

Abstract
Spontaneous calcium release from intracellular stores occurs during myofibrillogenesis, the process of sarcomeric protein assembly in striated muscle. Preventing these Ca2+ transients disrupts sarcomere formation, but the signal transduction cascade has not been identified. Here we report that specific blockade of Ca2+ release from the ryanodine receptor (RyR) activated Ca2+ store blocks transients and disrupts myosin thick filament (A band) assembly. Inhibition of an embryonic Ca2+/calmodulin-dependent myosin light chain kinase (MLCK) by blocking the ATP-binding site, by allosteric phosphorylation, or by intracellular delivery of a pseudosubstrate peptide, also disrupts sarcomeric organization. The results indicate that both RyRs and MLCK, which have well-described calcium signaling roles in mature muscle contraction, have essential developmental roles during construction of the contractile apparatus.
Transient elevations of intracellular calcium (Ca2+) are an information transfer mechanism (Berridge, 1997; Spitzer and Sejnowski, 1997) that may be conserved during cellular differentiation, because they can regulate cytoskeletal organization (Kater et al., 1988; Rees et al., 1989; Gomez et al., 1995) and gene expression (Sheng et al., 1990; Dolmetsch et al., 1997; Fields et al., 1997). For example, spontaneous Ca2+ elevations observed in Xenopus spinal neurons are necessary for normal differentiation in culture, since blocking these transients prevents normal extension of neurites, maturation of potassium current kinetics, and development of GABA immunoreactivity (Gu et al., 1994; Gu and Spitzer, 1997). Moreover, imposed Ca2+ transients are necessary and sufficient to promote these aspects of neuronal differentiation in a frequency-dependent manner (Gu and Spitzer, 1995).
Many developmental studies have focused on the role of Ca2+ signaling in early events—waves after fertilization (Busa and Nuccitelli, 1985; Galione et al., 1993; Gillot and Whitaker, 1993; Jaffe, 1995) or Ca2+ transients in blastomeres during cytokinesis (Reinhard et al., 1995; Muto et al., 1996; Silver, 1996; Webb et al., 1997). If Ca2+ transients are a signaling mechanism used throughout development, then many tissues undergoing primary differentiation should exhibit them, and distinct patterns of transients could be correlated with cell type. In support of this view, spontaneous Ca2+ transients occur in embryonic Xenopus myocytes both in culture and in vivo and have been shown to regulate myofibrillogenesis (Ferrari et al., 1996; Ferrari, M.B., and N.C. Spitzer. 1997. Dev. Biol. Abstr. 186:337a). Skeletal muscle is an excellent system for studying general mechanisms of Ca2+-dependent cytoskeletal processes; contractile and associated proteins are assembled in highly ordered units called sarcomeres during the process of myofibrillogenesis so that disruptions of this lattice are readily apparent.
Cell-free systems have been used to examine myofilament and myofibril dynamics and protein turnover (Bouche et al., 1988), demonstrating that some sarcomeric proteins are in dynamic equilibrium with the cytosol. However, myofibril construction in a cell-free system has not yet been achieved. This may indicate that formation of this complex apparatus does not obey simple rules of self-assembly used for first order processes such as G- to F-actin assembly or polymerization of tubulin to form microtubules. Our previous work supports the hypothesis that second messenger systems are involved in coordinating the spatial arrangement of contractile proteins and their subsequent organization, since the formation of myofibrils is significantly disrupted when Ca2+ transients are blocked (Ferrari et al., 1996).
How myocyte Ca2+ transients are generated and what downstream mechanisms facilitate formation of sarcomeres are currently unknown. At least a dozen Ca2+-dependent proteins may be involved in myofibrillogenesis (Epstein and Fischman, 1991), thus providing a framework for Ca2+-mediated sarcomere assembly. We focused on the mechanisms of Ca2+ release and the potential role of Ca2+-dependent kinases, and report here that ryanodine receptors (RyRs)1 and myosin light chain kinase (MLCK), in addition to their well-documented roles in the Ca2+-dependent contraction of adult muscle, play novel developmental roles during myofibrillogenesis.
Materials and Methods
Myocyte Cultures
Myocytes were cultured from neural plate (stage 15) Xenopus embryos; this stage is several hours after cells have completed their final cell cycle and acquired the ability to differentiate autonomously (Kato and Gurdon, 1993). Embryos were split mid-sagittally to be plated in paired dishes for experimental versus control conditions using established techniques (Spitzer and Lamborghini, 1976; Kidokoro et al., 1980; Ferrari et al., 1996). The posterior neural plate with adjacent lateral regions was excised and placed in a divalent cation-free medium ([mM] 117 NaCl, 0.7 KCl, 4.6 Tris, 0.4 EDTA, pH 7.8) for 20–30 min to promote disaggregation. Cells were gently aspirated and plated in 35-mm tissue culture dishes (Costar Corp., Cambridge, MA) in standard (control) saline ([mM] 117 NaCl, 0.7 KCl, 1.3 MgCl2, 2 CaCl2, 4.6 Tris, pH 7.8) or zero-calcium [0-Ca2+] saline (as above, no added CaCl2 with 2 mM EGTA). These mixed cultures contain myocytes, neurons, and morphologically undifferentiated cells.
