

Abstract
We have developed a permeabilized cell assay to study the nuclear export of the shuttling transcription factor NFAT, which contains a leucine-rich export signal. The assay uses HeLa cells that are stably transfected with NFAT fused to the green fluorescent protein (GFP). Nuclear export of GFP–NFAT in digitonin-permeabilized cells occurs in a temperature- and ATP-dependent manner and can be quantified by flow cytometry. In vitro NFAT export requires the GTPase Ran, which is released from cells during the digitonin permeabilization. At least one additional rate-limiting export factor is depleted from permeabilized cells by a preincubation at 30°C in the absence of cytosol. This activity can be provided by cytosolic or nucleoplasmic extracts in a subsequent export step. Using this assay, we have purified a second major export activity from cytosol. We found that it corresponds to CRM1, a protein recently reported to be a receptor for certain leucine-rich export sequences. CRM1 appears to be imported into the nucleus by a Ran-dependent mechanism that is distinct from conventional signaling pathways. Considered together, our studies directly demonstrate by fractionation and reconstitution that nuclear export of NFAT is mediated by multiple nucleocytoplasmic shuttling factors, including Ran and CRM1.
Nucleocytoplasmic trafficking is mediated by nuclear pore complexes (NPCs)1, large supramolecular structures that span the nuclear envelope (for reviews see Doye and Hurt, 1997; Nigg, 1997). Molecules smaller than ∼20–40 kD can passively diffuse through aqueous channels in the NPC. In contrast, most macromolecules traverse the NPC by energy-, temperature-, and signal-dependent mechanisms. The best characterized signals for nuclear import (nuclear localization sequences [NLSs]) are short stretches of amino acids enriched in basic residues that occur as either a single or a bipartite motif (for review see Dingwall and Laskey, 1991). The M9 domain of the hnRNP A1 protein contains a qualitatively distinct type of NLS, a 38-amino acid stretch that is enriched in aromatic and glycine residues (Siomi and Dreyfuss, 1995).
The nuclear import of proteins containing basic amino acid-rich (classical) NLSs is beginning to be characterized in molecular detail (for review see Görlich and Mattaj, 1996; Nigg, 1997). Studies with a transport assay using digitonin-permeabilized cells (Adam et al., 1990) have identified four conserved soluble factors directly involved in the nuclear import of proteins with classic NLSs: (a) importin α/karyopherin α/NLS receptor/Srp1α, (b) importin β/karyopherin β/p97, (c) the small GTPase Ran/TC4, and (d) NTF2/p10 (for reviews see Görlich and Mattaj, 1996; Nigg, 1997). Nuclear import appears to involve the stepwise movement of a complex containing the transport substrate, importin α, and importin β through the NPC (Nigg, 1997). Translocation through the NPC also requires RanGTP hydrolysis (Melchior et al., 1993; Moore and Blobel, 1993; Schlenstedt et al., 1995; Mahajan et al., 1997) and NTF2 (Moore and Blobel, 1994; Paschal and Gerace, 1995), but the exact functions of these components are not resolved. Nuclear import mediated by the M9 domain of hnRNP A1 involves a receptor called transportin (Pollard et al., 1996) that is distantly related to importin β, and also is suggested to involve Ran (Bonifaci et al., 1997; Izaurralde et al., 1997).
In comparison to nuclear protein import, considerably less is known about the mechanisms of nuclear export of proteins and RNA–protein complexes (for review see Izaurralde and Mattaj, 1995; Görlich and Mattaj, 1996). An important advance was the characterization of sequences in nuclear proteins that are sufficient and necessary for nuclear export, called nuclear export sequences (NESs; for review see Gerace, 1995). NESs consisting of short amino acid stretches enriched in leucine residues appear to be the most prevalent (e.g., Fischer et al., 1995; Wen et al., 1995; Richards et al., 1996), although a distinct type of NES is present within the M9 domain of hnRNP A1 (Michael et al., 1995) and in a sequence of the hnRNP K protein (Michael et al., 1997). It was recently reported that certain leucine-rich NESs interact with a member of the importin β superfamily called CRM1 (Fornerod et al., 1997b ; Fukuda et al., 1997). CRM1 has been implicated in nuclear export from in vivo studies involving temperature sensitive CRM1 mutants in yeast (Fukuda et al., 1997; Stade et al., 1997), and the overexpression of CRM1 in Xenopus oocytes (Fornerod et al., 1997b ). Other work has suggested that CRM1 targets the exported substrate to proteins of the NPC (Neville et al., 1997). Nuclear export of the Rev protein, which also contains a leucine-rich NES, appears to involve an additional factor, eIF-5A (Ruhl et al., 1993; Bevec et al., 1996). CRM1-independent nuclear export has been described for importin α (Kutay et al., 1997), which also contains a leucine-rich NES (Boche and Fanning, 1997).
Genetic studies in yeast have provided evidence that Ran has a role in RNA export (e.g., Schlenstedt et al., 1995). Recent work with vertebrate cells has expanded upon this, showing that the GTP-bound form of Ran is required for the nuclear export of a protein containing a leucine-rich NES (Richards et al., 1997) and of various RNAs (Izaurralde et al., 1997). However, in contrast to nuclear import, RanGTP hydrolysis does not appear to be necessary for export under these conditions. Although the precise role of RanGTP in nuclear export in these experiments remains to be elaborated, in vitro studies indicate that RanGTP stimulates the formation of complexes between certain export receptors and their cargos (Fornerod et al., 1997b ; Kutay et al., 1997), and suggests a direct role for Ran in nuclear export.
The availability of a cytosol-dependent in vitro assay for nuclear export would greatly facilitate the understanding of nuclear export by allowing the detailed biochemical characterization of soluble factors involved in this process. Arts et al. (1997) have described an in vitro assay for export of RNA from synthetic nuclei, but a cytosolic requirement for export in this system is unclear. Yang et al. (1997) have shown that the glucocorticoid receptor is exported from nuclei of digitonin-permeabilized cells in an ATP- and temperature-dependent manner, but the export is apparently independent of exogenous cytosol under their experimental conditions.
Here we report the development and characterization of a cytosol-dependent assay for the analysis of nuclear protein export in permeabilized cells. The model export substrate in our system is the shuttling transcription factor NFAT (nuclear factor of activated T cells; Northrop et al., 1994), which plays a crucial role in the activation of T lymphocytes by stimulating the transcription of cytokine genes (for review see Crabtree and Clipstone, 1994). NFAT contains two regulatable NLSs of the basic amino acid type (Beals et al., 1997a ), as well as a regulatable leucine-rich NES (Klemm et al., 1997). In resting cells, NFAT resides in the cytoplasm. Its nuclear import is triggered by elevated intracellular calcium (Flanagan et al., 1991; Shibasaki et al., 1996), which induces the activation of the phosphatase calcineurin, leading to the dephosphorylation of NFAT and subsequent exposure of its NLSs (Beals et al., 1997a ). Upon return of calcium to resting levels, NFAT rapidly reappears in the cytosol (Shibasaki et al., 1996) by an export process that requires rephosphorylation of NFAT by glycogen synthase kinase-3 (GSK-3; Beals et al., 1997b ).
Using our permeabilized cell assay, we have characterized the cytosolic requirements for nuclear export of NFAT. By biochemical fractionation and reconstitution, we found that Ran and CRM1 are the two major cytosolic activities involved in NFAT export under our conditions. Our studies further suggest that the nuclear import of CRM1 is mediated by a Ran-dependent pathway distinct from those involving conventional NLSs, and provide the basis for developing a detailed understanding of the nucleocytoplasmic shuttling of a major nuclear export receptor.
Materials and Methods
Molecular Cloning of the Green Fluorescent Protein–NFAT Transfection Vector pAD-4
In a first step, the green fluorescent protein (GFP) fragment of the eukaryotic expression vector pS65T-C1 (Clontech Laboratories, Inc., Palo Alto, CA) was replaced with the human codon usage-optimized SGFP– TYG (synthetic GFP containing the amino acid change 65SYG to 65TYG). SGFP–TYG was amplified from the vector blue-SGFP–TYG-nosKS (J. Sheen, Harvard Medical School, Boston, MA) by PCR and then primers were designed to introduce a NheI site and a HindIII site. This NheI–HindIII fragment encoding SGFP–TYG was introduced into the eukaryotic expression vector pS65T-C1 (replacing the original GFP sequence in the vector), creating pAD-3. In a second step, the coding sequence of human NFAT was then PCR amplified out of pSH107c (provided by G.R. Crabtree, Howard Hughes Medical Institute, Stanford, CA), inserted into the BamHI and XbaI sites of pAD-3 (resulting in pAD-4), and then verified by sequencing.
Cell Culture and Transfections
HeLa cells were grown either in DME on plastic dishes, or in suspension in Joklik's modified S-MEM (GIBCO BRL, Gaithersburg, MD). In both cases media contained 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Transfections were performed with lipofectamine (GIBCO BRL, Gaithersburg, MD) using 25 μg of linearized plasmid per ∼2 × 106 adherent cells. Approximately 50 G418-resistant clones (GIBCO BRL) were pooled and induced with 250 nM of trichostatin A (Wako BioProducts, Richmond, VA) to express GFP–NFAT. Cells expressing high levels were enriched by FACS® (Becton and Dickinson Co., Mountain View, CA). Positive cells were expanded and stored in aliquots in liquid nitrogen.
Preparation of Cytosol and Nuclear Extract
Approximately 4.5 × 109 HeLa cells (8 liters of cells growing in suspension) were collected by centrifugation at 300 g for 10 min, washed twice with ice-cold PBS, and then once with washing buffer (10 mM Hepes– KOH, pH 7.3, 110 mM KOAc, 2mM Mg[OAc]2, 2 mM DTT). After resuspension in one volume of lysis buffer (5 mM Hepes–KOH, pH 7.3, 10 mM KOAc, 2mM Mg[OAc]2, 2 mM DTT, 1 mM PMSF, 1 μg/ml each of leupeptin, pepstatin, and aprotinin) and swelling for 10 min on ice, the cells were lysed in a stainless steel Dounce homogenizer. After centrifugation at 1,500 g for 15 min, the supernatant was cleared by centrifugation at 120,000 g for 1 h and then dialyzed overnight against transport buffer (20 mM Hepes–KOH, pH 7.3, 110 mM KOAc, 2 mM Mg[OAc]2, 1mM EGTA). The resulting cytosol (∼10 mg/ml) was frozen in liquid nitrogen and stored at −80°C.
