

Abstract
The characterization of scully, an essential gene of Drosophila with phenocritical phases at embryonic and pupal stages, shows its extensive homology with vertebrate type II l-3-hydroxyacyl-CoA dehydrogenase/ERAB. Genomic rescue demonstrates that four different lethal mutations are scu alleles, the molecular nature of which has been established. One of them, scu3127, generates a nonfunctional truncated product. scu4058 also produces a truncated protein, but it contains most of the known functional domains of the enzyme. The other two mutations, scu174 and scuS152, correspond to single amino acid changes. The expression of scully mRNA is general to many tissues including the CNS; however, it is highest in both embryonic gonadal primordia and mature ovaries and testes. Consistent with this pattern, the phenotypic analysis suggests a role for scully in germ line formation: mutant testis are reduced in size and devoid of maturing sperm, and mutant ovarioles are not able to produce viable eggs. Ultrastructural analysis of mutant spermatocytes reveals the presence of cytoplasmic lipid inclusions and scarce mitochondria. In addition, mutant photoreceptors contain morphologically aberrant mitochondria and large multilayered accumulations of membranous material. Some of these phenotypes are very similar to those present in human pathologies caused by β-oxidation disorders.
Although energy storage and metabolism have been well-studied in Drosophila (Clark, 1989), the enzymes implicated in fatty acid oxidation have not been characterized, and the phenotypes associated with genetic alterations in this metabolic pathway have not been described. β-oxidation is a major metabolic process by which fatty acids are oxidized to provide a significant source of energy, while also generating acetyl-CoA, a metabolite that is located at the crossroads of many metabolic routes. In mammals, hepatic β-oxidation provides circulating ketone bodies. These ketone bodies are a very important fuel for other organs—especially the brain—when blood glucose levels are low, for example, during long-lasting exercise or starvation. By contrast, in muscles, β-oxidation is almost exclusively used to obtain energy from complete oxidation of acetyl-CoA. In animal cells, both mitochondria and peroxisomes are the subcellular organelles where β-oxidation takes place (reviewed by Mannaerts and Van Veldhoven, 1996; Eaton et al., 1996), but the mitochondrion is the main site of energy production. As a secondary product of mitochondrial aerobic respiration, reactive oxygen species are generated (Boveris et al., 1973). Also, mitochondria are important storage sites for intracellular calcium, and are necessary for intracellular calcium buffering (Gunter et al., 1994). Currently, mitochondria are considered a triggering factor in the onset of many neurodegenerative diseases (Beal et al., 1993; Sims, 1996).
During one passage through the β-oxidation pathway, saturated fatty acids with an even number of carbon atoms release a pair of carbon residues. This release is achieved by four consecutive reactions successively catalyzed by acyl-CoA dehydrogenase, enoyl-CoA hydratase, 3-hydroxyacyl-CoA dehydrogenase (HADH),1 and 3-ketoacyl-CoA thiolase. Over the last years, it has become clear that β-oxidation pathway enzymes consist of specificity groups of isoenzymes that catalyze the same reaction, but differ in their affinity for carbon chain length of the various substrates. Complexity of this metabolic pathway is further increased by tissue-specific isoenzymes. In mitochondria, the third step of the pathway was known to be catalyzed by two HADHs with overlapping substrate chain-length specificities. Long-chain HADH is a trifunctional protein that catalyzes the last three steps of β oxidation. It is tightly associated with the inner mitochondrial membrane, and is active with medium and long chain–length substrates (El-Fakhri and Middleton, 1982). In contrast, short-chain HADH is a monofunctional soluble enzyme located in the mitochondrial matrix that preferentially metabolizes short chain–length substrates (He et al., 1989). However, a new type of HADH has been recently characterized (Kobayashi et al., 1996) and cloned (Furuta et al., 1997) from bovine liver. Termed type II short chain HADH, it differs from the classical isozyme (type I) in its primary structure, and also in its molecular and catalytic properties. It is clear now that the β-oxidation pathway conceals a more elaborate specificity than previously thought.
Primary defects in mitochondrial function are implicated in a growing number of human diseases (Luft, 1994; Roe and Coates, 1995). Manifestation of these diseases are thought to result from oxidative stress derived from energy imbalance. Oxidative stress, perhaps partly glutamate-mediated, has also been implicated in some age- related neurodegenerative diseases such as Parkinson, Alzheimer, and Huntington diseases, and amyotrophic lateral sclerosis (Beal et al., 1993; Coyle and Puttfarcken, 1993). In several inherited enzymopathies of the mitochondrial fatty acid β-oxidation pathway (reviewed in Roe and Coates, 1995), the affected enzymatic activity remains unknown, partly as consequence of the emerging complexity of the enzymatic repertoire. Studies of patients with these genetic disorders suggest that mitochondrial β-oxidation may be essential only during periods of high energy demand such as fasting, febrile illness, or muscular exertion. In addition, the levels of some of the β-oxidation mitochondrial enzymes have been shown to increase only after birth (Lopaschuk et al., 1992; Hainline et al., 1993). During prenatal development, β-oxidation seems to represent a minor energy source, and thus the role of the β-oxidation enzymes in this period is not well understood.
In this study, we report the molecular characterization of a Drosophila gene, scully (scu), that encodes a protein with high structural homology to type II HADH. This is the first enzyme related to β-oxidation that has been reported in Drosophila. Recently, the human homologue ERAB has been shown to bind the amyloid-β peptide, and has consequently been related with Alzheimer's disease neuronal dysfunction (Yan et al., 1997). We also report the phenotypes associated with different lethal alleles of this gene. The mitochondrial phenotypes presented here suggest an identical localization for this new protein with that of its vertebrate counterparts. Striking similarities between the cellular phenotypes of scu mutants and the human pathologies associated with alterations in this metabolic pathway are discussed.
Materials and Methods
Fly Culture and Strains
Flies were grown and collected under standard conditions (Ashburner, 1989). We used Canton-S (CS) as the wild-type stock. Lethal mutations scu174 and scu4058 were induced by ethyl methanesulphonate (EMS) and x ray, respectively, on a f 5 os-marked chromosome (Ferrús et al., 1990). scu3127 was induced by EMS on a y w hdp3-marked chromosome. scuS152 was generated by EMS mutagenesis, and was kindly provided by Dr. N. Perrimon (Department of Genetics, Harvard Medical School, Boston, MA). All mutations were kept either balanced with chromosome FM6 or covered by duplication Dp(1,3)JC153 (stock was C[1]M3/scu*;Dp[1;3]JC153/TM3).
Biology of Mutants: Mosaics and Lethal Phase Analysis
To determine the lethality phase, scu*/+ females were crossed to CS males. We allowed fertilized females to lay eggs for a 24-h period at 25°C. Groups of 20–30 eggs were examined at 1-d intervals. For generating twin clones, progeny from the cross: y w scu*os/FM6 × f 5 os was x ray–treated 0–48 h after egg laying. The presence of somatic clones was monitored in the cuticle of adult y w scu*os/f 5 os F1-females. A twin clone is composed of two adjacent patches generated in the same event of recombination: one marked with forked and the other with yellow, where the yellow clone is homozygous for the lethal mutation scu*. Single forked clones were considered internal controls. Eye mosaics were generated in y w hdp3 scu3127/M(1)n flies that had been x ray–treated between 48 and 72 h of development.