Unless otherwise noted, all pharmacologicals were applied from 6 to 24 h in culture, 3–6 h before A band assembly begins (see Fig. 2 A), or 24–48 h in culture, when myocytes have functionally differentiated. Treatments for shorter periods were followed by 10 washes of 4 ml each (culture bath volume was 2 ml). Kinase inhibitors were used at concentrations only 2–10-fold above their reported inhibition constant (Ki) values to enhance the likelihood of specificity for individual kinases. Only bipolar myocytes, with length ≥4× width, were examined; multipolar cells and myoballs were not considered. Myocytes were screened for apoptosis using a DNA fragmentation detection system (ApopTag Plus; Oncor, Gaithersburg, MD).
Figure 2.
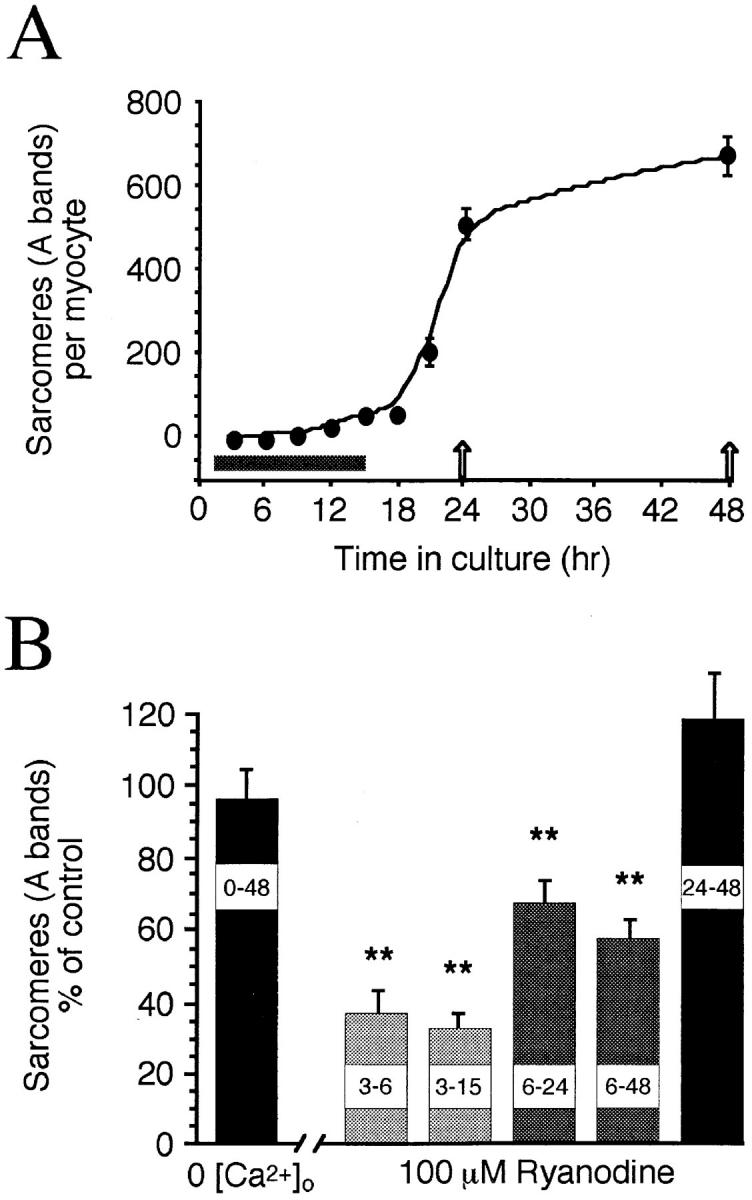
Time course and ryanodine sensitivity of A band assembly in Xenopus myocytes. (A) Mean number of sarcomeres per myocyte (n ≥ 30 myocytes per time point) versus time in culture. The period of spontaneous Ca2+ transient production is shown along the x axis (gray bar). After experimental perturbations, sarcomeres were assayed at 24 or 48 h in culture (arrows). (B) Normalized mean sarcomere numbers per myocyte from chronic 0-Ca2+ and ryanodine-treated cultures. 0-Ca2+ cultures were assayed at 48 h; ryanodine was applied at 100 μM in control saline, with 3–6, 3–15, and 6–24 h treatments assayed at 24 h. Note that the early period of ryanodine sensitivity corresponds to the period of Ca2+ transient production shown above. Asterisks, significantly different from controls in this and subsequent figures.
Calcium Imaging
Measures of resting Ca2+ and spontaneous transients were achieved with fura-2 and fluo-3, respectively, using established techniques (Tsien, 1980; Grynkiewicz et al., 1985; Gu et al., 1994; Ferrari et al., 1996). Cells were loaded with calcium indicator by bath application (2 μM, solubilized in <0.1% DMSO). Indicator loading time was 30–60 min, cultures were then washed in 5–10 volumes of standard saline, and images were collected at 10-s intervals. Zeiss Neofluor water immersion objectives (Carl Zeiss Inc., Thornwood, NY) were used for both calcium imaging and indirect immunofluorescence. Ratiometric measures of resting Ca2+ were made using fura-2 visualized with a Zeiss Photomicroscope and Dage 72SX ICCD camera. Cells were imaged between 3 and 9 h after plating in control, 100 μM ryanodine, and 0-Ca2+ cultures. Myocytes were imaged at 10-s intervals for 10 min, and these numbers were averaged for baseline values; details of data acquisition and analysis were previously described (Ferrari et al., 1996). Non-ratiometric measures of fluo-3 signals and immunofluorescence were made with an MRC 600 confocal laser system (Bio-Rad Laboratories, Hercules, CA). Images were digitized and saved using the COMOS program (Bio-Rad Laboratories), and analyzed using macros for the National Institutes of Health (NIH) Image program (version 1.47; W. Rasband, NIH, Bethesda, MD) as previously described (Ferrari et al., 1996). Myocytes were imaged for spontaneous transients 1–2 h after incubation in the same concentration of kinase inhibitor that disrupted myofibrillogenesis.
