

Abstract
Neurofilaments (NFs) are prominent components of large myelinated axons and probably the most abundant of neuronal intermediate filament proteins. Here we show that mice with a null mutation in the mid-sized NF (NF-M) subunit have dramatically decreased levels of light NF (NF-L) and increased levels of heavy NF (NF-H). The calibers of both large and small diameter axons in the central and peripheral nervous systems are diminished. Axons of mutant animals contain fewer neurofilaments and increased numbers of microtubules. Yet the mice lack any overt behavioral phenotype or gross structural defects in the nervous system. These studies suggest that the NF-M subunit is a major regulator of the level of NF-L and that its presence is required to achieve maximal axonal diameter in all size classes of myelinated axons.
Neurofilaments (NFs)1 are the most prominent cytoskeletal components in large myelinated axons and probably the most abundant and widely expressed of neuronal intermediate filament (IF) proteins. In mammals, NFs are composed of three proteins termed light (NF-L), mid-sized (NF-M), and heavy (NF-H) NFs. These proteins are encoded by separate genes (17, 21, 27) and have apparent molecular weights of ∼68,000, 150,000, and 200,000, respectively, when separated on SDS-PAGE gels.
Like all IFs, NF proteins contain a relatively well-conserved α helical rod domain of ∼310 amino acids with variable NH2-terminal and COOH-terminal regions (33). In NFs, the COOH-terminal domains are greatly extended relative to other IFs and contain a glutamic acid–rich region of unknown significance and in NF-M and NF-H a series of lysine-serine-proline-valine (KSPV) repeats (21, 27) which are major sites of phosphorylation in both proteins. In axons, NFs form bundles of 10-nm diameter “core filaments” with sidearms consisting of phosphorylated COOH-terminal tail sequences of NF-M and NF-H (12, 13, 26, 29) that have been thought to extend and maintain the spacing between filaments (4). Similar sidearm extensions are not found in IFs composed of other IF proteins such as desmin, glial fibrillary acidic protein, or vimentin. In NFs assembled in vitro, all three subunits appear to be incorporated into core filaments (12, 26). Thus, current models of NF assembly suggest that NF-M and NF-H are the major components of sidearm extensions and are anchored to a core of NF-L via their central rod domains.
Although much is known about NF structure and assembly, questions remain concerning NF function. A primarily structural role for NFs is suggested by their prominence in large axons (41). Small unmyelinated axons contain few NFs (9) and some small neurons lack morphologically identifiable NFs (3, 32, 38). Most dendrites contain few NFs and only in dendrites of large neurons such as motor neurons are NFs numerous (41).
A role for NFs as a major determinant of axonal diameter has long been suspected from the correlation between NF content in axonal cross sections and axonal caliber (16). This correlation persists during axonal degeneration and regeneration (14) and changes in NF transport correlate temporally with alterations in the caliber of axons in regenerating nerves (15). Additionally, fewer NFs occur at nodes of Ranvier where axonal diameter is reduced (1), and certain NF epitopes are found only in regions where maximal axonal caliber has developed (6).
Several animal models have supported a role for NFs in establishing axonal diameter. One is a Japanese quail (Quiverer) with a spontaneous mutation in NF-L that generates a truncated protein incapable of forming NFs (31). Homozygous mutants contain no axonal NFs and exhibit a mild generalized quivering. In these animals, radial growth of myelinated axons is severely attenuated (44) with a consequent reduction in axonal conduction velocity (37). In transgenic mice, Eyer and Petersen (8) expressed an NF-H/β-galactosidase fusion protein in which the COOH terminus of NF-H was replaced by β-galactosidase. NF inclusions were found in the perikarya of neurons and the resulting NF aggregates blocked all NF transport into axons resulting in axons with reduced calibers. More recently, Zhu et al. (45) have shown that mice lacking NFs due to a targeted disruption of the NF-L gene have diminished axonal calibers and delayed maturation of regenerating myelinated axons.
Although these models clearly suggest a role for NFs in establishing axonal diameter, they contribute only limited information concerning the roles of the individual NF subunits. During development, NF-L and NF-M are coexpressed initially whereas NF-H appears later (4). Studies in transgenic mice have found that overexpressing mouse NF-L leads to an increased density of NFs, but no increase in axonal caliber (25). More recently, Xu et al. (43) overexpressed each of the mouse NF subunits either individually or in various combinations. They found that only when NF-L was overexpressed in combination with either NF-M or NF-H was axonal growth significantly increased. Interestingly, when NF-M and NF-H were overexpressed alone or in combination with one another, radial axonal growth was inhibited.
It also remains incompletely understood how NF stoichiometries are regulated and the degree to which any one NF subunit is dominant in this regulation. Recently, conflicting data has appeared concerning the role of NF-M in regulating NF stoichiometries. We found that overexpression of human NF-M in transgenic mice increases the levels of endogenous mouse NF-L protein and decreases the extent of phosphorylation of NF-H (39). These results imply that NF-M may play a dominant role in regulating the levels of NF-L protein, the relative stoichiometry of NF subunits, and the phosphorylation status of NF-H. However different results were obtained by Wong et al. (40) who found that overexpression of mouse NF-M in transgenic mice did not effect the levels of axonal NF-L, and although it reduced NF-H, it did not effect its phosphorylation status.
To further address these issues we generated mice bearing a null mutation in the mouse NF-M gene. Here we describe the effects of this mutation on nervous system development with particular reference to the role of the NF-M subunit in specifying axonal diameter and its effect on levels of the remaining NF subunits.
Materials and Methods
Generation of Targeting Vectors
Isologous genomic DNA for mouse NF-M was isolated from a 129 Sv/Ev mouse genomic library prepared in λ Dash. Screening was performed with a 1,600-bp EcoRI–HindIII fragment containing the third exon of mouse NF-M (22). This probe was derived from an NF-M genomic clone λ18b (gift from P. Shneidman and W. Schlaeffer, University of Pennsylvania, Philadelphia, PA). An 11-kb clone was isolated. Restriction mapping confirmed that this clone was similar to the known structure of the mouse NF-M gene.
Targeting vectors were designed to use the positive and negative selection procedure described by Mansour et al. (23). A map of the relevant portion of the NF-M gene and the targeting strategy is shown in Fig. 1. The neomycin (Neo) resistance gene linked to the phosphoglycerol kinase-1 (PGK) promoter (540 bp) and PGK 3′ nontranslated sequence was derived from the plasmid pGEM7 (KJ1)Sal and the Herpes virus thymidine kinase gene also driven by the PGK promoter was derived from the plasmid pGEM 7 (TK) SalI (both provided by R. Jaenisch, Whitehead Institute, Cambridge, MA).
Figure 1.



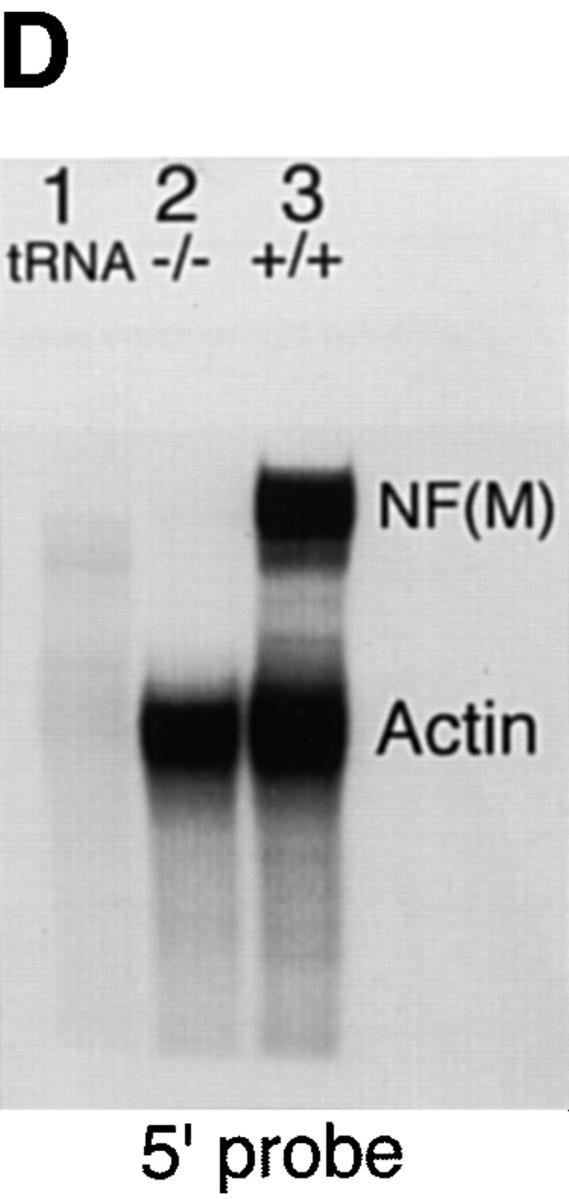
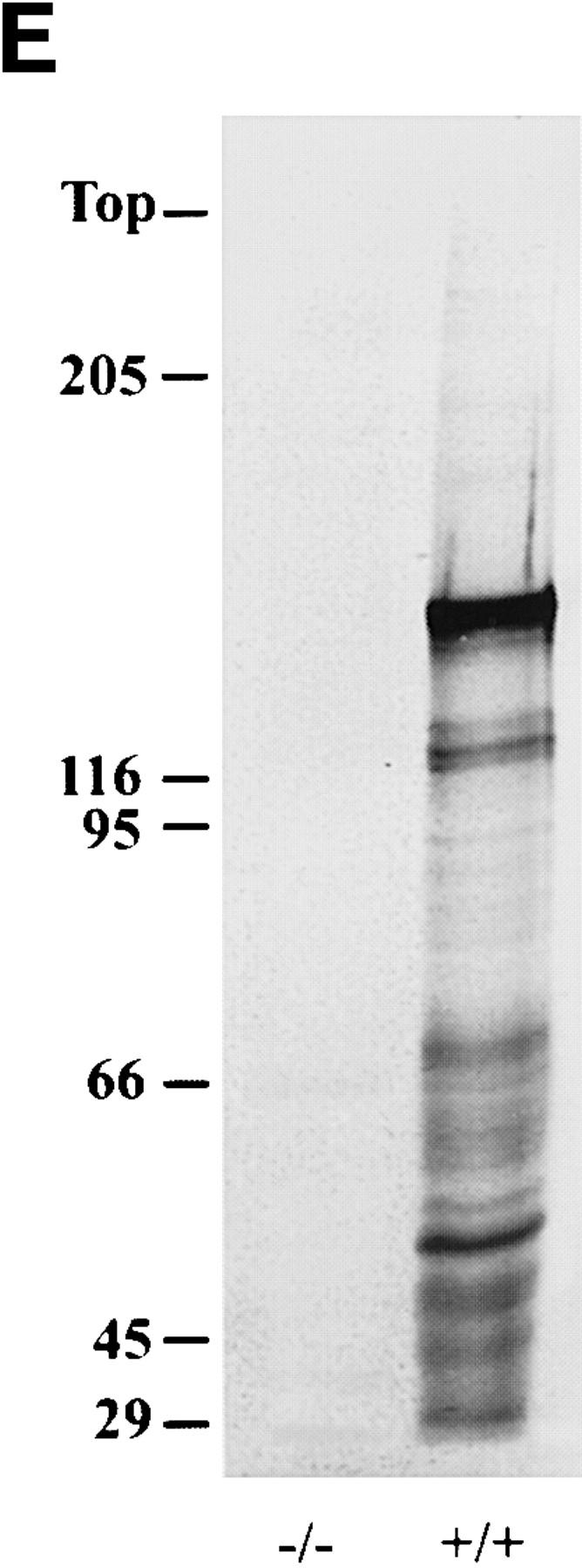
