

Abstract
We aimed to determine if and how endothelial cells (EC) recruit precursors of smooth muscle cells and pericytes and induce their differentiation during vessel formation. Multipotent embryonic 10T1/2 cells were used as presumptive mural cell precursors. In an under-agarose coculture, EC induced migration of 10T1/2 cells via platelet-derived growth factor BB. 10T1/2 cells in coculture with EC changed from polygonal to spindle-shaped, reminiscent of smooth muscle cells in culture. Immunohistochemical and Western blot analyses were used to examine the expression of smooth muscle (SM)-specific markers in 10T1/2 cells cultured in the absence and presence of EC. SM-myosin, SM22α, and calponin proteins were undetectable in 10T1/2 cells cultured alone; however, expression of all three SM-specific proteins was significantly induced in 10T1/2 cells cocultured with EC. Treatment of 10T1/2 cells with TGF-β induced phenotypic changes and changes in SM markers similar to those seen in the cocultures. Neutralization of TGF-β in the cocultures blocked expression of the SM markers and the shape change. To assess the ability of 10T1/2 cells to contribute to the developing vessel wall in vivo, prelabeled 10T1/2 cells were grown in a collagen matrix and implanted subcutaneously into mice. The fluorescently marked cells became incorporated into the medial layer of developing vessels where they expressed SM markers. These in vitro and in vivo observations shed light on the cell–cell interactions that occur during vessel development, as well as in pathologies in which developmental processes are recapitulated.
The vasculature is among the first organ systems to develop, and is vital for the distribution of nutrients and oxygen as well as removal of waste products. Blood vessels in general are composed of distinct cell layers. The intima, the innermost layer, is made up of a single cell type: endothelial cells (EC).1 The media is composed of layers of mural cells, smooth muscle cells (SMC) in large vessels, and pericytes in microvessels. The outermost layer of large vessels, the adventitia, consists of loose connective tissue containing smaller blood vessels and nerves.
Blood vessel assembly during embryogenesis, a process termed vasculogenesis, begins with clustering of primitive vascular cells also known as hemangioblasts (13) into tubelike endothelial structures that define the pattern of the vasculature (41). Mural cells become associated with the forming vessels at later stages of development (26), which has led to the suggestion that the EC may govern vessel layer acquisition. Although EC are thought to be a homogeneous population of cells derived entirely from mesodermal tissue (8, 48), SMC are locally derived from one of two sources: cardiac neural crest (i.e., ectoderm) or lateral mesenchyme (i.e., mesoderm; reference 28). Recruitment of pericytes to nascent capillaries during angiogenesis is likely to involve similar processes.
The exact mechanism(s) by which EC recruit mural cell precursors and induce their differentiation into mural cells during vessel formation is unknown, and is difficult to examine using in situ developmental models. There has been intense interest in understanding the regulation of muscle cell differentiation. Much progress has been made in elucidating the molecular regulators of skeletal and cardiac muscle differentiation (42, 43). These rapid advances have been facilitated by the availability of culture systems that can be used to model the differentiation process in vitro. Although the identification of a number of smooth muscle (SM)–specific proteins, including SM-myosin, calponin, and SM22α (14, 19) has enabled the description of SM development in vivo, understanding the molecular regulation of SM differentiation has been hindered by the lack of an appropriate in vitro system.
Thus, we have developed in vitro coculture systems in order to dissect and analyze the cellular interactions and potential mediators involved in SMC recruitment and differentiation. We have used the multipotent mouse embryonic 10T1/2 cells as presumptive mural cell precursors. These cells have been used in other systems, and are induced by various means to express markers of adipocyte (33), osteoblast (17), and myoblast cell lineages (10). We report a novel and biologically relevant coculture system in which we demonstrate recruitment and differentiation of a multipotent precursor toward a SMC lineage in response to EC.
The studies described herein demonstrate that growth factors released from EC induce migration of undifferentiated mesenchymal cells toward EC, and that, upon contact with the EC, the mesenchymal cells differentiate into SM-like cells. We hypothesize that these processes operate in vivo during vessel assembly via vasculogenesis and angiogenesis. We further postulate that contact and communication between EC and mural cells is important not only for establishing new vessels, but also for maintaining the differentiated quiescent vessel.
Materials and Methods
Tissue Culture
EC were isolated from bovine aortas by collagenase digestion as previously described (18). The cells were grown on uncoated tissue culture plastic in DME with 10% calf serum (CS) supplemented with penicillin, streptomycin, and glutamine, and were used through passage 20. 10T1/2 cells (CCL 226; American Type Culture Collection, Rockville, MD) were grown and maintained in DME with 10% FCS and 4.5 g/liter glucose. All cells were maintained at 37°C in a humidified atmosphere. DME with 2% CS was used in all coculture experiments. All tissue culture experiments were performed at least three times. Representative experiments are shown.
Cocultures
Cocultures of bovine aortic EC (BAE) and 10T1/2 cells were established in one of two ways: (a) an under-agarose system assay was conducted with modifications of a previously described assay (40). EC and mural cell precursors were plated into two 5-mm wells created 1–2 mm apart in 1% agarose/1% BSA in DME. 10T1/2 cells were plated in one well, and EC, a control vehicle, or growth factors were added to the other well. Cells were incubated for up to 6 d at 37°C; or (b) direct cocultures in which equal numbers of both cell types were plated simultaneously into the same plate and incubated for up to 48 h. This assay was used when a large number of interacting cells was needed (e.g., for Western blot analyses).
Migration Assay
BAE and 10T1/2 cells were plated in the under-agarose assay at 2 × 104 cells/well in 25 μl DME with 2% CS as described above, and were allowed to incubate for 48 h. The cells were then fixed with 2% paraformaldehyde in PBS for 30 min and stained with 0.1% Coomassie blue for 30 min. The distance migrated by each cell type was quantified by projecting the image of each well on a Profile Projector Model 6C-2™ (Nikon, Inc., Melville, NY) under a low-power objective fitted with a grid. In some experiments, neutralizing antibodies against PDGF-A or -B (1:200; Genzyme Corp., Cambridge, MA) or basic fibroblast growth factor (bFGF; provided by Dr. Michael Klagsbrun, Children's Hospital, Boston, MA; used at 1:100) were incorporated into the agarose before plating. In all experiments, the distance migrated by 10T1/2 cells cultured alone (background) was subtracted from the distances migrated by the cells under experimental conditions.
Immunocytochemistry
EC and 10T1/2 cells were cocultured in the under-agarose assay established in four-chamber culture slides (Lab-tek, Naperville, IL). In some cases, neutralizing antiserum against TGF-β was added at the initiation of the cocultures. Before plating, EC were labeled with 5 μg/ml Di-I-acetylated low density lipoprotein (Ac-LDL; Biomedical Technologies, Inc., Stoughton, MA) for 4–18 h at 37°C. After coculture for up to 6 d, the cells were fixed with 4% paraformaldehyde and immunostained for the presence of SMC-specific markers, including αSM-actin, SM-myosin, calponin, and SM22α. Anti-αSM-actin (mouse monoclonal antibody; DAKO Corp., Carpinteria, CA) was used at 1:50 in blocking buffer consisting of 4% normal goat serum/3% BSA/0.1% Triton X-100 in PBS. Anti-SM-myosin heavy chain mouse monoclonal antibody (9A9 used at 1:200) provided by Dr. Gary Owens (University of Virginia Medical School, Charlottesville, VA) has been shown to cross-react with both isoforms of SM myosin heavy chain (50). This antibody specifically recognized SMC, and not EC, in bovine aorta (data not shown). SMC specificity is further demonstrated in Fig. 8, where antibody labels only vascular SMC, and not surrounding skeletal muscle. Similar labeling patterns were obtained using a polyclonal SM myosin antisera from Biomedical Technologies, Inc. Anti-calponin (mouse monoclonal antibody provided by Dr. Marina Glukhova, Curie Institute, Paris, France) was used at 1:10. Affinity-purified rabbit polyclonal SM22α antisera was generously provided by Dr. Mario Gimona (Salzburg, Austria), and was used at 1:500. All antibody–antigen complexes were visualized using the Vectastain Elite ABC Kit (Vector Labs, Inc., Burlingame, CA), and the biotinylated secondary antibodies (1:250) were provided by the manufacturer.
