

Abstract
In polarized cells, signal transduction by cholera toxin (CT) requires apical endocytosis and retrograde transport into Golgi cisternae and perhaps ER (Lencer, W.I., C. Constable, S. Moe, M. Jobling, H.M. Webb, S. Ruston, J.L. Madara, T. Hirst, and R. Holmes. 1995. J. Cell Biol. 131:951–962). In this study, we tested whether CT's apical membrane receptor ganglioside GM1 acts specifically in toxin action. To do so, we used CT and the related Escherichia coli heat-labile type II enterotoxin LTIIb. CT and LTIIb distinguish between gangliosides GM1 and GD1a at the cell surface by virtue of their dissimilar receptor-binding B subunits. The enzymatically active A subunits, however, are homologous. While both toxins bound specifically to human intestinal T84 cells (K d ≈ 5 nM), only CT elicited a cAMP-dependent Cl− secretory response. LTIIb, however, was more potent than CT in eliciting a cAMP-dependent response from mouse Y1 adrenal cells (toxic dose 10 vs. 300 pg/well). In T84 cells, CT fractionated with caveolae-like detergent-insoluble membranes, but LTIIb did not. To investigate further the relationship between the specificity of ganglioside binding and partitioning into detergent-insoluble membranes and signal transduction, CT and LTIIb chimeric toxins were prepared. Analysis of these chimeric toxins confirmed that toxin-induced signal transduction depended critically on the specificity of ganglioside structure. The mechanism(s) by which ganglioside GM1 functions in signal transduction likely depends on coupling CT with caveolae or caveolae-related membrane domains.
In polarized epithelial cells, signal transduction by cholera toxin (CT)1 and the structurally related Escherichia coli heat-labile toxin type I (LTI) requires endocytosis of toxin–receptor complexes into the apical endosome, retrograde transport into Golgi cisternae or ER (Lencer et al., 1992, 1993, 1995a ; Orlandi et al., 1993; Majoul et al., 1996; Orlandi, 1997), and finally transport of toxin across the cell to its site of action on the basolateral membrane. In the human intestine, this process leads to intestinal salt and water secretion and the massive diarrhea seen in cholera.
CT and LTI are composed of five identical B polypeptides that bind GM1 at the cell surface, and a single A polypeptide. The A polypeptide is composed of two domains (A1 and A2) linked by an exposed loop containing a site for proteolytic cleavage and a disulfide bond that bridges the cleavage site (Sixma et al., 1991; Spangler, 1992; Zhang et al., 1995). The pentameric B subunit (55 kD) binds specifically five GM1 molecules with high affinity. Proteolytic cleavage within the exposed loop of the A subunit and reduction generates the enzymatically active A1 peptide (∼22 kD), which enters the cell and activates adenylyl cyclase by catalyzing the ADP-ribosylation of the α subunit of the heterotrimeric GTPase Gs (for review see Spangler, 1992). The A2 peptide (∼5 kD) forms the scaffolding that tethers the A and B subunits together and contains a COOH-terminal K(R)DEL motif. The KDEL-motif protrudes from the pentameric B subunit on the side that binds GM1 at the cell surface (Zhang et al., 1995).
Based on our published data, we have proposed that targeting of toxin into the cell may plausibly depend on the structure and function of the toxin's endogenous receptor ganglioside GM1 (Lencer et al., 1995a ). For example, we find that the endoplasmic reticulum targeting K(R)DEL motif on the toxin's A subunit is not absolutely required for toxin action (but in its absence, signal transduction is significantly less efficient), and that purified CT B subunit, in the absence of signals from A subunit, will also traverse a transcytotic pathway. Thus, simply binding or clustering GM1 by the toxin's pentameric B subunit appears to be sufficient for transport into all intracellular compartments necessary for signal transduction. Whether GM1 provides specific targeting information to this system (or whether other glycosphingolipids could perform the same function), and if so, how such a glycolipid may act as a sorting motif in vesicular transport, remains unknown.
In many cell types, a large fraction of GM1 clusters in caveolae. In fact, cholera toxin has already been used to identify caveolae in human A431 epidermoid cells, canine MDCK epithelial cells, and endothelium of rat lung (Parton, 1994; Parton et al., 1994; Schnitzer et al., 1995b ). Caveolae and related membrane structures display light density and resistance to detergent extraction by virtue of their lipid and possibly protein composition (Anderson, 1993a ; Schroeder et al., 1994; Simons and Ikonen, 1997). These specialized membrane domains exist on both intracellular (Brown and Rose, 1992) and plasma membranes (Anderson, 1993a ). Caveolae are thought to mediate key cellular functions that include ligand-induced signal transduction, protein and lipid sorting, endocytosis, and (in vascular endothelium) transcytosis (van Meer and Palade, 1989; Simons and Wandinger-Ness, 1990; Anderson et al., 1992; Dupree et al., 1993; van Meer, 1993; Lisanti et al., 1994; Parton et al., 1994; Schnitzer et al., 1994, 1995c ; Rodgers and Rose, 1996; Liu et al., 1997). Whether caveolae mediate toxin-induced signal transduction in human intestine, however, remains unknown.
Our aim in the present study was to test whether toxin action depends on binding or clustering GM1 specifically. To address this hypothesis, we took advantage of a member of the family of related type II heat-labile toxins produced by E. coli. The structures of type II toxins are similar but not identical to CT or LTI (Guth et al., 1986; Holmes et al., 1986; Chang et al., 1987; Fukuta et al., 1988). The major structural differences are found in the pentameric B subunits, and this is reflected functionally in that CT or LTI and type II toxins bind to different glycosphingolipids. In particular, the type II toxin LTIIb binds with highest affinity to ganglioside GD1a and not at all to GM1 (Fukuta et al., 1988). The structure and function of the enzymatic A subunits of CT or LTI and LTIIb, however, are homologous. They are ADP-ribosyltransferases with similar enzymatic activities in vitro (Chang et al., 1987; Lee et al., 1991). Like CT, the A2 peptide of LTIIb also contains a COOH-terminal KDEL-motif that protrudes from the B pentamer in a position where the KDEL-sequence may plausibly interact with membrane receptors (van den Akker et al., 1996).
The studies reported here show that signal transduction by heat-labile enterotoxins in the model polarized intestinal cell line T84 depends specifically on binding or clustering GM1. Binding to a closely related ganglioside, GD1a, renders the toxin inactive. We also find that ganglioside structure dictates toxin association with caveolae-like membrane domains. Thus, the mechanism(s) by which ganglioside GM1 functions in signal transduction likely depends on the ability of GM1 to couple cholera and related heat-labile enterotoxins with caveolae or caveolae-related membrane structures.
Materials and Methods
Materials, Toxins, and Antibodies
CT was obtained from Calbiochem (San Diego, CA), cholera toxin B subunit–colloidal gold was from List Biological Laboratories (Campbell, CA), and purified gangliosides GM1 and GD1a from Matreya, Inc. (Pleasant Gap, PA). All other commercially available reagents were from Sigma Chemical Co. (St. Louis, MO) unless otherwise stated.
Periplasmic extracts from recombinant E. coli containing CT and LTIIb and purified LTIIb were prepared as previously described (Lencer et al., 1995a ). Where indicated, LTIIb (20 nM) was activated by preincubation with 20 μg/ml trypsin for 30 min at 30°C as described (Lencer et al., 1997). Purified anthrax protective antigen was a gift from R.J. Collier (Harvard Medical School). Purified mutant LT G33D B subunit was a gift from T.R. Hirst (University of Bristol, Bristol, UK). Sulfo-NHS-biotin (Pierce Chemical Co., Rockford, IL) was used to label purified CT and LTIIb as previously described (Lencer et al., 1992). Antibodies used included polyclonal rabbit antiserum against CT A and B subunits (Lencer et al., 1995b ) and LTIIb holotoxin (Connell and Holmes, 1992) and monoclonal antibodies raised against CT B and A subunits (Holmes and Twiddy, 1983) and LTIIb B and A subunits (as described in Holmes and Twiddy, 1983). Monoclonal antibody 11E5 is specific for the B subunit of LtIIb, and 3C4 is specific for the A subunit of LTIIb. Neither 11E5 nor 3C4 cross-reacts with the A or B subunit of CT.
Cell Culture
T84 and Y1 cells obtained from American Type Culture Collection (Rockville, MD) were cultured and passaged as previously described (Lencer et al., 1995a ). Cells from passages 70–98 (T84) and 41–70 (Y1) were used for these experiments.
Preparation of Recombinant Wild-Type and Mutant CT and LTIIb Chimeric Toxins
All holotoxin-producing clones were made in the vector pBluescript SKII− (Stratagene, La Jolla, CA) and were expressed from the IPTG- inducible lac promoter of the vector. Table I identifies the toxin constructs used in this study. pMGJ148 encodes native CT holotoxin with the LTIIb-B gene leader expressing CT B (Jobling et al., 1997), and pMGJ176 encodes native LTIIb holotoxin. Chimeric holotoxin clones were made by reciprocal exchange of restriction fragments encoding the A1 polypeptide or A2 and B polypeptides using the unique ClaI restriction site naturally present in the DNA encoding the 11-residue loop between cys-187 of CT A1 and cys-199 of CT A2. A ClaI restriction site was introduced into the DNA encoding the loop between cys-185 of LTIIb A1 and cys-197 of LTIIb A2 in two steps. First, the LTIIb-A1 coding sequence was PCR-amplified from a native LTIIb holotoxin clone (MGJ176) with oligonucleotides IIbA1RC (5′GCCATCGATGCTTTATTATTTGGTAGACA3′) and M13 reverse primer and cloned into SmaI-digested pUC18 to produce pMGJ182. Second, the LTIIb-A2-B coding sequence was similarly amplified and cloned using oligonucleotides IIb-A2 (5′CGGGCATCGATGGATACCTGTGCCTC3′) and M13-20 primer to produce pMGJ178. An XbaI-ClaI fragment of pMGJ182 (LTIIb-A1) and a ClaI-SalI fragment of pMGJ178 (LTIIb-A2-B) were cloned into XbaI-SalI–digested pBluescript SKII− to create pMGJ183. This clone produced LTIIb-holotoxin with a single amino acid substitution (S193M) in the A1-A2 loop encoded by the ClaI site. A ClaI-KpnI fragment of pMGJ178 (LTIIb-A2-B) was cloned into a ClaI-KpnI–cut pMGJ148 to create pMGJ179 encoding the CT-A1/LTIIb-A2/LTIIb-B chimera. An NheI-ClaI fragment of pMGJ182 (LTIIb A1) was cloned into XbaI-ClaI–cut pMGJ148 to create pMGJ184, encoding the LTIIb-A1/CT-A2/CT-B chimera.