Isolation and Molecular Cloning of cDNA and Genomic Clones
An adult MATCHMAKER cDNA library from Drosophila melanogaster (CLONTECH Laboratories, Inc., Palo Alto, CA) was screened at high stringency with the 422E1 genomic fragment (Fig. 1). Recombinant DNA manipulations were performed using standard procedures (Sambrook et al., 1989). All purified cDNA positives were sequenced by the dideoxy chain termination method using a DNA sequencer (PE Applied Biosystems, Foster City, CA). DNA sequence data were analyzed using DNAstar software (DNASTAR Inc., Madison, WI), and comparisons were done using the BLASTX program (Gish et al., 1993; Altschul et al., 1990). The complete genomic sequence of scully gene has been submitted to the DDBJ/ EMBL/GenBank databases under accession No. Y15102.
Figure 1.

Molecular map of scully transcription unit. scully is located on the X chromosome 50 kb proximal to the Shaker gene (Baumann et al., 1987) and 14 kb distal to the Troponin I gene (Barbas et al., 1991). Lethality rescue was analyzed in transformants using the genomic EcoRI-fragment (2,4 kb) shown, termed 422E1. It contains the entire transcription unit, including the promoter region. Relevant nucleotide signal sequences are indicated (see text). Centromere is to the right.
Genomic Rescue
Genomic clones were previously obtained (described in Baumann et al., 1987). EcoRI fragments were subcloned into pBluescript KS+ vector (Stratagene, La Jolla, CA). For genomic rescue, EcoRI fragment 422E1 was inserted into the germ line transformation vector pCasper (Pirrotta, 1988). P element–mediated germline transformation was performed according to described methods (Spradling and Rubin, 1982). Df(1)w flies were the parental strain for all germline transformations.
Northern Blot Analysis
Poly(A)+ mRNA was isolated from different developmental stages of Drosophila melanogaster CS strain using a QuickPrep Micro™ mRNA purification kit (Pharmacia Biotech, Inc., Piscataway, NJ). The same amount of mRNA (5 μg) for each sample was loaded on 1.2% agarose formaldehyde gels and transferred onto nylon membranes (Nycomed Amersham Inc., Princeton, NJ). The hybridization was in 50% formamide, 6× SSC, 10× Denhardt's solution, and 100 μg/ml herring sperm DNA using 2 × 106 cpm/ml of random priming labeled probe (Feinberg and Vogelstein, 1983). Finally, the filter was washed in 0.2× SSC, 0.1% SDS at 65°C. Exposure times ranged from several hours to 3 d. Loading and transfer efficiency were monitored by staining the ribosomal RNA on the nylon membrane with 0.05% methylene blue in 0.5 M sodium salicylate (pH 5.2). Usually, filters were further hybridized with Dras-1 probe to quantify mRNA content in each lane (Neuman-Silberberg et al., 1984). Transcript sizes were estimated using mRNA markers (Boehringer Mannheim Corp., Indianapolis, IN).
In Situ Hybridization
For in situ hybridization, we used a probe corresponding to the longest cDNA. cRNA was labeled with digoxigenin and hybridized to 12-μm paraffin sections according to Schaeren-Wiemers and Gerfin-Moser (1993) with the following modification. Tissues were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.5) for 24 h at 4°C. After embedding and cutting, paraffin sections were deparaffinized in xylene, rehydrated through a series of ethanol/PBS solutions, permeabelized in 1% Tween for 30 min, and postfixed. Sections were prehybridized in 50% formamide, 1.3× SSC (pH 4.5), 0.5% CHAPS, 0.2% Tween, 100 μg/ml heparine, 50 μg/ml t-RNA, and 5 mM EDTA for 2 h at 65°C. Overnight hybridization was performed at the same temperature and in the same solution with the probe at a concentration of 500 ng/ml. Sections were then extensively washed in 0.2× SSC at 65°C, passed to 0.1 M Tris-HCl, pH 7.5, 0.15 M NaCl, and blocked with the addition of 10% normal goat serum and 0.1% Tween. After incubating with anti-DIG-alkaline phosphatase (1:1,000; Boehringer Mannheim) and washing several times in 100 mM maleic acid, 150 mM NaCl, 2 mM levamisole (Sigma Chemical Co., St. Louis, MO) at pH 7.5, alkaline phosphatase activity was developed with NBT/BCIP in 0.1 M Tris-HCl pH 9.5, 0.1 M NaCl, 50 mM MgCl2. Reaction was stopped in 1 mM EDTA.
Histological Procedures
Testes of white-to-light brown pupae (0–12 h after puparium formation) of CS or scully mutants and adult mosaic eyes were dissected and prefixed for 1 h in an ice-cold solution of 4% paraformaldehyde plus 1% glutaraldehyde in 0.1 M phosphate buffer, pH 7.2. After rinsing the specimens in phosphate buffer, postfixation was performed in 2% OsO4 for 90 min in the dark. Samples were dehydrated in an ascending alcohol series and embedded in SPURR resin. Semithin sections (2 mm) were stained with Toluidine blue. Ultrathin sections (60–70 nm) were contrasted with uranyl acetate and lead citrate, and were examined using an electron microscope (JEOL USA, Inc., Peabody, MA). For lipid staining, testes were dissected in 5 mM Hepes pH 7.4 containing 115 mM sucrose, 100 mM NaCl, 5 mM KCl, 20 mM MgCl2, and 0.15 mM CaCl2, fixed in 4% formaldehyde for 1 h, cryoprotected with 30% sucrose in 0.1 M phosphate buffer, pH 7.4, and embedded in Tissue Tek™ (Salavra, The Netherlands). 10-μm cryostat sections were stained with red oil O solution following a described protocol (Humason, 1972).
Enzymatic Assay
Testes were dissected as above and homogenized in 50 mM Tris-HCl, pH 7.4. Crude extracts were assayed for the HADH reverse reaction as described in Osumi and Hashimoto (1979) using acetoacetyl-CoA as substrate. One enzyme unit corresponds to the amount of enzyme protein that catalyzes the oxidation of 1 μM NADH/min. Reaction specificity was monitored, omitting the substrate in a control assay. Protein content in extracts was quantified using the Protein Assay™ reagent (Bio-Rad Laboratories, Hercules, CA), and equal amounts of protein was used for comparison of HADH activity in different extracts.
PCR Analysis of the Mutants
To find the molecular alterations of scu mutants, both larval genomic DNA and cDNA were PCR-amplified under standard conditions (Innis and Gelfand, 1990). The oligonucleotides used to amplify genomic DNA were: 5′-ccggatcctcactgtttgcctgctc-3′ (sense) and 5′-ccgaattccccgccagctgccctac-3′ (antisense). To amplify cDNA, we used the primers: 5′-caggatcctttcgcacccacaaca-3′ (sense) and 5′-ccgaattcgagccacttccacagg-3′ (antisense). The products of two independent amplifications were cloned in pBluescript and sequenced in each case to confirm any possible alteration found.