Figure 8.

Incorporation of 10T1/2 cells into developing vessels. 10T1/2 cells were prelabeled with PKH26, a permanent red fluorescent dye, and seeded onto a cross-linked collagen matrix. The matrix was then implanted subcutaneously into the backs of C57 mice and incubated for 10 d. Tissue from the experimental area was excised, fixed, and immunostained for αSM-actin, SM myosin, and calponin. Fluorescent cells in association with the abluminal vessel surface indicated that 10T1/2 cells (arrowhead) had become incorporated into the newly forming vessels. Counterstaining revealed that some of these cells expressed αSM-actin, SM myosin, and calponin. Importantly, mouse skeletal muscle did not stain with any of the three SM-specific antibodies. Bar, 80 μm.
Western Blot Analysis
EC and 10T1/2 cells were cultured alone (6 × 105 cells/100 mm dish) or in coculture (3 × 105 of each cell type/100 mm dish) for 48 h with or without a monoclonal anti-TGF-β antibody that recognizes TGF-β 1, 2, and 3 (10 μg/ml; generously provided by Genzyme Corp., Boston, MA). Protein was isolated from cells using a method previously described (53); 10 μg of protein from solo cultures and 20 μg of protein from cocultures were electrophoresed on SDS-PAGE minigels (Bio-Rad Laboratories, Hercules, CA) and electrophoretically transferred to Immobilon-P membranes (Millipore Corp., Bedford, MA). Membranes were blocked with 5% nonfat dry milk/1% BSA in PBS, and then incubated for 1–2 h with primary antibody (αSM-actin, 1:50; SM-myosin, 1:150; SM22α, 1:1,000; calponin, 1:10,000) diluted in blocking buffer. Mouse monoclonal antibodies against calponin were purchased from DAKO Corp. Similar results were obtained with a rabbit polyclonal antisera against SM-myosin (Biomedical Technologies, Inc.) and mouse monoclonal antibodies (9A9). Antibody– antigen complexes were revealed using the ECL detection system (Amersham Life Sciences, Arlington Heights, IL). Quantitation was performed using a Digital Imaging System™ (Alpha Innotech Corp., San Leandro, CA).
Integration of 10T1/2 Cells into Vessels In Vivo
These studies were conducted to determine if undifferentiated 10T1/2 cells would become incorporated into newly forming vessels and display a SM phenotype in a complex in vivo environment. 10T1/2 cells were permanently labeled with a fluorescent dye and implanted subcutaneously onto the backs of 6–8-wk-old C57 mice.
Permanent Labeling of 10T1/2 Cells.
10T1/2 cells were prelabeled with PKH26 (Sigma Chemical Co., St. Louis, MO), a permanent red fluorescent dye that is retained in the cells for up to 100 cell doublings (25). Cells were trypsinized to a single-cell suspension, rinsed with PBS, and counted using a Coulter Counter (Coulter Corp., Miami, FL). Cells were gently resuspended at 2 × 106 cells/100 μl of Diluent C (provided with the PKH26 dye). An equal volume of 40 μM PKH26 dye was added, followed by incubation for 2–3 min with gentle agitation. The labeling reaction was terminated by adding 2 vol of FCS and 7 ml of 0.1% BSA in PBS. The cells were then layered onto 3 ml of FCS and pelleted. After centrifugation, the cells were rinsed with DME/10% FCS, resuspended in fresh media, and incubated overnight at 37°C. Cells were then trypsinized, counted, and plated as needed.
Mouse Implantation Assay.
The fluorescently-labeled 10T1/2 cells were seeded at various densities (4–20 × 104 cells/cm2) onto 0.3-mm2 pieces of cross-linked collagen matrix (Instat™; Johnson & Johnson Medical, Inc., Arlington, TX) in the presence or absence of 30 ng bFGF and incubated for 3 d at 37°C. Cell/matrix preparations were implanted subcutaneously onto the backs of 6–8-wk-old C57 mice and incubated for 7–14 d. Tissue from the experimental area was subsequently excised, fixed in 4% paraformaldehyde, embedded, sectioned, and immunostained for αSM-actin (1:500), SM-myosin (9A9; 1:2,000) and calponin (1:10). Stained sections were analyzed with an Axiophot epifluorescent microscope (Carl Zeiss, Inc., Thornwood, NY) or an Odyssey XL confocal microscope with Intervision (Noran Instruments, Middleton, WI).
Results
Effects of EC on 10T1/2 Cell Migration
10T1/2 cells, a multipotent cell line, were used as presumptive mural cell precursors. EC and 10T1/2 cells were cocultured in the under-agarose assay to examine the effects of EC on migration of the presumptive mural cell precursors. 10T1/2 cells and BAE were plated into two wells created ∼2 mm apart in an agarose matrix. The 10T1/2 cells were cocultured for 48 h with either BAE or media (DME with 2% CS) as a control. At the end of the incubation period, the cells were fixed and visualized by staining with 0.1% Coomassie blue, and migration was measured using images cast with a Profile Projector. 10T1/2 cells cultured alone (in the absence of BAE) exhibited a pattern of random movement, migrating uniformly from all edges of the circle. In contrast, 10T1/2 cells cocultured with BAE migrated toward the BAE in a directional manner (Fig. 1 a). A higher magnification view revealed that the 10T1/2 cells at the front of the well closest to the BAE migrated as single cells, extending long pseudopodia in the direction of the EC (Fig. 1 b). 10T1/2 cells at the back of the well furthest from the BAE also migrated outward, but traveled a shorter distance. In addition, these 10T1/2 cells moved as a sheet, and were flat, spread cells (Fig. 1 c) and not at all elongated as in the case of the cells closer to the BAE.
Figure 1.

BAE-induced 10T1/2 cell migration. 10T1/2 cells and BAE were cocultured in an under-agarose assay for 48 h, and then fixed and stained with Coomassie blue. (a) 10T1/2 cells (right) migrated directionally toward the BAE (left). (b) Higher magnification of 10T1/2 cells closest to BAE. 10T1/2 cells were elongated, and extended processes toward the BAE. (c) 10T1/2 cells at the back side of the well, furthest from the BAE, migrated less and as a continuous sheet with no processes. Bars: (a) 800 μm; (b and c) 200 μm.
10T1/2 cells in the presence of BAE migrated ∼500–700 μm above the background migration (∼100 μm) occurring in the absence of EC (set to zero in Fig. 2). Parallel experiments, conducted using bovine capillary EC, yielded similar results. As a test of the specificity of this effect, 10T1/2 cells were cocultured with Madin-Darby canine kidney (MDCK) cells; 10T1/2 in coculture with MDCK cells were not stimulated to migrate further than the cells cultured alone (data not shown).
Figure 2.

Effects of neutralizing PDGF antibodies on EC-induced 10T1/2 cell migration. 10T1/2 cells were cocultured in the under-agarose assay with either BAE or media (DMEM/2% CS) as a control. In some experiments, neutralizing antibodies to PDGF-B (5 μg/ml) and PDGF-A (5 μg/ml) were incorporated into the agarose before plating cells at a density of 2 × 104 cells/well in 25 μl of media. Cells were incubated for 48 h and then fixed and stained with Coomassie blue. Distance migrated was measured using a Profile Projector™ (Nikon, Inc., Melville, NY). Baseline migration of 10T1/2 cells cultured alone (vs. media) was ∼100 μm. This value was subtracted from all experimental values and the control was set to zero.
