

Abstract
Reactive oxygen species (ROS) are thought to be involved in many forms of programmed cell death. The role of ROS in cell death caused by oxidative glutamate toxicity was studied in an immortalized mouse hippocampal cell line (HT22). The causal relationship between ROS production and glutathione (GSH) levels, gene expression, caspase activity, and cytosolic Ca2+ concentration was examined. An initial 5–10-fold increase in ROS after glutamate addition is temporally correlated with GSH depletion. This early increase is followed by an explosive burst of ROS production to 200–400-fold above control values. The source of this burst is the mitochondrial electron transport chain, while only 5–10% of the maximum ROS production is caused by GSH depletion. Macromolecular synthesis inhibitors as well as Ac-YVAD-cmk, an interleukin 1β–converting enzyme protease inhibitor, block the late burst of ROS production and protect HT22 cells from glutamate toxicity when added early in the death program. Inhibition of intracellular Ca2+ cycling and the influx of extracellular Ca2+ also blocks maximum ROS production and protects the cells. The conclusion is that GSH depletion is not sufficient to cause the maximal mitochondrial ROS production, and that there is an early requirement for protease activation, changes in gene expression, and a late requirement for Ca2+ mobilization.
The production of reactive oxygen species (ROS)1 is associated with many forms of apoptosis (Suzuki et al., 1997), as well as the cell death that occurs in stroke, ischemia, and many neurodegenerative diseases (Halliwell, 1992; Ames et al., 1993; Coyle and Puttfarcken, 1993; Jenner, 1994; Shigenaga et al., 1994). Glutamate toxicity is a major contributor to pathological cell death within the nervous system and appears to be mediated by ROS (Coyle and Puttfarcken, 1993). There are two forms of glutamate toxicity: receptor-initiated excitotoxicity (Choi, 1988) and non–receptor-mediated oxidative glutamate toxicity (Murphy et al., 1989). Oxidative glutamate toxicity is initiated by high concentrations of extracellular glutamate that prevent cystine uptake into the cells, followed by the depletion of intracellular cysteine and the loss of glutathione (GSH). With a diminishing supply of GSH, there is an accumulation of excessive amounts of ROS and ultimately cell death. Understanding the relationship between GSH depletion and ROS production should lead to a better understanding of all forms of programmed cell death in which ROS play a central role.
Oxidative glutamate toxicity has been observed in primary neuronal cell cultures (Murphy et al., 1989, 1990; Oka et al., 1993), neuronal cell lines (Miyamoto et al., 1989; Murphy et al., 1989), and tissue slices (Vornov and Coyle, 1991) and has been studied recently in the immortalized mouse hippocampal cell line, HT22 (Davis and Maher, 1994; Maher and Davis, 1996; Li et al., 1997a ,b; Sagara et al., 1998). In HT22 cells, glutamate induces a form of programmed cell death with characteristics of both apoptosis and necrosis (Tan et al., 1998). HT22 cells exposed to 5 mM glutamate do not change morphologically until after 7 h of glutamate exposure. At this time, cells become rounded and form apoptotic bodies before becoming smaller shrunken cells. After 10 h, most cells are very small and dead or close to death (Tan et al., 1998). The exposure of HT22 cells, cortical neurons, and neuroblastoma cells to glutamate results in the rapid depletion of GSH followed by an increase in ROS (Murphy et al., 1989; Oka et al., 1993; Li et al., 1997a ; Sagara et al., 1998). The assumption has been that the increase in ROS is a direct result of this GSH depletion, but the functional relationship between the two has not been defined. The experiments outlined below show that GSH depletion alone cannot account for the increase in ROS. Instead, high levels of ROS production require both interleukin 1β–converting enzyme (ICE) protease activity and early gene expression. We also provide the first evidence that the majority of the ROS in oxidative glutamate toxicity are generated from the mitochondrial electron transport chain. Finally, extracellular Ca2+ influx is shown to regulate ROS production.
Materials and Methods
The following supplies were purchased from Sigma Chemical Co. (St. Louis, MO): cycloheximide, actinomycin D, carbonyl cyanide p-trifluoromethoxyphenyl-hydrazone (FCCP), 4,4,4-trifluoro-1-[2-thienyl]-1,3- butanedione, 1-chloro-2,4-dinitrobenzene, antimycin A, rotenone, oligomycin, GSH, glutathione reductase, triethanolamine, sulfosalicylic acid, NADPH, 5,5′-dithiobis (2-nitrobenzoic acid), l-buthionine-[S,R]-sulfoximine (BSO), horseradish peroxidase (Sigma Type VI), p-hydroxyphenylacetic acid (p-HPAA), hydrogen peroxide (H2O2), and l-glutamic acid. 2′,7′-dichlorofluorescein diacetate (DCF), indo-acetoxymethylester (Indo-1), pluronic F-127, and propidium iodide were obtained from Molecular Probes (Eugene, OR). Ac-YVAD-cmk, also known as ICE inhibitor II, was from both Calbiochem (La Jolla, CA) and Bachem California (Torrance, CA).
Cell Culture
HT22 cells were maintained in DME supplemented with 10% fetal bovine serum. Cells were kept at no greater than 50% confluence.
Flow Cytometric Studies
Cells were plated at 2 × 105 cells per dish on 60-mm tissue culture dishes (Falcon, Lincoln Park, NJ) 12 h before glutamate exposure. Inhibitors were added either at the same time as 5 mM l-glutamic acid (glutamate) or at 2-h intervals after glutamate addition to determine how late the drug could be added and still prevent ROS production and/or protect the cells from death.
ROS Measurement
Time course experiments were performed to compare ROS production in HT22 cells after different lengths of glutamate exposure. ROS production was detected using the dye DCF. DCF is a nonfluorescent cell-permeant compound. Once inside the cell, it is cleaved by endogenous esterases and can no longer pass out of the cell. The de-esterified product becomes the fluorescent compound 2′,7′-dichlorofluorescein upon oxidation by ROS (Bass et al., 1983; Cathcart et al., 1983; Hirabayashi et al., 1985; Miyamoto et al., 1989; LeBel et al., 1992). 10 μM DCF was added to cells during dissociation with pancreatin (GIBCO BRL, Gaithersburg, MD) diluted 1:5 in serum-free DME. The cells were then incubated at 37°C for 10 min and washed once in Hepes buffer supplemented with 2% dialyzed fetal bovine serum. Washed cells were resuspended in 750 μl Hepes buffer, filtered through a 4-μm nylon mesh membrane (Small Parts Inc., Miami Lakes, FL), and kept on ice until flow cytometric analysis. Propidium iodide dye was used to gate for live cells and was added to each tube at a final concentration of 5 μg per ml. Data were collected with a FACScan® fluorescence accelerated cell scanner using the data acquisition program CELLQuest (Becton Dickinson, San Jose, CA). DCF data were collected with the following excitation and emission wavelengths: λexc = 475 nm, λem = 525 nm. 10,000 live cells, as determined by the lack of propidium iodide fluorescence, were analyzed per sample. To the best of our knowledge, ROS production leads directly to DCF oxidation. The dissociation of cells before measuring DCF fluorescence accurately reflects the amount of ROS present in the plated cells at the time just before dissociation. Visual inspection using fluorescence microscopy confirms the increase in fluorescence with lengthened exposures to glutamate in the plated cells. This late high intensity fluorescence was blocked by a caspase inhibitor, protein synthesis inhibitors, FCCP, ruthenium red, and cobalt (Figs. 2, A–C, 3 A, 7, and 8). In addition, DCF fluorescence was the same in attached cells loaded with DCF before dissociation and in cells loaded with DCF while dissociation took place. Similar results were also seen using the dye dihydrorhodamine 123.