Immunochemistry
Sarcomeric myosin was visualized with the mAb MF20 (Developmental Studies Hybridoma Bank, the University of Iowa, Iowa City, IA) and an FITC-conjugated secondary antibody. For immunoblots, antibodies to smooth MLCK isoforms (R57, K36; gifts of P. Gallagher, Indiana University School of Medicine, Indianapolis, IN) were used because they recognize a combination of smooth 130-kD, embryonic 208-kD, and 220-kD MLCK isoforms in avian and mammalian tissues and cell lines (Gallagher et al., 1995). MLCK isoforms were examined from homogenates of Xenopus embryonic and adult tissues; each lane was loaded with 100 μg of protein. Dilutions of the primary and secondary antibodies were determined empirically, and labeled bands were detected using the enhanced chemiluminescence detection system (Amersham Corp., Arlington Heights, IL).
Sarcomere Assays
Sarcomere (A band) numbers were counted at 400× in ≥45 myocytes in blind assays from ≥3 culture pairs for each condition at 24 or 48 h. Digital images of sarcomeric myosin immunofluorescence were captured on the Bio-Rad MRC 600 using a fluorescein filter set. Thin optical sections (minimum aperture; 12–16 sections per cell) were taken with a focus motor in 1.2-μm steps. Numbers of bipolar cells were counted in six to nine dishes. In all cases n is the same for control and experimental data. Raw data from paired controls were used for statistical tests.
Electrophysiology
Myocyte inward rectifier potassium current was measured in ≥12 cells grown for 24 h in culture under various conditions, using standard techniques (Spruce and Moody, 1992; Ferrari et al., 1996). Pipettes contained (mM): 100 KCl, 10 NaCl, 5 EGTA, 10 Hepes, 2 MgATP, 20 KOH, pH 7.4, and had resistances of 2–4 MΩ. External recording saline contained (mM): 117 NaCl, 3 KCl, 2 CaCl2, 5 Hepes, 2 NaOH, pH 7.4. Potassium currents were isolated using 0.2 μg/ml tetrodotoxin and 0.4 mM CdCl2 in the external solution to block Na+ and Ca2+ currents, respectively. Current was recorded with a series of 30-ms voltage steps (−130 to +40 mV) from a holding potential of −50 mV. Leak-subtracted current traces were filtered at 3–5 KHz and digitized at 20 KHz. Steady-state mean current amplitudes were measured over the last 5 ms of the voltage step. Currents are expressed as pA/pF to normalize for cell size.
Synthetic Peptides
The MLCK pseudosubstrate inhibitory amino acid sequence (MLCKi), AKKLSKDRMKKYMARRKWQKTG, is highly conserved, being identical at the amino acid level among the known vertebrate smooth MLCK isoforms (BlastP Search of GenBank). This inhibitory sequence has Ki values of 0.3–21 nM in vitro (Ikebe et al., 1992). A scrambled version of this peptide, KKDTQWMYLKMRKGRAKSAKRK, showed no significant homology to any GenBank sequences. MLCKi and the scrambled version were each fused to an internalizable peptide of the homeodomain antennapedia protein (pANT 43–58: RQIKIWFQNRRMKWKK) by a disulfide linkage (Theodore et al., 1995; Prochiantz, 1996). Peptides were synthesized, conjugated, and then analyzed at the Stanford Beckman Center's Protein & Nucleic Acid facility. Antennapedia peptide or the conjugates were applied at 100 nM for 1 h at 6 or 24 h in culture.
Statistics
Measurements are reported as mean ± SEM. Unpaired two-tailed t tests were used and values are considered significantly different for P < 0.05.
Results
Ryanodine Receptor Stores Are Necessary for Transients and Myofibrillogenesis
Transients are produced from intracellular Ca2+ stores and are required for normal sarcomere assembly (Ferrari et al., 1996), suggesting Ca2+ influx may not be necessary for myofibrillogenesis. In support of this hypothesis, myocytes grown in 0-Ca2+ for 24 or 48 h in culture have normal bipolar morphology as well as myofibrillar and sarcomeric structure (Fig. 1 A). This arrangement is clearest in the myocyte endfeet, which are free of yolk inclusions. The numbers of bipolar myocytes (0-Ca2+ 76 ± 11% of controls, P = 0.24) and the numbers of sarcomeres per myocyte in 0-Ca2+ cultures (0-Ca2+ 96 ± 8% of controls, P = 0.68; Figs. 1 A, and 2 B) do not differ from controls grown in 2 mM Ca2+. Since Ca2+ transients are blocked by ryanodine (Ferrari et al., 1996; Fig. 1 B), we tested whether blocking the RyR Ca2+ store results in myofibrillar defects. Ryanodine (100 μM) in standard 2 mM Ca2+ saline, applied from 6 to 24 or 6 to 48 h in culture, reduces the number of sarcomeres per cell (6–24 h ryanodine 66 ± 6% of controls, P = 0.006; Figs. 1 B, and 2 B) and disrupts myofibril alignment; most myofibrils do not span the length of the cell and are not in close parallel register.
