (arrows) were also associated with these microtubules (Fig. 6 b), even after stronger fixation protocol (2% paraformaldehyde plus 0.2% glutaraldehyde). These Golgi fragments were not associated with the ER fragments and ER protrusions (Fig. 6 c, arrowheads). Second, we examined cDHC−/− cells under electron microscope at higher magnification (Fig. 3 e). We found microtubules (arrowheads) frequently associated with the small Golgi stacks (arrows). Lysosomes were also found to be located close to microtubules (Fig. 5 d). Since Golgi fragments and lysosomes were similarly distributed throughout the cytoplasm and were both associated with microtubules, and since previous studies have shown the active transport of materials between the TGN and endosome/lysosomes (Goda and Pfeffer, 1989; Griffiths, 1989), we would like to determine whether the Golgi fragments and endosomes/lysosomes are located in close proximity or not. We compared them with respect to their distribution and found that the distributions of the Golgi fragments and endosomes/lysosomes were clearly different (Fig. 5, e and f).
Figure 5.

Immunofluorescence localization of the Golgi complex, ER, and lysosomes and their association with microtubules. (a–e) Double immunofluorescence was performed for detection of the Golgi complex (red) and microtubules (green) (a–c), lysosomes (red) and microtubules (green) (d), and the Golgi complex (red) and lysosomes (green) (e). Higher magnification views of the peripheral (b) and the perinuclear (c–e) cytoplasm are shown. (f) Triple immunofluorescence was performed for detection of the Golgi complex (red), lysosomes (blue), and microtubules (green). Bars, 10 μm.
Figure 6.
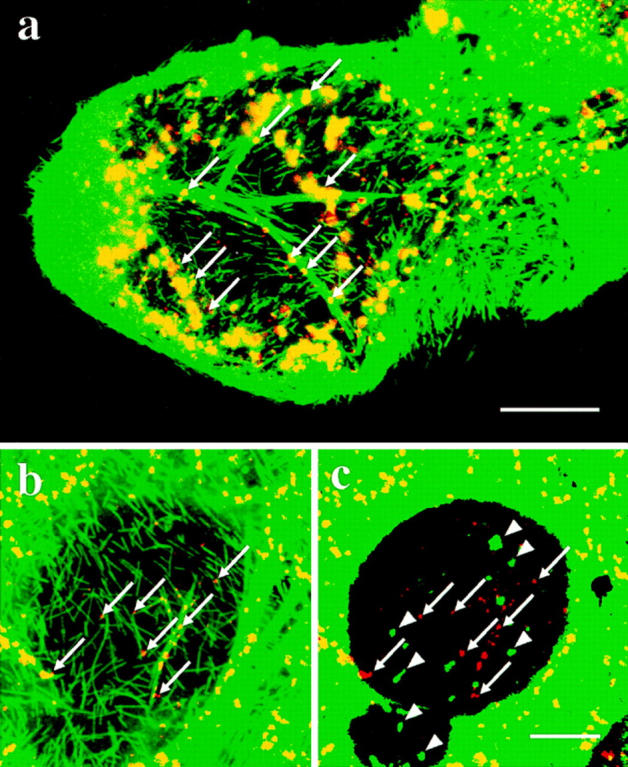
Effect of taxol on the distribution of microtubules and the Golgi fragments in cDHC−/− cells. cDHC−/− cells were treated for 24 h with 5 or 20 μM taxol. Double (a and b) immunostaining was performed with antitubulin antibody (green) and anti-GM130 antisera (red). (a) Yellowish staining indicates that the Golgi fragments were largely colocalized with microtubules. The Golgi fragments clearly localized on microtubule bundles were shown by arrows. (b) At higher magnification, the Golgi fragments (arrows) were also associated with the thin microtubule filaments even after stronger fixation. (c) Double staining of the same area with anti-GM130 antisera (red) and anti-PDI antibody (green) shows these Golgi fragments (red, arrows) were distinct from the ER fragments and protrusions (green, arrowheads). Bars: (a) 20 μm; (c) 25 μm.