Targeted disruption of the mouse NF-M gene. (A) Targeting strategy for disruption of the mouse NF-M gene. The structure of the endogenous mouse NF-M is shown in the top line. Exons are indicated by open boxes. A targeting vector designed to use positive and negative selection (23) is shown in the center line. A PGK/Neo resistance gene was cloned in an anti-sense orientation and replaces an ∼800-bp SacI–EcoRI fragment that includes the last 93 bp of exon 1 and most of intron 1. It contains 2 kb of 5′ and 1.5 kb of 3′ homologous sequence and was linearized with HindIII. A map of the targeted recombinant gene is shown in the bottom line of the panel. The overall targeting frequency was ∼1 in 150 clones. (B) Southern blot of DNA from mice with disrupted NF-M alleles. A Southern blot of tail DNA from offspring of a heterozygous/heterozygous mating is shown. Blotting was performed with the downstream HindIII probe indicated in A. Successful targeting converts a wild-type (WT) 7.2-kb BamHI fragment to a 5.5-kb mutant (M) band. Examples of wild-type (+/+), heterozygous (+/−), and null mutant animals (−/−) are indicated. In subsequent crosses mutant and wild-type NF-M alleles were also identified by PCR using primers (indicated by arrows in A) P1 (5′ ATCCAGGCGTCGCACATCACGGTA 3′) and P2 (5′ CTGCCGTTCCAGGGACTCCTTAGT 3′) derived from the wild-type NF-M gene and P3 (5′ GTTCTAAGTACTGTGGTTTCC 3′) derived from the PGK 3′ nontranslated region. (C and D) RNA analysis of mutant animals. In C an RNase protection assay was performed with 25,000 CPM of an exon 3 murine NF-M probe (3′ to the neomycin resistance gene) and 5,000 CPM of a GAPDH probe. Protected fragments were separated as double-stranded RNA on a 6% native polyacrylamide gel. Positions of the 240-bp GAPDH and 129-bp NF-M protected fragments are indicated. Lanes were hybridized with 10 μg of tRNA (lane 1) or with 10 μg of total brain RNA from an unrelated wild-type mouse (lane 2), a homozygous mutant (−/−, lane 3), and heterozygous (+/−, lane 4) or wild-type (+/+, lane 5) littermates. In D an RNase protection assay was performed with 25,000 CPM of an exon 1 murine NF-M probe (5′ to the neomycin resistance gene) and 25,000 CPM of a murine β-actin probe. Positions of the 65-bp actin and 150-bp NF-M protected fragments are indicated. Lanes were hybridized with 10 μg of tRNA (lane 1) or with 10 μg of total brain RNA from a homozygous mutant (−/−, lane 2) or wild-type (+/+, lane 3) animal. (E) No detection of NF-M protein in NF-M null mice. Western blotting was performed with a polyclonal rabbit antiserum (NFM-N) raised against the head domain of NF-M (18). No full-length or truncated NF-M protein could be detected in the spinal cord of NF-M−/− mice. Lower molecular weight bands in the wild-type (+/+) lane likely reflect degradation products.
Transfection and Screening of ES Cells
Electroporations were performed in the R1 cell line (28). Embryonic stem (ES) cells were maintained in DME (4,500 mg/ml glucose) supplemented with 15% fetal calf serum, 0.1 mM β-mercaptoethanol, 1 mM sodium pyruvate, 40 μg/ml gentamicin, and 1,000 units/ml LIF (GIBCO BRL, Gaithersburg, MD). Cells were split within 24 h of electroporation. Targeting vectors were linearized, extracted with phenol/chloroform, ethanol precipitated, and dissolved in sterile H2O. 20 μg of plasmid DNA was electroporated into 107 ES cells with a BioRad Gene Pulser at 125 μF and 400 V at room temperature. 2 × 106 cells were plated per 100-mm dish in a nonselective media for 2 d and then selected in 150 μg/ml G418 (GIBCO BRL) plus 2 μM ganciclovir (Syntex Research, Palo Alto, CA). Neo-resistant colonies were isolated after 10–14 d. Colonies were initially picked onto feeder layers of mitomycin C–treated mouse embryonic fibroblasts in 96-well microtiter plates. Embryonic fibroblasts were prepared from mid-gestation BALB/c mice as described in Robertson (36). Duplicate 96-well plates were initially established. One plate was frozen as described in Wurst and Joyner (42) until screening results were known. The second was expanded into 24-well plates without feeder layers and DNA was prepared (42). Potentially targeted clones were screened by Southern blotting as described in Fig. 1.
Generation of Chimeric Mice
Chimeras were generated essentially as described by Bradley (2). ES cells were injected into the blastocoele cavity of C57Bl/6 blastocysts at day 3.5 post coitum and blastocysts were reimplanted into the uteri of pseudopregnant Swiss-Webster recipients at day 2.5 post coitum. Chimeras were identified on the basis of agouti coat pigmentation. A male chimera was bred with a C57Bl/6 female. Offspring from this mating were subsequently bred with 129 Sv/J or Swiss-Webster females.
RNA Analysis
RNase protection assays with probes for mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH), β-actin, NF-L, and exon 3 of mouse NF-M were performed as previously described (7, 39) using uniformly labeled RNA probes synthesized with T3 or T7 RNA polymerase and 100 μCi of α-[32P]UTP. A probe for exon 1 of mouse NF-M was generated by PCR amplification of a 150-bp segment of exon 1 (22) with primers 5′ GGCAACCCGTCCGCCTACCG 3′ and 5′ GGACCGCGCTGCGCGTGTAGG 3′. The PCR product was cloned into the TA cloning vector pCRII (CLONTECH Laboratories, Inc., Palo Alto, CA) and subsequently recloned as a HindIII–XhoI fragment into the plasmid pBluescript II SK+ (Stratagene, La Jolla, CA). The final template was linearized with HindIII and probes were synthesized with T7 RNA polymerase. After overnight hybridization at 45°C with 10 μg of total cellular RNA, samples were digested with RNase A (80 μg/ml; Sigma Chemical Co., St. Louis, MO) and RNase T1 (700 units/ml; Boehringer-Mannheim Corp., Indianapolis, IN) for 1 h at 30°C and then digested with proteinase K (125 μg/ml), phenol/chloroform extracted, and ethanol precipitated. Protected fragments were separated as double-stranded RNA on 6% native polyacrylamide gels. After gel electrophoresis protected fragments were localized by autoradiography.
Quantitative RNase protection assays for mouse NF-L were performed as described in Tu et al. (39). Each reaction contained 5 μg brain RNA and 5 μg tRNA. Quantitation was done by densitometry. To ensure linearity, a set of RNA standards containing 0–10 μg wild-type brain RNA supplemented with tRNA as needed to give 10 μg in each sample was run in all experiments. NF-L levels were normalized to the expression of β-actin.
Quantitative Western Blot Analysis
Quantitative Western blots were performed as previously described with minor modifications (39). In brief, tissue was homogenized, sonicated in BUST buffer (50 mM Tris-HCl, pH 7.4, 8 M urea, 2% β-mercaptoethanol and 0.5% SDS), and centrifuged at 40 × 103 rpm, at 25°C for 30 min in a TL-100 ultracentrifuge (Beckman Instruments, Inc., Fullerton, CA). Protein concentrations in the supernatants were determined using the Coomassie protein assay method (Pierce Chemical Co., Rockford, IL) according to the manufacturer's instructions. Each sample was loaded in triplicate and each lane contained 40 μg of total protein from neocortex or hippocampus or 10 μg of total protein from spinal cord, brainstem, or sciatic nerve. Blots were cut into three parts. The top third was incubated overnight with RM024 for detection of NFHP+++ level or RMdO9 for NFHP− level; the middle third was incubated with RM0189 for total NFM level, or RM055 for NFMP+++; and the lower third, was incubated with a rabbit anti-NFL polyclonal antiserum for total NFL or a mouse mAb anti–β-tubulin (Amersham Pharmacia Biotech Inc., Piscataway, NJ) for tubulin levels. Each part was then incubated for 1 h with 10 μCi 125I-conjugated goat anti–mouse IgG for the mouse mAbs (RM024, RM055, RM0189, and anti–β-tubulin) or 125I-conjugated Protein A for the rabbit anti-NFL polyclonal antisera. The dried blots were exposed to PhosphorImager plates for various time periods and individual bands were visualized and quantified with ImageQuant software (Molecular Dynamics, Inc., Sunnyvale, CA). Western blotting to detect NH2-terminal epitopes of NF-M was performed with a polyclonal antiserum raised against the NF-M head domain (18).