Figure 1.
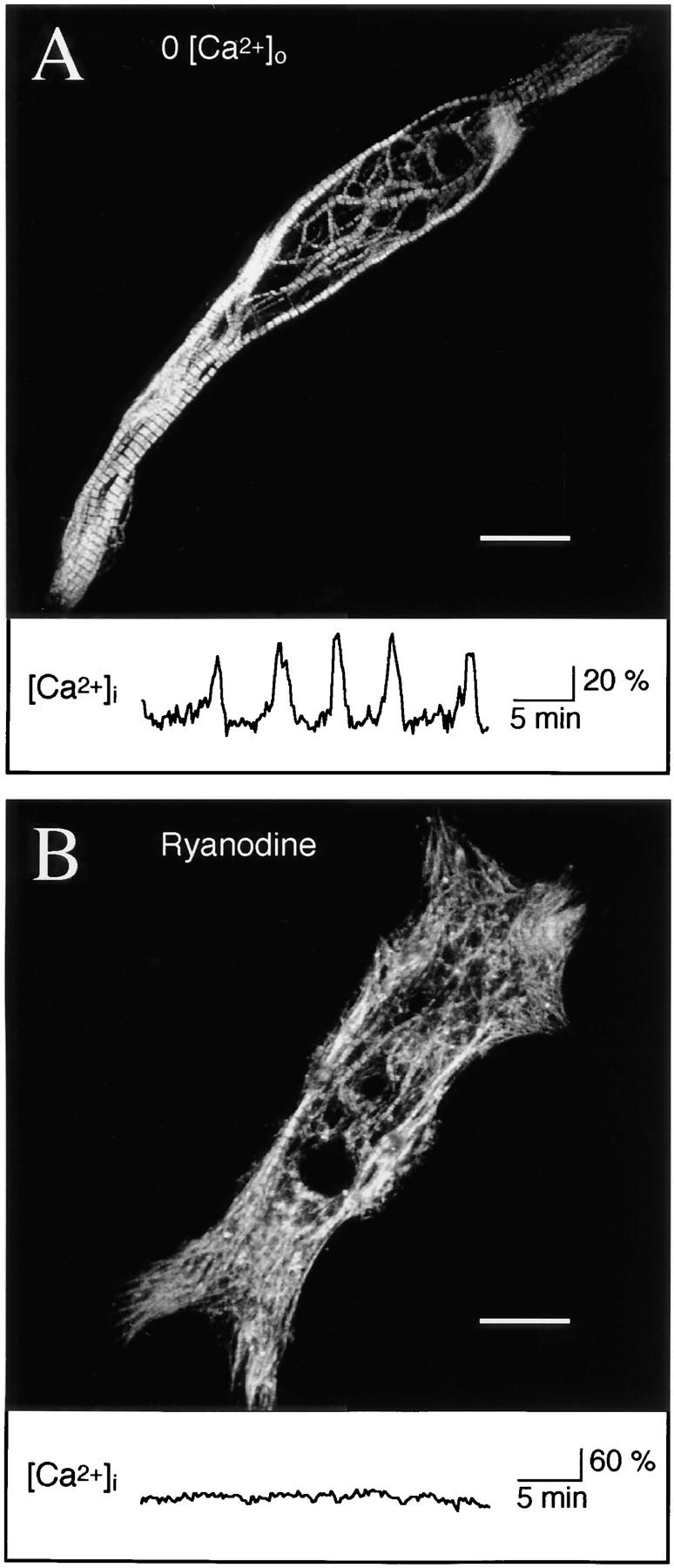
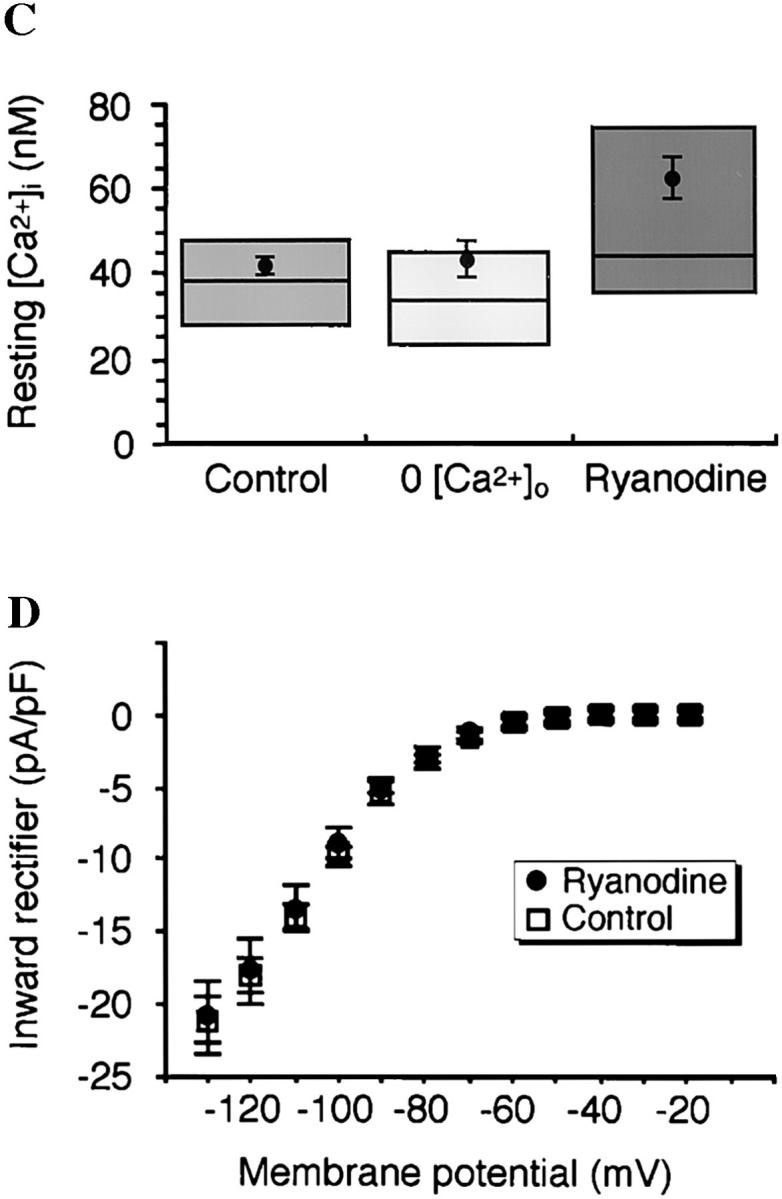
Release of intracellular Ca2+ from RyR stores is necessary for myofibrillogenesis. (A) Myocytes grown 48 h in 0-Ca2+ medium are indistinguishable from controls with respect to bipolar morphology, parallel myofibril alignment, and number and regularity of sarcomeres. Image is a Z-series maximum projection of sections encompassing the full thickness of the cell. In the central region, a loose meshwork of myofibrils surrounds normal cell inclusions including numerous yolk platelets. Trace below shows Ca2+ transients that occur in normal and 0-Ca2+ media up to 15 h in culture (Ferrari et al., 1996). (B) Myocytes grown in the presence of ryanodine (100 μM) at early times show disruption of myofibrils and reduced sarcomere numbers; cell shown was treated with ryanodine from 6 to 48 h in culture. Major phenotypic characteristics are myofibril misalignments, diffuse myosin immunoreactivity throughout the cell, fewer sarcomeres, and localized patches of dense myosin accumulation. Trace below shows Ca2+ transients are blocked by ryanodine (n = 109 cells examined for 30–60 min at 3–9 h in culture; Ferrari et al., 1996). (C) Resting Ca2+ levels are not significantly altered in 0-Ca2+ medium or with the application of 100 μM ryanodine when measured during 3–9 h in culture. Means are from ≥20 cells per condition, boxes enclose 50% of the data, and lines indicate median values. (D) Mean steady-state current–voltage relations of the inward rectifier potassium current at 24–28 h in culture are normal in control and 100 μM ryanodine-treated (6–24 h in culture) cells. Number of myocytes is ≥12 for each condition. Bars, 20 μm.
