

Abstract
OCI-5/GPC3 is a member of the glypican family. Glypicans are heparan sulfate proteoglycans that are bound to the cell surface through a glycosyl-phosphatidylinositol anchor. It has recently been shown that the OCI-5/GPC3 gene is mutated in patients with the Simpson-Golabi-Behmel Syndrome (SGBS), an X-linked disorder characterized by pre- and postnatal overgrowth and various visceral and skeletal dysmorphisms. Some of these dysmorphisms could be the result of deficient growth inhibition or apoptosis in certain cell types during development. Here we present evidence indicating that OCI-5/GPC3 induces apoptosis in cell lines derived from mesothelioma (II14) and breast cancer (MCF-7). This induction, however, is cell line specific since it is not observed in NIH 3T3 fibroblasts or HT-29 colorectal tumor cells. We also show that the apoptosis-inducing activity in II14 and MCF-7 cells requires the anchoring of OCI-5/GPC3 to the cell membrane. The glycosaminoglycan chains, on the other hand, are not required. MCF-7 cells can be rescued from OCI-5/GPC3–induced cell death by insulin-like growth factor 2. This factor has been implicated in Beckwith-Wiedemann, an overgrowth syndrome that has many similarities with SGBS. The discovery that OCI-5/GPC3 is able to induce apoptosis in a cell line– specific manner provides an insight into the mechanism that, at least in part, is responsible for the phenotype of SGBS patients.
The Simpson-Golabi-Behmel Syndrome (SGBS)1 is an X-linked condition characterized by pre- and postnatal overgrowth (Garganta and Bodurtha, 1992; Gurrieri et al., 1992; Terespolsky et al., 1995). Affected individuals have several dysmorphisms that can include a distinct facial appearance, macroglossia, cleft palate, cardiac defects, enlarged and dysplastic kidneys, cryptorchidism, hypospadia, hernias, supernumerary nipples, vertebral and rib anomalies, coccygeal bony appendage, syndactyly, and polydactyly (Garganta and Bodurtha, 1992; Gurrieri et al., 1992; Terespolsky et al., 1995). In addition, these patients have an increased risk for the development of Wilms' tumors (Hughes-Benzie et al., 1992). Premature death is also very frequent (Garganta and Bodurtha, 1992).
Recently, Pilia et al. (1996) reported that individuals with SGBS display mutations in the OCI-5/glypican 3 (OCI-5/GPC3) gene. OCI-5/GPC3 is a member of the glypican family. Glypicans are heparan sulfate proteoglycans that are bound to the cell surface through a glycosyl-phosphatidylinositol (GPI) anchor (David, 1993). Five members of the glypican family have been identified in mammals: GPC1 (glypican) (David et al., 1990), GPC2 (cerebroglycan) (Stipp et al., 1994), GPC3 (OCI-5) (Filmus et al., 1995), GPC4 (K-glypican) (Watanabe et al., 1995), and GPC5 (Saunders et al., 1997; Veugelers et al., 1997). Although the degree of similarity between the different glypicans varies from 20 to 50%, the positions of the 13 cysteines and the putative glycosaminoglycan attachment sites are well conserved across the family (Veugelers et al., 1997). Recently, the cloning of dally, a Drosophila gene with significant homology to the glypican family, was reported (Nakato et al., 1995). This gene also shares the structural features common to the vertebrate glypicans. Regulatory mutations of dally have a severe impact on the postembryonic development of the nervous system and produce morphological defects in several tissues, including the eyes, antennae, wings, and genitalia (Nakato et al., 1995).
The specific functions of glypicans are still not known, but experimental evidence has been provided indicating that (a) GPC1 can act in vitro as a co-receptor for “heparin binding” growth factors such as basic fibroblast growth factor (Steinfeld et al., 1996); and (b) dally controls cellular responses to Decapentaplegic, a transforming growth factor β–related morphogen (Jackson et al., 1997).
OCI-5, the rat homologue of GPC3, was originally identified as a gene that is developmentally regulated in the intestine (Filmus et al., 1988). OCI-5 is expressed at high levels in numerous tissues during development but is downregulated in most adult tissues (Filmus, J., unpublished observations). Functional studies on OCI-5/GPC3 have not yet been reported, but the phenotype of the SGBS patients suggests that this gene is involved in the control of cell proliferation and/or survival. For example, syndactyly in SGBS patients may be the consequence of deficient induction of growth inhibition or apoptosis in the mesenchymal cells forming the interdigital and interphalangeal spaces during development (Jacobson et al., 1997).
Here we present experimental evidence indicating that OCI-5/GPC3 can induce apoptosis and that this apoptosis-inducing capacity of OCI-5/GPC3 is cell line specific.
Materials and Methods
Cell Lines
The asbestos-induced rat mesothelioma cell line II14 and the rat mesothelial cell line NRM2 were kindly provided by Dr. C. Walker (M.D. Anderson, Smithville, TX). They were cultured in Ham's F-12: DME (1:1; GIBCO BRL, Gaithersburg, MD) with 10% heat-inactivated FBS and supplemented with 0.1 μg/ml hydrocortisone, 2.5 μg/ml insulin, 2.5 μg/ml transferrin, and 2.5 ng/ml sodium selenite. COS-1, NIH 3T3, MCF-7, HT-29, and HepG2 cell lines (American Type Culture Collection, Rockville, MD) were cultured in DME supplemented with 10% FBS. All cell lines were maintained at 37°C in a humidified atmosphere with 5% CO2.
Generation of OCI-5/GPC3 Mutants
To delete the GPI-anchoring domain, an AgeI site was introduced by changing bases 1765 and 1766 from TC to GG (Filmus et al., 1988) using a PCR-based site-directed mutagenesis method (Cadwell and Joyce, 1995). After mutation, the AgeI-EcoRI fragment containing the 3′ end of the OCI-5 cDNA was removed and replaced by a linker sequence (5′-CCGGAGCTGACTAACTGAATT-3′) that contains three stop codons.
To mutate the two potential glycosaminoglycan (GAG) attachment sites, serine residues 494 and 508 (Filmus et al., 1988) were mutated to alanine by site-directed mutagenesis (Cadwell and Joyce, 1995). Mutations were verified by DNA sequencing.
Transfections
For the colony forming efficiency assay, the wild-type or mutated hemagglutinin A (HA)-tagged OCI-5/GPC3 cDNA (Filmus et al., 1995) was introduced into the pEF-BOS vector (Mizushima and Nagata, 1990) and transfected into cells by using Lipofectin according to the manufacturer's instructions (GIBCO BRL). Cell cultures were then selected in the presence of 600 μg/ml of geneticin, and surviving colonies were counted 2 wk later. Experiments were performed in triplicate and repeated using different batches of DNA. In some cases, individual colonies were isolated using cloning cylinders and expanded in the presence of 200 μg/ml of geneticin.
To generate MCF-7 cells with Cd2+-inducible OCI-5/GPC3 expression, the cDNA was cloned into a vector under the control of the rat metallothionein promoter (Filmus et al., 1994), and transfection was performed with Lipofectin. Selection was performed in the presence of 600 μg/ml geneticin. Surviving clones were isolated and expanded. To induce expression, MCF-7 cells were incubated with 5 μM CdCl2 in serum-containing medium.