To prepare a nuclear extract, the pellet of the 1,500-g spin was washed in 4 ml of lysis buffer and resuspended in 10 ml of 20 mM Hepes–KOH, pH 7.9, 25% glycerol, 0.5 M NaCl, 1.5 M MgCl2, 0.2 mM EDTA, 1 mM PMSF, and 1 mM DTT. The nuclei were lysed by 10 strokes in a stainless steel Dounce homogenizer, left on ice for 15 min, and then centrifuged at 35,000 g for 30 min. The resulting supernatant was dialyzed overnight against transport buffer and then centrifuged again at 100,000 g for 30 min. The final supernatant (nuclear extract, ∼3.5 mg/ml) was frozen in liquid nitrogen and stored at −80°C.
SDS-PAGE and Western Blotting
Proteins were separated by SDS-PAGE and blotted onto nitrocellulose using standard methods. Blots were blocked with 5% milk powder in PBS overnight. NFAT was detected with the mouse monoclonal antibody 7A6 (1:5,000 in 5% milk in PBS) and HRP-coupled goat anti–mouse IgG (Pierce Chemical Co., Rockford, IL; 1:5,000 in 10 mM Tris-HCl, pH 8, 150 mM NaCl, 0.05% Tween 20 [Sigma Chemical Co.]). CRM1 was detected with different anti-CRM1 antibodies (see below; 1:5,000–1:3,000 in 5% milk in PBS) and HRP-coupled donkey anti–rabbit IgG (Pierce Chemical Co.; 1:10,000 in 10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.05% Tween 20). The enhanced chemiluminescence system (Pierce Chemical Co.) was used for visualization of proteins.
Antibodies
The mouse monoclonal anti-NFAT antibody (7A6) is described by Northrop et al. (1994). Anti-importin β antibody was obtained from Affinity BioReagents, Inc. (clone 3E9; Golden, CO). Before use in the export assay, the ascites fluid was diluted 1:4 with transport buffer containing 5 mg/ml BSA and then dialyzed against transport buffer. Two different polyclonal antibodies against CRM1 were used. The first is described by Fornerod et al. (1997a) and was provided by G. Grosveld (St. Jude Children's Research Hospital, Memphis, TN). The second was raised in rabbits by injecting a peptide corresponding to the COOH terminus of CRM1 (GIFNPHEIPEEMCD), coupled with glutaraldehyde to keyhole limpet hemocyanin (Calbiochem-Novabiochem Corp., La Jolla, CA).
Purification of GST-M9
pGEX-M9 provided by G. Dreyfuss (University of Pennsylvania School of Medicine, Philadelphia, PA) was transformed into DH5α cells. An overnight culture was diluted 1:50 in Luria-Bertani medium, grown to an OD600 of 0.7, and then induced with 1 mM isopropylthio-β-Δ-galactoside for 3 h. Cells were harvested by centrifugation at 5,000 g for 10 min, washed twice in ice-cold PBS, and then resuspended to 1% of the original volume in PBS with 5 μg/ml each of aprotinin, leupeptin, and pepstatin. After cell lysis, Triton X-100 was added to a final concentration of 1%. The lysate was centrifuged at 10,000 g for 10 min and the supernatant was incubated with glutathione–Sepharose 4B beads (Pharmacia Biotech. Inc., Piscataway, NJ) for 1 h at 4°C. The beads were collected by centrifugation at 800 g for 5 min and then washed in batch three times in PBS and once in 50 mM Tris-HCl, pH 8.8, 150 mM NaCl before being transferred to a column. GST-M9 was eluted with 50 mM Tris base, 150 mM NaCl, and 15 mM glutathione (reduced). Peak fractions were dialyzed against 100 mM Hepes–KOH, pH 9.6.
Recombinant Import Factors
Wild-type Ran was prepared as described (Melchior et al., 1995). RanQ69L and RanT24N were obtained from J. Becker (MPI für molekulare Physiologie, Dortmund, Germany) (Klebe et al., 1995). Importin α and importin β were prepared as in Hu et al. (1996). NTF2 was prepared as in Paschal and Gerace (1995).
Preparation of Fluorescent Import Substrates
To prepare Cy5-NLS-BSA, 2.5 mg of BSA in 1 ml 0.1 M Na2CO3 was coupled to Cy5™ (1 vial, Amersham Pharmacia Biotech. Inc., Piscataway, NJ) for 40 min at room temperature. The conjugate was separated from the free dye by chromatography on a PD-10 column (Pharmacia Biotech. Inc.). The NLS peptide derived from the sequence of the SV-40 large T antigen was reduced as described (Adam et al., 1990) and coupled to Cy5-BSA that had previously been activated with a 20-fold molar excess of sulfo-SMCC (Pierce Chemical Co.; Adam et al., 1990) by incubation at 4°C overnight. The Cy5–NLS-BSA conjugate was then separated from free peptide by chromatography on a PD-10 column equilibrated with transport buffer. Peak fractions were pooled and then aliquots were frozen in liquid nitrogen.
To prepare Cy5–GST-M9, 5 mg of GST-M9 was coupled with Cy5™ (1 vial, Amersham Pharmacia Biotech. Inc.) for 30 min at room temperature and then separated from free dye by chromatography on a PD-10 column (Pharmacia Biotech. Inc.). Peak fractions of Cy5–GST-M9 were pooled and diluted with transport buffer to a final concentration of 1 mg/ml. Aliquots were frozen in liquid nitrogen.
Nuclear Export Assay
To assay nuclear export of NFAT, its expression was induced by treating stably transfected HeLa cells growing on tissue culture dishes with 250 nM of trichostatin A (Wako BioProducts) overnight. To induce nuclear import, ionomycin (Calbiochem-Novabiochem Corp.) and LiOAc were added to final concentrations of 1 μM and 30 mM, respectively, and then the cells were incubated for 30 min at 37°C. They were trypsinized in the continued presence of ionomycin and LiOAc and then washed once with 10% FBS in transport buffer and once with transport buffer containing 80 mM KOAc, 30 mM LiOAc, 2 mM DTT, 1 mM PMSF, and 1 μg/ml each of leupeptin, pepstatin, and aprotinin. The cells were collected by centrifugation at 300 g for 5 min at 4°C and then resuspended in transport buffer (with 30 mM LiOAc) at ∼107/ml. After permeabilization with 60 μg/ml digitonin (Calbiochem-Novabiochem Corp.) for 5 min on ice, the cells were washed and centrifuged as described above and then resuspended to a concentration of 107/ml in transport buffer containing KOAc and LiOAc. A preincubation in the presence of an ATP-regenerating system was performed for 15 min at 0°C or 30°C. Cells were then washed in transport buffer lacking LiOAc but containing 110 mM KOAc and then resuspended at ∼3 x 107/ml. The complete transport mixture (40 μl in transport buffer) contained ∼300,000 cells, an ATP-regenerating system (1 mM ATP, 5 mM creatine phosphate, 20 U/ml creatine phosphokinase), 2.5 mg/ ml BSA, import ligand (Cy5–NLS-BSA at ∼2.5 μg/ml or Cy5–GST-M9 at ∼25 μg/ml), 1 μM of annealed oligonucleotide (5′ AGA GGA AAA TTT GTT TCA TA and 5′ TAT GAA ACA AAT TTT CCT CT, both prepared and HPLC purified by GIBCO BRL), and cytosol or nuclear extract at various concentrations, as indicated. For reactions in the absence of ATP, the ATP-regenerating system was replaced by 50 U/ml hexokinase (Sigma Chemical Co.) and 12.5 mM glucose. Reactions were incubated at 30°C for 30 min and stopped by the addition of ice-cold transport buffer. After centrifugation at 300 g for 5 min at 4°C, the cells were resuspended in a small volume of transport buffer. The average nuclear fluorescence emitted from GFP–NFAT, Cy5–NLS-BSA, or Cy5–GST-M9 in 10,000 cells was measured with a FACScan® flow cytometer (Becton and Dickinson Co.). Reactions were standardized by arbitrarily assigning a value of 100 to the nuclear fluorescence of GFP–NFAT in a 0°C control reaction and, unless otherwise indicated, to the maximal nuclear fluorescence of the imported ligands after incubation at 30°C. For export reactions in which permeabilized cells were not preincubated, treatment of cells was as described above, except that transport buffer always contained 110 mM KOAc and no LiOAc. For analysis of export by SDS-PAGE, nuclei were collected by centrifugation upon completion of the export reaction, washed once in transport buffer, and then resuspended in SDS-sample buffer. The supernatant of the first centrifugation was retained and mixed with an equal volume of 2× SDS-sample buffer and nuclei and supernatant fractions were analyzed by SDS-PAGE and immunoblotting. To perform the export assay with adherent cells growing on coverslips, cells were treated with trichostatin A and ionomycin as above, permeabilized with digitonin, and then subjected to export reactions in the same manner as previously described for nuclear import reactions (Adam et al., 1990). Cells were then fixed with 3.7% formaldehyde in PBS and analyzed by fluorescence microscopy using an Axiophot (Carl Zeiss, Inc., Thornwood, NY).
Purification of a Major Cytosolic Activity for Nuclear Export
6 ml of cytosol (containing 60 mg of protein) were dialyzed into triethanolamine buffer (20 mM triethanolamine, pH 7.4, 2 mM MgCl2) and saturated ammonium sulfate was added to a final concentration of 900 mM. The precipitate (6% of total protein) was collected by centrifugation (14,000 g for 5 min) and then resuspended in triethanolamine buffer. The remaining ammonium sulfate was removed by chromatography of the dissolved precipitate on a PD-10 column (Pharmacia Biotech. Inc.) equilibrated in triethanolamine buffer. The sample then was loaded on a FPLC Mono Q column (HR 5/5; Pharmacia Biotech. Inc.) and eluted with a 20-ml linear gradient of 100 to 350 ml NaCl in triethanolamine buffer using a flow rate of 0.4 ml/min and collecting 1-ml fractions. Maximal nuclear export activity was typically recovered at ∼230 mM NaCl. 900 μl of the peak fraction (as determined by the nuclear export assay and by immunoblotting) were concentrated to 300 μl by vacuum dialysis applied to a gel filtration column (Superdex 200 HR 10/30; Pharmacia Biotech. Inc.) equilibrated in transport buffer. The sample was chromatographed at 0.5 ml/ min, collecting 0.5-ml fractions. The nuclear export activity eluted with a molecular mass of ∼120 kD.