Table I.
Construction of Recombinant Wild-Type and Chimeric Toxins
| Clone | Toxin or chimera | A1 subunit | Junction | A2 subunit B subunit | ||||
|---|---|---|---|---|---|---|---|---|
| pMGJ148 | CT (native) | CTA1 | CGNAP R*SSMSNTC | CTA2-CTB | ||||
| 187 199 | ||||||||
| pMGJ179 | CTA/LTIIbB | CTA1 | CGNAP R*SSMDTTC | LTIIbA2-LTIIbB | ||||
| 187 197 | ||||||||
| pMGJ183 | LTIIb(S193M) | LTIIbA1 | CLPNN*KASMDTTC | LTIIb A2-LTIIbB | ||||
| 185 197 | ||||||||
| pMGJ184 | LTIIbA/CTB | LTIIbA1 | CLPNN*KASMSNTC | CT A2-CTB | ||||
| 185 199 | ||||||||
| pMGJ187 | LTIIbA/CTB | LTIIbA1 | KKYIKRQIFF | LTIIbA2::CTA2-CTB | ||||
| 213 224 |
Amino acid sequences of the junctions formed between A1 and A2 peptides are shown in letter code. Numbers beneath cysteines indicate the position in the native protein. In the native protein these cysteines are linked by disulfhydryl bond.
above the letter codes identifies the serine-protease cleavage site linking the A1 and A2 peptides. The cleavage site for pMGJ187 is the same as for native LTIIb and the fusion joint (underlined) is within the A2 domain instead of between the A1 and A2 domains.
Plasmid pMGJ187 was created to produce an LTIIbA-CTB chimera with a fusion joint in the middle of the A2 domain so as to maximize the homologous A1–A2 and A2–B domain interactions, predicted to enhance holotoxin formation. The dibasic KR peptide at the fusion joint is conserved between CT A2 and LTIIb A2 and is located at the point where the A2 domain penetrates the B subunit in both toxins. A hybrid LTIIbA2-CTA2 oligonucleotide IIBCTKR (KR coding sequence underlined; TTTAAGAAATATATAAAGAGACAAATATTTTCAGGC) was used in conjunction with a CTB reverse primer (SP4, CTTGAAAAGTTGCACCATTC) to PCR amplify the region encoding CT-A2-CT-B from a clone lacking T7 primer sequences. LTIIb-A1 sequences were added by using the PCR product to prime on a IIb holotoxin clone and reamplifying with a vector primer (T7; Stratagene) and SP4. The product was then digested with AccI and cloned into pMGJ184, replacing the A2 encoding sequences.
For chimeric toxins 179 (pMGJ179) and 184 (pMGJ184), junctions of the chimeric A subunits were made within the C-loop connecting the A1 and A2 peptides. This preserved the complete A2 peptide, including the KDEL-motif, and all interactions between the A2 peptide and its respective pentameric B subunit. For chimeric toxin 187 (pMGJ187), the junction of the chimeric A subunit was in the middle of the A2 domain, thereby conserving the entire loop region between A1 and A2 from LTIIb.
Preparation of Toxin Extracts
500 ml Luria Broth cultures of E. coli TE1 bearing each of the plasmids in Table I were grown at 37°C with 50 μg/ml each of ampicillin and methicillin and induced overnight with 200 μM IPTG. Extracts were prepared essentially according to French et al. (1996), except that cells were resuspended in 15 ml 20% sucrose, 0.2 M Tris, pH 7.5, 1 mM EDTA, 0.5 mg/ml lysozyme, incubated for 15 min at ambient temperature followed by osmotic shock with the addition of 15 ml cold sterile water, mixed, and incubated 15 min on ice. The supernatant was cleared by centrifugation at 16,000 g at 4°C and retained as crude toxin extract. Periplasmic extracts were dialyzed overnight at 4°C against 1,000-vol excess HBSS (containing in g/liter 0.185 CaCl2, 0.098 MgSO4, 0.4 KCl, 0.06 KH2PO4, 8 NaCl, 0.048 Na2HPO4, 1 glucose, to which 10 mM Hepes, pH 7.4, was added).
Electrophysiology, Mouse Y1 Adrenal Cell, and Toxin-binding Assays
HBSS was used for all assays unless otherwise stated. An antibody-based or ligand-binding ELISA was used to define binding isotherms of purified CT and LTIIb, and biotin-labeled LTIIb was used to define binding specificity as previously described (Lencer et al., 1995c ). For electrophysiological studies on T84 cells, measurements of short circuit current (Isc) and resistance (R) were performed with 0.33 cm2 monolayers as previously described (Lencer et al., 1992).
Toxin action on mouse Y1 adrenal cells was assessed as a cAMP- dependent shape change as described (Guth et al., 1986). Two methods were used to calibrate toxin activity. First, 100-μl samples of serial twofold dilutions in culture medium of periplasmic extracts containing toxins were applied to 96-well plates of Y1 cells grown to confluence for 2 d. After overnight incubation at 37°C, toxin-induced shape change was scored based on the fraction of cells exhibiting rounding, as assessed by light microscopy. Scores from 0 to 4+ were assigned as follows: 4+ (75–100% cells rounded), 3+ (50–75% cells rounded), 2+ (25–50% cells rounded), 1+ (<25% cells rounded), and 0 (no detectable cell rounding). The amount of toxin present in the last dilution giving 4+ rounding was assigned a value of 1 unit (U). Toxin titers were calculated as (dilution factor) × 10 U/ml original sample. In the second method, toxin titers were assessed in a 3-h time course and graded against standard curves constructed in parallel by serial dilutions of purified LTIIb (20–0.08 nM).
Detergent Extractions of Filter-grown Monolayers
T84 cells grown on 5-cm2 filters were incubated apically with 20 nM CT, LTIIb, or no toxin in HBSS for 20–30 min at 4°C, washed to remove all unbound toxin, immersed in 1.0 ml of ice-cold 1% TTBS (150 mM NaCl, 10 mM Tris, 1% Triton X-100, pH 7.5, 0.175 mM PMSF, and 20 μg/ml chymostatin), and tumbled end over end for 30 min at 4°C. Triton-soluble proteins were removed and saved. Triton-insoluble proteins were solubilized in 1% TTBS containing 1% N-octylglucoside at 4°C. Acetone precipitates of solubilized proteins were recovered by centrifugation at 15,000–20,000 g for 15 min and analyzed by SDS-PAGE and Western blotting (Lencer et al., 1995b ).
Sucrose Equilibrium Density Centrifugation
One or two confluent monolayers of T84 (45 cm2 each) or a 150-cm2 flask of confluent Y1 cells was used for isolation of detergent-insoluble membranes. All steps were performed at 4°C. Cells were scraped into 2 ml of ice-cold 1% TTBS and homogenized with five strokes in a tight-fitting Dounce homogenizer on ice. The homogenate was adjusted to 40% sucrose by addition of 2 ml of 80% sucrose in 1% TTBS, layered under a linear 5–30% sucrose gradient, and centrifuged at 39,000 rpm for 16–20 h in a swinging bucket rotor (model SW41; Beckman Instruments, Palo Alto, CA). The presence of a floating membrane fraction was noted visually. Sequential 0.5- or 1-ml fractions were collected from the top of the gradient. 20 μl from each fraction was analyzed by SDS-PAGE and Coomassie stain or Western/ligand blot. Sucrose density was monitored by refractometry. Light scattering was measured as absorbance at 600 nm (Spectra Max; Molecular Dynamics, Sunnyvale, CA).
Ganglioside Extraction and Analysis
Ganglioside-enriched lipid extracts and ligand blots using LTIIb and CT were prepared as modified from described methods (Fukuta et al., 1988; Schnaar and Needham, 1994). Total lipids were extracted from 1,500– 4,500 cm2 confluent T84 monolayers in chloroform/methanol/water (10:10:1) in a total volume of 10 ml. For quantitative thin layer chromatography, phospholipids and cholesterol were removed on silica gel. Thereafter, the glycosphingolipids were separated by ion exchange chromatography, and the gangliosides were eluted in a monosialoganglioside fraction and polysialoganglioside fraction. The fractions were separated on HPTLC plates (Silica Gel 60; E. Merck, Darmstadt, Federal Republic of Germany), and the gangliotetrose series (GM1, GD1a, GD1b, and GT1b) was detected by ligand blotting with CT B subunit (1 nM) and anti–CT B subunit antibodies in an ELISA performed on the HPTLC plate. Values are presented as total mass gangliosides (pmol) per mg cell protein. For “dot blots” and for qualitative thin layer chromatography, the concentrated total lipid extracts were enriched for gangliosides by Folch cut in chloroform/methanol/water (10:10:1). 100 ng each of ganglioside GM1 and GD1a and 10–15 μl representing 20–30% of total T84 cell ganglioside-enriched lipid extracts were applied to HPTLC strips or HPTLC plates and analyzed as described (Fukuta et al., 1988). For dot blots, GM1 and GD1a were detected by ligand blotting with CT (1 nM) and LTIIb (1 nM) and anti-CT or LTIIb antibodies in an ELISA performed on the HPTLC plate.