Transient transfection assays of MCF-7 cells were performed as previously described (Muzio et al., 1996). Briefly, cells were transfected with a β-galactosidase expression vector and a fivefold excess of the pEF-BOS vector or a vector containing wild-type or mutant OCI-5/GPC3. Serum was removed 12 h after transfection, and 36 h later cells were either fixed and stained with X-gal or lysed to prepare cytoplasmic extracts to be used in the Cell Death Elisa Assay. In addition, cells transfected in parallel plates were lysed, and the expression of OCI-5/GPC3 was assessed by Western blot of immunoprecipitated extracts as described below.
Transient transfection and [35S]sulfate labeling of COS-1 cells was performed as previously described (Filmus et al., 1995).
Characterization of OCI-5/GPC3 Protein in Transfected Clones
OCI-5/GPC3 expression was investigated by Western blotting. When indicated, OCI-5/GPC3 was immunoprecipitated before the Western blotting. Briefly, cells were lysed in RIPA buffer containing protease inhibitors (2 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 μg/ml pepstatin, and 10 mM EDTA). Conditioned media was concentrated using Centricon-10 filters (Amicon, Beverley, MA). Lysates and conditioned media were immunoprecipitated using the anti-HA monoclonal antibody 12CA5 (20 μg/ml) and protein A beads. Immunoprecipitates were rinsed three times with RIPA buffer, and an equal volume of 2× reducing sample buffer was added. After boiling for 5 min, proteins were run on a 4–9% polyacrylamide gradient gel or a 7.5% polyacrylamide gel. For Western blotting, proteins were transferred to a polyvinyl difluoride membrane (Dupont, Boston, MA). Membranes were blocked in PBS plus 5% nonfat dry milk for 1 h at room temperature and then incubated for 1 h with 1 μg/ ml 12CA5 antibody. After incubation with horseradish peroxidase anti– mouse or anti–rabbit secondary antibody for 1 h, the bands were detected using ECL reagents (Amersham Corp., Arlington Heights, IL). The gel containing [35S]sulfate-labeled proteins immunoprecipitated with the 12CA5 antibody was analyzed by autoradiography.
Analysis of Poly ADP Ribose Polymerase
Cells were washed with PBS, collected by scraping, and lysed with RIPA buffer containing protease inhibitors. Proteins were then transferred to a polyvinyl difluoride membrane and subjected to Western blotting as described above, using 2 μg of an anti–poly ADP ribose polymerase (PARP) antibody (Boehringer-Mannheim Corp., Indianapolis, IN).
Apoptosis Assays in the Mesothelioma Cell Line II14
2 × 105 cells were seeded in 3.5-cm plates in complete medium. After 24 h, cells were rinsed once with PBS, once with serum-free medium (1:1 F12/ DME), and incubated in the same medium for 48 h. Floating and attached cells were then counted using a hemocytometer.
Quantitation of Apoptosis by the Cell Death Elisa Assay
This assay was performed using the Cell Death Elisa kit from Boehringer-Mannheim following the manufacturer's instructions. Briefly, microwells were coated with the antihistone antibody, and cytoplasmic extract from 5 × 103 cells was added to each well. Binding of histone-DNA conjugates to the histone antibody was detected with an anti-DNA antibody conjugated to peroxidase.
Generation of Polyclonal Antibodies against OCI-5/GPC3
The polyclonal antibody against human GPC3 was generated by injecting sheep with a glutathione-S-transferase fusion protein containing the last 70 amino acids of GPC3. The polyclonal antibody was affinity purified against the immunogen. The rat polyclonal antibody against rat OCI-5 was generated by injecting rabbits with a glutathione-S-transferase fusion protein containing the last 70 amino acids of OCI-5. The polyclonal antibody was affinity purified against the immunogen.
Results
Effect of OCI-5/GPC3 Expression on Colony Forming Efficiency
The fact that SGBS patients display supernumerary nipples strongly suggests that OCI-5/GPC3 is involved in the regulation of cell survival in the mammary gland during development (Hughes-Benzie et al., 1996). We have hypothesized that OCI-5/GPC3 could also be a negative regulator of survival of mammary tumor cells, which, like mammary cells during development, are actively proliferating. To test this hypothesis we decided to transfect this glypican into the OCI-5/GPC3–negative MCF-7 breast cancer line. We also included in our transfection studies a rat mesothelioma cell line (II14), since OCI-5/GPC3 is highly expressed in normal mesothelial cells, but it is downregulated in mesotheliomas (Testa, J.R., unpublished observations).
In an initial attempt to investigate whether OCI-5/GPC3 has growth-suppressive activity, a colony forming efficiency assay was performed. This assay has been previously used to demonstrate, for example, the growth-suppressive activities of p53 and p16 (Baker et al., 1990; Lin et al., 1996). Thus, the MCF-7 breast cancer cell line and the II14 mesothelioma cell line were transfected with an expression vector with or without the OCI-5/GPC3 cDNA, and the number of colonies that grew in the presence of geneticin were counted after 14 d of selection. Table I shows that the number of colonies generated after OCI-5/ GPC3 transfection was reduced by 60 and 70% in II14 and MCF-7 cells, respectively, compared with transfection with vector control. Similar results were obtained by using at least two different batches of DNA. To test whether this inhibitory effect of OCI-5/GPC3 on colony forming efficiency is cell line specific, we used the same expression vectors to transfect NIH 3T3 mouse fibroblasts and the colorectal tumor cell line HT-29 (both cell lines are OCI-5/ GPC3 negative). Table I shows that OCI-5/GPC3 did not affect the colony forming efficiency of NIH 3T3 and HT-29 cells, since the number of colonies that grew after OCI-5/ GPC3 transfection was similar to the number obtained after transfection of the vector control.
Table I.
Colony Forming Efficiency Assay
| II14 | MCF-7 | NIH3T3 | HT-29 | |||||
|---|---|---|---|---|---|---|---|---|
| Vector alone | 100* | 100 | 100 | 100 | ||||
| OCI-5/GPC3 | 41.7 ± 2.7 | 29.5 ± 3.9 | 92.1 ± 11.9 | 104.9 ± 1.6 | ||||
| ΔOCI-5/GPC3 | 91.5 ± 3.3 | 91.5 ± 6.0 | nd | nd | ||||
| OCI-5/GPC3 (ΔGAG) | 37.4 ± 4.3 | 79.4 ± 1.7 | nd | nd |
Results are presented as percentage (mean ± standard error) with respect to number of geneticin-surviving colonies in cells transfected with vector alone. Similar results were obtained with at least two batches of DNA. nd, not done.
Colonies that grew after OCI-5/GPC3 transfection were expanded and screened for OCI-5/GPC3 expression. Only 3 out of 60 II14-derived clones and 3 out of 50 MCF-7–derived clones displayed detectable OCI-5/GPC3. In the case of NIH 3T3 cells and HT-29 cells, on the other hand, 90% of the colonies were OCI-5/GPC3 positive. This confirms the cell-specific nature of the effect of OCI-5/GPC3 in the colony forming assay.