Figure 2.


Inhibitors of macromolecular synthesis and ICE proteases prevent ROS production. Inhibitors were added to HT22 cells at 2-h intervals during a 10-h exposure to 5 mM glutamate, and DCF fluorescence and cell survival were measured. ROS production is presented as the ratiometric increase in median DCF fluorescence versus the control value (black circles). Cell survival was measured by trypan blue exclusion and is presented as the percent of the control value (black squares). Each set of data represents the mean of two trials that were repeated four times, plus or minus the standard error. (A) Effect of actinomycin D on ROS production and cell survival. 0.1 μg/ml actinomycin D was added to HT22 cells at 2-h intervals during a 10-h exposure to 5 mM glutamate. (B) Effect of cycloheximide on ROS production and cell survival. 300 nM cycloheximide was added to HT22 cells during a 10-h exposure to 5 mM glutamate. (C) Effect of Ac-YVAD-cmk on ROS production and cell survival. 50 μM Ac-YVAD-cmk was added to HT22 cells as in parts A and B.
Percent cell survival at each time point was determined by trypan blue staining after each sample was analyzed by flow cytometry. Cells were diluted 1:2 with 0.1% trypan blue (ICN Biomedicals, Costa Mesa, CA) in PBS. Live cells were then counted under a light microscope. Live cells were not permeable to trypan blue and remained clear and phase bright. Percent survival was calculated versus the negative control.
DCF data were plotted as histograms using the data analysis program CELLQuest (Becton Dickinson). The median fluorescence of each peak was obtained and multiplied by the number of events in that peak. Sample values were divided by the control value to yield the ratiometric increase in DCF fluorescence per time point.
Calcium Measurement
Cytosolic Ca2+ was measured using the dye Indo-1 acetoxymethylester (Indo-1) and flow cytometry. Once inside the cell, the Indo-1 acetoxymethylester is cleaved by endogenous esterases, and Indo-1 is trapped inside the cell. The dye then binds Ca2+, and upon excitation by UV light, it emits fluorescent light at two wavelengths: 410 nm (FL32) and 485 nm (FL4). The Ca2+ concentration present in the cell is proportional to the ratio of FL32/FL4, which reflects the fluorescence of Indo-1 bound to Ca2+ versus the fluorescence of Indo-1 that is not bound to Ca2+. The ratio of bound to unbound dye accounts for any changes that take place in the cell's volume upon glutamate exposure. HT22 cells were loaded with 1 μM Indo-1 in the presence of 0.005% Pluronic F-127 for 30 min at 37°C. Cells were then dissociated from the dishes in the presence of 10 μM DCF, washed, and analyzed as described above. Ca2+ data were plotted as dot plots, and the fluorescence above a certain wavelength was designated as high. The number of cells with high fluorescence was calculated for each sample, and the ratiometric increase of cells with high Ca2+ was determined with respect to the control value. The effects of the inhibitors CoCl2, ruthenium red, and FCCP on cytosolic Ca2+, ROS levels, and cell death were observed by adding glutamate to HT22 cells and then adding the inhibitors either at the same time as or at 2-h intervals after the glutamate addition.
Glutathione Chemical Assay
Total intracellular reduced (GSH) and oxidized (GSSG) glutathione was measured using a method by Tietze (1969), as modified by Griffith (1980). Cells were plated on 100-mm tissue culture dishes at 5 × 105 cells per dish 12 h before 5 mM glutamate or 50 μM BSO treatment. Cells were processed as described previously by Sagara et al. (1998). Pure GSH was used to obtain a standard curve.
In Vitro Determination of Mitochondrial H2O2 Release
The release of H2O2 by isolated mitochondria from HT22 cells was done by the method described by Liu et al. (1993). Briefly, p-HPAA (Guilbault et al., 1968) was used as a substrate that is converted by HRP to a fluorescent compound in the presence of H2O2. Mitochondria from HT22 cells were isolated according to the method of Moreadith and Fiskum (1984), with the exception that the second homogenization step was eliminated. Cells were grown to confluence in 12 150-mm tissue culture dishes to obtain a large quantity of mitochondria. To measure H2O2 release, 0.1 mg/ml mitochondria were incubated in KCl buffer (125 mM KCl, 2 mM K2HPO4, 1 mM MgCl2, and 20 mM Hepes, adjusted to pH 7.4 with KOH) and the H2O2 detection system (0.2 mM p-HPAA and 0.5 μM HRP) at 37°C. 6 mM succinate or 6 mM malate plus 6 mM glutamate were added as FAD-linked or NADH-linked substrates for the mitochondrial electron transport chain. To quantitate the amount of H2O2 released by the mitochondria, a known amount of H2O2 was used to compare the amount of fluorescence obtained in the experimental samples. All measurements were made using a Perkin-Elmer LS-50B fluorescence spectrometer (Norwalk, CT).
Exposure of Cells to Cystine-free Media
Cells were plated as described above. 12 h after plating, the medium was replaced with cystine-free DME supplemented with 10% dialyzed fetal bovine serum. After 4 and 8 h exposure to cystine-free media, ROS levels were determined with DCF as described above. The same experiment was done using 5 mM glutamate to compare the amount of ROS production caused by both conditions.
Statistics
Statistics were done using a Math-Stat program. Unless otherwise indicated, the results are the mean plus or minus the standard error of the mean of four independent determinations. For FACS® analysis, 10,000 cells were quantitated.
Results
GSH Depletion Causes Only a Partial Increase in ROS
Although oxidative glutamate toxicity is associated with the depletion of GSH (Murphy et al., 1989), the causal relationship between GSH depletion and ROS production has not been formally explored. Exposure of HT22 cells to glutamate leads to the increased production of ROS. This increase is concurrent with the depletion of GSH, which is caused by the inability of the cells to take up the cystine necessary for GSH production (Fig. 1 A). ROS production proceeds in two phases: an initial slow increase for the first 6 h, followed by a much higher rate (Fig. 1 B). By 10 h, the intracellular ROS levels are over 200 times greater than the control levels. The latter high rate of increase in ROS only begins after the cellular GSH levels drop below ∼20%.