Results
Genetic Mapping of scully
The gene scully (scu) is defined as the complementation group composed by the lethal mutations scu174, scu4058, scu3127, and scuS152. These alleles have been mapped by recombination and segmental aneuploidies to the interval between the Shaker (Sh; Baumann et al., 1987) and Troponin I (TnI; Barbas et al., 1991) genes in the 16F region of the X chromosome. The genomic DNA between the Sh and TnI transcription units extends over 65 kb, and has been previously cloned (Baumann et al., 1987). Six independent transcripts have been identified in this interval by Northern blot analysis using different genomic probes (not shown). To identify the genomic region containing the scu gene, we generated transgenic flies carrying different genomic fragments, and tested their ability to rescue scu lethal mutations (see Materials and Methods). The smallest fragment used, 422E1, spans 2.4 kb (Fig. 1) and completely rescued the male lethality of the four alleles, but it did not rescue any other complementation group present in the area (data not shown). Three independent insertions of this fragment were tested in the rescue experiments. These results demonstrate that the scu gene is fully contained within these 2.4 kb.
Molecular Characterization of the scully Transcription Unit
Northern blot analysis of this transcription unit using the genomic fragment 422E1 as a probe revealed a single 1.0-kb transcript. Screening of an adult cDNA library with the same fragment rendered four cDNA clones whose sequences were compared with that of the genomic counterpart, and provided the gene structure represented in Fig. 1. The transcription unit is composed of two exons separated by a 551-bp intervening sequence. The intron/exon borders precisely match consensus splice sequences (Mount et al., 1992). The four cDNAs start at different but nearby sites, suggesting that initiation of transcription occurs close to this region. A GCAGT sequence close to the first nucleotide of the longest cDNA and preceded by an AT-rich zone is a good candidate for a functional arthropod transcription initiator (Cherbas and Cherbas, 1993). Furthermore, 20–40 nucleotides downstream of position +1 there are three sequences—TCGA, AACA and ACAA—that have been reported as downstream elements important for initiation of transcription (Arkhipova, 1995). There is also a TATA box–like sequence in the upstream genomic region. Finally, three of the cDNAs are polyadenylated at the same site 18 nucleotides downstream of the polyadenylation signal AUUAAA (Wickens and Stephenson, 1984). Thus, our analysis predicts a full-length mRNA of 1026 nucleotides in agreement with the size of the band detected by Northern blot analysis.
scully Encodes a Short Chain l-3-hydroxyacyl-CoA Dehydrogenase
All four cDNAs comprise a complete and identical open reading frame of 765 nucleotides. The start codon 118ATG matches the consensus sequence described for Drosophila (Cavener and Ray, 1991). Translation of the cDNA predicted a 255–amino acid protein with an estimated Mr of 27 kg/mol. A PROSITE pattern search of this protein sequence detected the consensus motif for short-chain alcohol dehydrogenase between amino acids 149 and 177. In agreement, a basic local alignment search tool search revealed a high degree of homology with proteins of the short-chain dehydrogenase/reductase (SDR) family, identifying the protein Scully as a new member. Highest homology scores revealed a bovine protein (AB002156), biochemically characterized as a new type of HADH (Furuta et al., 1997), and its human counterpart (U73514; Zhuchenko et al., unpublished data). The last sequence is identical to human ERAB (U96132), a protein that binds amyloid-β peptide (ERAB) and mediates neurotoxicity in Alzheimer's disease (Yan et al., 1997). Fig. 2 shows the alignment of Scully with the consensus sequence derived from its three mammalian homologues. Based on the high degree of identity between Scully and this consensus (61% along the whole sequence), we propose that this new protein is the Drosophila homologue of the enzyme l-3-hydroxyacyl-CoA dehydrogenase type II, a new type of monofunctional enzyme involved in the mitochondrial β-oxidation of fatty acids. Scully and its homologues seem to define a new subfamily in the SDR family of proteins.
Figure 2.
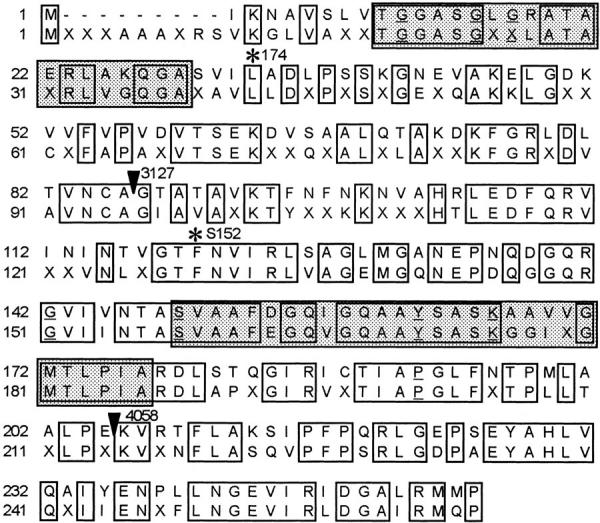
Scully and its mammalian homologues. Alignment of Scully (upper row) and the mammalian consensus derived from: Bos taurus type II l-3-hydroxyacyl-CoA dehydrogenase (AB002156; Furuta et al., 1997); Homo sapiens ERAB (U96132; Yan et al., 1997) and U73514 (Zhuchenko et al., unpublished data); and Mus musculus ERAB (U96116; Fu et al., unpublished data). Functional domains are boxed and shaded; the first is the cofactor-binding domain and the second is the catalytic domain. Single amino acid mutations scu174 and scuS152 are indicated with asterisks. Frameshift mutations scu3127 and scu4058 are indicated with arrowheads. Amino acids conserved in more than 90% of the short-chain alcohol dehydrogenases sequences (Jörnvall et al., 1995) are underlined. The complete genomic sequence of scully gene has been submitted to the DDBJ/EMBL/GenBank databases under accession No. Y15102.
Molecular Characterization of scully Mutations
Three alleles—scu174, scu3127, and scuS152—were induced by EMS mutagenesis; scu4058 was generated by x rays. Southern analysis detected an alteration only in scu3127. Sequencing of at least two independent PCR amplifications of genomic DNA and cDNA from male third instar mutant larvae identified the molecular alteration in each of the four scu alleles (Fig. 2). scu3127 is a 254-bp deletion within exon 2. The putative altered transcript encodes a truncated peptide that contains the first 86 amino acids followed by seven out-of-frame residues and a premature stop codon. The other three mutations are point mutations. In scu174 the codon change 1136CTG→ CAG results in the amino acid substitution L33Q, affecting a residue that is strictly conserved in all Scully homologues. In scuS152, the mutation 1397TTC → ATC produces the amino acid substitution F120I in another well-conserved residue. scu4058 is a deletion of two nucleotides (1651GG) that causes a frame shift at E205 and generates a stop codon 60 nucleotides farther downstream. Note that in scu4058 the truncated putative protein has both the catalytic center and the site for cofactor binding, but lacks the carboxy terminal region that is supposed to confer substrate specificity (see Discussion).