Transient Expression of OCI-5/GPC3 in MCF-7 Cells
As shown previously in colony forming assays with p53 and p16 (Baker et al., 1990; Lin et al., 1996), the inhibitory effect of OCI-5/GPC3 in colony formation could be due to an apoptotic-inducing activity or to an ability to inhibit cell proliferation. To distinguish between these two possibilities, the apoptotic-inducing activity of OCI-5/GPC3 was evaluated by transient expression in MCF-7 cells. This type of assay has been extensively used in MCF-7 cells to identify molecules that are able to induce apoptosis (Chinnaiyan et al., 1995; Muzio et al., 1996) and is based on the observation that MCF-7 cells undergoing apoptosis acquire a characteristic rounded morphology before detaching from the dish. To identify cells that have taken up the transfected DNA, a β-galactosidase expression vector is included in the transfection mixture. When MCF-7 cells were transfected with the OCI-5/GPC3 expression vector, >80% of blue cells displayed the typical apoptotic morphology. After transfection of vector control, on the other hand, only 30% of blue cells became rounded (Fig. 1 A). This experiment was repeated several times with similar results. We have also quantitated the apoptotic-inducing activity of OCI-5/GPC3 by using the Cell Death Elisa Assay, which measures the amount of cytoplasmic histone- associated DNA fragments. Fig. 1 B shows that this assay also demonstrates that OCI-5/GPC3 induces apoptosis of MCF-7 cells after transient transfection. To verify that OCI-5/GPC3 is being expressed in the transiently transfected MCF-7 cells, a Western blot was performed. Fig. 2 shows that indeed this is the case. These results indicate, therefore, that OCI-5/GPC3 has the capacity to induce apoptosis in MCF-7 cells upon transient expression.
Figure 1.

Transient expression assay in MCF-7 cells. MCF-7 cells were transiently transfected with RSV β-galactosidase and a fivefold excess of (a) vector control, (b) OCI-5/ GPC3, (c) ΔOCI-5/GPC3, and (d) OCI-5/ GPC3Δ (GAG). (A) 48 h after transfection, cells were fixed and stained with X-gal. Every vector was transfected into triplicate plates. The results (mean ± standard error) were expressed as percentage of rounded (apoptotic) cells among the blue-stained cells. The experiment was repeated three times with similar results. (B) 48 h after transfection, cytoplasmic extracts from equal number of cells were prepared, and the amount of DNA–histone complexes was measured by the Cell Death Elisa Assay. Every vector was transfected into triplicate plates. The results (mean ± standard error) are expressed as fold increase in apoptosis (as measured by OD at 405 nm) compared with cells transfected with vector control.
Figure 2.

Expression of OCI-5/ GPC3 in transiently transfected MCF-7 cells. Equal numbers of cells were transfected with the indicated HA-tagged OCI-5/GPC3 expression vectors. 48 h later cells were lysed, and OCI-5/ GPC3 was immunoprecipitated with an anti-HA 12CA5 antibody. The immunoprecipitates were then analyzed by Western blot with the same antibody. Lane a, untransfected controls; lane b, OCI-5/ GPC3; lane c, OCI-5/GPC3 ΔGAG; lane d, ΔOCI-5/GPC3. The bracket indicates glycanated OCI-5/GPC3, the arrow the core protein, and the arrowhead a cleaved fragment of the core protein. Numbers on the left represent molecular mass markers expressed in kD.
Effect of Inducible Expression of OCI-5/GPC3 in MCF-7 Cells
To confirm the apoptosis-inducing activity of OCI-5/ GPC3 in MCF-7 cells, we generated a clone expressing OCI-5/GPC3 in an inducible manner. Thus, MCF-7 cells were transfected with a vector in which the expression of OCI-5/GPC3 was driven by the metallothionein promoter. More than 30 clones resistant to geneticin were screened, and three of them displayed inducible expression of OCI-5/ GPC3. Fig. 3 shows that significant levels of OCI-5/GPC3 are induced in one of these clones (MT3) after incubation with 5 μM Cd2+ overnight, although low levels of expression are also observed in uninduced cells. If the induction is maintained for 3 d in serum-containing medium, MT3 cells start to round up and detach from the plate. Fig. 4 d shows that after 4 d of incubation with Cd2+, a significant number of MT3 cells are floating in the medium. This has also been observed with the other two inducible clones (data not shown). Similar treatment of parental MCF-7 cells, on the other hand, does not affect their viability (Fig. 4 b). To verify that OCI-5/GPC3–expressing MT3 cells are undergoing apoptosis, the status of PARP was investigated by Western blot. Fig. 3 shows that after induction of OCI-5/GPC3 expression for 4 d, MT3 cells display the typical pattern of apoptotic cells with regard to the cleavage of PARP. No cleavage is observed in the parental MCF-7 cells treated with 5 μM Cd2+ for the same amount of time. We also confirmed that inducible expression of OCI-5/ GPC3 triggers apoptosis in MCF-7 cells by performing a Cell Death Elisa Assay after 4 d of induction (Fig. 5).
Figure 3.
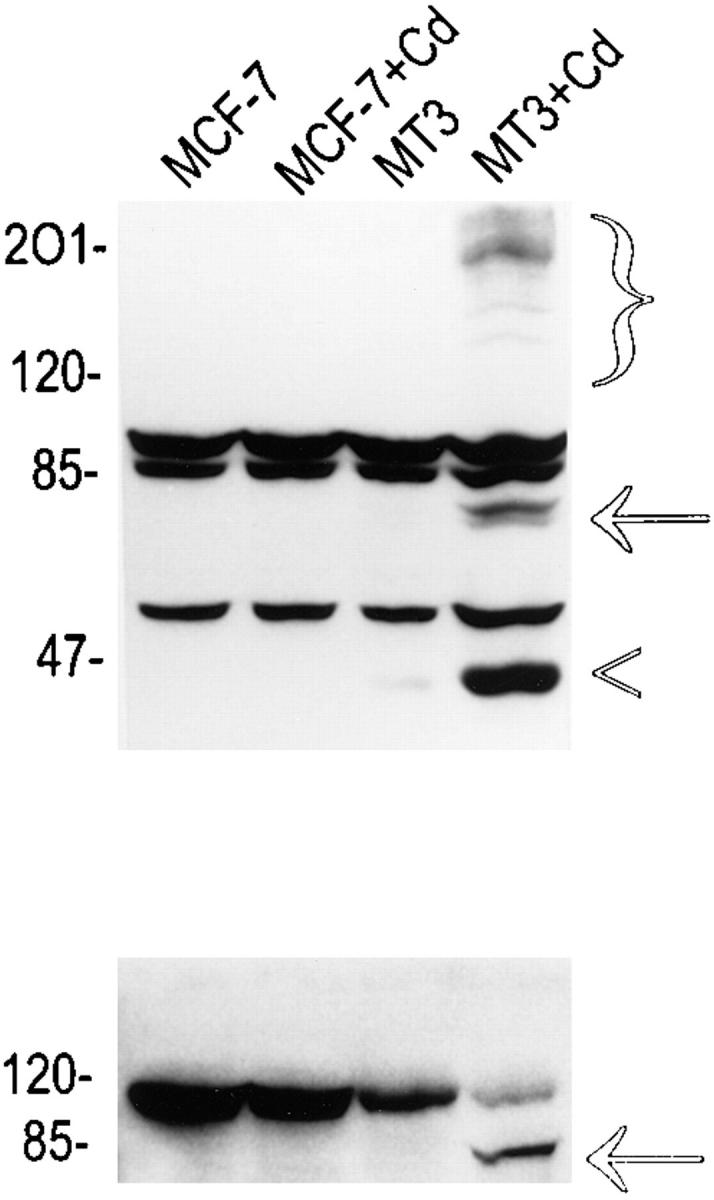
Western blot analysis of MCF-7 cells expressing inducible OCI-5/GPC3. (Top) OCI-5/GPC3. Bracket, glycanated OCI-5/GPC3; arrow, core protein; arrowhead, cleavage product of core protein. (Bottom) PARP. The arrow indicates the cleavage product of PARP. Numbers on the left represent molecular mass markers expressed in kD.