Statistics
Data were analyzed using Statview 512+ software (Brainpower, Inc., Calabasas, CA).
Results
We used CT and the type II heat-labile toxin from E. coli LTIIb to test the hypothesis that GM1 provides specificity to toxin action. LTIIb and CT are structurally and functionally homologous. LTIIb, however, binds GD1a and displays no detectable binding to GM1 (Fukuta et al., 1988; Lee et al., 1991).
CT and LTIIb Bind Specifically to T84 Cell Apical Membranes
Initial experiments showed that both CT and LTIIb bound to receptors on T84 cells with high affinity (2–5 nM) (Fig. 1 A). Competition experiments showed that receptor binding was specific (Fig. 1 B). Competition with LTIIb (1 μM) inhibited by 70% the binding of biotin-labeled LTIIb to T84 cell membranes. Binding was not inhibited, however, by competition with anthrax protective antigen (200 nM), chicken IgY (1 μM), bovine serum albumin (5 mg/ml), or rLTI B subunit(G33D) (500 nM). Similarly for CT, competition with rCT B subunit inhibited CT binding by 93%.
Figure 1.

CT and LTIIb bind specifically with high affinity to intact T84 monolayers. All binding assays were performed in HBSS containing 5% bovine serum albumin. Nonspecific signals in the absence of applied toxins were nearly identical to background (0.09 O.D. units). (A) Binding isotherms for CT and LTIIb applied at 4°C to T84 cells grown on plastic. Polyclonal anti-CT or anti-LTIIb antibodies were used to detect toxin bound at the cell surface. (B) Steady-state binding of biotin-labeled LTIIb (5 nM) or CT (5 nM) to T84 cells at 4°C in the presence of excess unlabeled LTIIb (1 μM), LTG33D B subunit (500 nM), purified anthrax protective antigen (200 nM), chicken IgY (1 μM), or unlabeled CT (1 μM) as indicated. Biotin-labeled toxins are indicated by *. In these studies, enzyme-linked avidin was used to detect toxin bound at the cell surface.
To demonstrate that T84 cells express GD1a and GM1, ganglioside-enriched lipid extracts of confluent monolayers of T84 cells (1,500–4,500 cm2) were prepared and dotted onto HPTLC strips, dried, blocked for nonspecific binding, and assessed for the presence of GM1 and GD1a by ligand affinity-blot using purified CT (20 nM) or rLTIIb (20 nM). Purified GM1 and GD1a were used as standards (300 ng each). Fig. 2 A shows that T84 cells express lipids to which both CT and LTIIb bind specifically. Anthrax toxin protective antigen (12 nM) did not label the lipid dot blots. Neither CT nor LTIIb bound proteins of total T84 cell extracts (50–100 μg) as assessed by SDS-PAGE and ligand blot (not shown). Analysis of glycolipid composition by thin layer chromatography confirmed the presence of GM1 and GD1a in polarized T84 cell membranes (Fig. 2 B). T84 cells contained per microgram total protein 0.82 pmol GM1, 2.41 pmol GD1a, 0.38 pmol GD1b, and 2.7 pmol GT1b (data from Pam Fredman and Jan Holmgren, University of Göteborg, Sweden). Thus, T84 cells express specific high-affinity receptors for LTIIb and CT, which represent ganglioside GD1a and GM1, respectively.
Figure 2.
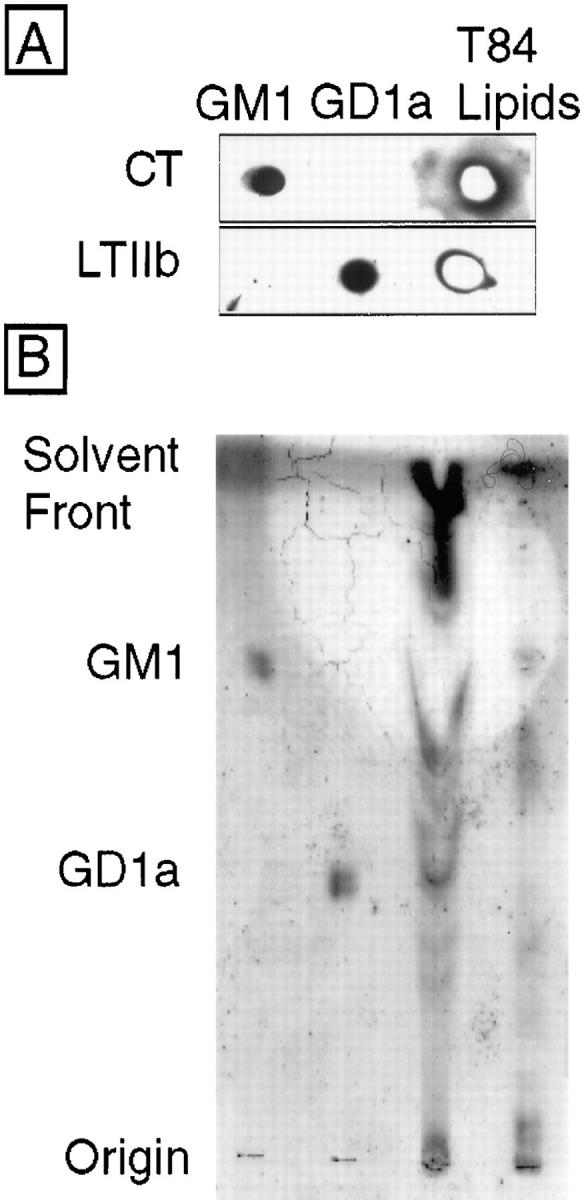
Ganglioside- enriched lipid extracts of T84 cell monolayers contain both GM1 and GD1a. (A) Lipid extracts prepared from 1,500 cm2 confluent T84 cells (third lane) and purified GM1 (first lane) and GD1a (second lane) standards (300 ng each) were applied to activated silica gel plates, processed as described in Materials and Methods, and ligand blotted with CT (1 nM, top) or LTIIb (1 nM, bottom). Toxin binding to ganglioside extracts was assessed by application of specific antitoxin antibodies, followed by HRP-conjugated secondary antibody, and development using enhanced chemiluminescence. (B) Size resolution of T84 cell gangliosides by thin layer chromatography and resorcinol spray. GM1 standard (first lane), GD1a standard (second lane), T84 cell (4,500 cm2) lipid extract lower phase Folch cut (third lane), and upper phase Folch cut (fourth lane). In this preparation, the bulk of acidic glycolipids including GD1a remained associated with the lower lipid soluble phase because of conditions of Folch extraction (low salt and pH). Gangliosides migrating with GD1a are apparent in the third lane (and faintly in the fourth). Gangliosides migrating with GM1 are apparent in the fourth lane.
Toxin Binding to GM1 but Not GD1a Leads to a Physiological Response
We next examined the ability of CT and LTIIb to elicit cAMP-dependent Cl− secretion. Fig. 3 A shows the time course of Cl− secretion elicited by CT (20 nM) or LTIIb (20 nM) applied to apical reservoirs of T84 cell monolayers. After a characteristic “lag” phase, CT elicited a brisk secretory response. LTIIb, however, was inactive. The addition of vasoactive intestinal peptide (VIP, 3 nM) to basolateral reservoirs at 125 min elicited a rapid response, demonstrating the viability of these monolayers. The inability of LTIIb to elicit a response from T84 cells was not dose dependent, as 500 nM LTIIb was still inactive. (A small signal, Isc = 4.8 μA/cm2 was obtained at 10 μM LTIIb, n = 2.) Structural differences between CT and LTIIb in the C-loop connecting the A1 and A2 peptides did not explain these results since proteolytic activation of the LTIIb A subunit by trypsin pretreatment in vitro had no effect on signal transduction by LTIIb (n = 2). LTIIb was also not active when applied to basolateral membranes of T84 cells, indicating that toxin function did not depend on cell polarity (data not shown). To demonstrate that LTIIb was nonetheless as equally functional as an ADP-ribosyl transferase, we applied both CT and LTIIb to mouse Y1 adrenal cells. Y1 adrenal cells respond to cAMP-agonists, including both CT and LTIIb with a characteristic shape change (Fig. 3, B–E). In this assay, LTIIb displayed greater potency than CT (toxic dose 10 vs. 300 pg/well, LTIIb vs. CT).
Figure 3.


Signal transduction by CT and LTIIb in polarized human intestinal T84 monolayers and mouse Y1 adrenal cells. (A) Time course of Cl− secretion induced by application of CT (20 nM) or LTIIb (20 nM) to apical reservoirs of T84 cell monolayers. The viability of monolayers exposed to LTIIb was demonstrated by application of the cAMP-agonist VIP to basolateral reservoirs at 120 min. (B–E) Bright field micrographs of mouse Y1 adrenal cells treated for 180 min at 37°C with the cAMP-agonist forskolin (10 μM, C), CT (20 nM, D), LTIIb (20 nM, E), or buffer alone (B).
Data from 12 independent experiments on T84 cells are summarized in Table II. The results of these studies show that high-affinity binding of LTIIb to the cell surface of T84 cells is not sufficient for signal transduction. These results provide evidence that signal transduction by CT in T84 cells depends on toxin binding or clustering GM1 specifically—a function of the toxin's B subunit.
Table II.