Figure 1.

GSH depletion and ROS production after exposure to glutamate or BSO. (A) Glutamate (5 mM) or BSO (50 μM) were added to cells, and GSH and ROS were measured as a function of time. The amount of GSH was calculated as nmoles GSH per mg protein and is presented as a percentage of the control value. ROS are presented as the ratiometric increase in median DCF fluorescence versus the control sample. Both GSH and ROS were quantified at 2-h intervals up to 10 h after the addition of glutamate. GSH data are the average of five trials. GSH depletion by BSO is an average of four trials. ROS data are averages of eight trials. (black squares) GSH, glutamate; (black circles) ROS, glutamate; (white squares) GSH, BSO; (white circles) ROS, BSO. (B) The glutamate data from the above and similar experiments were plotted in terms of GSH concentration (% maximum) vs. log ROS (% maximum).
There are three alternatives that could explain the apparent temporal coupling between GSH depletion and ROS production: (a) The increase in ROS is exclusively due to GSH depletion. (b) Glutamate may cause an elevation in ROS levels independently of GSH depletion. (c) One phase of ROS production is due to GSH depletion, and the other is not directly coupled to this event. To distinguish between these possibilities, GSH levels were depleted with BSO, an irreversible inhibitor of γ-glutamylcysteine synthetase. ROS levels were examined during BSO-induced GSH depletion and compared with the levels after glutamate-induced GSH depletion (Fig. 1 A). While 50 μM BSO is capable of depleting cellular GSH to the same levels as glutamate after 8–10 h of exposure, the ROS level in BSO-treated cells at 10 h is only 20% of that in glutamate-treated cells. In contrast, the initial rate of ROS production is about the same with BSO and glutamate. It can be concluded that while the depletion of GSH is necessary for the initial increase in ROS production, its decrease is not sufficient to generate the late exponential increase in ROS, which occurs after 6 h of glutamate treatment.
Gene Expression and ICE Protease Activity Are Necessary for ROS Production
Oxidative glutamate toxicity in HT22 cells is a form of programmed cell death (Tan et al., 1998). Most forms of programmed cell death, including that of HT22 cells, are blocked by macromolecular synthesis inhibitors as well as by caspase inhibitors. It was therefore important to determine whether ROS production is a signal for changes in macromolecular synthesis and protease activation or if it is dependent on gene expression and caspase activity. To answer this question, specific inhibitors were used to examine the roles of RNA and protein synthesis and ICE protease activity in ROS production after glutamate exposure. Glutamate was added to cells, and at 2-h intervals the inhibitors were applied. At 10 h, GSH and ROS levels were determined, and cell viability was scored. Surprisingly, these inhibitors all block the late second phase of ROS production, but only when added early in the cell death cascade (Fig. 2). However, even when added immediately before glutamate, they do not block the first phase of ROS production (data not presented).
Actinomycin D prevents the increase in ROS and protects the cells from glutamate toxicity only when it is added within 2 h of the glutamate exposure (Fig. 2 A). Similarly, cycloheximide is able to protect HT22 cells and keeps ROS levels low during a 10-h glutamate exposure when the inhibitor is added up to 6 h after glutamate (Fig. 2 B). If the inhibitor is added to the cells later than 6 h, ROS levels increase, and cell survival is less than 15% of that in the control. Cell survival is also increased and ROS production is decreased by the addition of the ICE protease inhibitor, Ac-YVAD-cmk (Fig. 2 C). Ac-YVAD-cmk must be added within the first 4 h of glutamate exposure to keep ROS levels low and protect the cells. Therefore, the requirement for ICE proteases is very early in the death cascade. None of the above inhibitors prevents the depletion of GSH (data not shown). Thus, the rapid rise of ROS production is dependent on both protein synthesis and protease activation.
The Mitochondrial Electron Transport Chain Is the Major Source of ROS
The above data suggest that the large increase in ROS production is not simply a consequence of GSH depletion. There are many possible sources of ROS. They include mitochondria and a wide array of enzymes such as the monoamine oxidases, tyrosine hydroxylase, l-amino oxidase, the lipoxygenases, cyclo-oxygenase, and xanthine oxidase (Coyle and Puttfarcken, 1993). To determine if the source of ROS is the mitochondrial electron transport chain, transport chain inhibitors were tested for their effects on ROS production and cell survival. The mitochondrial uncoupler, FCCP, dissipates the mitochondrial membrane potential by transporting protons across the membrane and down the proton gradient (Voet and Voet, 1990). Fig. 3 A shows that 5 μM FCCP protects the cells from glutamate toxicity and prevents the massive increase in ROS production, allowing only a 10-fold increase, which is similar to that observed before the exponential rise at 6 h (Fig. 1). The complex III inhibitor, antimycin A, also protects cells and keeps the ROS levels low (data not shown). In contrast, both rotenone, an inhibitor of complex I, and oligomycin, an inhibitor of the ATP synthase, cause an increase in ROS in the absence of glutamate, and glutamate addition increases ROS production over that seen with the drugs alone (data not shown). These inhibitor data were confirmed in vitro using mitochondria isolated from HT22 cells and a fluorometric method of H2O2 detection (Liu et al., 1993). FCCP prevents 97% of the H2O2 production in isolated mitochondria through the succinate pathway (Fig. 3 B). Antimycin A also lowers the in vitro production of H2O2 through this pathway (data not shown). Mitochondria isolated from HT22 cells do not produce detectable H2O2 in the presence of glutamate or malate (data not shown).
Figure 3.


Effect of uncoupling mitochondria on ROS production. (A) 5 μM of the protonophore FCCP was added to the cells at 2-h intervals after glutamate exposure to see how late the inhibitor could be added and still protect cells. ROS levels are expressed as the ratiometric increase in median DCF fluorescence versus the control value (black circles). Cell survival is the percent of the control (black squares). (B) In vitro production of H2O2 by mitochondria from HT22 cells was determined by fluorescence spectrometry using the fluorescent substrate, p-HPAA. Basal H2O2 production was initiated by providing mitochondria with 6 mM succinate. Once H2O2 production was established, 0.2 μM FCCP was added, resulting in inhibition of H2O2 production. 0.1 μM H2O2 was added to indicate the increase in relative fluorescence with a known concentration of H2O2. (C) In vitro production of H2O2 by mitochondria from HT22 cells was established as in B. Basal H2O2 production was initiated by providing the mitochondria with 6 μM succinate. 100 μM pargyline had no effect on H2O2 production, while 100 μM clorgyline decreased H2O2 production to less than 10% of the basal level. 0.1 μM H2O2 was added to indicate the increase in relative fluorescence with a known concentration of H2O2.