Figure 4.
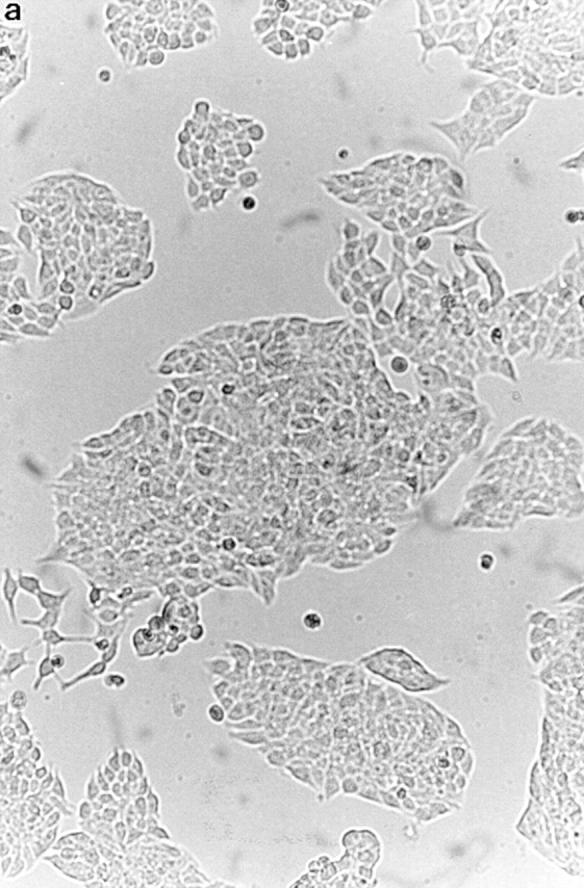
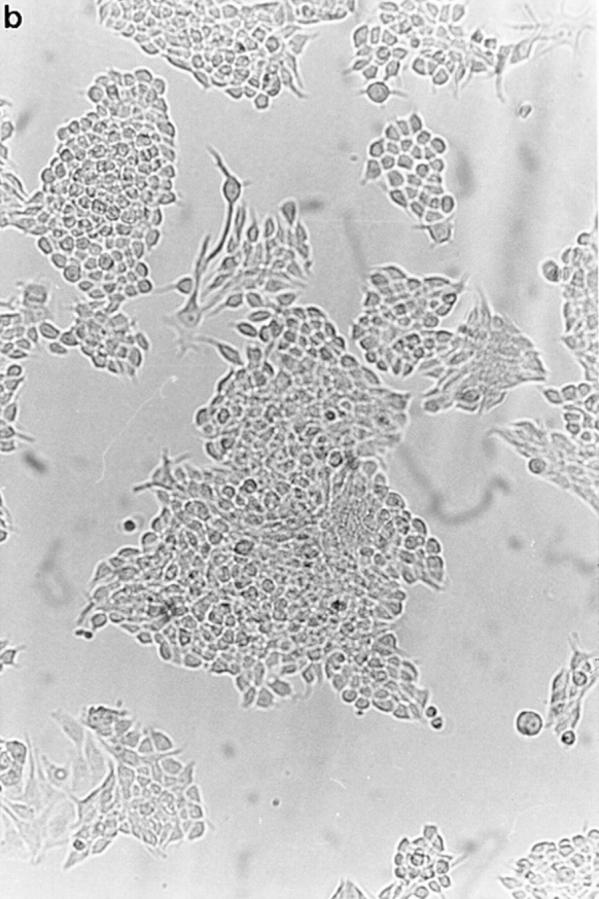

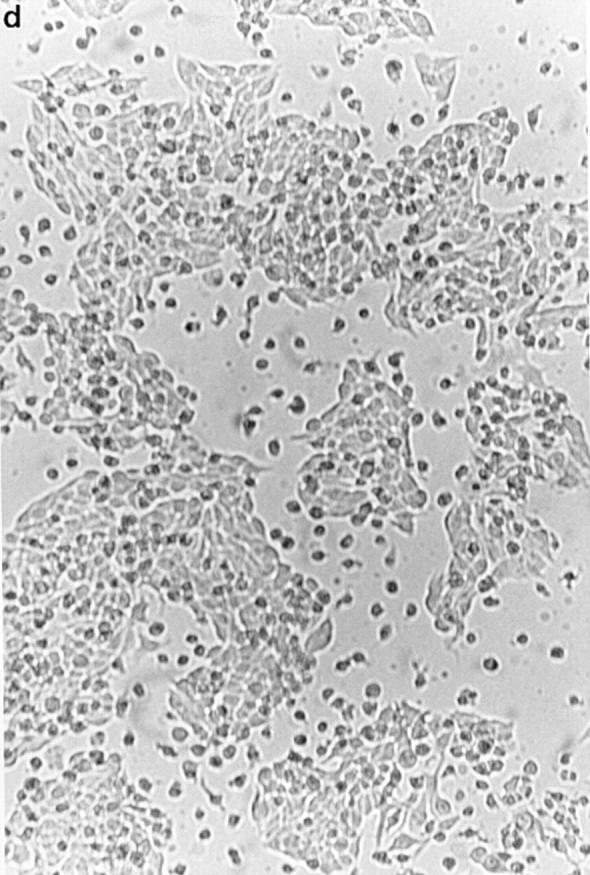
Effect of inducible OCI-5/GPC3 expression on MCF-7 cells. Cells were incubated in regular medium with (b and d) and without (a and c) 5 μM CdCl2 for 4 d and photographed. (a and b) MCF-7 cells. (c and d) MT3 cells.
Figure 5.