Results
We have developed a method for rapid, quantitative analysis of nuclear export of the shuttling transcription factor NFAT in permeabilized HeLa cells. For this, we prepared cells that were stably transfected with a cDNA coding for NFAT fused to the GFP. The expression of GFP–NFAT was induced to high levels by overnight treatment of the cells with the histone deacetylase inhibitor trichostatin A (Fig. 1) or with sodium butyrate (data not shown; Gorman et al., 1983; Arts et al., 1995). The actual level of expression of the GFP–NFAT in the induced cell population varied from cell to cell, although typically about two-thirds of the cells expressed high levels of GFP–NFAT (Fig. 2 a, − ion). The range of expression levels in the cell population was most clearly seen in cells analyzed by flow cytometry (see Fig. 3 b, 0°C).
Figure 1.

Detection of GFP–NFAT in total cell lysates (in vivo) or after export reactions (in vitro). In vivo: transfected HeLa cells were treated with (+) or without (−) 250 nM trichostatin A (TSA) overnight and with (+) or without (−) 1 μM ionomycin (ion) for 30 min. GFP–NFAT from total cell lysates (∼20,000 cells) was detected by immunoblotting. In vitro: cells were treated with trichostatin A and ionomycin as above. After standard nuclear export reactions at 0°C or 30°C, GFP–NFAT in the permeabilized cell/nuclear fraction (N) or in the supernatant (S) was detected by immunoblotting.
Figure 2.
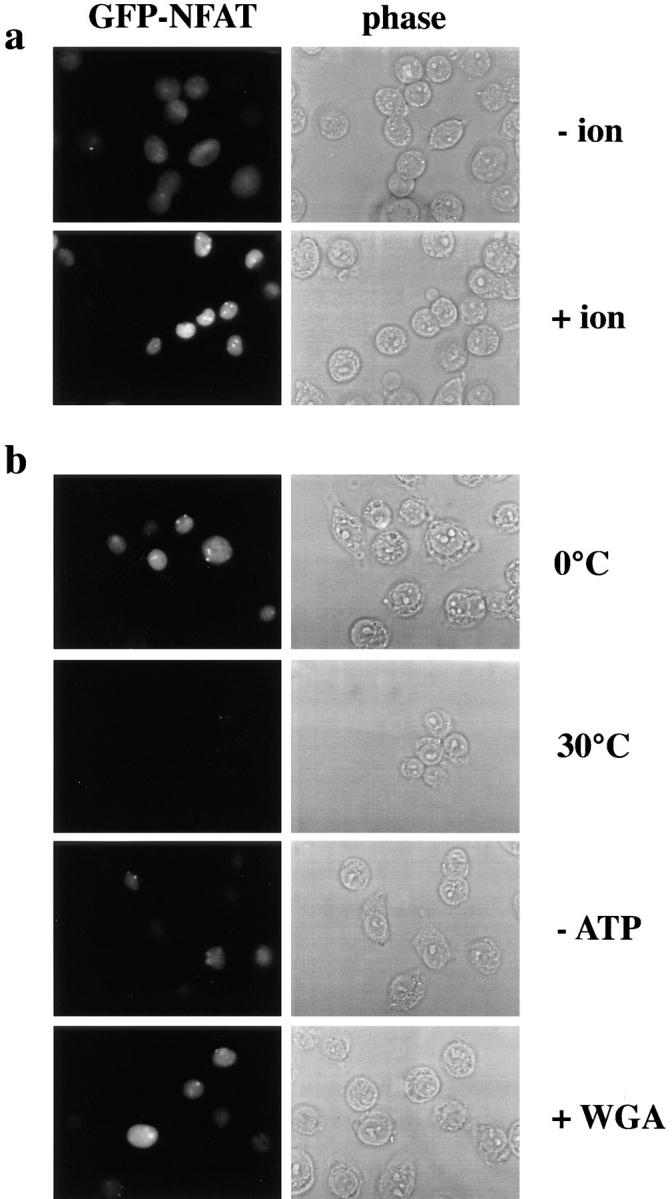
Nuclear trafficking of GFP–NFAT in vivo and in vitro. (a) Transfected HeLa cells were treated overnight with trichostatin A and incubated for 30 min plus (+ ion) or minus (− ion) ionomycin. (b) Standard nuclear export reactions were performed on coverslips. Permeabilized cells were incubated at 0°C or 30°C in the presence of 2 mg/ml of cytosol and 25 μg/ml of Ran. For the reaction without ATP (− ATP), the ATP-regenerating system was replaced by an ATP-depleting system. WGA was added to 200 μg/ml as indicated. Note that some cells express very little, if any, GFP–NFAT (see panels for − ion, + ion, and 0°C incubation).
Figure 3.
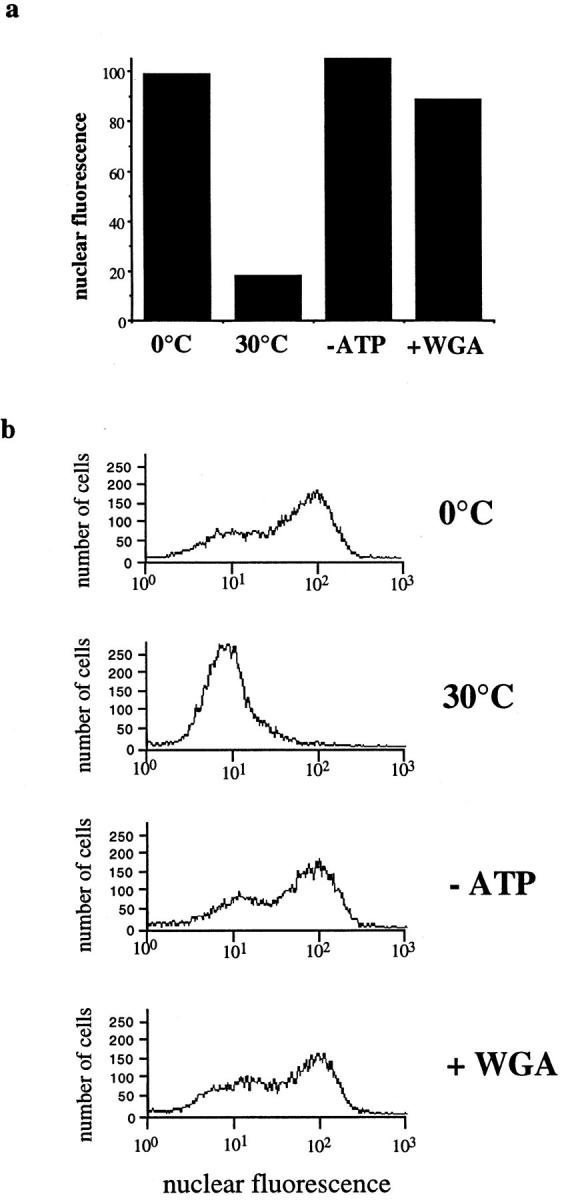
Characterization of temperature and ATP requirements of nuclear export by flow cytometry. Standard nuclear export reactions were performed in the presence of cytosol. For reactions in the absence of ATP (− ATP), the ATP-regenerating system was replaced by an ATP-depleting system. WGA was used at 200 μg/ml. All reactions contained recombinant Ran at 25 μg/ml. (b) Original FACS® profiles used to obtain the data in a and distribution of fluorescence values in the population of permeabilized cells. The absolute level of export of GFP–NFAT varied from assay to assay, although in a single experiment duplicate reactions gave very similar results. Very similar profiles were obtained with cells before permeabilization and in permeabilized cells after incubation at 0°C (data not shown).
The GFP–NFAT in the transfected cells exhibited the same nuclear transport behavior and responses to inhibitors as originally described for wild-type NFAT in vivo. The calcium ionophore ionomycin induced rapid nuclear import of GFP–NFAT (Flanagan et al., 1991; Shibasaki et al., 1996), which was predominantly cytoplasmic in untreated cells (Fig. 2 a, compare − ion with + ion). GFP–NFAT from ionomycin-treated cells had a higher electrophoretic mobility on an SDS gel than the protein from untreated cells (Fig. 1, in vivo, compare − ion and + ion lanes), reflecting a partial dephosphorylation of the protein (Ruff and Leach, 1995). The ionomycin-induced import of GFP– NFAT into the nucleus was inhibited by simultaneously adding cyclosporin A, a specific inhibitor of calcineurin (Flanagan et al., 1991; Liu et al., 1991; data not shown). When the ionomycin-treated cells were washed and incubated at 37°C in the presence of cyclosporin A, GFP– NFAT was rapidly exported from the nucleus (Shibasaki et al., 1996; data not shown). Finally, export of GFP– NFAT was inhibited by the addition of 30 mM of lithium acetate to the medium, which inhibits its rephosphorylation by inactivating GSK-3 (Klein and Melton, 1996; data not shown). Considered together, these in vivo experiments indicate that HeLa cells expressing GFP–NFAT are appropriate for use in an in vitro nuclear export assay.
Nuclear Export of GFP–NFAT In Vitro
After the expression of GFP–NFAT was induced in stably transfected HeLa cells, the nuclear import of the reporter was triggered by treatment with ionomycin. Next, the cells were removed from the dish by trypsinization, permeabilized with digitonin, and then washed to remove the endogenous cytosol. In our standard export assay, the permeabilized cells were first preincubated for 15 min at 30°C in the absence of cytosol and in the presence of ATP. To retain most of the GFP–NFAT in the nucleus during the preincubation step, we included lithium in the preincubation buffer to inhibit GSK-3 (refer to Introduction and Materials and Methods). Typically, between 80 and 90% of the GFP–NFAT remained in the nucleus after the preincubation (data not shown). Subsequently, the cells were incubated under various export conditions in the absence of lithium and were analyzed by flow cytometry. As shown below, the 30°C preincubation step depletes rate-limiting export factors from the permeabilized cells, thereby allowing the detection of these activities in the subsequent export assay.