Electron Microscopy
Mice were anesthetized and fixed by vascular perfusion with a solution containing 2% formaldehyde (from paraformaldehyde), 1% glutaraldehyde, and 0.12 M sodium phosphate buffer, pH 7.4. The brain, spinal cord, optic nerves, sciatic nerve, and L5 lumbar roots were dissected out, postfixed in buffered osmium tetroxide, and embedded in Epon by routine methods (10). Thin sections were examined using a JEOL 100CX electron microscope.
To count neurofilaments and microtubules, cross-sections of axons were photographed at a magnification of 20,000 and then enlarged an additional two and one-half fold during printing. NF densities were determined using methods similar to those described by Price et al. (35) by laying a template of hexagons on each print. Hexagons had 11-mm sides, equivalent to an actual print area of 0.10 μm2. NFs in all hexagons that fell completely within axonal borders were counted. Hexagons were excluded only if vesicular organelles filled more than ∼10% of the hexagon. Nearest neighbor distances were computed from the x/y coordinates of NFs in representative electron micrographic prints.
Measurement of Axonal Diameters
For measuring axonal diameters, 1-μm thick transverse sections of L5 ventral root, sciatic nerve, or spinal cord were stained with toluidine blue and photographed through a Zeiss Axiophot microscope with a 10× or 20× objective. 2 × 2 slide images were scanned into the program Adobe Photoshop 3.05 using a Kodak 35 mm rapid film scanner. Images were enlarged three- to fourfold and printed. Optimal brightness and gray scale pixel values were adjusted so as to provide the sharpest discrimination of the myelin/axon border. Axon profiles were traced in nonoverlapping contiguous fields using a digitizing tablet. The area of myelinated axons was then measured using the program NIH-Image and axons were assumed to be circular for purposes of diameter calculations. In measuring the sciatic nerves, all myelinated axons in the largest trunk of the nerve were numbered and every fifth axon was sampled chosen by a set of random numbers. In optic nerve, random fields were photographed in the electron microscope at a magnification of 4,800 and then enlarged an additional two and one-half fold during printing. Every third myelinated axon was sampled in five randomly chosen fields. Statistical analysis (unpaired t test or Mann-Whitney U test) was performed using the program StatView (Abacus Concepts, Inc., Berkeley, CA).
Results
Production of Mice Bearing a Null Mutation in the NF-M Subunit
The targeting strategy for generating NF-M null mutant mice is illustrated in Fig. 1. Lines were established by breeding a male chimera with C57Bl/6 females. Male heterozygotes from these matings were subsequently bred with 129 Sv/J or Swiss-Webster females. On all genetic backgrounds the mutant allele was transmitted in a Mendelian fashion. We have studied animals both on a mixed 129/C57Bl background and 129/C57Bl hybrids bred to outbred Swiss-Webster mice. We have not detected any qualitative effects of genetic background on the phenotype of NF-M null mutation.
RNase protection assays with probes either 5′ (exon 1) or 3′ (exon 3) to the neomycin resistance gene (Fig. 1, C and D) revealed that no NF-M mRNA could be detected in the NF-M homozygous null mutants. Western blotting (see Figs. 1 E, 2, and 4) confirmed the absence of NF-M protein in the null mutant. We were also unable to detect NF-M protein in sections of brain from NF-M homozygous animals stained immunocytochemically with monoclonal anti–NF-M antibodies even though in sections double labeled with a polyclonal anti NF-L antisera, NF-L staining was present (data not shown).
Figure 2.
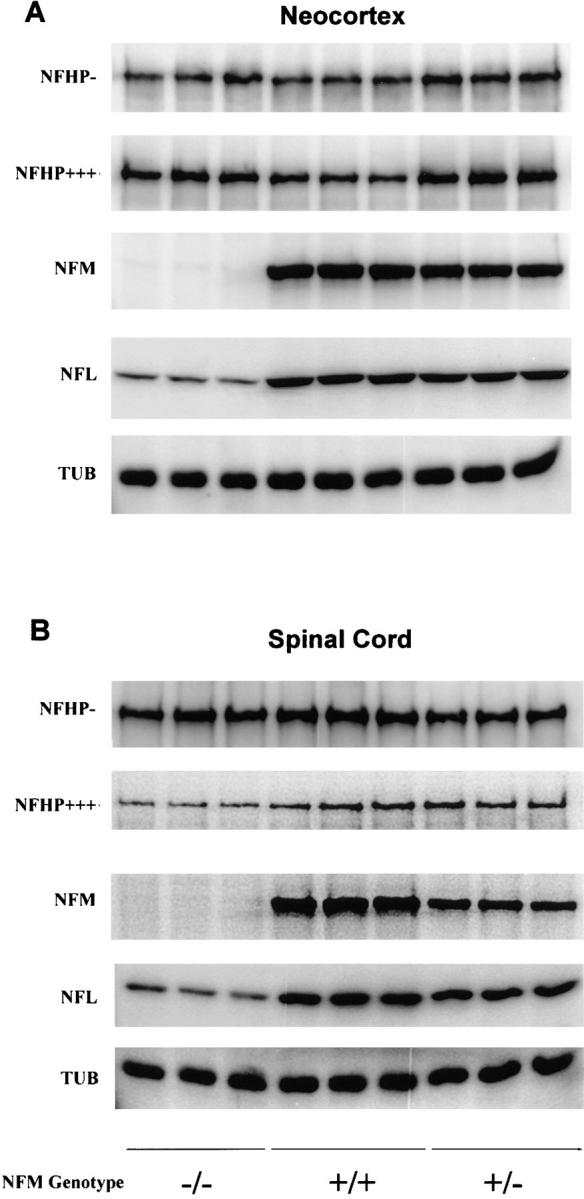
Quantitative Western blots of neocortex and spinal cord of NF-M heterozygous (+/−), NF-M null (−/−), and wild-type mice (+/+). Each sample was loaded in triplicate. The NF-M immunoreactivities are decreased in both neocortex and spinal cord of the heterozygous mice and are undetectable in the null mice. A concomitant decrease in NF-L is also observed in both neocortex and spinal cord of the heterozygous and null mice. The NF-H signals increase in the neocortex of both heterozygous and null mice but not in the spinal cord. NFHP− is measured with RMdO9, a mAb against poorly or nonphosphorylated NF-H epitopes; NFHP+++, with RM024 a mAb specific for highly phosphorylated epitopes; NF-M, with RM0189, a mAb against the rod domain of NF-M; NF-L, with a polyclonal rabbit anti-NFL antiserum; TUB, with a mAb specific for β-tubulin. All animals were 3 mo old.
Figure 4.
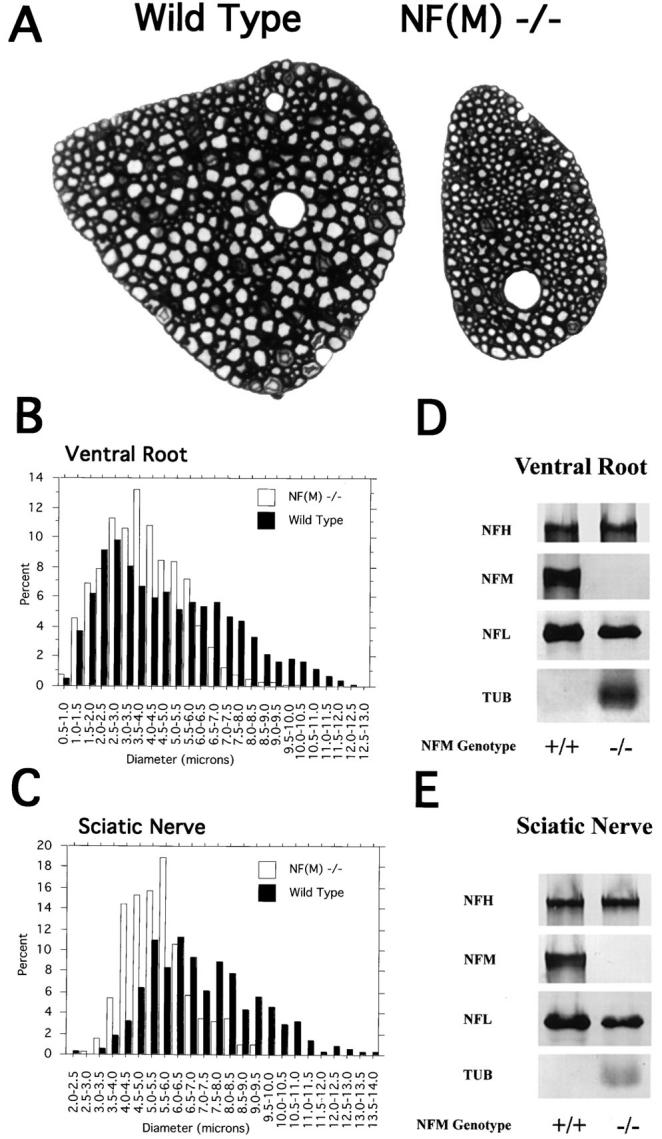
Axon calibers in L5 ventral roots from wild-type and NF-M null mutant animals. (A) Light microscopy of toluidine blue–stained L5 ventral roots from a 4-mo-old wild-type and NF-M null mutant mouse. Note the reduced size of the NF-M mutant (−/−) root as well as the absence in the mutant of axons with calibers comparable to the largest present in the control. (B) Diameters of all myelinated axons were measured in L5 roots (n = 4 wild type, n = 3 mutant). Note the marked reduction of axons >8 μm in diameter in the mutant accompanied by an increase in smaller diameter fibers. (C) Axon diameters were measured in the sciatic nerve of a 4-mo-old wild-type and mutant animal. Quantitation was performed by sampling every fifth myelinated axon in the largest trunk of a proximal portion of the nerve. Data is presented for all axons >2 μm in diameter (n = 374, wild type, 313 NF-M−/−). Note the absence of any axons >9.0 μm in diameter in the null mutant accompanied by a shift towards smaller diameter fibers. (D and E) Western blots of neurofilament content in the ventral roots (D) and sciatic nerves (E) of 4-mo-old wild-type and NF-M null mice. In D the total protein recovered from one L5 ventral root was loaded per lane. For sciatic nerve (E) 10 μg of total protein per lane was loaded for neurofilament studies and 50 μg of total protein per lane was used for β-tubulin immunoreactivity. NF-M was not detected in the null mouse. The level of NF-L protein decreased by ∼50% in both the ventral roots and sciatic nerve in the null mutant animal. An increase in the level of β-tubulin immunoreactivity was observed in both nerves in the NF-M null mouse.
