

Abstract
Protease protection assays of apolipoprotein B100 (apoB) in digitonin-permeabilized HepG2 cells indicated that multiple domains of apoB are exposed to the cytosol through an extensive portion of the secretory pathway. The intracellular orientation of apoB in the secretory pathway was confirmed by immunocytochemistry using antibodies recognizing specific domains of apoB in streptolysin-O (STP-O)– and saponin-permeabilized HepG2 cells. Lumenal epitopes on marker proteins in secretory pathway compartments (p63, p53, and galactosyltransferase) were not stained by antibodies in STP-O–treated cells, but were brightly stained in saponin-treated cells, confirming that internal membranes were not perforated in STP-O–treated cells. An anti-apoB peptide antibody (B4) recognizing amino acids 3221–3240 caused intense staining in close proximity to the nuclear membrane, and less intensely throughout the secretory pathway in STP-O–permeabilized cells. Staining with this antibody was similar in STP-O– and saponin-treated cells, indicating that this epitope in apoB is exposed to the cytosol at the site of apoB synthesis and throughout most of the remaining secretory pathway. Similar results indicating a cytosolic orientation were obtained with monoclonal antibody CC3.4, which recognizes amino acids 690–797 (79–91 kD) in apoB. Two polyclonal antibodies made to human LDL and two monoclonal antibodies recognizing amino acids 1878–2148 (D7.2) and 3214–3506 (B1B6) in apoB did not produce a strong reticular signal for apoB in STP-O–treated cells. The anti-LDL and B1B6 antibodies produced almost identical punctate patterns in STP-O–treated cells that overlapped with LAMP-1, a membrane marker for lysosomes. These observations suggest that the B1B6 epitope of apoB is exposed on the surface of the lysosome. The results identify two specific regions in apoB that are exposed to the cytosol in the secretory pathway.
Plasma low density lipoprotein (LDL)1 particles are derived from very low density lipoprotein (VLDL) particles secreted by liver. In humans both VLDL and LDL use apolipoprotein B100 (apoB) as the primary structural protein. ApoB is a large secretory protein (540 kD) that contains hydrophillic domains that help solubilize the lipoprotein in aqueous plasma and hydrophobic amphipathic domains that bind lipids and enclose a neutral core lipid droplet (Segrest et al., 1994). The human intestine and the livers of rats, mice, and dogs synthesize a shorter form of apoB called apoB48, which consists of the NH2-terminal 2,152 amino acids of apoB (Innerarity et al., 1996). As a secretory protein, apoB contains a cleavable signal sequence, is synthesized on membrane-bound ribosomes, and is cotranslationally translocated through the membrane of the ER (Dixon and Ginsberg, 1993; Innerarity et al., 1996; Yao et al., 1997). ApoB-containing lipoprotein assembly then occurs in a putative two-step process in which an initial lipoprotein particle is formed in the rough ER (Borén et al., 1993; Borén et al., 1994) and expansion of the lipid core then occurs in a second step in the smooth ER (Alexander et al., 1976) or possibly the Golgi (Higgins, 1988).
Regulation of the secretion of apoB is primarily posttranslational (Borchardt and Davis, 1987; Dixon and Ginsberg, 1993; reviewed in Yao et al., 1997). If lipid substrates are limiting, a large percentage of newly synthesized apoB molecules will be efficiently and rapidly degraded, either during or shortly after translation or after formation of an immature nascent lipoprotein particle within the secretory pathway (Dixon et al., 1991; Borén et al., 1994). When neutral lipid availability is increased, more apoB will be assembled into lipoprotein particles and secreted, and less apoB will be degraded intracellularly. To understand the transport of apoB in the secretory pathway, it is necessary to study the location of apoB in the cell and its orientation in the ER membrane. Although there is consensus that apoB becomes associated with the inner leaflet of the ER membrane, there is controversy concerning whether apoB is a transmembrane protein in the ER or remaining secretory pathway. Identifying apoB as a transmembrane protein represents a biological paradox because apoB contains a cleavable signal peptide and becomes glycosylated at multiple sites throughout the entire polypeptide. Additionally, although apoB is highly hydrophobic, it does not have even one classical membrane-spanning domain (Knott et al., 1986).
ApoB was identified as a cytosolically exposed protein in the studies of Davis et al. (1990), who reported that 56% of apoB particles and 70% of apoB48 particles in rough ER vesicles isolated from rat liver were sensitive to exogenous trypsin. Dixon et al. (1992) showed that 60– 75% of newly labeled apoB in rough ER vesicles isolated from chicken liver was susceptible to digestion with exogenous proteinase K. Other studies, for the most part using microsomes isolated through cell fractionation, have also observed that regions of apoB are exposed on the cytosolic surface of the ER (Rusiñol et al., 1993a ; Furukawa et al., 1992; Verkade et al., 1993; Wilkinson et al., 1993; Borén et al., 1993; Du et al., 1994; Wang et al., 1995; Bonnardel and Davis, 1995; McLeod et al., 1996). Du et al. (1994) reported that during translocation arrest, the 69-kD NH2 terminus of apoB resided in the lumen of microsomes, whereas the remaining domains of apoB were on the cytoplasmic surface of the ER membrane in HepG2 cells. Newly synthesized apoB was shown to be 60–80% cytosolically exposed in digitonin-permeabilized HepG2 cells (Macri and Adeli, 1997). Recently, apoB was found to be associated with heat shock protein 70 (Zhou et al., 1995), conjugated with ubiquitin (Yeung et al., 1996), and targeted for degradation to the cytosolic proteasome (Fisher et al., 1997). All of these observations strengthen the likelihood that apoB becomes transmembrane at some point in its transport through the secretory pathway.
However, other investigators have not observed a transmembrane orientation for apoB. A study in COS-1 cells transfected with the entire amino-terminal 50% of apoB indicated a complete lumenal localization of apoB in these nonhepatic cells (Shelness et al., 1994). ApoB did not assume an exposed cytosolic orientation after translation of mRNA coding for apoB30 in the presence of dog pancreas microsomes (Pease et al., 1995). In recent studies, a majority of endogenous apoB was protected from exogenous protease in membrane vesicles isolated from HepG2 cells (Ingram and Shelness, 1996; Leiper et al., 1996). Also, the extent of apoB glycosylation suggested that apoB was fully translocated into the lumen of the ER (Wong and Torbati, 1994; Leiper et al., 1996).
The experiments described in the current report have used both protease protection assays and immunocytochemistry to explore the location and orientation of apoB in the ER and in other compartments of the secretory pathway. The combined data provide convincing evidence that apoB, although most probably largely translocated into the lumen of the ER during translation, does assume a transmembrane orientation in the ER membrane of HepG2 cells. Information on the transmembrane nature of apoB may help in understanding regulation of apoB secretion by lipid substrates and other relevant signals in hepatocytes.
Materials and Methods
Materials
l-[4,5-3H]leucine (135 Ci/mmol, catalog no. TRK.683) was purchased from Nycomed Amersham Inc. (Princeton, NJ). Protein A-Sepharose CL-4B was obtained from Pharmacia LKB Biotechnology, Inc. (Piscataway, NJ). Minimum essential medium, nonessential amino acids, sodium pyruvate, streptolysin-O, and penicillin/streptomycin were from GIBCO BRL (Gaithersburg, MD). Labeling of cells was performed with a leucine-free medium generated from a minimum essential medium select amine kit (catalog no. 300–9050AV; GIBCO BRL). Leupeptin and pepstatin A were from Peninsula Laboratories, Inc. (Belmont, CA). BSA (essentially fatty acid–free), FBS (F6761), bovine thrombin, and apoferritin from horse spleen (A3641) were purchased from Sigma Chemical Co. (St. Louis, MO). Digitonin (high purity, no. 300410) was from Calbiochem-Novabiochem Corp. (San Diego, CA). Proteinase K (no. 745723, ∼20 U/mg lyophilized powder, with 1 U equal to 1 μmol of tyrosine liberated per min from hemoglobin at 37°C), and Pefabloc were from Boehringer Mannheim Corp. (Indianapolis, IN). Monospecific anti–human LDL antibody was raised in a rabbit. Sheep anti-human LDL was purchased from Serotec Ltd. (Raleigh, NC). The CC3.4, D7.2, and B1B6 monoclonal antibodies to apoB (domains shown in Fig. 1) were gifts of Dr. Gustav Schonfeld, Washington University, St. Louis, MO. Monoclonal antibodies to p63 (G1/296) and p53 (G1/93) were gifts of Dr. Hans-Peter Hauri, Department of Pharmacology, Biocenter of the University of Basel, Switzerland. Rabbit polyclonal antiserum to human galactosyltransferase was a gift from Dr. Eric G. Berger, Physiolog. Institut, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. Monoclonal antibody (H4A3) to lysosomal-associated membrane protein-1 (LAMP-1) was purchased from the Developmental Studies Hybridoma Bank, Department of Biological Sciences, The University of Iowa, Iowa City, IA. Antibodies conjugated with horseradish peroxidase (AffiniPure™ goat anti–rabbit IgG and AffiniPure™ rabbit anti–sheep IgG) were from Jackson Immuno Research Laboratories, Inc. (West Grove, PA). All other chemicals were of the highest purity available.
Figure 1.

Structure of apoB100. The structure shows the locations of the B1–B5 epitopes and the CC3.4, D7.2, and B1B6 monoclonal antibody-binding domains (Krul et al., 1988). Also shown are putative transmembrane helices (TM), hydrophobic domains of fifteen amino acids or more (▪), and amphipathic helices (boxed loops; Segrest et al., 1994). The top scale is the amino acid number while the bottom scale represents apoB using the centile system.
Synthesis of Human apoB Peptides and Raising Antipeptide Antisera in Rabbits
The SOAP program in PCGene was used to identify hydrophillic peptides in human apoB that are adjacent to highly hydrophobic regions (Fig. 1). Peptide names (amino acids in human apoB) are as follows: B1 (532–542): -arg-lys-met-glu-pro-lys-asp-lys-asp-glu-glu; B2 (1113–1122): -glu-thr-lys-asp-asp-arg-lys-ile-lys-gly; B3 (2059–2069) -lys-try-asp-lys-asn-gln-asp-val-his-ser-ile; B4 (3221–3240): -lys-ser-tyr-asn-glu-thr-lys-ile-lys-phe-asp-lys-tyr-lys-ala-glu-lys–ser-his-asp; and B5 (4010–4020): -gly-met-asp-met-asp-glu-asp-asp-asp-phe-ser.
