

Abstract
FUS7 was previously identified by a mutation that causes a defect in cell fusion in a screen for bilateral mating defects. Here we show that FUS7 is allelic to RVS161/END6, a gene implicated in a variety of processes including viability after starvation, endocytosis, and actin cytoskeletal organization. Two lines of evidence indicate that RVS161/END6's endocytic function is not required for cell fusion. First, several other endocytic mutants showed no cell fusion defects. Second, we isolated five function-specific alleles of RVS161/FUS7 that were defective for endocytosis, but not mating, and three alleles that were defective for cell fusion but not endocytosis. The organization of the actin cytoskeleton was normal in the cell fusion mutants, indicating that Rvs161p's function in cell fusion is independent of actin organization. The three to fourfold induction of RVS161 by mating pheromone and the localization of Rvs161p-GFP to the cell fusion zone suggested that Rvs161p plays a direct role in cell fusion. The phenotypes of double mutants, the coprecipitation of Rvs161p and Fus2p, and the fact that the stability of Fus2p was strongly dependent on Rvs161p's mating function lead to the conclusion that Rvs161p is required to interact with Fus2p for efficient cell fusion.
Cell fusion is the process by which the plasma membranes of two cells fuse to establish cytoplasmic continuity. The fusion of egg and sperm to form a zygote and the fusion of muscle cell precursors to generate multinucleate syncytia of muscle fibers are two examples wherein cell fusion is a key process. The mating pathway in the yeast Saccharomyces cerevisiae is an excellent system in which to study cell fusion (for reviews see Konopka and Fields, 1992; Sprague and Thorner, 1992; Herskowitz, 1995; Marsh and Rose, 1997). Each haploid cell produces a mating type–specific pheromone (a-factor or α-factor) and expresses a surface receptor that is able to bind the pheromone secreted by the opposite cell type. Binding of the pheromone to the receptor activates a mitogen-activated protein (MAP)1 kinase signal transduction pathway leading to G1 cell cycle arrest and to the transcriptional induction of several genes required for efficient mating (e.g., STE2, STE3, MFA1, MFA2, STE6, FUS1, FUS2, KAR3, KAR4 and CIK1). Cells initiate directional cell growth toward the mating partner, resulting in development of a mating projection sometimes called a shmoo. After contact between the partner cells is achieved, cell fusion occurs between them, establishing cytoplasmic continuity. Finally, the nuclei are brought together via microtubule-dependent movement, and the nuclear membranes fuse at the spindle pole body to form a single diploid nucleus (Rose, 1991; Rose, 1996; Marsh and Rose, 1997).
In contrast to our general understanding of the early steps leading to cell–cell contact and the late steps of nuclear fusion, little is known about the mechanism and regulation of cell fusion. To fuse, the partner cells must establish cell–cell contact, seal the junction area, remove the intervening cell wall, and finally fuse the two plasma membranes. Initial cell contact between mating partners is mediated by the cell surface agglutinins (Lipke and Kurjan, 1992), followed by irreversible attachment of cell walls and formation of a seal resistant to osmotic pressure at the periphery of cell contact. Electron microscopy showed that soon after cell contact is established, removal of the intervening cell wall starts with a thinning of the cell walls in the middle point of the contact region and proceeds gradually toward the edges (Osumi et al., 1974; Gammie et al., 1998). In addition, we also observed the presence of vesicles tightly aligned with respect to each other on either side of the cell fusion zone (Gammie et al., 1998). However, the molecular mechanism by which the intervening cell wall is removed as well as the fusion machinery that mediates the plasma membrane fusion process still remains largely unknown. It seems likely that localized release of cell wall–degrading enzymes at the site of cell contact contributes to removal of the intervening cell wall. In addition, portions of the cell wall material could be removed by endocytosis. Given the likely changes that occur to the cell wall during fusion, it is reasonable to think that inappropriate activation of the cell fusion machinery could be hazardous for the cells. Therefore, it is likely that a mechanism must exist to regulate the onset of cell fusion during mating. However, the nature of this regulation is not fully understood.
Several cell fusion mutants have been identified in which zygote formation is blocked after cell–cell contact, but before removal of the intervening cell wall. The characterization of one group of mutants (including, fus3, fus5/ AXL1, fus8/RAM1, cef1/STE6, and fps1) has given some clues regarding the signal and/or regulation for cell fusion (Elion et al., 1990; Brizzio et al., 1996; Elia and Marsh, 1996; Philips and Herskowitz, 1997). Based on the study of FUS5/AXL1 and FUS8/RAM1, two genes involved in processing a-factor, we proposed that high levels of pheromone are required to promote cell fusion (Brizzio et al., 1996). Interestingly, mutations in FUS3, a MAP kinase with several functions in the pheromone response pathway, also result in cell fusion defects. These results suggest direct involvement of the MAP kinase pathway in regulating cell fusion (Elion et al., 1990; Elion et al., 1993). However, even in the presence of excess amounts of mating pheromone, cells do not become osmotically sensitive. Therefore, some aspect of cell fusion seems to be triggered by the presence of the partner cell once contact between the two mating cells is achieved. Philips and Herskowitz (1997) proposed the existence of a checkpoint for cell fusion mediated by the protein kinase C (PKC) pathway based on the study of cells defective in the glycerol transporter FPS1 (Philips and Herskowitz, 1997). In this model, activation of the PKC pathway inhibits cell wall degradation of pheromone-stimulated cells until cell–cell contact is achieved (Philips and Herskowitz, 1997).
Mutations in several genes involved in cell polarity and/ or actin cytoskeleton reorganization also lead to cell fusion defects (TMP1, BNI1, PEA2, and SPA2). These results suggest that cell fusion depends on the cells' ability to polarize efficiently (Liu and Bretscher, 1992; Dorer et al., 1997; Gammie et al., 1998). In addition, mutations in CHS5 required to target the catalytic subunit of chitin synthase III to sites of polarized growth were also shown to result in cell fusion defects (Dorer et al., 1997; Santos et al., 1997). Finally, mutations in FUS1 and FUS2 result in zygotes with a strong defect in cell fusion (McCaffrey et al., 1987; Truehart et al., 1987; Berlin et al., 1991). In contrast to the rest of the genes mentioned here, FUS1 and FUS2 seem to be specifically required for cell fusion. Both genes are strongly induced by pheromone, and mutations in these genes do not cause mutant phenotypes other than prezygote accumulation. Fus1p is an O-glycosylated type I membrane protein that localizes to the shmoo projection (Truehart and Fink, 1989). Fus2p is also tightly associated with membranes or insoluble particles, and localizes to punctate structures under the surface of the shmoo projection (Elion et al., 1995). Both proteins localize to the cell fusion zone, suggesting a direct role in cell fusion (Truehart and Fink, 1989; Elion et al., 1995). Fus1p and Fus2p may function in parallel pathways since fus1Δ fus2Δ double mutant strains are more defective for cell fusion than either single mutant alone (Truehart et al., 1987). However, overexpression of either gene can partially compensate for the absence of the other (Truehart et al., 1987). Interactions between Fus1p and Fus2p and other cell fusion genes have not been described, and the exact role of these proteins in cell fusion is not known.
In this paper, we show that FUS7 is identical to RVS161, a gene previously implicated in endocytosis, organization of the actin cytoskeleton, and a variety of other cellular functions. However, we found that RVS161 is likely to play a direct role in cell fusion that it is different from both its role in endocytosis and in actin organization. We also found that Rvs161p is induced by mating pheromone and localized to the cell fusion zone. Genetically, Rvs161p and Fus2p appear to act in the same pathway. Rvs161p and Fus2p are components of the same complex, and Rvs161p is required for Fus2p's stability. This is the first example of a physical interaction between two components of the cell fusion pathway.
Materials and Methods
Microbial Techniques, General Methods, and Strains
Yeast media and genetic techniques were as described previously (Rose et al., 1990). Yeast and Escherichia coli plasmid DNA minipreps were performed as described elsewhere (Rose et al., 1990). Yeast transformations were done by the lithium acetate method (Ito et al., 1993).
Limited plate matings were performed as described previously (Brizzio et al., 1996). In brief, patches of cells were replica-printed onto prewarmed yeast extract/peptone/dextrose (YEPD) plates containing lawns of the opposite mating type. The mating plates were incubated at 30°C for 2.5–3 h, followed by replica printing to appropriate media to select for diploids. Filter matings for the microscopic analysis of zygotes were performed essentially as described previously (Brizzio et al., 1996). 1 ml of each of the MAT a and MATα strain in the early exponential phase of growth was filtered together onto 0.45-μm nitrocellulose filter discs (Millipore Corp., Bedford, MA). The filters were placed cell-side up onto a YEPD plate and incubated at 30°C for 2.5–3 h. The mating mixtures were fixed in methanol/acetic acid (3:1) for 60 min on ice, and then washed several times in PBS. The cells were stained with DAPI (4′, 6′-diamidino-2-phenylindole; 1 mg/ml in PBS) for 5 min and washed once with PBS. The mating mixtures were then examined by differential interference contrast (DIC) and fluorescence microscopy using an Axiophot microscope (Carl Zeiss Inc., Thornwood, NY).
Isolation of yeast RNA for Northern analysis was done as described previously (Kurihara et al., 1996). Northern blots were performed as described elsewhere (Rose et al., 1990). We used a 878-bp SalI/SacII restriction fragment from pMR3151 to detect RVS161 mRNA and a 280-bp HindIII/EcoRI fragment to detect ACT1 mRNA.
The strains used in this study are listed in Table I. Unless stated otherwise, all strains are isogenic to S288C.
Table I.
Yeast Strains Used in This Study
| Strain | Genotype | |
|---|---|---|
| MS2746 | matΔ::LEU2 his3::TRP1 fus7-1811 ura3-52 trp1-Δ1 leu2-3, leu2-112 | |
| MS2745 | matΔ::LEU2 his3::TRP1 fus7-1811 ura3-52 trp1-Δ1 leu2-3, leu2-112 pB1131 | |
| MY3371 | MAT a ura3-52 leu2-Δ1 | |
| MY2792 | MATα ura3-5 leu2-Δ1 his3-Δ200 | |
| MY3905 | MATα ura3-5 leu2-Δ1 his3-Δ200 rvs161-Δ1::LEU2 | |
| MY2788 | MAT a ura3-52 leu2-Δ1 trp1-Δ63 | |
| MY3722 | MAT a fus7-1811 ura3-52, leu2 | |
| MY3784 | MATα fus7-1811 ura3-5 leu2 lys2-Δ202 | |
| MY3909 | MAT a rvs161Δ1::LEU2 ura3-52 leu2-Δ1 his3-Δ200 trp1-Δ63 | |
| MY4495 | MATα rvs161-Δ1::LEU2 ura3-5 leu2 his3-Δ200 lys2-801 | |
| MY4661 | MAT a ura3-52 | |
| MY4662 | MATα leu2,3-112 | |
| MY4208 | MAT a end3-1 ura3-52 | |
| MY4209 | MATα end3-1 ura3-52, leu2, his4 | |
| MY4678 | MAT a end4-1 ura3-52 leu2-3, leu2-112 his | |
| MY4680 | MATα end4-1 leu2-3, leu2-112 | |
| MY4665 | MAT a end5-1 ura3-52 | |
| MY4666 | MATα end5-1 leu2-3, leu2-112 his | |
| MY4682 | MAT a end6-1(rvs161-R59K) leu2-3, leu2-112 his | |
| MY4683 | MATα end6-1(rvs161-R59K) ura3-52, his | |
| MY4684 | MAT a end7-1 leu2-3, leu2-112 his | |
| MY4685 | MATα end7-1 ura3-52, his | |
| RH2079 | MATα end6-1 ura3-52, his4, leu2, bar1* | |
| MY4533 | MAT a rvs167-Δ1::HIS3 ura3-52 leu2 his3-Δ200 trp1-Δ63 | |
| MY4535 | MATα rvs167-Δ1::HIS3 ura3-52 leu2 his3-Δ200 lys2-801 | |
| MY4500 | MAT a ura3-52 leu2 his3-Δ200 trp1-Δ63 | |
| MY4529 | MATα ura3-52 leu2 his3-Δ200 lys2-801 | |
| MY4545 | MAT a rvs161-Δ1::LEU2 rvs167-Δ1::HIS3 ura3-52 leu2 his3-Δ200 trp1-Δ63 | |
| MY4546 | MATα rvs161-Δ1::LEU2 rvs167-Δ1::HIS3 ura3-52 leu2 his3-Δ200 lys2-801 | |
| MY3377 | MAT a ura3-52 leu2-Δ1 his3-Δ200 trp1-Δ63 | |
| JY429 | MATα fus1-Δ1 fus2-Δ3 ura3-52 trp1-Δ1 ‡ | |
| MY5301 | MAT a rvs161-R35C ura3-52 leu2-Δ1 his3-Δ200 trp1-Δ63 | |
| MY5303 | MAT a rvs161-R113K ura3-52 leu2-Δ1 his3-Δ200 trp1-Δ63 | |
| MY5300 | MAT a rvs161-P158S ura3-52 leu2-Δ1 his3-Δ200 trp1-Δ63 | |
| MY5227 | MAT a rvs161-A175P ura3-52 leu2-Δ1 his3-Δ200 trp1-Δ63 | |
| MY5224 | MAT a rvs161-P203Q ura3-52 leu2-Δ1 his3-Δ200 trp1-Δ63 | |
| MY5322 | MAT a rvs161-P158S ura3-52 leu2-Δ1 his3-Δ200 trp1-Δ63 | |
| RH2635 | MATα his4 leu2 ura3-52 bar1 * | |
| RH2151 | MATα his4 leu2 ura3-52 act1-1 bar1 * | |
| MY4663 | MAT a ura3-52 his act1-1 | |
| MY4664 | MATα leu2-3, leu2-112 his act1-1 | |
| MY3375 | MAT a ura3-52 leu2-Δ1 his3-Δ200 | |
| MY3468 | MAT a ura3-52 leu2-Δ1 his3-Δ200 cdc28-4 | |
| JY424 | MAT a fus2-Δ3 ura3-52 leu2-3, leu2-112 his4-34 ‡ | |
| MY4858 | MAT a fus2-Δ::URA3 ura3-52 leu2-3, leu2-112 | |
| JY427 | MAT a fus1-Δ1 ura3-52 leu2-3, leu2-112 ‡ | |
| MY4160 | MAT a fus1-Δ1 fus2-Δ3 ura3-52 lys2-801 | |
| MY3378 | MATα ura3-52 leu2-Δ1 trp1-Δ63 | |
| MY4164 | MATα fus1-Δ1 ura3-52 trp1-Δ1 | |
| JY428 | MATα fus2-Δ3 ura3-52 trp1-Δ1 his4-34 ‡ | |
| MY4161 | MAT a fus1-Δ1 ura3-52 trp1-Δ1 | |
| MY4905 | MAT a fus1-Δ1 rvs161-Δ1::LEU2 ura3-52 leu2-Δ1 his3-Δ200 trp1-Δ63 | |
| MY2787 | MATα ura3-52 lys2-Δ202 | |
| MY4907 | MATα fus1-Δ1 rvs161-Δ1::LEU2 ura3-5 leu2 his3-Δ200 lys2-801 | |
| MY4801 | MAT a fus2-Δ3 rvs 161-Δ2::URA3 ura3-52 5-FOAR leu2-3, leu2-112 his4-34 | |
| MY4802 | MATα fus2-Δ3 rvs161-Δ2::URA3 ura3-52 5-FOAR trp-Δ1 his4-34 |
Reizman laboratory;
Fink laboratory.