Developmental and Tissue Expression of scully
Expression of scu transcripts was analyzed by Northern blot and in situ hybridization to paraffin sections. Northern analysis detected a single 1.0-kb band at all developmental stages studied that displayed developmental quantitative differences. mRNA was low in unfertilized oocytes, and increased during embryonic development (Fig. 3 a). In adults, expression was higher in females than in males (Fig. 3 a). mRNA in situ hybridization to late embryos showed a very intense signal in the gonadal primordium (Fig. 3 b). The high expression in gonads persisted through all larval stages, during which other tissues, such as third instar larvae imaginal discs, central nervous system, and salivary glands also showed scu expression (Fig. 3 c). A weaker signal is also observed in the gut epithelial cells and malphigian tubules (not shown). In adults, the transcript was especially abundant in female abdomen where signal appeared associated with nurse cells in the ovaries (Fig. 3 d). In males, testes also maintained high levels of mRNA (not shown).
Figure 3.

scully expression pattern. (a) Northern blot showing the 1.0-kb scu mRNA band detected with the genomic probe 422E1 (top). Total mRNA per lane was controlled by subsequent hybridization with a probe for d-ras 1 mRNA. In situ hybridization to sections of (b) late embryo; (c) third instar larva; and (d) adult female. u, unfertilized eggs; e, 0–48 h embryos; (female symbol) adult females; (male symbol) adult males; gp, gonadal primordia; cns, central nervous system; sg, salivary gland; id, imaginal discs; nc, nurse cells. (b–d) Anterior is to the right, and dorsal is up.
scully Mutant Phenotypes
The developmental effects of scu mutations were analyzed in different cell types in either entirely mutant or mosaic flies. Analysis of the lethal phases revealed interallelic differences (Table I). These differences were studied in detail for three alleles: scu174, scu4058, and scu3127. scu174, which encodes a protein with a single amino acid substitution, and scu4058, which contains a small carboxy-terminal deletion, exhibit an early lethal phase during the embryo stage, followed by a major period of lethality around the onset of metamorphosis. Interestingly, scu3127, which encodes a protein truncated at its amino terminus, is completely viable during embryonic development, and becomes lethal only when larvae reach the late third instar. The molecular alteration of scu3127 indicates that this is a functionally null allele, and mutant survival during embryonic and larval stages must result from the maternal normal product accumulated during the oogenesis. By contrast, scu174 and scu4058 behave as dominant negative (antimorphic) alleles; they show earlier lethality that cannot be completely rescued by maternal protein.
Table I.
Lethal Phase of scu Alleles
| Allele | N | % D | % E | % LIII | % LP | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| scu174 | 77 | 35 | 20 | — | 80 | |||||
| scu4058 | 150 | 26 | 25 | — | 75 | |||||
| scu3127 | 113 | 24 | — | 75 | 25 |
Female scu
/+ were crossed to CS males, and the F1 was analyzed. N, number of analyzed individuals; D, dead individuals; E, embryos; LIII, third instar larvae; LP, late pupae. Percentages of lethality at each developmental stage refer the total number of dead individuals.
We analyzed the in vivo effects of scully mutations in mosaic adult flies with patches of mutant cells. Previously it was reported that recovery of gynandromorphs for scu174 was extremely rare (10%), and male/mutant territories were small and with few and short bristles (Ferrús et al., 1990). Small somatic mosaics were produced in the cuticle of individuals heterozygous for scu4058 or scu3127. Bristle markers allowed to recognize the wild-type (+/+) and homozygous mutant (scu*/scu*) areas derived from a single heterozygotic cell (twin mosaic analysis; see Materials and Methods). Table II shows that the number of mutant clones obtained was reduced with respect to the wild-type controls in both alleles tested. This result indicates that scully activity is required for cell survival, and that this requirement is cell-autonomous. As in the previous trait, there are differences between the two alleles. scu4058 was incapable of generating mutant clones with more than one cell, and, similar to scu174, the bristles were short and thin. scu3127, however, was able to generate larger clones of normal morphology. Thus, the null mutation (e.g., scu3127) also showed the mildest phenotype in mosaic analysis.
Table II.
Twin Mosaic Analysis of scu Alleles
| Allele | N | single bristle | several bristles | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| f | y | f | y | |||||||
| scu4058 | 328 | 43 | 9 | 14 | 0 | |||||
| scu3127 | 315 | 17 | 4 | 20 | 6 | |||||
The thoracic cuticle of females y w scu
os/f os, that had been irradiated between 0 and 48 h of development (see Materials and Methods), was examined in search for somatic recombination clones. Adjacent yellow (y) and forked (f) clones represent the derivatives of a single recombination event: the y patch is homozygous for scu
* mutation; the f patch is the scu+ control. Isolated f clones indicate that the scu mutant cells have not been able to grow and form the adult structures. Note the severe reduction in the number of mutant clones obtained with respect to the twin control. N, number of females analyzed.
To analyze the effects of scully mutations on cell morphology, we attempted to obtain larger patches of mutant cells in the form of Minute+ clones (see Materials and Methods). Eye mosaics of the allele scu3127 were recovered at a frequency three times lower than that of controls. Photoreceptor cells did not differentiate normally, and ommatidia were not recognizable in the mutant territory (Fig. 4 a). Ultrastructural analysis of these mosaics revealed putative photoreceptor cells that were unable to form a proper rhabdomere, and usually contained large multilayered accumulations of membranous material (Fig. 4 c). Particularly interesting was the abnormality detected in the mitochondria. They appeared smaller and with fewer and swollen crestae when compared with neighboring wild-type cells. This mitochondrial phenotype (pleomorphism) is a typical pathology in humans with inherited β oxidation deficiencies (reviewed in Roe and Coates, 1995).
Figure 4.

scully mutant phenotypes in eye mosaics. (a) electron photomicrograph showing a tangential section of a mosaic eye at the border of mutant (left) and wild-type territories. Note the normal ommatidia (om) showing the normal complement of seven photoreceptors in the wild-type region, while this number is altered in other ommatidia near the border with the mutant area. In the mutant territory the normal organization of ommatidia, and even the normal morphology of photoreceptors, is lost. (b) Higher magnification of normal photoreceptor; and (c) higher magnification of mutant putative photoreceptor. Note the morphological abnormalities of mitochondria (mt) and the multilayered membrane bodies (mm) in the mutant cell. rb, rhabdomere. (a) Bar, 5 μm; (b and c) bar, 250 nm.