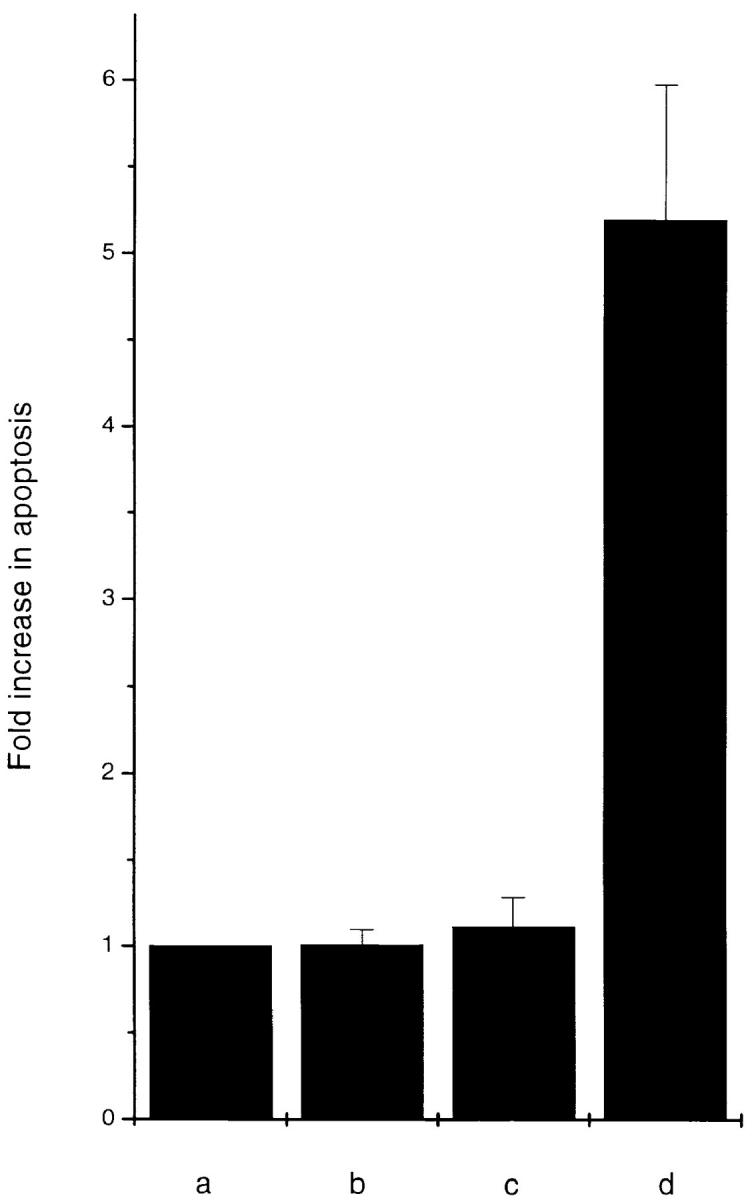
Cell Death Elisa Assay of MCF-7 cells expressing inducible OCI-5/ GPC3. (a) MCF-7 cells, (b) MCF-7 cells + 5 μM Cd2+, (c) MT3 cells, (d) MT3 + 5 μM Cd2+. Every cell line was tested in triplicate plates. The results (mean ± standard error) are expressed as fold increase in apoptosis (as measured by OD at 405 nm) compared with noninduced MCF-7 cells.
Effect of OCI-5/GPC3 Expression on II14 Mesothelioma Cells
It proved difficult to transfect II14 cells with high enough efficiency for transient expression assays. In addition, we have not yet been able to generate II14 mesothelioma clones with inducible OCI-5/GPC3. However, we have expanded the few clones that survived after the transfection of constitutive OCI-5/GPC3. Fig. 6 shows a Western blot analysis of OCI-5/GPC3 expression in three of them. This Western blot had to be overexposed to be able to detect the smear corresponding to the glycanated form of OCI-5/ GPC3. Still, it is obvious that in these cells there is strong expression of the nonglycanated form of OCI-5/GPC3. However, this is not an artifact caused by exogenous expression of OCI-5/GPC3 in II14 mesothelioma cells; when Western blot analysis was performed on NRM2 normal mesothelial cells and HepG2 hepatoma cells using polyclonal antibodies against OCI-5/GPC3, a similar strong expression of the nonglycanated form was observed (Fig. 7). When the OCI-5/GPC3–containing mesothelioma clones were subconfluent, no obvious indication of apoptosis was observed. However, we noticed that when these three clones become confluent, there is an increase in the number of cells that detach from the plate, compared with II14 parental cells or clones transfected with vector control. We decided, therefore, to grow OCI-5/GPC3–positive and –negative II14 clones in the absence of serum to determine whether in this apoptosis-prone condition, a difference between OCI-5/GPC3–positive and –negative clones can be observed. Indeed, after 2 d in serum-free conditions, a significant proportion of cells in the OCI-5/GPC3–expressing clones rounded up and detached. In control clones, on the other hand, the number of floating cells was much lower (Fig. 8). When the floating cells were stained with acridine orange, they showed the typical pattern of apoptotic cells with condensed and fragmented nuclei (data not shown). This result indicates that the inhibitory activity of OCI-5/ GPC3 on II14 cells in the colony forming assay is, at least in part, due to OCI-5/GPC3–induced apoptosis. The OCI-5/ GPC3–expressing clones that survive probably express levels of the glypican that are insufficient to induce apoptosis in the presence of serum in subconfluent cultures but are enough to do so in serum-free conditions.
Figure 6.

Western blot analysis of OCI-5/GPC3 expression in transfected II14 cells. Lane a, parental II14 cells; lanes b–d, clones transfected with OCI-5/GPC3. Bracket, glycanated OCI-5/GPC3; arrow, core protein; arrowhead, cleavage product of core protein. Numbers on the left represent molecular mass markers.
Figure 7.

Western blot analysis of endogenous OCI-5/GPC3 expression. (A) Normal NRM2 rat mesothelial cells (lane a) and II14 mesothelioma cells (lane b) were lysed, and OCI-5 was immunoprecipitated with 10 μg/ml of affinity-purified anti–OCI-5 polyclonal antibody. The immunoprecipitated material was analyzed by Western blot using the same antibody (1 μg/ml). The arrow indicates the OCI-5/GPC3 core protein, and the arrowhead indicates the Ig band. The glycanated form of OCI-5 could not be detected in the conditions used here. (B) HepG2 hepatoma cells (lane a) and COS-1 fibroblasts (lane b) were lysed, and expression of OCI-5/GPC3 was analyzed by Western blot with 1 μg/ml of affinity-purified anti-GPC3 polyclonal antibody. The arrow indicates the GPC3 core protein, and the bracket indicates the glycanated GPC3.
Figure 8.

Effect of OCI-5/GPC3 expression in II14 cells. Cells were incubated in serum-free conditions for 48 h, and the number of floating cells was counted. Results are the mean value ± standard error of triplicates. (a) II14 cells. (b and c) Clones transfected with vector alone. (d–f) Clones expressing OCI-5/GPC3.
Role of the GPI Anchor and the Glycosaminoglycan Chains in the Apoptosis-inducing Activity of OCI-5/GPC3
Since glypicans can be secreted into the medium (David et al., 1990; Watanabe et al., 1995), it was important to investigate whether the apoptotic activity of OCI-5/GPC3 requires the anchorage of this glypican to the cell membrane. A deletion mutant was then generated by removing the last 25 amino acids of OCI-5/GPC3. This fragment includes the sequences required for the addition of the GPI tail, but not the consensus sequences required for the attachment of the GAG chains (Filmus et al., 1988). To verify that this mutated glypican (ΔOCI-5/GPC3) was released into the medium, COS-1 cells were transiently transfected with wild-type and mutant OCI-5/GPC3. Fig. 9 shows that, whereas most of the wild-type glycanated OCI-5/GPC3 is found in the cell lysate, the mutant is predominantly present in the medium. The small amount of mutant OCI-5/GPC3 that can be seen in the cell lysate is probably peripheral and bound to the cell surface by electrostatic interactions. It has already been shown that a significant proportion of GPC1 remains bound to the cell surface after cleavage of the GPI tail (Carey and Evans, 1989; Bonneh-Barkay et al., 1997). It is also possible that we are detecting OCI-5/GPC3 that is in transit from the Golgi apparatus to the cell surface. When ΔOCI-5/GPC3 was transfected into II14 and MCF-7 cells, the number of geneticin-surviving colonies was similar to the number observed with the vector control (Table I). This result clearly indicates that the inhibitory activity of OCI-5/GPC3 in the colony forming efficiency assay requires its anchorage to the cell surface. Several II14 clones transfected with the mutant OCI-5/GPC3 were pooled and analyzed by Western blot. As expected, most of the OCI-5/GPC3 was found in the supernatant (data not shown). The requirement of the GPI tail for the apoptosis-inducing activity of OCI-5/ GPC3 was confirmed by transient expression assays in MCF-7 cells. As shown in Fig. 1, the percentage of OCI-5/ GPC3–transfected cells that undergo apoptosis is similar to that obtained by transfection with vector control, despite the fact that the transfected glypican is highly expressed in the transfected cells (Fig. 2).