Proliferating EC have been shown to synthesize various diffusible factors, including PDGF and FGF, which can act as chemoattractants for SMC and other mesenchymally derived cells (64, 67). To determine if PDGF was mediating the EC-directed migration of the 10T1/2 cells, we first assessed its direct effects on 10T1/2 cell migration. 10T1/2 cells were plated in the presence of 20 ng/ml of each of the three PDGF isoforms. PDGF-BB was the most potent chemoattractant, followed by PDGF-AB and PDGF-AA; the 10T1/2 cells migrated 1559 ± 62, 1013 ± 36, and 512 ± 5 μm above background, respectively.
We then examined the role of PDGF in the EC-10T1/2 cell cocultures. When neutralizing antibodies against PDGF-B were added to the agarose before establishing the EC-10T1/2 cell coculture, EC-stimulated migration of 10T1/2 cells was completely inhibited. Neutralizing antibodies against PDGF A or bFGF (not shown) had no effect on EC-induced 10T1/2 cell recruitment (Fig. 2). Although in this experiment PDGF-B–neutralizing antisera caused some suppression of background 10T1/2 cell migration, and PDGF-A antisera caused mild stimulation, neither of these effects was reproducible among numerous experiments.
There is obviously an important role for proliferation in vessel formation that may also be mediated by EC-derived PDGF; we are examining this possibility in a separate series of studies. However, to eliminate the possible contribution of EC-induced 10T1/2 cell proliferation to the migration observed in the under-agarose assay, the assay was also conducted with 10T1/2 cells that had been growth- arrested with mitomycin C (45). The magnitude of EC-stimulated 10T1/2 cell migration was equivalent to that observed in assays conducted with proliferating 10T1/2 cells (data not shown).
Effects of EC on 10T1/2 Cell Phenotype
We hypothesized that EC might induce differentiation of mesenchymal cells into mural cells, that is, SMC and pericytes. To assess the effects of EC on the phenotype of 10T1/2 cells, 10T1/2 cells and BAE were cultured alone or together in the under-agarose assay for 3–6 d, allowing the two cell types to come into contact. For these experiments, BAE were prelabeled with the fluorescent molecule Di-I-Ac-LDL so that the EC in the cocultures could be distinguished from the 10T1/2 cells. All cocultures were subsequently immunostained for αSM-actin, SM-myosin, calponin, or SM22α. The SM myosin results shown were obtained with monoclonal antibody 9A9; staining with a rabbit polyclonal anti–SM-myosin antisera yielded similar results. Importantly, none of the antibodies used to assess the SMC phenotype cross-reacted with skeletal muscle. This result is demonstrated in Fig. 8, where the antibodies specifically recognized vascular SMC and not the surrounding skeletal muscle.
BAE exhibited negligible staining for αSM-actin (Fig. 3 a), SM-myosin (Fig. 3 b), calponin (Fig. 3 c), and SM22α (Fig. 3 d). 10T1/2 cells cultured alone exhibited a low but detectable level of αSM-actin staining (Fig. 3 e) and less labeling for SM-myosin (Fig. 3 f), calponin (Fig. 3 g), and SM22α (Fig. 3 h). However, when the 10T1/2 cells contacted the BAE, the levels of immunoreactivity for αSM-actin (Fig. 3 i), SM-myosin (Fig. 3 j), calponin (Fig. 3 k), and SM22α (Fig. 3 l) were significantly increased. Furthermore, in the presence of EC, the 10T1/2 cells underwent a dramatic shape change from polygonal to spindle-shaped (Fig. 3, i–l). BAE in the coculture were distinguished from the 10T1/2 cells by fluorescent visualization of EC-associated Di-I-Ac-LDL (Fig. 3, m–p; BAE are indicated by arrows), and in many cases were seen to lie underneath the 10T1/2 cells.
Figure 3.

Expression of SM-specific markers in BAE-10T1/2 cell coculture. 10T1/2 cells were incubated alone or in coculture in the under-agarose assay for 4 d with BAE that had been prelabeled with Di-I-Ac-LDL. The cultures were then fixed and immunostained for SM-specific markers. BAE exhibited negligible amounts of (a) αSM-actin, (b) SM-myosin, (c) calponin and (d) SM22α. 10T1/2 cells had low but detectable levels of (e) αSM- actin, and lesser amounts of (f) SM-myosin, (g) calponin, and (h) SM22α. 10T1/2 cells cocultured with BAE had significantly higher levels of (i) αSM-actin, (j) SM-myosin, (k) calponin, and (l) SM22α. BAE (arrows) were visualized within each coculture using fluorescent microscopy (m–p). Bar, 100 μm.
Western blot analyses revealed that αSM-actin, SM-myosin, SM22α, and calponin proteins are all expressed specifically by SMC (Fig. 4 a). A faint SM-myosin band was occasionally observed in BAE with both the monoclonal (9A9) and polyclonal (BTI) antibodies, and has been reported elsewhere (6). Changes in SM-specific protein expression were examined by Western blot analyses in experiments in which 10T1/2 cells and BAE were plated alone or simultaneously in direct cocultures (1:1) for 48 h. There was a 20-fold increase in αSM-actin protein level in 10T1/2 cells cocultured with BAE over that in 10T1/2 cells cultured alone (Fig. 4 b). SM-myosin, SM22α, and calponin were undetectable in 10T1/2 cells cultured alone, but were significantly induced in 10T1/2 cells cocultured with EC (Fig. 4 b). 10T1/2 cells cocultured with BAE did not express skeletal muscle actin or type II collagen, and did not demonstrate lipid accumulation, indicating that under these coculture conditions, 10T1/2 cells were not induced toward skeletal muscle, chondrocyte, or adipocyte lineages (data not shown).
Figure 4.
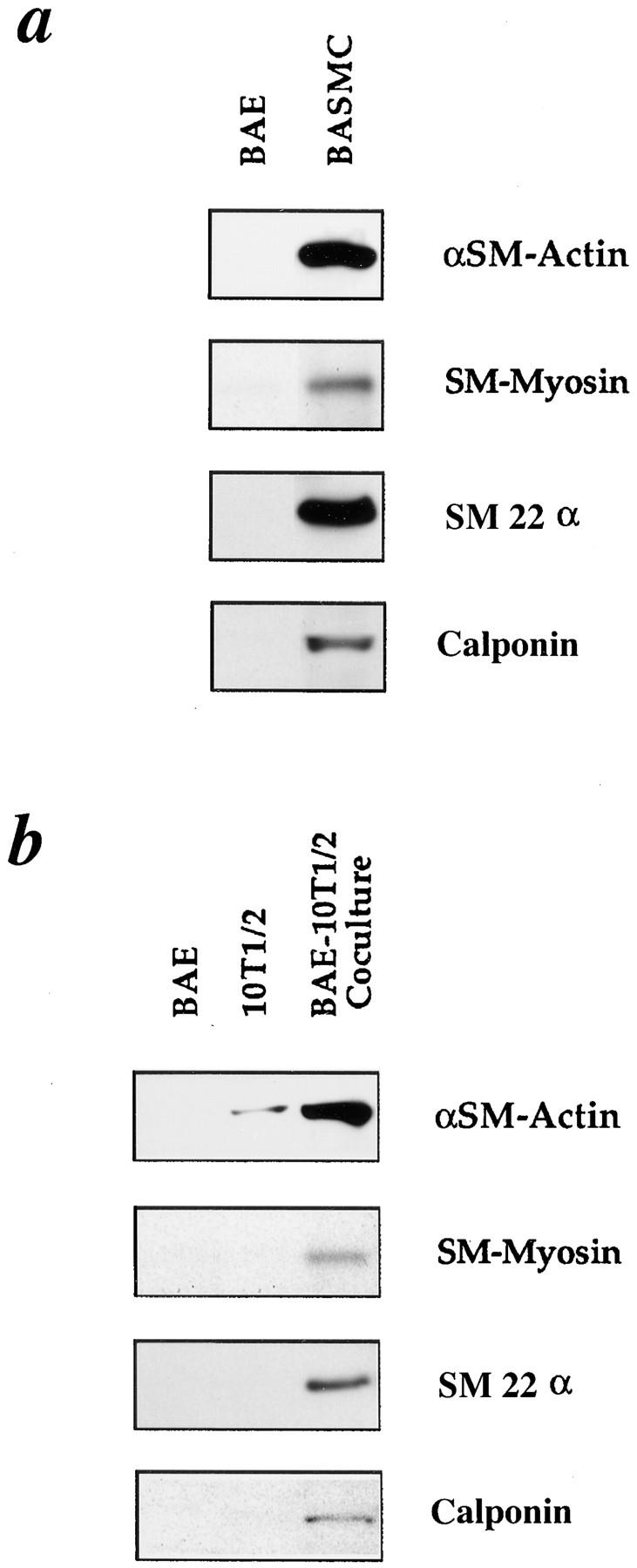
Western analyses of SM markers in BAE-10T1/2 cell cocultures. (a) Comparison of SM-specific protein expression in aortic EC and SMC. (b) Comparison of SM-specific protein expression in 10T1/2 cells in solo culture and 10T1/2 cells in coculture with BAE. 10T1/2 cells and BAE were plated alone or simultaneously in coculture (1:1) and incubated for 48 h. Protein was isolated from all cell populations and subjected to Western blot analyses. 10 μg of total protein was loaded into lanes for solo cultures of BAE, 10T1/2, and bovine aortic SMC (BASMC), and 20 μg of total protein was loaded for the cocultures. Blots were probed with antibodies to αSM-actin, SM-myosin, SM22α, or calponin, and then quantified using scanning computer densitometry.