We also examined the morphological phenotypes in scu mutant males before onset of lethality. In third instar larvae, we observed a clear reduction of the salivary glands (not shown). Most striking, however, was the dramatic reduction of testes size in all four scu alleles. The degree of reduction varied in the following order: scu174 > scu4058 > scuS152 > scu3127. This striking phenotype in testes prompted a functional assay for HADH activity in these organs. We analyzed crude extracts from wild-type and mutant-dissected male gonads, and we detected up to 10−2 U/mg of total protein (see Materials and Methods) in the wild-type extracts, while scu3127 yielded no detectable activity. This observation is further evidence that scully encodes a HADH activity, and that testes have a specific requirement for this enzyme. To further study the morphological features of the mutant phenotype, we analyzed the ultrastructure of testes from either mutant or rescued early pupae. In wild-type pupal testes when early stages of spermatogenesis are occurring, spermatocytes are organized in cysts of 16 cells derived from four mitotic divisions (Fuller, 1993). However, in all scu mutants, spermatocytes were generally organized in smaller groups of ∼2–6 cells. Normally, the first cohort of developing spermatocytes enters meiosis at about the white prepupal stage, and forms sperm bundles. Except for scu3127, which contained few sperm bundles (Fig. 5 a), we did not detect any bundle of maturing sperm in any of the other scu alleles, even at the later brown pupal stage. Degenerating spermatocytes are only occasionally observed in normal testes, but we detected numerous degenerating necrotic cells in scu174 (Fig. 5 c). However, the most remarkable phenotype at the light microscope was the high amount of clear cytoplasmic vesicles observed in mutant testes cells (Fig. 5, a and c). The lipid nature of these inclusions was demonstrated after oil red staining of cryostat sections (Fig. 5 d). The large accumulation of small fat-containing vesicles in the cytoplasm of mutant spermatocytes was more clearly observed at the electron microscope (Fig. 6). At this magnification, the normal eccentric position of nuclei in primary spermatocytes was conserved in scu3127 mutants, but the nucleoli of mutant cells were not as compact as in wild-type cells, and had many cavities. Moreover, the cytoplasm of the spermatocytes normally has a large number of mitochondria, whereas this number was dramatically reduced in all scu mutants (Fig. 6 a). Rescued scu3127; T(2)422E1/T(2)422E1 displayed wild-type characteristics. Most strikingly, mitochondria were abundant, and steatosis was absent (Figs. 5 b and 6 b).
Figure 5.

scully mutant phenotypes in testes. Semithin sections of early brown pupal testis from (a) scu3127: note that only few sperm bundles (sb) and abundant cytoplasmic clear inclusions are observed; (b) rescued scu3127: note that T(2)422E1/T(2) 422E1 displays wild-type morphology and size (see below the different magnification of sections in a–c); and (c) scu174: note that it is severely affected and shows several necrotic cells (nc) and also numerous lipid inclusions. (d) cryostat section of scu3127 testes stained with oil red O, showing the lipid composition of the cytoplasmic inclusions. (a and c) Bar, 40 μm; (b) bar, 70 μm; (d) bar, 15 μm.
Figure 6.

Ultrastructure of scu spermatocytes. Electron photomicrographs of spermatocytes sections from (a) scu3127; and (b) rescued scu3127; T(2)422E1/T(2)422E1. Note the abundance of lipid inclusions (li) and the lack of mitochondria (mt) in the mutant spermatocyte. Bar, 2 μm.
By mosaic analysis, scu174 has been shown to be lethal in the female germ line (Ferrús et al., 1990). This together with the high level of scully expression in the nurse cells of the ovaries, indicates a requirement for this protein during oogenesis.
Discussion
scully as a New Member of the Type II HADH Family
In this study we present the molecular and phenotypical characterization of a Drosophila gene named scully. The high degree of structural homology between the encoded product and the biochemically characterized bovine mitochondrial 3-l-hydroxyacyl-CoA dehydrogenase type II (type II HADH) strongly suggests a functional conservation between these two proteins. The involvement of Scully in β-oxidation of fatty acids is further supported by the absence of HADH activity in mutant testes extracts and the steatosis trait of the mutant phenotype. Other sequences of still uncharacterized function in human, mouse, and Caenorhabditis elegans show strong identity with both Scully and its bovine homologue, indicating that they may be counterparts of this enzyme. The human protein ERAB has been related with Alzheimer's disease, although its pathological mechanism remains unknown (Yan et al., 1997). The primary sequence of Scully and its homologues identifies these proteins as members of the SDR family. This family includes a growing number of proteins from prokaryotes to mammals, with diverse substrate specificity but with conserved tertiary structure (reviewed in Krozowski, 1994 and Jörnvall et al., 1995). Two other known members of the SDR family in Drosophila melanogaster are encoded by the Adh and the Fbp2 genes. While these genes are closely related to one another, scully displays low homology with them, indicating a very distant evolutionary relationship.
scu Mutations and Their Structural Implications
The amino-terminal 16 amino acids of bovine type II HADH has been proposed as a noncleaved signal that targets the enzyme into mitochondria (Furuta et al., 1997) by a mechanism similar to that used by another enzyme of mitochondrial β-oxidation, the short-chain 3-oxoacyl-CoA thiolase (Arakawa et al., 1990, Amaya et al., 1988). Comparison of the amino-terminal sequence of these proteins reveals only a loose consensus, but a high number of hydrophobic and basic amino acids and a lack of acidic residues are in common. Although the putative import signal of Scully is significantly shorter than that of its homologues (10 vs. 15–19 aa, see Fig. 2), it is rich in basic and hydrophobic residues, suggesting that it might have a similar function. Furthermore, morphological defects observed in mitochondria of scu mutants support a mitochondrial localization of Scully.
Downstream from the targeting signal in the primary sequences of all members of the SDR family, there is a segment with three glycine residues. These residues are characteristic of the cofactor (NAD[P]+)-binding fold that is common to dehydrogenases in general (Rossman et al., 1975). Structural and mutational analysis indicates that the adjacent region is also important for cofactor affinity and specificity (Grimshaw et al., 1992; Tanaka et al., 1996; Nakanishi et al., 1997). Interestingly, the scu174 substitution L33N is located in this area, most likely affecting the cofactor-binding capacity of the enzyme, and therefore strongly diminishing its activity. Alternatively, the substitution in scu174 might have a more general effect by disrupting the tertiary structure of the protein. In fact, crystallographic studies of two members of the SDR family have shown that these proteins form almost identical one-domain structures in which single substitutions might have profound effects (Ghosh et al., 1991; Ghosh et al., 1994; and Varughese et al., 1992).
Several members of the SDR family are known to form homomultimers. Two α-helices have been identified in each of two SDR proteins as the subunit-interacting area that gives strong dimer interactions (Ghosh et al., 1991; Varughese et al., 1992). Kobayashi et al. (1996) have shown that bovine type II HADH is a tetramer. The antimorphic behavior of three scu mutations (scu174, scu4058, and scuS152) is consistent with the Scully protein also forming homomultimers. Mutation scu3127 gives rise to a truncated transcript encoding a peptide that contains only the 86 amino-terminal amino acids. This peptide includes the coenzyme-binding regions, but lacks both the putative interacting domain and the catalytic region. However, although scu3127 should be a functionally null mutation, it displays the mildest phenotype: absence of embryonic lethal phase, larger somatic clones, and weaker testes reduction. In contrast, the other scu alleles, two of which have only a single amino acid substitution, and the other of which has only a small carboxy-terminal deletion, show more extreme phenotypes. In these latter cases, the hypothetical formation of multimers composed of mutant and wild-type maternal subunits would result in effective sequestering of functional monomers into enzymatic complexes with reduced or null activity. The amino acid change F120I in scuS152 affects an α-helix putatively involved in multimer formation. Therefore, this mutation may impair formation of the multimer, and may explain why scuS152 is the least antimorphic allele.
Photoaffinity labeling studies indicate that substrate specificity of the SDR enzymes is determined to a large extent by the carboxy-terminal portion of the protein (Murdock et al., 1986). In fact, this portion is the most variable region in the SDR family, reflecting the wide variety of substrates for the different enzymes. Biochemical analysis of bovine type II HADH have shown that the specific activity of this enzyme is much higher with longer carbon chain substrates than that of type I enzyme (apparent Km values of type II enzyme for substrates other than 3-hydroxyhexadecanoyl-CoA are higher; Kobayashi et al., 1996). The strong similarity between Scully and the bovine type II HADH in their carboxy-terminal regions suggests similar substrate specificities. scu4058 generates a putative truncated protein lacking this region, and may have lost at least substrate specificity.