Figure 9.

Western blot analysis of GPI domain-deletion mutant. cell, immunoprecipitated cell lysates: (lane a) Cells transfected with vector alone; (lane b) cells transfected with wild-type OCI-5/ GPC3; (lane c) cells transfected with ΔOCI-5/GPC3. sup, conditioned media: (lane a) Cells transfected with wild-type OCI-5/GPC3; (lane b) cells transfected with ΔOCI-5/GPC3. Numbers on the middle represent molecular mass markers.
It has been clearly established that the GAG chains play an important role in the interactions of cell surface proteoglycans with growth factors and cell adhesion molecules (Wight et al., 1992; Rapraeger et al., 1994; Salmivirta et al., 1996). In the particular case of glypicans, however, it has been proposed that the core proteins have functions in addition to acting as anchors for GAG chains. This hypothesis is based on the observation that the sequence of glypicans is highly conserved during evolution and that the GAG insertion sites are all located close to the COOH terminus (Stipp et al., 1994; Saunders et al., 1997). It was decided, therefore, to investigate whether the GAG chains of OCI-5/GPC3 are required for its apoptosis-inducing activity. To do this, the serines at the two potential GAG attachment sites were converted into alanines. To verify that these mutations inhibited the addition of GAG chains, the mutated glypicans were transiently transfected into [35S]sulfate-labeled COS-1 cells and analyzed by immunoprecipitation and autoradiography. Fig. 10 shows that, as expected, cells transfected with OCI-5/GPC3 in which both serines have been mutated do not show any signal after immunoprecipitation. The glypicans with only one serine mutated, on the other hand, show bands whose mobility indicate that they correspond to proteoglycans that are smaller than the wild-type OCI-5/GPC3.
Figure 10.

Analysis of GAG-deleted mutants by immunoprecipitation of [35S]sulfate-labeled cells. Cells were transfected with: lane a, vector alone; lane b, wild-type OCI-5/GPC3; lane c, serine 494 to alanine mutant; lane d, serine 508 to alanine mutant; lane e, serines 494 and 508 to alanines mutant. Numbers on the left represent molecular mass markers.
The GAG-deficient mutant was then tested for its apoptosis-inducing ability. Both the colony-forming efficiency assay and the transient expression assay showed that the ΔGAG chains are not required for the OCI-5/GPC3 apoptosis-inducing activity (Table I and Fig. 1). In the case of MCF-7 cells, the efficiency of the apoptosis-inducing activity of ΔOCI-5/GPC3 seems to be lower than wild type. We have assessed the level of expression of OCI-5/GPC3 ΔGAG by Western blot analysis of transiently transfected cells (Fig. 2). This type of analysis, however, is not quantitative enough to allow us to establish whether the lower apoptosis-inducing activity of the mutant is due to lower levels of expression or to the fact that GAG chains of OCI-5/GPC3 play a role in the induction of cell death in MCF-7 cells.
Insulin-like Growth Factors Can Rescue MCF-7 Cells from GPC3-induced Apoptosis
SGBS shares many clinical features with the Beckwith-Wiedemann Syndrome (Verloes et al., 1995; Weksberg et al., 1996). This disorder is thought to be in most cases the result of the overexpression of insulin-like growth factor (IGF-2), a growth factor that can also act as a survival factor (Harrington et al., 1994). It has been proposed, therefore, that OCI-5/GPC3 is involved in the downregulation of IGF-2 activity (Hughes-Benzie et al., 1996; Pilia et al., 1996). Although our studies have indicated that OCI-5/GPC3 does not interact directly with IGF-2 (Song et al., 1997), it is possible that OCI-5/GPC3 regulates IGF-2 activity by interacting with other molecules involved in IGF-2 signaling. We decided, therefore, to test whether this factor can rescue MCF-7 cells from OCI-5/GPC3– induced apoptosis. Thus, physiological concentrations of IGF-2 were added to the MT3 clone in which OCI-5/ GPC3 expression had been induced for 2 d, and the degree of apoptosis was quantitated 2 d later by the Cell Death Elisa Assay. It can be seen in Fig. 11 that IGF-2 was able to completely rescue the MT3 cells from cell death. In parallel, we also tried to rescue the MT3 cells with IGF-1, FGF-1, and EGF. These factors are also known to be able to act as survival factors in certain cell types (Guillonneau et al., 1997; Hague et al., 1997; Rodeck et al., 1997). Only IGF-1 was able to inhibit OCI-5/GPC3–induced apoptosis of MT3 cells (Fig. 11).
Figure 11.

Effect of growth factors on OCI-5/GPC-3–induced apoptosis of MCF-7 cells. OCI-5/GPC3 expression was induced by adding 5 μM Cd2+ to the MT3 cells. 2 d later, 50 ng/ml of IGF-1, IGF-2, EGF, and FGF-1 were added, and cells were incubated for two more days in the presence of the inducer and the added factors. Every factor was tested in triplicate plates. The results (mean ± standard error) are expressed as fold increase in apoptosis (as measured by OD at 405 nm) compared with noninduced MT-3 cells (Cont.).
Discussion
We have shown here that OCI-5/GPC3, a heparan sulfate proteoglycan that is bound to the cell surface through a GPI tail, can induce apoptosis in the II14 rat mesothelioma cell line and in the MCF-7 breast cancer cell line. This conclusion is based on results obtained with three different experimental approaches: colony forming efficiency assays, transient expression, and the generation of cell lines with inducible and constitutive expression of OCI-5/ GPC3. With regard to the last approach, it is important to note that we have recently generated MCF-7 clones in which the expression of OCI-5/GPC3 is regulated by the tetracycline-inducible system (Gossen et al., 1993). In these clones we have also verified that OCI-5/GPC3 induces apoptosis (data not shown).
To our knowledge, this is the first report demonstrating that a cell surface heparan sulfate proteoglycan can induce programmed cell death. Some GPI-anchored proteins, on the other hand, have already been shown to be involved in apoptosis (Brodsky et al., 1997). OCI-5/GPC3 has been implicated as the gene whose loss of function mutations is responsible for the generation of SGBS (Hughes-Benzie et al., 1996; Pilia et al., 1996). Interestingly, some of the features of SGBS, such as syndactyly, and the presence of supernumerary nipples and coccygeal bony appendage, could be the consequence of defective apoptosis during development (Hughes-Benzie et al., 1996; Jacobson et al., 1997). The results presented here, therefore, provide important clues regarding the mechanism that, at least in part, is involved in the generation of SGBS. Another important finding of this work is that the apoptotic-inducing activity of OCI-5/GPC3 is cell line specific, since we failed to observe such activity in NIH 3T3 fibroblasts and HT-29 colorectal tumor cells. This is also consistent with the phenotype of SGBS patients since only certain organs at particular stages of development are affected in this syndrome.