NF-L Levels Are Decreased and NF-H Levels Are Increased in the NF-M Null Mutant
Since rodent NFs are obligate heteropolymers (5, 20) and are assembled into filaments with a defined stoichiometry in vivo, we determined how the lack of NF-M expression would affect levels of the other NF subunits by Western blotting. Representative Western blots of neocortex and spinal cord of NF-M null, heterozygous, and control mice are shown in Fig. 2. Quantitations (see Table I) showed that the immunoreactivities of both the phosphorylation-independent (NFMPi) and highly phosphorylation dependent (NFMP+++) epitopes of NF-M decreased in the neocortices of heterozygous mice and were undetectable in the NF-M null mice. Both NFMPi and NFMP+++ decreased in the heterozygous mice by ∼40%. Thus, the decreases in NF-M immunoreactivities were not affected by the phosphorylation state of NF-M suggesting that these decreases result from reduced levels of total NF-M rather than a change in the phosphorylation state.
Table I.
Summary of Quantitative Western Blot Analysis of NF-M Heterozygous and Null Mutant Animals
| Cortex | Spinal Cord | |||||||
|---|---|---|---|---|---|---|---|---|
| Hetero/WT | Null/WT | Hetero/WT | Null/WT | |||||
| NFHP− | 1.26 ± 0.13 | 1.00 ± 0.12 | 0.90 ± 0.07 | 0.92 ± 0.06 | ||||
| NFHP+++ | 1.62 ± 0.21 | 1.40 ± 0.23 | 1.06 ± 0.15 | 0.90 ± 0.14 | ||||
| NFMPi | 0.64 ± 0.02 | − | 0.58 ± 0.04 | − | ||||
| NFMP+++ | 0.62 ± 0.04 | − | 0.58 ± 0.07 | − | ||||
| NFL | 0.47 ± 0.02 | 0.13 ± 0.006 | 0.67 ± 0.07 | 0.23 ± 0.02 | ||||
| Tubulin | 0.93 ± 0.04 | 0.91 ± 0.06 | 0.97 ± 0.09 | 0.88 ± 0.11 | ||||
Three pairs of 3-mo-old wild-type, heterozygous, or null mutant animals were analyzed in quantitative Western blots as shown in Fig. 2. Each value represents the average ± SEM of nine observations from three separate experiments on each animal. Data is presented as a ratio of heterozygous (Hetero) or null mutants relative to wild type (WT). NFHP− was measured with RMdO9, a mAb against poorly or nonphosphorylated NF-H epitopes; NFHP+++ with RM024, a mAb specific for highly phosphorylated epitopes of NF-H; NFMPi with RM0189, a mAb against the rod domain of NF-M; NFMP+++ with RMO55, a mAb specific for highly phosphorylated epitopes of NF-M; NFL with a polyclonal rabbit anti-NF-L antiserum; tubulin with a mAb specific for β-tubulin.
Interestingly, a concomitant decrease in the level of NF-L was also detected in the NF-M heterozygous and NF-M null mice. Quantitations (see Fig. 2 and Table I) showed that the extent of decrease in NF-L levels were comparable to those of NF-M. For example in neocortex, the level of NF-L decreased in NF-M heterozygous mice by ∼50%, similar to the decrease in NF-M. Furthermore, in neocortex, the NF-M null mice that lacked all NF-M protein contained only 13% of the level of NF-L found in control mice. Dramatic decreases in NF-L were also observed in other regions including spinal cord (see Fig. 2 and Table I), hippocampus, brainstem, and cerebellum (data not shown). These data are consistent with our previous observations that NF-M levels regulate the level of NF-L protein to maintain the relative stoichiometry between NF subunits (39).
By contrast, immunoreactivities of antibodies to the heavily phosphorylated (NFHP+++) and nonphosphorylated (NFHP−) epitopes of NF-H increased in the neocortices of both NF-M heterozygous and null mice (Fig. 2). Quantitations (Table I) showed that the levels of NF-H in neocortex increased 20–50% in both the NF-M heterozygous and NF-M null mice. An increase in the level of NF-M has been shown to downregulate the phosphorylation state of NF-H by ∼20% (39). Thus, the increases in NF-H levels in the NF-M mutants are likely to be due, at least in part, to the changes in NF-M levels although a secondary effect of altered NF-L levels on NF-H is also possible. Interestingly, unlike neocortex, NF-H was not increased in spinal cord (Fig. 2 and Table I). This is consistent with our previous observation that overexpression of human NF-M in transgenic mice does not affect NF-H levels in spinal cord (39). Taken together, these data clearly demonstrate that expression of NF-L and NF-M is coordinately regulated in many CNS regions and that levels of NF-M and NF-H are coregulated in an inverse manner in neocortex but not in spinal cord. As shown in Fig. 2 and Table I, the level of β-tubulin in brain and spinal cord was not affected by the changes in NF subunits in the NF-M heterozygous and null mice, remaining comparable to that of control mice.
Previously, when human NF-M was overexpressed in transgenic mice, the change in NF-L protein level appeared to reflect changes in posttranscriptional regulation since no change in NF-L mRNA levels occurred (39). To quantitate NF-L mRNA levels in the NF-M null animals we performed quantitative RNase protection assays as previously described (39). As in the overexpression experiments reported earlier, mRNA levels for NF-L were unchanged in the null mutant (Fig. 3) implying that NF-L protein levels are being regulated posttranscriptionally.
Figure 3.

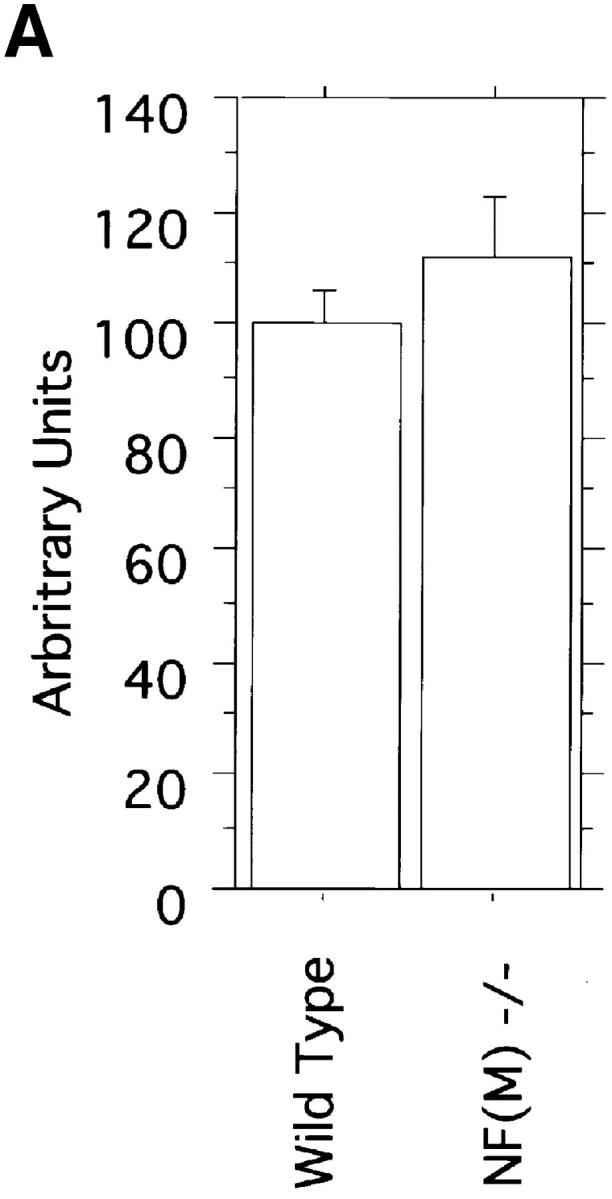
NF-L mRNA levels in NF-M null mutant animals. (A) Quantitative RNase protection assays were performed on total brain RNA from two wild-type and two NF-M−/− animals each 2 mo old. NF-L levels were normalized to the expression of β-actin. Data from three independent determinations for each sample is shown. Results are presented as arbitrary units with wild-type NF-L levels set as 100. NF-L levels in wild type were 100 ± 6 (SEM) and in NF-M−/− animals 111 ± 11 (P = 0.35, unpaired t test). (B) A sample RNase protection assay is shown. 5 μg of total brain RNA from a wild-type (lane 1) or NF-M null mutant (lane 2) were hybridized with 20,000 cpm of a mouse NF-L probe and 10,000 cpm of a murine β-actin probe. Protected fragments were separated as double stranded RNA on a 6% native polyacrylamide gel. Positions of the NF-L and actin bands are indicated.
Diminished Axonal Diameters in NF-M Null Mutant Mice
A role for NFs in establishing axonal diameter has long been suspected from the correlation between NF content in axonal cross sections and axonal caliber (16). This view has been reinforced by several recent animal models that have shown that radial growth of myelinated axons is severely inhibited in axons lacking all NFs (8, 31, 44, 45). To examine the effects of the NF-M null mutation on axonal development, we measured axon sizes in PNS (L5 ventral root and sciatic nerve) and CNS (spinal cord and optic nerve) structures.