Mutant Phenotype and β-oxidation
One striking observation of the scully subcellular phenotype is its similarity with the human pathologies associated with defective β-oxidation of fatty acids (reviewed in Roe and Coates, 1995). One of the most characteristic of these pathologies, mitochondrial pleomorphism, is also observed in mutant scu photoreceptors. Blockade of the HADH activity leads to strong inhibition of mitochondrial β-oxidation by direct and feedback effects of the precursors on most enzymatic reactions of the chain (review in Eaton et al., 1996). As a direct consequence, depletion of the mitochondrial CoA pool results in arrest of mitochondrial fatty acid import and accumulation of lipids in the cytoplasm. This phenotype is observed in the testes of all scu mutants (Fig. 5), and resembles steatosis, another common pathology caused by β-oxidation deficiencies. Studies in both invertebrates (Geer et al., 1972) and vertebrates (Risley, 1990) have shown that there are important metabolic changes in testicular enzymatic activities during sexual maturation. In Drosophila, spermatogonia and spermatocytes obtain energy mainly from fatty acids oxidation, while spermatozoa primarily use carbohydrates and amino acids as energy sources (Geer et al., 1972). The high requirement for β-oxidation during the first developmental stages in testes accounts for the phenotypes observed mostly in the spermatocyte stages in scu mutants. The requirement of Scully for gonad development is also consistent with the high mRNA expression observed in these organs since early developmental stages.
Differential metabolic patterns and energy demands can also explain both the tissue distribution of the scu transcript and the different phenotypes of various cell types. For example, scu photoreceptors do not show lipid inclusions; instead, rhabdomeres are absent, and cytoplasmic multi-lamellar bodies are often observed. During photoreceptor differentiation, formation of rhabdomeres requires very active synthesis of lipids; this process needs ATP and acetyl-CoA, both produced by the β-oxidation pathway. In this case, the primary defect of scu deficiency could be related more to phospholipid synthesis alteration than to energy imbalance.
Several human inherited disorders have been related to alterations in mitochondrial β-oxidation (see Roe and Coates, 1995). They are a common cause of exercise- induced rhabdomyolisis and myoglobinuria (Tonin et al., 1990). These pathologies, together with other peripheral neuropathies, have been associated with alterations of the long-chain HADH (Schaefer et al., 1996). Hepato and cardiomegalies (Tyni et al., 1997), and retino and acute encephalopathies (Pons et al., 1996) have also been related with deficiencies of this enzyme. Short-chain type I HADH alterations have been described in a few cases (Bennett et al., 1996, Tein et al., 1991), but the enzymatic defect in several cases with altered oxidation remains uncharacterized. Our results indicate a more stringent requirement of Scully function. Scully is absolutely necessary for normal development in Drosophila, and point mutations in this protein can have dominant negative effects. Studies in mammals will be required to understand the role of this enzyme in mammalian development and physiology. In this regard, it was recently shown that the human homologue ERAB mediates neurotoxicity induced by amyloid-β peptide, and is overexpressed in neurons affected by Alzheimer's disease (AD; Yan et al., 1997). Defects in mitochondrial function have been reported in AD, and it has been proposed that mitochondrial dysfunction is a primary causal factor for AD (Sims, 1996). Generation of reactive oxygen species and disturbances in calcium homeostasis can ultimately lead to cell damage and neuronal loss in AD. Also, the reduction in the activity of mitochondrial pyruvate dehydrogenase observed in AD has been suggested as a possible cause for the disease-associated cholinergic deficit through a decrease in acetyl-coA production (Hoshi et al., 1997). Therefore, an attractive model for the role of ERAB in Alzheimer's pathology is that the effect of amyloid-β peptide on its enzymatic activity may result in energy imbalance and/or acetyl-coA depletion, leading to mitochondrial dysfunction and, finally, neuronal damage.
An intriguing question is that the activity of several enzymes of the β-oxidation of fatty acids in the brain increase during the postnatal period in mammals (Reichmann et al., 1988; Kelly et al., 1989), even though there are no evidences that the brain can produce energy from systemic fatty acids. Analysis of scu phenotypes demonstrates a role of this enzyme, at least in the development of the photoreceptors, and findings about human ERAB also support a role in neuronal physiology. Further analysis of the function of this enzyme in neurons will help us to understand the role of fatty acid β-oxidation in brain and to gain new insights into the pathology of some neurodegenerative diseases.
Acknowledgments
This work has been funded by grants PB93-149 and PB96-006 from the Dirección General de Ciencia y Tecnologia.
Abbreviations used in this paper
- AD
Alzheimer's disease
- CS
Canton-S
- EMS
ethyl methanesulphonate
- HADH
3-hydroxyacyl-CoA dehydrogenase
- SDR
short-chain dehydrogenase/reductase
Footnotes
Generation of transgenic flies was done in the laboratory of Dr. Kalpana White. We are especially grateful to her for her scientific advice and experimental support. The authors wish to thank Dr. A. Prado for generating reduced genomic duplications that first located the scully mutants. We are indebted to Drs. A. Rodríguez-Tébar, P. Bovolenta, G. Marqués, and M. Gordon, who criticized and improved this manuscript. D. Ortuño- Sahagún was on leave of absence from Universidad de Guadalajara (México).
Laura Torroja and Daniel Ortuño-Sahagún contributed equally to this work. The present address of Laura Torroja is Biology Department, Brandeis University, 415 South Street, Waltham, MA 02254-9110.