These peptides were synthesized on multiple antigenic peptides (MAP) resins on a model 432A synthesizer (PE Applied Biosystems, Foster City, CA) using Fmoc chemistry. Peptides were synthesized, cleaved, and purified according to the manufacturer's protocol (PE Applied Biosystems).
Polyclonal antibodies to MAPs were prepared in New Zealand White rabbits. After collecting preimmunized sera, each rabbit received a subcutaneous injection of a mixture of an individual peptide (0.5–1 mg) and Freund's adjuvant. The rabbits were boosted at 3–4-wk intervals with half the quantity of peptide and Freund's incomplete adjuvant. Before use in immunocytochemistry, the B4 antipeptide antibody and the rabbit and sheep polyclonal anti–human LDL antibodies were affinity-purified on columns containing either purified peptide or human LDL, respectively. For the antiB4 antibody, 3 ml of antiserum (diluted to 6 ml with 0.1 M Na2HPO4 buffer, pH 7.0) was loaded onto a column consisting of 3 ml of aminolink-gel coupled with 2.78 mg of the B4 MAP. The column was washed with 23 ml of 0.1 M Na2HPO4 buffer, pH 7.0, and eluted with 0.1 M glycine buffer, pH 2.8. The B4 antibody was eluted in a peak of 3 ml total volume. There was almost complete recovery of apoB immunoprecipitation activity. Both rabbit and sheep polyclonal anti-human LDL were purified in the same way on columns containing aminolink gel coupled with human LDL.
Metabolic Labeling of Cells, Permeabilization with Digitonin, and Immunoprecipitation
HepG2 cells were grown in collagen-coated six-well cluster culture dishes (9.6 cm2/well) in complete growth medium (MEM with 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% heat-inactivated FBS) in a CO2 incubator at 37°C. For protease protection assay in permeabilized cells, medium was removed from confluent wells, and the cells were washed twice with PBS and preincubated for 1 h in serum-free medium containing 1.5% BSA. For pulse labeling, the medium was removed, and the cells were washed with PBS and incubated in leucine-free medium containing [3H]leucine (75 μCi/ml) for 10 min. The labeling medium was removed, the cells were washed twice with PBS, and serum-free chase medium was added for the indicated time. At the end of the chase period, medium was removed and cells were either harvested immediately or permeabilized with digitonin. For harvesting, 1 ml of lysis buffer (150 mM NaCl, 5 mM EDTA, 50 mM Tris, pH 7.4, 0.0625 M sucrose, 0.5% Triton X-100, and 0.5% sodium deoxycholate) containing a protease inhibitor mixture (final concentrations: 1 mM benzamidine, 5 mM EDTA, 0.86 mM phenylmethylsulfonyl fluoride, 100 kallikrein-inactivating U/ml aprotinin, 10 mM 4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid [pH 8.0], 50 μg/ml leupeptin, 50 μg/ml pepstatin A and 239 μg/ml pefabloc) was added per well, and cells were scraped off of the dish. For permeabilization, 1 ml of intracellular buffer I (described by Plutner et al., 1992), 75 mM potassium acetate, 2.5 mM magnesium acetate, 5 mM EGTA, 1.8 mM CaCl2, 1 mM ATP, and 25 mM Hepes buffer, pH 7.2 (designated BB I) containing 60 μg/ml digitonin was added to the cells at 4°C. After 5 min, BB I containing digitonin was removed, and the cells were washed three times with BB I alone. The cells were then incubated on a rotating platform at 80–100 rpm for 40 min at 4°C with either BB I alone or BB I containing proteinase K (PK). After incubation, PK was removed and the cells (which remained attached during incubations and washes) were washed three times with BB I (1 ml). ApoB was then extracted from the cells at 4°C with 1 ml of lysis buffer plus protease inhibitors as described above. Immunoprecipitation of [3H]- labeled protein from cell extracts and medium was carried out at 4°C by the method previously described (Yu et al., 1983; Dixon et al., 1991). A small aliquot of each immunoprecipitate was taken for scintillation counting, and samples were separated by SDS-PAGE (3–15% gradient; Laemmli, 1970). The gels were treated with Autofluor (National Diagnostic, Inc., Atlanta, GA) for 20 min, dried, and exposed to film (XAR-2; Eastman Kodak Co., Rochester, NY).
Immunoblotting
Cells were grown to 90% confluence and washed with PBS, and proteins were extracted with 2 ml of lysis buffer plus protease inhibitors. The proteins were concentrated in a Centricon-10 concentrator (Amicon Corp., Eastman, TX), boiled in electrophoresis sample buffer, separated by SDS-PAGE (3–15% gel), and transferred overnight from gels to polyvinyl difluoride Immobilon P membranes (Towbin et al., 1979). After blocking with 5% nonfat dried milk in Tris-buffered saline (0.02 M Trizma base, 0.137 M sodium chloride, pH 7.6) plus 0.02% Tween 20, membranes were immunoblotted with antibodies (affinity-purified rabbit anti-human apoB B4 peptide, 1/5,000 final dilution; sheep anti-human LDL, 1/10,000 final dilution), and immunoreactive protein was visualized with horseradish peroxidase–conjugated second antibodies (AffiniPure goat anti–rabbit IgG and AffiniPure™ (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) rabbit anti–sheep IgG, 1/5,000 final dilution) and luminol reagent (ECL kit; Nycomed Amersham).
Culture of HepG2 cells for Immunocytochemistry and Permeabilization with Streptolysin-O
HepG2 cells were seeded onto collagen-coated coverslips and grown for 48 h to 20–30% confluence in complete growth medium in a CO2 incubator at 37°C. 1 h before fixing cells, the medium was changed to a serum-free medium. For certain controls, cells were grown in serum-free medium for 4 h before fixing. After 1 h in serum-free medium, the medium was aspirated and the cells were washed three times with intracellular buffer II (designated BB II; 75 mM potassium acetate, 25 mM Hepes buffer, pH 7.2) that was modified after the buffer described by Plutner et al. (1992). The cells were fixed in 2% paraformaldehyde for 30 min at 22°C, washed three times with BB II, and incubated for 15 min at 4°C with 500 U/ml streptolysin-O (STP-O) dissolved in BB II. STP-O was removed, and the cells were washed once and incubated in BB II for 15 min at 37°C to allow for pore formation. STP-O permeabilizes the plasma membrane of hepatocytes, allowing access of IgG molecules to domains of proteins exposed on the cytosolic side of intracellular membranes without rupturing these internal membranes (Ahnert-Hilger et al., 1989; Esparís-Ogando et al., 1994; Pimplikar et al., 1994). Cells were washed three times with BB II at 22°C, and were blocked for 1 h with BB II plus 0.1% BSA. Permeabilized cells were incubated for 4 h or overnight with primary antibodies (monoclonal, polyclonal, or antipeptide) to apoB or to control resident secretory pathway proteins, followed by incubation for 4 h with the appropriate anti–mouse, –rabbit, or –sheep IgG secondary antibody conjugated with fluorochromes (Cy 3, indocarbocyanine or Cy 5, indodicarbocyanin; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). After washing and mounting coverslips onto glass slides, cells were examined with a MRC-600 confocal microscope (Bio-Rad Laboratories, Hercules, CA) located in the Molecular Cytology Core Laboratory supported by the University of Missouri.
Saponin Permeabilization of Cells
In addition to STP-O permeabilization, cells were also permeabilized with saponin in order to expose lumenal epitopes of both apoB and marker proteins that remained hidden in STP-O–permeabilized cells. Saponin is an agent that forms holes in membranes, but does not extract membrane proteins. Saponin allows access of antibody molecules to epitopes that are lumenal and accessible. If epitopes are hidden by lipids or by protein conformation, they may not be recognized by an antibody in saponin-treated cells. After fixing cells with 2% paraformaldehyde and washing three times with BB II, the cells were treated with 0.1% saponin in BB II plus 0.1% BSA for 30 min at 22°C. The cells were then washed with 0.1% saponin in BB II plus 0.1% BSA. All incubations of cells with primary and secondary antibodies and washes were done with 0.1% saponin in BB II plus 0.1% BSA.
Analysis of Digitized Images
To compare and determine quantitative differences in fluorescence images obtained between treatments with the confocal microscope, it was necessary to compare the total spectrum of pixel intensity in specific areas of cells in fields of cells of similar cell density. All other experimental conditions were kept constant, including antibody titre, total cell confluence, laser strength, magnification, and basal instrument settings. In the current study, the approximate signal strength for apoB in the ER of STP-O–permeabilized cells was compared with that in saponin-permeabilized cells. The digitized images of HepG2 cells (see Fig. 7, C and D) were subjected to image analysis by selecting a 2-μm–wide area around the nucleus of each cell and obtaining a histogram of pixel intensity for all pixels within this region. The fluorescence signal was calculated by obtaining the integrated sum of all pixel values in the histogram. This sum of all pixel values for each cell was then divided by the area of the 2-μm bounded region to obtain total integrated pixel intensity/μm2 (TIPI/μm2). The TIPI/μm2 for cells with different permeabilization treatments were compared in order to determine whether the relative signal intensities differed between treatments. The signal obtained in saponin-treated cells was taken as the total epitope exposed both to the cytosol and lumen, and was set at 100%. The signal obtained in STP-O–permeabilized cells was considered to be only exposed to the cytosol. The fluorescence signal due to lumenal epitopes should increase greatly in saponin-treated cells (>10×) compared with STP-O alone, but the fluorescence due to cytosolic epitopes should change little or only slightly.
Figure 7.

Immunocytochemistry of apoB in HepG2 cells. HepG2 cells were fixed in 2% paraformaldehyde and permeabilized with either STP-O (A, C, and E) or saponin (B, D, and F) and probed with the following antibodies: (A and B) monoclonal anti-p63 (1/1000); (C and D) affinity-purified rabbit anti-human apoB peptide B4 (1/100; recognizes apoB between amino acids 3221 and 3240); and (E and F) affinity-purified polyclonal sheep anti-human LDL (1/ 250). The appropriate secondary antibodies were used at a dilution of 1/100.