The monoamine oxidase-A (MAO-A) inhibitor clorgyline protects HT22 cells from glutamate-induced toxicity and prevents the major glutamate-induced increase in ROS (Maher and Davis, 1996). In vitro data using mitochondria isolated from HT22 cells demonstrate that both pargyline, an inhibitor of MAO-B, and clorgyline are able to prevent H2O2 production when the MAO substrates tryptamine or tyramine are added to mitochondria (data not shown). However, only clorgyline is able to prevent H2O2 production when the mitochondrial electron transport chain substrate, succinate, is supplied to mitochondria (Fig. 3 C). In vivo data indicate that H2O2 production is reduced by clorgyline in a similar fashion as is seen with FCCP (data not shown).
Because glutamate is a substrate for the mitochondrial electron transport chain, it is possible that the increase in ROS levels is caused simply by the presence of excess glutamate in the cells. To rule out this possibility, cells were placed in cystine-free media, and ROS production was monitored. Depleting the cells of cysteine produces the same result as was achieved with glutamate (Fig. 4). The cells die within 24 h, and ROS levels at 4 and 8 h are similar to those with 5 mM glutamate.
Figure 4.

ROS production in cystine-free medium. Cells were exposed to either cystine-free medium supplemented with 10% dialyzed fetal bovine serum or 5 mM glutamate. The ratiometric increase in median DCF fluorescence versus the control value was determined after 4 and 8 h exposure for each sample.
Cytosolic Ca2+
Since an increase in cytosolic Ca2+ is a common feature of many forms of cell death (McConkey and Orrenius, 1997), including oxidative glutamate toxicity (Murphy et al., 1989; Davis and Maher, 1994), the relationship between calcium and the two phases of ROS production was explored. To determine the temporal relationship between Ca2+ and ROS levels, both were measured in the same cell at 2-h intervals after the addition of glutamate. Fig. 5 shows that there is very little increase in cytoplasmic Ca2+ until the cells enter the second (late) phase of ROS production, and then Ca2+ is high only in cells that have accumulated a high ROS load. These data suggest that the late phase mitochondrial ROS may be necessary for Ca2+ accumulation. To test this possibility, ROS production from the electron transport chain was eliminated by FCCP, and intracellular Ca2+ was monitored. Fig. 6 shows that Ca2+ levels increase to less than 15% of that in cells exposed to glutamate alone. Although FCCP itself is slightly toxic to cells after 8–10 h, it blocks glutamate-induced cell death (Fig. 3 A and Fig. 6) as well as the normal increase in Ca2+. Therefore, late-phase ROS production has a major effect on intracellular Ca2+ concentration.
Figure 5.

ROS production and intracellular Ca2+ changes. ROS were monitored (DCF fluorescence, horizontal axis) at the same time as intracellular Ca2+ levels (Indo-1, vertical axis) during a 10-h exposure to 5 mM glutamate. Data from 10,000 live cells were collected for each graph.
Figure 6.

Ca2+ changes and the mitochondrial electron transport chain. Ca2+ levels were monitored at 2-h intervals in cells treated with 5 μM FCCP + glutamate (black squares) or with glutamate alone (black circles) during a 10-h time course. Ca2+ increase is expressed as the ratiometric increase in the number of cells with high fluorescence (Fig. 5, top) with respect to the control. Cell survival is expressed as a percent of the control (dotted lines) and is normalized to the number of cells surviving after the exposure to 5 μM FCCP in the absence of glutamate. Survival in FCCP alone at 10 h was 40%.
To further examine the relationship between Ca2+ and ROS, we asked if there were a reciprocal relationship between the two. Does the inhibition of Ca2+ uptake block ROS production? CoCl2 is a Ca2+ channel blocker that inhibits the influx of extracellular Ca2+ through most plasma membrane Ca2+ channels (McFarlane et al., 1993). To determine when in the toxicity cascade extracellular Ca2+ enters the cell and the extent to which it modifies ROS production, CoCl2 was added to cells at 2-h intervals after the addition of glutamate. ROS and Ca2+ levels were monitored, and cell viability was determined 10 h after the addition of glutamate. When CoCl2 is added up to 6 h after the glutamate addition, HT22 cells are protected from cell death (Fig. 7). Previous work has also shown that cobalt completely protects cells from oxidative glutamate toxicity (Murphy et al., 1989; Davis and Maher, 1994; Li et al., 1997b ). When cells are protected by CoCl2, the late phase of ROS increase is reduced to about 30% of the maximum, and cytosolic Ca2+ levels only increase to about 10% of the maximum level (Fig. 7). Since the inhibition of mitochondrial ROS production reduces intracellular Ca2+ to 15% of its maximum in the presence of glutamate alone (Fig. 6), and ROS levels only reach 30% of their maximum in the presence of cobalt (Fig. 7), it follows that there is a mutual requirement for Ca2+ and ROS for each to reach their maximal level.
Figure 7.

Cytosolic Ca2+ changes that are due to the influx of Ca2+. Glutamate was added to all samples for 10 h, while 20 μM CoCl2 was added at 2-h intervals after glutamate to determine how late into the glutamate exposure CoCl2 could be added and still protect cells from death. Cell viability was determined after 24 h. Ca2+ changes were detected as an increase in Indo-1 fluorescence over a wavelength designated as high (Fig. 5) and presented as a ratiometric increase with respect to the control (black circles). ROS production is measured using DCF as in previous figures (black squares). Cell survival is shown as a percentage of the control (bars). The * symbol indicates that P < 0.05 between the 6- and the 8-h time points.
The above data show that the major source of intracellular free Ca2+ is from outside the cell via cobalt-sensitive channels. How can this Ca2+ influence mitochondrial ROS production? Ruthenium red is a potent inhibitor of both the mitochondrial Ca2+ uptake uniporter and the ryanodine receptor–mediated Ca2+ channel on the ER (Gunter and Pfeiffer, 1990; McPherson and Campbell, 1993; Kohda et al., 1995; Park et al., 1996). If mitochondrial Ca2+ uptake is required for ROS production, then ruthenium red should block the late phase of ROS. Fig. 8 shows that ruthenium red protects the cells when added up to 6 h after glutamate. In addition, ruthenium red keeps cytosolic Ca2+ levels at baseline levels and allows only the early increase in ROS production (Fig. 8). When ruthenium red is added to the cells after they have been exposed to glutamate for more than 6 h, cell survival is very low, and ROS and Ca2+ increase to much higher levels. Since mitochondria are responsible for late-phase ROS production and since the mitochondrial Ca2+ uptake inhibitor ruthenium red inhibits this process, it follows that Ca2+ uptake into mitochondria is a necessary step for the high rate of ROS formation.
Figure 8.