Intracellular Ca2+ was monitored in response to ryanodine treatment. Ryanodine blocks transients without affecting baseline Ca2+; fura-2 measures in 100 μM ryanodine-treated myocytes are not significantly different from controls ([nM] ryanodine 62 ± 9, control 42 ± 4, P = 0.06; Fig. 1 C), in agreement with previous control values (24–64 nM; Ferrari et al., 1996). In addition to these steady-state values, measurements of Ca2+ during acute ryanodine application show that resting levels are not altered, but a single spontaneous transient can still occur before complete block (data not shown). A use-dependent block is expected for this agent, as high concentrations of ryanodine antagonize by binding a low affinity site only when the receptor is in the open configuration (Meissner, 1986).
Ryanodine is a highly specific toxin (Coronado et al., 1994), and does not affect the total number of bipolar myocytes per dish (6–24 h ryanodine 122 ± 16% of controls, P = 0.35; 6–48 h ryanodine 103 ± 24% of controls, P = 0.94). Moreover, ryanodine has no effect on the normal maturation of inward rectifier potassium current during the period of transient production (Fig. 1 D). We chose the inward rectifier as a positive control because it is the earliest developing voltage-gated current in Xenopus myocytes, with an increase in current density occurring in parallel with the period of myofibrillogenesis (Spruce and Moody, 1992).
The period of transient production precedes A band formation (Fig. 2 A). Brief ryanodine treatment during this time, from 3 to 6 h, or 3 to 15 h in culture, also disrupts thick filament assembly (Fig. 2 B). These results indicate that later synthesis and insertion of new RyRs does not compensate for blocking calcium release during this sensitive period. Since myofibrillogenesis is perturbed by ryanodine during transient production and initial assembly, but not at later times (Fig. 2 B), ryanodine does not exert its effects by promoting disassembly.