Effect of CT vs. LTIIb on cAMP-dependent Cl− Secretion in Human Intestinal T84 Cells
| Purified native toxin | LAG | dlsc/dt | Peak Isc | |||||
|---|---|---|---|---|---|---|---|---|
| min | μA/cm/min | μA/cm2 | ||||||
| CT | 20 nM | 26 ± 1.3 | 1.2 ± 0.2 | 59 ± 5 | ||||
| n = 9 | ||||||||
| LTIIb | 20 nM | >128* | Not Present | 3.9 ± 0.2* | ||||
| n = 12 | ||||||||
Values are means ± SE.
Peak Isc was obtained at 120 min. ANOVA, P = 0.0001.
Functional Association of Ganglioside GM1 with Detergent-insoluble Membrane Domains
The requirement for binding GM1 specifically in toxin action implies that GM1 can distinguish between membrane components in T84 cells that function in toxin-induced signal transduction. Based on available evidence, the membrane structures recognized by GM1 (or clusters of GM1) and likely to subserve such a function are caveolae or “caveolae-like” detergent-insoluble membrane domains (Parton, 1994; Parton and Simons, 1995; Schnitzer et al., 1995b , 1996). Thus, to test whether CT and LTIIb may partition differentially into caveolae-like membranes, we examined the detergent-solubility of CT–GM1 and LTIIb–GD1a complexes in T84 cells.
Initial studies showed that the CT–GM1 complex fractionated with Triton-insoluble membrane fractions isolated from T84 cell monolayers at 4°C (Fig. 4, A and B). These membrane fractions were enriched >100-fold in a subset of cellular proteins that included CD73 (the GPI-linked 5′-ectonucleotidase) and caveolin 1 (Strohmeier et al., 1997) but were depleted in B1 integrins and the Na+ K+ 2Cl− cotransporter (data not shown). The interactions between CT–GM1 and Triton-insoluble membranes were stable and resisted six successive rounds of extraction in detergent (as assessed biochemically). While some variability was observed in the fraction of CT–GM1 solubilized by 1% Triton X-100, CT–GM1 always fractionated with detergent-insoluble membranes. In six of nine studies, the overwhelming mass of CT–GM1 fractionated with these membranes (as in Fig. 4 A). In contrast, nearly all of the LTIIb–GD1a complex was solubilized by 1% Triton X-100, and both A and B subunits of LTIIb were found at the bottom of the gradient with all other solubilized proteins (Fig. 4 B, n = 9). In some studies (especially when concentrations >50 nM were used), a small amount of LTIIb could be detected in detergent-insoluble light-density membrane fractions (estimated <1–2%). In all studies on T84 cells, the differential association of LTIIb and CT with Triton-insoluble membranes was readily apparent.
Figure 4.

Association of the CT–GM1 and LTIIb–GD1a complexes with detergent-insoluble apical membrane domains in human T84 and mouse Y1 cells. Polarized human intestinal T84 or nonpolarized mouse Y1 adrenal cell monolayers were exposed apically to CT (20 nM) or LTIIb (20 nM) at 4°C for 30 min, and unbound toxins were removed by washing. The monolayers were homogenized in 1% TTBS at 4°C, adjusted to 40% sucrose, and layered under a 5–30% continuous sucrose gradient as described in Materials and Methods. Fractions from left to right (top to bottom) represent linear sucrose gradient from 5–32% sucrose. (A) SDS-PAGE and Coomassie stain (top) and Western blot (bottom) of sucrose gradient fractions from T84 cells. Fractions 2–15 represent linear sucrose gradient from 5–32% sucrose. Less than 1% total cellular protein floats into the gradient (fractions 6–9, representing 18.2–22.8% sucrose). This fraction is highly enriched for the CT–GM1 complex as assessed by Western blot. Fractions 16–19 represent Triton-soluble proteins at 40% sucrose. (B) Sucrose gradient of T84 cells exposed apically to biotin-labeled CT or LTIIb as described above and analyzed for CT or LTIIb B subunits by avidin blot. For gradients labeled CT and LTIIb, fractions 10 and 11 represent 19.4 –21% sucrose. Fractions 18–25 represent solubilized proteins at 40% sucrose. (C) Sucrose gradient of Y1 cells exposed to CT or LTIIb as described above and analyzed for CT or LTIIb A subunits by Western blot. For CT, fractions 11–13 represent 19.5–23.9% sucrose. For LTIIb, fractions 9–11 represent 19–24% sucrose. In both gradients, fractions 18–25 represent solubilized proteins at 40% sucrose. Lanes C indicate 1 ng of purified toxins run as controls. In all studies, A and B subunits for CT and LTIIb fractionated together.
As CT elicited a physiological response from T84 cells but LTIIb did not, these data raised the distinct possibility that toxin association with detergent-insoluble membranes may be required for signal transduction. To test this idea, mouse Y1 adrenal cells were examined. As defined above, both CT and LTIIb elicit a cAMP-dependent shape change in this cell type, and LTIIb is more potent than CT. Thus, if toxin action depends on association with detergent-insoluble membranes, studies on Y1 cells should show that both CT–GM1 and LTIIb–GD1a fractionate with membranes that float up into the sucrose gradient after detergent extraction. These studies showed the predicted results. In Y1 cells, both CT–GM1 and LTIIb– GD1a complexes fractionated with detergent-insoluble membranes (Fig. 4 C, n = 2). Thus, in human T84 and mouse Y1 cells, the ability of each toxin to associate with this specialized membrane fraction correlated with the ability of each toxin to elicit a cAMP-dependent intracellular signal.
LTIIb-CT Chimeric Toxins
To verify this idea, recombinant chimeric toxins were prepared by combining the enzymatic A1 domains of CT with the A2 domains and B subunit of LTIIb, and vice versa (Table I). In doing so, LTIIb A subunits with CT A2 domains were targeted into the cell by CT B subunits, which bind GM1 (chimeras 187 and 184, termed LTIIbA/CTB), and CT A subunits with LTIIb A2 domains were targeted into the cell by LTIIb B subunits, which bind GD1a (chimera 179, termed CTA/LTIIbB). Recombinant LTIIb S193M (see Materials and Methods) was used to construct the chimeric toxins described above, and this toxin predicted to exhibit wild-type phenotype was also prepared. Trypsin-cleavage sites between A1 and A2 domains were present in all of the chimeric toxins. The chimeras 184 and 187 containing alternative fusion sites within the A polypeptide were generated to test for possible effects of the fusion joint location on A and B subunit assembly and function. Western blots of each preparation confirmed the predicted structures of the chimeric toxins (data not shown).
The function of each recombinant toxin in periplasmic extracts prepared from E. coli TE1 was examined by applying samples of the extracts to apical membranes of polarized human intestinal T84 cell monolayers or to mouse Y1 adrenal cells. Preparations of all wild-type and chimeric toxins elicited a potent cAMP-dependent shape change from mouse Y1 adrenal cells, demonstrating that all toxins were functional. Thus, in Y1 cells, toxin binding to either GM1 or GD1a led to cAMP-dependent shape change. In contrast, signal transduction by chimeric toxins applied to T84 cells was dependent on toxin binding to GM1 specifically.
Chimeric Toxins That Enter the Cell via Binding or Clustering GM1
Fig. 5 A shows that wild-type LTIIb and LTIIb S193M toxins, which bind GD1a (5,000 U/ml), were inactive when applied to apical membranes of T84 monolayers (as expected). The chimeric toxin 184 (LTIIbA/CTB, 5,000 U/ml), however, which was predicted to enter the cell via binding GM1, elicited a distinct Cl− secretory response. Nearly identical results were obtained with chimeric toxin 187 (also LTIIbA/CTB, 5,000 U/ml), indicating that the failure of wild-type LTIIb to elicit a secretory response in T84 cells cannot be due solely to the structure of the peptide loop between C185 to C197 that joins the A1 and A2 domains of LTIIb. We also found that currents induced by LTIIbA/CTB chimeric toxins were not as great as those induced by wild-type (wt) CT, suggesting that CT and LTIIb A subunits may display differential potencies for intact human T84 cells. These data are summarized in Fig. 6 A.
Figure 5.
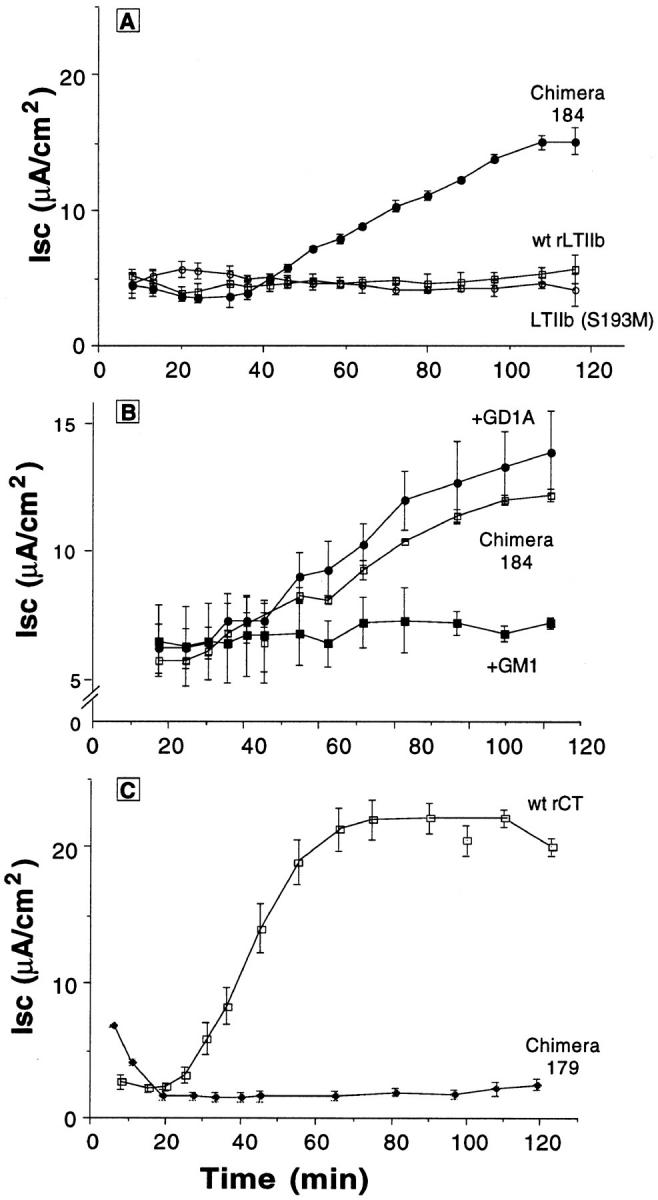
Time course of CT and LTIIb chimeric toxins on cAMP-dependent Cl− secretion in polarized T84 monolayers. (A) Time courses of Cl− secretion (Isc) induced by application of purified LTIIb, LTIIb(S193M), or chimeric toxin 184 (LTIIbA/ CTB) to apical reservoirs of T84 cell monolayers. (B) Time courses of Cl− secretion induced by application of chimeric toxin 184 (LTIIbA/CTB) to apical reservoirs of T84 cell monolayers in the absence or presence of purified GM1 (10 μM) or GD1a (10 μM) as competitive inhibitors. (C) Time courses of Cl− secretion induced by application of rCT, or chimeric toxin 179 (CTA/ LTIIbB) to apical reservoirs of T84 cell monolayers. All toxins were applied in equivalent concentrations of 5,000 U/ml as assessed by bioassay on Y1 cells.