In control studies not shown, increased expression of SM-specific proteins was also seen in 10T1/2 cells cocultured with either bovine or rodent capillary EC, indicating that these cell–cell interactions are neither species- nor vessel size–specific. Similar phenotypic changes, including increased expression of mural cell-specific markers and altered cell shape, occurred in bovine adventitial fibroblasts, human dermal fibroblasts, and human placental fibroblasts cocultured with aortic EC. However, well-differentiated cell types such as MDCK did not express SMC-specific markers when cocultured with EC. Conversely, MDCK cells did not induce a mural cell phenotype in either the 10T1/2 cells or the fibroblasts.
Mechanism of EC-Induced 10T1/2 Cell Differentiation
We were next interested in determining the factor(s) involved in the EC-induced SMC differentiation. TGF-β has been shown to influence αSM-actin expression in myofibroblasts (11), pericytes (66), and SMC (44), and we have previously shown that cocultures of EC and SMC activate TGF-β (2). Therefore, we hypothesized that TGF-β was involved in induction of the SMC phenotype in the EC-10T1/2 cocultures. To test this hypothesis, 10T1/2 cells were treated with TGF-β to determine whether the phenotypic changes observed in coculture could be mimicked by exogenous addition of this regulatory factor. Indeed, treatment of 10T1/2 cells with 1 ng/ml TGF-β1 for 24 h led to morphological changes similar to those seen in the BAE-10T1/2 cell cocultures: a change from a flat, polygonal to a polarized and elongated shape (Fig. 5). To determine if changes in SM-specific protein expression accompanied the shape change in 10T1/2 cells, Western analysis was performed on similarly treated cells. Importantly, we found that TGF-β–treated 10T1/2 cells exhibited increased expression of αSM-actin, SM-myosin proteins (Fig. 5, b and d), and SM22α (data not shown) compared with untreated controls (Fig. 5, a and c). These TGF-β–induced alterations in cell shape and differentiation were not accompanied by a change in cell proliferation.
Figure 5.

Effects of TGF-β on 10T1/2 cell phenotype. 10T1/2 cells were cultured in 4-well chamber slides in the absence (a and c) or presence (b and d) of 1 ng/ml TGF-β1 for 24 h. The cells were fixed with 4% paraformaldehyde and immunostained for αSM-actin (a and b) and SM-myosin (c and d). Bar, 40 μm.
Next, neutralizing antibodies against TGF-β (10 μg/ml) were added to the agarose before the coculture of prelabeled BAE and 10T1/2 cells in an under-agarose assay. The cocultures were allowed to incubate for 4 d followed by staining for αSM-actin and SM-myosin. The intensity of immunostaining for both proteins was significantly less in the 10T1/2 cell-EC cocultures cultivated in the presence of neutralizing TGF-β antisera (Fig. 6, b and e, respectively), compared with untreated cocultures (Fig. 6, a and d, respectively). The fluorescently-labeled EC of the cocultures incubated with anti-TGF-β antibodies are visualized in Fig. 6, c and f. Western blot analyses revealed that the neutralization of TGF-β suppressed the levels of αSM-actin by 95% (Fig. 7 a), and SM-myosin protein expression by 87% (Fig. 7 b). The specificity of this effect was demonstrated in parallel studies in which PDGF-B–neutralizing antisera were shown to have no effect on 10T1/2 cell differentiation. Cellular toxicity due to the neutralizing antibody to TGF-β was ruled out in related studies (Hirschi et al., manuscript submitted for publication) showing that the antibody did not affect the growth of either BAE or 10T1/2 cells.
Figure 6.

Immunohistochemical analysis of the effect of anti-TGF-β on αSM-actin and SM-myosin in BAE-10T1/2 cell cocultures. 10T1/2 and BAE cells were cocultured in the under-agarose assay for 4 d in the presence (b, c, e, and f) or absence (a and d) of a neutralizing antibody to TGF-β (10 μg/ml) and then stained for αSM-actin (a and b) or SM-myosin (e and d). BAE prelabeled with fluorescent Di-I-Ac-LDL for identification in the anti-TGF-β-treated cocultures are shown in c and f. Bar, 40 μm.
Figure 7.
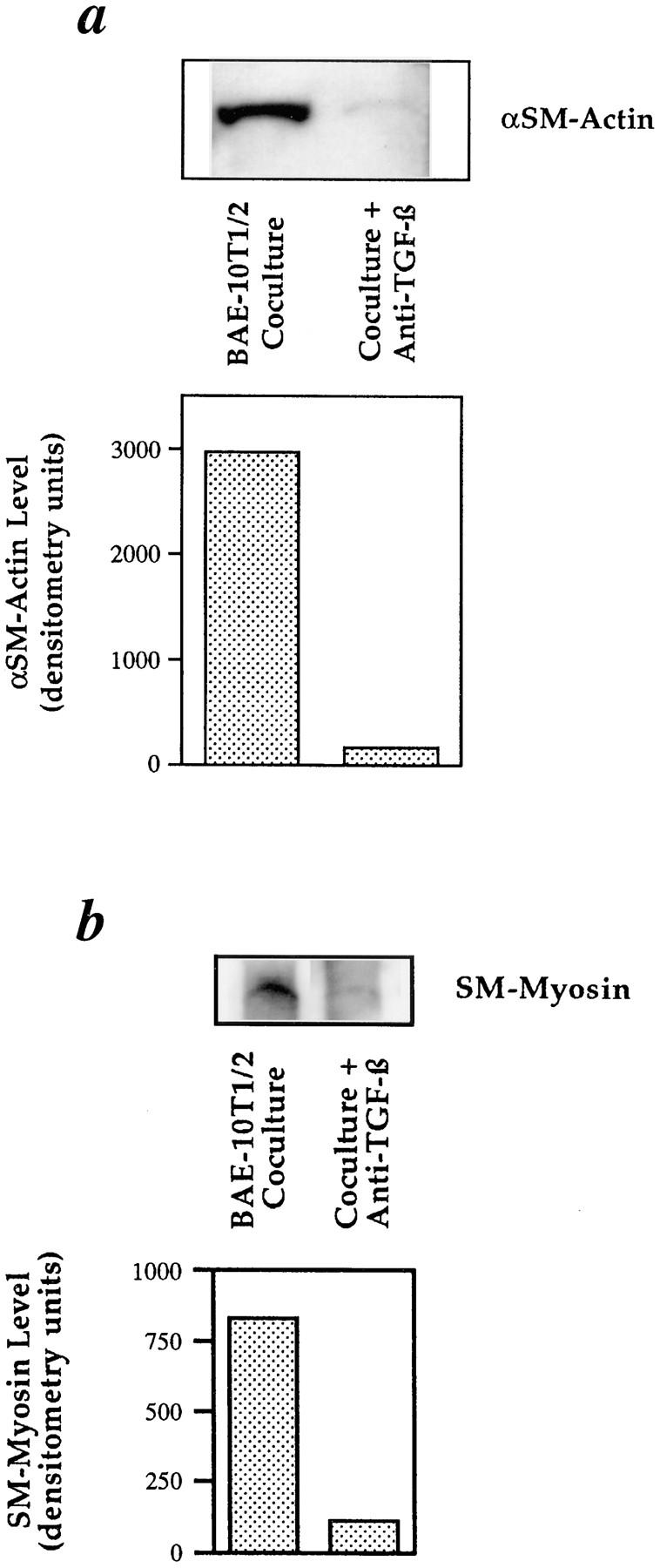
Western blot analyses of the effect of TGF-β neutralization on expression of αSM-actin and SM-myosin in BAE-10T1/2 cell cocultures. 10T1/2 and BAE cells were cocultured (1:1) for 48 h in the presence or absence of a neutralizing antibody to TGF-β (10 μg/ml). Protein was isolated from all treatment groups and subjected to Western blot analyses for (a) αSM-actin and (b) SM-myosin and quantified using scanning computer densitometry.