Results
Synthesis of apoB Peptides as MAP Conjugates and Production of Antipeptide Antibodies
A hydrophobicity plot of the amino acid sequence of human apoB was examined in order to identify hydrophillic segments of apoB that are adjacent to highly hydrophobic regions (Fig. 1). Five hydrophillic segments were chosen (B1–B5), and these peptides were synthesized as MAP conjugates as described in Materials and Methods. Rabbit antisera to these peptides were produced and evaluated by immunoprecipitation of [3H]leucine-labeled HepG2 proteins. Only one of the five peptides was able to immunoprecipitate [3H]apoB (Fig. 2) with high specificity and titre. The B2 antibody could be purified on a B2 peptide column, but did not recognize the corresponding domain in the apoB molecule (data not shown). Fig. 2 shows that the antibody to the B4 peptide (later referred to as the B4 antibody) immunoprecipitated apoB (lower band) with good efficiency compared with a rabbit polyclonal antibody made to human LDL. The protein that migrated slower than the apoB standard has been identified as aggregated apoB, and was immunoprecipitated by both polyclonal and B4 antibodies.
Figure 2.

Comparison of anti-apoB peptide antibodies to the conventional polyclonal anti-LDL in ability to immunoprecipitate apoB. Five hydrophilic peptides of apoB (B1–B5) were synthesized as described in Materials and Methods. Polyclonal antisera to these peptides were raised in rabbits and evaluated by immunoprecipitation of [3H]leucine-labeled HepG2 proteins. [3H]apoB from 500 μl of HepG2 lysis buffer cell extracts was immunoprecipitated with 1 μl of sheep polyclonal anti-human LDL (poly, leftmost lane) or with 20 μl of each antipeptide antisera under normal immunoprecipitation conditions (lanes designated with a 1), or when the immunoprecipitation binding and washing buffers contained final concentrations of 2 mM dithiothreitol and 0.5% deoxycholic acid (lanes designated with a 2). In the lane marked 1′, apoB was immunoprecipitated with 5 μl of the B4 antiserum under normal immunoprecipitation conditions. ApoB was immunoprecipitated and analyzed by SDS-PAGE (3–15% gel) and fluorography. Markers show the location of Coomassie-stained standard apoB (isolated from human plasma) and molecular weight standards (kD). Antiserum from the rabbit injected with the B4 peptide was affinity-purified on an aminolink-B4 peptide affinity column before use in subsequent immunocytochemistry studies.
Immunoblot Analysis of the B4 Antibody
HepG2 cell proteins solubilized by lysis buffer were electrophoresed and blotted to a polyvinyl difluoride membrane and probed by an affinity-purified preparation of B4 antibody (Fig. 3). The results show that the B4 antibody was highly specific for apoB. Light bands at other molecular weights were shown by comparison with other anti-apoB antibodies to be breakdown products of full-size apoB (data not shown). The polyclonal antibody made to human LDL gave main bands at apoB100 (540 kD) and at ∼70 kD, the major small degradation product of apoB seen in cells and medium (Adeli, 1994; Du et al., 1994).
Figure 3.

Western blot analysis of HepG2 cell proteins using the B4 antibody. HepG2 cell proteins were solubilized in lysis buffer, separated by SDS-PAGE (3–15% gradient), blotted to membranes, and probed by the sheep polyclonal anti-human LDL antibody (P) or the affinity-purified B4 antibody (B4). Markers show the location of Coomassie-stained standard apoB (isolated from human plasma) and molecular weight standards.
Protease Protection Assay of [3H]apoB in Digitonin-permeabilized Cells
The topology of newly synthesized [3H]apoB in digitonin-permeabilized HepG2 cells was investigated by protease protection assay after a 10-min pulse/10-min chase (Fig. 4). As it takes ∼40 min to see significant secretion of newly synthesized apoB (Dixon et al., 1991), most labeled apoB was still probably located in the ER. Digitonin was used to permeabilize cells in this experiment as STP-O (GIBCO BRL) was only able to permeabilize at most ∼50% of cells in confluent monolayers of HepG2 cells (data not shown). Apoferritin (100 μg/ml) was included in the assay buffer as a carrier protein in lieu of BSA. Certain wells of cells were preincubated with 40 μg/ml (104 μM) acetyl-leucyl-leucyl-norleucinal (ALLN) for 1 h before labeling in order to prevent endogenous degradation of large apoB peptides formed when apoB in permeabilized cells was initially cut with PK (data not shown). Lanes 1 and 5 (Fig. 4) show that apoB immunoprecipitated from intact cells immediately after the 10-min chase migrated at the same mobility as plasma apoB. Cells incubated with ALLN (compare lane 5 to lane 1) had a greater apoB signal intensity due to inhibition of very early apoB degradation during the pulse/chase period (Sakata et al., 1993). There was no loss of [3H]apoB due to the permeabilization process or subsequent 40-min incubation period (Fig. 4, lanes 2 and 6). Unlike when isolated microsomes are used to determine sidedness, close to 100% of labeled apoB can be probed in permeabilized cells. After the incubation with PK, protease activity was inhibited by adding protease inhibitors including phenylmethylsulfonyl fluoride. ApoB and degradation peptides from apoB were immunoprecipitated with the sheep polyclonal anti-LDL antibody. Incubation of permeabilized cells for 40 min at 4°C with 1.4 or 4.2 μg PK/ml (lanes 7 and 8) decreased the full-length apoB signal by ∼70 and 90%, respectively. ApoB cleavage products were observed at ∼450–470 (arrow 2), 387 (dark band at arrow 3), 200– 300 (arrow 4), 153 (dark band at arrow 5), 120 (arrow 6), and 60–70 kD (arrow 7). More consistent degradation band patterns were observed after PK digestion in cells that had been pretreated with ALLN compared with cells not treated with ALLN (data not shown). Increasing the PK concentration to 10 and 50 μg/ml led to partial loss of the primary apoB digestion products (lanes 9 and 10), most likely due to an inability to adequately inhibit protease activity during sample processing for immunoprecipitation (data not shown). When permeabilized cells were incubated with thrombin, a large thrombin breakdown product of apoB was observed (arrow 8, lane 4) only in the presence of Triton X-100. This product was identified as 387 kD based on the major thrombin site in apoB, T3/T2 (Krul et al., 1988). One of the major large apoB degradation peptides resulting from PK digestion (arrow 3; seen in lanes 7 and 8) was identical in mobility to the major thrombin cleavage product (arrow 8, lane 4). The production of a distinct pattern of mainly large peptides in the presence of ALLN suggests that PK had access to nascent apoB at only a few sites, and that cleavage at one of the sites caused the major peptides at 387 and 150 kD. The results indicate that immediately after its complete biosynthesis, apoB is a transmembrane protein with only a limited number of sites exposed on the cytosolic face of ER membrane.
Figure 4.

Sensitivity of apoB to proteinase K in digitonin-permeabilized cells. HepG2 cells were labeled with [3H]leucine and chased as described in Materials and Methods. Cells in lanes 5–10 were preincubated for 1 h with 40 μg/ml (104 μM) ALLN, and were also treated with ALLN during the pulse/chase periods. After the chase cells were harvested immediately in lysis buffer (lanes 1 and 5), or were permeabilized with 60 μg/ml digitonin (DIG; lanes 2–4 and 6–10). Permeabilized cells were incubated for 40 min at 4°C with either BB I + 100 μg/ml apoferritin (lanes 2 and 6), BB I + 100 μg/ml apoferritin + proteinase K (PK; lane 7: 1.4 μg/ml; lane 8: 4.2 μg/ml; lane 9: 10 μg/ml; lane 10: 50 μg/ ml), or BB I + 100 μg/ml apoferritin + bovine thrombin (Th; 7.56 U/ml or 0.14 mg/m; lanes 3 and 4). Cells in lane 4 were also incubated in 1% Triton X-100 (TX). After removing PK, the cells were washed three times and harvested in lysis buffer containing protease inhibitors, and [3H]apoB was immunoprecipitated and analyzed by SDS-PAGE (3–15% gel) and fluorography. Markers show the location of Coomassie-stained standard apoB (isolated from human plasma) and molecular weight standards. The right-facing arrow to the left of lane 4 (arrow 8) shows the location of the major thrombin cleavage product at 387 kD. Arrow 3 shows the PK cleavage product that has the same mobility as the major thrombin cleavage product. ApoB peptides formed by PK were observed at ∼450–470 (arrow 2), 387 (dark band at arrow 3), 200–300 (arrow 4), 153 (dark band at arrow 5), 120 (arrow 6), and 60–70 kD (arrow 7).
ApoB Topology Through the Secretory Pathway
Studies on apoB orientation (Fig. 5) were carried out at longer chase times in order to test the hypothesis that apoB translocates through the membranes of the secretory pathway or changes its orientation in the membrane upon formation of nascent lipoprotein particle in the ER. An alternate hypothesis is that apoB retains a transmembrane orientation even after partial lipidation and extensive transport through the secretory pathway. In the experiment depicted in Fig. 5, cells were treated with 40 μg/ml (104 μM) ALLN in order to slow rapid intracellular apoB degradation (Sakata et al., 1993) and to preserve the large fragments of apoB formed by PK digestion of permeabilized cells as seen in Fig. 4. Cells were labeled with [3H]leucine as in Fig. 4, except that cells were chased for either 10, 20, 30, 40, or 60 min before cell harvest or permeabilization. Additionally, certain cells destined for longer chase periods were treated with 0.4 mM oleate (OA) after the 10-min chase time point in order to test whether increased lipid synthesis alters the conformation of apoB in secretory pathway membranes. The permeabilization protocol in the current experiment was similar to that used for lanes 5–7 of the previous experiment (Fig. 4). Although cells were treated with ALLN, there was endogenous posttranslational degradation of apoB during the first 30 min of chase in nonpermeabilized cells (Fig. 5, A and C). The decay in cellular [3H]apoB was much slower in ALLN-treated cells compared with cells not treated with ALLN (Fig. 5 C). Taking the amount of [3H]apoB in cells after 10 min of chase as 100%, the decay in cellular [3H]apoB was similar in ALLN-treated control and oleate-treated cells. Decay in cellular apoB between 10 and 30 min was due to intracellular degradation. The decay in apoB in oleate-treated cells between 30 and 60 min represented loss of cellular apoB primarily due to secretion, whereas the decay in control cells was due to both degradation and secretion during this period. After 60 min of chase, 45–55% of labeled apoB was still present intracellularly (Fig. 5 C). The dashed line in Fig. 5 C represents data from a previous experiment (Dixon et al., 1991) in which apoB decay was monitored in control cells that were not treated with ALLN. Only 15 ± 3% of cellular [3H]apoB remained after 60 min of chase in control cells not treated with ALLN (Dixon et al., 1991). In the current experiment there was 54% total recovery (cellular + medium) of [3H]apoB in control cells, and 75% total recovery in oleate-treated cells. This translates into 46% total degradation of [3H]apoB in control cells and 25% total degradation in oleate cells. The apoB degradation observed occurred largely by an ALLN-independent proteolytic system, with more degradation occurring in control cells.