Discussion
We examined the role of cDHC in the formation and distribution of organelles for the first time by gene targeting, and the results were surprising in that only one species among various DHCs (Tanaka et al., 1995; Vaisberg et al., 1996) is essential for the movement of various organelles in the centrosomal orientation and in cell proliferation. Regarding cell proliferation, our results are in agreement with those of Drosophila cDHC mutant analysis (Gepner et al., 1996), suggesting that cDHC is essential in higher eukaryotes, while in yeast (Eshel et al., 1993; Li et al., 1993) and fungi (Plamann et al., 1994; Xiang et al., 1994), it is dispensable.
It is intriguing that, without cDHC, the Golgi complex breaks apart into numerous fragments and disperses throughout the cytoplasm. It seems that, in the steady state, the Golgi complex is in dynamic equilibrium between the anterograde force mediated by kinesin superfamily motors and the retrograde force mediated by cytoplasmic dyneins rather than simply being associated with microtubules near their minus end (or centrosomes). On reduction in the concentration of cDHC, this equilibrium favors the anterograde force, causing the Golgi complex to be fragmented and dispersed throughout the cytoplasm. Fragmentation of the Golgi complex is easily recognized during mitosis or upon treatment of cells with a microtubule-depolymerizing agent. Even in the fragmented state, the Golgi complex can serve its function (Cole et al., 1996) and maintain its polarity (Shima et al., 1997). Therefore, fragmentation caused by a reduction in the concentration of cDHC likely reflects a physiological phenomenon rather than being simply caused by a mechanical force.
To date, fragmentation of the Golgi during mitosis has been considered to result from a lack of cytoplasmic microtubules due to microtubule depolymerization (Wehland et al., 1983; Warren, 1993). Since the microtubule network was not substantially affected in cDHC−/− cells in this study, we consider Golgi fragmentation to have resulted from the loss of activity of cytoplasmic dynein. Since cDHC is thought to be important in mitosis and is localized to the mitotic spindle and kinetochores (Pfarr et al., 1990; Steuer et al., 1990), translocation of cDHC from the cytoplasm to the nucleus might occur before the formation of the mitotic spindle, which may cause a reduction in the level of cDHC activity in the cytoplasm and thus lead to fragmentation of the Golgi complex. Alternatively, the inactivation of cDHC in the cytoplasm may occur by some unknown mechanism such as phosphorylation/dephosphorylation and thus lead to the same results.
Previous studies have shown that injection of anti-DHC2 leads to dispersion of the Golgi complex (Vaisberg et al., 1996). As suggested by depletion studies indicated by this paper and the paper of Vaisberg et al., cDHC and DHC2 appear to have similar functions in maintenance of the Golgi complex. However, as shown in the study of Vaisberg et al., the localization of DHC1 (cDHC in this paper) and DHC2 is clearly different. Recently, Vaisberg et al. indicated that DHC2 is localized to the ER–Golgi intermediate compartment (Vaisberg, E.A., P.M. Grissom, S.R. Gill, T.A. Schroer, and J.R. McIntosh. 1996. Mol. Biol. Cell. 7:403a). Since the intermediate compartment and Golgi complex actively exchange materials and are known to have similar morphology and distributions under various conditions (e.g., in the presence of brefeldin A or nocodazole, under temperature change) (Lippincott-Schwartz et al., 1990; Cole et al., 1996), it is conceivable that, under physiological conditions, the two species of dynein heavy chains are localized to different compartments that reside somewhere between the intermediate compartment and Golgi complex, and that depletion of either molecule leads to a similar phenotype (i.e., dispersion of the Golgi complex). This can explain the results of recent dynamitin overexpression studies (Burkhardt et al., 1997; Presley et al., 1997) because its overexpression is likely to affect the interactions between both species of cytoplasmic dyneins and their target organelles.