Endothelial–Mesenchymal Interactions In Vivo
Our in vitro observations indicated that EC can recruit undifferentiated mesenchymal cells and direct their differentiation into smooth muscle. To determine if mesenchymal cells can become recruited toward, and incorporated into, newly forming vessels in a more complex in vivo environment, fluorescently-labeled 10T1/2 cells embedded in a cross-linked collagen matrix were implanted subcutaneously onto the backs of mice.
After 10 d in the mouse, the experimental tissue was excised, and the newly formed vessels were examined for the presence of the labeled 10T1/2 cells. Examination of the sections by fluorescent microscopy revealed that the fluorescently-labeled 10T1/2 cells had become associated with vessels in tissue from the back of the mouse; red fluorescent labeling is specifically seen in cells associated with the vessel wall (arrowhead; Fig. 8). Furthermore, once incorporated into the vessel, the prelabeled cells stained strongly for αSM-actin, SM-myosin, and calponin. Importantly, none of the SM-specific antisera cross-reacted with skeletal muscle tissue seen in these sections, demonstrating the specificity of these reagents for vascular SMC. Interestingly, in parallel studies conducted in the chick chorioallantoic membrane assay, prelabeled 10T1/2 were observed to become incorporated into other nonvascular portions of the chorioallantoic membrane, but did not express SM markers at these sites (data not shown).
Discussion
Development of the vascular system is a complex process that begins with the formation of simple tubes. In vasculogenesis, endothelial precursors called angioblasts associate to form vessel tubes. Major axial vessels such as the aorta, and the vascular plexus of endodermal organs such as the spleen, are formed by vasculogenesis (8, 48). In angiogenesis, vessels form by sprouting from pre-existing vessels. Vascularization of neural and mesodermal tissues, like the brain and limb buds, occurs by angiogenesis (15, 47, 61). In both cases, the primitive vessel tubes then remodel, forming the more complex architecture of the adult vasculature. Developmental studies suggest that the EC in these nascent tubes may govern the acquisition of additional vessel layers (23, 38). Knowledge of the mechanism(s) by which EC recruit mural cell precursors and enable their differentiation remains unknown, and would significantly add to the understanding not only of blood vessel development, but of pathological conditions such as atherosclerosis or vascular malformations, in which these basic processes appear to be deregulated.
Our studies demonstrate that EC do indeed recruit undifferentiated mesenchymal cells. Furthermore, EC-derived PDGF-BB mediates the mesenchymal movement. We found that the ability of BAE as well as bovine or rodent capillary EC to recruit 10T1/2 cells (presumptive mural cell-precursors) was totally blocked by a neutralizing antibody to PDGF-B. PDGF has been shown in previous in vitro studies to act as a mitogen and chemoattractant for mesenchymal cells (67). Neutralizing antibodies to PDGF-A or to bFGF had no effect on EC recruitment of 10T1/2 cells, even though these factors have been shown to be mitogenic and chemotactic for SMC and fibroblasts. These findings suggest that EC-derived PDGF-B specifically exerts a paracrine effect on undifferentiated mesenchymal cells during vascular development in vivo. Observations by Holmgren et al. (24) on the expression of PDGF ligand and receptors in forming blood vessels of human placenta, support this concept. They found that EC of developing blood vessels express the mRNA and protein for PDGF-B, but not the PDGF-β receptor, whereas PDGF-β receptor mRNA was detectable in fibroblast-like cells and SMC surrounding intermediate and large blood vessels.
The phenotypes of mice deficient for components of the PDGF system lend further support to our hypothesis. Patch mice that have a spontaneous mutation deleting the PDGF-α receptor are embryonic lethal, and have cardiovascular defects characterized by reduced numbers of SMC (56). Mice lacking PDGF-B also exhibited severe cardiovascular and renal abnormalities (31). The lack of mesangial cells in the kidneys of PDGF-B null mice is of particular interest, since mesangial cells are considered to be specialized pericytes and developmentally related to SMC (57). Indeed, more recently, PDGF-B null mice have been reported to lack pericytes (32).
Our studies show that once undifferentiated mesenchymal cells are recruited to EC, they are induced to become SMC-like. Mature SMC and pericytes in intact vessels express a series of proteins that define the SMC lineage (for review see reference 58). These include, but are not limited to, αSM-actin (46, 65), SM-myosin (29, 52), calponin (3, 63), SM22α (14, 30, 58), h-caldesmon (59), and desmin (39). A variety of cells including astrocytes (20) and myofibroblasts (11) have been reported to express αSM-actin in culture. Therefore, induction of αSM-actin in an otherwise undifferentiated cell is not evidence for differentiation towards an SMC lineage. Furthermore, a few of these proteins expressed specifically in adult SMC are also transiently expressed in other cell types (e.g., cardiac muscle) during embryogenesis (36, 37). However, simultaneous expression of multiple SM-specific proteins is indicative of a SM phenotype. Hence, simultaneous expression of αSM-actin, SM-myosin, SM22α, and calponin in 10T1/2 cells in culture with EC, along with a dramatic change in cell shape, provides strong evidence that these mesenchymal cells are truly induced toward the smooth muscle lineage. Certainly it is possible that this coculture system will have limitations and will not reproduce all aspects of differentiated SMC function. However, the biologically relevant induction of SM-specific protein expression in 10T1/2 cells, which can be easily genetically manipulated, make this an ideal system in which to investigate the molecular aspects of SM-specific gene expression.
We demonstrated that induction of 10T1/2 to an SMC phenotype occurs only after the mesenchymal cells come into close proximity with the EC. This sequence of events has been shown to occur during development of the aorta in the quail. Hungerford et al. found that mesodermal cells become associated with the developing aorta, beginning at the ventral surface, and that they express αSM-actin only after associating with the EC tube (26). By recreating aspects of vessel assembly in vitro, we have begun to investigate directly the mechanisms involved in this EC–mesenchymal interaction. We have found that the induction of the SMC phenotype in this model is mediated at least in part by TGF-β. In addition to showing that direct treatment of 10T1/2 cells with TGF-β1 induces expression of SM-specific markers, we have demonstrated that neutralizing antibodies against TGF-β blocks induction of SM-specific markers in the cocultures.