Strain Construction and Plasmids
To generate an rvs161Δ allele (rvs161-Δ1::LEU2), plasmid pMR3245 (Fig. 1) was constructed by cloning a 270-bp HpaI/PstI and a 397-bp HindIII/HpaI restriction fragment from pMR3234 and pMR3151, respectively, into the pRS405 YIp-LEU2 vector (Sikorski and Hieter, 1989) cut with PstI/HindIII. Plasmid pMR3245 was then linearized with HpaI before transformation of MY2792. This construct results in an 800-bp deletion that removes the RVS161 promoter region and 207 amino acids of the RVS161 coding region, leaving 58 COOH-terminal amino acids. Confirmation of the rvs161Δ (MY3905) was done by PCR analysis using the following primers: RVS161U: ATA TGT ACT GGC TCG TCC; RVS161D: GGC TGA TTA CGG ATC ACG; and T3 and T7 from the pBluescript polylinker region. The MAT a rvs161-Δ1::LEU2 (MY3909) was generated by crossing strains MY3905 and MY2788. An analogous construct, pMR3234, designed to generate a rvs161Δ marked with URA3, rvs161-Δ2::URA3, was made by subcloning the 270-bp HpaI/PstI and the 397-bp HindIII/HpaI into pRS406 YIp-URA3 vector (Sikorski and Hieter, 1989). Plasmid pMR3234 was linearized with HpaI before transformation of JY428 and JY424. Confirmation of the rvs161Δ was done by PCR as described above.
Figure 1.

Restriction map of FUS7/RVS161 and surrounding regions of chromosome III. The figure depicts one of the original plasmids (pMR3124) able to rescue the fus7-1811 mating defect and several subclones generated. Bars represent the DNA fragments present in the different plasmids. The ability (+) or inability (−) of the different plasmids to suppress the mating defect is indicated to the right. Shown at the bottom is the structure of plasmid pMR3245 that was used to generate rvs161Δ:: LEU2 by one-step gene replacement.
The rvs167-Δ1::HIS3 strains (MY4533 and MY4535) were generated by PCR. Primers RVS167HISU: CTG ACT AAA TTA TCA ATC CGA CCG ATG TTC GGA TGT TGC GCG TTT TAA GAG CTT GGT GAG; and RVS167HISD: ATA GAA GGT AAT GAA TAC AGA GGG ATG CAG GGG CCT CCT CAT CCG TCG AGT TCA AGA GAA; having 40 bases of homology to 5′ and 3′ regions of RVS167, and 20 bp homologous to HIS3, were used to amplify the HIS3 gene from pRS403 (Sikorski and Hieter, 1989). This PCR fragment was then used to transform MY4500 and MY4529 to create the rvs167-Δ1::HIS3 allele by gene replacement. Confirmation of the rvs167Δ::HIS3 allele was done by PCR using the following primers: RVS167U: TCC GAC GCT TGT ACT GG; RVS167D: ACG GAA GGA CTG AGG AG; and HIS3D: GGC AGT GAC TCC TAG CG.
Generation of fus1-Δ1 in the rvs161Δ strains MY4905 and MY4907 was done by two-step gene replacement (Scherer and Davis, 1979). Plasmid pSB281 (gift from G. Fink, Massachusetts Institute of Technology, Cambridge, MA) was linearized with KpnI and used to transform MY3909 and MY4495. Confirmation of the fus1Δ was done by PCR as described (Gammie et al., 1998).
The mutation end6-1 was recovered by gap repair (Rothstein, 1991). Plasmid pMR3149 (Fig. 1) digested with SacII/Bsu36I was used to transform RH2079, and transformants were selected on synthetic complete media lacking uracil. Plasmids were recovered from several transformants and introduced in Escherichia coli. Plasmid pMR3550 containing the end6-1 mutation was then transformed into a rvs161Δ strain (MY3909). The phenotype of end6-1 was checked by inability to grow on yeast extract/peptone/glycerol (YPG) and YEPD + NaCl (yeast extract/peptone/ dextrose containing 1 M NaCl).
Generation of pMR3510 (containing the RVS161-GFP fusion) was done by cloning a 750-bp PCR fragment encoding GFP into the 3′ region of RVS161. Primer GFPAvaIU: GCG CTC GGG ATA AAA AAG ATG AGT AAA GGA GAA G; and primer GFPAvaID: CGC CCC GAG TTA TTT GTA TAG TTC ATC CAT G; were used to amplify GFP. The PCR product was cloned into the TA vector (Invitrogen Corp., San Diego, CA) cut with AvaI and subcloned into pMR3246 (pMR3234 with KpnI/EcoRI deletion in the polylinker) linearized with AvaI to generate an in-frame fusion of RVS161-GFP. The ability of pMR3510 to complement the rvs161Δ fusion defects was tested by microscopic analysis of zygotes from a filter mating of MY3909 and MY4495 transformed with pMR3510, pRS416, or pMR3246. Mating MY3909 × MY4495, (where both mating partners contain pMR3510) yielded 41% wild-type, 38% partial Fus−, and 21% full Fus− zygotes. Mating where both strains contained the negative control (pRS416) yielded 9% wild-type, 16% partial Fus−, and 75% full Fus− zygotes. The mating with both mating partners containing wild-type RVS161 (pMR3246) showed 60% wild-type, 26% partial Fus−, and 14% full Fus− zygotes.
Generation of pMR3462, a plasmid encoding a PGAL-GFP-RVS161 fusion, was done by cloning an 800-bp PCR fragment that contained the entire RVS161 sequence at the 3′ of the GFP coding region. Primer RVS161XhoU: GTC TCG AGA ATG AGT TGG GAA GGT TTT AAG; and primer RVS161XhoD: CAC TCG AGT TAT TTT ATC CCG AGC GCA C; were used to amplify RVS161. The PCR product was digested with XhoI and cloned into a SalI-linearized LEU-CEN-PGAL-GFP plasmid pCD-GAL-GFP (gift from C. Davis and J. Broach, Princeton University, Princeton, NJ) to generate an in-frame fusion between GFP and RVS161.
Cloning of FUS7 by Complementation
Complementation of the mating defect of fus7-1811 was used to clone FUS7. A yeast centromere–based (YCp50) genomic library (Rose et al., 1987) was transformed into a matΔ fus7-1811 strain (MS2746). 5,500 Ura+ transformants were screened for restoration of mating ability when mated with a MATα fus7-1811 strain (MS2745). Three candidate plasmids were identified that suppressed the mating defect. After reisolation in E. coli, the plasmids were all found to share DNA fragments in common (judged by restriction enzyme analysis). Retransformation of MS2746 confirmed that all of the plasmids complemented the mating defect. The sequence of the ends of the insert in plasmid pMR3124 was determined using Sequenase (United States Biochemical Corp., Cleveland, OH) and YCp50 primers (gift from J. Broach, Princeton University, Princeton, NJ) following the manufacturer's instructions. Examining the DNA sequence and consulting GeneBank showed that the complementing DNA contained a 17-kb region to the right of CENIII. Deletion analysis and subcloning were used to identify the complementing gene (Fig. 1). Plasmids pMR3147, pMR3148, pMR3149, pMR3150, and pMR3151 were created by cutting the original genomic plasmid pMR3234 with EagI, NruI, SphI, BamHI, and BssHI, respectively (Fig. 1). Plasmid pMR3234 was constructed by subcloning an EcoRI/SacII 1.7-kb fragment into the pRS416 YCp-URA3 vector (Sikorski and Hieter, 1989). By these means, the complementing gene was identified as RVS161.
To confirm the identity of FUS7, linkage analysis among fus7-1181, CENIII, and the MAT locus was performed by crossing MY3371 × MS2745. After tetrad dissection, the plasmid pB1311 containing the MATα gene was segregated away by growing the spore colonies on 5-fluoro-orotic acid medium (5-FOA) media, which resulted in all four spore colonies being phenotypically MAT a. The mating phenotypes of the spore colonies were then tested by mating to MS2745. 22 tetrads were analyzed, indicating a FUS7-to-CEN distance of ∼7 centiMorgans (cM), and a FUS7-to-MAT distance of 22 cM, consistent with the identification of FUS7 as RVS161. Finally, the identity of FUS7 as being RVS161 was confirmed by demonstrating that an rvs161Δ allele failed to complement the mutant defect of fus7-1811. Strain MY3909 was mated with MS2745, and plasmid pB1311 containing the MATα gene was subsequently segregated away by growing the diploid on 5-FOA media. The fus7-1181/rvs161Δ mating diploid was then tested for complementation by mating to MS2745 in limited plate mating assays.
In Vitro Mutagenesis of RVS161
Plasmid pMR3234 (RVS161-URA3-CEN) was mutagenized in vitro using hydroxylamine basically as described in Rose et al. (1990). The mutagenized DNA was then used to transform a MAT a rvs161Δ strain (MY3909). 15,000 Ura+ transformants were replica-plated onto media containing glycerol as a carbon source (YPG) onto rich media containing 1 M NaCl (YEPD + NaCl) and onto a lawn of MATα fus1Δfus2Δ (JY429) for 4 h at 30°C to test for mating. We screened for colonies that were unable to grow on YPG or YEPD + NaCl, but that mated like wild-type. We also screened for mutants that showed reduced mating efficiency, but grew on YPG or YEPD + NaCl. Mutants defective for all three properties were discarded. Petite colonies (that would score as possible mutant candidates on YPG) were identified by their small colony size on YEPD after 2 d at 30°C. Plasmid linkage was tested by isolation of the plasmids and retransformation into MY3909. The mutant plasmids were sequenced using an automated sequencer at the Princeton University Sequencing Facility (Princeton, NJ) with the following primers: RVS161UB: CTG GAC GAT CCA AAT GCG, RVS161XhoU (see below), and T3 from pBluescript polylinker region. Subsequently, the EcoRI/SacII restriction fragments isolated from the plasmids were subcloned into the pRS406 YIp-URA3 vector. Integration of the different alleles was achieved by transforming MY3377 with the respective plasmids linearized with AflII or EspI. Transformants in which the plasmid had been lost by recombination were selected by growth on 5-FOA media. Several colonies were tested for growth on YPG and YEPD + NaCl, and mating. Lucifer yellow assays were done as described in Munn et al. (1995). Cell fusion was scored by microscopic analysis of the zygotes from filter matings as described above.
For the act1-1 unlinked noncomplementation analysis, wild-type (MY3377), rvs161-Δ1::LEU2 (MY3909), rvs161-R35C (MY5301), rvs161-R59K (MY4682), rvs161-R113K (MY5303), rvs161-P158S (MY5322), rvs161-A175P (MY5227), and rvs161-P203Q (MY5224) were mated to act1-1 (RH2151) and to wild-type (RH2635). The diploid strains together with an act1-1/act1-1 strain (MY4663 × MY4664) were streaked onto YEPD and YEPD + NaCl and incubated at 30°C. The act1-1/act1-1 diploid strain grew very poorly on YEPD + NaCl at 30°C. Likewise, all the diploids containing the End−Fus+ alleles grew poorly on YEPD + NaCl (rvs161-R35C/RVS161 ACT1/act1-1, rvs161-R59K/RVS161 ACT1/act1-1, rvs161-R113K/RVS161 ACT1/act1-1 and rvs161-P158S/RVS161 ACT1/ act1-1). In contrast, the diploids containing the End+Fus− alleles grew like wild-type on YEPD + NaCl (rvs161-A175P/RVS161 ACT1/act1-1 and rvs161-P203Q/RVS161 ACT1/act1-1). All the control strains were able to grow well on YEPD + NaCl (RVS161/RVS161 ACT1/act1-1, rvs161-Δ1:: LEU2/RVS161 ACT1/act1-1, rvs161-R35C/RVS161 ACT1/ACT1, rvs161-R59K/RVS161 ACT1/ACT1, rvs161-R113K/RVS161 ACT1/ACT1, rvs161-P158S/RVS161 ACT1/ACT1, rvs161-A175P/RVS161 ACT1/ACT1, and rvs161-P203Q/RVS161 ACT1/ACT1).
Anti-Rvs161p Antibody Production and Purification
Plasmid pMR3548 coding for a His6x-Rvs161p fusion was created by cloning an 800-bp PCR fragment containing the entire RVS161 coding sequence into an XhoI-linearized pET-30c (+) bacterial expression vector (Novagen, Madison, WI). Primers RVS161XhoU and RVS161XhoD (described above) were used to amplify RVS161 from pMR3234. The His6x– Rvs161p fusion protein was purified from 1 l culture of the BL21(DE) bacterial strain containing pMR3548. Preparation and purification of His6x-Rvs161p was carried out as suggested in the pET System Manual (Novagen) with some modifications. Protein induction was done with 1 mM IPTG for 3 h at 37°C. 100 μg/ml lysozyme and 0.1% Triton X-100 were added to the cells resupended in 80 ml of binding buffer (5 mM imidazole, 500 mM NaCl, 20 mM Tris-HCl, pH 7.9), and the cells were incubated at 30°C for 15 min before sonication. The inclusion bodies were solubilized in 10 ml of binding buffer containing 8 M urea. Activated Ni-NTA resin (QIAGEN Inc., Chastworth, CA) was used to affinity-purify His6x– Rvs161p by chromatography. Elution was carried out using a linear 0–500-mM immidazole elution gradient. Finally, the eluted protein was dialyzed into PBS. Antisera were generated in two females New Zealand white rabbits at the Princeton University Animal Facility (Princeton, NJ).
Affinity purification of anti-Rvs161p was carried out using nitrocellulose blots as described previously (Pringle et al., 1991) with some modifications. 100 μg of purified His6x–Rvs161p were electrophoretically separated by SDS-PAGE and transferred to nitrocellulose. The membrane was blocked twice with 5% nonfat dry milk and 0.05% Tween-20 in PBS for 15 min, and was then washed twice with binding solution (3% BSA, 0.05% Tween-20 in PBS and 0.02%NaN3). 1 ml of crude rabbit anti-Rvs161p serum was diluted 1:2 with binding solution and incubated for 2 h at room temperature. The membrane was then washed four times with PBS, 0.1% Tween-20, and one time with 50 mM Tris-Cl pH 7.5 for 5 min. 0.5 ml of 200 mM glycine-Cl pH 2.2 was used to elute the antibodies. Two 250-μl rinses of the membrane with 100 mM Tris-Cl pH 8 were pooled with the elution. Finally, the eluted antibody solution was neutralized with 25 μl of a 2 M Tris base solution.