Figure 6.

Effect of CT and LTIIb chimeric toxins on cAMP-dependent Cl− secretion in polarized T84 monolayers. (A) ΔIsc (peak Isc mean ± SEM, 120 min after toxin addition corrected for baseline currents) for wt LTIIb(S193M), chimeric toxin 187, or chimeric toxin 184 (LTIIbA/CTB) applied to apical reservoirs of T84 cell monolayers in the presence or absence of purified GM1, GD1a, rCT B subunit, and rLTIIB as competitive inhibitors. ANOVA, P = 0.0001. * indicates conditions significantly different from chimera 184 alone (third column) at P ≤ 0.05 by multiple comparison procedures. (B) ΔIsc (peak Isc mean ± SEM, 120 min after toxin addition corrected for baseline currents) for wt rCT and chimeric toxin 179 (CTA/LTIIbB) to apical reservoirs of T84 cell monolayers in the presence or absence of GM1 or rCT B subunit as competitive inhibitors. ANOVA, P = 0.0001. * indicates conditions significantly different from rCT alone (third column) at P ≤ 0.05 by multiple comparison procedures. In all studies, mean ± SEM baseline currents were 1.1 ± 0.2 μA/cm2, n = 25.
To show that the Cl− secretory response induced by chimeric toxin 184 (LTIIbA/CTB) was dependent on the specificity of B subunit binding to GM1, we examined the time course of toxin action in the presence or absence of excess soluble GM1 or GD1a as competitive and specific inhibitors of B subunit function. Fig. 5 B shows that preincubation with 10 μM GM1 blocked completely the secretory response elicited by chimera 184 (LTIIbA/CTB, 5,000 U/ml). Preincubation with GD1a (10 μM), however, had no effect. Furthermore, competition with rCT B subunit to block binding of holotoxin also inhibited (dose dependently) the Cl− secretory response induced by chimera 184 (LTIIbA/ CTB). Competition with 10 μM LTIIb had no inhibitory effect, but these studies (though still interpretable) were confounded by a small secretory response induced by LTIIb at this high dose (4.8 μA/cm2, n = 2). These data (summarized in Fig. 6 A) show that the secretory response elicited by chimera 184 (LTIIbA/CTB) was dependent on binding GM1, a function of its chimeric B subunit.
Chimeric Toxins That Enter the Cell via Binding or Clustering GD1a
Fig. 5 C shows the secretory response elicited by the complementary chimeric toxin 179 (CTA/LTIIbB). While recombinant wild-type CT elicited a strong secretory response (as expected), toxin-induced Cl− secretion was greatly attenuated when the CT A subunit was forced to enter the cell via binding GD1a (data summarized in Fig. 6 B). In two of three independent preparations, periplasmic extracts containing chimeric toxin 179 were completely inactive on T84 cells, even though the chimeric toxin elicited a potent cAMP-dependent response from Y1 cells (5,000 U/ml). In all studies, periplasmic extracts from E. coli TE1 not harboring toxin-producing plasmids were physiologically inactive when applied to either cell type (human T84 or mouse Y1, data not shown). Thus, to show that the action of chimeric toxin 179 (CTA/LTIIbB) on Y1 cells was dependent on the specificity of B subunit binding to GD1a (as predicted), we examined the cAMP-dependent shape change induced by chimeric toxin 179 in the presence or absence of excess soluble GM1 or GD1a as competitive and specific inhibitors of B subunit function. Both wt rCT and chimeric toxin 179 (CTA/LTIIbB) (5,000 U/ml) elicited maximal cell rounding (75–100% cells rounded) after incubation at 37°C for 100 min. Preincubation with 10 μM GD1a almost completely inhibited the cell rounding response to chimera 179 (0–25% cells rounded at 100 min, n = 2 in quadruplicate). In contrast, preincubation with 10 μM GM1 had no effect (75–100% cells rounded at 100 min). The converse was found for wt rCT. As expected, preincubation with GM1 (10 μM) inhibited the shape change induced by rCT, but preincubation with GD1a (10 μM) had no effect.
In parallel experiments, signal transduction by wt LTIIb and chimeric toxin 184 (LTIIbA/CTB) was also examined in mouse Y1 cells. GM1 (10 μM) completely inhibited the response of Y1 cells to chimeric toxin 184 (LTIIbA/CTB) (0–25% rounded cells at 140 min, n = 2 in quadruplicate), but GD1a (10 μM) had no effect (75–100% cells rounded at 140 min). Likewise, GD1a inhibited the response to wt LTIIb (25–50% rounded cells at 140 min, n = 2 in quadruplicate), but GM1 had no effect (75–100% cells rounded at 140 min). Thus, signal transduction by chimeric toxin 184 and wt LTIIb in Y1 cells was also dependent on the specificity of ganglioside binding.
Taken together, these studies show that LTIIb A subunits were rendered functional in T84 cells by targeting them into T84 cells via binding GM1 (chimeric toxins 184 and 187, LTIIbA/CTB), and conversely, CT A subunits were rendered inactive by targeting them into the cell by binding GD1a (chimeric toxin 179, CTA/LTIIbB) (summarized in Fig. 6, A and B). Thus, the cAMP-dependent responses elicited by both chimeric toxins 179 (CTA/LTIIbB) and 184 (LTIIbA/CTB) in mouse Y1 or human T84 cells were dependent on binding GD1a or GM1, respectively, functions of their chimeric B subunits.
Ganglioside Structure Dictates Association with Caveolae-like Membrane Domains
To strengthen the correlation between toxin-induced signal transduction and association with caveolae-like detergent-insoluble membranes, chimeric toxins 184 (LTIIbA/ CTB) and 179 (CTA/LTIIbB) bound to apical receptors on T84 cell monolayers were solubilized in Triton X-100 and fractionated on sucrose gradients. Fig. 7 shows that chimeric toxin 184 (LTIIbA/CTB) floated into the sucrose gradient as predicted. In contrast, the complementary chimeric toxin 179 (CTA/LTIIbB) was completely solubilized and remained at the bottom of the gradient (n = 2). Thus, in T84 cells, binding (or clustering) GM1 dictates both toxin association with detergent-insoluble membrane domains and toxin-induced signal transduction.
Figure 7.

Association of chimeric toxins 184 LTIIbA/CTB (A) and 179 CTA/LTIIbB (B) with detergent-insoluble apical membrane domains in human T84 cells. Sucrose gradient of T84 cells exposed apically to chimeric toxins 184 and 179 and analyzed for CT or LTIIb B subunits by Western blot. For gradient labeled chimera 184, fractions 13–16 represent 19.2–24.8% sucrose. Fractions 20–25 represent solubilized proteins at 40% sucrose. For gradient labeled chimera 179, fractions 12–16 represent 19–28% sucrose. Fractions 20–25 represent solubilized proteins at 40% sucrose. Association of toxin with detergent-insoluble membranes depends on binding GM1—a function of the toxin's B subunit.
Discussion
Functional Specificity of GM1 in CT-induced Signal Transduction
The results of these studies show that specific functional properties of GM1 that are not shared by GD1a are essential for cholera toxin action in human intestinal T84 cells. Ganglioside function was assessed using two related enterotoxins, CT and LTIIb. These toxins distinguish between gangliosides GM1 and GD1a at the cell surface by virtue of their dissimilar receptor binding B subunits. Their enzymatically active A subunits, however, are homologous in structure and function. Both toxins activate adenylyl cyclase inside the cell via the same mechanism of action: ADP-ribosylation of the heterotrimeric GTPase Gsα. Our data show that signal transduction by CT and LTIIb depends on the specificity of receptor binding, a function of the toxin's B subunit.
Two lines of evidence support this view. First, CT and type I E. coli heat-labile toxin LTI bound GM1 in T84 cells and induced a physiological cAMP-dependent Cl− secretory response (this study and Lencer et al., 1995a , 1997). Unlike CT and LTI, however, type II E. coli heat-labile toxin LTIIb displayed no detectable binding to GM1, and LTIIb did not elicit a physiological response in T84 cells. Even so, LTIIb bound with high affinity to GD1a on human T84 cells, and the enzymatic A subunit of LTIIb was capable of activating adenylyl cyclase, as evidenced by the cAMP-dependent shape change elicited by LTIIb in cultured mouse Y1 adrenal cells. That toxin action depended critically on the specificity of ganglioside binding (a function of the B subunit) was confirmed by using recombinant CT and LTIIb chimeric toxins. Thus, LTIIb A subunits were rendered functional in T84 cells by targeting the A subunit into the cell via binding GM1 (chimeric toxin LTIIbA/CTB), and conversely the function of CT A subunits were rendered inactive by targeting the A subunit into the cell by binding GD1a (chimeric toxin CTA/LTIIbB).