A double-stranded oligonucleotide corresponding to an NFAT DNA-binding site (Northrop et al., 1994) was included in all export reactions. This oligonucleotide stimulated in vitro ATP-dependent nuclear export of GFP– NFAT approximately twofold at a concentration of 1 μM (data not shown), apparently by promoting release of the protein from chromatin (see below). Export reactions in the absence of the oligonucleotide gave qualitatively similar results, although the absolute levels of export were lower. Interestingly, the addition of lithium to export reactions containing oligonucleotide did not significantly decrease the level of GFP–NFAT export (data not shown). This indicates that the inclusion of the oligonucleotide abrogates the requirement for phosphorylation by GSK-3 for efficient export. Thus, the lithium-sensitive phosphorylation of NFAT appears to stimulate export by releasing NFAT from chromatin, not by activating an NES on NFAT per se. Decreased DNA binding activity of NFAT upon phosphorylation has been described previously (Park et al., 1995).
Fluorescence micrographs of permeabilized cells that had been incubated in the standard export assay are shown in Fig. 2 b. When cells were incubated at 0°C, the amount of GFP–NFAT retained in the nucleus (Fig. 2 b; 0°C) was essentially identical to the amount retained in the nuclei of cells examined immediately after permeabilization (data not shown). In contrast, incubation at 30°C in the presence of cytosol and an ATP-regenerating system led to the loss of most nuclear fluorescence (Fig. 2 b; 30°C). The export of GFP–NFAT, as seen by the loss of fluorescence, was largely prevented if the reaction included an ATP-depleting system (Fig. 2 b; −ATP) or wheat germ agglutinin (WGA; Fig. 2 b; +WGA), which binds to a group of O-glycosylated proteins of the NPC (Hanover et al., 1987) and inhibits mediated nuclear import and export in vivo (Yoneda et al., 1987; Dargemont and Kühn, 1992). As shown below, nuclear export of GFP–NFAT was largely dependent on the addition of cytosol (see Figs. 4 b and 5 b).
Figure 4.
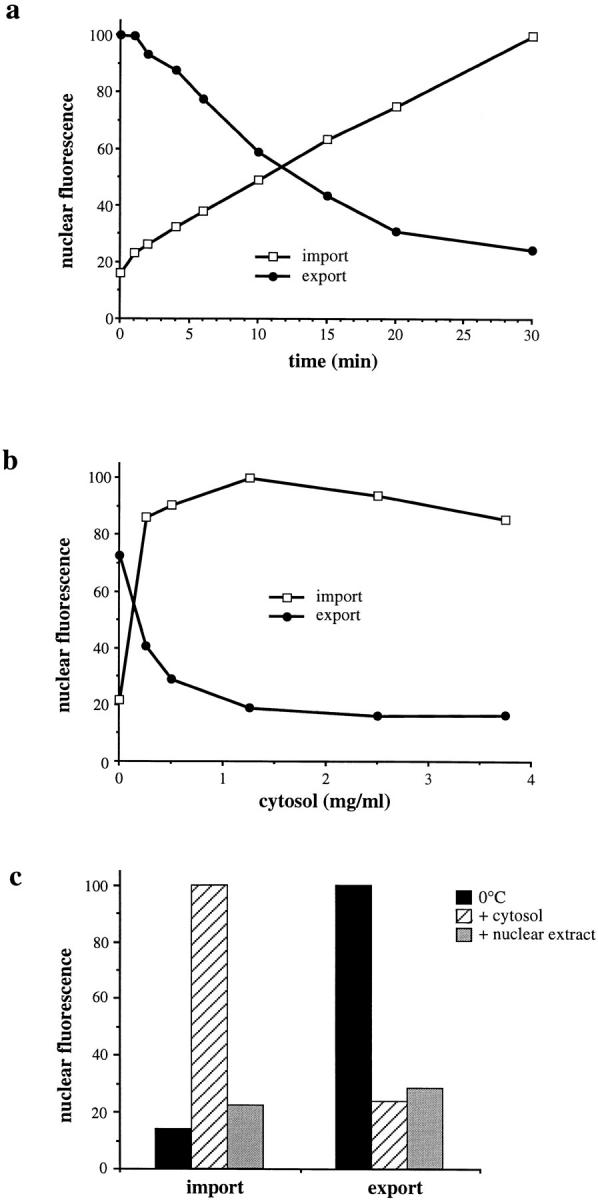
General characteristics of nuclear export under standard assay conditions. (a) Time course of standard nuclear export/import reactions (2 mg/ml of cytosol and 25 μg/ml of Ran in all reactions). (b) Cytosol dependence of standard nuclear export/import reactions (50 μg/ml of Ran in all reactions). (a and b) Closed circles, export; open squares, import. (c) Standard nuclear export/import reactions were performed at 0°C (black bars) or at 30°C with either 2 mg/ml of cytosol (hatched bars) or 2 mg/ml of nuclear extract (grey bars) and 25 μg/ml of Ran in all reactions. The import substrate was Cy5–NLS-BSA in all cases.
Flow cytometry provided a rapid and quantitative method to analyze nuclear export in a large number of cells. Fig. 3 shows the result of a standard nuclear export assay carried out in the presence of cytosol that was analyzed by this method. The original flow cytometry data used to generate the graph in Fig. 3 a is shown in Fig. 3 b. It should be noted that in assays measuring nuclear export, low values of nuclear fluorescence indicate high levels of transport, whereas in assays measuring nuclear import (see below), high fluorescence values represent high levels of transport. Incubation at 30°C resulted in the retention of only 18.2 units of nuclear fluorescence as compared with 100 units retained in the 0°C control (Fig. 3 a). Export was strongly inhibited by depleting ATP or by including the lectin WGA, both of which gave export values similar to the 0°C control (Fig. 3 a).
Several controls indicated that the loss of fluorescence during the export incubations was due to physiologically relevant export of GFP–NFAT from intact nuclei. When we included rhodamine-labeled dextrans of different molecular weights in the export reactions (Melchior et al., 1993), we found that a small dextran (4.5 kD), but not a large one (155 kD) was able to enter the nucleus (data not shown). This demonstrated that the permeability barrier of the nuclear envelope remained normal. Additional validation for the integrity of the nuclear envelope was obtained by incubating permeabilized cells with Cy5-labeled NLS-BSA and cytosol in the standard export assay. In this case, efficient nuclear accumulation of the NLS-BSA occurred at the same time as export of the GFP–NFAT (see Fig. 4), and both the import and export were inhibited by WGA (data not shown). A further control involved centrifuging the assay mixture after the export reaction and examining the resulting supernatants and pellets by Western blotting to detect GFP–NFAT (refer to Fig. 1). The total amount of GFP–NFAT in a reaction incubated at 30°C was similar to that in a reaction incubated at 0°C. However, in the 0°C incubation, most of the GFP–NFAT was found in the permeabilized cell pellet, whereas in the 30°C incubation, most of the GFP–NFAT was found in the supernatant. Thus, the loss of GFP fluorescence at 30°C does not result from degradation of GFP–NFAT, but instead reflects its release from the permeabilized cells into the supernatant. It should be noted that after incubation at 30°C, the electophoretic mobility of GFP–NFAT decreases and resembles that of the protein before nuclear import in vivo (refer to Fig. 1, compare in vivo with in vitro panels). This apparently reflects rephosphorylation of the NFAT, which activates the export of NFAT and inactivates its NLS in vivo (Beals et al., 1997). Since calcineurin-mediated dephosphorylation of GFP–NFAT cannot occur in vitro due to the presence of EGTA in our assay buffer, the exported GFP–NFAT is trapped outside the nucleus and cannot be reimported.
Further Characterization of the Standard Nuclear Export Assay
To further characterize nuclear transport in the standard export assay, we simultaneously measured export of GFP– NFAT and import of Cy5–NLS-BSA in the same cells (Fig. 4). Fig. 4 a shows a concurrent time course of nuclear import and export over a range of 30 min. Nuclear import of Cy5–NLS-BSA was linear for at least 30 min, yielding a final value that was approximately sixfold greater than the 0°C background (100 versus 16.4 units). Examination of nuclear export of GFP–NFAT in the same cells showed that the fluorescent signal of GFP–NFAT was reduced from 100 units at the beginning of the reaction to a final typical value of 24.5 units after a 30-min incubation. Nuclear export was roughly linear for 15–20 min and then started to level off, which was probably due to a limitation of the export substrate after this time. In cells expressing much lower levels of GFP–NFAT, export was linear for only ∼10 min (data not shown).
Fig. 4 b shows a comparison of the cytosol requirement of nuclear import and nuclear export. The reactions were carried out in the presence of excess recombinant Ran so that it would not be rate limiting. We found that 1.25 mg/ml cytosol stimulated nuclear export of GFP–NFAT to almost the maximal level (from 72.9 to 19 units) and also gave maximal stimulation of import of Cy5–NLS-BSA into the nucleus (from 21.7 to 100 units). Thus, the cytosol requirement for nuclear import and export is quantitatively very similar under these conditions.
Under steady-state conditions, nuclear export factors are expected to be somewhat concentrated in the nucleus. Therefore, we tested the ability of a HeLa cell nuclear extract to stimulate nuclear export. Fig. 4 c shows that the nuclear extract stimulated export of GFP–NFAT to roughly the same extent as did cytosol when used at the same protein concentration (from 100 units at 0°C to 28.6 units for nuclear export, compared with 23.6 units for cytosol). In contrast, the nuclear extract had only a minor stimulatory effect on protein import into the nucleus as compared with cytosol (from 14 units at 0°C, to 22.6 and 100 units at 30°C for the nuclear extract and cytosol, respectively). A nuclear extract prepared with a low-salt extraction buffer (50 mM NaCl) was able to stimulate nuclear export at least as well as the nuclear extract prepared with a high-salt buffer (500 mM NaCl, refer to Materials and Methods), suggesting that the extracted factors are not tightly bound to NPCs and are probably intranuclear (data not shown). These results suggest that factors distinct from those required for the nuclear import of NLS-containing substrates are involved in the export of GFP–NFAT from the nucleus. The recombinant factors other than Ran that support the nuclear import of cargoes with classical NLS in permeabilized cells (NTF2, importin α, and importin β) strongly stimulated import of Cy5–NLS-BSA into the nucleus without affecting export of GFP–NFAT (data not shown). Thus, whereas some or all of these import factors are rate-limiting for the nuclear import of a protein containing a classical NLS in our system, none of these components are rate-limiting in the assay measuring export of GFP–NFAT.