Immunological Techniques
Actin staining was performed by indirect immunofluorescence using polyclonal affinity-purified antiactin antibodies kindly provided by Tongtong Wang and Anthony Bretscher (Cornell University, Ithaca, NY). Cells grown in YEPD at 30°C were treated with or without α-factor at 6 μM for 90 min, were fixed by adding formaldehyde to 4%, and were incubated for 1 h at 30°C. The cells were then permeabilized by incubation with 25 μg/ml Zymolyase 100,000 (ICN Immunobiologicals, Lisle, IL) in 1.2 M sorbitol, 0.1 M potassium phosphate, pH 7.5, and 25 mM β-mercaptoethanol for 30 min at 30°C. Staining of the cells was done as described previously (Kilmartin and Adams, 1984). Antiactin antibodies were used at a 1:50 dilution, and fluorescein-conjugated goat anti–rabbit antiserum (Boehringer Mannheim, Indianapolis, IN) at a 1:25 dilution was used as secondary antibody.
Total protein extracts were prepared as described previously (Ohashi et al., 1982). Typically 10 ml of early exponential cultures (5 × 106–1.5 × 107 cells/ml) in YEPD low pH (pH 3.5) media were treated or not with α-factor. For Western blotting, the proteins were electrophoretically separated in 10% or 7.5% SDS-PAGE gels and transferred to a nitrocellulose membrane. A 6.5−7.5% SDS-PAGE gel was more suitable to distinguish the multiple forms of Fus2p. All the steps for Western blotting were carried out at room temperature. Blocking of the membranes was done for 1 h with 5% nonfat dry milk in buffer A (10 mM Tris-HCl pH 7.4, 150 mM NaCl, and 0.02% Tween-20). Incubations with primary and secondary antibody were done for 1 h in buffer A. Membranes were washed three times after primary and secondary antibody incubations with Buffer A for 15 min. Affinity-purified anti-Rvs161p was used at 1:500. Crude rabbit anti-Fus2p antibody, a gift from E. Elion (Harvard University, Cambridge, MA), was affinity-purified using the nitrocellulose method described above. Plasmid pMR3680 encoding His6x fused to 387 amino acids of Fus2p was created by subcloning a 1162-bp HindIII/HindIII restriction fragment into pET-30b (+). To isolate His6x–Fus2 antigen, part of the insoluble fraction of a 100-ml IPTG-induced culture of bacterial strain MR3683 (pMR3680 in BL21 DE with argU+-CamR-TetS) was electrophoretically separated by SDS-PAGE and transferred to a nitrocellulose membrane. Affinity-purified anti-Fus2p was used at 1:2,000. In addition, preabsortion of the affinity-purified anti-Fus2p onto a nitrocellulose membrane containing fus2Δ protein extracts for 1 h was needed to reduce background binding to nitrocellulose. Crude rabbit anti-Fus1p antibody was used at 1:1,000 (Gift from J. Trueheart, Cadus Pharmaceuticals, Tarrytown, NY). Rabbit anti–glutathione-S-transferase (GST) antibody (Sigma Chemical Co., St. Louis, MO) was used at 1:1,000. HRP-conjugated goat anti–rabbit secondary antibody was used at a 1:2,500 dilution (Amersham Corp., Arlington Heights, IL). Proteins were visualized by ECL chemiluminescent system (Amersham Corp.).
Cell extracts for immunoprecipitation experiments were prepared from 100 ml of early exponential cultures (5 × 106–1.5 × 107 cells/ml). MY3371, MY4858, MY3909 transformed with pMR3397 (2 μ RVS161 URA3), and MY3909 transformed with pRS426 (Sikorski and Hieter, 1989) were induced with α-factor for 120 min at 30°C. Cells were collected, washed once with 20 ml of cold breaking buffer (50 mM Tris pH 7.4, 50 mM NaCl, 0.5% Triton X-100) and resuspended in 1 ml of breaking buffer containing protease inhibitors (5 μg/ml chymostatin, 5 μg/ml leupeptin, 5 μg/ml aprotinin, 5 μg/ml pepstatin, and 1 mM PMSF). The cells were transferred to microfuge tubes containing 0.8 g of acid-washed glass beads and agitated by 2 pulses of 3 min in a minibead beater (Biospec Products, Inc., Bartlesville, OK). The extracts were then centrifuged twice for 5 min at 14 K rpm at 4°C. The supernatant was transferred to a new tube between centrifugations. 10 μl of affinity-purified anti-Fus2p or 15 μl of affinity-purified anti-Rvs161p was added to half of the extract. The volume was brought to 1 ml with breaking buffer, and NaCl was added to 100 mM. The extracts were incubated for 1 h at 4°C with constant agitation. The extracts were centrifuged for 10 min at 14 K rpm, and the supernatant was transferred to a new tube containing 100 μl of 50% Protein A-sepharose beads in PBS (Pharmacia LKB Biotechnology, Inc., Piscataway, NJ). The extracts were then incubated at 4°C for 1 h with constant agitation, and were then centrifuged for 20 s at 14 K rpm. The beads were washed 5× with 1 ml of breaking buffer containing protease inhibitors and resuspended in 30 μl of sample buffer (80 mM, Tris pH 6.8, 10% SDS, 12.5% glycerol, 4% β-mercaptoethanol, 0.1% bromphenol blue). Samples were electrophoretically separated in SDS-PAGE, transferred to nitrocellulose membranes, and incubated with either anti-Rvs161p or anti-Fus2p antibodies as described above. To estimate the amount of Fus2p and Rvs161p precipitated, 30-μl aliquots (∼3%) of total extract were analyzed together with the precipitate samples. To quantify the percentage of precipitated protein, the intensity of the corresponding bands (measured by densitometry) for precipitates and total extracts were compared. These values were normalized to the total volumes of the samples, and the ratio between the precipitate and the total extract was calculated.
Pulse-chase experiments were performed as described previously (Gammie et al., 1995) with some modifications. In brief, MY3371, MY4858, and MY3909 cells were grown to OD600 0.2–0.3 (1–1.5 × 107 cells/ml) in synthetic complete media and induced with α-factor for 35 min at 30°C. Cultures were washed, resuspended at 2 OD/ml in synthetic media lacking cysteine, and methionine in the presence of α-factor. Cells were incubated for 30 min at 30°C. 200 μCi/OD of 35S-Translabel™ (ICN Biomedicals Inc., Irvine, CA) were added, and the cells were incubated for 7.5 min at 30°C. The chase was performed by adding 10× chase solution to achieve a final concentration of 0.1% cysteine, 0.1% methionine, and 0.1 M (NH4)2 S04. At 0, 5, 10, 20, 60, and 120 min, 1-ml aliquots were transferred to microfuge tubes containing 500 μl of 40 μM cold NaN3 and placed on ice for 15 min to stop the reaction. Cell lysis, immunoprecipitations, and autoradiography were done as described previously (Scidmore, 1993). 5 and 4 μl of the affinity-purified anti-Rvs161p and anti-Fus2p were used for each sample, respectively.
GST-Fus2p Coprecipitation Experiments
To create a GST-FUS2 in-frame fusion, a 2.2-kb FUS2 fragment was first amplified by PCR using primer FUS2GSTU: GGA CTA GTG ATG TTT AAG ACT TCA; and primer FUS2GSTD: CCC TCG AGA TAG CGT CAA CCA TC; and subsequently cloned into a TA vector (Invitrogen Corp.). A SpeI/AvaI fragment was then subcloned into pEG-KT (2 μ URA leu2d PGAL-GST), linearized with XbaI/SalI at the multiple cloning site 3′ of GST (Mitchell et al., 1993). The function of the fusion protein was tested by transforming this plasmid into JY424 and examining the complementation of the fus2Δ when mated to JY429 in a limited plate mating analysis.
To prepare cell extracts for GST–glutathione bead precipitation, 100 ml of early exponential phase cultures (5 × 106–1.5 × 107 cells/ml) of MY5299 and MY5297 in synthetic complete media lacking uracil with 2% raffinose were induced by adding 3% galactose for 1 h at 30°C. α-factor was then added to 6 μM, and the cultures were incubated for another 2 h at 30°C. Cell extracts were prepared as described for immunoprecipitation experiments described above. Before adding glutathione-sepharose beads, NaCl was added to 200 mM, and the extracts were mixed for 1 h at 4°C with constant agitation. The extracts were then centrifuged for 10 min at 14 K rpm, and the supernatants were transferred to a new tube containing 100 μl of 50% gluthathione-sepharose beads (Pharmacia LKB Biotechnology, Inc.) in PBS. The extracts were incubated for 1 h at 4°C. The beads were washed five times with breaking buffer containing protease inhibitors, and were finally resuspended in sample buffer. Samples were electrophoretically separated in SDS-PAGE and transferred to nitrocellulose membranes. The membranes were incubated with anti-GSTp, anti-Fus2p, or anti-Rvs161p antibodies as described above. The primary antibodies were visualized using ECL chemiluminescent system (Amersham Corp., Arlington Heights, IL) after incubations with HRP-conjugated goat anti–rabbit secondary antibody (Amersham Corp.). Quantification of the amount of precipitated protein was done as described in the Fus2p and Rvs161p immunoprecipitation experiments.
Results
FUS7 is Allelic to RVS161
The fus7-1811 mutation was identified in a screen for bilateral karyogamy and cell fusion mating mutants (Kurihara et al., 1994). Diploid formation in matings between MAT a and MATα fus7 mutant parents is reduced 50-fold compared with wild-type matings (Kurihara et al., 1994). In contrast to wild-type zygotes (Fig. 2, A and B), fus7 zygotes showed a pronounced septum between the two partner cells (Fig. 2, C–F), suggestive of the presence of residual cell walls. Two types of Fus− zygotes were observed. One type called full Fus− showed a complete septum and two unfused nuclei (Fig. 2, E and F). The second type, called partial Fus−, showed a partial septum that would allow for cytoplasmic mixing and nuclear fusion (Fig. 2, C and D). The morphology of fus7 zygotes was very similar to that described for other previously identified cell fusion mutants. Table II presents a quantitative analysis of the cell fusion phenotype analyzed by microscopic examination of the zygotes. As expected, a wild-type mating (i.e., wild-type × wild-type) resulted in the great majority of zygotes with wild-type morphology (94%; Table II). In contrast, the fus7-1181 × wild-type mating showed a significant number of zygotes with partial Fus− (36%) and full Fus− (22%) morphology (Table II). The cell fusion defect of fus7-1181 was more severe when both mating partners were mutant (Table II). In the fus7-1181 × fus7-1181 mating, most of the zygotes showed the full Fus− morphology (72%). Many fewer showed partial Fus− (23%) and wild-type morphology (5%). Therefore, fus7-1181 caused a significant cell fusion defect when only one of the mating partners has the mutation. However, fus7-1181 was strongly bilateral because the phenotype was much more severe when both mating partners were mutant. In addition, fus7-1181 showed no cell type specificity.
Figure 2.

Phenotype of the cell fusion defective zygotes. A–F are images of zygotes from a mating between a MAT a rvs161Δ (MY3909) and a MATα rvs161Δ (MY4495) strain. Each image shows the nucleus by DAPI fluorescence and the zygote morphology by DIC. A and B are examples of wild-type zygotes. C and D are examples of partial Fus− zygotes. E and F are examples of full Fus− zygotes.
Table II.
Cell Fusion Defect of fus7-1181 and rvs161Δ by Microscopic Analysis of Zygotes
| Wild-type | Partial Fus− | Full Fus− | Zygotes formed* | |||||
|---|---|---|---|---|---|---|---|---|
| Wild-type × wild-type | 94 | 5 | 1 | 50 | ||||
| fus7-1811 × wild-type | 42 | 36 | 22 | ND | ||||
| fus7-1811 × fus7-1811 | 5 | 23 | 72 | 51 | ||||
| rvs161Δ × rvs161Δ | 7 | 32 | 61 | ND |
Zygotes from filter matings of wild-type × wild-type (MY2788 × MY2787), fus7-1811 × wild-type (MY3722 × MY2787), fus7-1811 × fus7-1811 (MY3722 × MY3784) and rvs161Δ × rvs161Δ (MY3909 × MY 4495) were analyzed microscopically. At least 200 zygotes were analyzed in each experiment. The numbers represent the percentages of wild-type, partial Fus−, and full Fus− zygotes.
Number of mating pairs formed/total cells × 100. ND, not done. Part of the data presented in this table appears in Gammie et al. (1998).
The FUS7 gene was cloned by complementation of the mating defect (see Materials and Methods). Three candidate plasmids were isolated, of which the smallest contained a 17-kb region immediately adjacent to CENIII (Fig. 1). Deletion and subcloning analysis showed that a plasmid (pMR3234) containing the previously identified gene RVS161 was able to complement the mating defect (see Fig. 1). Linkage analysis showed that the FUS7 gene mapped ∼7 and 22 cM from the CEN and MAT loci, respectively (see Materials and Methods), consistent with the possibility that FUS7 could be allelic to RVS161. We generated MAT a and MATα rvs161Δ strains, and found that they exhibited a cell fusion defect identical to that of fus7-1811 (Table II). Furthermore, the rvs161Δ allele failed to complement fus7-1811's mating defect (see Materials and Methods). Therefore, we concluded, that FUS7 is identical to RVS161.
RVS161 was originally identified by a mutation that causes reduced viability upon starvation (Crouzet et al., 1991). Although mutations in RVS161 are highly pleiotropic, RVS161 is not essential for viability (Crouzet et al., 1991; Desfarges et al., 1993). Mutations in RVS161 result in a delocalization of the actin cytoskeleton (Sivadon et al., 1995), high salt sensitivity (Crouzet et al., 1991), random budding pattern in diploid cells (Durrens et al., 1995), and defects in endocytosis (Munn et al., 1995). The end6-1 allele of RVS161 was isolated in a screen for mutants defective in endocytosis, and was shown to affect the internalization step of endocytosis (Munn et al., 1995). Rvs161p belongs to a family of proteins that includes another yeast protein, Rvs167p, and a group of proteins called amphiphysins (Lichte et al., 1992; Bauer et al., 1993; Butler et al., 1997). They all share homology in one domain, called Rvs, defined by the entire Rvs161p (David et al., 1994; Sivadon et al., 1995). In addition, Rvs167p and the amphiphysins also share a glycine-proline-alanine (GPA)–rich domain and an SH3 domain (David et al., 1994; Sivadon et al., 1995). RVS167 has been reported to have the same functions as RVS161 since mutations in either gene result in the same phenotypes (Bauer et al., 1993; Sivadon et al., 1995). Human amphiphysin has been implicated in endocytosis due to its interaction with dynamin and its homology to RVS161 and RVS167. It is thought to be important in recycling the plasma membrane at the synaptic terminals (Lichte et al., 1992; David et al., 1996; Grabs et al., 1997; Shupliakov et al., 1997).