Toxin Action Depends on Association with Detergent-insoluble Membrane Domains
We also find that the structure of the ganglioside receptor determines association of toxin with caveolae-like detergent-insoluble membrane domains. This was closely correlated with toxin function as assessed by the activities of wt CT, LTIIb, and chimeric toxins CTA/LTIIbB and LTIIbA/ CTB on human intestinal T84 and mouse Y1 adrenal cells. That partitioning into detergent-insoluble membranes in T84 cells depended critically on the specificity of ganglioside binding was confirmed by using recombinant CT and LTIIb chimeric toxins as described above. Thus, in human intestinal T84 cells, binding or clustering GM1, a function of the B subunit, directs CT into a specialized detergent-insoluble membrane fraction. In this respect, GM1 acts as a sorting motif. As glycosphingolipids such as GM1 are luminally oriented and cannot span the membrane bilayer to interact with cytoplasmic proteins, these data imply that the functional membrane receptor for CT and LT must involve a membrane structure of greater complexity than ganglioside GM1 alone.
Based on current evidence, we believe the membrane structure(s) most likely to subserve such a sorting function are caveolae or “caveolae-like” detergent-insoluble membranes (Parton et al., 1995; Schnitzer et al., 1996; and this study). The fact that GM1 represents a structural component of caveolae in many other cell types (Parton, 1994; Parton et al., 1994; Schnitzer et al., 1995b ) provides evidence in support of this hypothesis. Furthermore, that glycosphingolipids can self-assemble with other similarly predisposed cellular components is well described (Sharom and Grant, 1978; Hakomori, 1983; Thompson and Tillack, 1985; Simons and van Meer, 1988; van Meer et al., 1989; van Genderen and van Meer, 1993; van Meer, 1993). Such a propensity for self-assembly may contribute to the driving force for lipid sorting and caveolae biogenesis in many if not all cell types (Bretscher and Munro, 1993; Schroeder et al., 1994; Thomas et al., 1994; Gorodinsky and Harris, 1995; Hanada et al., 1995). On the other hand, detergent-insoluble membranes from T84 cells exclude GD1a in preference to GM1, but detergent-insoluble membranes from Y1 cells do not. These data provide evidence that membrane components other than gangliosides contribute to caveolae biogenesis, structure, and function, and this likely varies among differentiated cell types and/or species as previously suggested (Stan et al., 1997).
How specific gangliosides may function as discrete sorting motifs for partitioning ligands into membrane subdomains, however, remains undefined. The structures of GM1 and GD1a differ most significantly in their respective carbohydrate head groups, and even these are closely related (Hakomori, 1983). Nonetheless, detergent-insoluble membrane microdomains of human intestinal T84 cells clearly distinguish between these similar glycolipids. Thus, one possibility is that carbohydrate head groups may define specificity in this system. If so, the results of these studies would imply that epitopes on proteins or lipids resident in the exoplasmic bilayer leaflet provide motifs for sorting of membrane components at the cell surface and possibly within endosomes and other transport vesicles (as previously hypothesized; Fiedler et al., 1994; Simons and Ikonen, 1997). Alternatively, glycosphingolipids exhibit heterogeneity in structure of their lipid tails (Hakomori, 1983), and this may also impart specificity to glycolipid function (Sandvig et al., 1994; Mayor and Maxfield, 1995). When tested experimentally, replacing the ceramide moiety on GM1 with aliphatic amines, cholesterol, or phospholipid enhanced or reduced CT-induced signal transduction in rat glioma C6 cells (Pacuszka et al., 1991).
Mechanism of Action: Signal Transduction by CT in Human Intestinal T84 Cells
Taken together, the results of these studies show that human intestinal T84 cells discriminate between toxin bound to GM1 and GD1a by sorting GM1–toxin complexes into caveolae-like detergent-insoluble plasma membrane subdomains. This appears to be critical for toxin function. Based on available data, we can envision two plausible consequences of such sorting on the mechanism of toxin-induced signal transduction.
First, by analogy with the biology of GPI-linked (Rothberg et al., 1990; Anderson, 1993b ; Deckert et al., 1996) and dual-acylated proteins (Shenoy-Scaria et al., 1994; Bhatnagar and Gordon, 1997; Rehm and Ploegh, 1997), CT may ensure its own delivery to its target enzyme adenylyl cyclase by binding GM1 and thus partitioning itself into caveolae-like membrane structures that also cluster Gsα and adenylyl cyclase into a functional unit (Huang et al., 1997).
Alternatively, as signal transduction by CT in polarized cells requires endocytosis and transport of toxin through multiple intracellular compartments (Lencer et al., 1992, 1993, 1995a ,b; Orlandi et al., 1993; Majoul et al., 1996; Orlandi, 1997), the results of these studies raise the possibility that GM1 may sort CT into caveolae or caveolae-like membrane structures, which then mediate endocytosis of toxin-receptor complexes. If so, this would fit nicely with morphological evidence that CT enters hepatocytes, fibroblasts, A431, and endothelial cells via smooth, non–clathrin-coated membrane invaginations characteristic of caveolae (Montesanto et al., 1982; Tran et al., 1987; Parton et al., 1994; Schnitzer et al., 1996). The biological plausibility of such a hypothesis is emphasized by evidence that caveolae mediate endocytosis of albumin, alkaline phosphatase, protectin (CD59), and SV-40 virus in endothelial, epidermoid A431, Jurkat T-lymphocyte, and CV1 cell systems (Parton et al., 1994; Schnitzer et al., 1994; Deckert et al., 1996; Stang et al., 1997); that endothelial caveolae contain the transport machinery for vesicular traffic (Schnitzer et al., 1995a , 1996); and that both GM1 and caveolin-1 likely recycle between the plasma membrane and trans-Golgi in murine NCTC 2071 and rat glioma C6 cells (GM1) (Fishman et al., 1983) and in canine MDCK cells and human fibroblasts (VIP-21/caveolin-1) (Kurzchalia et al., 1992, 1994; Smart et al., 1994; Conrad et al., 1995).
These data also provide reason to suggest that caveolae-like membrane domains may carry the CT–GM1 complex sequentially from the site of toxin binding on the apical membrane through endosomes to trans-Golgi of human intestinal T84 cells, where the toxin may plausibly encounter KDEL-receptors. We do not envision, however, that caveolae or caveolae-like detergent-insoluble membranes in T84 cells necessarily form unique transport vesicles after endocytosis. Evidence from other cell systems indicates that ligands internalized by non–clathrin-coated and clathrin-coated mechanisms ultimately merge in common intracellular compartments (Tran et al., 1987; Hansen et al., 1993). That detergent-insoluble membrane subdomains are present on intracellular organelles and transport vesicles (Brown and Rose, 1992; Harder et al., 1997) provides further evidence that such structures may function alongside classic transmembrane receptors to target membrane components to specific sites within the vacuolar network of eukaryotic cells. Whether CT–GM1 complexes in T84 cells remain associated with detergent-insoluble membrane subdomains during transport into or beyond the apical endosome, however, remains unknown.
In summary, the results of these studies show that signal transduction by CT in polarized intestinal epithelial cells depends on binding or clustering specifically the ganglioside GM1. Remarkably, toxin binding to the closely related ganglioside GD1a renders the toxin inactive in such polarized epithelial cells. We also find that ganglioside structure dictates toxin association with caveolae-like membrane domains, and this is closely correlated with toxin function. Thus, the mechanism(s) by which gangliosides GM1 and GD1a function in signal transduction likely depend on their ability to couple cholera and related heat-labile enterotoxins with caveolae or caveolae-related membrane subdomains in specific cell types.
Acknowledgments
We thank Pam Fredman and Jan Holmgren for providing data on ganglioside expression in T84 cells; Martin Carey and Karen Nibbering (Harvard University Medical School, Boston, MA), and Randall Mrsny (Genentech, South San Francisco, CA) for helpful discussions on lipid biochemistry; Focco van den Akker (University of Washington, Seattle, WA) for advice on where to place the fusion joint in chimera 187; and J. Casanova (Harvard University Medical School) and J. Holmgren for critical reading of the manuscript.
This work was supported by T32-DK07477 (A. Wolf), National Institute of Health research grants DK 48106, DK/AI 53056 (W.I. Lencer), DK 35932, DK 33506 (J.L. Madara), AI 31940, AI14107 (R. Holmes), and the Harvard Digestive Diseases Center DK 34854. Dr. Lencer is a recipient of the Miles and Shirley Fitterman Basic Research Award from the American Digestive Health Foundation.