Although the cells in these cocultures make contact, it is not clear if physical contact between the cells is necessary for the inductive event. Speculation regarding the source of the TGF-β in the cocultures is based on previous studies from our lab and others, showing that both EC and mural cells, when grown separately, produce a latent form of TGF-β that is activated in EC-mural cell cocultures (2, 55). The demonstration that local activation of TGF-β may be involved in directing undifferentiated mesenchymal cells to an SMC lineage in the vessel wall is consistent with previous observations of elevated TGF-β in mesenchymal remodeling, and at sites of epithelial–mesenchymal interactions (22, 49).
An increasing body of evidence indicates roles for members of the TGF-β family in regulating muscle differentiation in general. TGF-β has been implicated in the regulation of both cardiac and skeletal muscle differentiation (for review see references 42 and 43). The epithelial–mesenchymal transformation in embryonic heart can be mimicked by TGF-β (49), and antisense against TGF-β3 blocks epithelial–mesenchymal transformation of cardiac endothelial cells (49). Expression of a dominant-negative form of the type II TGF-β receptor suppressed myogenic differentiation in a culture model (16). Furthermore, data from gene disruption studies suggest that a new member of the TGF-β family, growth/differentiation factor-8, acts as a negative regulator of skeletal muscle growth (35).
Whether all of the effects of TGF-β on SM-specific gene expression in this model are direct remains to be determined. To date, regulatory regions identified as conferring responsiveness to TGF-β have been found within the 5′ flanking sequences of αSM-actin (21). The 5′ and 3′ regulatory regions of other SMC-specific genes, including SM-myosin and SM22α, are under investigation (27, 68). In addition, some of the effects of TGF-β on vascular wall cell differentiation may be due to TGF-β–induced changes in the extracellular matrix (34). It is well known that the matrix can influence cell growth, polarity, and organization as well as differentiation (1, 62). In fact, the interaction of presumptive SMC and pericytes with the abluminal EC surface are temporally associated with deposition of a basement membrane (9). In this regard, it is interesting to speculate that TGF-β, activated when mural cells associate with EC, may serve several functions, all aimed at establishing a mature vessel. These include inhibition of endothelial proliferation (45) and migration (54), stimulation of SMC/pericyte differentiation (our present observations), and induction of basement membrane assembly (34).
Observations of mice with a targeted disruption of TGF-β support this concept. Fifty percent of TGF-β1 null mice die in utero from a defect in yolk sac vasculogenesis, which is thought to be due to improper interactions between epithelial cells and mesenchymal cells, and may be the result of altered cell–matrix interactions (12). This observation also suggests that local activation of TGF-β, resulting from such cell–cell interactions, may occur widely throughout development. Whether TGF-β activation is a constitutive event at the site of EC–mural cell contact is not known. The reversibility of the SMC phenotype (4, 7) and the concept that differentiation may require constant signaling, at least under some circumstances (5), leads us to suspect that local activation of TGF-β is an ongoing process. In fact, it is well-documented that the endothelium makes frequent contacts with SMC and pericytes throughout the vasculature (51, 60).
Although we have shown that 10T1/2 cells are capable of becoming SMC-like in vitro in response to EC, we wanted to confirm that 10T1/2 cells have the capacity to become incorporated into developing vessels in vivo. Therefore, undifferentiated 10T1/2 cells were permanently labeled with a fluorescent dye, and were placed in the proximity of developing vessels in collagen matrices subcutaneously in the mouse. Not only did these cells become incorporated into the medial layers of the newly forming vessels (Fig. 8), but once associated with the vessel, simultaneously expressed αSM-actin, SM-myosin, and calponin, reflective of a SM phenotype. The resolution was not sufficient to comment on the role of contact in the induction of the SMC phenotype in these vessels. However, the 10T1/2 cells expressing SM markers usually comprised the innermost layer of cells in the vessel wall.
In summary, we have developed coculture systems to identify potential regulators of vessel formation, and to elucidate their relative contributions. A complimentary in vivo system was established, and observations of developing vessels in the model corroborate our tissue culture observations. We believe that the information gained from this system regarding the cellular and molecular regulation of vessel formation will be important in understanding not only developmental regulation, but also pathophysiological processes such as atherosclerosis and vascular malformations, where there appear to be defects in the normal control mechanisms.
Acknowledgments
The authors thank Dr. Shmuel Ben-Sasson, Dr. Diane Darland, Laurence Beck, and Sandra Smith for their critical reading of the manuscript. We gratefully acknowledge the contribution of Evelyn Flynn in immunohistochemistry of the mouse tissue. We thank Dr. Gary Owens, Dr. Marina Glukhova, and Dr. Mario Gimona for their generosity with their antibody reagents.
This work was supported by National Institutes of Health grants EY05318 and CA45548 to P.A. D'Amore. Dr. Karen Hirschi is supported by a National Institutes of Health Postdoctoral Fellowship (HL09037), and Dr. Stephanie Rohovsky was supported by the Harvard-Longwood Research Training Program Grant in Vascular Surgery (HL 07734).
Abbreviations used in this paper
- Ac-LDL
acetylated low density lipoprotein
- BAE
bovine aortic EC
- CS
calf serum
- EC
endothelial cells
- SM
smooth muscle
- SMC
smooth muscle cells
Footnotes
Address all correspondence to Patricia A. D'Amore, Laboratory for Surgical Research, Children's Hospital, Enders 1061, 300 Longwood Ave., Boston, MA 02115. Tel.: 617-355-8377. Fax: 617-355-7043. E-mail: damore_p@a1.tch.harvard.edu
The current address of Karen K. Hirschi is Children's Nutrition Research Center, Baylor College of Medicine, 1100 Bates Street, Houston, TX 77030. The current address of Stephanie A. Rohovsky is Department of Surgery, Beth Israel-Deaconess Hospital, Boston, Massachusetts 02115.
References
- 1.Adams JC, Watt FM. Regulation of development and differentiation by the extracellular matrix. Development. 1993;117:1183–1198. doi: 10.1242/dev.117.4.1183. [DOI] [PubMed] [Google Scholar]
- 2.Antonelli-Orlidge A, Saunders KB, Smith SR, D'Amore PA. An activated form of transforming growth factor β is produced by cocultures of endothelial cells and pericytes. Proc Natl Acad Sci USA. 1989;86:4544–4548. doi: 10.1073/pnas.86.12.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Applegate D, Feng W, Green RS, Taubman MB. Cloning and expression of a novel acidic calponin isoform from rat aortic vascular smooth muscle. J Biol Chem. 1994;269:10683–10690. [PubMed] [Google Scholar]
- 4.Blank RS, Thompson MM, Owens GK. Cell cycle versus density dependence of smooth muscle alpha actin expression in cultured rat aortic smooth muscle cells. J Cell Biol. 1988;107:299–306. doi: 10.1083/jcb.107.1.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blau HM, Baltimore D. Differentiation requires continuous regulation. J Cell Biol. 1991;112:781–783. doi: 10.1083/jcb.112.5.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borrione A, Zanellato AMC, Giuriato L, Scannapieco G, Pauletto P, Saltore S. Nonmuscle and smooth muscle myosin isoforms in bovine endothelial cells. Exp Cell Res. 1990;190:1–10. doi: 10.1016/0014-4827(90)90136-x. [DOI] [PubMed] [Google Scholar]
- 7.Chamley-Campbell JH, Campbell GR, Ross R. Phenotype-dependent response of cultured aortic smooth muscle to serum mitogens. J Cell Biol. 1981;89:379–383. doi: 10.1083/jcb.89.2.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coffin JD, Poole TJ. Embryonic vascular development: immunohistochemical identification of the origin and subsequent morphogenesis of the major vessel primordia in quail embryo. Development. 1988;102:1–14. doi: 10.1242/dev.102.4.735. [DOI] [PubMed] [Google Scholar]
- 9.Crocker DJ, Murad TM, Greer JC. Role of the pericyte in wound healing. An ultrastructural study. Exp Mol Pathol. 1970;13:51–65. doi: 10.1016/0014-4800(70)90084-5. [DOI] [PubMed] [Google Scholar]
- 10.Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51:987–1000. doi: 10.1016/0092-8674(87)90585-x. [DOI] [PubMed] [Google Scholar]
- 11.Desmouliere A, Genioz A, Gabbiani F, Gabbiani G. Transforming growth factor-β1 induces α-smooth muscle cell actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122:103–111. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-β1 knock-out mice. Development. 1995;121:1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- 13.Doetschman, T.A., A. Gossler, and R. Kemler. 1987. Blastocyst-derived embryonic stem cells as a model for embryogenesis. In Future Aspects in Human In Vitro Fertilization. W. Feichtingen and P. Kemeter, editors. Springer-Verlag, Berlin. 187–195.