Figure 5.



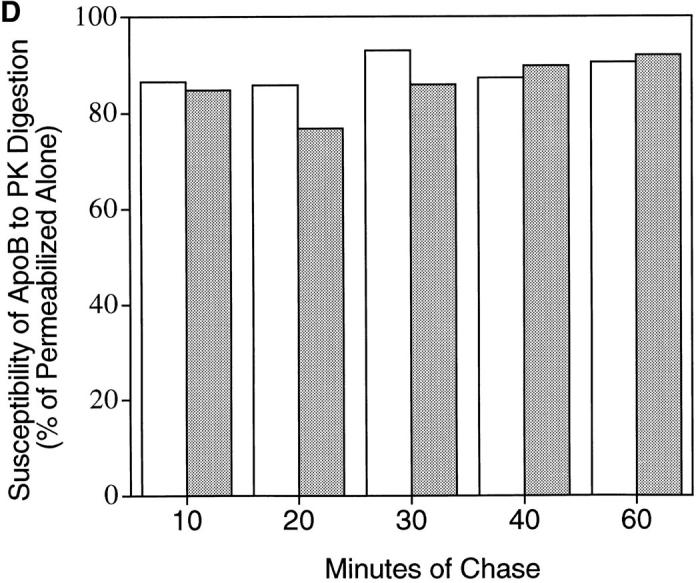
Time course of apoB topology in the secretory pathway of HepG2 cells. (A and B) Cells were preincubated for 1 h in serum-free medium containing 40 μg/ml (104 μM) ALLN. ALLN was also present in the medium of all cells during the pulse and chase periods. Cells were pulsed with [3H]leucine for 10 min as described in Materials and Methods. After the pulse, all cells were chased for 10 min with serum-free medium containing 1.5% BSA. At this point, the 10-min chase cells were harvested, and the medium on all of the other plates was changed to serum-free medium containing either 1.5% BSA (C, control; lanes 1–3 and 7–9) or 0.4 mM oleate complexed to 1.5% BSA (OA; lanes 4–6 and 10–12). The remaining cells were then chased for a total of 20, 30, 40 or 60 min. After each chase period the cells were either harvested immediately in lysis buffer (S, lanes 1, 4, 7, or 10) or permeabilized with 60 μg/ml digitonin (B or PK, lanes 2, 3, 5, 6, 8, 9, 11, and 12) as described in Materials and Methods. After washing away the digitonin, permeabilized cells were incubated for 40 min at 4°C with either buffer (BB I) + 100 μg/ml apoferritin (B; lanes 2, 5, 8, and 11) or with BB I + 100 μg/ml apoferritin + 2.8 μg/ml proteinase K (PK, lanes 3, 6, 9, and 12). After 40 min of incubation, the buffer was removed, the cells were washed three times with PBS and harvested in lysis buffer containing protease inhibitors. ApoB (A) or albumin (B) were immunoprecipitated and analyzed by SDS-PAGE (3–15% gel) followed by fluorography. The lower series of lanes 7–12 in each panel are apoB (A) and albumin (B) immunoprecipitated from medium of cells that were chased for 60 min (corresponding cell extracts are in lanes 1–6 of each lower series). Markers show the location of Coomassie-stained human plasma apoB and molecular weight standards. (C) Analysis of time course of apoB decay. The fluorograms shown in A were scanned by laser densitometry, and the cellular apoB signal remaining after each chase period is plotted in C. ApoB signal in nonpermeabilized cells (S in Fig. 5 A) is denoted for control cells (▪) and for cells treated with oleate (O). For permeabilized cells incubated with buffer alone (B in Fig. 5 A), the data is denoted for control cells (♦) and for cells treated with oleate (▵). The data denoted (encircled +) is the decay of apoB in control cells not treated with ALLN as reported previously (Dixon et al., 1991). The bars on the right side of Fig. 5 C represent the amount of apoB secreted by control cells (closed bar) or oleate-treated cells (open bar) after 60 min of chase (as a % of the initial cellular [3H]apoB after 10 min of chase). (D) The percentage of apoB that was susceptible to PK digestion in control (open bar) and OA-treated cells (closed bar) after each chase period (expressed as percent of the apoB signal at each time point). This experiment was repeated several times with similar results.
The percentage of cellular full-size apoB that was susceptible to PK in permeabilized cells remained constant at each of the chase time points (Fig. 5 D). At each of the chase points, the pattern of apoB peptides observed after PK digestion (Fig. 5 A; lanes 3, 6, 9, and 12) was similar to the pattern observed in lane 7 of Fig. 4. One possible exception was that there may have been a slightly lower signal for full-size apoB (540 kD) and peptide 2 (450–470 kD) in control cells compared with oleate-treated cells after 30 or 40 min of chase (Fig. 5 A). That access of PK to apoB was not the result of disruption of secretory pathway membranes was most clearly evident at 10, 20, and 30 min of chase when there was minimal loss of [3H]albumin from the secretory pathway (4.5–9.7%; Fig. 5 B). Loss of [3H]albumin due to the permeabilization process (Fig. 5 B) was 9.7% at 10 min (lanes 2 and 5 compared with lanes 1 and 4), 4.5% at 20 min (lanes 8 and 11 compared with lanes 7 and 10), and 6% at 30 min (lanes 2 and 5 compared with lanes 1 and 4). In contrast, loss of [3H]albumin from permeabilized cells was 29.5% at 40 min (lanes 8 and 11 compared with lanes 7 and 10) and 43% at 60 min (lanes 2 and 5 compared with lanes 1 and 4). These results indicate that membranes of the later secretory pathway (possibly trans Golgi or secretory vesicles) were more sensitive to treatment with digitonin than were membranes of the earlier secretory pathway (RER/SER), possibly because downstream membranes contain more cholesterol (Bretscher and Munro, 1993). In cells that were chased for 60 min, close to 100% of the labeled albumin that was initially present in the cells was secreted into the medium (Fig. 5 B, compare medium lanes 7–12 at 60 min with cellular lanes 1–6 at 10 min). The results show that membrane damage can be precisely monitored in digitonin-permeabilized cells by following loss of [3H]albumin from the lumen of the secretory pathway to washes of the permeabilized cells. The results also show that there was little difference in the conformation of apoB in secretory pathway membranes between control and oleate-treated cells between 10 and 30 min of chase. This was the case even though there was a large stimulation in apoB secretion due to adding oleate at the 10-min chase time point (Fig. 5, A and C). The results indicate that apoB maintains its transmembrane orientation through at least 30 min of chase, and that formation of a nascent lipoprotein particle does not alter its transmembrane nature.
Confirmation of the Transmembrane Nature of apoB by Immunocytochemistry
The sensitivity of apoB to exogenous protease in protease protection assays is the primary basis for claims that apoB becomes a transmembrane protein at some point after its complete biosynthesis (for review see Yao et al., 1997). One of the criticisms of such studies is that apoB is highly susceptible to proteolysis, and that problems such as membrane permeability, overdigestion, or incomplete inhibition of exogenous protease are difficult to control adequately (Ingram and Shelness, 1996). To confirm that domains of apoB are present on the cytosolic side of ER membranes, cells were permeabilized with STP-O and then probed with specific antibodies to apoB and examined with the confocal microscope. First, experiments were performed to show that internal membranes were not perforated in HepG2 cells treated with STP-O. HepG2 cells were fixed in paraformaldehyde, permeabilized with either STP-O (Fig. 6, left) or saponin (Fig. 6, right), and probed with antibodies to lumenal domains of resident marker proteins of different secretory pathway compartments. The marker proteins were as follows: p63 (a marker for the ER; Schweizer et al., 1995), p53 (a marker for the ER-to-Golgi intermediate compartment; ERGIC; Schweizer et al., 1995), and galactosyltransferase (a marker for the Golgi; Berger et al., 1993). None of the lumenal epitopes were accessible in STP-O–permeabilized cells (1a, 2a, and 3a), but all were exposed in cells permeabilized with saponin (1a, 2b, and 3b). To demonstrate that the cells were in focal plane, STP-O–permeabilized cells were also observed under bright-field conditions (1a, left). P63 is a type II integral membrane protein that is predominantly localized in the rough ER, but is absent from the nuclear membrane and the cis-Golgi region (Schweizer et al., 1995). In saponin-treated HepG2 cells (Fig. 6, 1b), the p63 antibody stained an area around the nucleus as well as a vast reticular network throughout the cell. The p63 antibody did not label the nuclear outer membrane as there was no bright continuous line of staining surrounding the nucleus. The p53 antibody stained a finite area of the cell that resembled a crown (Fig. 6, 2b). This area was usually located on one side of the nucleus and extended from the nuclear region to the vicinity of the plasma membrane. P53 is a marker for the ERGIC (usually located in the tubulovesicular membranes at the cis face of the Golgi apparatus). It is also present in the rough ER as p53 is postulated to cycle between the ER and ERGIC (Schweizer et al., 1995). The antibody to galactosyltransferase stained a reticulum network that was removed from the nuclear area, and that was in close proximity to the plasma membrane. Galactosyltransferase is predominantly localized in trans cisternae of the Golgi apparatus (Berger et al., 1993). In the current experiments, the Golgi appeared to be located in a more peripheral area of the cell. In agreement with published reports (Ahnert-Hilger et al., 1989; Esparís-Ogando et al., 1994; Pimplikar et al., 1994), observations with all three markers indicated that intracellular membranes were not perforated in STP-O–permeabilized HepG2 cells.
Figure 6.

Membranes of the ER, ER-to-Golgi intermediate compartment, and the Golgi are left intact when the plasma membrane of HepG2 cells is permeabilized with STP-O. HepG2 cells were fixed in paraformaldehyde and permeabilized with either STP-O (1a, 2a, and 3a) or saponin (1b, 2b, and 3b). The cells were then probed with a 1/1,000 dilution of a monoclonal antibody to p63 (a marker for the ER; 1a and 1b), a 1/1,000 dilution of a monoclonal antibody to p53 (a marker for the ER to Golgi intermediate compartment; 2a and 2b), and a 1/50 dilution of a rabbit anti-human galactosyltransferase (a marker for the Golgi; 3a and 3b). The appropriate secondary antibodies were used at a dilution of 1/100. The left side of 1a is a bright-field image.