Abbreviations used in this paper
- CT
V. cholerae cholera toxin
- Isc
short circuit current
- LTI
E. coli heat-labile toxin type I
- LTIIb
E. coli heat-labile toxin type IIb
- VIP
vasoactive intestinal peptide
- wt
wild-type
Footnotes
Address all correspondence to W.I. Lencer, M.D., GI Cell Biology, Combined Program in Pediatric Gastroenterology and Nutrition, Children's Hospital, 300 Longwood Ave., Boston, MA 02115. Tel.: (617) 355-8599. Fax: (617) 730-0404. E-mail: lencer@a1.tch.harvard.edu
References
- Anderson RG. Caveolae: where incoming and outgoing messengers meet. Proc Natl Acad Sci USA. 1993a;90:10909–10913. doi: 10.1073/pnas.90.23.10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RG. Plasmalemmal caveolae and GPI-anchored membrane proteins. Curr Opin Cell Biol. 1993b;5:647–652. doi: 10.1016/0955-0674(93)90135-d. [DOI] [PubMed] [Google Scholar]
- Anderson RG, Kamen BA, Rothberg KG, Lacey SW. Potocytosis: sequestration and transport of small molecules by caveolae. Science. 1992;255:410–411. doi: 10.1126/science.1310359. [DOI] [PubMed] [Google Scholar]
- Bhatnagar RS, Gordon JI. Understanding covalent modifications of proteins by lipids: where cell biology and biophysics merge. Trends Cell Biol. 1997;7:14–20. doi: 10.1016/S0962-8924(97)10044-7. [DOI] [PubMed] [Google Scholar]
- Bretscher MS, Munro S. Cholesterol and the Golgi apparatus. Science. 1993;261:1280–1281. doi: 10.1126/science.8362242. [DOI] [PubMed] [Google Scholar]
- Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68:533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- Chang PP, Moss J, Twiddy EM, Holmes RK. Type II heat-labile enterotoxin of Escherichia coliactivates adenylate cyclase in human fibroblasts by ADP ribosylation. Infect Immun. 1987;55:1854–1858. doi: 10.1128/iai.55.8.1854-1858.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell TD, Holmes RK. Characterization of hybrid toxins produced in Escherichia coliby assembly of A and B polypeptides from Type I and Type II heat-labile enterotoxins. Infect Immun. 1992;60:1653–1661. doi: 10.1128/iai.60.4.1653-1661.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad PA, Smart EJ, Ying Y-S, Anderson RGW, Bloom GS. Caveolin cycles between plasma membrane caveolae and the Golgi complex by microtubule-dependent and microtubule-independent steps. J Cell Biol. 1995;131:1421–1433. doi: 10.1083/jcb.131.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckert M, Ticchioni M, Bernard A. Endocytosis of GPI-anchored proteins in human lymphocytes: role of glycolipid-based domains, actin cytoskeleton, and protein kinases. J Cell Biol. 1996;133:791–799. doi: 10.1083/jcb.133.4.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree P, Parton RG, Raposo G, Kurzchalia TV, Simons K. Caveolae and sorting in the trans-Golgi network of epithelial cells. EMBO (Eur Mol Biol Organ) J. 1993;12:1597–1605. doi: 10.1002/j.1460-2075.1993.tb05804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler K, Parton RG, Kellner R, Etzold T, Simons K. VIP36, a novel component of glycolipid rafts and exocytic carrier vesicles in epithelial cells. EMBO (Eur Mol Biol Organ) J. 1994;13:1729–1740. doi: 10.1002/j.1460-2075.1994.tb06437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman PH, Bradley RM, Hom BE, Moss J. Uptake and metabolism of exogenous gangliosides by cultured cells: effect of choleragen on the turnover of GM1. J Lipid Res. 1983;24:1002–1011. [PubMed] [Google Scholar]
- French C, Keshavarz-Moore E, Ward JM. Development of a simple method for the recovery of recombinant proteins from the Escherichia coliperiplasm. Enzyme Microb Technol. 1996;19:332–338. [Google Scholar]
- Fukuta S, Magnani JL, Twiddy EM, Holmes RK, Ginsburg V. Comparison of the carbohydrate-binding specificities of cholera toxin and Escherichia coliheat-labile enterotoxins LTh-I, LT-IIa, and LT-IIb. Infect Immun. 1988;56:1748–1753. doi: 10.1128/iai.56.7.1748-1753.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorodinsky A, Harris DA. Glycolipid-anchored proteins in neuroblastoma cells form detergent-resistant complexes without caveolin. J Cell Biol. 1995;129:619–627. doi: 10.1083/jcb.129.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guth BEC, Twiddy EM, Trabulsi LR, Holmes RK. Variation in chemical properties and antigenic determinants among type II heat-labile enterotoxins of Escherichia coli. . Infect Immun. 1986;54:529–536. doi: 10.1128/iai.54.2.529-536.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakomori, S. 1983. Chemistry of glycosphingolipids. In Sphingolipid Biochemistry. J.N. Kanfer and S. Hakomori, editors. Plenum Press, New York. 1–166.
- Hanada K, Nishijima M, Akamatsu Y, Pagano RE. Both sphingolipids and cholesterol participate in the detergent insolubility of alkaline phosphatase, a glycosylphosphatidylinositol-anchored protein, in mammalian membranes. J Biol Chem. 1995;270:6254–6260. doi: 10.1074/jbc.270.11.6254. [DOI] [PubMed] [Google Scholar]
- Hansen SH, Sandvig K, van Deurs B. Molecules internalized by clathrin-independent endocytosis are delivered to endosomes containing transferrin receptors. J Cell Biol. 1993;123:89–97. doi: 10.1083/jcb.123.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder T, Kellner R, Parton RG, Gruenberg J. Specific release of membrane-bound annexin II and cortical cytoskeleton elements by sequestration of membrane cholesterol. Mol Biol Cell. 1997;8:533–545. doi: 10.1091/mbc.8.3.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes RK, Twiddy EM. Characterization of monoclonal antibodies that react with unique and cross-reacting determinants of cholera enterotoxin and its subunits. Infect Immun. 1983;42:914–923. doi: 10.1128/iai.42.3.914-923.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes RK, Twiddy EM, Pickett CL. Purification and characterization of type II heat-labile enterotoxin of Escherichia coli. . Infect Immun. 1986;53:464–473. doi: 10.1128/iai.53.3.464-473.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Hepler JR, Chen LT, Gilman AG, Anderson RGW, Mumby SM. Organization of G proteins and adenylyl cyclase at the plasma membrane. Mol Biol Cell. 1997;8:2365–2378. doi: 10.1091/mbc.8.12.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobling MG, Palmer LM, Erbe JL, Holmes RK. Construction and characterization of versatile cloning vectors for efficient delivery of native foreign proteins to the periplasm of Escherichia coli. . Plasmid. 1997;38:158–173. doi: 10.1006/plas.1997.1309. [DOI] [PubMed] [Google Scholar]
- Kurzchalia TV, Dupree P, Monier S. VIP21-Caveolin, a protein of the trans-Golgi network and caveolae. FEBS Lett. 1994;346:88–91. doi: 10.1016/0014-5793(94)00466-8. [DOI] [PubMed] [Google Scholar]
- Kurzchalia TV, Dupree P, Parton RG, Kellner R, Virta H, Lehnert M, Simons K. VIP21, a 21-kD membrane protein is an integral component of trans-Golgi-network–derived transport vesicles. J Cell Biol. 1992;118:1003–1014. doi: 10.1083/jcb.118.5.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C-M, Chang PP, Tsai S-C, Adamik R, Price SR, Kunz BC, Moss J, Twiddy EM, Holmes RK. Activation of Escherichia coliheat- labile enterotoxins by native and recombinant adenosine diphosphate-ribosylation factors, 20-kD guanine nucleotide-binding proteins. J Clin Invest. 1991;87:1780–1786. doi: 10.1172/JCI115197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencer WI, Delp C, Neutra MR, Madara JL. Mechanism of cholera toxin action on a polarized human epithelial cell line: role of vesicular traffic. J Cell Biol. 1992;117:1197–1209. doi: 10.1083/jcb.117.6.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencer WI, de Almeida JB, Moe S, Stow JL, Ausiello DA, Madara JL. Entry of cholera toxin into polarized human intestinal epithelial cells: identification of an early brefeldin A sensitive event required for A1-peptide generation. J Clin Invest. 1993;92:2941–2951. doi: 10.1172/JCI116917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencer WI, Constable C, Moe S, Jobling M, Webb HM, Ruston S, Madara JL, Hirst T, Holmes R. Targeting of cholera toxin and E. coliheat labile toxin in polarized epithelia: role of COOH-terminal KDEL. J Cell Biol. 1995a;131:951–962. doi: 10.1083/jcb.131.4.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencer WI, Moe S, Rufo PA, Madara JL. Transcytosis of cholera toxin subunits across model human intestinal epithelia. Proc Natl Acad Sci USA. 1995b;92:10094–10098. doi: 10.1073/pnas.92.22.10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencer WI, Strohmeier G, Moe S, Carlson SL, Constable CT, Madara JL. Signal transduction by cholera toxin: processing in vesicular compartments does not require acidification. Am J Physiol. 1995c;269:G548–G557. doi: 10.1152/ajpgi.1995.269.4.G548. [DOI] [PubMed] [Google Scholar]
- Lencer WI, Constable C, Moe S, Rufo PA, Wolf A, Jobling MG, Ruston SP, Madara JL, Holmes RK, Hirst TR. Proteolytic activation of cholera toxin and Escherichia colilabile toxin by entry into host epithelial cells: signal transduction by a protease-resistant toxin variant. J Biol Chem. 1997;272:15562–15568. doi: 10.1074/jbc.272.24.15562. [DOI] [PubMed] [Google Scholar]