Defects in Endocytosis Do Not Generally Lead to Defects in Cell Fusion
Given the previously characterized role of RVS161/END6 in endocytosis, we wanted to address whether endocytosis played a role in cell fusion. Fig. 3 A shows the analysis of mating mixtures using wild-type, rvs161Δ, and various mutants defective for endocytosis (Raths et al., 1993; Munn et al., 1995). Matings where both the MAT a and MATα parents were mutant were performed, and the zygotes were analyzed by microscopy. Less than 25% of the rvs161Δ zygotes were either wild-type or partial Fus− (Fig. 3 A). In contrast, the wild-type mating produced >91% wild-type and very few Fus− zygotes (Fig. 3 A). Most of the end mutants including end3-1, end4-1, end6-1, and end7-1 behaved like the wild-type (Fig. 3 A). end5-1, however, did show considerably fewer wild-type and more Fus− zygotes than did the wild-type mating (Fig. 3 A). Because the overall mating efficiency of end5-1 was greatly reduced (i.e., end5-1 formed very few mating pairs), we believed that the apparent cell fusion defect observed is a consequence of an underlying growth defect. From these results we concluded that defects in endocytosis do not generally lead to defects in cell fusion.
Figure 3.

Cell fusion phenotypes of various endocytic mutants. A represents the % of wild-type (dark gray) and partial Fus− (lighter gray) zygotes from matings of: rvs161Δ (MY3909 × MY4495), wild-type (MY4661 × MY4662), end3-1 (MY4208 × MY4209), end4-1 (MY4678 × MY4680), end5-1 (MY4665 × MY4666), end6-1 (MY4682 × MY4683), end7-1 (MY4684 × MY4685), rvs167Δ (MY4533 × MY4535), and rvs161Δ rvs167Δ (MY4545 × MY4546). In each case, both parents contained the indicated mutation. Zygote formation was analyzed microscopically. B represents the % of wild-type (dark gray) and partial Fus− (lighter gray) zygotes formed using MATα rvs161Δ (MY4495) as one of the parents, and MAT a rvs161Δ (MY3909) transformed with vector pRS416 (Sikorski and Hieter, 1989), pMR3149 (containing RVS161; see Fig. 1), or pMR3234 (containing the end6-1 allele) as the other parent. More than 144 fixed zygotes were analyzed for each of the above matings.
Interestingly, deletion of RVS167 also did not show a cell fusion defect (Fig. 3 A). Furthermore, the rvs161Δ rvs167Δ double mutant was identical to rvs161Δ (Fig. 3 A). Therefore, we concluded that RVS161 is required for cell fusion whereas RVS167 is not.
Surprisingly, we also found that the end6-1 allele of RVS161 behaved like wild-type for cell fusion (Fig. 3 A). To rule out differences in strain background, the end6-1 allele was recovered by gap repair on a CEN plasmid, introduced into the rvs161Δ, and checked for its ability to rescue the rvs161Δ cell fusion defect. Fig. 3 B shows that rvs161Δ containing pCENend6-1 is indistinguishable from wild-type when mated with a rvs161Δ strain. Because end6-1 was able to restore rvs161Δ's cell fusion ability, we concluded that end6-1 does not result in a cell fusion defect.
The Role of RVS161 in Cell Fusion is Separable From Its Role in Endocytosis
To see if we could further separate the role of RVS161 in endocytosis from its role in mating, we performed in vitro mutagenesis of RVS161 and screened for alleles that only affected one or the other of the two functions. Both rvs161Δ and end6-1 are unable to grow on nonfermentable carbon sources such as glycerol (YPG; Crouzet et al., 1991; Munn et al., 1995). Other end mutants (including end3, end4, end5, act1, and rvs167Δ) do not grow well under these conditions (Bauer et al., 1993; and our unpublished observations), suggesting a strong correlation between defects in endocytosis and the inability to grow on YPG. In addition, rvs161Δ and end6-1 as well as other mutants defective for endocytosis (act1, sac6, sla2/end4, and end3) are unable to grow under hypertonic conditions such as 1 M NaCl (Desfarges et al., 1993; Munn et al., 1995; J. O'Dell and A. Adams, personal communication). Therefore, there is also a strong correlation between defects in endocytosis and the inability to grow under hypertonic conditions. We used the failure to grow on YPG or on media containing 1 M NaCl (YEPD + NaCl) to screen for mutant alleles of RVS161 that affect endocytosis. A CEN-URA3-RVS161 plasmid was mutagenized with hydroxylamine and introduced into a rvs161Δ strain. We screened for colonies that were unable to grow on YPG or on YEPD + NaCl, but mated like wild-type. We also screened for mutants that showed reduced mating efficiency but did grow on YPG or YEPD + NaCl. Mutants defective for all three properties were discarded, as there were likely to be null alleles. We found a good correlation between reduced growth on YPG and inability to grow on YEPD+NaCl. In general, we found that the mutants grew worse on YEPD + NaCl than on YPG. We isolated five mutants that were defective for growth on YPG and YEPD + NaCl, but not for mating (YPG/YEPD+NaCl− Mating+). We also isolated three mutants that were defective for mating, but did not show any growth defects on YPG and YEPD + NaCl (YPG/YEPD+NaCl+ Mating−). All but two of the various alleles were integrated into the genome.
Fig. 4, A–C shows a summary of the endocytic and cell fusion phenotypes of the different rvs161 alleles isolated. Fluid phase endocytosis was measured using Lucifer yellow (LY) uptake. Internalized LY is transported to the vacuole where it accumulates. Cells defective in endocytosis do not internalize LY, and therefore show no vacuolar fluorescence (Dulic et al., 1991; Munn and Riezman, 1994). The cell fusion phenotype was measured by microscopic analysis of matings to a rvs161Δ strain. Wild-type cells showed the typical vacuolar staining in the endocytosis assay, and gave mostly wild-type (40%) or partial Fus− zygotes (47%; Fig. 4, A–C). In contrast, the null strain showed no vacuolar staining, and the majority of the zygotes were full Fus− (73%). The mutants selected on the basis of their YPG/YEPD + NaCl− phenotypes did not accumulate LY in the vacuole (rvs161-R35C, rvs161-R59K, rvs161-R113K, and rvs161-P158S), consistent with a defect in endocytosis (Fig. 4 A). However, these mutants behaved like the wild-type control for cell fusion because they showed mostly wild-type (42–51%) or partial Fus− (36–46%) zygotes (Fig. 4 C). In contrast, two mutants selected to be YPG/YEPD + NaCl+ Mating− (rvs161-A175P and rvs161-P203Q) showed a cell fusion defect comparable to that of the null mutant (59–65% full Fus− zygotes), but did not display an endocytosis defect (Fig. 4 C). We concluded from these results that RVS161's role in endocytosis is separate from its role in cell fusion during mating.
Figure 4.

Endocytic and cell fusion phenotype of RVS161 function-specific mutations. A shows the Lucifer yellow uptake to analyze endocytosis. To visualize the LY, FITC filter sets were used (first column). The overall shape of the cells and the presence of the vacuole were assessed by using DIC optics (second column). B lists rvs161 alleles. The MAT a strains analyzed were wild-type (MY3377), rvs161Δ (MY3909), rvs161-R35C (MY5301), rvs161-R59K (MY4682), rvs161-R113K (MY5303), rvs161-P158S (MY5300), rvs161-A175P (MY5227), and rvs161-P203Q (MY5224). C shows the results of the microscopic analysis of zygotes to characterize the cell fusion phenotype. In each case, the mutants were mated with a MATα rvs161Δ strain (MY4495). At least 200 zygotes were analyzed in each experiment. The numbers represent the percentages of wild-type, partial Fus−, and full Fus− zygotes. D is a representation of the RVS161 gene showing the location of the different rvs161 alleles. Above the gene are shown the locations and identities of the wild-type residues in RVS161. Below the gene are the mutant changes from each allele. Mutations indicated with double lines or a single thick line correspond to the alleles that are End−Fus+ and End+Fus−, respectively.
Fig. 4 D shows the location and the specific changes of the different mutations in RVS161. The 5 YPG/YEPD + NaCl− mating+ alleles, now called End–Fus+, mapped within the NH2-terminal 65% of RVS161. The three YPG/ YEPD+NaCl+ mating− alleles, designated End+Fus−, mapped within the COOH-terminal 35% of RVS161 (Fig. 4 D). Two of the cell fusion mutants affected the same residue; in one case proline 203 was changed to tyrosine, and in the other case to glutamine (Fig. 4 D). Sequencing the end6-1 allele recovered by gap repair showed that it corresponded to one of the End−Fus+ rvs161 alleles that we had isolated, rvs161-R59K. Based on the location of the End− Fus+ and End+Fus− rvs161 alleles, we propose that RVS161 has two domains: an NH2-terminal involved in endocytosis, and a COOH-terminal involved in cell fusion.
The rvs161 End− Mutants Exhibited Defects in Actin Organization While the Fus− Mutants Were Normal
Mutations in RVS161 (rvs161Δ and end6-1) have been shown to cause defects in the organization of the actin cytoskeleton (Munn et al., 1995; Sivadon et al., 1995). We wanted to test if defects in the actin cytoskeleton were correlated with the rvs161 alleles defective for endocytosis, cell fusion, or both. Therefore, we examined the effects of the various rvs161 alleles in vegetative and in pheromone-induced cells on actin localization. In yeast, the actin cytoskeleton is organized in cortical patches and a network of cytoplasmic cables (Adams and Pringle, 1984). The distribution of the actin cytoskeleton changes throughout the cell cycle (Kilmartin and Adams, 1984). During bud emergence and growth, cortical actin patches concentrate in the bud, with cables in the mother oriented along the mother– daughter cell axis (Adams and Pringle, 1984; Kilmartin and Adams, 1984). During development of the shmoo projection, cortical actin patches concentrate at the shmoo tip with cables running towards the tip (Ford and Pringle, 1986; Hasek et al., 1987; Read et al., 1992). At cytokinesis, cortical actin patches are distributed over the surface of mother and daughter cells, and cables are no longer visible, but a ring of filamentous actin forms between the two cells where the septum forms (Kilmartin and Adams, 1984).
Fig. 5, A–H shows examples of actin localization in wild-type and in rvs161 cells. A quantitative analysis of the localization of cortical actin patches in small-budded cells and in shmoos is presented in Table III. As expected, most of the wild-type cells (94%) showed actin patches concentrated at the growing bud (Fig. 5 A and Table III) or at the tip of the shmoo in pheromone-induced cells (97%; Fig. 5 E and Table III). In contrast, only a minor fraction of the rvs161Δ cells showed a wild-type concentration of cortical patches in the bud or shmoo tip. The majority of the cells showed actin patches that were more randomly distributed over the mother cell and the bud (41%) or heavily concentrated at the mother-bud neck (41%; Fig. 5 B and Table III). In addition, 72% of the rvs161Δ shmoos showed many actin patches that were not concentrated at the tip of the shmoo (Fig. 5 F and Table III). All of the End−Fus+ mutants showed some defects in actin localization (i.e., small-budded cells and shmoos that failed to concentrate actin patches at the zone of polarized growth) although none of the alleles was as strong as rvs161Δ (Fig. 5, C and G, and Table III). Furthermore, there was a correlation between the strength of the growth defect on YPG or YEPD + NaCl and the severity of the actin delocalization phenotype. The rvs161-R35C and rvs161-R59K mutants, which were the most impaired for growth on YPG/YEPD + NaCl, also showed the strongest defect in actin organization. The rvs161-R113K and rvs161-P158S mutants, which were the weakest, were most like wild-type, but still showed some actin defects. In contrast, the End+Fus− mutants showed no observable defects in the organization of actin patches (Fig. 5, D and H, and Table III).
Figure 5.

(A–H) Actin localization in wild-type and various rvs161 cells. Vegetatively growing cells (A–D) and pheromone-induced cells (E–H) were fixed and prepared for indirect immunofluorescence using anti-actin antibodies. Shown are examples of the actin localization(A and E) in wild-type cells (MY3377); (B and F) in rvs161Δ (MY3909); (C and G) in an End−Fus+ mutant, rvs161-R35C (MY5301); and (D and H) in an End+Fus− mutant, rvs161-A175P (MY5227). (I–K) Unlinked noncomplementation analysis of the RVS161 function-specific mutations with act1-1. The following diploid strains were tested for growth on YEPD (I) and on YEPD+NaCl (J): +/+ +/act1, RVS161/RVS161 ACT1/ act1-1 (MY3377 × RH2151); R35C/+ +/act1, rvs161-R35C/ RVS161 ACT1/ACT1 (MY5301 × RH2151); R59K/+ +/act1, rvs161-R59K/RVS161 ACT1/act1-1 (MY4682 × RH2151); R113K/+ +/act1, rvs161-R113K/RVS161 ACT1/act1-1 (MY5303 × RH2151); P158S/+ +/act1, rvs161-P158S/RVS161 ACT1/act1-1 (MY5322 × RH2151); A175P/+ +/act1, rvs161-A175P/RVS161 ACT1/act1-1 (MY5227 × RH2151); P203Q/+ +/act1, rvs161-P203Q/RVS161 ACT1/act1-1 (MY5224 × RH2151) and Δ/+ +/ act1, rvs161-Δ1::LEU2/RVS161 ACT1/act1-1 (MY3909 × RH2151). K indicates the position of the diploid strains in the plate. Photographs were taken after 3 d of incubation at 30°C.
Table III.
Cortical Actin Localization in rvs161 Alleles
| Vegetative | Shmoos | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| In the bud | Random | At the neck | Tip and neck | Random | ||||||
| WT | 94 | 6 | 0 | 97 | 3 | |||||
| rvs161Δ | 18 | 41 | 41 | 28 | 72 | |||||
| rvs161-R35C | 78 | 8 | 14 | 61 | 39 | |||||
| rvs161-R59K | 61 | 31 | 8 | 52 | 48 | |||||
| rvs161-R113K | 84 | 12 | 4 | 84 | 16 | |||||
| rvs161-P158S | 96 | 0 | 4 | 71 | 29 | |||||
| rvs161-A175P | 93 | 7 | 0 | 89 | 11 | |||||
| rvs161-P203Q | 93 | 7 | 0 | 91 | 9 | |||||
Vegetatively growing cells (vegetative) or cells treated with α-factor (shmoos) were fixed and prepared for indirect immunofluorescence using polyclonal anti-actin antibodies as described in Materials and Methods. The strains analyzed were wild type (MY3377), rvs161Δ (MY3909), rvs161-R35C (MY5301), rvs161-R59K (MY4682), rvs161-R113K (MY5303), rvs161-P158S (MY5300), rvs161-A175P (MY5227), and rvs161-P203Q (MY5224). About 50 small-budded vegetative cells and 40 shmoos were analyzed in each experiment. The numbers represent the percentages of each class. The overall patterns of actin localization for the End−Fus+ mutants were significantly different from wild type (P < 0.001) for all classes with the possible exception of rvs161-P158S. The overall patterns of actin localization for the End+Fus− mutants did not differ significantly from wild-type.