Immunocytochemistry of apoB in HepG2 Cells
HepG2 cells were permeabilized with either STP-O (Fig. 7, A, C, and E) or saponin (Fig. 7, B, D, and F), blocked with buffer plus 0.1% BSA, and incubated with the following antibodies: (A, B) monoclonal anti-p63; (C, D) affinity-purified rabbit anti-B4 peptide; (E, F) affinity-purified polyclonal sheep anti-human LDL). After washing and additional blocking, cells were treated with the appropriate secondary antibodies conjugated with either Cy-3 or Cy-5 and examined with a confocal microscope. In preliminary experiments, it was shown that there was extremely low background fluorescence in cells that were: (a) nonpermeabilized and treated with antibody B4 and secondary antibody; (b) permeabilized and treated with preimmune serum and secondary antibody; or (c) permeabilized and treated with primary antibodies alone (data not shown). The lack of significant signal for p63 in STP-O–permeabilized cells (Fig. 7 A) indicates that membranes of the endoplasmic reticulum were not permeabilized in this experiment. The p63 signal was greatly increased when cells were permeabilized with saponin (Fig. 7 B). In contrast, the signal for apoB probed with the B4 antibody was similar in both STP-O–permeabilized cells and saponin-treated cells (compare C and D). Image analysis of the fluorescent signals for apoB in the perinuclear region of cells (defined as a 2 μm–wide band starting with the transition from relative blackness of the nuclear area to fluorescence and radiating out toward the periphery of the cell) was performed, and the mean and standard deviation of total integrated pixel intensities/μm2 (TIPI/μm2) for three cells from each treatment group was calculated. STP-O–treated cells (Fig. 7 C) had a TIPI/μm2 = 3178 ± 155 (mean ± SD) compared with 3564 ± 83 for saponin-treated cells (Fig. 6 D). Therefore, the relative signal intensity for the B4 antibody in STP-O–treated cells was 89% that of saponin-treated cells, indicating that the B4 antibody had access to most B4 epitopes regardless of whether internal membranes were permeabilized or not. Equivalent signal intensities for apoB with the B4 antipeptide antibody were observed in STP-O– and saponin-treated cells in six separate experiments. These data indicate that the B4 epitope in this region is exposed primarily on the cytosolic surface of the ER membrane. In addition to giving a very strong fluorescence in a bright continuous line surrounding the nucleus, the B4 antibody stained the entire reticular network throughout the cell in STP-O–treated cells (Fig. 7 C). The brighter fluorescence observed in the area in close proximity to the nucleus in both STP-O– and saponin-treated cells probably represents a higher concentration of apoB at the primary location of apoB synthesis. We have previously established that there is rapid degradation of apoB in control HepG2 cells such that more than 50% of newly synthesized [3H]apoB was degraded between 10 and 20 min of chase without isotope after a 10-min pulse period with [3H]leucine (Dixon et al., 1991). The less intense fluorescent signal that is observed in areas of the cell away from the nuclear membrane represents apoB in the secretory pathway (ER, ERGIC, and Golgi compartments). We interpret the decrease in fluorescence signal in membranes removed from the perinuclear region to result from a lower concentration of apoB in these regions because of degradation of apoB in the ER. The weaker signals away from the perinuclear area may also be consistent with some translocation of apoB across the ER on the way to secretion.
Unexpectedly, there was no intense region of staining in the perinuclear area of STP-O–treated cells probed with the sheep anti-LDL antibody (Fig. 7 E). Instead, the polyclonal anti-LDL antibody gave a punctate staining pattern with bright focused areas of signal scattered throughout the cytoplasm of the cell (Fig. 7 E). Similar observations were made in cells grown in serum-free medium for 4 h. When interior membranes were permeabilized with saponin, an apoB signal was seen throughout the extensive secretory pathway (Fig. 7 F). The labeling pattern obtained with sheep anti-LDL in saponin-treated cells was similar to that observed with the B4 antibody in saponin-treated cells (Fig. 7 D), except for the absence of the intense, narrow band of signal in the perinuclear region. A lack of a defined perinuclear signal by the polyclonal anti-LDL antibody in saponin-treated cells may have resulted from the absence of mature epitope sites in nascent apoB molecules recognized by this antibody (see Discussion). These results demonstrate that none of the epitopes recognized by the polyclonal anti-LDL antibody are exposed to the cytosol in the perinuclear region of STP-O–treated cells, and only become exposed after saponin permeabilization. These data also confirm the earlier observation that the membranes of the secretory pathway were not ruptured in STP-O-permeabilized cells.
Orientation of Other Sites in apoB
The orientation of two other sites in apoB was tested by immunocytochemistry. The CC3.4 monoclonal antibody (Krul et al., 1988) that recognizes apoB within the region of amino acids 690–797 (79–91 kD) and the D7.2 monoclonal antibody (Krul et al., 1988) that recognizes apoB within the region of amino acids 1878–2148 (216–247kD) were used to probe apoB orientation in STP-O– and saponin-permeabilized cells. The CC3.4 antibody produced bright staining in close proximity to the nucleus and in peripheral areas in STP-O–permeabilized cells (Fig. 8 A). The bright staining near the nucleus was less defined compared with the staining produced by the B4 antibody (Fig. 7 C). The signal produced by the CC3.4 antibody was not greatly increased in saponin-permeabilized cells (Fig. 8 B). There was almost a complete absence of intracellular signal when STP-O–permeabilized cells were probed with the D7.2 antibody (Fig. 8 C). In contrast, a strong fluorescent signal was seen throughout saponin-treated cells (Fig. 8 D). These results indicate that the region of apoB identified by the CC3.4 antibody is exposed to the cytosol in the perinuclear region and throughout the secretory pathway, whereas the region identified by the D7.2 monoclonal antibody most likely is not cytosolically exposed but located in the lumen of the secretory pathway. Except for areas of punctate staining in CC3.4-treated cells, the signals produced by the B4 and CC3.4 antibodies overlapped in both perinuclear and cytosolic areas of STP-O–permeabilized HepG2 cells (Fig. 9). These results show that two regions of apoB that are widely separated are exposed to the cytosol in extensive regions of the secretory pathway but that a domain in between (the D7.2 region) is lumenal.
Figure 8.

Cellular orientation of CC3.4 and D7.2 domains of apoB in HepG2 cells. Hep G2 cells were fixed and permeabilized with either STP-O (A and C) or saponin (B and D) and probed with the following antibodies: A and B: monoclonal anti-human apoB CC3.4 (1/ 100); C and D: monoclonal anti-human apoB D7.2 (1/ 500). The appropriate secondary antibodies were used at final dilution of 1/100.
Figure 9.

Dual localization of B4 and CC3.4 domains in apoB in STP-O–permeabilized cells. HepG2 cells were permeabilized with STP-O and probed simultaneously with monoclonal CC3.4 (1/ 100) and affinity-purified rabbit anti-B4 peptide antibody (1/25). After washing, the cells were incubated with the appropriate secondary antibodies conjugated with different fluorochromes and examined at 570 nm (A, green; CC3.4 monoclonal) and 680 nm (B, red; anti-B4 peptide). The digital images were superimposed, and significant overlap is denoted by yellow (C).
Colocalization of apoB with Monoclonal Antibody B1B6 and Sheep Polyclonal Anti-human apoB
The punctate labeling observed for apoB in STP-O–permeabilized cells using affinity-purified sheep polyclonal anti-human LDL (Fig. 7 E) may have been due to a nonspecific interaction. Therefore, other antibodies to apoB were used to confirm the labeling pattern of the sheep polyclonal anti-LDL. STP-O–permeabilized HepG2 cells were probed simultaneously with the sheep polyclonal anti-LDL (Fig. 10 A) and with monoclonal antibody B1B6 (Fig. 10 B) that recognizes amino acids 3214–3506 in apoB (Krul et al., 1988). After washing, the cells were incubated with the appropriate secondary antibodies. Both the sheep polyclonal anti-LDL antibody and monoclonal B1B6 produced signals in a punctate pattern in identical intracellular locations in STP-O–permeabilized cells (Fig. 10, A and B). The digital images were compared, and significant overlap is shown as yellow in Fig. 10 C. An overlapping punctate pattern was also obtained when cells were probed simultaneously with affinity purified rabbit and sheep polyclonal anti-LDL antibodies (data not shown).
Figure 10.

Localization of a COOH-terminal region of apoB in lysosomes. HepG2 cells were permeabilized with STP-O (A–F). (A–C) STP-O–treated cells were probed simultaneously with the affinity-purified sheep polyclonal anti-human LDL antibody, and with monoclonal B1B6 anti-apoB. After washing, the cells were incubated with the appropriate secondary antibodies conjugated with different fluorochromes and examined at 570 nm (A, green; sheep poly anti-human LDL) or at 680 nm (B, red; monoclonal B1B6). The digital images were superimposed, and significant overlap is colored yellow (C). (D–F) STP- O–treated cells were probed simultaneously with the affinity-purified sheep polyclonal anti-human LDL antibody and monoclonal H4A3 to LAMP-1. After washing, the cells were incubated with the appropriate secondary antibodies conjugated with different fluorochromes, and were examined at both 570 nm (D, green; sheep polyclonal anti-human LDL) or at 680 nm (E, red; monoclonal anti-LAMP-1). The digital images were superimposed, and significant overlap is yellow (F).
Colocalization of Punctate apoB Staining with the Lysosomal Marker LAMP-1
The punctate staining pattern observed with both the polyclonal anti-LDL and B1B6 monoclonal anti-apoB antibodies did not colocalize with antibodies to catalase or an antibody to peroxisomal membrane proteins (data not shown), ruling out that these signals of apoB were in peroxisomes. The punctate staining pattern did colocalize with LAMP-1, a lysosomal membrane glycoprotein (Mane et al., 1989). There was almost total overlap (Fig. 10 F) in the signals for the sheep polyclonal anti-human LDL (Fig. 10 D) and for anti-LAMP-1 (Fig. 10 E) in cells that were permeabilized with STP-O. The punctate pattern that was observed for the antibodies may represent apoB or portions of apoB that have been delivered to lysosomes. Additionally, as observed for epitopes of apoB recognized by the polyclonal anti-LDL antibody (Fig. 7 E), the epitope of apoB that binds the B1B6 monoclonal antibody (Fig. 10 B) was hidden throughout most of the secretory pathway, and only became exposed on or near the surface of the lysosome.