- Lisanti MP, Scherer PE, Tang Z, Sargiacomo M. Caveolae, caveolin and caveolin-rich membrane domains: a signalling hypothesis. Trends Cell Biol. 1994;4:231–235. doi: 10.1016/0962-8924(94)90114-7. [DOI] [PubMed] [Google Scholar]
- Liu J, Oh P, Horner T, Rodgers RA, Schnitzer JE. Organized endothelial cell surface signal transduction in caveolae distinct from glycosylphosphatidylinositol-anchored protein microdomains. J Biol Chem. 1997;272:7211–7222. doi: 10.1074/jbc.272.11.7211. [DOI] [PubMed] [Google Scholar]
- Majoul IV, Bastiaens PIH, Soling H-D. Transport of an external Lys-Asp-Glu-Leu (KDEL) protein from the plasma membrane to the endoplasmic reticulum: studies with cholera toxin in Vero cells. J Cell Biol. 1996;133:777–789. doi: 10.1083/jcb.133.4.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor S, Maxfield FR. Insolubility and redistribution of GPI- anchored proteins at the cell surface after detergent treatment. Mol Biol Cell. 1995;6:929–944. doi: 10.1091/mbc.6.7.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesanto R, Roth J, Orci L. Non-coated membrane invaginations are involved in binding and internalization of cholera and tetanus toxins. Nature. 1982;296:651–653. doi: 10.1038/296651a0. [DOI] [PubMed] [Google Scholar]
- Orlandi PA. Protein-disulfide isomerase-mediated reduction of the A subunit of cholera toxin in a human intestinal cell line. J Biol Chem. 1997;272:4591–4599. [PubMed] [Google Scholar]
- Orlandi PA, Curran PK, Fishman PH. Brefeldin A blocks the response of cultured cells to cholera toxin: implications for intracellular trafficking in toxin action. J Biol Chem. 1993;268:12010–12016. [PubMed] [Google Scholar]
- Pacuszka Y, Bradley RM, Fishman PH. Neoglycolipid analogues of ganglioside GM1as functional receptors of cholera toxin. Biochemistry. 1991;30:2563–2570. doi: 10.1021/bi00224a001. [DOI] [PubMed] [Google Scholar]
- Parton RG. Ultrastructural localization of gangliosides: GM1 is concentrated in caveolae. J Histochem Cytochem. 1994;42:155–166. doi: 10.1177/42.2.8288861. [DOI] [PubMed] [Google Scholar]
- Parton RG, Joggerst B, Simons K. Regulated internalization of caveolae. J Cell Biol. 1994;127:1199–1215. doi: 10.1083/jcb.127.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton RG, Simons K. Digging into caveolae. Science. 1995;269:1398–1399. doi: 10.1126/science.7660120. [DOI] [PubMed] [Google Scholar]
- Rehm A, Ploegh HL. Assembly and intracellular targeting of the βγ subunits of heterotrimeric G proteins. J Cell Biol. 1997;137:305–317. doi: 10.1083/jcb.137.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers W, Rose JK. Exclusion of CD45 inhibits activity of p56lck associated with glycolipid-enriched membrane domains. J Cell Biol. 1996;135:1515–1523. doi: 10.1083/jcb.135.6.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothberg KG, Ying Y-S, Kamen BA, Anderson RGW. Cholesterol controls the clustering of the glycospholipid-anchored membrane receptor for 5-methyltetrahydrofolate. J Cell Biol. 1990;111:2931–2938. doi: 10.1083/jcb.111.6.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandvig K, Ryd M, Garred Ø, Schweda E, Holm PK, van Deurs B. Retrograde transport from the Golgi complex to the ER of both shiga toxin and the nontoxic shiga B-fragment is regulated by buteric acid and cAMP. J Cell Biol. 1994;126:53–64. doi: 10.1083/jcb.126.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaar RL, Needham LK. Thin-layer chromatography of glycosphingolipids. Methods Enzymol. 1994;230:371–389. doi: 10.1016/0076-6879(94)30025-9. [DOI] [PubMed] [Google Scholar]
- Schnitzer JE, Oh P, Pinney E, Allard J. Filipin-sensitive caveolae-mediated transport in endothelium: reduced transcytosis, scavenger endocytosis, and capillary permeability of select macromolecules. J Cell Biol. 1994;127:1217–1232. doi: 10.1083/jcb.127.5.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnitzer JE, Liu J, Oh P. Endothelial caveolae have the molecular transport machinery for vesicle budding, docking, and fusion including VAMP, NSF, SNAP, annexins, and GTPases. J Biol Chem. 1995a;270:14399–14404. doi: 10.1074/jbc.270.24.14399. [DOI] [PubMed] [Google Scholar]
- Schnitzer JE, McIntosh DP, Dvorak AM, Liu J, Oh P. Separation of caveolae from associated microdomains of GPI-anchored proteins. Science. 1995b;269:1435–1439. doi: 10.1126/science.7660128. [DOI] [PubMed] [Google Scholar]
- Schnitzer JE, Oh P, Jacobson BS, Dvorak AM. Caveolae from luminal plasmalemma of rat lung endothelium: microdomains enriched in caveolin, Ca2+-ATPase, and inositol trisphosphate receptor. Proc Natl Acad Sci USA. 1995c;92:1759–1763. doi: 10.1073/pnas.92.5.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnitzer JE, Oh P, McIntosh DP. Role of GTP hydrolysis in fission of caveolae directly from plasma membranes. Science. 1996;274:239–242. doi: 10.1126/science.274.5285.239. [DOI] [PubMed] [Google Scholar]
- Schroeder R, London E, Brown D. Interactions between saturated acyl chains confer detergent resistance on lipids and glycosylphosphatidylinositol (GPI)-anchored proteins: GPI-anchored proteins in liposomes and cells show similar behavior. Proc Natl Acad Sci USA. 1994;91:12130–12134. doi: 10.1073/pnas.91.25.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharom FJ, Grant CWM. A model for ganglioside behavior in cell membranes. Biochim Biophys Acta. 1978;507:280–293. doi: 10.1016/0005-2736(78)90423-6. [DOI] [PubMed] [Google Scholar]
- Shenoy-Scaria AM, Dietzen DJ, Kwong J, Link DC, Lublin DM. Cysteine3 of Src family protein tyrosine kinases determines palmitoylation and localization in caveolae. J Cell Biol. 1994;126:353–363. doi: 10.1083/jcb.126.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Simons K, van Meer G. Lipid sorting in epithelial cells. Biochemistry. 1988;27:6197–6202. doi: 10.1021/bi00417a001. [DOI] [PubMed] [Google Scholar]
- Simons K, Wandinger-Ness A. Polarized sorting in epithelia. Cell. 1990;62:207–210. doi: 10.1016/0092-8674(90)90357-k. [DOI] [PubMed] [Google Scholar]
- Sixma TK, Pronk SE, Kalk HH, Wartna ES, van Zanten BAM, Witholt B, Hol WGJ. Crystal structure of a cholera toxin-related heat-labile enterotoxin from E. coli. . Nature. 1991;351:371–377. doi: 10.1038/351371a0. [DOI] [PubMed] [Google Scholar]
- Smart EJ, Ying Y-S, Conrad PA, Anderson RG. Caveolin moves from caveolae to the Golgi apparatus in response to cholesterol oxidation. J Cell Biol. 1994;127:1185–1197. doi: 10.1083/jcb.127.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangler BD. Structure and function of cholera toxin and the related Escherichia coliheat-labile enterotoxin. Microbiol Rev. 1992;56:622–647. doi: 10.1128/mr.56.4.622-647.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stan R-V, Roberts WG, Predescu D, Ihida K, Saucan L, Ghitescu L, Palade GE. Immunoisolation and partial characterization of endothelial plasmalemmal vesicles (caveolae) Mol Biol Cell. 1997;8:595–605. doi: 10.1091/mbc.8.4.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stang E, Kartenbeck J, Parton RG. Major histocompatibility complex class I molecules mediate association of SV40 with caveolae. Mol Biol Cell. 1997;8:47–57. doi: 10.1091/mbc.8.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohmeier GR, Lencer WI, Patapoff TW, Thompson LF, Carlson SL, Moe SJ, Carnes D, Mrsny RJ, Madara JL. Surface expression, polarization, and functional significance of CD73 in human intestinal epithelia. J Clin Invest. 1997;99:2588–2601. doi: 10.1172/JCI119447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JL, Holowka D, Baird B, Webb WW. Large–scale co- aggregation of fluorescent lipid probes with cell surface proteins. J Cell Biol. 1994;125:795–802. doi: 10.1083/jcb.125.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson TE, Tillack TW. Organization of glycosphingolipids in bilayers and plasma membranes of mammalian cells. Annu Rev Biophys Bioeng. 1985;14:361–386. doi: 10.1146/annurev.bb.14.060185.002045. [DOI] [PubMed] [Google Scholar]
- Tran D, Carpentier J-L, Sawano F, Gorden P, Orci L. Ligands internalized through coated or noncoated invaginations follow a common intracellular pathway. Proc Natl Acad Sci USA. 1987;84:7956–7961. doi: 10.1073/pnas.84.22.7957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Akker F, Sarfaty S, Twiddy EM, Connell TD, Holmes RK, Hol WGJ. Crystal structure of a new heat-labile enterotoxin, LTIIb. Structure (Lond) 1996;4:665–678. doi: 10.1016/s0969-2126(96)00073-1. [DOI] [PubMed] [Google Scholar]
- van Genderen IL, van Meer G. Lipid sorting—measurement and interpretation. Biochem Soc Trans. 1993;21:235–239. doi: 10.1042/bst0210235. [DOI] [PubMed] [Google Scholar]
- van Meer G. Transport and sorting of membrane lipids. Curr Opin Cell Biol. 1993;5:661–673. doi: 10.1016/0955-0674(93)90137-f. [DOI] [PubMed] [Google Scholar]
- van Meer G, Palade GE. Lipid traffic in animal cells. Annu Rev Cell Biol. 1989;5:247–275. doi: 10.1146/annurev.cb.05.110189.001335. [DOI] [PubMed] [Google Scholar]
- Zhang R-G, Scott DL, Westbrook ML, Nance S, Spangler BD, Shipley GG, Westbrook EM. The three-dimensional crystal structure of cholera toxin. J Mol Biol. 1995;251:563–573. doi: 10.1006/jmbi.1995.0456. [DOI] [PubMed] [Google Scholar]