Identification of a Downstream Kinase Necessary for Myofibrillogenesis
Do Ca2+ transients regulate myofibrillogenesis via activation of kinases? Staurosporine (100 nM), a general serine/ threonine kinase inhibitor, reduces numbers of A bands (56 ± 6% of controls, P = 0.001; Fig. 3, A and C) and results in a diffuse distribution of sarcomeric myosin in the cell soma. Thin processes and lamellipodial-like structures emanate from the myocyte endfeet with dense punctate myosin at the edge of these membranes. This result encouraged use of more specific blockers that inhibit Ca2+-dependent kinases. Convergent results with multiple inhibitors of MLCK—ML-7, ML-9, and KT5926—implicate this Ca2+/calmodulin-dependent enzyme as an effector in myofibrillogenesis (Fig. 3 C). These inhibitors also produce a diffuse distribution of myosin with some dense accumulations, and sarcomere numbers are significantly reduced (ML-7 [1 μM] 47 ± 15% of controls, P = 0.0001; Figs. 3, B and C). Inhibitors of protein kinase A, protein kinase C (PKC), and Ca2+/calmodulin kinase II (CaMK II) are without effect (Fig. 3 C). ML-7, the most specific of the MLCK inhibitors tested, disrupts myofibrillogenesis without affecting development of the inward rectifier potassium current (Fig. 3 D). ML-7, ML-9, KT5926, and staurosporine also do not affect normal generation of Ca2+ transients when examined between 3 and 9 h in culture (data not shown), nor do these inhibitors affect the number of bipolar myocytes differentiating per culture (e.g., ML-7 [1 μM] 78 ± 16% of controls, P = 0.39).
Figure 3.


Pharmacological inhibition of MLCK disrupts A band formation. All data are from 24 h cultures. (A) Staurosporine (100 nM), a general kinase inhibitor, inhibits thick filament assembly without disrupting bipolar morphology. Myosin is diffusely distributed throughout the cells, with regions of dense accumulation. (B) ML-7 (1 μM), a specific MLCK inhibitor, disrupts myosin incorporation into A bands without affecting bipolar morphology. Myosin is distributed diffusely throughout the cells with localized dense patches. (C) Summary of kinase inhibitor effects on A band assembly. Inhibitors were applied at the concentration indicated (μM) to block the listed kinase(s). Note that KT5926, which inhibits CaMK II with high specificity (10 nM), has no effect, whereas a higher concentration (100 nM) of this agent inhibits MLCK as well, producing the same effects as ML-7 and ML-9. KT, KT 5926; Stauro, staurosporine; Bis I, bisindolylmaleimide 1. (D) Development of the inward rectifier potassium current is unaffected by 6–24 h, 1 μM ML-7 when assayed at 24 h in culture. Bars, 20 μm.
Myocytes treated with ryanodine or kinase inhibitors appear normal under phase optics, having phase dark endfeet, birefringent yolk inclusions, clear nuclei, and no membrane blebbing. Since cytoskeletal disruptions could result from pharmacologically triggered apoptosis, myocytes treated with ryanodine or ML-7 from 6 to 48 h in culture were screened for DNA fragmentation, a late marker of apoptosis. Although some pyknotic cells and cellular fragments in these mixed cultures had labeled nuclei, neither the number of bipolar myocytes, nor the number of labeled nuclei in myocytes, differed from controls (<3% labeled nuclei in n > 100 myocytes for each condition).
MLCK Activity Is Suppressed by Activation of PKC
Activation of PKC inhibits MLCK activity in vitro by phosphorylating its only known substrate, the regulatory light chain (RLC), preventing MLCK access to the RLC (Nishikawa et al., 1984; Turbedsky et al., 1997). To test the hypothesis that activation of PKC generates the phenotype produced by MLCK inhibitors, we stimulated PKC with phorbol 12-myristate, 13-acetate (PMA). Application of 10 nM PMA from 6 to 24 h in culture disrupts myofibrillogenesis and reduces sarcomere numbers (PMA 30 ± 7% of controls, P = 0.0001; Fig. 4). This disruption is not seen with PMA application from 24 to 48 h in culture (Fig. 4 C). Moreover, PMA-activated inhibition of sarcomere assembly is blocked by co-application of PMA with the PKC inhibitor bisindolylmaleimide I (Bis I; 100 nM) (PMA/Bis I 85 ± 10% of controls, P = 0.35; Figs. 4, B and C), rescuing normal differentiation.
Figure 4.


Activation of PKC disrupts A band assembly. (A) Phorbol 12-myristate, 13-acetate (PMA; 10 nM), a potent activator of PKC, disrupts myofibrillogenesis in the same manner as MLCK inhibitors. Myosin is diffusely distributed throughout these cells, with dense punctate staining located centrally. (B) Co-application of the PKC inhibitor Bis I (100 nM) blocks the action of PMA. Myofibril and sarcomeric structure are normal. (C) PMA application from 6 to 24 h, but not 24 to 48 h, disrupts A band assembly and is rescued by Bis I. Bars, 20 μm.
Molecular Inhibition of MLCK Activity In Situ by a Pseudosubstrate Peptide
Vertebrate MLCKs contain a conserved autoinhibitory domain (Gallagher et al., 1997), and pseudosubstrate peptides from this region inhibit MLCK activity in biochemical assays (Ikebe et al., 1992; Knighton et al., 1992). We treated myocytes with a 22–amino acid synthetic inhibitory peptide (MLCKi) from this region conjugated to a 16–amino acid homeopeptide of the antennapedia gene (pANT) to transport it into cells (Theodore et al., 1995; Prochiantz, 1996). As with pharmacological inhibitors, myofibril arrays are disrupted and sarcomere numbers are reduced with pANT-MLCKi ([100 nM] 34 ± 6% of controls, P = 0.0001; Figs. 5, A and C). Some normal A bands are visible within disrupted myofibrils and regions of dense myosin accumulation are evident. Treatments with pANT alone, pANT coupled to a scrambled MLCKi sequence, or later application of pANT-MLCKi have no significant effect on myofibril alignment or sarcomere numbers, showing specificity both with respect to the inhibitory sequence of amino acids and the time of application (Fig. 5 C).