Address all correspondence to Julio A. Barbas, Instituto Cajal, C.S.I.C., Ave. Dr. Arce, 37. 28002 Madrid, Spain. Tel.: 34-1-585-47-25; Fax: 34-1-585-47-54; E-mail: jbarbas@cajal.csic.es
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Amaya Y, Arakawa H, Takiguchi M, Ebina Y, Yokota S, Mori M. A noncleavable signal for mitochondrial import of 3-oxoacyl-CoA thiolase. J Biol Chem. 1988;263:14463–14470. [PubMed] [Google Scholar]
- Arakawa H, Amaya Y, Mori M. The NH2-terminal 14–16 amino acids of mitochondrial and bacterial thiolases can direct mature ornithine carbamoyltransferase into mitochondria. J Biochem. 1990;107:160–164. doi: 10.1093/oxfordjournals.jbchem.a123001. [DOI] [PubMed] [Google Scholar]
- Arkipova IR. Promoter elements in Drosophila melanogasterrevealed by sequence analysis. Genetics. 1995;139:1359–1369. doi: 10.1093/genetics/139.3.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner, M. 1989. Drosophila. A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- Barbas JA, Galceran J, Krah-Jentgens I, de la Pompa JL, Canal I, Pongs O, Ferrús A. Troponin I is encoded in the haplolethal region of the Shaker gene complex of Drosophila. . Genes Dev. 1991;5:132–140. doi: 10.1101/gad.5.1.132. [DOI] [PubMed] [Google Scholar]
- Barbas JA, Galceran J, Torroja L, Prado A, Ferrús A. Abnormal muscle development in the heldup3 mutant of Drosophila melanogaster is caused by a splicing defect affecting selected troponin I isoforms. Mol Cell Biol. 1993;13:1433–1439. doi: 10.1128/mcb.13.3.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann A, Krah-Jentgens I, Muller R, Muller-Holtkamp F, Seidel R, Kecskemethy N, Casal J, Ferrús A, Pongs O. Molecular organization of the maternal effect region of the Shaker complex of Drosophila: characterization of an Ia channel transcript with homology to vertebrate Na+ channel. EMBO (Eur Mol Biol Organ) J. 1987;6:3419–3429. doi: 10.1002/j.1460-2075.1987.tb02665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MT, Hyman BT, Koroshetz W. Do defects in mitochondrial energy metabolism underlie the pathology of neurodegenerative diseases? . Trends Neurosci. 1993;16:125–131. doi: 10.1016/0166-2236(93)90117-5. [DOI] [PubMed] [Google Scholar]
- Bennett MJ, Weinberger MJ, Kobori JA, Rinaldo P, Burlina AP. Mitochondrial short-chain l-3-hydroxyacyl-coenzyme A dehydrogenase deficiency: a new defect of fatty acid oxidation. Pediatr Res. 1996;39:185–188. doi: 10.1203/00006450-199601000-00031. [DOI] [PubMed] [Google Scholar]
- Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effects on hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavener DR, Ray SC. Eukaryotic start and stop translation sites. Nucleic Acids Res. 1991;19:3185–3192. doi: 10.1093/nar/19.12.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherbas L, Cherbas P. The arthropod initiator: the cap site consensus plays an important role in transcription. Insect Biochem. Mol Biol. 1993;23:81–90. doi: 10.1016/0965-1748(93)90085-7. [DOI] [PubMed] [Google Scholar]
- Clark AG. Causes and consequences of variation in energy storage in Drosophila melanogaster. . Genetics. 1989;123:131–144. doi: 10.1093/genetics/123.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:679–685. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- Eaton S, Bartlett K, Pourfarzam M. Mammalian mitochondrial beta-oxidation. Biochem J. 1996;320:345–357. doi: 10.1042/bj3200345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Fakhri M, Middleton B. The existence of an inner-membrane-bound, long acyl-chain-specific 3-hydroxyacyl-CoA dehydrogenase in mammalian mitochondria. Biochim Biophys Acta. 1982;713:270–279. doi: 10.1016/0005-2760(82)90244-2. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B. A technique for radiolabeling DNA restriction endonucleases fragments to high specific activity. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- Ferrús A, Llamazares S, de la Pompa JL, Tanouye MA, Pongs O. Genetic analysis of the Shakergene complex of Drosophila melanogaster. Genetics. 1990;125:383–398. doi: 10.1093/genetics/125.2.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller, M.T. 1993. Spermatogenesis. In The Development of Drosophila melanogaster. M. Bate and A. Martínez-Arias, editors. Cold Spring Harbor Laboratory Press, Plainview, NY. 71–148.
- Furuta S, Kobayashi A, Miyazawa S, Hashimoto T. Cloning and expression of cDNA for a newly identified isozyme of bovine liver 3-hydroxyacyl-CoA dehydrogenase and its import into mitochondria. Biochim Biophys Acta. 1997;1350:317–324. doi: 10.1016/s0167-4781(96)00171-6. [DOI] [PubMed] [Google Scholar]
- Geer BW, Martensen DV, Downing BC, Muzyka GS. Metabolic changes during spermatogenesis and thoracic tissue maturation in Drosophilahydei. Dev Biol. 1972;28:390–406. doi: 10.1016/0012-1606(72)90022-x. [DOI] [PubMed] [Google Scholar]
- Ghosh D, Weeks CM, Grochulski P, Duax WL, Erman M, Rimsay RL, Orr JC. Three-dimensional structure of holo 3 alpha, 20 beta-hydroxysteroid dehydrogenase: a member of a short-chain dehydrogenase family. Proc Natl Acad Sci USA. 1991;88:10064–10068. doi: 10.1073/pnas.88.22.10064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D, Wawrzak Z, Weeks CM, Duax WL, Erman M. The refined three-dimensional structure of 3 alpha, 20 beta-hydroxy steroid dehydrogenase and possible roles of the residues conserved in short-chain dehydrogenases. Structure. 1994;2:629–640. doi: 10.1016/s0969-2126(00)00064-2. [DOI] [PubMed] [Google Scholar]
- Gish W, States DJ. Identification of protein coding regions by database similarity search. Nat Genet. 1993;3:266–272. doi: 10.1038/ng0393-266. [DOI] [PubMed] [Google Scholar]
- Grimshaw CE, Matthews DA, Varughese KI, Skinner M, Xuong NH, Bray T, Hoch J, Whiteley JM. Characterization and nucleotide binding properties of a mutant dihydropteridine reductase containing an aspartate 37-isoleucine replacement. J Biol Chem. 1992;267:15334–15339. [PubMed] [Google Scholar]
- Gunter TE, Gunter KK, Sheu SS, Gavin CE. Mitochondrial calcium transport: physiological and pathological relevance. Am J Physiol. 1994;267:C313–C339. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- Hainline BE, Kahlenbeck DJ, Grant J, Strauss AW. Tissue specific and developmental expression of rat long- and medium-chain acyl-CoA dehydrogenases. Biochim Biophys Acta. 1993;1216:460–468. doi: 10.1016/0167-4781(93)90015-6. [DOI] [PubMed] [Google Scholar]
- He XY, Yang SY, Schulz H. Assay of l-3-hydroxyacyl-coenzyme A dehydrogenase with substrates of different chain lengths. Anal Biochem. 1989;180:105–109. doi: 10.1016/0003-2697(89)90095-x. [DOI] [PubMed] [Google Scholar]
- Hoshi M, Takashima A, Murayama M, Yasutake K, Yoshida N, Ishiguro K, Hoshino T, Imahori K. Nontoxic amyloid beta peptide 1–42 suppresses acetylcholine synthesis. Possible role in cholinergic dysfunction in Alzheimer's disease. J Biol Chem. 1997;272:2038–2041. doi: 10.1074/jbc.272.4.2038. [DOI] [PubMed] [Google Scholar]
- Humason, G.L. 1972. Animal Tissue Techniques. W.H. Freeman and Company, San Francisco. 641 pp.
- Innis, M.A., and D.H. Gelfand. 1990. PCR Protocols: A Guide to Methods and Applications. Academic Press Inc., San Diego, CA. 482 pp.