Toluidine blue–stained sections of L5 ventral roots from a 4-mo-old control and null mutant are shown in Fig. 4 A. Myelinated axons appeared generally smaller in the mutant roots with the largest diameter fibers in the mutant failing to reach a caliber similar to the largest axons in control. Morphometric analysis, measuring the area of every myelinated axon within the L5 ventral root confirmed this initial impression (Fig. 4 B). Average axonal diameter was decreased from 4.9 ± 2.5 (SD) μm in wild type to 3.9 ± 1.6 μm in the NF-M mutant (P < 0.0001, Mann-Whitney U test). Examination of the frequency distribution of axonal diameters in control and mutant roots (Fig. 4 B) revealed that >28% of myelinated axons in wild-type roots were >6.5 μm compared with only 6.4% in the null mutant (P = 0.028, unpaired t test). Only rare axons in the mutant reached diameters >8 μm even though >9.0% fell into this class in wild-type roots. The loss of large diameter myelinated axons was accompanied by a shift towards medium and small diameter fibers. Although the number of myelinated axons was slightly reduced from 697 ± 42 in wild-type animals to 630 ± 66 in NF-M null mutants, the decrease was not statistically significant (P = 0.4111 unpaired t test). Thus, the decreased size of the mutant L5 root appears to be primarily the result of a general reduction in size of all myelinated axons.
Equally dramatic changes were seen in a morphometric analysis of axonal diameters in sciatic nerves from NF-M mutant and control animals (Fig. 4 C). Average diameters in the proximal segment of the nerve decreased from 7.2 ± 2.2 (SD) μm in wild type to 5.5 ± 1.2 μm in NF-M null mutants (P < 0.0001, Mann-Whitney U test). As in the L5 roots, axonal diameters in sciatic nerve were shifted towards small and medium sized axons and no axons >9.0 μm in diameter were found in the null mutant.
As shown in Fig. 4, D and E, neither ventral root nor sciatic nerve contained detectable amounts of NF-M protein, whereas levels of NF-L were reduced by ∼50% and levels of tubulin appeared increased. The latter observation is consistent with the relatively increased numbers of microtubules found in axons in the L5 roots (see below).
To determine whether CNS axons were also effected we examined axon sizes in the spinal cord. We measured all axons >5 μm in diameter in a 1.9 × 105 μm2 area of the ventral medial portion of the third cervical cord segment (Fig. 5 B). This region was chosen since comparable areas could be easily identified in different animals and because this region contains many large axons. As shown in Fig. 5 A, axons in this region were significantly smaller in the null mutant than in control (P < 0.0001, Mann-Whitney U test). Lost large diameter axons (>8 μm) appeared to have been replaced by medium diameter fibers (5–8 μm) in the NF-M null mutant.
Figure 5.
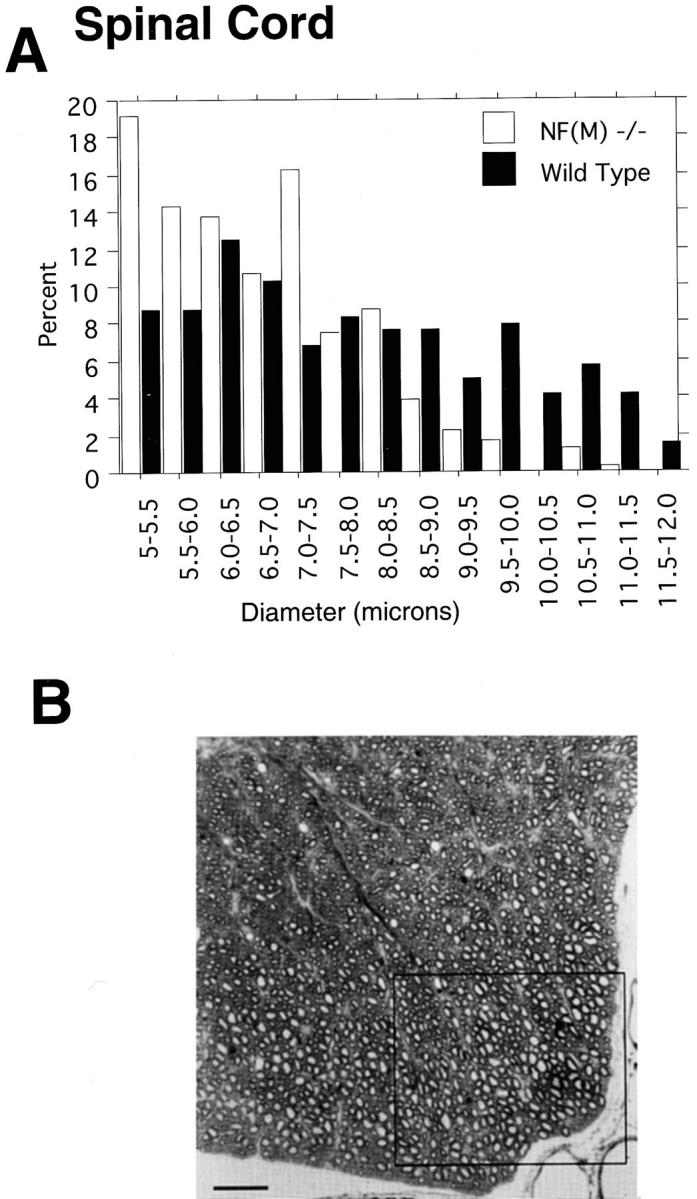
Axonal calibers in ventral spinal cord of wild-type and NF-M mutant mice. (A) Axon sizes were measured in a 1.9 × 105 μm2 area of the ventral medial portion of C3 (boxed area shown in B). Data is presented for all axons >5 μm in diameter (n = 263 for wild type and 307 for NF-M−/−) from a 5-mo-old wild-type and mutant NF-M animal. Note the dramatic reduction in large diameter fibers accompanied by a shift to smaller diameter fibers in the null mutant. (B) Light microscopy of a toluidine blue– stained section of ventral cervical cord (C3) from a wild-type animal. Box indicates the region used to generate the frequency distributions shown in A. Bar, 50 μm.
Thus, the NF-M null mutation appears to reduce the diameter of myelinated axons in both PNS and CNS. However, since the regions examined (L5 ventral root, sciatic nerve, spinal cord) contain mainly medium and large sized axons it was less clear if the mutation was effecting all sizes of myelinated axons. To determine the effects of the mutation on smaller caliber axons we measured axon sizes in the optic nerves, an area of the CNS containing relatively small myelinated axons nearly all <2 μm in diameter. In electron micrographs of optic nerve, myelinated axons were visibly smaller in the NF-M null mutant than control animals (Fig. 6, A and B). Average diameters decreased from 1.16 ± 0.42 μm in wild type to 0.93 ± 0.29 μm in NF-M null mutants (P < 0.0001, Mann-Whitney U test). As in the other regions examined axonal diameters were shifted towards smaller diameter fibers in the mutant (Fig. 6 C). Thus, the NF-M subunit appears to be required to achieve maximal axonal diameter in all size classes of myelinated axons in both the CNS and PNS.
Figure 6.
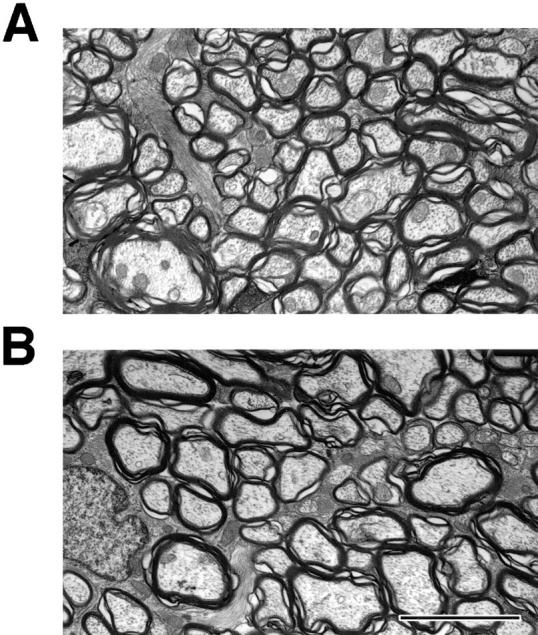

Axon calibers in optic nerves. (A and B) Electron micrographs from optic nerves of 5-mo-old NF-M null mutant (A) and wild-type (B) mice. Note the generally reduced size of myelinated axons in the NF-M animal. (C) Axon sizes were determined in the optic nerves of a 5-mo-old wild type and NF-M null mutant. Quantitation was performed by sampling every third myelinated axon in five randomly selected fields (n = 193 wild type, 322 NF-M−/−). Note the shift towards smaller diameter fibers in the mutant. Bar, 3 μm.
Reduced Neurofilament Content in Mice Lacking an NF-M Subunit
It remains unclear how NFs contribute to the specification of axonal diameter. One popular idea for the role of the larger NF subunits has been that the phosphorylated tail domains of NF-M and NF-H determine interfilament distance and that spacing between filaments in turn specifies axonal diameter (4, 24). If so, then loss of NF-M from NF sidearms should result in more tightly packed NFs and the NF-M null mutant should require more NFs than wild type to produce an axon of equivalent diameter.
To look for an ultrastructural basis for the diminution of axonal diameters in the null mutant we examined electron micrographs of L5 ventral roots from mutant and control animals. NFs were readily apparent in both the null mutant and control (Fig. 7). However, filaments in the mutant animal appeared to be reduced in number although otherwise of normal configuration in both transverse and longitudinal sections (Fig. 7). Microtubules also appeared to be normal in appearance, although in many axons their numbers seemed to be increased.
Figure 7.