It is also surprising that, even in cDHC−/− cells, the Golgi fragments and endosomes/lysosomes are attached to microtubules. This association raises the possibility that other molecules such as the dynactin complex (Hirokawa, 1993; Holzbaur and Vallee, 1994; Schroer, 1994; Hirokawa 1998) or other anterograde/retrograde motors (Hirokawa, 1996) might mediate attachment of these organelles to microtubules. In particular, dynactin, known as a binding protein of cytoplasmic dynein (Gill et al., 1991; Paschal et al., 1993), has intriguing characteristics because one of its complexes, p150Glued, has its own microtubule-binding domain (Waterman-Storer et al., 1993) and cytoplasmic dynein depleted of p150Glued is inactive in promoting organelle movement in vitro (Gill et al., 1991). Therefore, it is quite likely that in the absence of cDHC, the Golgi complex and endosomes/lysosomes are attached via p150Glued to microtubules. Previous analyses of the amino acid sequence of p150Glued revealed a motif that is homologous to a microtubule-binding motif in the endosome–microtubule linker protein CLIP170 (Pierre et al., 1992). Thus, for endosomes/lysosomes, CLIP170, together with p150Glued, may link these organelles to microtubules through this common microtubule-binding domain.
Acknowledgments
We would like to thank Dr. T. Noda (Cancer Institute, Tokyo, Japan) for his advice and generous gifts of various plasmids. We would also like to thank: Drs. M. Fujiwara (University of Tokyo School of Medicine), S. Matsuyama, and S. Tanaka (University of Tokyo School of Agricultural Sciences) for their advice on animal care; E.A. Vaisberg and J.R. McIntosh (University of Colorado Department of Molecular, Cellular, and Developmental Biology, Boulder, CO), M. Bornens (Institut Curie, Paris, France), N. Nakamura, G. Warren (Imperial Cancer Research Fund Cell Biology Laboratory, London, UK), and T. Schroer (Johns Hopkins University, Baltimore, MD) for their generous gifts of antibodies; and R. Sato-Harada, Y. Okada, Y. Yonekawa, Z. Zhang, H. Sato, C. Zhao, and other members of Hirokawa's lab for help and discussions. The anti-LAMP-2 antibody ABL-93 was obtained from the Developmental Studies Hybridoma Bank maintained by the Department of Pharmacology and Molecular Sciences, Johns Hopkins University School of Medicine and the Department of Biological Sciences, University of Iowa (Iowa City, IA) under contract NO1-HD-2-3144 from the National Institue of Child Health and Human Development.
This work was supported by a Special Grant-in-Aid for Center of Excellence from the Japan Ministry of Education, Science and Culture to N. Hirokawa.
Footnotes
1. Abbreviations in this paper: cDHC, cytoplasmic dynein heavy chain; dpc, days postcoitum; ES, embryonic stem; ICM, inner cell mass; RT, room temperature.