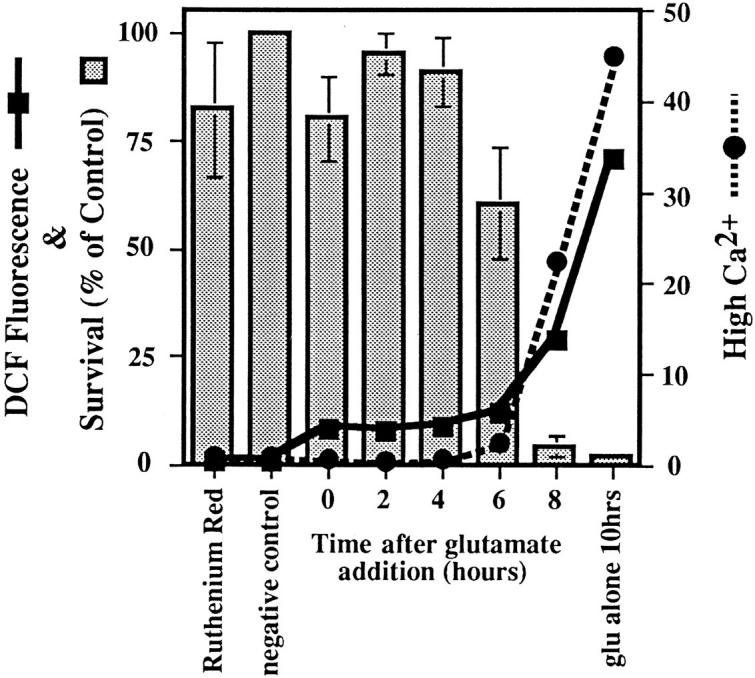
Intracellular Ca2+ cycling during glutamate exposure. Glutamate was added to all samples for 10 h, and 150 μM ruthenium red was added to the cells at 2-h intervals after the addition of glutamate. Ca2+ changes were detected as an increase in Indo-1 fluorescence over a wavelength designated as high, and data are presented as a ratiometric increase with respect to the control (black circles). ROS were detected using DCF and are expressed as the ratio of the median DCF fluorescence with respect to the control (black squares). Cell survival at 10 h is expressed as a percentage of the control (bars).
Discussion
Previous work has defined oxidative glutamate toxicity in terms of the depletion of intracellular cysteine and the subsequent decrease in GSH (Murphy et al., 1989). In the hippocampal cell line HT22, these events lead to a form of programmed cell death that is similar to, but distinct from, classical apoptosis (Tan et al., 1998). The data presented here outline the temporal and functional relationships between glutamate, GSH, Ca2+, ROS, and cell death. ROS production is divided into two phases: an initial 5–10-fold increase followed by a massive 200–400-fold increase. The source of ROS in the latter phase is identified as the mitochondrial electron transport chain. The large second phase increase in ROS production is not due solely to the depletion of GSH. ICE protease activity, macromolecular synthesis, and Ca2+ fluxes all contribute to the mitochondria-generated large increase in ROS but are not required for the initial increase.
There Are Two Phases of ROS Production
When glutamate is added to cells and the accumulation of ROS followed as a function of time, there is a gradual increase in ROS for the first 6 h, followed by an explosive increase in the rate of production after 6 h (Fig. 1). The slow accumulation of ROS occurs until GSH levels fall below 20%. After this juncture, ROS begin to accumulate at a much greater rate (Fig. 1 B). To determine the contribution of GSH to the increase in ROS, cells were depleted of GSH by the γ-glutamylcysteine synthetase inhibitor, BSO. BSO completely depletes cellular GSH but causes only a 20–25% increase in ROS relative to glutamate (Fig. 2). Therefore, the loss of GSH alone cannot account for the greater than 200-fold increase in ROS that is seen with glutamate. Mitochondria contain ∼15–20% of the total intracellular GSH (Jocelyn, 1975; Ravindranath and Reed, 1990), and it has been suggested that ROS increase uncontrollably when mitochondrial GSH stores are depleted (Reed and Savage, 1995). The data with BSO show that the severe depletion of intracellular GSH does not necessarily lead to maximal ROS production. The differences in ROS production between these two methods of GSH depletion indicate that there is a specific event that takes place in the presence of glutamate and/or cysteine depletion, which leads to extreme ROS production when GSH levels drop below 20%. In the case of BSO depletion, intracellular cysteine remains high and may prevent the second burst of ROS production by acting as an antioxidant. Only after the loss of the antioxidants cysteine and GSH is there the second large rise in ROS. Therefore, the majority of the ROS production in oxidative glutamate toxicity is not simply a by-product of GSH depletion.
The Majority of the ROS Is Produced by the Mitochondrial Electron Transport Chain
Evidence that the late burst of ROS formation comes from the mitochondrial electron transport chain is most clearly seen when FCCP is used to uncouple oxidative phosphorylation, resulting in the loss of ROS formation (Fig. 3, A and B). FCCP dissipates the proton gradient that is generated for the production of ATP. During uncoupling, the electron transport chain works more efficiently to reestablish the proton gradient, while ATP synthesis becomes dependent upon glycolysis. The increase in efficiency leads to less leakage of electrons and therefore much lower levels of ROS are generated (Boveris et al., 1972; Boveris and Chance, 1973; Cino and Del Maestro, 1989).
Other inhibitors of the electron transport chain, rotenone and antimycin A, as well as one inhibitor of oxidative phosphorylation, oligomycin, were also used to identify the source of ROS production. Antimycin A decreases the rate of ROS production in cells and isolated mitochondria. Because this drug itself causes ROS production at a lower rate than glutamate-induced ROS production, it lowers the ROS produced by glutamate and protects the cells. The fact that it can prevent the effects of glutamate confirms that the majority of the late-phase ROS in oxidative glutamate toxicity come from the electron transport chain. Rotenone and oligomycin are unable to protect the cells from glutamate because they cause extreme increases in ROS production by themselves.
Glutamate acts as a substrate for complex I of the mitochondrial electron transport chain through the glutamate– malate shuttle. It is therefore possible that excess glutamate could increase electron flow into the transport chain, leading to elevated ROS production. However, the same increase in ROS production is seen with cystine-free medium (Fig. 4), indicating that increased ROS production is not simply due to more substrate. In addition, mitochondria isolated from HT22 cells do not produce detectable ROS in the presence of glutamate (data not presented). The fact that the ROS production during cystine deprivation is so similar to glutamate-induced ROS production supports the data showing that glutamate acts by blocking the cysteine/glutamate antiporter in HT22 cells.