One of the main phenotypic characteristics of SGBS patients is overgrowth. Although we have shown here that OCI-5/GPC3 induces apoptosis in II14 and MCF-7 cells, we do not exclude the possibility that in other cell types this glypican could induce growth inhibition. A typical example of this is p53, a tumor suppressor that, depending on the cell type, can induce growth arrest or apoptosis (Polyak et al., 1996).
As discussed above, the similarities between SGBS and Beckwith-Wiedemann Syndrome strongly suggest that OCI-5/GPC3 is a negative regulator of IGF-2 signaling. In addition, it has been recently reported that transgenic mice that express very high levels of IGF-2 display several features that resemble SGBS (Eggenschwiler et al., 1997). Thus, our finding that IGFs can rescue MCF-7 cells from OCI-5/GPC3–induced apoptosis is consistent with the idea that, in some way, the signaling pathway that IGF-2 uses while acting as a survival factor intersects with the pathway used by OCI-5/GPC3 for its apoptotic-inducing activity. In this respect, this study provides two important clues regarding the molecular interactions that could explain the mechanism by which OCI-5/GPC3 is able to induce apoptosis. First, we have shown that OCI-5/GPC3 has to be anchored to the cell membrane. This suggests that OCI-5/ GPC3 activity requires interaction with molecule(s) located in the cell membrane. Second, we have found that the GAG chains are not required for the apoptosis-inducing activity. Our second finding is particularly important, since we have previously reported that OCI-5/GPC3, like other heparan sulfate proteoglycans, has the ability to interact directly with basic fibroblast growth factor through its GAG chains (Song et al., 1997). The fact that the GAG chains are not required suggests that, at least for the induction of apoptosis in these cells, the interaction with basic fibroblast growth factor (or any other heparin-binding growth factor) is not important. Our finding demonstrates, on the other hand, that as suspected, the protein core of OCI-5/GPC3 has functions that do not depend on the GAG chains. Moreover, given the high abundance of the nonglycanated form of (endogenously or exogenously transfected) OCI-5/GPC3 in several cell types, one possibility to be explored is that only the nonglycanated form of OCI-5/ GPC3 is responsible for this apoptosis-inducing activity.
Acknowledgments
We thank Ms. Cassandra Cheng for her assistance in the preparation of the manuscript and Dr. Danielle Cano-Gauci for critically reviewing it.
This work has been supported by grants from the Medical Research Council of Canada and the National Cancer Institute (CA-45745).
Abbreviations used in this paper
- GAG
glycosaminoglycan
- GPI
glycosylphosphatidylinositol
- GPC
glypican
- HA
hemagglutinin A
- IGF2
insulin-like growth factor 2
- PARP
poly ADP ribose polymerase
- SGBS
Simpson-Golabi-Behmel Syndrome
Footnotes
Alfonso Dueñas Gonzalez and Mitsunori Kaya contributed equally to this work.
Address all correspondence to Dr. Jorge Filmus, Division of Cancer Biology Research, S-218 Research Building, Sunnybrook Health Science Centre, 2075 Bayview Avenue, Toronto, Ontario M4N 3M5, Canada. Tel.: (416) 480-6100 ext. 3350. Fax: (416) 480-5703. E-mail: filmus@srcl.sunnybrook.utoronto.ca.
References
- Baker SJ, Markowitz S, Fearon ER, Willson JKV, Vogelstein B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science. 1990;249:912–915. doi: 10.1126/science.2144057. [DOI] [PubMed] [Google Scholar]
- Bonneh-Barkay D, Shlissel M, Berman B, Shaoul E, Admon A, Vlodavsky I, Carey DJ, Asundi VK, Reich-Slotky R, Ron D. Identification of glypican as a dual modulator of the biological activity of fibroblast growth factors. J Biol Chem. 1997;272:12415–12421. doi: 10.1074/jbc.272.19.12415. [DOI] [PubMed] [Google Scholar]
- Brodsky RA, Vala MS, Barber JP, Medof ME, Jones RJ. Resistance to apoptosis caused by PIG-A gene mutations in paroxysmal nocturnal hemoglobinuria. Proc Natl Acad Sci USA. 1997;94:8756–8760. doi: 10.1073/pnas.94.16.8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell, R.C., and G.F. Joyce. 1995. Mutagenesis by PCR. In PCR Primer, a Laboratory Manual. C.W. Dieffenbach and G.S. Dveksler, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 581–613.
- Carey DJ, Evans DM. Membrane anchoring of heparan sulfate proteoglycans by phosphatidylinositol and kinetics of synthesis of peripheral and detergent-solubilized proteoglycans in Schwann cells. J Cell Biol. 1989;108:1891–1897. doi: 10.1083/jcb.108.5.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- David G. Integral membrane heparan sulfate proteoglycans. FASEB (Fed Am Soc Exp Biol) J. 1993;7:1023–1030. doi: 10.1096/fasebj.7.11.8370471. [DOI] [PubMed] [Google Scholar]
- David G, Lories V, Decock B, Marynen P, Cassiman J, Van Den Berghe H. Molecular cloning of a phosphatidylinositol-anchored membrane heparan sulfate proteoglycan from human lung fibroblasts. J Cell Biol. 1990;111:3165–3176. doi: 10.1083/jcb.111.6.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggenschwiler J, Ludwig T, Fisher P, Leighton PA, Tilghman SM, Efstratiadis A. Mouse mutant embryos overexpressing IGF-II exhibit phenotypic features of the Beckwith-Wiedemann and Simpson-Golabi-Behmel syndromes. Genes Dev. 1997;11:3128–3142. doi: 10.1101/gad.11.23.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filmus J, Church J, Buick RN. Isolation of a cDNA corresponding to a developmentally regulated transcript in rat intestine. Mol Cell Biol. 1988;8:4243–4249. doi: 10.1128/mcb.8.10.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filmus J, Robles AI, Shi W, Wong MJ, Colombo LL, Conti CJ. Induction of cyclin D1 overexpression by activated ras. Oncogene. 1994;9:3627–3633. [PubMed] [Google Scholar]
- Filmus J, Shi W, Wong ZM, Wong MJ. Identification of a new membrane-bound heparan sulfate proteoglycan. Biochem J. 1995;311:561–565. doi: 10.1042/bj3110561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garganta CL, Bodurtha JN. Report of another family with Simpson-Golabi-Behmel syndrome and a review of the literature. Am J Med Genet. 1992;44:129–135. doi: 10.1002/ajmg.1320440202. [DOI] [PubMed] [Google Scholar]