Discussion
The ER of cells has been subdivided into three distinct but continuous compartments: the nuclear envelope, the ribosome-containing rough ER, and the smooth ER (Palade, 1975). The current studies using an anti-apoB peptide antibody (B4) show that there is strong staining for apoB in close proximity to the nucleus in both STP-O– and saponin-permeabilized HepG2 cells. The stronger signal for apoB in this location (compared with that in the remaining secretory pathway) suggests that this region is the site for apoB synthesis. A location for apoB synthesis close to the nuclear membrane was also observed in the immunocytochemical studies of Keller et al. (1986), who transiently depleted rat hepatocytes of apoB and other secretory proteins by incubating cells with cycloheximide. After cycloheximide washout, strong labeling of apoB that did not overlap with albumin labeling was observed in the vicinity of the nuclear envelope (Keller et al., 1986). Therefore, it appears that a significant portion of apoB is synthesized in the area of the ER that is localized close to the outer membrane of the nuclear envelope. An intriguing hypothesis is that the very large apoB mRNA does not diffuse far from nuclear membrane pores. This hypothesis is supported by observations that the mRNA for apoB gives a nonclassical distribution among polysomes/monosomes in sucrose gradients (Chen et al., 1993). The widespread staining for apoB in the extensive reticular network represents apoB that has escaped degradation in the ER and is now located in other compartments of the secretory pathway where additional lipids are acquired and lipoprotein particle maturation occurs (Alexander et al., 1976; Higgins, 1988; Bamberger and Lane, 1990).
Another finding in this study was the identification of two domains of apoB that are exposed to the cytosol throughout a large portion of the secretory pathway: in ER membranes in close proximity to the nuclear outer membrane, in other ER membranes, and in other compartments of the secretory system. The B4 antibody (recognizing amino acids 3221–3240) was specifically raised to a strongly hydrophillic domain in apoB juxtaposed next to a highly hydrophobic domain (Fig. 1). We hypothesized that such a hydrophilic region in apoB may be exposed on the cytosolic face of the ER membrane after nonclassical spanning of the ER membrane by multiple hydrophobic domains of apoB. The similar intensity of staining of apoB obtained with the B4 antibody in both STP-O– or saponin-permeabilized cells is strong evidence that this domain is exposed to the cytosol throughout the secretory pathway. Any additional detergent or disrupting activity afforded by saponin only slightly increased the signal due to the binding of the B4 antibody to this domain of apoB by ∼10% (Fig. 7). The domain recognized by the CC3.4 monoclonal antibody was also cytosolic (Fig. 8). The region in apoB identified by this antibody is a region of apoB that has been previously proposed to be cytosolic (Du et al., 1994).
Unlike a typical secretory protein, apoB contains pause-transfer sequences that cause apoB to pause transiently as it translocates during translation through the aqueous protein–conducting channel of the ER (Chuck et al., 1990; Hegde and Lingappa, 1996). Domains of apoB become exposed to the cytosol when the ER membrane–ribosome junction opens during translocation pausing (Hegde and Lingappa, 1996). Pause transfer sequences are asymmetrically distributed throughout apoB (Kivlen et al., 1997). Of the 23 pause transfer sequences noted, nine are located in the first 20% of the apoB molecule, four are near the apoB48 junction, and ten are found between apoB65 and apoB95. Three (B167.6, B169.5, and B173.1) occur near the B4 site in apoB at B171.2. Translocation pausing may be the mechanism by which certain domains of apoB remain exposed to the cytosol, allowing access to cytosolic chaperones (Zhou et al., 1995) or the components of the proteasome degradation system (Yeung et al., 1996; Fisher et al., 1997; Benoist and Grandperret, 1997).
In contrast to the anti-B4 and CC3.4 antibodies, neither the polyclonal anti-human LDL antibody (Fig. 7 E), the D7.2 monoclonal (Fig. 8 C), or the monoclonal antibody B1B6 (Fig. 10 B) produced a signal for apoB in the perinuclear region or a reticular ER staining pattern in the intermediate regions of the secretory pathway in STP-O–permeabilized cells. The region identified by the D7.2 monoclonal was not exposed to the cytosol, but was most probably lumenal as strong signals were observed in saponin-treated cells (Fig. 8 D). The anti-LDL and B1B6 monoclonal antibodies produced similar punctate patterns throughout the cytoplasm in STP-O–permeabilized cells. The regions of apoB identified by these antibodies are either not exposed to the cytosol, or if on the cytosolic face of membranes of the secretory pathway, are not available for antibody binding. The lack of staining for apoB by either polyclonal anti-human LDL antibodies tested (one raised in a sheep; the other in a rabbit) in membranes of STP-O–permeabilized cells was at first difficult to understand. Additional studies with this antibody involving probing for apoB in a diverse series of nonhepatic cell lines indicated that the sheep polyclonal anti-LDL antibody recognizes mature domains of apoB that are properly processed (data not shown). It was concluded that all of the epitopes in apoB recognized by the sheep anti-LDL antibody are lumenally exposed as they are only observed when cell membranes are perforated with saponin. The relatively low signal produced by the polyclonal anti-LDL antibody throughout the entire interior of STP-O–treated cells (except for the punctate areas) is additional evidence that membranes of the secretory pathway were not perforated in STP-O–permeabilized cells. Interestingly, the B1B6 antibody, unlike the D7.2 antibody, did not produce a reticular staining pattern in saponin-permeabilized cells, indicating that the epitope in apoB recognized B1B6 is not available for antibody binding throughout the entire secretory pathway, even when internal membranes are perforated by saponin. The identical punctate patterns of staining observed with both polyclonal anti-LDL antibodies (one raised in a rabbit, the other raised in a sheep) and the B1B6 monoclonal anti-apoB antibody are strong evidence that these antibodies are binding to apoB, and are not binding nonspecifically to other proteins located in the lysosome. Additional evidence for this conclusion is that the B1B6 antibody did not produce an equivalent punctate, lysosomal staining pattern in human HeLa and 1321N1J astrocytoma cells, indicating that B1B6 is not binding to a constitutive lysosomal membrane protein (data not shown). The punctate signal produced by the B1B6 antibody suggests that this region of apoB in some apoB molecules may be eventually degraded in lysosomes.
The observations in the current study and in previous reports can be used to derive partially the transmembrane structure of apoB in the secretory pathway of HepG2 cells. Previous studies (Du et al., 1994) provided data that the amino terminal of apoB was lumenal, and that apoB could be cleaved by exogenous trypsin at a cytosolic site at ∼70 kD. This led to a model of apoB conformation where only 70 kD of the amino terminus of apoB was translocated, while the remaining portion of the apoB polypeptide was exposed to the cytosol (Du et al., 1994).
Our protease protection assays using ALLN to stabilize exogenous protease-cleaved fragments of apoB from endogenous proteases indicated that there are probably two major regions of apoB susceptible to proteinase K on the cytosolic surface of the ER membrane. The most prominent bands observed were at ∼387 (size deduced from comigration with the major thrombin cleavage product as shown in Fig. 4) and 150 kD, polypeptide fragments that fit with a cytosolically exposed region of apoB at the epitope site of the B4 peptide at amino acids 3221–3240 (approximately B71 in the centile nomenclature). The lighter bands observed (Fig. 4) could be the result of another cleavage site at ∼70 kD, agreeing with the immunocytochemistry data (Fig. 8 A) showing that the CC3.4 site is cytosolically exposed, and the previous data of Du et al. (1994) showing an exposed site in apoB at ∼70 kD. Using the pattern of peptides generated by PK and the results from the antibodies used in the immunocytochemistry study, a putative model for the conformation of apoB in the ER membrane can be drawn (Fig. 11). During early translation, a region of apoB is exposed on the cytosolic side of the ER membrane, and may provide the opportunity for cotranslational degradation; possibly through a ubiquitin proteasome–dependent process (Fig. 11 [1]). Recent studies by Benoist and Grandperret (1997) suggested that apoB can be degraded by the proteasome cotranslationally when an apoB molecule of about the size of B65 is attained. The current study shows that after complete translation, most of apoB is lumenal except for two relatively short regions at ∼70–90 kD (including the CC3.4 site) and at the B4 epitope site (at about B71; Fig. 11 [2]). The D7.2 and B1B6 epitopes and multiple epitopes (★) recognized by the affinity-purified polyclonal antibody are not available from the cytosol or, as shown in the diagram, are located on the luminal side of the ER. ApoB that is made into a nascent lipoprotein leaves the vicinity of certain ER proteases (Sakata et al., 1993; Adeli et al., 1997) and traverses the secretory pathway (Fig. 11 [3a]). Pulse-chase studies indicated that full-sized apoB can be rapidly degraded in the ER compartment of HepG2 cells (Furukawa et al., 1992). This degradation may be accomplished by a retrograde transport mechanism that directs apoB to the proteasome (Fig. 11 [3b]). Export of proteins from the ER to the cytosol for degradation by proteasomes has recently been shown to involve the translocon in both mammalian cells (Wiertz et al., 1996) and in yeast (Pilon et al., 1997). The mechanism used by apoB is not known. It appears that a region of apoB that in part comprises the B1B6 epitope, may escape degradation by the proteasome (Fig. 11 [4]), and is transported to the lysosome where it can be observed cytosolically exposed on the lysosomal membrane (Fig. 11 [5]). In this model, early degradation of a majority of the apoB molecules in the ER may at first involve a specific protease (Adeli, 1994; Adeli et al., 1997) and later the cytosolic proteasome (Fisher et al., 1997). The depiction of apoB as cytosolically exposed at only two small regions may require modification as more information on cytosolic sites in apoB becomes available. Studies using an ELISA to probe exposed apoB in isolated cell fractions (Wilkinson et al., 1993) indicated that the NH2-terminal 48%, NH2-terminal 48–74%, and the COOH- terminal 25% regions of membrane-bound apoB were fairly prominently exposed to the cytosol in RER, SER, and Golgi fractions. The current results generally agree with these observations, but more precisely localize the exposed domains in apoB. The previous results (Wilkinson et al., 1993) indicate that a region of apoB more COOH-terminal than the B4 and B1B6 domains is also transmembrane.
Figure 11.

Model of apoB structure in the membrane of the endoplasmic reticulum. The diagram shows a putative model of nascent apoB in the ER membrane. Pathways are explained in the text. Putative epitopes for the polyclonal anti-LDL antibodies are shown (★). The shaded ovals represent chaperon proteins, including heat shock protein 70. The circles with a missing wedge represent putative proteases in the ER.