Figure 5.


Molecular inhibition of MLCK blocks A band assembly. (A) Intracellular delivery via antennapedia peptide (pANT) of a pseudosubstrate inhibitory peptide (MLCKi) results in disruption of myofibrillogenesis. (B) Later application of MLCKi at 24 h in culture has no effect on sarcomere assembly. (C) Effects of treatment with synthetic peptides. In addition to late application of MLCKi, pANT alone or pANT-scrambled MLCKi have no significant effect on myofibrillogenesis. Bars, 20 μm.
Detection and Developmental Regulation of MLCK Isoforms
Which MLCK is present in myocytes during the period of A band assembly? An embryonic MLCK is expressed in developing skeletal muscle and other tissues, and R57 polyclonal antiserum recognizes both smooth and embryonic isoforms (Gallagher et al., 1995). This antiserum identifies a single band at 225 kD on immunoblots of Xenopus myotomal tissue during the period of sarcomere assembly (Fig. 6 A). This Xenopus MLCK isoform shows developmental upregulation during the period of myofibrillogenesis corresponding to production of transients, but is absent in adult skeletal tissue. These blots also show the presence of a 130-kD Xenopus smooth MLCK in adult skeletal, but not embryonic, tissue. The mAb K36 also recognizes these 130- and 225-kD isoforms, giving the same pattern on immunoblots seen with R57 (data not shown). Since only one embryonic MLCK isoform is recognized by these antibodies, we determined its cellular distribution in cultured myocytes using the K36 mAb. While immunofluorescence was weak compared with myosin immunoreactivity with MF20, either because of lower levels of the epitope and/or lower affinity of the primary antibody, the 225-kD embryonic MLCK is localized in a striated pattern (Fig. 6 B).
Figure 6.



Detection and developmental regulation of an embryonic MLCK isoform. (A) An embryonic isoform of MLCK is developmentally upregulated during the period of Ca2+ transient production. Western blot of Xenopus embryonic and adult skeletal muscle tissue with R57 antiserum shows that a single isoform running ∼225 kD is upregulated in embryonic myotomal tissue from stage 15 (lane 2) to stage 27 (lane 3), corresponding to 0 and 15 h in culture, respectively. This isoform is absent in adult skeletal muscle (lane 4), where a strong single band at 130 kD is detected. MLCK isoforms at 130, 208, and 220 kD are recognized in the rat A10 cell line (ATCC CRL1476) lysate (lane 1). (B) Striated pattern of labeling with the K36 smooth muscle MLCK mAb is visible in myocyte endfeet at 24 h in culture. The width and spacing of these bands matches the normal A band geometry. (C) Schematic model of Ca2+ transient–dependent MLCK activation and A band assembly in embryonic skeletal muscle. Inhibiting this cascade (dashed lines) at multiple points (boxes) disrupts formation of thick filaments. Bar, 20 μm.
Our findings are summarized by a signal transduction cascade schematic that includes the components we have identified (Fig. 6 C). Intervention at the different points indicated suppresses the assembly of sarcomere A bands. These results are expected to be useful in decoding the algorithm by which Ca2+ transients enable cytoskeletal organization.
Discussion
Our previous work led to the hypothesis that Ca2+ transients direct some aspects of myofibrillogenesis in embryonic Xenopus myocytes (Ferrari et al., 1996). Since Ca2+ transient production does not depend on extracellular Ca2+, a corollary is that removal of extracellular Ca2+ should not disrupt construction of the contractile apparatus. We show here that myocytes grown for ≤48 h in the absence of Ca2+ have no defects in myofibril orientation and sarcomere assembly. This suggests that voltage-dependent Ca2+ channels and other Ca2+ influx pathways do not play a role in the early stages of myofibrillogenesis, although they may participate later in the accelerated formation of striations observed in twitching cells (Kidokoro and Saito, 1988).
Ryanodine disrupts myofibril alignment and reduces sarcomere numbers in a manner both qualitatively and quantitatively similar to that achieved by BAPTA-AM blockade of transients (Ferrari et al., 1996). Since ryanodine blocks transients without affecting the total number of bipolar myocytes per culture, resting Ca2+ levels, or the development of the potassium inward rectifier current, it is a highly specific tool for examining the role of transients in muscle development. Structural defects similar to those reported here have been observed in two vertebrate RyR mutants and in wild-type chicken muscle cultured in the presence of chronic ryanodine (Airey et al., 1993a ,b; Takekura et al., 1995), suggesting that RyRs may have developmental roles (Airey et al., 1991). In this regard, it is notable that the sarcoplasmic reticulum develops in close physical association with the formation of myofibrils (Huang and Hockaday, 1988; Flucher et al., 1993; Takekura et al., 1994). While not excluding the possibility that RyRs play a structural role during development, our results indicate that RyRs play a physiological role, suggesting that release of Ca2+ from this store is not only important for E-C coupling in mature muscle, but also for proper construction of the contractile apparatus.