Table IV presents a quantitative analysis of the percentage of cells with and without actin cables. While the majority of wild-type cells showed actin cables running along the mother–daughter or mating projection axes, most of the rvs161Δ cells did not show actin cables. The End−Fus+ mutants showed a significant reduction in the number of cells with actin cables, particularly the rvs161-R35C and rvs161-R59K alleles. In contrast, the End+Fus− mutants behaved like wild-type. Based on these results, we concluded that the degree of defects in endocytosis of the End−Fus+ mutants (as measured by poor growth on YPG and YEPD + NaCl) was correlated with the severity of defects in the actin cytoskeleton. We also concluded that Rvs161p's function in cell fusion is likely to be independent of its role in the organization of the actin cytoskeleton.
Table IV.
Presence of Actin Cables in rvs161 Alleles
| Vegetative | Shmoos | |||||||
|---|---|---|---|---|---|---|---|---|
| with cables | without cables | with cables | without cables | |||||
| WT | 96 | 4 | 89 | 11 | ||||
| rvs161Δ | 20 | 80 | 41 | 49 | ||||
| rvs161-R35C | 53 | 47 | 76 | 24 | ||||
| rvs161-R59K | 53 | 47 | 54 | 46 | ||||
| rvs161-R113K | 83 | 17 | 67 | 33 | ||||
| rvs161-P158S | 90 | 10 | 84 | 16 | ||||
| rvs161-A175P | 93 | 7 | 98 | 2 | ||||
| rvs161-P203Q | 85 | 15 | 97 | 3 | ||||
See legend to Table III.
To further investigate the relationship between the two roles of Rvs161p and actin organization, we performed genetic analyses of the interaction between the function-specific alleles of RVS161 and act1-1. Munn et al. (1995) reported that end6-1, but not rvs161Δ, showed unlinked noncomplementation with act1-1 for growth at 37°C. That is, diploids of the form end6-1/RVS161 ACT1/act1-1 were temperature-sensitive, even though both mutations were recessive. We tested if any other rvs161 allele besides end6-1 also showed unlinked noncomplementation with act1-1. Like the End−Fus+ mutants, the act1-1 and act1-1/ act1-1 strains were unable to grow on YEPD + NaCl at 30°C (data not shown). Fig. 5, I and J shows that the heterozygous diploids containing the End−Fus+ alleles and act1-1 (i.e., rvs161-R35C, rvs161-R59K, rvs161-R113K, and rvs161-P158S) grew well on YEPD, but very poorly on YEPD + NaCl. Therefore, we concluded that, like end6-1, the End−Fus+ alleles showed unlinked noncomplementation with act1-1. Furthermore, the severity of growth defects of each of the double heterozygotes was correlated with the severity of the respective haploid growth defects. Importantly, the heterozygous diploids containing the End+Fus− alleles and act1-1 (rvs161-A175P and rvs161-P203Q) grew like the wild-type control (Fig. 5 J). Likewise, the heterozygous diploid containing rvs161Δ and act1-1 grew like the wild- type (Fig. 5 J). In all cases, the diploids obtained by mating the rvs161 mutants strains and act1-1 with wild-type did not show any growth defects (see Materials and Methods).
In summary, the localization data and the genetic analysis showed a strong correlation between Rvs161p's role in endocytosis and actin organization. In contrast, Rvs161p's role in cell fusion is separated and independent of its role in both endocytosis and actin organization.
RVS161 Expression is Pheromone-induced
One of the hallmarks of a gene that is specifically required during mating is its transcriptional induction by the mating pheromones. We therefore wanted to investigate if RVS161 was induced by pheromone. Northern blot analysis showed that RVS161 produced two mRNA transcripts (Fig. 6 A). These two transcripts were not present in rvs161Δ, proving that they both corresponded to RVS161 (data not shown). RVS161 was induced between three to fourfold higher levels by α-factor within 90 min of treatment (Fig. 6 A). Transcription of RVS161 was not induced in cdc28-4 G1 arrested cells at 37°C in the absence of pheromone (Fig. 6 A). Therefore, RVS161 transcriptional induction is specific to pheromone, and it is not a consequence of the G1 cell cycle arrest that occurs when the cells respond to pheromone. Actin mRNA levels used as a control were not significantly changed regardless of the experimental conditions (Fig. 6 A).
Figure 6.

Pheromone induction of RVS161. (A) Northern blot analysis of RVS161 mRNA expression. Wild-type (MY3375) and cdc28-4 (MY3468) strains were incubated with (+) or without (−) α-factor at 30°C or 37°C for 90 min before analysis. An 878-bp DNA fragment from RVS161 (see Materials and Methods) was used as a probe in this experiment (top). Analysis of actin mRNA was used as a loading control (bottom). (B) Western blot analysis of Rvs161p. Wild-type (MY3371) and rvs161Δ (MY3909) strains were incubated with (+) or without (−) α-factor before preparing the yeast protein extracts. The blot was probed with affinity-purified rabbit anti-Rvs161p antibody as described in Materials and Methods. Molecular weight standards (in kD) are shown to the left of the panel.
Western analysis of total yeast proteins using anti-Rvs161p polyclonal antibodies is shown in Fig. 6 B. The antibodies recognized a single 31-kD band that was absent in the rvs161Δ control strain. The levels of Rvs161p were increased in pheromone treated cells (Fig. 6 B), consistent with the increased transcription in the presence of α-factor observed by Northern blots.
Rvs161p Localizes to the Region of Cell Fusion in Prezygotes
To determine the localization of Rvs161p in vegetative, pheromone-induced, and mating cells, we used a Rvs161-GFP fusion protein. In this construct GFP is fused to the 3′ region of RVS161, and the gene is expressed under RVS161 promoter regulation. The fusion protein was able to complement the mating defects of rvs161 mutants as analyzed by microscopic examination of zygotes (see Materials and Methods). We found that Rvs161-GFP concentrates at distinct and precise locations in vegetative, pheromone-induced, and mating cells. During vegetative growth, unbudded cells showed cytoplasmic fluorescence with some dots distributed randomly within the cell (14% of the cells). In small budded cells, Rvs161-GFP concentrated mainly at the mother-bud neck (58% of the cells) with some punctate fluorescence also present in the bud (27% of the cells; Fig. 7, A–D). In contrast, large budded cells showed mostly diffuse fluorescence (68% of the cells) with some punctate fluorescence at the neck (32% of the cells; Fig. 7, E and F). In pheromone-induced cells, Rvs161-GFP concentrated at the tip of the shmoo (84% of the shmoos; Fig. 7, G and H). More importantly, in mating cells we found that Rvs161-GFP localized mainly to the cell fusion zone in early zygotes (92% of early zygotes; Fig. 7, I and J). Interestingly, Rvs161-GFP was no longer concentrated at the former cell fusion zone in mature zygotes (98% of mature zygotes). These results suggest that once cell fusion occurs, Rvs161p no longer localizes to the intersection between mating cells (Fig. 7, M and N). We found the same pattern of localization using an NH2-terminal GFP fusion under the regulation of the GAL10 promoter, suggesting that this localization truly reflects the localization of Rvs161p, and does not depend on the location of GFP in the protein (Fig. 7, K and L). Taking these results together, we concluded that the spatial and temporal localization of Rvs161p is consistent with a direct role of Rvs161p in cell fusion.
Figure 7.
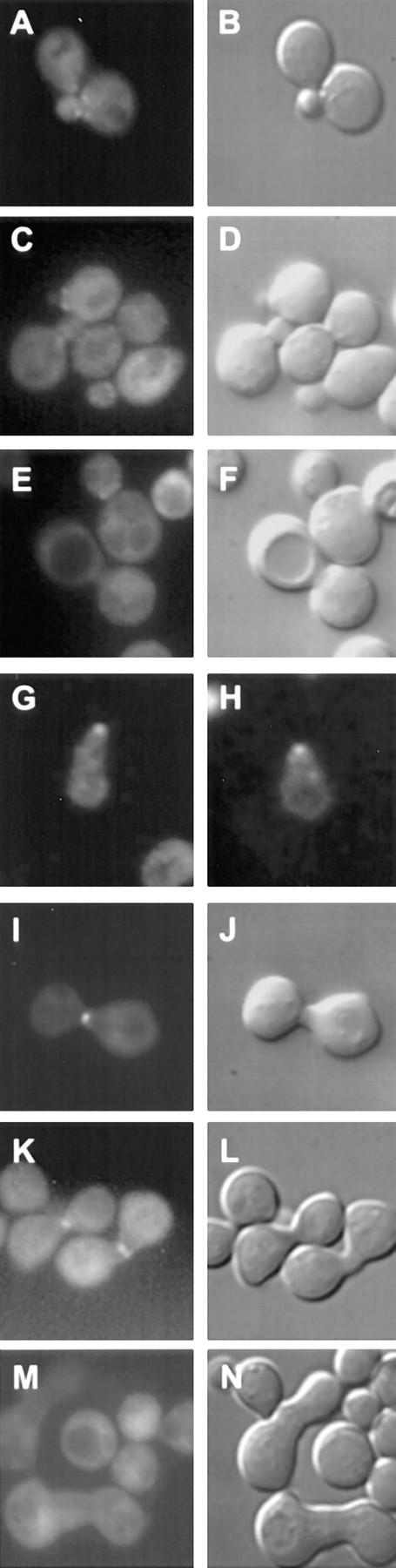
Localization of Rvs161-GFP. A–F are examples of vegetative cells expressing Rvs161-GFP fusion (MY3909 transformed with pMR3510). A, C, and E show the GFP fluorescence visualized using a High Q FITC set (#41001 from Chroma technology Corp., Brattleboro, VT). B, D, and F are the respective DIC images. G and H are examples of pheromone-induced cells expressing the Rvs161-GFP fusion (MY3909 transformed with pMR3510) visualized using FITC filter set. I, J, M, and N are examples of mating cells (MY3909 mated with MY4495 transformed with pMR3510). I and M show the GFP fluorescence, and J and N show the respective DIC images. K and L show examples of mating cells expressing PGAL-GFP-Rvs161p (MY3372 mated with MY3784 transformed with pMR3462). K and L show GFP fluorescence and DIC images, respectively.
rvs161Δ is Synthetically Sterile with fus1Δ, but Not With fus2Δ
To investigate further the role of RVS161 in cell fusion, we analyzed genetic interactions with two other genes involved in cell fusion. Like rvs161Δ, mutations in FUS1 or in FUS2 show no cell-type specificity, and the mutant phenotype is stronger when both partner cells are mutant (Trueheart et al., 1987). The fus1Δ fus2Δ double mutant shows a more drastic cell fusion phenotype than either single mutant (Trueheart et al., 1987). From this result, it was proposed that FUS1 and FUS2 might act in partially redundant parallel pathways. Therefore, we analyzed genetic interactions among rvs161Δ, fus1Δ, and fus2Δ to determine if RVS161 may act in either pathway. Fig. 8 shows pictures of plate-mating assays of rvs161Δ in combination with fus1Δ (Fig. 8 B) and fus2Δ (Fig. 8 C). As a control, we also included the analysis of the fus1Δ fus2Δ double mutant (Fig. 8 A). We found that, like fus1Δ fus2Δ, the fus1Δ rvs161Δ double mutant has a more severe defect than either single mutant alone (Fig. 8 B). First, unlike the single mutants, the fus1Δ rvs161Δ double mutant has a visible mating defect when mated with the wild-type (Fig. 8 B). Second, the fus1Δ rvs161Δ double mutant defect is much worse than either single mutant defect (Fig. 8 B). Finally, the fus1Δ rvs161Δ double mutants mated together gave the most severe mating defect. Even after 19 h of mating there was no diploid formation (data not shown). Therefore, in an analogous manner to FUS1 and FUS2, we propose that FUS1 and RVS161 might also act in parallel pathways. In contrast, we found that the fus2Δ rvs161Δ double mutant mated no worse than either single mutant alone (Fig. 8 C). In fact, the fus2Δ rvs161Δ strain behaved in all the crosses exactly like fus2Δ or rvs161Δ single mutants. We reasoned that once either protein is defective, the pathway is inactive and further mutation does not lead to a greater defect. Therefore, in this context we concluded that FUS2 and RVS161 most likely act in the same pathway.
Figure 8.