The current results with protease protection assays in permeabilized cells (Fig. 5) agree with the results obtained in a previous study (Macri and Adeli, 1997) at earlier chase times (5–10 min), but differ at a longer chase time (30 min). In digitonin-permeabilized HepG2 cells that had been treated with 10 μM ALLN and oleate, apoB was highly susceptible to exogenous trypsin after a 5-min chase, but became largely resistant to trypsin after 30 min of chase. These studies (Macri and Adeli, 1997) showed that the translocation of apoB was mostly complete after 30 min of chase time. It is not clear why our results differ at 30 min, but the immunocytochemical observations support the time course protease protection assay experiment (Fig. 5), indicating that apoB maintains a transmembrane orientation throughout extensive parts of the secretory pathway.
The current results suggest that small regions of most apoB molecules are transmembrane, and remain so until dislodgement for secretion or degradation later in the secretory pathway. This concept/model is different from previous models that indicated that all apoB molecules are fully cotranslationally translocated across the ER membrane, or that only apoB molecules destined for secretion are cotranslationally translocated across the ER membrane. The current model is consistent with the following processes observed experimentally: (a) ApoB is predominately a transmembrane protein in the membranes of the secretory pathway (reviewed in Yao et al., 1997). (b) ApoB in certain hepatic cell models can be rapidly degraded in an early secretory compartment (Sato et al., 1990; Dixon et al., 1991; Furukawa et al., 1992; White et al., 1992), most likely through several process including one that involves tagging by a cytosolic chaperone followed by degradation using the ubiquitin/proteasome system (Zhou et al., 1995; Yeung et al., 1996; Fisher et al., 1997; Benoist and Grandperret, 1997). (c) In the presence of ALLN, completely translated apoB molecules that would have been destined for degradation without ALLN retain the capacity to form an apoB lipoprotein that is secreted when lipid synthesis is increased at a later time period (Sakata et al., 1993). This capacity would reinforce the concept that apoB molecules that are transmembrane after translation are not totally marked for degradation. (d) ApoB can also be degraded later in the secretory pathway, especially in rat hepatocytes (Wang et al., 1995; Cartwright and Higgins, 1996; Sparks et al., 1996). (e) A transmembrane region in apoB at the B4 epitope (B71) would explain the differences observed in the susceptibility of rat apoB100 and apoB48 to exogenous trypsin. In a series of studies (Rusinũl et al., 1993b; Wang et al., 1995; McLeod et al., 1996), apoB48 was almost totally resistant to trypsin, but apoB100 was almost 100% susceptible. These observations lead to the conclusion that only apoB100 exists as a transmembrane protein, and that membranes in the prepared microsomes were not perforated because B48 was not degraded in the same protease protection assay incubation.
In summary, the results identify two specific regions in apoB that are exposed to the cytosol in the secretory pathway. A transmembrane orientation may be important in the regulation of apoB in the following ways: ApoB would remain in close proximity to the ER membrane so that it could be efficiently lipidated by microsomal triglyceride transfer protein, and routed correctly to the appropriate ER to Golgi secretory vesicle. The transmembrane nature of poorly lipidated apoB molecules would aid in targeting these molecules for degradation both early (ER) and late (Golgi or lysosomal compartments) in the secretory pathway.
Acknowledgments
We thank Kurt Strutz for technical assistance, Thomas Phillips and Nobuhiro Sakata for reviewing the manuscript, Elizabeth Norton and Paul Fell for preparing photographs and JoAnn Lewis for typing the manuscript.
This work was supported by Grant HL-47586 to Joseph L. Dixon from the National Heart, Lung, and Blood Institute, National Institutes of Health.
Abbreviations used in this paper
- ALLN
acetyl-leucyl-leucyl-norleucinal
- ApoB
apolipoprotein B-100
- ERGIC
ER-to-Golgi intermediate compartment
- LAMP-1
lysosomal-associated membrane protein-1
- LDL
low-density lipoprotein
- MAP
multiple antigenic peptide
- OA
oleate
- PK
proteinase K
- STP-O
streptolysin-O
- TIPI
total integrated pixel intensity
Footnotes
Preliminary versions of this work were presented at the Experimental Biology 95 meeting (Atlanta, GA, April 1995, abstract 4460) and the American Heart Association meeting (Anaheim, CA, November 1995, abstract 0787).
Address all correspondence to Joseph L. Dixon, Department of Food Science & Human Nutrition, University of Missouri-Columbia, 122 Eckles Hall, Columbia, MO 65211. Tel.: 573-882-4113; Fax: 573-882-0596; E-mail: jdixon@showme.missouri.edu
References
- Adeli K. Regulated intracellular degradation of apolipoprotein B in semipermeable HepG2 cells. J Biol Chem. 1994;269:9166–9175. [PubMed] [Google Scholar]
- Adeli K, Macri J, Mohammadi A, Kito M, Urade R, Cavallo D. Apolipoprotein B is intracellularly associated with an ER-60 protease homologue in HepG2 cells. J Biol Chem. 1997;272:22489–22494. doi: 10.1074/jbc.272.36.22489. [DOI] [PubMed] [Google Scholar]
- Ahnert-Hilger G, Bader M-F, Bhakdi S, Gratzl M. Introduction of macromolecules into bovine adrenal medullary chromaffin cells and rat pheochromocytoma cells (PC12) by permeabilization with streptolysin O: inhibitory effect of tetanus toxin on catecholamine secretion. J Neurochem. 1989;52:1751–1758. doi: 10.1111/j.1471-4159.1989.tb07253.x. [DOI] [PubMed] [Google Scholar]
- Alexander CA, Hamilton RL, Havel RJ. Subcellular localization of B apoprotein of plasma lipoproteins in rat liver. J Cell Biol. 1976;69:241–263. doi: 10.1083/jcb.69.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamberger MJ, Lane MD. Possible role of the Golgi apparatus in the assembly of very low density lipoprotein. Proc Natl Acad Sci USA. 1990;87:2390–2394. doi: 10.1073/pnas.87.7.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoist F, Grandperret T. Co-translational degradation of apolipoprotein B100 by the proteasome is prevented by microsomal triglyceride transfer protein: synchronized translation studies on HepG2 cells treated with an inhibitor of microsomal triglyceride transfer protein. J Biol Chem. 1997;272:20435–20442. doi: 10.1074/jbc.272.33.20435. [DOI] [PubMed] [Google Scholar]
- Berger EG, Grimm K, Bächi T, Bosshart H, Kleene R, Watzele M. Double immunofluorescent staining of alpha-2,6 sialyltransferase and beta-1,4 galactosyltransferase in monensin-treated cells: evidence for different Golgi compartments? . J Cell Biochemistry. 1993;52:275–288. doi: 10.1002/jcb.240520304. [DOI] [PubMed] [Google Scholar]
- Bonnardel JA, Davis RA. InHepG2 cells, translocation, not degradation, determines the fate of the de novo synthesized Apolipoprotein B. J Biol Chem. 1995;270:28892–28896. doi: 10.1074/jbc.270.48.28892. [DOI] [PubMed] [Google Scholar]
- Borchardt RA, Davis RA. Intrahepatic assembly of very low density lipoproteins. Rate of transport out of the endoplasmic reticulum determines rate of secretion. J Biol Chem. 1987;262:16394–16402. [PubMed] [Google Scholar]
- Borén J, Rustaeus S, Olofsson S-O. Studies on the assembly of apolipoprotein B-100- and B-48-containing very low density lipoproteins in McA-RH7777 cells. J Biol Chem. 1994;269:25879–25888. [PubMed] [Google Scholar]
- Borén J, Rustaeus S, Wettesten M, Andersson M, Wiklund A, Olofsson S-O. Influence of triacylglycerol biosynthesis rate on the assembly of apoB-100-containing lipoproteins in HepG2 Cells. Arterioscler Thromb. 1993;13:1743–1754. doi: 10.1161/01.atv.13.12.1743. [DOI] [PubMed] [Google Scholar]
- Bretscher MS, Munro S. Cholesterol and the Golgi apparatus. Science. 1993;261:1280–1281. doi: 10.1126/science.8362242. [DOI] [PubMed] [Google Scholar]
- Cartwright IJ, Higgins JA. Intracellular degradation in the regulation of secretion of apolipoprotein B-100 by rabbit hepatocytes. Biochem J. 1996;314:977–984. doi: 10.1042/bj3140977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Sparks JD, Yao Z, Fisher EA. Hepatic polysomes that contain apoprotein-B mRNA have unusual physical properties. J Biol Chem. 1993;268:21007–21013. [PubMed] [Google Scholar]
- Chuck SL, Yao Z, Blackhart BD, McCarthy BJ, Lingappa VR. New variation on the translocation of proteins during early biogenesis of apolipoprotein B. Nature. 1990;346:382–385. doi: 10.1038/346382a0. [DOI] [PubMed] [Google Scholar]
- Davis RA, Thrift RN, Wu CC, Howell KE. Apolipoprotein B is both integrated into and translocated across the endoplasmic reticulum membrane. Evidence for two functionally distinct pools. J Biol Chem. 1990;265:10005–10011. [PubMed] [Google Scholar]
- Dixon JL, Chattapadhyay R, Huima T, Redman CM, Banerjee D. Biosynthesis of lipoprotein: location of nascent ApoAI and ApoB in the rough endoplasmic reticulum of chicken hepatocytes. J Cell Biol. 1992;117:1161–1169. doi: 10.1083/jcb.117.6.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JL, Furukawa S, Ginsberg HN. Oleate stimulates secretion of apolipoprotein B-containing lipoproteins from HepG2 cells by inhibiting early intracellular degradation of apolipoprotein B. J Biol Chem. 1991;266:5080–5086. [PubMed] [Google Scholar]
- Dixon JL, Ginsberg HN. Regulation of hepatic secretion of apolipoprotein B-containing lipoproteins: Information obtained from cultured liver cells. J Lipid Res. 1993;34:167–179. [PubMed] [Google Scholar]