- 14.Duband J-L, Gimona M, Scatena M, Sartore S, Small JV. Calponin and SM22 as differentiation markers of smooth muscle: spatiotemporal distribution during avian embryonic development. Differentiation. 1993;55:1–11. doi: 10.1111/j.1432-0436.1993.tb00027.x. [DOI] [PubMed] [Google Scholar]
- 15.Ekblom P, Sariola H, Karkinen-Jaaskelainen M, Saxen L. The origin of the glomerular endothelium. Cell Differ. 1982;11:35–39. doi: 10.1016/0045-6039(82)90014-8. [DOI] [PubMed] [Google Scholar]
- 16.Filvaroff EH, Ebner R, Derynck R. Inhibition of myogenic differentiation in myoblasts expressing a truncated type ii TGF-β receptor. Development. 1994;120:1085–1095. doi: 10.1242/dev.120.5.1085. [DOI] [PubMed] [Google Scholar]
- 17.Gazit D, Ebner R, Kahn AJ, Derynck R. Modulation of expression and cell surface binding of members of the transforming growth factor-beta superfamily during retinoic acid-induced osteoblastic differentiation of multipotential mesenchymal cells. Mol Endocrinol. 1993;7:189–198. doi: 10.1210/mend.7.2.8385738. [DOI] [PubMed] [Google Scholar]
- 18.Gimbrone MA, Cotran RS, Folkman J. Human vascular endothelial cells in culture. J Cell Biol. 1974;60:673–684. doi: 10.1083/jcb.60.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gimona M, Herzog M, Vandekerckhove J, Small JV. Smooth muscle specific expression of calponin. FEBS. 1990;274:159–162. doi: 10.1016/0014-5793(90)81353-p. [DOI] [PubMed] [Google Scholar]
- 20.Guidry C. Isolation and characterization of porcine Muller cells. Invest Ophthamol Vis Sci. 1996;37:740–752. [PubMed] [Google Scholar]
- 21.Hautmann MB, Madsen CS, Owens GK. A transforming growth factor-beta (TGF-beta) control element drives TGF-β-induced stimulation of smooth-muscle alpha-actin gene expression in concert with 2 CARG elements. J Biol Chem. 1997;272:948–956. doi: 10.1074/jbc.272.16.10948. [DOI] [PubMed] [Google Scholar]
- 22.Heine UI, Munoz EF, Flanders KC, Ellingworth LR, Lam H-YP, Thompson NL, Roberts AB, Sporn MB. Role of transforming growth factor-β in the development of the mouse embryo. J Cell Biol. 1987;105:2861–2876. doi: 10.1083/jcb.105.6.2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmgren L. Potential dual roles of PDGF-B during human placental blood vessel formation. Endothelium. 1993;1:167–171. [Google Scholar]
- 24.Holmgren L, Glaser A, Pfeifer-Ohlsson S, Ohlsson R. Angiogenesis during human extraembryonic development involves the spatiotemporal control of PDGF ligand and receptor gene expression. Development. 1991;113:749–754. doi: 10.1242/dev.113.3.749. [DOI] [PubMed] [Google Scholar]
- 25.Horan PK, Slezak SE. Stable cell membrane labeling. Nature. 1989;340:167–168. doi: 10.1038/340167a0. [DOI] [PubMed] [Google Scholar]
- 26.Hungerford JE, Owens GK, Argraves WS, Little CD. Development of the aortic vessel wall as defined by vascular smooth muscle and extracellular matrix markers. Dev Biol. 1996;178:375–392. doi: 10.1006/dbio.1996.0225. [DOI] [PubMed] [Google Scholar]
- 27.Kim S, Ip HS, Lu M, Clendenin C, Parmacek MS. A serum response factor-dependent transcriptional regulatory program identified distinct smooth muscle cell sublineages. Mol Cell Biol. 1997;17:2266–2278. doi: 10.1128/mcb.17.4.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirby ML, Waldo KL. Neural crest and cardiovascular patterning. Circ Res. 1995;77:211–215. doi: 10.1161/01.res.77.2.211. [DOI] [PubMed] [Google Scholar]
- 29.Kuro-o M, Nagai R, Nakahara K-I, Katoh H, Tsai R-C, Tsuchimochi H, Yazaki Y, Ohkubo A, Takaku F. cDNA cloning of a myosin heavy chain isoform in embryonic smooth muscle and its expression during vascular development and in arteriosclerosis. J Biol Chem. 1991;266:3766–3773. [PubMed] [Google Scholar]
- 30.Lees-Miller JP, Heeley DH, Smillie LB, Kay CM. Isolation and characterization of an abundant and novel 22-kDa protein (SM22α) from chicken gizzard smooth muscle. J Biol Chem. 1987;262:2988–2993. [PubMed] [Google Scholar]
- 31.Leveen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E, Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994;8:1875–1887. doi: 10.1101/gad.8.16.1875. [DOI] [PubMed] [Google Scholar]
- 32.Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 33.Lu Y, Raptis L, Anderson S, Corbley MJ, Zhou YC, Pross H, Haloitis T. Ras modulates commitment and maturation of 10T1/2 fibroblasts to adipocytes. Biochem Cell Biol. 1992;70:1249–1257. doi: 10.1139/o92-171. [DOI] [PubMed] [Google Scholar]
- 34.Madri JA, Pratt BM, Tucker AM. Phenotypic modulation of endothelial cells by transforming growth factor-beta depends upon the composition and organization of the extracellular matrix. J Cell Biol. 1988;106:1375–1384. doi: 10.1083/jcb.106.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McPherron AC, Lawler AM, Lee S-J. Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 36.Miano J, Cserjesi P, Ligon K, Periasamy M, Olson E. Smooth muscle myosin heavy chain marks the smooth muscle lineage during mouse embryogenesis. Circ Res. 1994;75:803–812. doi: 10.1161/01.res.75.5.803. [DOI] [PubMed] [Google Scholar]
- 37.Miano JM, Olson EN. Expression of the smooth muscle cell calponin gene marks the early cardiac and smooth muscle cell lineages during mouse embryogenesis. J Biol Chem. 1996;271:7095–7103. doi: 10.1074/jbc.271.12.7095. [DOI] [PubMed] [Google Scholar]
- 38.Nakamura H. Electron microscopic study of the prenatal development of the thoracic aorta in the rat. Am J Anat. 1988;181:406–418. doi: 10.1002/aja.1001810409. [DOI] [PubMed] [Google Scholar]
- 39.Nanaev AK, Shirinsky VP, Birukov KG. Immunofluorescent study of heterogeneity in smooth muscle cells of human fetal vessels using antibodies to myosin, desmin, and vimentin. Cell Tissue Res. 1991;266:535–540. doi: 10.1007/BF00318595. [DOI] [PubMed] [Google Scholar]
- 40.Nelson RD, Quie PQ, Simmons RL. Chemotaxis under agarose: a new and simple method for measuring chemotaxis and spontaneous migration of human polymorphonuclear leukocytes and monocytes. J Immunol. 1975;115:1650–1656. [PubMed] [Google Scholar]
- 41.Noden DM. Embryonic origins and assembly of blood vessels. Am Rev Respir Dis. 1989;140:1097–1103. doi: 10.1164/ajrccm/140.4.1097. [DOI] [PubMed] [Google Scholar]
- 42.Olson EN, Srivastava D. Molecular pathways controlling heart development. Science. 1996;272:671–675. doi: 10.1126/science.272.5262.671. [DOI] [PubMed] [Google Scholar]