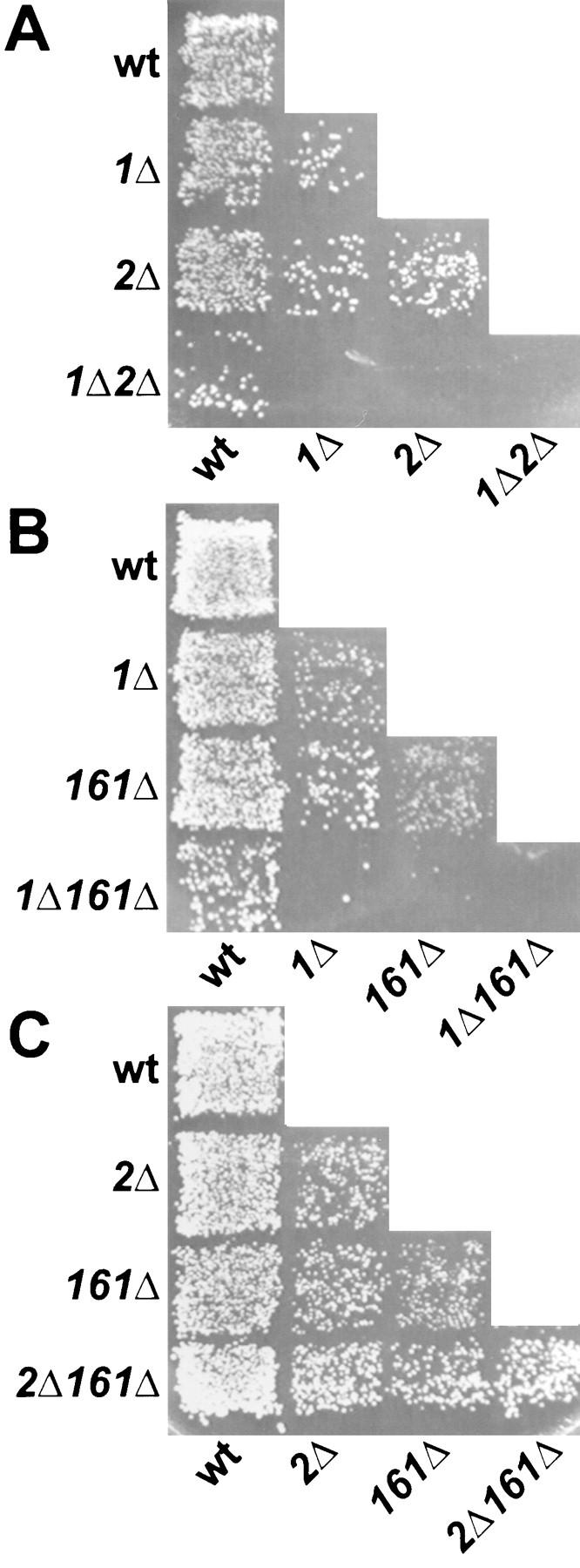
Limited plate-mating analysis of double mutants. (A) Diploid selective plate from the fus1Δ fus2Δ limited mating analysis. Mating was performed on YEPD for 3 h at 30°C and then replica-printed onto synthetic complete media lacking uracil and tryptophan to select for diploid formation. The MAT a strains were as follows: wt, wild-type (MY3377); 1Δ, fus1Δ (JY427); 2Δ, fus2Δ (JY424); and 1Δ2Δ, fus1Δ fus2Δ (MY4160). The MATα strains transformed with pRS426 were as follows: wt, wild-type (MY3378); 1Δ, fus1Δ (MY4164); 2Δ, fus2Δ (JY428); and 1Δ2Δ, fus1Δ fus2Δ (JY429). (B) Diploid selective plate from the fus1Δ rvs161Δ limited mating analysis. Mating was performed on YEPD for 3 h at 30°C, and was then replica-printed onto synthetic complete media lacking uracil and tryptophan to select for diploid formation. The MAT a strains transformed with pRS426 were as follows: wt, wild-type (MY2788); 1Δ, fus1Δ (MY4161); 161Δ, rvs161Δ (MY3909); and 1Δ161Δ, fus1Δ rvs161Δ (MY4097). The MATα strains were as follows: wt, wild-type (MY2787); 1Δ, fus1Δ (MY4161); 161Δ, rvs161Δ (MY4495); and 1Δ161Δ, fus1Δ rvs161Δ (MY4905) transformed with pRS424. (C) Diploid selective plate from the fus2Δ rvs161Δ limited mating analysis. Mating was performed on YEPD for 3 h at 30°C, and was then replica-printed onto synthetic complete media lacking uracil and leucine to select for diploid formation. The MAT a strains transformed with pRS426 were as follows: wt, wild-type (MY2788); 2Δ, fus2Δ (JY424); 161, fus7-1811 (MY3722); and 2Δ161Δ, fus2Δ rvs161Δ (MY4801). The MATα strains were as follows: wt, wild-type (MY2787); 2Δ, fus2Δ (JY428); 161Δ, rvs161Δ (MY4495); and 2Δ161Δ, fus2Δ rvs161Δ (MY4802).
Rvs161p Coprecipitates with Fus2p
The genetic evidence that Fus2p and Rvs161p act in the same pathway suggested that these two proteins might physically interact. To determine if Rvs161p and Fus2p show any direct or indirect physical interaction, we tested Rvs161p's ability to coprecipitate with Fus2p and vice-versa (Fig. 9). First, we tested if Rvs161p coprecipitates with Fus2p using a GST-Fus2p fusion. We created a functional GST-Fus2p fusion under the regulation of the GAL10 promoter that fully complemented the mating defect of a fus2Δ strain (data not shown). As a control, a strain that expressed GST alone was used. Proteins were probed with specific antibodies to identify GST, GST-Fus2p, and Rvs161p (Fig. 9 A). The first two lanes in Fig. 9 A show the proteins that bound to glutathione–sepharose beads. The third and fourth lanes are samples of total protein extracts from strains expressing GST or GST-Fus2p. Both GST and GST-Fus2p were able to bind to glutathione-sepharose beads (Fig. 9 A). Approximately 10% of total GST-Fus2p precipitated under the conditions used. We found that a fraction of Rvs161p (∼3% of total Rvs161p) was able to coprecipitate specifically with GST-Fus2p, but not with the GST control alone (Fig. 9 A). Therefore, as much as 30% of Rvs161p was bound to GST-Fus2p. The fourth panel in Fig. 9 A shows that Rvs161p was nevertheless present in both GST and GST-Fus2p extracts.
Figure 9.

Interaction between Rvs161p and Fus2p. (A) Rvs161p coprecipitated with Fus2p-GST fusion protein. GST and Fus2p-GST were precipitated from a fus2Δ (JY429) strain transformed with pEG-KT (GST) or pMR3715 (GST-Fus2p) using glutathione-sepharose beads. Aliquots of proteins from the precipitates (ppt; lanes 1 and 2) or from total protein extracts (total; lanes 3 and 4) were analyzed by Western blot using antibodies against GST (α-GST), Fus2p (α-Fus2p), and Rvs161p (α-Rvs161p) as indicated to the right of the panel. The identity of the bands is indicated to the left of the panel. (B) Immunoprecipitation experiments using anti-Fus2p and anti-Rvs161p antibodies. (Left) Fus2p was immunoprecipitated from a wild-type strain (wt; MY3371) or a fus2Δ strain (fus2Δ; MY4858) with anti-Fus2p antibodies. Proteins from precipitates (ppt α-Fus2p; lanes 1 and 2) or total protein extracts (total; lanes 3 and 4) were analyzed by Western blot with anti-Fus2p or anti-Rvs161p. Asterisks at the left of the panel indicate the bands corresponding to Fus2p and Rvs161p. In addition to Rvs161p, two nonspecific bands precipitated by Protein A-sepharose beads were recognized by the anti-Rvs161p antibodies. (Right) Rvs161p was immunoprecipitated from an rvs161Δ strain (MY3909) transformed with pMR3336 (RVS161 2μ) or with pRS426 (vector) using anti-Rvs161p antibodies as described in Materials and Methods. Proteins from precipitates (ppt α-Rvs161p; lanes 1 and 2) or total protein extracts (total; lanes 3 and 4) were analyzed by Western blot with anti-Fus2p or anti-Rvs161p. Asterisks at the left of the panel indicate the bands corresponding to Fus2p and Rvs161p.
Second, we tested whether Rvs161p coprecipitated with Fus2p in immunoprecipitation experiments using polyclonal anti-Fus2p antibodies. The immunoprecipitates were then probed with anti-Fus2p antibodies and anti-Rvs161p antibodies to detect coprecipitation. The first two lanes in Fig. 9 B show immunoprecipitates from a wild-type or a fus2Δ control strain, using anti-Fus2p antibodies. The third and fourth lanes are samples of total protein extracts (Fig. 9 B). As shown in Fig. 9 B, a fraction of Rvs161p did coprecipitate with Fus2p by this method. No Rvs161p was detected in the immunoprecipitate from the control fus2Δ strain, even though Rvs161p was present at comparable levels in both wild-type and fus2Δ strains (Fig. 9 B, lanes 3 and 4). Finally, we tested whether Fus2p would coprecipitate with Rvs161p using polyclonal anti-Rvs161p as the first antibody. Fig. 9 B shows that a fraction of Fus2p did coprecipitate with Rvs161p in immunoprecipitation using anti-Rvs161p antibodies. In this experiment, ∼9% of total Rvs161p precipitated. We found that ∼1.5% of the total Fus2p coprecipitated with Rvs161p, suggesting that ∼17% of the Fus2p was bound to Rvs161p. Similar amounts were precipitated using an rvs161Δ strain transformed with either a 2 μ RVS161 plasmid or a CEN–RVS161 plasmid, but not when using a rvs161Δ strain transformed with the vector (Fig. 9 B and data not shown). Taking these results together, we concluded that Rvs161p and Fus2p interact as part of a protein complex. Combining the genetic and the biochemical data, we concluded that Rvs161p and Fus2p might act in the same pathway to promote cell fusion, as part of the same protein complex.
Fus2p's Stability Depends on RVS161p's Mating Function
To investigate if the cell fusion defect of rvs161Δ was due to reduced levels of Fus2p or vice versa, we examined the levels of the two proteins in the mutant strains. We performed Western blot analysis of wild-type, fus2Δ, and rvs161Δ protein extracts, using anti-Fus2p and anti-Rvs161p (Fig. 10 A). We found that the levels of Fus2p in rvs161Δ were greatly reduced compared with wild-type (Fig. 10 A). The reduction of Fus2p levels in rvs161Δ was specific for Fus2p since Fus1p levels were normal compared with wild type (Fig. 10 A). In addition, Rvs161p and Fus1p levels in fus2Δ were the same as wild type (Fig. 10 A).
Figure 10.

Fus2p's stability is dependent on Rvs161p. (A) Western blot analysis of Fus2p in a rvs161Δ strain. Wild-type (MY3371), fus2Δ (MY4858) and rvs161Δ (MY3909) strains were treated with α-factor for 90 min. Total protein extracts were analyzed by Western blot using antibodies against Fus2p, Rvs161p, or Fus1p. The forms of Fus2p are indicated with arrows. (B) Pulse-chase analysis of Fus2p. Wild-type (wt; MY3371) and rvs161Δ (MY3909) cells were pulsed for 7.5 min using 35S and chased for 0, 5, 10, 20, 60, and 120 min as described in Materials and Methods. The modified forms of Fus2p are indicated by an asterisk. (C) Quantitation of Fus2p stability from the pulse-chase analysis. Plotted are the logarithms from the percentages of Fus2p remaining at various times after the chase in wild-type (MY3371; black symbols) and in rvs161Δ (MY3909; open symbols). (D) Western blot analysis of Fus2p in various rvs161 alleles. Wild-type (MY3377), rvs161-P203Q (MY5224), rvs161-A175P (MY5227), rvs161-R59K (MY4682), and rvs161-R35C (MY5301) strains were treated with α-factor for 90 min. Total protein extracts were analyzed by Western blot using antibodies against Fus2p or Rvs161p. (E) Coimmunoprecipitation of Fus2p and Rvs161p in various rvs161 alleles. Fus2p was immunoprecipitated from a wild-type strain (wt; MY3371) and three rvs161 mutants: rvs161-P203Q (MY5224), rvs161-A175P (MY5227), and rvs161-R59K (MY4682), using anti-Fus2p antibodies. Western blots of the precipitates were probed for Fus2p and Rvs161p as described in Figure 9. To compensate for the reduced levels of Fus2p in rvs161-P203Q (lane 2) and rvs161-A175P strains (lane 3), for these mutants sixfold larger samples were loaded relative to wt (lane 1) and rvs161-R59K (lane 4).
To analyze if the reduction in Fus2p levels in rvs161Δ was due to instability of Fus2p rather than a reduction in the levels of FUS2 induction, we performed a pulse-chase analysis of Fus2p (Fig. 10, B and C). In a wild-type strain, Fus2p was a fairly stable protein having a half-life of ∼120 min (Fig. 10, B and C). In contrast, the stability of Fus2p in rvs161Δ was greatly reduced to a half-life of 15 min (Fig. 10, B and C). From these results we concluded that the stability of Fus2p is dependent on Rvs161p.
To test if Fus2p's stability depends on Rvs161p's mating function, we analyzed the levels of Fus2p in several of the rvs161 function-specific alleles. We found that Fus2p levels were reduced specifically in the rvs161 End+ Fus− mutants, but not in the End−Fus+ mutants (Fig. 10 D). These results demonstrate that Fus2p's stability depends on Rvs161p's mating function, and suggests that the cell fusion defect in these mutants could be due to reduced Fus2p levels.
To investigate whether the defect in Fus2p stability might be due to reduced interaction with the mutant forms of Rvs161p, we immunoprecipitated Fus2p and analyzed Rvs161p's ability to coprecipitate (Fig. 10 E). Extracts were prepared from wild-type, two End+Fus−, and two End−Fus+ mutants. To compensate for the reduced levels of Fus2p in the End+Fus− mutants, we loaded six times as much of the immunoprecipitate (Fig. 10 E) such that levels of Fus2p comparable to those of the wild-type were observed. Similar amounts of Rvs161p coprecipitated from the wild-type, and the two End−Fus+ mutants (data for rvs161-R35C not shown). In contrast, we found that only a barely detectable amount of Rvs161p coprecipitated from the End+Fus− mutants. Therefore, we conclude that these mutations severely impair the interaction between Rvs161p and Fus2p.
Interestingly, in the Western and pulse-chase analyses of Fus2p, we noticed the presence of two or more bands corresponding to Fus2p, which were particularly evident in a low percentage of polyacrylamide gels (Fig. 10, A and B). In rvs161Δ the higher molecular weight bands of Fus2p were absent (Fig. 10, A and B), suggesting that these forms of Fus2p also depend on the mating function of Rvs161p. In the pulse-chase analysis the higher molecular weight forms of Fus2p appeared 10 min after the chase, and after ∼1 h of pheromone induction (Fig. 10 B). This result shows that Fus2p is modified posttranslationally, and suggests that the modification is a rather late event in the pheromone induction of the cells.
Discussion
Conjugation in Saccharomyces cerevisiae involves fusing two cells of opposite mating type. Conjugation is thought to require a complex interplay of pheromone response, spatial organization, and alteration of the cell wall and plasma membrane. In this paper we describe the characterization of a cell fusion mutant, fus7-1811, that blocks cell fusion. We identified FUS7 as being allelic to RVS161, a gene previously implicated in viability upon starvation, actin organization, and endocytosis (Crouzet et al., 1991; Durrens et al., 1995; Munn et al., 1995; Sivadon et al., 1995). We tested whether endocytosis per se plays a role in cell fusion, and found that most of the endocytic mutants examined, including end3-1, end4-1, end6-1 (an allele of RVS161), end7-1, and rvs167Δ, showed no defect in cell fusion. Using in vitro mutagenesis, we isolated mutant alleles of RVS161/FUS7 that separate the endocytic function from the mating function. Mapping of the function-specific alleles defined an NH2-terminal region required for endocytosis and a COOH-terminal region required for cell fusion. Therefore, we propose that RVS161 is a bifunctional gene with a role in endocytosis that is different from its role in cell fusion during mating. Interestingly, we found that in contrast to the alleles that affect endocytosis, the cell fusion–defective alleles showed no defects in the actin cytoskeletal organization. We conclude that Rvs161p's role in cell fusion is likely to be independent of its role in actin organization.
Like other genes required for cell fusion (e.g., FUS1 and FUS2), we found that RVS161 was induced by mating pheromone. Using a functional Rvs161-GFP hybrid protein, we found that Rvs161p concentrated at the region of cell fusion, consistent with a role in cell fusion. Analysis of double mutants indicated that rvs161Δ behaved like fus2Δ genetically (i.e., rvs161Δ fus1Δ is more severe than either fus1Δ or rvs161Δ single mutant alone). Furthermore, the rvs161Δ fus2Δ double mutant showed the same phenotype as either single mutant. Therefore, we propose that Rvs161p and Fus2p might act in the same pathway for cell fusion. In agreement with this hypothesis, we showed that Rvs161p coprecipitates with Fus2p, and that the stability of Fus2p depends on Rvs161p's mating function in the cell. Therefore, we concluded that Rvs161p interacts with Fus2p to promote cell fusion.