Ran and Other Nucleocytoplasmic Shuttling Factors Are Involved in Nuclear Export of NFAT
Ran has a well-established role in nuclear import and recent studies have suggested a direct role for Ran in nuclear export as well (refer to Introduction). To investigate a possible involvement of Ran and other nucleocytoplasmic shuttling factors in nuclear export in our assay, we analyzed the stimulation of nuclear export of GFP–NFAT by Ran alone or by cytosol, after preincubation of the permeabilized cells either at 0°C or at 30°C in the presence of ATP and absence of cytosol (Fig. 5, a and b). The preincubation at 30°C, as used in our standard assay, might be expected to deplete rate-limiting export factors whose exit from the nucleus is ATP- and temperature-dependent, whereas the preincubation at 0°C would deplete only factors that are small enough to exit the nucleus by passive diffusion. Previous work has shown that Ran is almost completely depleted from the nucleus of permeabilized cells during digitonin treatment and washing at 0°C (Melchior et al., 1995).
Figure 5.
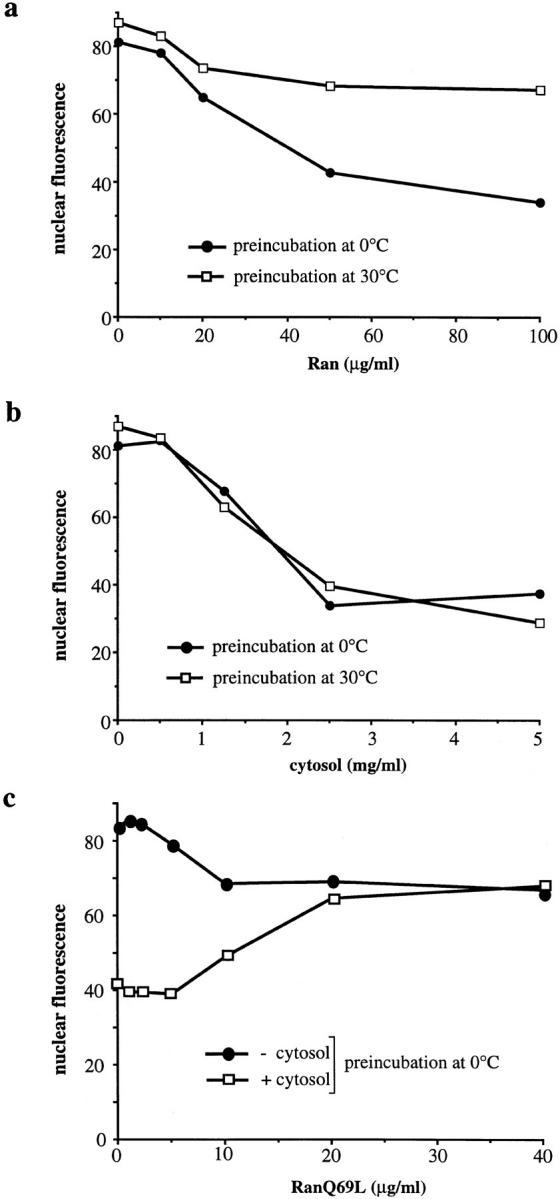
Preincubation leads to a loss of rate-limiting factors for nuclear export. Standard nuclear export reactions were performed with increasing amounts of Ran (a) or cytosol (b) after preincubation of the permeabilized cells at either 0°C (closed circles) or 30°C (open squares). (c) Nuclear export reactions after preincubation at 0°C were performed with increasing amounts of RanQ69L in the absence (− cytosol, closed circles) or presence (+ cytosol, open squares) of cytosol (2.5 mg/ml).
In permeabilized cells that had been preincubated at 0°C, Ran alone strongly stimulated export to a degree that was similar to the export stimulation obtained with complete cytosol (Fig. 5, compare a with b): the fluorescence decreased from 81.2 units after incubation with buffer to 34.3 units with Ran (100 μg/ml) or to 37.6 units with cytosol (5 mg/ml). By contrast, in cells that had been preincubated at 30°C, Ran by itself only weakly stimulated nuclear export (reducing the fluorescence from 87.2 to 67.4 units), whereas cytosol still stimulated export to the same extent as in cells preincubated at 0°C (reducing fluorescence from 87.2 to 28.9 U; Fig. 5, a and b). These results indicate that Ran is a rate-limiting export factor that is depleted from cells that have been preincubated at 0°C, whereas additional rate-limiting factors are depleted from the nucleus by preincubating the permeabilized cells at 30°C.
We next investigated the effect of the RanQ69L and RanT24N mutants on nuclear export in cells that had been preincubated at 0°C to deplete Ran only but not other rate-limiting export factors. RanQ69L cannot hydrolyze GTP and thus, is predominantly in the GTP-bound form (Klebe et al., 1995). RanT24N is either in the nucleotide-free state or bound to GDP (Klebe et al., 1995). These two mutants are well-characterized inhibitors of nuclear protein import, both in vivo (Dickmanns et al., 1996) and in permeabilized cells (Palacios et al., 1996). Interestingly, increasing concentrations of RanQ69L progressively stimulated nuclear export of GFP–NFAT in the absence of cytosol (Fig. 5 c; from 83.9 units without addition to 67.2 units with 40 μg/ml of RanQ69L), albeit to a lower extent than the stimulation obtained with cytosol (from 83.9 to 41.9 units) or with wild-type Ran (data not shown; also compare Fig. 5, a with b). However, in the presence of cytosol, RanQ69L inhibited export from 41.9 to 68.2 units, a value that is very similar to that observed with RanQ69L alone (67.2 units). In cells that had been preincubated at 30°C, RanQ69L had no effect on nuclear export in the absence of cytosol and had an inhibitory effect in the presence of cytosol (data not shown). RanT24N had no effect on nuclear export in the absence of cytosol and inhibited export in the presence of cytosol in cells that had been preincubated at 0°C (data not shown).
These data indicate that Ran has a dual effect on nuclear export of GFP–NFAT in vitro. One effect reflects the ability of GTP-bound Ran to stimulate nuclear export in the absence of nuclear protein import and of RanGTP hydrolysis, resulting in a moderate increase in export when RanQ69L is added to the export assay. This effect is consistent with recent results indicating that RanGTP, but not GTP hydrolysis, is required for nuclear export of several substrates (Izaurralde et al., 1997; Richards et al., 1997). A second effect involves the additional stimulation of export by wild-type Ran, which allows GTP hydrolysis. We suggest that this stimulatory effect reflects the RanGTP hydrolysis-dependent import of rate-limiting export factors into the nucleus. This Ran-dependent import appears to be inhibited by either RanQ69L or RanT24N, resulting in an inhibition of export by these mutants in the presence of cytosol.
Well-characterized Pathways for Mediated Nuclear Protein Import Are Not Required for Nuclear Export of NFAT In Vitro
Since our data indicate that our standard nuclear export assay requires the nuclear import of export factors by a Ran-dependent–mediated pathway, we analyzed whether the well-characterized pathways for import of proteins containing either a basic amino acid-rich NLS or the M9-type NLS are involved. To address this we performed kinetic competition experiments with either classical NLS-BSA or GST-M9 transport substrates, and also carried out antibody inhibition experiments to inactivate importin β, which is involved in import of proteins containing basic amino acid-type NLSs (refer to Introduction).
Fig. 6 a shows that a high concentration of NLS-BSA inhibited the nuclear import of Cy5–NLS-BSA (from 100 fluorescent to 18 units, with a 0°C background of 14 units). In contrast, nuclear export was not significantly affected by NLS-BSA (resulting in 27 units in the presence of NLS-BSA, as compared with 23.6 units without NLS-BSA). We next examined the effects of a monoclonal antibody against importin β that inhibits nuclear import of NLS-containing substrates (Chi et al., 1995). In the presence of cytosol, this antibody reduced the level of import of Cy5– NLS-BSA into the nucleus approximately fourfold without affecting export of GFP–NFAT (data not shown). In the presence of a nuclear extract, which supports import to a much lower extent than cytosol (see above), the anti-importin β antibody completely blocked import (Fig. 6 b): fluorescence was reduced from 41.6 units with buffer to 16.1 units with antibody; a 0°C control resulted in 16.3 units. Again, the antibody had only a minor effect on the nuclear export of GFP–NFAT (30.2 units with antibody compared with 25.4 units with buffer).
Figure 6.



Import of export factors by a nonconventional pathway. (a and c) Competition of import and export: reactions were performed in the absence (0°C, black bars and 30°C, − competitor, hatched bars) or presence (30°C, white bars) of 0.5 mg/ml unlabeled NLS–BSA (a, + NLS-BSA) or 0.5 mg/ml GST-M9 (c, + GST-M9). Cy5–NLS-BSA (a) or Cy5–GST-M9 (c) was used as a fluorescent import substrate. (b) Anti-importin β antibody abolishes import but not export. Reactions were performed in the presence of 2 mg/ml of cytosol (0°C, black bars and + cytosol, 30°C, hatched bars) or 1.5 mg/ml nuclear extract (+ nuclear extract, 30°C, grey bars and + nuclear extract + anti-importin β, 30°C, white bars). 4 μl of the monoclonal antibody against importin β was included as indicated.
Similar to the results with NLS-BSA, unlabeled GST-M9 competed for the import of Cy5–GST-M9 (Fig. 6 c). The fluorescence was reduced from 100 to 45.6 units with a 0°C background of 23.2 units (in this case the applied concentration of GST-M9 was not high enough to achieve complete inhibition). In contrast, the export of GFP–NFAT remained essentially unchanged under these conditions (resulting in a fluorescence of 17.8 units with competitor, compared with 16.9 units without competitor). These results indicate that the nuclear protein import pathways specified by the two well-characterized NLSs are not involved in nuclear export of GFP–NFAT. Thus, the nuclear export factors provided by exogenously added cytosol or nuclear extract appear to reenter the nucleus by a nonconventional-mediated import pathway that is dependent on Ran.