- 43.Olson EN, Sternberg E, Hu JS, Spizz G, Wilcox C. Regulation of myogenic differentiation by type beta transforming growth factor. J Cell Biol. 1986;103:1799–1805. doi: 10.1083/jcb.103.5.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Orlandi A, Ropraz P, Gabbiani G. Proliferative activity and α-smooth muscle actin expression in cultured rat aortic smooth muscle cells are differently modulated by transforming growth factor-β1 and heparin. Exper Cell Res. 1994;214:528–536. doi: 10.1006/excr.1994.1290. [DOI] [PubMed] [Google Scholar]
- 45.Orlidge A, D'Amore PA. Inhibition of capillary endothelial cell growth by pericytes and smooth muscle cells. J Cell Biol. 1987;105:1455–1462. doi: 10.1083/jcb.105.3.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Owens GK, Loeb A, Gordon D, Thompson MM. Expression of smooth muscle-specific α-isoactin in cultured vascular smooth muscle cells: relationship between growth and cytodifferentiation. J Cell Biol. 1986;102:343–352. doi: 10.1083/jcb.102.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pardanaud L, Dieterlen-Lièvre F. Expression of C-ETS1 in early chick embryo mesoderm: Relationship to the hemangioblastic lineage. Cell Adhes Commun. 1993;1:151–160. doi: 10.3109/15419069309095691. [DOI] [PubMed] [Google Scholar]
- 48.Pardanaud L, Yassine F, Dieterlen-Lièvre F. Relationship between vasculogenesis, angiogenesis and haemopoiesis during avian ontogeny. Development. 1989;105:473–485. doi: 10.1242/dev.105.3.473. [DOI] [PubMed] [Google Scholar]
- 49.Potts JD, Runyan RB. Epithelial-mesenchymal cell transformation in the embryonic heart can be mediated, in part, by transforming growth factor β. Dev Biol. 1989;134:392–401. doi: 10.1016/0012-1606(89)90111-5. [DOI] [PubMed] [Google Scholar]
- 50.Price R, Owens G, Skalak T. Immunohistochemical identification of arteriolar development using markers of smooth muscle differentiation. Circ Res. 1994;75:520–527. doi: 10.1161/01.res.75.3.520. [DOI] [PubMed] [Google Scholar]
- 51.Robison WG, Jr, Magata M, Tillis TN, Laver N, Kinoshita JH. Aldose reductase and pericyte-endothelial cell contacts in retina and optic nerve. Invest Ophthalmol Vis Sci. 1989;30:2293–2299. [PubMed] [Google Scholar]
- 52.Rovner AS, Murphy RA, Owens GK. Expression of smooth muscle and nonmuscle myosin heavy chains in cultured vascular smooth muscle cells. J Biol Chem. 1986;261:14740–14745. [PubMed] [Google Scholar]
- 53.Ruch RJ, Bonney WJ, Sigler K, Guan X, Matesic D, Schafer LD, Dupont E, Trosko JE. Loss of gap junctions from DDT-treated rat liver epithelial cells. Carcinogenesis. 1994;15:301–306. doi: 10.1093/carcin/15.2.301. [DOI] [PubMed] [Google Scholar]
- 54.Sato Y, Rifkin DB. Inhibition of endothelial cell movement by pericytes and smooth muscle cells: activation of a latent transforming growth factor-beta 1-like molecule by plasmin during co-culture. J Cell Biol. 1989;109:309–315. doi: 10.1083/jcb.109.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sato Y, Tsuboi R, Lyons R, Moses H, Rifkin DB. Characterization of the activation of latent TGF-β by co-cultures of endothelial cells and pericytes or smooth muscle cells: a self-regulating system. J Cell Biol. 1990;111:757–763. doi: 10.1083/jcb.111.2.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schatteman GC, Motley ST, Effmann EL, Bowen-Pope DF. Platelet-derived growth factor receptor alpha subunit deleted patchmouse exhibits severe cardiovascular dysmorphogenesis. Teratology. 1995;51:351–366. doi: 10.1002/tera.1420510602. [DOI] [PubMed] [Google Scholar]
- 57.Schlondorff D. The glomerular mesangial cell: an expanding role for a specialized pericyte. FASEB J. 1987;1:272–281. doi: 10.1096/fasebj.1.4.3308611. [DOI] [PubMed] [Google Scholar]
- 58.Shanahan CM, Weissberg PL, Metcalfe JC. Isolation of gene markers of differentiated and proliferating vascular smooth muscle cells. Circ Res. 1993;73:193–204. doi: 10.1161/01.res.73.1.193. [DOI] [PubMed] [Google Scholar]
- 59.Sobue K, Muramoto Y, Fujita M, Kakiuchi S. Purification of a calmodulin-binding protein from chicken gizzard that interacts with F-actin. Proc Natl Acad Sci USA. 1981;78:5652–5655. doi: 10.1073/pnas.78.9.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spagnoli LG, Villaschi S, Neri L, Palmieri G. Gap junctions in myo-endothelial bridges of rabbit carotid arteries. Experientia. 1982;38:124–125. doi: 10.1007/BF01944566. [DOI] [PubMed] [Google Scholar]
- 61.Stewart PA, Wiley MJ. Developing nervous tissue induces formation of blood-brain barrier characteristics in invading endothelial cells: a study using quail-chick transplantation chimeras. Dev Biol. 1981;84:183–192. doi: 10.1016/0012-1606(81)90382-1. [DOI] [PubMed] [Google Scholar]
- 62.Streuli CH, Bailey N, Bissell MJ. Control of mammary epithelial differentiation: Basement membrane induces tissue-specific gene expression in the absence of cell-cell interaction and morphological polarity. J Cell Biol. 1991;115:1383–1395. doi: 10.1083/jcb.115.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takeuchi K, Tskahashi K, Abe M, Nishida W, Hiwada K, Nabeya T, Maruyama K. Co-localization of immunoreactive forms of calponin with actin cytoskeleton in platelets, fibroblasts, and vascular smooth muscle. J Biochem. 1991;109:311–316. [PubMed] [Google Scholar]
- 64.Terranova VP, DiFlorio R, Lyall RM, Hic S, Freisel R, Maciag R. Human endothelial cells are chemotactic to endothelial cell growth factor and heparin. J Cell Biol. 1985;101:2330–2334. doi: 10.1083/jcb.101.6.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vanderkerckhove J, Weber K. The complete amino acid sequence of actins from bovine aorta, bovine heart, bovine fast skeletal muscle, and rabbit slow skeletal muscle. A protein-chemical analysis of muscle actin differentiation. Differentiation. 1979;14:123–133. doi: 10.1111/j.1432-0436.1979.tb01021.x. [DOI] [PubMed] [Google Scholar]
- 66.Verbeek MM, Otte-Höller I, Wesseling P, Ruiter DJ, de Waal RMW. Induction of α-smooth muscle actin expression in cultured human brain pericytes by transforming growth factor-β1. Am J Pathol. 1994;144:372–382. [PMC free article] [PubMed] [Google Scholar]
- 67.Westermark B, Siegbahn A, Heldin C-H, Claesson-Welsh L. B-type receptor for platelet-derived growth factor mediates a chemotactic response by means of ligand-induced activation of the receptor protein-tyrosine kinase. Proc Natl Acad Sci USA. 1990;87:128–132. doi: 10.1073/pnas.87.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.White SL, Low RB. Identification of promoter elements involved in cell-specific regulation of smooth muscle myosin heavy chain gene transcription. J Biol Chem. 1996;271:15008–15017. doi: 10.1074/jbc.271.25.15008. [DOI] [PubMed] [Google Scholar]