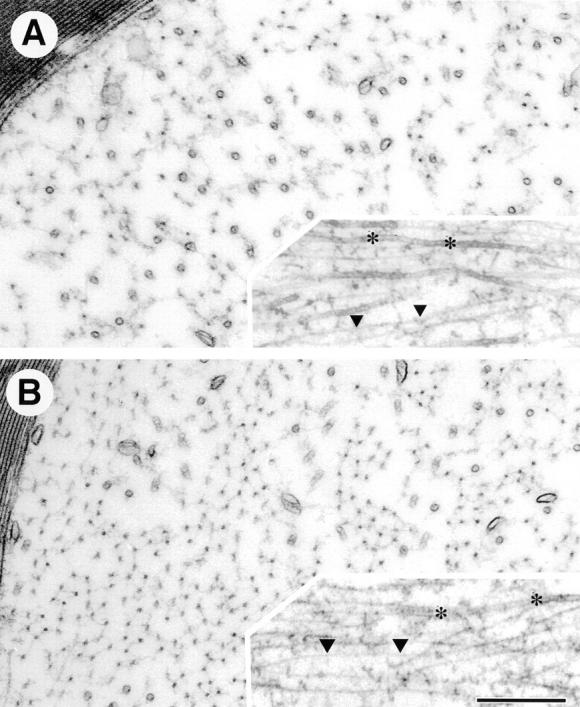
Appearance of neurofilaments in mice with an NF-M null mutation. (A and B) NFs in axons of L5 ventral root are viewed in cross section and longitudinally (insets) from 4-mo-old NF-M null mutant (A) or wild-type mice (B). NFs (triangles) are reduced in the NF-M null mutant (A) as compared with control (B), while microtubules (asterisks) are increased. Bar, 300 nm.
To determine if NF content was actually altered in the null mutants, NFs were counted in the internodal regions of axons over a range of sizes and NF counts were plotted against axonal area. As shown in Fig. 8 A, axons in the null mutant consistently contained fewer NFs than comparably sized axons in controls. By contrast, these same axons contained more microtubules (Fig. 8, C and D) increasing the average ratio of microtubules to NFs from 0.22 ± 0.8 (SD) in wild type to 0.83 ± 0.41 in the mutant axons (P < 0.0001, Mann-Whitney U test).
Figure 8.

Neurofilament and microtubule content in NF-M–deficient animals. (A) NFs were counted in the internodal regions of L5 ventral root axons of 4-mo-old mutant and control animals. The number of NFs in each axon was plotted against axonal size (area in μm2). Note that in axons of similar size, the wild type has more NFs than the NF-M null mutant. (B) NF densities were determined using methods similar to those described by Price et al. (35). A template of hexagons was applied over each electron micrograph and the number of NFs per hexagon counted. At least 300 hexagons (n = 314 wild type, 322 NF-M mutant) each equivalent to an area of 0.10 μm2 were counted and a frequency distribution plot was generated showing the number of NFs per hexagon. Note the dramatically reduced density of NFs in the NF-M mutant. (C) Microtubules were counted in the same axons as in A. In contrast to NFs, axons in NF-M mutant animals have more microtubules than axons of comparable size in wild type. (D) The ratio of microtubules (MT) to NFs is shown for the axons in A and C.
Since axons in the null mutant contain fewer NFs, NF densities in the mutant should also be decreased. We measured NF densities in mutant and wild-type axons using methods similar to those described by Price et al. (35). NFs densities were determined by applying a template of hexagons (every hexagon equivalent to an area of 0.10 μm2) over each electron micrograph and counting the number of NFs in each hexagon. A frequency distribution plot was then generated showing the number of NFs per hexagon (Fig. 8 B). The average number of NFs per hexagon was reduced from 17.4 ± 5.8 (SD) in control axons to 7.5 ± 4.1 in the mutant (P < 0.0001, unpaired t test) and as shown in Fig. 7 C the frequency distribution was dramatically shifted in the mutant towards hexagons containing fewer NFs, demonstrating that NFs are less densely packed in the NF-M mutant.
To determine the effect of the reduced NF density on interfilament spacing in the mutant we measured nearest neighbor distances in these same axons (Fig. 9). Mean interfilament distances increased from 46 ± 17 (SD) nm in control to 62 ± 33 in the mutant (P < 0.0001, Mann-Whitney U test) although the modal interfilament distance was identical between mutant and control (47 nm). Indeed an analysis of those filaments with nearest neighbors of 60 nm or less revealed that interfilament spacing in the mutant (43 ± 10) was little changed from control (41 ± 10, P < 0.0001). Thus, although average interfilament distances were increased (as would be expected due to the decreased filament number) when filaments are closely spaced in the mutant they assume an interfilament distance that is similar to wild type.
Figure 9.
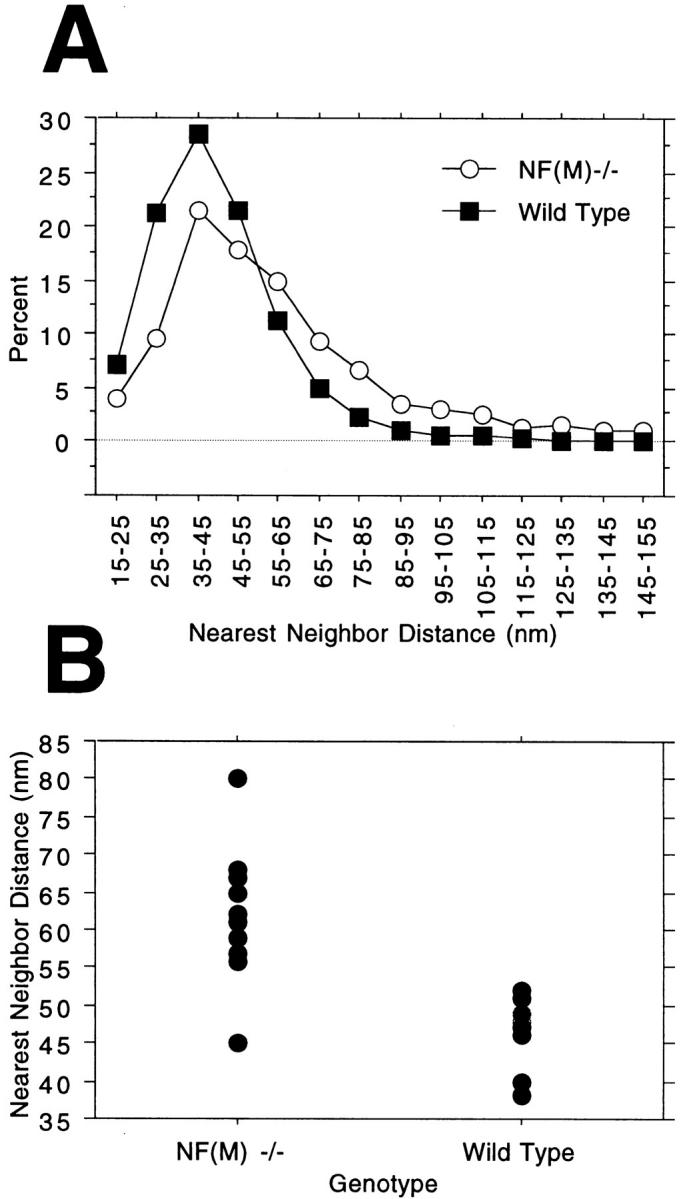
Nearest neighbor analysis of neurofilament spacing in NF-M–deficient animals. (A) Interfilament spacing was analyzed in 10 mutant (range 0.78–8.72 μm2, average 3.91 ± 3.10 SD) and 10 wild-type (range 1.63–8.58, average 3.99 ± 2.50) axons from the L5 ventral roots of 4-mo-old animals. The positions of all NFs in each axon (n = 1,683 mutant and 4,709 wild type) were determined and nearest neighbor distances computed. Note that although the decreased NF density in the mutant results in an increased average interfilament distance, the modal distance is similar in both mutant and control. (B) Values for the individual axons measured in A are shown.
Taken together these data are not consistent with the prediction that axons in mutant animals would require more NFs, packed at a closer density in order to produce axons of comparable size to wild type. However, these findings are consistent with suggestions that levels of NF-L determine the number of NFs (25, 30).
Lack of Overt Phenotype or Major Structural Defects in Mice Carrying a Disrupted NF-M Gene
No overt phenotype was associated with a null mutation in the NF-M gene. Animals appeared normal at birth and were indistinguishable from littermates. 4-mo-old mutant animals appeared the same size as littermates. Both male and female homozygous mutants were fertile. No striking behavioral changes were apparent and null mutants appeared to have normal motor strength and coordination. NF-M−/− animals have shown no obvious health problems up to one year of age.
A comparison by light microscopy of coronal sections of the brain from 4-mo-old mutant and wild-type animals revealed no obvious differences between null mutants and wild-type animals. Both cortical and subcortical structures appeared normal. In particular large neurons such as the Purkinje cells of the cerebellum and anterior horn cells in the spinal cord that contain large numbers of NFs appeared to have developed normally.
Discussion
The highly conserved nature of NF proteins suggests that NFs serve some important developmental function. Suggestions for the in vivo function of NF-L has come from a Japanese quail (Quiverer) with a spontaneous mutation in the NF-L gene (31) and a recent targeted disruption of the gene in mice (45). In the homozygous state, the Quiverer quail contains no axonal NFs and the radial growth of myelinated axons is severely attenuated. Mice with a targeted disruption of the NF-L gene (45) also lack axonal NFs and, although they exhibit no behavioral phenotype, have diminished axonal calibers, and delayed maturation of regenerating myelinated axons. The absence of axonal NFs with both mutations is consistent with suggestions that at least in some species NFs are obligate heteropolymers (5, 20) requiring the presence of NF-L plus either NF-M or NF-H if filaments are to form at all. Thus, both the Quiverer quail and the NF-L knockout show that in these species NF-L is required for NF formation. However, these mutants cannot address the functional roles of the larger subunits NF-M (the subject of the present paper) or NF-H since no filaments are formed.
This study demonstrates the dominant role that NF-M levels play in regulating the levels of NF-L protein. An NF-M null mutation resulted in ∼40% less NF-L in heterozygotes and an 80–90% reduction in homozygotes. These findings are consistent with our previous observations that overexpression of human NF-M in transgenic mice increases levels of mouse NF-L protein (39).