The monoamine oxidase A inhibitor clorgyline inhibits oxidative glutamate toxicity in HT22 cells, but only at concentrations 100-fold higher than those required to inhibit the MAO activity (Maher and Davis, 1996). Since the late robust ROS production originates from mitochondria, it was asked if clorgyline is also able to block this event. Fig. 3 C shows that clorgyline blocks mitochondrial H2O2 production at high concentrations, suggesting that this is how it prevents cell death. More work must be done to understand the mechanism by which clorgyline protects HT22 cells. However, according to Fig. 3 C it is possible that clorgyline binds to a flavin-binding protein of the mitochondrial electron transport chain, thus inhibiting ROS production by blocking electron transport.
Gene Expression and ICE Protease Activity Are Necessary for ROS Production
Fig. 2 demonstrates that proteases and newly synthesized proteins are used to signal late ROS production when cells are exposed to glutamate. The effectiveness of the three inhibitors, actinomycin D, cycloheximide, and Ac-YVAD-cmk, in blocking cell death and late ROS production emphasizes the role of macromolecular synthesis and caspases in regulating ROS. A combination of several signals may lead to the 200-fold increase in ROS production seen in Fig. 1. For example, GSH depletion, the initial 5–10-fold increase in ROS, and newly synthesized proteins may all act synergistically to stimulate mitochondrial ROS formation. Previous work suggests that ROS can act as signals in apoptotic cell death (Chaudhri et al., 1986, 1988; Nakamura et al., 1993; Staal et al., 1994; Russo et al., 1995; Slater et al., 1995; Kazzaz et al., 1996; Abe et al., 1997; Esposito et al., 1997; Goldstone and Hunt, 1997). Therefore, the initial small increase in ROS may be the signal that leads to the changes in gene expression required to initiate the death program. The depletion of cysteine and GSH, which are antioxidants, may be directly responsible for the initial ROS accumulation.
The time course experiments of Fig. 2 indicate that changes in gene expression are required within the first 6 h of the cell death pathway. Both actinomycin D and cycloheximide prevent almost all ROS production when added at the same time as the glutamate, but they do not prevent GSH depletion. Ac-YVAD-cmk allows an ∼10-fold increase in ROS when it is added with glutamate, has no effect on GSH depletion, and yet still totally protects the cells when added at the same time as glutamate. One possible role of the ICE proteases may be to prevent an initial protective role of ROS. Evidence that a small increase in ROS can signal a protective mechanism is shown by the observation that if glutamate treatment is less than 6 h, the cells survive and continue dividing, in spite of the activation of genes that contribute to the increase in ROS production. Because ICE proteases seem to be activated within the first 2 h of exposure to glutamate, it is likely that they are already present in the cell but are only activated with the addition of glutamate. This conclusion is consistent with the observation that glutamate-induced macromolecular synthesis seems to occur both during and after the ICE protease activation. Although the signals that lead to gene expression and ICE protease activation are unknown, they are necessary to generate the large increase in ROS.
In addition to macromolecular synthesis changes and protease activation, 12-lipoxygenase (12-LOX) activity is required for maximal late-phase mitochondrial ROS production and cell death (Li et al., 1997a ). In the presence of glutamate, low intracellular GSH activates 12-LOX, generating an unidentified product that is required for maximum ROS levels but has no effect on the initial small increase in ROS. The 12-LOX product may interact with mitochondria directly to increase ROS production, but it also activates guanylate cyclase and the production of cGMP. cGMP, in turn, activates a cobalt-sensitive Ca2+ channel, allowing for the large increase in intracellular Ca2+ near the point of cell lysis (Li et al., 1997b ). As outlined in the following section, there is a reciprocal interaction between Ca2+ elevation and ROS formation, so it cannot be stated with certainty where in the death cascade 12-LOX products are influencing ROS formation. It is clear, however, that a series of parallel biochemical events are required to achieve the final burst of mitochondrial ROS that precedes cell lysis. Each appears to be necessary, but none sufficient to carry out the death program.
The Role of Ca2+ in ROS Production and Cell Death
A major event during programmed cell death is the increase in cytosolic Ca2+ (for review see McConkey and Orrenius, 1997). Although the interaction of Ca2+ with the ROS-generating systems is not well understood, an outline of the temporal and functional interactions between Ca2+ metabolism and ROS formation in HT22 cells after glutamate exposure can be made. Fig. 5 shows that the late-phase increase in mitochondrial-derived ROS precedes cytosolic Ca2+ elevation. When late-phase ROS production is blocked by FCCP, there is no increase in intracellular Ca2+ (Fig. 6). Conversely, if Ca2+ influx is blocked by cobalt, there is little increase in ROS (Fig. 7). Therefore, the influx of Ca2+ and mitochondrial ROS production appear to be tightly coupled. Since ruthenium red inhibits both the uptake of Ca2+ into mitochondria and glutamate toxicity (Fig. 8), it is likely that Ca2+ uptake into mitochondria is necessary for maximal ROS production. Because ruthenium red also regulates Ca2+ metabolism from the endoplasmic reticulum, it is possible that the early, low-level increases in Ca2+ are derived from intracellular stores, either in the ER or mitochondria, and that these stores may be required for the signaling leading to the later mitochondrial burst of ROS. More work is required, however, to firmly establish this relationship.
It has been shown that Ca2+ is a common signal in many different cell death pathways and that there is an intricate relationship between mitochondrial function, ROS production, Ca2+, and cell death (Randall and Thayer, 1992; Reynolds and Hastings, 1995; Richter et al., 1995; Budd and Nicholls, 1996a ,b; Isaev et al., 1996; Macho et al., 1997). Depletion of the ER Ca2+ pool can lead to apoptosis (He et al., 1997), while loss of mitochondrial function causes an increase in cytosolic Ca2+ (Luo et al., 1997). Like HT22 cells undergoing oxidative glutamate toxicity, receptor-mediated glutamate toxicity is blocked by CoCl2 and ruthenium red (Dessi et al., 1995; Thayer and Wang, 1995; Isaev et al., 1996; Korkotian and Segal, 1996). It is therefore likely that there is a common pathway of programmed cell death that uses ROS and Ca2+ in spite of differences in the initial cytotoxic insults (Richter et al., 1995).
In summary, these data show that there are two phases of ROS formation after exposure to glutamate: an early 5–10-fold increase coupled to GSH depletion and a later 200–400-fold increase derived from mitochondria. Early gene activation and caspase activity are required for both maximal ROS production and subsequent cell death. This knowledge provides a conceptional framework on which to delineate the molecular mechanisms that lead to oxidative glutamate toxicity and other forms of programmed cell death in which ROS are involved.
Acknowledgments
This work was supported by grants from the National Institutes of Health to D. Schubert (R01NS09658), Y. Sagara (2F32NS10032), Y. Liu (1F32NS10279-2), P. Maher and D. Schubert (5P01NS2812), and a fellowship from the Bundy Foundation to S. Tan.