Address all correspondence to N. Hirokawa, Department of Cell Biology and Anatomy, Graduate School of Medicine, University of Tokyo, Hongo, Bunkyo-ku, Tokyo, 113, Japan. Tel.: 81-3-3812-2111, ext. 3326. Fax: 81-3-5802-8646. E-mail: hirokawa@m.u-tokyo.ac.jp
References
- Blocker A, Severin FF, Burkhardt JK, Bingham JB, Yu H, Olivo JC, Schroer TA, Hyman AA, Griffiths G. Molecular requirements for bidirectional movement of phagosomes along microtubules. J Cell Biol. 1997;137:113–129. doi: 10.1083/jcb.137.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt JK, Echeverri CJ, Nilsson T, Vallee RB. Overexpression of the dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. J Cell Biol. 1997;139:469–484. doi: 10.1083/jcb.139.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JW, Chen GL, D'Souza MP, Murphy TL, August JT. Lysosomal membrane glycoproteins: properties of LAMP-1 and LAMP-2. Biochem Soc Symp. 1986;51:97–112. [PubMed] [Google Scholar]
- Cole NB, Sciaky N, Marotta A, Song J, Lippincott-Schwartz J. Golgi dispersal during microtubule disruption: regeneration of Golgi stacks at peripheral endoplasmic reticulum exit sites. Mol Biol Cell. 1996;7:631–650. doi: 10.1091/mbc.7.4.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corthesy-Theulaz I, Pauloin A, Pfeffer SR. Cytoplasmic dynein participates in the centrosomal localization of the Golgi complex. J Cell Biol. 1992;118:1333–1345. doi: 10.1083/jcb.118.6.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshel D, Urrestarazu LA, Vissers S, Jauniaux J C, van Vliet-Reedijk JC, Planta RJ, Gibbons IR. Cytoplasmic dynein is required for normal nuclear segregation in yeast. Proc Natl Acad Sci USA. 1993;90:11172–11176. doi: 10.1073/pnas.90.23.11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fath KR, Trimbur GM, Burgess DR. Molecular motors are differentially distributed on Golgi membranes from polarized epithelial cells. J Cell Biol. 1994;126:661–675. doi: 10.1083/jcb.126.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich G, Soriano P. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 1991;5:1513–1523. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- Gepner J, Li M, Ludmann S, Kortas C, Boylan K, Iyadurai SJ, McGrail M, Hays TS. Cytoplasmic dynein function is essential in Drosophila melanogaster. . Genetics. 1996;142:865–878. doi: 10.1093/genetics/142.3.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons BH, Asai DJ, Tang WJ, Hays TS, Gibbons IR. Phylogeny and expression of axonemal and cytoplasmic dynein genes in sea urchins. Mol Biol Cell. 1994;5:57–70. doi: 10.1091/mbc.5.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SR, Schroer TA, Szilak I, Steuer ER, Sheetz MP, Cleveland DW. Dynactin, a conserved, ubiquitously expressed component of an activator of vesicle motility mediated by cytoplasmic dynein. J Cell Biol. 1991;115:1639–1650. doi: 10.1083/jcb.115.6.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda Y, Pfeffer SR. Cell-free systems to study vesicular transport along the secretory and endocytic pathways. FASEB (Fed Am Soc Exp Biol) J. 1989;3:2488–2495. doi: 10.1096/fasebj.3.13.2680705. [DOI] [PubMed] [Google Scholar]
- Griffiths G. The structure and function of a mannose 6-phosphate receptor-enriched, pre-lysosomal compartment in animal cells. J Cell Sci. 1989;11(Suppl.):139–147. doi: 10.1242/jcs.1989.supplement_11.11. [DOI] [PubMed] [Google Scholar]
- Harada A, Sobue K, Hirokawa N. Developmental changes of synapsin I subcellular localization in rat cerebellar neurons. Cell Struct Funct. 1990;15:329–342. doi: 10.1247/csf.15.329. [DOI] [PubMed] [Google Scholar]
- Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, Hirokawa N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369:488–491. doi: 10.1038/369488a0. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. Axonal transport and the cytoskeleton. Curr Opin Neurobiol. 1993;3:724–731. doi: 10.1016/0959-4388(93)90144-n. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. Organelle transport along microtubules-the role of KIFs . Trends Cell Biol. 1996;6:135–141. doi: 10.1016/0962-8924(96)10003-9. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Sato-Yoshitake R, Yoshida T, Kawashima T. Brain dynein (MAP1C) localizes on both anterogradely and retrogradely transported membranous organelles in vivo. J Cell Biol. 1990;111:1027–1037. doi: 10.1083/jcb.111.3.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzbaur EL, Vallee RB. DYNEINS: molecular structure and cellular function. Annu Rev Cell Biol. 1994;10:339–372. doi: 10.1146/annurev.cb.10.110194.002011. [DOI] [PubMed] [Google Scholar]
- Jasmin BJ, Cartaud J, Bornens M, Changeux JP. Golgi apparatus in chick skeletal muscle: changes in its distribution during end plate development and after denervation. Proc Natl Acad Sci USA. 1989;86:7218–7222. doi: 10.1073/pnas.86.18.7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonce MP, Grissom PM, McIntosh JR. Dynein from Dictyostelium: primary structure comparisons between a cytoplasmic motor enzyme and flagellar dynein. J Cell Biol. 1992;119:1597–1604. doi: 10.1083/jcb.119.6.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YY, Yeh E, Hays T, Bloom K. Disruption of mitotic spindle orientation in a yeast dynein mutant. Proc Natl Acad Sci USA. 1993;90:10096–10100. doi: 10.1073/pnas.90.21.10096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SX, Collins CA. Immunolocalization of cytoplasmic dynein to lysosomes in cultured cells. J Cell Sci. 1992;101:125–137. doi: 10.1242/jcs.101.1.125. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Donaldson JG, Schweizer A, Berger EG, Hauri HP, Yuan LC, Klausner RD. Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell. 1990;60:821–836. doi: 10.1016/0092-8674(90)90096-w. [DOI] [PubMed] [Google Scholar]
- Lye RJ, Porter ME, Scholey JM, McIntosh JR. Identification of a microtubule-based cytoplasmic motor in the nematode C. elegans. . Cell. 1987;51:309–318. doi: 10.1016/0092-8674(87)90157-7. [DOI] [PubMed] [Google Scholar]
- McBurney MW, Sutherland LC, Adra CN, Leclair B, Rudnicki MA, Jardine K. The mouse Pgk-1 gene promoter contains an upstream activator sequence. Nucleic Acids Res. 1991;19:5755–5761. doi: 10.1093/nar/19.20.5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikami A, Paschal BM, Mazumdar M, Vallee RB. Molecular cloning of the retrograde transport motor cytoplasmic dynein (MAP 1C) Neuron. 1993;10:787–796. doi: 10.1016/0896-6273(93)90195-w. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis TE, Warren G. Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol. 1995;131:1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschal BM, Shpetner HS, Vallee RB. MAP 1C is a microtubule-activated ATPase which translocates microtubules in vitro and has dynein-like properties. J Cell Biol. 1987;105:1273–1282. doi: 10.1083/jcb.105.3.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschal BM, Holzbaur EL, Pfister KK, Clark S, Meyer DI, Vallee RB. Characterization of a 50-kD polypeptide in cytoplasmic dynein preparations reveals a complex with p150GLUED and a novel actin. J Biol Chem. 1993;268:15318–15323. [PubMed] [Google Scholar]
- Pfarr CM, Coue M, Grissom PM, Hays TS, Porter ME, McIntosh JR. Cytoplasmic dynein is localized to kinetochores during mitosis. Nature. 1990;345:263–265. doi: 10.1038/345263a0. [DOI] [PubMed] [Google Scholar]
- Pierre P, Scheel J, Rickard JE, Kreis TE. CLIP-170 links endocytic vesicles to microtubules. Cell. 1992;70:887–900. doi: 10.1016/0092-8674(92)90240-d. [DOI] [PubMed] [Google Scholar]