How NF-M regulates NF-L levels remains unclear although the effects must be mediated post-transcriptionally since NF-L mRNA levels are little if at all changed by either overexpression (39) or underexpression of NF-M (this paper). It is interesting that although NF-L protein was reduced to 10–20% of wild type in brain and spinal cord, levels were reduced by only 50% in the sciatic nerves and lumbar roots. The concomitant 50% reduction in filaments in the lumbar roots suggests that NF-L protein that reaches the axon is incorporated into stable filaments. Conversely in the perikarya NF-L mRNA may be either less translated or NF-L protein in excess of that which can be incorporated into filaments degraded.
The effects of alterations in NF-M on the levels of NF-H and the extent of its phosphorylation are more complex. NF-H levels either increased or remained unchanged depending on the region of the nervous system sampled. In neocortex, a 20–50% increase in the phosphorylation state of NF-H accompanied the decrease in NF-M protein levels. This observation complements our previous finding that overexpression of NF-M reduces the levels of highly phosphorylated NF-H by 25% (39). Collectively, these data demonstrate that in the neocortex, the levels of NF-M and NF-H as well as their phosphorylation state are coregulated. However, the change in NF-M levels does not seem to affect the level of NF-H nor its phosphorylation state in the spinal cord in transgenic mice overexpressing NF-M (39) or in the NF-M knockout mice described here. This suggests that NF-M and NF-H are not coregulated in the spinal cord. The mechanism responsible for the differential effects of NF-M on the levels of NF-H in different brain regions is unknown. One possible explanation could be different ratios of large versus small caliber axons in these two brain regions. Since NF-H serves as a better spacer between individual NFs to maintain axonal caliber than NF-M, NF-H is highly enriched in large diameter axons. Our data suggest that in the spinal cord, NF-H may have reached maximum levels whereas in neocortex, the levels of NF-H may be submaximal and this could reflect the relative abundance of small versus large axons in these two regions. Thus, any alteration in the levels of NF-M in the neocortex may produce compensatory changes in NF-H but may have no effect in the spinal cord.
The only obvious structural effect of the NF-M null mutation was to decrease the diameter of myelinated axons in both the CNS and PNS. In each region examined, reduced numbers of relatively large diameter axons were accompanied by increased numbers of small- and medium-sized fibers. This shift, along with the observation that axon numbers were not significantly altered in the mutant, argues that the mutation is limiting the radial growth of all myelinated axons rather than causing a class of large diameter axons to fail to develop.
How NFs contribute to controlling axonal diameter remains unclear. Two different theories have been proposed previously to explain how NFs regulate axonal caliber. One popular notion for the role of the larger NF subunits has been that the tail domains of NF-M and/or NF-H form cross-bridges or cross-linkers that determine interfilament distance and that spacing between filaments in turn specifies axonal diameter (11). A second theory suggests that the highly charged phosphate groups on NF-H, and to a lesser extent NF-M, tend to repel rather than mediate a cohesive interaction (4, 24). Thus, in this model an increase in NF-M or NF-H phosphorylation serves a space-filling function by increasing the repulsion of adjacent filaments through their negatively charged sidearms.
The results presented here could be interpreted as supporting the latter model since the packing density of NFs in axons of the null mutant are much lower than those of control animals. In this interpretation NFs in the mutant would be moving further away from each other as more space becomes available in axons of the null mutant. Such expansion would suggest that NF sidearms do not form rigid cross-bridges or cross-linkers as proposed by the first model. However, it is interesting that filaments in the mutant most frequently occur 47 nm from their nearest neighbor (the modal distance) identical to that found in control. Additionally an analysis of those filaments with nearest neighbors of 60 nm or less revealed that interfilament spacing in the mutant (43 nm) was little changed from control (41 nm). Thus, when filaments are closely spaced in the mutant they assume an interfilament distance that is similar to wild type arguing that associative interactions (as suggested by the first model) may be occurring between filaments.
It is also difficult to reconcile a charge repulsion model with the surprising observation that axons in the NF-M null mutant contain fewer filaments than comparably sized axons in the wild type. A charge repulsion model would predict that loss of NF-M from sidearms should have resulted in more tightly packed NFs in the NF-M null mutant with the mutant requiring more NFs than wild type to produce an axon of equivalent diameter. However, axons in NF-M null mutant contain less than half the filaments found in comparably sized axons in the wild type. Even if interfilament distances are mainly set by NF-H, the predicted effect should still have been that an axon in the NF-M null mutant would have the same number of filaments as a comparably sized axon in the control. Thus, it seems unlikely that any current model can adequately explain the relationship between NF number and axonal diameter, a relationship itself which may be more complex than previously believed.
These same axons did contain a slightly increased ratio of microtubules to NFs. A similar change also occurs in the Quiverer quail (44), in mice with a targeted mutation of the NF-L gene (45), and the NF-H β-galactosidase transgenic mice (8), suggesting that increases in microtubules may be a general response to decreased numbers of NFs. The relative roles of microtubules and NFs in determining axonal diameter has been long discussed (19). In large caliber axons, microtubule content does not correlate with axonal diameter as closely as does NF content (9, 14). Microtubules likely have more significance in maintaining the diameter of medium- and small-sized axons where they are the major cytoskeletal component. Thus, the shift in the NF-M null mutant from large to medium and small-caliber axons as a result of a decrease in axonal NFs suggests that increased microtubules may partially compensate to maintain caliber.
Despite the loss of all NF-M protein and 90% of NF-L, the null mutants lacked any overt behavioral phenotype consistent with other studies (8, 31, 45) showing that NF proteins are not essential for survival. It is interesting to note that large neurons (e.g., the Purkinje cells of the cerebellum and anterior horn cells in the spinal cord) that contain large numbers of NFs developed normally. This is perhaps not surprising since during the initial phases of axonal elongation, NFs are minor components of the cytoskeleton (1, 34). Only as axons reach their targets and their diameters increase do NFs accumulate in numbers. It is during this second phase, referred to as radial growth, that NFs become the most abundant cytoskeletal element, exceeding microtubules in number by up to an order of magnitude in large axons (9).
It remains unclear whether the generally reduced sizes of myelinated axons will have other functional consequences. A decrease in axonal caliber has been shown to reduce the rate of nerve conduction velocity in the Quiverer quails (37). A similar reduction in nerve conduction velocity is expected to occur in our NF-M null mice. The reduced NF content might result in destabilization of axons with aging resulting in altered axonal transport and dying back of the axon. Finally, insights into the mechanisms that coregulate NF subunit synthesis and phosphorylation are likely to lead to further clarification of the normal function of NFs as well as their role in neurodegenerative diseases.
Acknowledgments
We thank Chi Li, Zuozong Liang, and Honor O'Sullivan for technical assistance. We also thank Drs. Paul Shneidman and William Schlaeffer for gift of a mouse genomic NF-M clone, Drs. Rudolf Jaenisch and Konstantin Andrikopoulos for gift of PGK/TK and PGK/Neo plasmids, Dr. Konstantin Andrikopoulos for the gift of a 129 Sv/Ev mouse genomic library, Dr. Andras Nagy for gift of R1 cells, and Dr. Ron Liem for gift of antibodies. Dr. Ron Gordon is thanked for assistance with electron microscopy, Dr. Tom Lufkin for help with microinjection of ES cells, and Dr. John Reinitz for assistance with nearest neighbor analysis. We would also like to thank Dr. Jean-Pierre Julien for communicating data concerning the NF-L knockout before publication.
This work was supported by the American Health Assistance Foundation (G.A. Elder) and National Institute on Aging grant P50 AGO 5138-11 (R.A. Lazzarubu).
Abbreviations used in this paper
- CNS
central nervous system
- ES
embryonic stem
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- IF
intermediate filament
- Neo
neomycin
- NF-H
heavy neurofilament
- NF-L
light neurofilament
- NF-M
mid-sized neurofilament
- NF
neurofilament
- PGK
phosphoglycerol kinase
- PNS
peripheral nervous system
Footnotes
Address all correspondence to Dr. Robert A. Lazzarini, Brookdale Center for Developmental and Molecular Biology, Box 1126, Mount Sinai School of Medicine, New York, NY 10029. Tel.: (212) 241-4272. Fax: (212) 860-9279. E-mail: rlazzar@smtplink.mssm.edu
References
- 1.Berthold, C.-H. 1978. Morphology of normal peripheral axons. In Physiology and Pathobiology of Axons. S.G. Waxman, editor. Raven Press, New York. 3–63.
- 2.Bradley, A. 1987. Production and analysis of chimaeric mice. In Teratocarcinomas and Embryonic Stem Cells: A Practical Approach. E.J. Robertson, editor. IRL Press, Washington, DC. 113–151.
- 3.Burgoyne RD, Cambray-Deakin MA. The cellular neurobiology of neuronal development: the cerebellar granule cell. Brain Res Rev. 1988;13:77–101. doi: 10.1016/0165-0173(88)90006-9. [DOI] [PubMed] [Google Scholar]
- 4.Carden MJ, Trojanowski JQ, Schleapfer WW, Lee VM-Y. Two-stage expression of neurofilament polypeptides during rat neurogenesis with early establishment of adult phosphorylation. J Neurosci. 1987;7:3489–3504. doi: 10.1523/JNEUROSCI.07-11-03489.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ching G, Liem R. Assembly of type IV neuronal intermediate filaments in nonneuronal cells in the absence of preexisting cytoplasmic intermediate filaments. J Cell Biol. 1993;122:1323–1335. doi: 10.1083/jcb.122.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Waegh S, Lee VM, Brady ST. Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell. 1992;68:451–463. doi: 10.1016/0092-8674(92)90183-d. [DOI] [PubMed] [Google Scholar]
- 7.Elder GA, Friedrich VL, Liang Z, Li X, Lazzarini RA. Enhancer trapping by a human mid-sized neurofilament transgene reveals unexpected patterns of neuronal enhancer activity. Mol Brain Res. 1994;26:177–188. doi: 10.1016/0169-328x(94)90089-2. [DOI] [PubMed] [Google Scholar]
- 8.Eyer J, Peterson A. Neurofilament-deficient axons and perikaryal aggregates in viable transgenic mice expressing a neurofilament-β-galactosidase fusion protein. Neuron. 1994;12:389–405. doi: 10.1016/0896-6273(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 9.Friede R, Sarnorajski T. Axon caliber related to neurofilaments and microtubules in sciatic nerve. Anat Rec. 1970;167:379–388. doi: 10.1002/ar.1091670402. [DOI] [PubMed] [Google Scholar]
- 10.Friedrich, V.L.J., and E. Mugnaini. 1981. Preparation of neural tissues for electron microscopy. In A Handbook of Neuroanatomical Tract Tracing Techniques. Plenum Press, New York. 377–406.