Abbreviations used in this paper
- BSO
l-buthionine-[S,R]-sulfoximine
- DCF
dichlorofluorescin diacetate
- FCCP
carbonyl cyanide p-trifluoromethoxyphenyl-hydrazone
- GSH
glutathione
- ICE
interleukin 1β–converting enzyme
- Indo-1
indo-acetoxymethylester
- 12-LOX
12-lipoxygenase
- MAO
monoamine oxidase
- p-HPAA
p-hydroxyphenylacetic acid
- ROS
reactive oxygen species
Footnotes
The authors are especially grateful to Dr. David Chambers (Salk Institute) for his help with the flow cytometry data collection and analysis.
Address all correspondence to Professor David Schubert, Cellular Neurobiology Lab, The Salk Institute for Biological Studies, 10010 N. Torrey Pines Road, La Jolla, CA 92037. Tel.: (619) 453-4100, ext. 1562. Fax: (619) 535-9062.
References
- Abe J, Takahashi M, Ishida M, Lee J-D, Berk BC. c-Src is required for oxidative stress-mediated activation of big mitogen-activated protein kinase 1 (BMK1) J Biol Chem. 1997;272:20389–20394. doi: 10.1074/jbc.272.33.20389. [DOI] [PubMed] [Google Scholar]
- Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci USA. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass DA, Parce JW, Dechatelet LR, Szejda P, Seeds MC, Thomas M. Flow cytometric studies of oxidative product formation by neutrophils: a graded response to membrane stimulation. J Immunol. 1983;130:1910–1917. [PubMed] [Google Scholar]
- Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J. 1972;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1996a;67:2282–2291. doi: 10.1046/j.1471-4159.1996.67062282.x. [DOI] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. A reevaluation of the role of mitochondria in neuronal Ca2+homeostasis. J Neurochem. 1996b;66:403–411. doi: 10.1046/j.1471-4159.1996.66010403.x. [DOI] [PubMed] [Google Scholar]
- Cathcart R, Schwiers E, Ames BN. Detection of picomole levels of hydroperoxides using a fluorescent dichlorofluorescein assay. Anal Biochem. 1983;134:111–116. doi: 10.1016/0003-2697(83)90270-1. [DOI] [PubMed] [Google Scholar]
- Chaudhri G, Clark IA, Hunt NH, Cowden WB, Ceredig R. Effect of antioxidants on primary alloantigen-induced T cell activation and proliferation. J Immunol. 1986;137:2646–2652. [PubMed] [Google Scholar]
- Chaudhri G, Hunt NH, Clark IA, Ceredig R. Antioxidants inhibit proliferation and cell surface expression of receptors for interleukin-2 and transferrin in T lymphocytes stimulated with phorbol myristate acetate and ionomycin. Cell Immunol. 1988;115:204–213. doi: 10.1016/0008-8749(88)90174-8. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Cino M, Del Maestro RF. Generation of hydrogen peroxide by brain mitochondria: the effect of reoxygenation following postdecapitative ischemia. Arch Biochem Biophys. 1989;269:623–638. doi: 10.1016/0003-9861(89)90148-3. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- Davis JB, Maher P. Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell line. Brain Res. 1994;652:169–173. doi: 10.1016/0006-8993(94)90334-4. [DOI] [PubMed] [Google Scholar]
- Dessi F, Ben-Ari Y, Charriaut-Marlangue C. Ruthenium red protects against glutamate-induced neuronal death in cerebellar culture. Neurosci Lett. 1995;201:53–56. doi: 10.1016/0304-3940(95)12128-q. [DOI] [PubMed] [Google Scholar]
- Esposito F, Cuccovillo F, Vanoni M, Cimino F, Anderson CW, Appella E, Russo T. Redox-mediated regulation of p21waf/cip1expression involves a post-transcriptional mechanism and activation of the mitogen-activated protein kinase pathway. Eur J Biochem. 1997;245:730–737. doi: 10.1111/j.1432-1033.1997.00730.x. [DOI] [PubMed] [Google Scholar]
- Goldstone SD, Hunt NH. Redox regulation of the mitogen-activated protein kinase pathway during lymphocyte activation. Biochim Biophys Acta. 1997;1355:353–360. doi: 10.1016/s0167-4889(96)00150-4. [DOI] [PubMed] [Google Scholar]
- Griffith OW. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem. 1980;106:207–212. doi: 10.1016/0003-2697(80)90139-6. [DOI] [PubMed] [Google Scholar]
- Guilbault, G.G., J. Brignac, P.I., and M. Juneau. 1968. New substrates for the fluorometric determination of oxidative enzymes. Anal. Chem. 40:1256– 1263. [DOI] [PubMed]
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- He H, Lam M, McCormick TS, Distelhorst CW. Maintenance of calcium homeostasis in the endoplasmic reticulum by Bcl-2. J Cell Biol. 1997;138:1219–1228. doi: 10.1083/jcb.138.6.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi Y, Taniuchi S, Kobayashi Y. A quantitative assay of oxidative metabolism by neutrophils in whole blood using flow cytometry. J Immunol Methods. 1985;82:253–259. doi: 10.1016/0022-1759(85)90357-6. [DOI] [PubMed] [Google Scholar]
- Isaev NK, Zorov DB, Stelmashook EV, Uzbekov RE, Kozhemyakin MB, Victorov IV. Neurotoxic glutamate treatment of cultured cerebellar granule cells induces Ca2+-dependent collapse of mitochondrial membrane potential and ultrastructural alterations of mitochondria. FEBS Lett. 1996;392:143–147. doi: 10.1016/0014-5793(96)00804-6. [DOI] [PubMed] [Google Scholar]
- Jenner P. Oxidative damage in neurodegenerative disease. Lancet. 1994;344:796–798. doi: 10.1016/s0140-6736(94)92347-7. [DOI] [PubMed] [Google Scholar]
- Jocelyn PC. Some properties of mitochondrial glutathione. Biochim Biophys Acta. 1975;396:427–436. doi: 10.1016/0005-2728(75)90148-6. [DOI] [PubMed] [Google Scholar]
- Kazzaz JA, Xu J, Palaia TA, Mantell L, Fein AM, Horowitz S. Cellular oxygen toxicity. Oxidant injury without apoptosis. J Biol Chem. 1996;271:15182–15186. doi: 10.1074/jbc.271.25.15182. [DOI] [PubMed] [Google Scholar]
- Kohda K, Inoue T, Mikoshiba K. Ca2+ release from Ca2+ stores, particularly from ryanodine-sensitive Ca2+stores, is required for the induction of LTD in cultured cerebellar Purkinje cells. J Neurophysiol. 1995;74:2184–2188. doi: 10.1152/jn.1995.74.5.2184. [DOI] [PubMed] [Google Scholar]
- Korkotian E, Segal M. Lasting effects of glutamate on nuclear calcium concentration in cultured rat hippocampal neurons: regulation by calcium stores. J Physiol (Lond) 1996;496:39–48. doi: 10.1113/jphysiol.1996.sp021663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBel CP, Ischiropoulos H, Bondy SC. Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol. 1992;5:227–231. doi: 10.1021/tx00026a012. [DOI] [PubMed] [Google Scholar]
- Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997a;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- Li Y, Maher P, Schubert D. Requirement for cGMP in nerve cell death caused by glutathione depletion. J Cell Biol. 1997b;139:1–8. doi: 10.1083/jcb.139.5.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Rosenthal RE, Fiskum G. H2O2release by brain mitochondria in a canine cerebral ischemia/reperfusion model. FASEB (Fed Am Soc Exp Biol) J. 1993;7:A424. [Google Scholar]
- Luo X, Bond JD, Ingram VM. Compromised mitochondrial function leads to increased cytosolic calcium and to activation of MAP kinases. Proc Natl Acad Sci USA. 1997;94:9705–9710. doi: 10.1073/pnas.94.18.9705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macho A, Hirsch T, Marzo I, Marchetti PD, Dallaporta B, Susin SA, Zamzami N, Kroemer G. Glutathione depletion is an early and calcium elevation is a late event of thymocyte apoptosis. J Immunol. 1997;158:4612–4619. [PubMed] [Google Scholar]
- Maher P, Davis JB. The role of monoamine metabolism in oxidative glutamate toxicity. J Neurosci. 1996;16:6394–6401. doi: 10.1523/JNEUROSCI.16-20-06394.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConkey DJ, Orrenius S. The role of calcium in the regulation of apoptosis. Biochem Biophys Res Commun. 1997;239:357–366. doi: 10.1006/bbrc.1997.7409. [DOI] [PubMed] [Google Scholar]
- McFarlane MB, Bruhn TO, Jackson IM. Postsomatostatin hypersecretion of growth hormone from perfused rat anterior pituitary cells is dependent on calcium influx. Neuroendocrinology. 1993;57:496–502. doi: 10.1159/000126397. [DOI] [PubMed] [Google Scholar]
- McPherson PS, Campbell KP. Characterization of the major brain form of the ryanodine receptor/Ca2+release channel. J Biol Chem. 1993;268:19785–19790. [PubMed] [Google Scholar]
- Miyamoto M, Murphy TH, Schnaar RL, Coyle JT. Antioxidants protect against glutamate-induced cytotoxicity in a neuronal cell line. J Pharmacol Exp Ther. 1989;250:1132–1140. [PubMed] [Google Scholar]
- Moreadith RW, Fiskum G. Isolation of mitochondria from ascites tumor cells permeabilized with digitonin. Anal Biochem. 1984;137:360–367. doi: 10.1016/0003-2697(84)90098-8. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB (Fed Am Soc Exp Biol) J. 1990;4:1624–1633. [PubMed] [Google Scholar]
- Nakamura K, Hori T, Sato N, Sugie K, Kawakami T, Yodoi J. Redox regulation of a src family protein tyrosine kinase p56lckin T cells. Oncogene. 1993;8:3133–3139. [PubMed] [Google Scholar]
- Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ. Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J Neurosci. 1993;13:1441–1453. doi: 10.1523/JNEUROSCI.13-04-01441.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YB, Herrington J, Babcock DF, Hille B. Ca2+clearance mechanisms in isolated rat adrenal chromaffin cells. J Physiol (Lond) 1996;492:329–346. doi: 10.1113/jphysiol.1996.sp021312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall RD, Thayer SA. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J Neurosci. 1992;12:1882–1895. doi: 10.1523/JNEUROSCI.12-05-01882.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindranath V, Reed DJ. Glutathione depletion and formation of glutathione-protein mixed disulfide following exposure of brain mitochondria to oxidative stress. Biochem Biophys Res Commun. 1990;169:1075–1079. doi: 10.1016/0006-291x(90)92004-j. [DOI] [PubMed] [Google Scholar]
- Reed DJ, Savage MK. Influence of metabolic inhibitors on mitochondrial permeability transition and glutathione status. Biochim Biophys Acta. 1995;1271:43–50. doi: 10.1016/0925-4439(95)00008-r. [DOI] [PubMed] [Google Scholar]
- Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter C, Gogvadze V, Laffranchi R, Schlapbach R, Schweizer M, Suter M, Walter P, Yaffee M. Oxidants in mitochondria: from physiology to diseases. Biochim Biophys Acta. 1995;1271:67–74. doi: 10.1016/0925-4439(95)00012-s. [DOI] [PubMed] [Google Scholar]
- Russo T, Zambrano N, Esposito F, Ammendola R, Cimino F, Fiscella M, Jackman J, O'Connor PM, Anderson CW, Appella E. A p53- independent pathway for activation of WAF1/CIP1 expression following oxidative stress. J Biol Chem. 1995;270:29386–29391. doi: 10.1074/jbc.270.49.29386. [DOI] [PubMed] [Google Scholar]
- Sagara, Y., R. Dargusch, D. Chambers, J. Davis, D. Schubert, and P. Maher. 1998. Cellular mechanisms of resistance to chronic oxidative stress. Free Radical Biol. Med. In press. [DOI] [PubMed]
- Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater AF, Nobel CS, Maellaro E, Bustamante J, Kimland M, Orrenius S. Nitrone spin traps and a nitroxide antioxidant inhibit a common pathway of thymocyte apoptosis. Biochem J. 1995;306:771–778. doi: 10.1042/bj3060771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal FJ, Anderson MT, Staal GE, Herzenberg LA, Gitler C, Herzenberg LA. Redox regulation of signal transduction: tyrosine phosphorylation and calcium influx. Proc Natl Acad Sci USA. 1994;91:3619–3622. doi: 10.1073/pnas.91.9.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki YS, Forman HJ, Sevanian A. Oxidants as stimulators of signal transduction. Free Radical Biol Med. 1997;22:269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- Tan, S., M. Wood, and P. Maher. 1998. Oxidative stress induces a form of programmed cell death with characteristics of both apoptosis and necrosis in neuronal cells. J. Neurochem. In press. [DOI] [PubMed]
- Thayer SA, Wang GJ. Glutamate-induced calcium loads: effects on energy metabolism and neuronal viability. Clin Exp Pharmacol Physiol. 1995;22:303–304. doi: 10.1111/j.1440-1681.1995.tb02004.x. [DOI] [PubMed] [Google Scholar]
- Tietze F. Enzymatic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- Voet, D., and J.G. Voet. 1990. Biochemistry. Wiley Liss, New York. 1223 pp.
- Vornov JJ, Coyle JT. Glutamate neurotoxicity and the inhibition of protein synthesis in the hippocampal slice. J Neurochem. 1991;56:996–1006. doi: 10.1111/j.1471-4159.1991.tb02020.x. [DOI] [PubMed] [Google Scholar]