CRM1 Is a Major Cytosolic Export Factor In Vitro
Since Ran alone was unable to support export after preincubation at 30°C to the same extent as cytosol (refer to Fig. 5, a and b), we carried out biochemical fractionation of cytosol to isolate a major activity other than Ran that promotes nuclear export in our assay. As an initial purification step, we enriched approximately half of the export activity by a selective ammonium sulfate precipitation step (data not shown; refer to Materials and Methods). The sample from ammonium sulfate precipitation then was loaded onto a Mono Q ion exchange column and then individual fractions were tested for their ability to stimulate nuclear export in our standard assay in the presence of an excess of exogenous Ran (Fig. 7 a). A single strong peak of export-stimulating activity was obtained from this column. Fraction 22, which had the highest activity, reduced the nuclear fluorescence from a background of 69.2 to 39.2 units. A saturating amount of total cytosol supported nuclear export to the same extent as fraction 22 (data not shown). Since CRM1 has been implicated as an export receptor for certain leucine-rich NESs (refer to Introduction), we also analyzed the column fractions for the presence of CRM1 using immunoblotting (Fig. 7 a, inset). Interestingly, the peak of export activity in this column precisely coincided with the elution profile of CRM1. The peak fraction from the Mono Q column was subsequently chromatographed onto a gel filtration column, fractions were tested for their ability to stimulate nuclear export under standard conditions in the presence of excess Ran, and were also immunoblotted to detect CRM1 (Fig. 7 b). A single peak of export activity was detected in this column profile, and again, the peak coincided with the elution of CRM1. The export activity peak and CRM1 eluted at the position of a monodisperse protein at ∼120 kD (Fig. 7 b, inset), a size very similar to the calculated molecular mass (123 kD) of CRM1 (Fornerod et al., 1997a ). Fraction 13, which had the highest activity, stimulated nuclear export of NFAT from 63.8 to 47.3 units. In comparison, cytosol reduced the nuclear fluorescence to 23.4 units in this assay. As shown by silver staining, CRM1 appeared to be the only protein in the most active fractions (Fig. 7 c). In other experiments, we were able to recover larger amounts of export activity (and also of CRM1 antigen) by including 100 μg/ml BSA in the gel filtration column buffer. With Ran and the CRM1 obtained in this preparation, we obtained a level of nuclear export that was comparable to that seen with cytosol (data not shown).
Figure 7.


CRM1 is a nuclear export factor in vitro. (a) Activity profile of fractions from the Mono Q column. Standard nuclear export reactions were performed using 25 μl of Mono Q fractions that had been dialyzed against transport buffer plus 50 μg/ml of Ran. Inset, immunoblot of the indicated fractions with anti-CRM1 antibodies. CRM1 was not detected in any other fractions. (b) Activity profile of fractions from the Superdex 200 column. Standard nuclear export reactions were performed using 30 μl of Superdex 200 fractions and 50 μg/ml of Ran. Inset, immunoblot of the indicated fractions with anti-CRM1 antibodies. CRM1 was undetectable in the other fractions. (c) Silver-stained SDS gel (7.5%) of selected fractions from the Superdex 200 column in the region of the activity peak. Whereas certain background bands are seen in all fractions, the band migrating at the position of CRM1 was detected mainly in fractions 12–14. Note that the ∼100-kD band in fractions 9 and 10 migrates distinctly faster than CRM1. A 10– 20% gradient gel did not reveal any protein bands in the range between 20 and 50 kD in fractions 12–14 (data not shown). (d) Standard nuclear export reactions were performed by incubating permeabilized cells with or without partially purified CRM1 obtained from a Mono Q fraction, with or without 50 μg/ml of Ran as indicated. (e) HeLa cells were permeabilized with digitonin and equivalent amounts of fractions containing permeabilized cells/nuclei (N, lane 1) and released cytosol (C, lane 2) before preincubation, or permeabilized cell/nuclei (N) and supernatant (S) after preincubation either at 0°C (lanes 3 and 4) or 30°C (lanes 5 and 6) were analyzed by immunoblotting. In contrast to CRM1, the vast majority of proteins was recovered in the permeabilized cell/nuclei fraction after preincubation at either temperature (lanes 3–6) as seen by Ponceau staining (data not shown).
Fig. 7 d depicts the export activity of a partially purified CRM1 fraction in the absence and presence of exogenous Ran. Whereas little export was obtained with CRM1 alone or with Ran alone, strong stimulation was obtained with a combination of CRM1 and Ran (from 79.3 units of nuclear fluorescence in the absence of both proteins to 36.6 units). This demonstrates that Ran is required together with CRM1 to reconstitute in vitro export of NFAT in our standard export assay.
To further evaluate the role of CRM1 as an export factor in our assay, we analyzed the distribution of endogenous CRM1 during cell permeabilization and preincubation to determine whether its fractionation behavior paralleled the presence of nuclear export activity (Fig. 7 e). After digitonin permeabilization, the majority of CRM1 was recovered in the cytosolic fraction (Fig. 7 e, lane 2), although a significant amount of the protein remained in the permeabilized cells (Fig. 7 e, lane 1). Some additional CRM1 protein was removed from permeabilized cells by a 0°C incubation (Fig. 7 e, lanes 3 and 4). However, preincubation of cells at 30°C in the absence of cytosol led to the loss of the great majority of the remaining CRM1 from the permeabilized cells (Fig. 7 e, lane 6), leaving only a small amount behind (Fig. 7 e, lane 5). Thus, most CRM1 is released from the permeabilized cells during the 30°C preincubation step when the loss of export activity occurs, whereas substantial CRM1 remains after a 0°C preincubation step when the permeabilized cells still support strong export. In further experiments, we found that leptomycin B, a drug that inhibits the formation of a complex between CRM1 and NES substrates and thus blocks nuclear export in vivo (Fornerod et al., 1997b , Fukuda et al., 1997, Ossareh-Nazari et al., 1997), inhibited nuclear export in our in vitro assay at submicromolar concentrations (data not shown). Moreover, when we immunodepleted CRM1 from cytosol, we found that the antibody-depleted cytosol had substantially reduced nuclear export activity when compared with cytosol that had been treated with a nonspecific IgG (data not shown). Considering these observations together with the results of our biochemical fractionation and reconstitution studies, we conclude that CRM1 and Ran are nucleocytoplasmic shuttling factors that are required for the nuclear export of NFAT.
Discussion
An In Vitro Assay for Nuclear Export of NFAT
We have used the shuttling transcription factor NFAT as a transport substrate to develop a rapid, quantitative in vitro assay to study nuclear protein export. In our standard export assay, the permeabilized cells are subjected to a preincubation step at 30°C in the absence of cytosol. This step depletes shuttling export factors from the nucleus under conditions in which the nuclear export of GFP–NFAT is reversibly arrested. During a subsequent incubation, efficient export of GFP–NFAT can be achieved if nuclear or cytosolic extracts are added to the permeabilized cells. Numerous criteria demonstrate that the assay reflects physiologically relevant in vitro export of GFP–NFAT. Nuclear export occurs in a time-, temperature-, and ATP-dependent manner, and can be blocked by WGA, a reagent that is known to inhibit signal-mediated import and export through NPCs (Yoneda et al., 1987; Dargemont and Kühn, 1992). Moreover, the nuclei retain their normal diffusional permeability barrier and can simultaneously import proteins containing NLSs.
Reimport of the GFP–NFAT into the nucleus during the export assay, which would substantially hinder quantitative analysis of nuclear export, does not seem to occur in our system to any significant level. This is because GFP– NFAT becomes partially rephosphorylated during the export assay in a manner that apparently inactivates its NLSs and traps it outside the nucleus. The activity of calcineurin, the calcium-dependent protein phosphatase that is responsible for NFAT dephosphorylation, is likely to be inhibited by the EGTA present in our assay buffer. Consistent with this possibility is our observation that cyclosporin A, which inhibits the activity of calcineurin in vivo (Flanagan et al., 1991; Liu et al., 1991), has no further stimulatory effect on nuclear export of GFP–NFAT in vitro.
Effects of the GTPase Ran on Nuclear Export
Moroianu and Blobel (1995) previously found that a protein containing a classic NLS was released from the nucleus of permeabilized cells in a Ran-stimulated fashion. The meaning of these results is not entirely clear, because the presence of a basic type NLS does not appear to specify shuttling between the nucleus and cytoplasm in vivo after nuclear import (Michael et al., 1995). The GFP–NFAT that we have used as a nuclear export substrate in these studies is a nucleocytoplasmic shuttling protein with a well-characterized NES, and thus serves as a good model for in vitro nuclear export studies.
Our results clearly demonstrate that the GTPase Ran is required for nuclear export of GFP–NFAT in vitro. When in vitro nuclear export of GFP–NFAT is analyzed in permeabilized cells that have not been subjected to a 30°C preincubation step, Ran alone stimulates the nuclear export of GFP–NFAT to the level achieved with cytosol. Analysis of the effects of Ran mutants in this assay suggest that Ran stimulates nuclear export by two different mechanisms that have additive effects. In the first of these mechanisms, nuclear export is stimulated by RanQ69L, which cannot hydrolyze its bound GTP, but is unaffected by RanT24N, which is either free of nucleotides or in a GDP-bound state. Thus, this mechanism appears to require GTP-bound Ran but not RanGTP hydrolysis. A second mechanism through which Ran stimulates nuclear export is seen with wild-type Ran and appears to require RanGTP hydrolysis, since it is inhibited by RanQ69L. We suggest that this requirement reflects an involvement of Ran in the reimport of CRM1 (see below) and possibly other export factors that are released from the nucleus during the preincubataion. This dual effect of Ran can explain the higher stimulation of nuclear export in the absence of cytosol by wild-type Ran versus RanQ69L.