- Plamann M, Minke PF, Tinsley JH, Bruno K S. Cytoplasmic dynein and actin-related protein Arp1 are required for normal nuclear distribution in filamentous fungi. J Cell Biol. 1994;127:139–149. doi: 10.1083/jcb.127.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Saunders WS, Koshland D, Eshel D, Gibbons IR, Hoyt MA. Saccharomyces cerevisiaekinesin- and dynein-related proteins required for anaphase chromosome segregation. J Cell Biol. 1995;128:617–624. doi: 10.1083/jcb.128.4.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DA, Gill SR, Cooper JA, Heuser JE, Schroer TA. Ultrastructural analysis of the dynactin complex: an actin-related protein is a component of a filament that resembles F-actin. J Cell Biol. 1994;126:403–412. doi: 10.1083/jcb.126.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnapp BJ, Reese TS. Dynein is the motor for retrograde axonal transport of organelles. Proc Natl Acad Sci USA. 1989;86:1548–1552. doi: 10.1073/pnas.86.5.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroer TA. Structure, function and regulation of cytoplasmic dynein. Curr Opin Cell Biol. 1994;6:69–73. doi: 10.1016/0955-0674(94)90118-x. [DOI] [PubMed] [Google Scholar]
- Schroer TA, Steuer ER, Sheetz MP. Cytoplasmic dynein is a minus end-directed motor for membranous organelles. Cell. 1989;56:937–946. doi: 10.1016/0092-8674(89)90627-2. [DOI] [PubMed] [Google Scholar]
- Shima DT, Haldar K, Pepperkok R, Watson R, Warren G. Partitioning of the Golgi apparatus during mitosis in living HeLa cells. J Cell Biol. 1997;137:1211–1228. doi: 10.1083/jcb.137.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steuer ER, Wordeman L, Schroer TA, Sheetz MP. Localization of cytoplasmic dynein to mitotic spindles and kinetochores. Nature. 1990;345:266–268. doi: 10.1038/345266a0. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Zhang Z, Hirokawa N. Identification and molecular evolution of new dynein-like protein sequences in rat brain. J Cell Sci. 1995;108:1883–1893. doi: 10.1242/jcs.108.5.1883. [DOI] [PubMed] [Google Scholar]
- Vaisberg EA, Koonce MP, McIntosh JR. Cytoplasmic dynein plays a role in mammalian mitotic spindle formation. J Cell Biol. 1993;123:849–858. doi: 10.1083/jcb.123.4.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisberg EA, Grissom PM, McIntosh JR. Mammalian cells express three distinct dynein heavy chains that are localized to different cytoplasmic organelles. J Cell Biol. 1996;133:831–842. doi: 10.1083/jcb.133.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren G. Membrane partitioning during cell division. Annu Rev Biochem. 1993;62:323–348. doi: 10.1146/annurev.bi.62.070193.001543. [DOI] [PubMed] [Google Scholar]
- Watakabe A, Tanaka K, Shimura Y. The role of exon sequences in splice site selection. Genes Dev. 1993;7:407–418. doi: 10.1101/gad.7.3.407. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer CM, Sanger JW, Sanger JM. Dynamics of organelles in the mitotic spindles of living cells: membrane and microtubule interactions. Cell Motil Cytoskel. 1993;26:19–39. doi: 10.1002/cm.970260104. [DOI] [PubMed] [Google Scholar]
- Wehland J, Henkart M, Klausner R, Sandoval IV. Role of microtubules in the distribution of the Golgi apparatus: effect of taxol and microinjected anti-α-tubulin antibodies. Proc Natl Acad Sci USA. 1983;80:4286–4290. doi: 10.1073/pnas.80.14.4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang X, Beckwith SM, Morris NR. Cytoplasmic dynein is involved in nuclear migration in Aspergillus nidulans. . Proc Natl Acad Sci USA. 1994;91:2100–2104. doi: 10.1073/pnas.91.6.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T, Nada S, Watanabe N, Tamemoto H, Kohmura N, Ikawa Y, Aizawa S. A novel negative selection for homologous recombinants using diphtheria toxin A fragment gene. Anal Biochem. 1993;214:77–86. doi: 10.1006/abio.1993.1459. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Tanaka Y, Nonaka S, Aizawa H, Kawasaki H, Nakata T, Hirokawa N. The primary structure of rat brain (cytoplasmic) dynein heavy chain, a cytoplasmic motor enzyme. Proc Natl Acad Sci USA. 1993;90:7928–7932. doi: 10.1073/pnas.90.17.7928. [DOI] [PMC free article] [PubMed] [Google Scholar]