- 11.Hirokawa N. Cross-linker system between neurofilaments, microtubules, and membranous organelles in frog axons revealed by the quick-freeze, deep-etching method. J Cell Biol. 1982;94:129–142. doi: 10.1083/jcb.94.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirokawa N, Glicksman MA, Willard MB. Organization of mammalian neurofilament polypeptides within the neuronal cytoskeleton. J Cell Biol. 1984;98:1523–1536. doi: 10.1083/jcb.98.4.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hisanaga S, Hirokawa N. Structure of the peripheral domains of neurofilaments revealed by low angle rotary shadowing. J Mol Biol. 1988;202:297–305. doi: 10.1016/0022-2836(88)90459-7. [DOI] [PubMed] [Google Scholar]
- 14.Hoffman PN, Griffin JW, Price DL. Control of axonal caliber by neurofilament transport. J Cell Biol. 1984;99:705–714. doi: 10.1083/jcb.99.2.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffman PN, Thompson GW, Griffin JW, Price DL. Changes in neurofilament transport coincide temporally with alterations in the caliber of axons in regenerating motor fibers. J Cell Biol. 1985;101:1332–1340. doi: 10.1083/jcb.101.4.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoffman, P.N., E.H. Koo, N.A. Muma, J.W. Griffin, and D.L. Price. 1988. Role of neurofilaments in the control of axonal caliber in myelinated nerve fibers. In Intrinsic Determinants of Neuronal Form and Function. Vol. 37. R.J. Lasek and M.M. Black, editors. Alan R. Liss, New York, NY. 389–402.
- 17.Julien J-P, Meyer D, Flavell D, Hurst J, Grosveld F. Cloning and developmental expression of the murine neurofilament gene family. Mol Brain Res. 1986;1:243–250. doi: 10.1016/0169-328x(86)90030-6. [DOI] [PubMed] [Google Scholar]
- 18.Kaplan MP, Chin SS, Macioce P, Srinawasan J, Hashim G, Liem RK. Characterization of a panel of neurofilament antibodies recognizing N-terminal epitopes. J Neurosci Res. 1991;30:545–554. doi: 10.1002/jnr.490300312. [DOI] [PubMed] [Google Scholar]
- 19.Lasek RJ, Oblinger MM, Drake PF. Molecular biology of neuronal geometry: expression of neurofilament genes influences axonal diameter. Cold Spring Harbor Symp Quant Biol. 1983;48:731–744. doi: 10.1101/sqb.1983.048.01.076. [DOI] [PubMed] [Google Scholar]
- 20.Lee M, Xu Z, Wong P, Cleveland D. Neurofilaments are obligate heteropolymers in vivo. J Cell Biol. 1993;122:1337–1350. doi: 10.1083/jcb.122.6.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lees JF, Shneidman PS, Skuntz SF, Carden MJ, Lazzarini RA. The structure and organization of the human heavy neurofilament subunit (NF-H) and the gene encoding it. EMBO (Eur Mol Biol Organ) J. 1988;7:1947–1955. doi: 10.1002/j.1460-2075.1988.tb03032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levy E, Liem RKH, D'Eustachio P, Cowan NJ. Structure and evolutionary origin of the gene encoding mouse NF-M, the middle-molecular-mass neurofilament protein. Eur J Biochem. 1987;166:71–77. doi: 10.1111/j.1432-1033.1987.tb13485.x. [DOI] [PubMed] [Google Scholar]
- 23.Mansour SL, Thomas KR, Capecchi MR. Disruption of the proto-oncogene int-2 in mouse-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature. 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 24.Matus A. Neurofilament protein phosphorylation-where, when and why. Trends Neurosci. 1988;11:291–292. doi: 10.1016/0166-2236(88)90086-0. [DOI] [PubMed] [Google Scholar]
- 25.Monteiro MJ, Hoffman PN, Gearhart JD, Cleveland DW. Expression of NF-L in both neuronal and nonneuronal cells of transgenic mice: increased neurofilament density in axons without affecting caliber. J Cell Biol. 1990;111:1543–1557. doi: 10.1083/jcb.111.4.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mulligan L, Balin BJ, Lee VM-Y, Ip WP. Antibody labeling of bovine neurofilaments: implications on the structure of neurofilament sidearms. J Struct Biol. 1991;106:145–160. doi: 10.1016/1047-8477(91)90084-a. [DOI] [PubMed] [Google Scholar]
- 27.Myers MW, Lazzarini RA, Lee VM-Y, Schlaepfer WW, Nelson DL. The human mid-size neurofilament subunit: a repeated protein sequence and the relationship of its gene to the intermediate filament gene family. EMBO (Eur Mol Biol Organ) J. 1987;6:1617–1626. doi: 10.1002/j.1460-2075.1987.tb02409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagy A, Rossant J, Nagy R, Abranow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakagawa T, Chen J, Zhang Z, Kanai Y, Hirokawa N. The distinct functions of the carboxyl-terminal tail domain of NF-M upon neurofilament assembly: cross-bridge formation and longitudinal elongation of filaments. J Cell Biol. 1995;129:411–429. doi: 10.1083/jcb.129.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nixon RA. The regulation of neurofilament protein dynamics by phosphorylation: clues to neurofibrillary pathology. Brain Pathol. 1993;3:29–38. doi: 10.1111/j.1750-3639.1993.tb00723.x. [DOI] [PubMed] [Google Scholar]
- 31.Ohara O, Gahara Y, Miyake T, Teraoka H, Kitamura T. Neurofilament deficiency in quail caused by nonsense mutation in neurofilament-lgene. J Cell Biol. 1993;121:387–395. doi: 10.1083/jcb.121.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palay, S.L., and V. Chan-Palay. 1974. Cerebellar Cortex Cytology and Organization. Springer, Berlin. 348 pp.
- 33.Parry, D.A.D. 1990. Primary and secondary structure of IF protein chains and models of molecular aggregation. In Cellular and Molecular Biology of Intermediate Filaments. R.D. Goldman and P.M. Steinert, editors. Plenum Press, New York. 175–204.
- 34.Peters A, Vaughn JE. Microtubules and filaments in the axons and astrocytes of early postnatal rat optic nerves. J Cell Biol. 1967;32:113–119. doi: 10.1083/jcb.32.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Price RL, Paggi P, Lasek RJ, Katz MJ. Neurofilaments are spaced randomly in the radial dimension of axons. J Neurocytol. 1988;17:55–62. doi: 10.1007/BF01735377. [DOI] [PubMed] [Google Scholar]
- 36.Robertson, E.J. 1987. Embryo derived stem cell lines. In Teratocarcinomas and Embryonic Stem Cells: A Practical Approach. E.J. Robertson, editor. IRL Press, Washington, DC. 71–112.
- 37.Sakaguchi T, Okada M, Kitarnura T, Kawasaki K. Reduced diameter and conduction velocity of myelinated fibers in the sciatic nerve of a neurofilament-deficient mutant quail. Neurosci Lett. 1993;153:65–68. doi: 10.1016/0304-3940(93)90078-y. [DOI] [PubMed] [Google Scholar]
- 38.Trojanowski JQ, Walkenstein N, Lee AM-Y. Expression of neurofilament subunits in neurons of the central and peripheral nervous system: an immunohistochemical study with monoclonal antibodies. J Neurosci. 1986;6:650–660. doi: 10.1523/JNEUROSCI.06-03-00650.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tu P-H, Elder G, Lazzarini RA, Nelson D, Trojanowski JQ, Lee VM-Y. Overexpression of the human NFM subunit in transgenic mice modifies the level of endogenous NFL and the phosphorylation state of NFH subunits. J Cell Biol. 1995;129:1629–1640. doi: 10.1083/jcb.129.6.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wong PC, Marszalek J, Crawford TO, Xu Z, Hsieh S-T, Griffin JW, Cleveland DW. Increasing neurofilament subunit NF-M expression reduces axonal NF-H, inhibits radial growth, and results in neurofilamentous accumulation in motor neurons. J Cell Biol. 1995;130:1413–1422. doi: 10.1083/jcb.130.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wuerker RB, Kirkpatrick JB. Neurofilaments and microtubules in anterior horn cells of the rat. Int Rev Cytol. 1972;33:45–75. doi: 10.1016/s0074-7696(08)61448-5. [DOI] [PubMed] [Google Scholar]
- 42.Wurst, W., and A.L. Joyner. 1993. Production of targeted embryonic stem cell clones. In Gene Targeting: A Practical Approach. A.L. Joyner, editor. IRL Press, New York. 33–61.
- 43.Xu Z, Marszalek JR, Lee MK, Wong PC, Folmer J, Crawford TO, Hsieh S-T, Griffin JW, Cleveland DW. Subunit composition of neurofilaments specifies axonal diameter. J Cell Biol. 1996;133:1061–1069. doi: 10.1083/jcb.133.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamasaki H, Itakura C, Mizutani M. Hereditary hypotrophic axonopathy with neurofilament deficiency in a mutant strain of the Japanese quail. Acta Neuropathol. 1991;82:427–434. doi: 10.1007/BF00293376. [DOI] [PubMed] [Google Scholar]
- 45.Zhu Q, Couillard-Depres S, Julien J-P. Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp Neurol. 1997;148:299–316. doi: 10.1006/exnr.1997.6654. [DOI] [PubMed] [Google Scholar]