Our observation that GTP-bound Ran but not RanGTP hydrolysis is required for (an intermediate level) of nuclear export of NFAT is consistent to the results of in vivo studies indicating that nuclear export of a leucine-rich NES substrate (Richards et al., 1997) and of various RNAs (Izaurralde et al., 1997) requires GTP-bound Ran. This requirement for RanGTP probably relates to the cooperative binding of RanGTP with export substrate to export receptors to form putative export complexes (Fornerod et al., 1997b ; Kutay et al., 1997).
CRM1 Is a Nucleocytoplasmic Shuttling Factor Involved in NFAT Export
With the exception of Ran, all rate-limiting factors required for efficient nuclear export of NFAT are retained in the permeabilized cells after digitonin treatment. However, by preincubating cells at 30°C in the absence of cytosol, additional rate-limiting factor(s) are released, thereby making the assay dependent on exogenous cytosol or nuclear extract, rather than on Ran alone. This provides the basis for isolating these activities by biochemical fractionation. We have purified a major cytosolic activity involved in the stimulation of nuclear export in our standard export assay, and have found that this activity corresponds to CRM1 (Fornerod et al., 1997b ; Fukuda et al., 1997; Ossareh-Nazari et al., 1997; Stade et al., 1997). Stimulation of nuclear export by purified CRM1 depends on the presence of Ran. This requirement probably reflects a need for Ran for the import of CRM1 into the nucleus, as well as the involvement of Ran in the formation of a CRM1-RanGTP-NFAT complex (Fornerod et al., 1997b ) that can be exported from the nucleus (see above). Considered together, our results involving fractionation and reconstitution contribute strong direct evidence that CRM1 functions as a nucleocytoplasmic shuttling receptor for nuclear export of NFAT, and complements the results of earlier in vivo studies involving temperature-sensitive mutants (Fukuda et al., 1997; Stade et al., 1997) and overexpression of CRM1 (Fornerod et al., 1997b ). The ability to reconstitute nuclear export in this assay with CRM1 and Ran will allow a detailed structure-function analysis of these components.
We believe that it will be possible to identify additional factors that regulate NFAT export using this assay. Indeed, we have partially purified a potent activity from cytosol that inhibits NFAT export in permeabilized cells (our unpublished observations). Additional positively acting export factors may be revealed under different preincubation and assay conditions.
Nuclear Export Factors for NFAT Enter the Nucleus by a Nonconventional Import Pathway
Shuttling nuclear export factors like CRM1 have to reenter the nucleus after one round of export. The strong inhibition of nuclear export by RanQ69L in our standard, cytosol-dependent export assay indicates that these factors are imported into the nucleus by a Ran-dependent transport pathway. Whereas importin β and transportin can be imported into the nucleus when added to permeabilized cells in the absence of Ran (Kose et al., 1997; Nakielny and Dreyfuss, 1997), we consider it very likely that the nuclear import of these proteins under more physiological conditions (i.e., in the presence of cytosol and Ran) involves RanGTP hydrolysis, since they both strongly bind RanGTP and their import (and that of their cargos) is efficiently inhibited by RanQ69L and nonhydrolyzable GTP analogues (Melchior and Gerace, 1995; Bonifaci et al., 1997; Izarraulde et al., 1997).
We determined that neither the importin β nor transportin pathways are involved in the import of export factors (including CRM1) for NFAT in our assay because inhibiting these pathways with an excess of competing import substrates or with a monoclonal antibody against importin β does not diminish nuclear export of GFP–NFAT. Taken together, our results indicate that the recycling of nuclear export factors for NFAT involves a nonconventional nuclear import pathway. If this pathway were selective for shuttling export factors, it would provide a means to regulate nuclear export in a manner that is independent of the nuclear protein import pathways used to transport the bulk of nuclear proteins. Furthermore, this would allow the recycling of nuclear export factors without competition by other major import substrates. Understanding the ability of the NPC to accommodate multiple different signaling pathways for import as well as for export represents an important future challenge.
Acknowledgments
We wish to thank G. Crabtree for providing the cDNA clone for NFAT, the anti-NFAT antibody, and for helpful advice, G. Grosveld for the gift of anti-CRM1 antibody, G. Dreyfuss for providing a cDNA clone for GST-M9, J. Becker for the RanQ69L and RanT24N proteins, C. Delphin (Scripps Research Institute, La Jolla, CA) for importin α and β, and D. Sweet (Trends in Cell Biology, Cambridge, UK) for the preparation of NTF2. The manuscript was improved by critical comments from S. Lyman, F. Melchior, A. Kehlenbach, and C. Fritze (all four from The Scripps Research Institute). We also gratefully acknowledge J. Hauber (Institute for Clinical and Molecular Virology, University Erlangen, Nürnberg, Germany) for his support of this project.
This work was supported by a grant from Novartis Pharmaceuticals to L. Gerace and by fellowships from the Human Frontier Science Program to R.H. Kehlenbach (LT-400/96) and the Deutsche Forschungsgemeinschaft to A. Dickmanns (Di 676/1-1).
Abbreviations used in this paper
- FACS®
fluorescence activated cell sort(er)ing
- GFP
green fluorescent protein
- GSK-3
glycogen synthase kinase-3
- NES
nuclear export sequence
- NFAT
nuclear factor of activated T cells
- NLS
nuclear localization sequence
- NPC
nuclear pore complex
- WGA
wheat germ agglutinin
Footnotes
Address all correspondence to Larry Gerace, Department of Cell Biology and Molecular Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037. Tel.: (619) 784-8514. Fax: (619) 784-9132. E-mail: lgerace@scripps.edu
References
- Adam SA, Marr RS, Gerace L. Nuclear protein import in permeabilized mammalian cells requires soluble cytoplasmic factors. J Cell Biol. 1990;111:807–816. doi: 10.1083/jcb.111.3.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts G-J, Englmeier L, Mattaj IW. Energy- and temperature-dependent in vitroexport of RNA from synthetic nuclei. Biol Chem. 1997;378:641–649. doi: 10.1515/bchm.1997.378.7.641. [DOI] [PubMed] [Google Scholar]
- Arts J, Lansink M, Grimbergen J, Toet KH, Kooistra T. Stimulation of tissue-type plasminogen activator gene expression by sodium butyrate and trichostatin A in human endothelial cells involves histone acetylation. Biochem J. 1995;310:171–176. doi: 10.1042/bj3100171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beals CR, Clipstone NA, Ho SN, Crabtree GR. Nuclear localization of NF-ATc by a calcineurin-dependent, cyclosporin-sensitive intramolecular interaction. Genes Dev. 1997a;11:824–834. doi: 10.1101/gad.11.7.824. [DOI] [PubMed] [Google Scholar]
- Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 1997b;275:1930–1934. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- Bevec D, Jaksche H, Oft M, Wohl T, Himmelspach M, Pacher A, Schebesta M, Koettnitz K, Dobrovnik M, Csonga R, et al. Inhibition of HIV-1 replication in lymphocytes by mutants of the Rev cofactor eIF-5A. Science. 1996;271:1858–1860. doi: 10.1126/science.271.5257.1858. [DOI] [PubMed] [Google Scholar]
- Boche I, Fanning E. Nucleocytoplasmic recycling of the nuclear localization signal receptor α subunit in vivois dependent on a nuclear export signal, energy and RCC1. J Cell Biol. 1997;139:313–325. doi: 10.1083/jcb.139.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifaci N, Moroianu J, Radu A, Blobel G. Karyopherin β2 mediates nuclear import of a mRNA binding protein. Proc Natl Acad Sci USA. 1997;94:5055–5060. doi: 10.1073/pnas.94.10.5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi NC, Adam EJ, Adam SA. Sequence and characterization of cytoplasmic nuclear protein import factor p97. J Cell Biol. 1995;130:265–274. doi: 10.1083/jcb.130.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree GR, Clipstone NA. Signal transmission between the plasma membrane and nucleus of T lymphocytes. Annu Rev Biochem. 1994;63:1045–1083. doi: 10.1146/annurev.bi.63.070194.005145. [DOI] [PubMed] [Google Scholar]
- Dargemont C, Kühn LC. Export of mRNA from microinjected nuclei of Xenopus laevisoocytes. JCell Biol. 1992;118:1–9. doi: 10.1083/jcb.118.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickmanns A, Bischoff FR, Marshallsay C, Lührmann R, Ponstingl H, Fanning E. The thermolability of nuclear protein import in tsBN2 cells is suppressed by microinjected Ran-GTP or Ran-GDP, but not by RanQ69L or RanT24N. J Cell Sci. 1996;109:1449–1457. doi: 10.1242/jcs.109.6.1449. [DOI] [PubMed] [Google Scholar]
- Dingwall C, Laskey RA. Nuclear targeting sequences—a consensus? . Trends Biochem Sci. 1991;16:478–481. doi: 10.1016/0968-0004(91)90184-w. [DOI] [PubMed] [Google Scholar]
- Doye V, Hurt E. From nucleoporins to nuclear pore complexes. Curr Opin Cell Biol. 1997;9:401–411. doi: 10.1016/s0955-0674(97)80014-2. [DOI] [PubMed] [Google Scholar]
- Fischer U, Huber J, Boelens WC, Mattaj IW, Lührmann R. The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell. 1995;82:475–483. doi: 10.1016/0092-8674(95)90436-0. [DOI] [PubMed] [Google Scholar]
- Flanagan WM, Corthesy B, Bram RJ, Crabtree GR. Nuclear association of a T-cell transcription factor blocked by FK-506 and cyclosporin A. Nature. 1991;352:803–807. doi: 10.1038/352803a0. [DOI] [PubMed] [Google Scholar]