Endocytosis Does Not Play a Direct Role in Cell Fusion
After initial cell–cell contact, partner cells remodel their cell wall to achieve cell fusion. Because mating can occur in hypo-osmotic media, it seems likely that the cells make a seal to ensure their integrity as the cells fuse. During fusion, the cells must remove the intervening cell wall at the point of cell contact. After cell wall removal the cells must then fuse their plasma membranes. The mechanisms responsible for removing the intervening cell wall and for fusing the plasma membranes are not known. During vegetative growth, yeast cells constantly remodel their cell wall to accommodate bud growth. This process requires delivery of glucanases and glucan-synthases through the secretory pathway to the growing site in the bud. The net result of the action of these enzymes in this case is the new addition of cell wall material. It seems likely that the removal of the intervening cell wall is accomplished through the localized delivery of glucanases to the cell fusion zone. EM analysis of mating cells showed clusters of vesicles at either side of the cell fusion zone in cells that are about to fuse (Gammie et al., 1998). The origin of the vesicles is not known, but it is tempting to speculate that these are secretory vesicles that carry hydrolytic enzymes required to remove the intervening cell wall. Alternatively, cell wall removal could be achieved by an endocytic mechanism or by a combination of both secretion and endocytosis.
Several mutants that affect endocytosis have been identified in yeast (Riezman et al., 1996). Several of the mutants (including end3-1, end4-1, end5-1, end6-1, end7-1, and rvs167Δ) that affect endocytosis at the internalization step also exhibit defects in actin organization (Raths et al., 1993; Benedetti et al., 12994; Munn et al., 1995). Cloning and identification of these genes showed that they all affect some aspect of actin cytoskeleton organization. END3 is a 40-kD protein with putative Ca2+ and PIP2 binding sites and a region of homology to the mammalian epidermal growth factor receptor tyrosine kinase substrate Eps15 (EH domain; Benedetti et al., 1994; Fang et al., 1997). END4 is allelic to SLA2, a 109-kD protein with homology to talin, a mammalian focal adhesion protein (Holtzman et al., 1993). END5 is allelic to VPR1, which encodes verprolin, a gene required for proper organization of the actin cytoskeleton, and for the proper mitochondrial/cytoplasmic protein distribution (Donnelly et al., 1993; Munn et al., 1995; Zoladek et al., 1995). Finally, end7-1 was found to be an allele of ACT1 (Munn et al., 1995). Given that this group of end mutants did not exhibit defects in cell fusion, we conclude that endocytosis, in general, is not required for cell fusion.
We were surprised to find that rvs167Δ did not result in any cell fusion defects. The Rvs domain of Rvs161p is also present in Rvs167p. Several lines of evidence have suggested that Rvs161p and Rvs167p are involved in the same cellular process. First, before this work, mutations in either gene had been shown to result in the same phenotypes (Bauer et al., 1993; Sivadon et al., 1995). Second, alleles from each gene have common suppressors (Desfarges et al., 1993; Revardel et al., 1995; Sivadon et al., 1997b ). Third, Rvs167p and Rvs161p were shown to interact physically (Sivadon et al., 1997a ). However, in spite of this evidence and the sequence homology between Rvs161p and Rvs167p, it seems that each of these proteins has a distinct role in a common cellular process. The Rvs domain of both Rvs161p and Rvs167p cannot be functionally exchanged with each other (Sivadon et al., 1997a ), and RVS161 and RVS167 in high copy number plasmids were not able to suppress each other's mutations (Bauer et al., 1993). Not only do Rvs161p and Rvs167p have distinct roles in endocytosis, but we have found that Rvs167p is not required for cell fusion. This is the first time that a cellular function is attributed to just one of the two Rvs proteins. Thus, it is possible that Rvs161p interacts with Rvs167p to fulfill vegetative functions (i.e., endocytosis/actin organization) and with other proteins during mating.
Endocytosis/Actin Organization and Cell Fusion: Two Genetically Separable Functions of Rvs161p
Based on our findings that the end6-1 allele of RVS161 did not exhibit a cell fusion defect, we hypothesized that the role of RVS161 in cell fusion must be separate from its role in endocytosis. In support of this hypothesis, we isolated alleles of RVS161 that affected cell fusion without showing endocytic defects. We also isolated several more alleles that were like end6-1 in that they affected endocytosis without affecting cell fusion. Therefore, RVS161 has two genetically separate cellular functions; endocytosis and cell fusion, which map to two separate domains: an NH2-terminal domain required for endocytosis and a COOH-terminal domain required for cell fusion. One plausible model to explain the bifunctional characteristic of Rvs161p is that it interacts with proteins involved in endocytosis through the NH2-terminal domain and with cell fusion proteins through the COOH-terminal domain.
Given the previously reported observation that mutations in RVS161 result in a disorganized actin cytoskeleton (Munn et al., 1995; Sivadon et al., 1995), we analyzed the actin organization in various rvs161 alleles isolated in this study. Interestingly, we found that only the alleles that affect endocytosis had defects in the organization of the actin cytoskeleton. Our findings support the suggestion of a tight link between actin and endocytosis (Munn et al., 1995; Riezman et al., 1996).
A connection between Rvs161p and actin has also been shown genetically. Munn et al. (1995) discovered that the end6-1 mutation of RVS161, but not rvs161Δ, exhibits unlinked noncomplementation with act1-1. This result indicates that the protein encoded by the end6-1 allele of RVS161 has a detrimental effect in the double heterozygous diploid, and suggests that Rvs161p and actin interact in vivo (Munn et al., 1995). We found that all the rvs161 End−-specific alleles showed some degree of unlinked noncomplementation with act1-1. In contrast, the double-heterozygous diploids containing rvs161Δ or any of the Fus− specific alleles did not show any growth defects. From this result and the observation that the rvs161 cell fusion alleles showed a normal morphology of the actin cytoskeleton, we concluded that Rvs161p's role in cell fusion does not affect the organization of the actin cytoskeleton.
Rvs161p Might be Directly Involved in Cell Fusion
Two other genes required for cell fusion, FUS1 and FUS2, are highly pheromone-induced and encode proteins that localize to the region of cell fusion (Trueheart et al., 1987; Elion et al., 1995). We found that RVS161 was also induced by mating pheromone to three to fourfold higher levels. In addition, RVS161 was recently identified in a screen for pheromone-induced genes (Erdman et al., 1998). Consistent with being a gene induced by mating pheromone, the DNA sequence upstream of RVS161 contains two putative pheromone-response elements that differ from the consensus sequence by 1 bp. The relatively high levels of RVS161 basal expression probably reflect the fact that this gene has a vegetative function.
During vegetative growth, Rvs161p localized mostly to the bud neck and bud in small-budded cells, consistent with the actin interaction. In pheromone-induced cells, we found that Rvs161-GFP localized at the tip of the shmoo. In spite of the fact that Rvs161p and Fus2p interact (see below), these proteins have apparently different localization patterns in shmoos. Elion et al. (1995) reported that Fus2p primarily localizes to punctate structures under the surface of the shmoo projection. One possible explanation for the apparent discrepancy is that different methods for localization were used. Elion et al. (1995) used immunofluorescence techniques with strains containing either a Fus2-lacZ fusion (which stabilizes Fus2p) or a high-copy FUS2 plasmid. Alternatively, a failure to colocalize completely might not be surprising considering that only a fraction of these two proteins interact by coprecipitation experiments (see below).
In zygotes, Rvs161p concentrated at the zone of cell fusion in cells that are about to or in the process of fusing (early zygotes). Early zygotes can be distinguished from mature zygotes because they have a small zone of cell fusion (Gammie et al., 1998). As mating proceeds and the zygote reenters the mitotic cycle, the waist of the zygote widens. We found that in later zygotes (i.e., zygotes with a wide zone of cell fusion or budding zygotes) Rvs161-GFP was no longer concentrated in the former region of cell fusion. Similarly, in mating cells that are about to fuse, Fus2p was present mainly at the cell fusion zone and in early zygotes, and was no longer detected in cells that had undergone nuclear fusion (Elion et al., 1995). In contrast to the apparent discrepancy between the Rvs161p and Fus2p localization in shmoos, these two proteins showed the same localization pattern in zygotes.
Taken together, Rvs161p's regulation and localization are consistent with it playing a direct role in cell fusion. It is likely that when cells are stimulated to undergo mating, regulatory signals dictate Rvs161p's relocalization to the cell fusion zone. This relocalization could be achieved by interactions with mating specific proteins that localize at the cell fusion zone.
Rvs161p Interacts with Fus2p to Promote Cell Fusion
FUS2 encodes a novel 677–amino acid protein with some homology to myosin-related proteins including the yeast Mlp1 (Elion et al., 1995). Although not predicted to be a membrane protein, Fus2p was shown to be tightly associated with an insoluble fraction in cell fractionation experiments. Based on this result and the punctate localization of Fus2p, it was proposed that Fus2p is associated with a novel vesicle or cytoskeletal structure that plays a specific role in cell fusion (Elion et al., 1995). However, Fus2p's role in cell fusion and the proteins that interact with Fus2p were not identified. Several lines of evidence suggest that Rvs161p interacts with Fus2p to promote cell fusion. First, rvs161Δ fus2Δ double mutant was not more defective than either single mutant, indicating that Rvs161p and Fus2p act in the same cell fusion pathway. Second, Rvs161p coprecipitated with Fus2p, suggesting that they are components of a common complex. Third, we found that the stability of Fus2p was dependent upon the presence of Rvs161p. Furthermore, Fus2p levels were specifically reduced in the cell fusion–defective alleles of RVS161. The allele specificity of the stability of Fus2p suggests that the rvs161Δ cell fusion defect is due, at least in part, to reduction in the level of Fus2p.
How does Rvs161p stabilize Fus2p and what is the relevance of the Rvs161–Fus2p interaction? One possibility is that Rvs161p directly interacts with Fus2p to prevent its degradation. In fact, we found that the cell fusion–defective rvs161 mutants showed not only a reduction in Fus2p levels but also a reduction in the ability of the mutant proteins to interact with Fus2p. However, even in wild-type, only a small fraction of Fus2p coprecipitates with Rvs161p. Therefore, the Rvs161p–Fus2p interaction might not be stable under these conditions, or the interaction might be transient. One possibility is that Rvs161p is necessary to deliver Fus2p to other components of the cell fusion machinery. In this model, in the absence of Rvs161p, Fus2p would fail to localize to the correct place and therefore be more susceptible to degradation.
Alternatively, Rvs161p could bind Fus2p to allow for a modification that would stabilize and/or activate Fus2p. Indeed, both by Western blot and in pulse-chase experiments, we observed two or sometimes three bands corresponding to Fus2p, indicating that Fus2p is modified posttranslationally. In the pulse-chase analysis, the higher molecular band only appeared upon longer incubations with α-factor. Significantly, the higher molecular band was absent in rvs161Δ, suggesting that the Fus2p modification is dependent on Rvs161p. However, it is also possible that the modification does not occur because of the shorter half-life of Fus2p in this strain.
Is Rvs161p's sole function in cell fusion to stabilize Fus2p? In other experiments we have shown that FUS2 can suppress the cell fusion defect of rvs161Δ strain when present in a high copy plasmid (Gammie et al., 1998). However, the suppression was only partial, even though the levels of Fus2p in the rvs161Δ strain containing FUS2 on a high copy plasmid were higher than in wild-type (V. Brizzio and M. Rose, unpublished data). These results suggest that the Rvs161p-dependent modification of Fus2p might be crucial for its role in cell fusion. Alternatively, Rvs161p might have other functions independent of Fus2p. One intriguing possibility is that several proteins required for cell fusion interact with Rvs161p and Fus2p, and that all of them are destabilized in rvs161Δ. Consistent with both of these possibilities, we found that high copy RVS161 did not suppress the fus2Δ cell fusion defect (Gammie et al., 1998). Further analysis of the Fus2p/ Rvs161p complex should elucidate the nature of the rvs161Δ defect.
Acknowledgments
We thank the members of this laboratory for helpful discussions. We are also grateful to Andreas Wesp, Howard Riezman, Gerry Fink, Joshua Trueheart, Elaine Elion, Tongtong Wang, Anthony Bretscher, Corey Davis and Jim Broach for providing us with strains, antibodies and reagents.
Abbreviations used in this paper
- 5-FOA
5-fluoro-orotic acid medium
- DIC
differential interference contrast
- cM
centiMorgans
- GST
glutathione-S-transferase
- LY
Lucifer yellow
- MAP
mitogen-activated protein
- YEPD
yeast extract/peptone/dextrose
- YPG
yeast extract/peptone/ glycerol
Footnotes
This research was supported by the National Institutes of Health grant to M.D. Rose (GM37739). V. Brizzio was supported by an institutional National Institutes of Health training Grant on genetics, Cell, and Molecular Biology. A.E. Gammie was partially supported by a Fellowship from the Jane Coffin Child Memorial Fund for Cancer Research.