- Jörnvall H, Persson B, Krook M, Atrian S, González-Duarte R, Jeffery J, Ghosh D. Short-chain dehydrogenases/reductases (SDR) Biochemistry. 1995;34:6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- Kelly DP, Gordon JI, Alpers R, Strauss AW. The tissue-specific expression and developmental regulation of two nuclear genes encoding rat mitochondrial proteins. Medium chain acyl-CoA dehydrogenase and mitochondrial malate dehydrogenase. J Biol Chem. 1989;264:18921–18925. [PubMed] [Google Scholar]
- Kobayashi A, Jiang LL, Hashimoto T. Two mitochondrial 3-hydroxyacyl-CoA dehydrogenases in bovine liver. J Biochem. 1996;119:775–782. doi: 10.1093/oxfordjournals.jbchem.a021307. [DOI] [PubMed] [Google Scholar]
- Krozowski Z. The short-chain alcohol dehydrogenase superfamily: variations on a common theme. J Steroid Biochem Mol Biol. 1994;51:125–130. doi: 10.1016/0960-0760(94)90084-1. [DOI] [PubMed] [Google Scholar]
- Lopaschuk GD, Collins-Nakai RL, Itoi T. Developmental changes in energy substrate use by the heart. Cardiovasc Res. 1992;26:1172–1180. doi: 10.1093/cvr/26.12.1172. [DOI] [PubMed] [Google Scholar]
- Luft R. The development of mitochondrial medicine. Proc Natl Acad Sci USA. 1994;91:8731–8738. doi: 10.1073/pnas.91.19.8731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannaerts GP, van Veldhoven PP. Functions and organization of peroxisomal beta-oxidation. Ann NY Acad Sci. 1996;804:99–115. doi: 10.1111/j.1749-6632.1996.tb18611.x. [DOI] [PubMed] [Google Scholar]
- Mount SM, Burks C, Hertz G, Stormo GD, White O, Fields C. Splicing signals in Drosophila: intron size, information content, and consensus sequences. Nucleic Acids Res. 1992;20:4255–4262. doi: 10.1093/nar/20.16.4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdock GL, Chin CC, Warren JC. Human placental estradiol 17 beta-dehydrogenase: sequence of a histidine-bearing peptide in the catalytic region. Biochemistry. 1986;25:641–646. doi: 10.1021/bi00351a019. [DOI] [PubMed] [Google Scholar]
- Nakanishi M, Matsuura K, Kaibe H, Tanaka N, Nonaka T, Mitsui Y, Hara A. Switch of coenzyme specificity of mouse lung carbonyl reductase by substitution of threonine 38 with aspartic acid. J Biol Chem. 1997;272:2218–2222. doi: 10.1074/jbc.272.4.2218. [DOI] [PubMed] [Google Scholar]
- Neuman-Silberberg FS, Schejter E, Hoffmann FM, Shilo BZ. The Drosophila ras oncogenes: structure and nucleotide sequence. Cell. 1984;37:1027–1033. doi: 10.1016/0092-8674(84)90437-9. [DOI] [PubMed] [Google Scholar]
- Osumi T, Hashimoto T. Occurrence of two 3-hydroxyacyl-CoA dehydrogenases in rat liver. Biochim Biophys Acta. 1979;574:258–267. [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrotta V. Vectors for P-mediated transformation in Drosophila. Biotechnology. 1988;10:437–456. doi: 10.1016/b978-0-409-90042-2.50028-3. [DOI] [PubMed] [Google Scholar]
- Pons R, Roig M, Riudor E, Ribes A, Briones P, Ortigosa L, Baldellou A, Gil-Gibernau J, Olesti M, Navarro C, Wanders RJ. The clinical spectrum of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Pediatr Neurol. 1996;14:236–243. doi: 10.1016/0887-8994(96)00021-5. [DOI] [PubMed] [Google Scholar]
- Reichmann H, Maltese WA, de Vivo DC. Enzymes of fatty acid beta-oxidation in developing brain. J Neurochem. 1988;51:339–344. doi: 10.1111/j.1471-4159.1988.tb01044.x. [DOI] [PubMed] [Google Scholar]
- Risley MS. Support of Xenopus laevis spermatogenesis in vitro by different energy substrates. Biol Reprod. 1990;42:511–522. doi: 10.1095/biolreprod42.3.511. [DOI] [PubMed] [Google Scholar]
- Roe, C.R., and P.M. Coates. 1995. The Metabolic Basis of Inherited Disease. C.R. Scriver, L. Beaudet, W.S. Sly, and D. Vaye, editors. McGraw-Hill Inc., New York. 889–914.
- Rossman, M.G., A. Liljas, C.I. Brändén, and L.J. Banaszak. 1975. The Enzymes. 3rd ed. Volume 11. P.D. Boyer, editor. Academic Press, Plainview, New York. 61–102.
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, New York.
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser A. A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labeled cRNA probes. Histochemistry. 1993;100:431–440. doi: 10.1007/BF00267823. [DOI] [PubMed] [Google Scholar]
- Schaefer J, Jackson S, Dick DJ, Turnbull DM. Trifunctional enzyme deficiency: adult presentation of a usually fatal beta-oxidation defect. Ann Neurol. 1996;40:597–602. doi: 10.1002/ana.410400409. [DOI] [PubMed] [Google Scholar]
- Sims NR. Energy metabolism, oxidative stress and neuronal degeneration in Alzheimer's disease. Neurodegeneration. 1996;5:435–440. doi: 10.1006/neur.1996.0059. [DOI] [PubMed] [Google Scholar]
- Spradling AC, Rubin GM. Transposition of cloned P elements into Drosophila germ line chromosomes. Science. 1982;218:341–347. doi: 10.1126/science.6289435. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Nonaka T, Tanabe T, Yoshimoto T, Tsuru D, Mitsui Y. Crystal structures of the binary and ternary complexes of 7 alpha- hydroxysteroid dehydrogenase from Escherichia coli. Biochemistry. 1996;35:7715–7730. doi: 10.1021/bi951904d. [DOI] [PubMed] [Google Scholar]
- Tein I, de Vivo DC, Hale DE, Clarke JT, Zinman H, Laxer R, Shore A, di Mauro S. Short-chain l-3-hydroxyacyl-CoA dehydrogenase deficiency in muscle: a new cause for recurrent myoglobinuria and encephalopathy. Ann Neurol. 1991;30:415–419. doi: 10.1002/ana.410300315. [DOI] [PubMed] [Google Scholar]
- Tonin P, Lewis P, Servidei S, di Mauro S. Metabolic causes of myoglobinuria. Ann Neurol. 1990;27:181–185. doi: 10.1002/ana.410270214. [DOI] [PubMed] [Google Scholar]
- Tyni T, Rapola J, Paetau A, Palotie A, Pihko H. Pathology of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation. Pediatr Pathol Lab Med. 1997;17:427–447. [PubMed] [Google Scholar]
- Varughese KI, Skinner MM, Whiteley JM, Matthews DA, Xuong NH. Crystal structure of rat liver dihydropteridine reductase. Proc Natl Acad Sci USA. 1992;89:6080–6084. doi: 10.1073/pnas.89.13.6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickens M, Stephenson P. Role of the conserved AAUAAA sequence: four AAUAAA point mutants prevent messenger RNA 3′ end formation. Science. 1984;226:1045–1051. doi: 10.1126/science.6208611. [DOI] [PubMed] [Google Scholar]
- Yan SD, Fu J, Soto C, Chen X, Zhu H, Al-Mohanna F, Collison K, Zhu A, Stern E, Saido T, et al. An intracellular protein that binds amyloid-β peptide and mediates neurotoxicity in Alzheimer's disease. Nature. 1997;389:689–695. doi: 10.1038/39522. [DOI] [PubMed] [Google Scholar]