- Fornerod M, van Deursen J, van Baal S, Reynolds A, Davis D, Murti KG, Fransen J, Grosveld G. The human homologue of yeast CRM1 is in a dynamic subcomplex with CAN/Nup214 and a novel nuclear pore component Nup88. EMBO (Eur Mol Biol Organ) J. 1997a;16:807–816. doi: 10.1093/emboj/16.4.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucin-rich nuclear export signals. Cell. 1997b;90:1051–1060. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, Nishida E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature. 1997;390:308–311. doi: 10.1038/36894. [DOI] [PubMed] [Google Scholar]
- Gerace L. Nuclear export signals and the fast track to the cytoplasm. Cell. 1995;82:341–344. doi: 10.1016/0092-8674(95)90420-4. [DOI] [PubMed] [Google Scholar]
- Görlich D, Mattaj IW. Nucleocytoplasmic transport. Science. 1996;271:1513–1518. doi: 10.1126/science.271.5255.1513. [DOI] [PubMed] [Google Scholar]
- Gorman CM, Howard BH, Reeves R. Expression of recombinant plasmids in mammalian cells is enhanced by sodium butyrate. Nucleic Acids Res. 1983;11:7631–7648. doi: 10.1093/nar/11.21.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanover JA, Cohen CK, Willingham MC, Park MK. O-linked N-acetylglucosamine is attached to proteins of the nuclear pore. Evidence for cytoplasmic and nucleoplasmic glycoproteins. J Biol Chem. 1987;262:9887–9894. [PubMed] [Google Scholar]
- Hu T, Guan T, Gerace L. Molecular and functional characterization of the p62 complex, an assembly of nuclear pore complex glycoproteins. J Cell Biol. 1996;134:589–601. doi: 10.1083/jcb.134.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izaurralde E, Mattaj IW. RNA export. Cell. 1995;81:153–159. doi: 10.1016/0092-8674(95)90323-2. [DOI] [PubMed] [Google Scholar]
- Izaurralde E, Kutay U, von Kobbe C, Mattaj I, Görlich D. The asymmetric distribution of the constituents of the Ran system is essential for transport into and out of the nucleus. EMBO (Eur Mol Biol Organ) J. 1997;16:6535–6547. doi: 10.1093/emboj/16.21.6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebe C, Bischoff FR, Ponstingl H, Wittinghofer A. Interaction of the nuclear GTP-binding protein Ran with its regulatory proteins RCC1 and RanGAP1. Biochemistry. 1995;34:639–647. doi: 10.1021/bi00002a031. [DOI] [PubMed] [Google Scholar]
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm JD, Beals CR, Crabtree GR. Rapid targeting of nuclear proteins to the cytoplasm. Curr Biol. 1997;7:638–644. doi: 10.1016/s0960-9822(06)00290-9. [DOI] [PubMed] [Google Scholar]
- Kose S, Imamoto N, Tachibana T, Shimamoto T, Yoneda Y. Ran-unassisted nuclear migration of a 97-kD component of nuclear pore-targeting complex. J Cell Biol. 1997;139:841–849. doi: 10.1083/jcb.139.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutay U, Bischoff FR, Kostka S, Kraft R, Görlich D. Export of importin α from the nucleus is mediated by a specific nuclear transport factor. Cell. 1997;90:1061–1071. doi: 10.1016/s0092-8674(00)80372-4. [DOI] [PubMed] [Google Scholar]
- Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP- FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- Mahajan R, Delphin C, Guan T, Gerace L, Melchior F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell. 1997;88:97–107. doi: 10.1016/s0092-8674(00)81862-0. [DOI] [PubMed] [Google Scholar]
- Melchior F, Gerace L. Mechanisms of nuclear protein import. Curr Opin Cell Biol. 1995;7:310–318. doi: 10.1016/0955-0674(95)80084-0. [DOI] [PubMed] [Google Scholar]
- Melchior F, Sweet DJ, Gerace L. Analysis of Ran/TC4 function in nuclear protein import. Methods Enzymol. 1995;257:279–291. doi: 10.1016/s0076-6879(95)57032-2. [DOI] [PubMed] [Google Scholar]
- Melchior F, Paschal B, Evans J, Gerace L. Inhibition of nuclear protein import by nonhydrolyzable analogues of GTP and identification of the small GTPase Ran/TC4 as an essential transport factor. J Cell Biol. 1993;123:1649–1659. doi: 10.1083/jcb.123.6.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael WM, Choi M, Dreyfuss G. A nuclear export signal in hnRNP A1: A signal-mediated, temperature-dependent nuclear protein export pathway. Cell. 1995;83:415–422. doi: 10.1016/0092-8674(95)90119-1. [DOI] [PubMed] [Google Scholar]
- Michael WM, Eder PS, Dreyfuss G. The K nuclear shuttling domain: A novel signal for nuclear import and nuclear export in the hnRNP K protein. EMBO (Eur Mol Biol Organ) J. 1997;16:3587–3598. doi: 10.1093/emboj/16.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MS, Blobel G. The GTP-binding protein Ran/TC4 is required for protein import into the nucleus. Nature. 1993;365:661–663. doi: 10.1038/365661a0. [DOI] [PubMed] [Google Scholar]
- Moore MS, Blobel G. Purification of a Ran-interacting protein that is required for protein import into the nucleus. Proc Natl Acad Sci USA. 1994;91:10212–10216. doi: 10.1073/pnas.91.21.10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroianu J, Blobel G. Protein export from the nucleus requires the GTPase Ran and GTP hydrolysis. Proc Natl Acad Sci USA. 1995;92:4318–4322. doi: 10.1073/pnas.92.10.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakielny S, Dreyfuss G. Import and export of the nuclear protein import receptor transportin by a mechanism independent of GTP hydrolysis. Curr Biol. 1997;8:89–95. doi: 10.1016/s0960-9822(98)70039-9. [DOI] [PubMed] [Google Scholar]
- Neville M, Stutz F, Lee L, Davis LI, Rosbash M. The importin-beta family member CRM1p bridges the interaction between Rev and the nuclear pore complex during nuclear export. Curr Biol. 1997;7:767–775. doi: 10.1016/s0960-9822(06)00335-6. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Nucleocytoplasmic transport: Signals, mechanisms and regulation. Nature. 1997;386:779–787. doi: 10.1038/386779a0. [DOI] [PubMed] [Google Scholar]
- Northrop JP, Ho SN, Chen L, Thomas DJ, Timmerman LA, Nolan GP, Admon A, Crabtree GR. NF-AT components define a family of transcription factors targeted in T-cell activation. Nature. 1994;369:497–502. doi: 10.1038/369497a0. [DOI] [PubMed] [Google Scholar]
- Ossareh-Nazari B, Bachelerie F, Dargemont C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science. 1997;278:141–144. doi: 10.1126/science.278.5335.141. [DOI] [PubMed] [Google Scholar]
- Palacios I, Weis K, Klebe C, Mattaj IW, Dingwall C. RAN/TC4 mutants identify a common requirement for snRNP and protein import into the nucleus. J Cell Biol. 1996;133:485–494. doi: 10.1083/jcb.133.3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Yaseen NR, Hogan PG, Rao A, Sharma S. Phosphorylation of the transcription factor NFATp inhibits its DNA binding activity in cyclosporin A-treated human B and T cells. J Biol Chem. 1995;270:20653–20659. doi: 10.1074/jbc.270.35.20653. [DOI] [PubMed] [Google Scholar]
- Paschal BM, Gerace L. Identification of NTF2, a cytosolic factor for nuclear import that interacts with nuclear pore complex protein p62. J Cell Biol. 1995;129:925–937. doi: 10.1083/jcb.129.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard VW, Michael WM, Nakielny S, Siomi MC, Wang F, Dreyfuss G. A novel receptor-mediated nuclear protein import pathway. Cell. 1996;86:985–994. doi: 10.1016/s0092-8674(00)80173-7. [DOI] [PubMed] [Google Scholar]
- Richards SA, Lounsbury KM, Carey KL, Macara IG. A nuclear export signal is essential for the cytosolic localization of the Ran binding protein, RanBP1. J Cell Biol. 1996;134:1157–1168. doi: 10.1083/jcb.134.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards SA, Carey KL, Macara IG. Requirement of guanosine triphosphate-bound Ran for signal-mediated nuclear protein export. Science. 1997;276:1842–1844. doi: 10.1126/science.276.5320.1842. [DOI] [PubMed] [Google Scholar]
- Ruff VA, Leach KL. Direct demonstration of NFATp dephosphorylation and nuclear localization in activated HT-2 cells using a specific NFATp polyclonal antibody. J Biol Chem. 1995;270:22602–22607. doi: 10.1074/jbc.270.38.22602. [DOI] [PubMed] [Google Scholar]
- Ruhl M, Himmelspach M, Bahr GM, Hammerschmid F, Jaksche H, Wolff B, Aschauer H, Farrington GK, Probst H, Bevec D, Hauber J. Eukaryotic initiation factor 5A is a cellular target of the human immunodeficiency virus type 1 Rev activation domain mediating trans-activation. JCell Biol. 1993;123:1309–1320. doi: 10.1083/jcb.123.6.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenstedt G, Saavedra C, Loeb JDJ, Cole CN, Silver PA. The GTP-bound form of the yeast Ran/TC4 homologue blocks nuclear protein import and appearance of poly(A)+ RNA in the cytoplasm. Proc Natl Acad Sci USA. 1995;92:225–229. doi: 10.1073/pnas.92.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki F, Price ER, Milan D, McKeon F. Role of kinases and the phosphatase calcineurin in the nuclear shuttling of transcription factor NF-AT4. Nature. 1996;382:370–373. doi: 10.1038/382370a0. [DOI] [PubMed] [Google Scholar]
- Siomi H, Dreyfuss G. A nuclear localization domain in the hnRNP A1 protein. J Cell Biol. 1995;129:551–560. doi: 10.1083/jcb.129.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stade K, Ford CS, Guthrie C, Weis K. Exportin 1 (CRM1p) is an essential nuclear export factor. Cell. 1997;90:1041–1050. doi: 10.1016/s0092-8674(00)80370-0. [DOI] [PubMed] [Google Scholar]
- Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995;82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]
- Yang J, Liu J, DeFranco DB. Subnuclear trafficking of glucocorticoid receptors in vitro: Chromatin recycling and nuclear export. J Cell Biol. 1997;137:523–538. doi: 10.1083/jcb.137.3.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda Y, Imamoto-Sonobe N, Yamaizumi M, Uchida T. Reversible inhibition of protein import into the nucleus by wheat germ agglutinin injected into cultured cells. Exp Cell Res. 1987;173:586–595. doi: 10.1016/0014-4827(87)90297-7. [DOI] [PubMed] [Google Scholar]