- Du EZ, Kurth J, Wang S-L, Humiston P, Davis RA. Proteolysis-coupled secretion of the N terminus of apolipoprotein B. Characterization of a transient, translocation arrested intermediate. J Biol Chem. 1994;269:24169–24176. [PubMed] [Google Scholar]
- Esparís-Ogando A, Zurzolo C, Rodriguez-Boulan E. Permeabilization of MDCK cells with cholesterol binding agents: Dependence on substratum and confluency. Am J Physiol. 1994;267:C166–C176. doi: 10.1152/ajpcell.1994.267.1.C166. [DOI] [PubMed] [Google Scholar]
- Fisher EA, Zhou MY, Mitchell DM, Wu XJ, Omura S, Wang HX, Goldberg AL, Ginsberg HN. The degradation of apolipoprotein B100 is mediated by the ubiquitin-proteasome pathway and involves heat shock protein 70. J Biol Chem. 1997;272:20427–20434. doi: 10.1074/jbc.272.33.20427. [DOI] [PubMed] [Google Scholar]
- Furukawa S, Sakata N, Ginsberg HN, Dixon JL. Studies of the sites of intracellular degradation of apolipoprotein B in HepG2 cells. J Biol Chem. 1992;267:22630–22638. [PubMed] [Google Scholar]
- Hegde RS, Lingappa VR. Sequence-specific alteration of the ribosome-membrane junction exposes nascent secretory proteins to the cytosol. Cell. 1996;85:217–228. doi: 10.1016/s0092-8674(00)81098-3. [DOI] [PubMed] [Google Scholar]
- Higgins JA. Evidence that during very low density lipoprotein assembly in rat hepatocytes most of the triacylglycerol and phospholipid are packaged with apolipoprotein B in the Golgi complex. FEBS Lett. 1988;232:405–408. doi: 10.1016/0014-5793(88)80780-4. [DOI] [PubMed] [Google Scholar]
- Ingram MF, Shelness GS. Apolipoprotein B-100 destined for lipoprotein assembly and intracellular degradation undergoes efficient translocation across the endoplasmic reticulum membrane. J Lipid Res. 1996;37:2202–2214. [PubMed] [Google Scholar]
- Innerarity TL, Borén J, Yamanaka S, Olofsson S-O. Biosynthesis of apolipoprotein B48-containing lipoproteins. Regulation by novel post-transcriptional mechanisms. J Biol Chem. 1996;271:2353–2356. doi: 10.1074/jbc.271.5.2353. [DOI] [PubMed] [Google Scholar]
- Keller G-A, Glass C, Louvard D, Steinberg D, Singer SJ. Synchronized synthesis and intracellular transport of serum albumin and apolipoprotein B in cultured rat hepatocytes as studied by double immunofluorescence. J Histochem Cytochem. 1986;34:1223–1230. doi: 10.1177/34.9.3525668. [DOI] [PubMed] [Google Scholar]
- Kivlen MH, Dorsey CA, Lingappa VR, Hegde RS. Asymmetric distribution of pause transfer sequences in apolipoprotein B-100. J Lipid Res. 1997;38:1149–1162. [PubMed] [Google Scholar]
- Knott TJ, Pease RJ, Powell LM, Wallis SC, Rall SC, Jr, Innerarity TL, Blackhart B, Taylor WH, Marcel Y, Milne R, et al. Complete protein sequence and identification of structural domains of human apolipoprotein B. Nature. 1986;323:734–738. doi: 10.1038/323734a0. [DOI] [PubMed] [Google Scholar]
- Krul ES, Kleinman Y, Kinoshita M, Pfleger B, Oida K, Law A, Scott J, Pease R, Schonfeld G. Regional specificities of monoclonal anti- human apolipoprotein B antibodies. J Lipid Res. 1988;29:937–947. [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Leiper JM, Harrison GB, Bayliss JD, Scott J, Pease RJ. Systematic expression of the complete coding sequence of apoB-100 does not reveal transmembrane determinants. J Lipid Res. 1996;37:2215–2231. [PubMed] [Google Scholar]
- Macri J, Adeli K. Studies on intracellular translocation of apolipoprotein B in a permeabilized HepG2 system. J Biol Chem. 1997;272:7328–7337. doi: 10.1074/jbc.272.11.7328. [DOI] [PubMed] [Google Scholar]
- Mane SM, Marzella L, Bainton DF, Holt VK, Cha Y, Hildreth JEK, August JT. Purification and characterization of human lysosomal membrane glycoproteins. Arch Biochem Biophys. 1989;268:360–378. doi: 10.1016/0003-9861(89)90597-3. [DOI] [PubMed] [Google Scholar]
- McLeod RS, Wang Y, Wang S, Rusiñol A, Links P, Yao Z. Apolipoprotein B sequence requirements for hepatic very low density lipoprotein assembly. Evidence that hydrophobic sequences within apolipoprotein B48 mediate lipid recruitment. J Biol Chem. 1996;271:18445–18455. doi: 10.1074/jbc.271.31.18445. [DOI] [PubMed] [Google Scholar]
- Palade G. Intracellular aspects of the process of protein synthesis. Science. 1975;189:347–358. doi: 10.1126/science.1096303. [DOI] [PubMed] [Google Scholar]
- Pease RJ, Leiper JM, Harrison GB, Scott J. Studies on the translocation of the amino terminus of apolipoprotein B into the endoplasmic reticulum. J Biol Chem. 1995;270:7261–7271. doi: 10.1074/jbc.270.13.7261. [DOI] [PubMed] [Google Scholar]
- Pilon M, Schekman R, Römisch K. Sec61p mediates export of a misfolded secretory protein from the endoplasmic reticulum to the cytosol for degradation. EMBO J. 1997;16:4540–4548. doi: 10.1093/emboj/16.15.4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimplikar SW, Ikonen E, Simons K. Basolateral protein transport in streptolysin O-permeabilized MDCK cells. J Cell Biol. 1994;125:1025–1035. doi: 10.1083/jcb.125.5.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plutner H, Davidson HW, Saraste J, Balch WE. Morphological analysis of protein transport from the ER to Golgi membranes in digitonin-permeabilized cells: role of the p58 containing compartment. J Cell Biol. 1992;119:1097–1116. doi: 10.1083/jcb.119.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusiñol A, Verkade H, Vance JE. Assembly of rat hepatic very low density lipoproteins in the endoplasmic reticulum. J Biol Chem. 1993a;268:3555–3562. [PubMed] [Google Scholar]
- Rusiñol AE, Chan EYW, Vance JE. Movement of apolipoprotein-B into the lumen of microsomes from hepatocytes is disrupted in membranes enriched in phosphatidylmonomethylethanolamine. J Biol Chem. 1993b;268:25168–25175. [PubMed] [Google Scholar]
- Sakata N, Wu X, Dixon JL, Ginsberg HN. Proteolysis and lipid-facilitated translocation are distinct but competitive processes that regulate secretion of apolipoprotein B in Hep G2 cells. J Biol Chem. 1993;268:22967–22970. [PubMed] [Google Scholar]
- Sato R, Imanaka T, Takatsuki A, Takano T. Degradation of newly synthesized apolipoprotein B-100 in a pre-Golgi compartment. J Biol Chem. 1990;265:11880–11884. [PubMed] [Google Scholar]
- Schweizer A, Rohrer J, Slot JW, Geuze HJ, Kornfeld S. Reassessment of the subcellular localization of p63. J Cell Sci. 1995;108:2477–2485. doi: 10.1242/jcs.108.6.2477. [DOI] [PubMed] [Google Scholar]
- Segrest JP, Jones MK, Mishra VK, Anantharamaiah GM, Garber DW. ApoB-100 has a pentapartite structure composed of three amphipathic alpha-helical domains alternating with two amphipathic beta-strand domains. Detection by the computer program LOCATE. Arterioscler Thromb. 1994;14:1674–1685. doi: 10.1161/01.atv.14.10.1674. [DOI] [PubMed] [Google Scholar]
- Shelness GS, Morris-Rogers KC, Ingram MF. Apolipoprotein B48-membrane interactions. Absence of transmembrane localization in nonhepatic cells. J Biol Chem. 1994;269:9310–9318. [PubMed] [Google Scholar]
- Sparks JD, Phung TL, Bolognino M, Sparks CE. Insulin-mediated inhibition of apolipoprotein B secretion requires an intracellular trafficking event and phosphatidylinositol 3-kinase activation: studies with brefeldin A and wortmannin in primary cultures of rat hepatocytes. Biochem J. 1996;313:567–574. doi: 10.1042/bj3130567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkade HJ, Fast DG, Rusiñol AE, Scraba DG, Vance DE. Impaired biosynthesis of phosphatidylcholine causes a decrease in the number of very low density lipoprotein particles in the Golgi but not in the endoplasmic reticulum of rat liver. J Biol Chem. 1993;268:24990–24996. [PubMed] [Google Scholar]
- Wang C-N, Hobman TC, Brindley DN. Degradation of apolipoprotein B in cultured rat hepatocytes occurs in a post-endoplasmic reticulum compartment. J Biol Chem. 1995;270:24924–24931. doi: 10.1074/jbc.270.42.24924. [DOI] [PubMed] [Google Scholar]
- White AL, Graham DL, LeGros J, Pease RJ, Scott J. Oleate-mediated stimulation of apolipoprotein B secretion from rat hepatoma cells. A function of the ability of apolipoprotein B to direct lipoprotein assembly and escape presecretory degradation. J Biol Chem. 1992;267:15657–15664. [PubMed] [Google Scholar]
- Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA, Ploegh HL. SEC61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 1996;384:432–438. doi: 10.1038/384432a0. [DOI] [PubMed] [Google Scholar]
- Wilkinson J, Higgins JA, Groot P, Gherardi E, Bowyer D. Topography of apolipoprotein-B in subcellular fractions of rabbit liver probed with a panel of monoclonal antibodies. J Lipid Res. 1993;34:815–825. [PubMed] [Google Scholar]
- Wong L, Torbati A. Differentiation of intrahepatic membrane-bound and secretory apolipoprotein B by monoclonal antibodies: membrane-bound apolipoprotein B is more glycosylated. Biochemistry. 1994;33:1923–1929. doi: 10.1021/bi00173a040. [DOI] [PubMed] [Google Scholar]
- Yao Z, Tran K, McLeod RS. Intracellular degradation of newly synthesized apolipoprotein B. J Lipid Res. 1997;38:1937–1953. [PubMed] [Google Scholar]
- Yeung SJ, Chen SH, Chan L. Ubiquitin-proteasome pathway mediates intracellular degradation of apolipoprotein B. Biochemistry. 1996;35:13843–13848. doi: 10.1021/bi9618777. [DOI] [PubMed] [Google Scholar]
- Yu S, Sher B, Kudryk B, Redman CM. Intracellular assembly of human fibrinogen. J Biol Chem. 1983;258:13407–13410. [PubMed] [Google Scholar]
- Zhou M, Wu X, Huang L-S, Ginsberg HN. Apoprotein B100, an inefficiently translocated secretory protein, is bound to the cytosolic chaperone, heat shock protein 70. J Biol Chem. 1995;270:25220–25224. doi: 10.1074/jbc.270.42.25220. [DOI] [PubMed] [Google Scholar]