Address all correspondence to Mark D. Rose, Department of Molecular Biology, Princeton University, Princeton, New Jersey 08544-1014. Tel.: 609-258-2804; Fax: 609-258-6175; E-mail: mrose@molbio.princeton.edu
References
- Adams AE, Pringle JR. Relationship of actin and tubulin distribution to bud growth in wild-type and morphogenetic-mutant Saccharomyces cerevisiae. . J Cell Biol. 1984;98:934–945. doi: 10.1083/jcb.98.3.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer F, Urdaci M, Aigle M, Crouzet M. Alteration of a yeast SH3 protein leads to conditional viability with defects in cytoskeletal and budding patterns. Mol Cell Biol. 1993;13:5070–5084. doi: 10.1128/mcb.13.8.5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti H, Raths S, Crausaz F, Riezman H. The END3gene encodes a protein that is required for the internalization step of endocytosis and for actin cytoskeleton organization in yeast. Mol Biol Cell. 1994;5:1023–1037. doi: 10.1091/mbc.5.9.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin V, Brill J, Trueheart J, Boeke J, Fink G. Genetic screens and selections for cell and nuclear fusion mutants. Methods Enzymol. 1991;194:774–792. doi: 10.1016/0076-6879(91)94058-k. [DOI] [PubMed] [Google Scholar]
- Brizzio V, Gammie AE, Nijbroek G, Michaelis S, Rose MD. Cell fusion during yeast mating requires high levels of a-factor mating pheromone. J Cell Biol. 1996;135:1727–1739. doi: 10.1083/jcb.135.6.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MH, David C, Ochoa GC, Freyberg Z, Daniell L, Grabs D, Cremona O, De Camilli P. Amphiphysin II (SH3P9; BIN1), a member of the amphiphysin/Rvs family, is concentrated in the cortical cytomatrix of axon initial segments and nodes of ranvier in brain and around T tubules in skeletal muscle. J Cell Biol. 1997;137:1355–1367. doi: 10.1083/jcb.137.6.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouzet M, Urdaci M, Dulau L, Aigle M. Yeast mutant affected for viability upon nutrient starvation: characterization and cloning of the RVS161gene. Yeast. 1991;7:727–743. doi: 10.1002/yea.320070708. [DOI] [PubMed] [Google Scholar]
- David C, McPherson PS, Mundigl O, de Camilli P. A role of amphiphysin in synaptic vesicle endocytosis suggested by its binding to dynamin in nerve terminals. Proc Natl Acad Sci USA. 1996;93:331–335. doi: 10.1073/pnas.93.1.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David C, Solimena M, De Camilli P. Autoimmunity in stiff-man syndrome with breast cancer is targeted to the C-terminal region of human amphiphysin, a protein similar to the yeast proteins, Rvs167 and Rvs161. FEBS Lett. 1994;351:73–79. doi: 10.1016/0014-5793(94)00826-4. [DOI] [PubMed] [Google Scholar]
- Desfarges L, Durrens P, Juguelin H, Cassagne C, Bonneu M, Aigle M. Yeast mutants affected in viability upon starvation have a modified phospholipid composition. Yeast. 1993;9:267–277. doi: 10.1002/yea.320090306. [DOI] [PubMed] [Google Scholar]
- Donnelly SF, Pocklington MJ, Pallotta D, Orr E. A proline-rich protein, verprolin, involved in cytoskeletal organization and cellular growth in the yeast Saccharomyces cerevisiae. . Mol Microbiol. 1993;10:585–596. doi: 10.1111/j.1365-2958.1993.tb00930.x. [DOI] [PubMed] [Google Scholar]
- Dorer R, Boone C, Kimbrough T, Kim J, Hartwell LH. Genetic analysis of default mating behavior in Saccharomyces cerevisiae. . Genetics. 1997;146:39–55. doi: 10.1093/genetics/146.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulic V, Egerton M, Elguindi I, Raths S, Singer B, Riezman H. Yeast endocytosis assays. Methods Enzymol. 1991;194:697–710. doi: 10.1016/0076-6879(91)94051-d. [DOI] [PubMed] [Google Scholar]
- Durrens P, Revardel E, Bonneu M, Aigle M. Evidence for a branched pathway in the polarized cell division of Saccharomyces cerevisiae. . Curr Genet. 1995;27:213–216. doi: 10.1007/BF00326151. [DOI] [PubMed] [Google Scholar]
- Elia L, Marsh L. Role of the ABC transporter Ste6 in cell fusion during yeast conjugation. J Cell Biol. 1996;135:741–751. doi: 10.1083/jcb.135.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elion EA, Grisafi PL, Fink GR. FUS3 encodes a cdc2/CDC28- related kinase required for the transition from mitosis into conjugation. Cell. 1990;60:649–664. doi: 10.1016/0092-8674(90)90668-5. [DOI] [PubMed] [Google Scholar]
- Elion EA, Satterberg B, Kranz JE. FUS3 phosphorylates multiple components of the mating signal transduction cascade: evidence for STE12 and FAR1. Mol Biol Cell. 1993;4:495–510. doi: 10.1091/mbc.4.5.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elion EA, Trueheart J, Fink GR. Fus2 localizes near the site of cell fusion and is required for both cell fusion and nuclear alignment during zygote formation. J Cell Biol. 1995;130:1283–1296. doi: 10.1083/jcb.130.6.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdman S, Lin L, Malczynski MD, Snyder M. Pheromone-regulated genes required for yeast mating differentiation. J Cell Biol. 1998;140:461–483. doi: 10.1083/jcb.140.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford S, Pringle J. Development of spatial organization during the formation of zygotes and shmoos in Saccharomyces cerevisiae. . Yeast. 1986;2(Suppl.):114. [Google Scholar]
- Gammie AE, Kurihara LJ, Vallee RB, Rose MD. DNM1, a dynamin-related gene, participates in endosomal trafficking in yeast. J Cell Biol. 1995;130:553–566. doi: 10.1083/jcb.130.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammie, A.E., V. Brizzio, and M.D. Rose. 1998. Distinct morphological phenotypes of cell fusion mutants. Mol. Biol. Cell. In press. [DOI] [PMC free article] [PubMed]
- Grabs D, Slepnev VI, Songyang Z, David C, Lynch M, Cantley LC, De Camilli P. The SH3 domain of amphiphysin binds the proline-rich domain of dynamin at a single site that defines a new SH3 binding consensus sequence. J Biol Chem. 1997;272:13419–13425. doi: 10.1074/jbc.272.20.13419. [DOI] [PubMed] [Google Scholar]
- Hasek J, Rupes I, Svobodova J, Streiblova E. Tubulin and actin topology during zygote formation of Saccharomyces cerevisiae. . J Gen Microbiol. 1987;133:3355–3363. doi: 10.1099/00221287-133-12-3355. [DOI] [PubMed] [Google Scholar]
- Herskowitz I. MAP kinase pathways in yeast: for mating and more. Cell. 1995;80:187–197. doi: 10.1016/0092-8674(95)90402-6. [DOI] [PubMed] [Google Scholar]
- Holtzman DA, Yang S, Drubin DG. Synthetic-lethal interactions identify two novel genes, SLA1 and SLA2, that control membrane cytoskeleton assembly in Saccharomyces cerevisiae. J Cell Biol. 1993;122:635–644. doi: 10.1083/jcb.122.3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilmartin JV, Adams AE. Structural rearrangements of tubulin and actin during the cell cycle of the yeast Saccharomyces. J Cell Biol. 1984;98:922–933. doi: 10.1083/jcb.98.3.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka JB, Fields S. The pheromone signal pathway in Saccharomyces cerevisiae. . Antonie Leeuwenhoek. 1992;62:95–108. doi: 10.1007/BF00584465. [DOI] [PubMed] [Google Scholar]
- Kurihara LJ, Beh CT, Latterich M, Schekman R, Rose MD. Nuclear congression and membrane fusion: two distinct events in the yeast karyogamy pathway. J Cell Biol. 1994;126:911–923. doi: 10.1083/jcb.126.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara LJ, Stewart BG, Gammie AE, Rose MD. Kar4p, a karyogamy-specific component of the yeast pheromone response pathway. Mol Cell Biol. 1996;16:3990–4002. doi: 10.1128/mcb.16.8.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichte B, Veh RW, Meywer HE, Kiliman MW. Amphiphysin, a novel protein associated with synaptic vesicles. EMBO (Eur Mol Biol Organ) J. 1992;11:2521–2530. doi: 10.1002/j.1460-2075.1992.tb05317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipke PN, Kurjan J. Sexual agglutination in budding yeasts: structure, function, and regulation of adhesion glycoproteins. Microbiol Rev. 1992;56:180–194. doi: 10.1128/mr.56.1.180-194.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Bretscher A. Characterization of TPM1disrupted yeast cells indicates an involvement of tropomyosin in directed vesicular transport. J Cell Biol. 1992;118:285–299. doi: 10.1083/jcb.118.2.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh, L., and M.D. Rose. 1997. The pathway of cell and nuclear fusion during mating in S. cerevisiae. In The Molecular and Cellular Biology of the Yeast Saccharomyces: Cell Cycle and Cell Biology. J.R. Pringle, J.R. Broach, and E.W. Jones, editors. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 827–888.
- McCaffrey G, Clay FJ, Kelsay K, Sprague GF., Jr Identification and regulation of a gene required for cell fusion during mating of the yeast Saccharomyces cerevisiae. . Mol Cell Biol. 1987;7:2680–2690. doi: 10.1128/mcb.7.8.2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DA, Marshall TK, Deschenes RJ. Vectors for the inducible overexpression of glutathione S-transferase fusion proteins in yeast. Yeast. 1993;9:715–722. doi: 10.1002/yea.320090705. [DOI] [PubMed] [Google Scholar]
- Munn AL, Riezman H. Endocytosis is required for the growth of vacuolar H(+)-ATPase-defective yeast: identification of six new ENDgenes. J Cell Biol. 1994;127:373–386. doi: 10.1083/jcb.127.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn AL, Stevenson BJ, Geli MI, Riezman H. end5, end6, and end7: mutations that cause actin delocalization and block the internalization step of endocytosis in Saccharomyces cerevisiae. . Mol Biol Cell. 1995;6:1721–1742. doi: 10.1091/mbc.6.12.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi A, Gibson J, Gregor I, Schatz G. Import of proteins into mitochondria. The precursor of cytochrome c1 is processed in two steps, one of them heme-dependent. J Biol Chem. 1982;257:13042–13047. [PubMed] [Google Scholar]
- Osumi M, Shimoda C, Yanagishima N. Mating reaction in Saccharomyces cerevisiae. V. Changes in the fine structure during the mating reaction. Arch Microbiol. 1974;97:27–38. [PubMed] [Google Scholar]
- Philips J, Herskowitz I. Osmotic balance regulates cell fusion during mating in Saccharomyces cerevisiae. . J Cell Biol. 1997;138:961–974. doi: 10.1083/jcb.138.5.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle JR, Adams AE, Drubin DG, Haarer BK. Immunofluorescence methods for yeast. Methods Enzymol. 1991;194:565–602. doi: 10.1016/0076-6879(91)94043-c. [DOI] [PubMed] [Google Scholar]
- Raths S, Rohrer J, Crausaz F, Riezman H. end3 and end4: two mutants defective in receptor-mediated and fluid-phase endocytosis in Saccharomyces cerevisiae. . J Cell Biol. 1993;120:55–65. doi: 10.1083/jcb.120.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read EB, Okamura HH, Drubin DG. Actin- and tubulin-dependent functions during Saccharomyces cerevisiaemating projection formation. Mol Biol Cell. 1992;3:429–444. doi: 10.1091/mbc.3.4.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revardel E, Bonneau M, Durrens P, Aigle M. Characterization of a new gene family developing pleiotropic phenotypes upon mutation in Saccharomyces cerevisiae. . Biochim Biophys Acta. 1995;1263:261–265. doi: 10.1016/0167-4781(95)00124-y. [DOI] [PubMed] [Google Scholar]
- Riezman H, Munn A, Geli MI, Hicke L. Actin-, myosin- and ubiquitin-dependent endocytosis. Experientia. 1996;52:1033–1041. doi: 10.1007/BF01952099. [DOI] [PubMed] [Google Scholar]
- Rose MD. Nuclear fusion in yeast. Annu Rev Microbiol. 1991;45:539–567. doi: 10.1146/annurev.mi.45.100191.002543. [DOI] [PubMed] [Google Scholar]
- Rose MD. Nuclear fusion in the yeast, Saccharomyces cerevisiae. . Annu Rev Cell Dev Biol. 1996;12:663–693. doi: 10.1146/annurev.cellbio.12.1.663. [DOI] [PubMed] [Google Scholar]
- Rose MD, Novick P, Thomas JH, Botstein D, Fink GR. A Saccharomyces cerevisiaegenomic plasmid bank based on a centromere-containing shuttle vector. Gene. 1987;60:237–243. doi: 10.1016/0378-1119(87)90232-0. [DOI] [PubMed] [Google Scholar]
- Rose, M.D., F. Winston, and P. Hieter. 1990. Methods of Yeast Genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 198.
- Rothstein R. Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol. 1991;194:281–301. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- Santos B, Duran A, Valdivieso MH. CHS5, a gene involved in chitin synthesis and mating in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:2485–2496. doi: 10.1128/mcb.17.5.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer S, Davis RW. Replacement of chromosome segments with altered DNA sequences constructed in vitro. Proc Natl Acad Sci USA. 1979;76:4951–4955. doi: 10.1073/pnas.76.10.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scidmore, M. 1993. A genetic analysis of KAR2, the yeast homologue of mammalian BiP. Ph.D. thesis. Department of Molecular Biology. Princeton University, Princeton, NJ. 255.
- Shupliakov O, Low P, Grabs D, Gad H, Chen H, David C, Takei K, De Camilli P, Brodin L. Synaptic vesicle endocytosis impaired by disruption of dynamin-SH3 domain interactions. Science. 1997;276:259–263. doi: 10.1126/science.276.5310.259. [DOI] [PubMed] [Google Scholar]
- Sikorski R, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivadon P, Bauer F, Aigle M, Crouzet M. Actin cytoskeleton and budding pattern are altered in the yeast rvs161mutant: the Rvs161 protein shares common domains with the brain protein amphiphysin. Mol Gen Genet. 1995;246:485–495. doi: 10.1007/BF00290452. [DOI] [PubMed] [Google Scholar]
- Sivadon P, Crouzet M, Aigle M. Functional assessment of the yeast Rvs161 and Rvs167 protein domains. FEBS Lett. 1997a;417:21–27. doi: 10.1016/s0014-5793(97)01248-9. [DOI] [PubMed] [Google Scholar]
- Sivadon P, Peypouquet MF, Doignon F, Aigle M, Crouzet M. Cloning of the multicopy suppressor gene SUR7: evidence for a functional relationship between the yeast actin-binding protein Rvs167 and a putative membranous protein. Yeast. 1997b;13:747–761. doi: 10.1002/(SICI)1097-0061(19970630)13:8<747::AID-YEA137>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Sprague, G.F., and J.W. Thorner. 1992. Pheromone response and signal transduction during the mating process of Saccharomyces cerevisiae. In The Molecular and Cellular Biology of the Yeast Saccharomyces: Gene Expression. Vol. 2. E.W. Jones, J.R. Pringle, and J.R. Broach, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 657–744.
- Tang HY, Munn A, Cai M. EH domain proteins Pan1p and End3p are components of a complex that plays a dual role in organization of the cortical actin cytoskeleton and endocytosis in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:4294–4304. doi: 10.1128/mcb.17.8.4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trueheart J, Boeke JD, Fink GR. Two genes required for cell fusion during yeast conjugation: evidence for a pheromone-induced surface protein. Mol Cell Biol. 1987;7:2316–2328. doi: 10.1128/mcb.7.7.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trueheart J, Fink GR. The yeast cell fusion protein FUS1 is O-glycosylated and spans the plasma membrane. Proc Natl Acad Sci USA. 1989;86:9916–9920. doi: 10.1073/pnas.86.24.9916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoladek T, Vaduva G, Hunter LA, Boguta M, Go BD, Martin NC, Hopper AK. Mutations altering the mitochondrial-cytoplasmic distribution of Mod5p implicate the actin cytoskeleton and mRNA 3′ ends and/or protein synthesis in mitochondrial delivery. Mol Cell Biol. 1995;15:6884–6894. doi: 10.1128/mcb.15.12.6884. [DOI] [PMC free article] [PubMed] [Google Scholar]