- Gossen M, Bonin AL, Bujard H. Control of gene activity in higher eukaryotic cells by prokaryotic regulatory elements. Trends Biol Sci. 1993;18:471–475. doi: 10.1016/0968-0004(93)90009-c. [DOI] [PubMed] [Google Scholar]
- Guillonneau X, Regnier-Ricard F, Dupuis C, Courtois Y, Mascarelli F. FGF2-stimulated release of endogenous FGF1 is associated with reduced apoptosis in retinal pigmented epithelial cells. Exp Cell Res. 1997;233:198–206. doi: 10.1006/excr.1997.3542. [DOI] [PubMed] [Google Scholar]
- Gurrieri F, Cappa M, Neri G. Further delineation of the Simpson-Golabi-Behmel (SGB) syndrome. Am J Med Genet. 1992;44:136–137. doi: 10.1002/ajmg.1320440203. [DOI] [PubMed] [Google Scholar]
- Hague A, Hicks DJ, Bracey TS, Paraskeva C. Cell-cell contact and specific cytokines inhibit apoptosis of colonic epithelial cells: growth factors protect against c-myc-independent apoptosis. Br J Cancer. 1997;75:960–968. doi: 10.1038/bjc.1997.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington EA, Bennett MR, Fanidi A, Evan GI. c-Myc- induced apoptosis in fibroblasts is inhibited by specific cytokines. EMBO (Eur Mol Biol Organ) J. 1994;13:3286–3295. doi: 10.1002/j.1460-2075.1994.tb06630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes-Benzie RM, Hunter AGW, Allanson JE, Mackenzie AE. Simson-Golabi-Behmel syndrome associated with renal dysplasia and embryonal tumor: localization of the gene to Xqcen-q21. Am J Med Genet. 1992;43:428–435. doi: 10.1002/ajmg.1320430165. [DOI] [PubMed] [Google Scholar]
- Hughes-Benzie RM, Pilia G, Xuan JY, Hunter AGW, Chen E, Golabi M, Hurst JA, Kobori J, Marymee K, Pagon RA, et al. Simpson-Golabi-Behmel Syndrome: genotype/phenotype analysis of 18 affected males from 7 unrelated families. Am J Med Genet. 1996;66:227–234. doi: 10.1002/(SICI)1096-8628(19961211)66:2<227::AID-AJMG20>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Jackson SM, Nakato H, Sugiura M, Jannuzi A, Oakes R, Kaluza V, Golden C, Selleck SB. dally, a Drosophila glypican, controls cellular responses to the TGF-β-related morphogen Dpp. Development (Camb) 1997;124:4113–4120. doi: 10.1242/dev.124.20.4113. [DOI] [PubMed] [Google Scholar]
- Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Lin J, Reichner C, Wu X, Levine AJ. Analysis of wild-type and mutant p21WAF-1 gene activities. Mol Cell Biol. 1996;16:1786–1793. doi: 10.1128/mcb.16.4.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima S, Nagata S. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- Nakato H, Futch TA, Selleck SB. The division abnormally delayed (dally) gene: a putative integral membrane proteoglycan required for cell division patterning during postembryonic development of the nervous system in Drosophila. Development (Camb) 1995;121:3687–3702. doi: 10.1242/dev.121.11.3687. [DOI] [PubMed] [Google Scholar]
- Pilia G, Hughes-Benzie RM, MacKenzie A, Baybayan P, Chen EY, Huber R, Neri G, Cao A, Forabosco A, Schlessinger D. Mutations in GPC3, a glypican gene, cause the Simpson-Golabi-Behmel overgrowth syndrome. Nat Genet. 1996;12:241–247. doi: 10.1038/ng0396-241. [DOI] [PubMed] [Google Scholar]
- Polyak K, Waldman T, He TC, Kinzler KW, Vogelstein B. Genetic determinants of p53-induced apoptosis and growth arrest. Genes Dev. 1996;10:1945–1952. doi: 10.1101/gad.10.15.1945. [DOI] [PubMed] [Google Scholar]
- Rapraeger AC, Guimond S, Krufka A, Olwin BB. Regulation by heparan sulfate in fibroblast growth factor signaling. Methods Enzymol. 1994;245:219–240. doi: 10.1016/0076-6879(94)45013-7. [DOI] [PubMed] [Google Scholar]
- Rodeck U, Jost M, Kari C, Shih DT, Lavker RM, Ewert DL, Jensen PJ. EGF-R dependent regulation of keratinocyte survival. J Cell Sci. 1997;110:113–121. doi: 10.1242/jcs.110.2.113. [DOI] [PubMed] [Google Scholar]
- Salmivirta M, Lidholt K, Lindahl U. Heparan sulfate: a piece of information. FASEB (Fed Am Soc Exp Biol) J. 1996;10:1270–1279. doi: 10.1096/fasebj.10.11.8836040. [DOI] [PubMed] [Google Scholar]
- Saunders S, Paine-Saunders S, Lander AD. Expression of the cell surface proteoglycan glypican-5 is developmentally regulated in kidney, limb, and brain. Dev Biol. 1997;190:78–93. doi: 10.1006/dbio.1997.8690. [DOI] [PubMed] [Google Scholar]
- Song HH, Shi W, Filmus J. OCI-5/rat glypican-3 binds to fibroblast growth factor-2 but not to insulin-like growth factor-2. J Biol Chem. 1997;272:7574–7577. doi: 10.1074/jbc.272.12.7574. [DOI] [PubMed] [Google Scholar]
- Steinfeld R, Van Den Berghe H, David G. Stimulation of fibroblast growth factor receptor-1 occupancy and signaling by cell surface-associated syndecans and glypican. J Cell Biol. 1996;133:405–416. doi: 10.1083/jcb.133.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipp CS, Litwac ED, Lander AD. Cerebroglycan: an integral membrane heparan sulfate proteoglycan that is unique to the developing nervous system and expressed specifically during neuronal differentiation. J Cell Biol. 1994;124:149–160. doi: 10.1083/jcb.124.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terespolsky D, Farrell SA, Siegel-Bartelt J, Weksberg R. Infantile lethal variant of Simpson-Golabi-Behmel syndrome associated with hydrops fetalis. Am J Med Genet. 1995;59:329–333. doi: 10.1002/ajmg.1320590310. [DOI] [PubMed] [Google Scholar]
- Verloes A, Massart B, Dehalleux I, Langhendries JP, Koulischer L. Clinical overlap of Beckwith-Wiedemann, Perlman and Simpson- Golabi-Behmel syndromes: a diagnostic pitfall. Clin Genet. 1995;47:257–262. doi: 10.1111/j.1399-0004.1995.tb04307.x. [DOI] [PubMed] [Google Scholar]
- Veugelers M, Vermeesch J, Reekmans G, Steinfeld R, Marynen P, David G. Characterization of glypican-5 and chromosomal localization of human GPC5, a new member of the glypican gene family. Genomics. 1997;40:24–30. doi: 10.1006/geno.1996.4518. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Yamada H, Yamaguchi Y. K-glypican: a novel GPI-linked heparan sulfate proteoglycan that is highly expressed in developing brain and kidney. J Cell Biol. 1995;130:1207–1218. doi: 10.1083/jcb.130.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weksberg R, Squire JA, Templeton DM. Glypicans: a growing trend. Nat Genet. 1996;12:225–227. doi: 10.1038/ng0396-225. [DOI] [PubMed] [Google Scholar]
- Wight TN, Kinsella MG, Qwarnstrom E. The role of proteoglycans in cell adhesion, migration and proliferation. Curr Opin Cell Biol. 1992;4:793–801. doi: 10.1016/0955-0674(92)90102-i. [DOI] [PubMed] [Google Scholar]