We initially used a pharmacological approach to determine if Ca2+-dependent kinases play a role in myofibrillogenesis. The results of this approach indicated that MLCK, a Ca2+/calmodulin-dependent enzyme, is involved in myofibrillogenesis. Inhibiting this kinase resulted in a more severe phenotype than that produced by ryanodine. Since ryanodine does not alter the resting Ca2+ concentration, one possibility is that basal MLCK activity persists in ryanodine-treated cells. This is not unlikely, given the activation constant for MLCK by Ca2+/calmodulin is 1 nM (Gallagher et al., 1997). This residual activity would be blocked by MLCK inhibitors.
Further support for MLCK involvement was achieved by activation of PKC, which indirectly inhibits MLCK. The RLC is the only known substrate for MLCK, and when phosphorylated by PKC, access to serine 19 of the RLC by MLCK is blocked (Nishikawa et al., 1984; Turbedsky et al., 1997). PKC activators were thus predicted to disrupt myofibrillogenesis, as observed with PMA. This result also indicates that the absence of an effect with the PKC inhibitor Bis I was not due to the lack of PKC in these cells. In fact, PKC-mediated inhibition of sarcomere assembly was blocked by co-application of PMA with Bis I, indicating that Bis I is an effective inhibitor of PKC in these cells. These observations support the idea that PKC activity acts as a negative regulator of MLCK activity, and implies that basal PKC activity remains low during striated muscle development to allow for MLCK-mediated sarcomere (A band) assembly.
Despite the strength of the pharmacological results, we sought more specific means of blocking MLCK activity to verify its role in myofibrillogenesis. We used a synthetic peptide from the conserved autoinhibitory domain of smooth MLCK (Ikebe et al., 1992; Knighton et al., 1992), using an intracellular delivery method shown to be effective in blocking PKC activity in neurons (Theodore et al., 1995; Prochiantz, 1996). Sarcomere assembly was affected only by the inhibitory MLCKi peptide construct, whereas a scrambled version had no effect. This result indicates that inhibition was due to MLCKi instead of a nonspecific consequence of the antennapedia transport peptide or cleavage by-products. Furthermore, later application of MLCKi had no effect, consistent with the sensitive window already established with ryanodine and pharmacological inhibitors of MLCK.
The three major vertebrate MLCK isoforms that have been described (smooth, skeletal, and embryonic) are all Ca2+ dependent (for reviews see Trybus, 1994; Gallagher et al., 1997). Adult skeletal muscle expresses smooth muscle as well as skeletal muscle MLCK isoforms, whereas developing skeletal muscle expresses a newly discovered embryonic MLCK (Gallagher et al., 1995). Our results suggest amphibian skeletal muscle also expresses the embryonic MLCK isoform, since antibodies to smooth and embryonic forms recognize only a single band of appropriate size in embryonic tissue. In addition, the inverse developmental regulation of the 130-kD Xenopus smooth muscle MLCK and embryonic 225-kD isoforms is similar to that observed in chicken and mammalian tissues (Gallagher et al., 1995).
Structural motifs in smooth MLCK are similar to those found in proteins of the N-CAM superfamily associated with thick filaments, including the giant sarcomeric protein titin (Herring et al., 1990; Epstein and Fischman, 1991; Labeit and Kolmerer, 1995), and smooth MLCK localizes to myosin-containing structures (Guerriero et al., 1981; de Lanerolle et al., 1981). If the embryonic MLCK contains the same motifs, it is likely targeted to developing A bands. The striated labeling produced by an MLCK antibody which recognizes a single embryonic isoform on immunoblots is consistent with this idea.
The myosin II isoform(s) that must be activated by MLCK for A band assembly in skeletal muscle remain to be determined. In cardiac myocytes, myosin IIB appears in premyofibrils and is gradually replaced by sarcomeric myosin (Rhee et al., 1994). Since MLCK activates nonmuscle myosin II and is responsible for assembly of “thick filaments” in activated smooth muscle (Scholey et al., 1980), this process may be a conserved early step in striated muscle A band assembly.
Spontaneous Ca2+ transients occur with a characteristic mean frequency (Ferrari et al., 1996), raising the possibility that macromolecular assembly is encoded by this parameter. Because phosphorylation of the RLC by MLCK occurs in seconds and dephosphorylation in minutes (Somlyo and Somlyo, 1994; Trybus, 1994; Sobieszek et al., 1997), the RLC phosphorylation state has been suggested to serve as a molecular form of short term “muscle memory” for twitch potentiation in adult skeletal muscle (Levine et al., 1996). Since a myofibrillar phosphatase isoform complexes directly with calmodulin and MLCK (Somlyo and Somlyo, 1994; Sobieszek et al., 1997), periodic activation of MLCK by Ca2+ transients may be required to maintain myosin in the conformation necessary for assembly, implying a threshold level of activity below which A bands would fail to form.
Acknowledgments
We thank D. Dagan, T. Gomez, X. Gu, and M.-M. Poo for comments on the manuscript; A. Smith and A. Sanchez at Stanford's Beckman Center for peptides; P. Gallagher for MLCK antisera and A10 cell line lysate; and P. Kassner and W. Conroy for advice.
This work was supported by NIH grants to N.C. Spitzer and a Muscular Dystrophy Association postdoctoral research fellowship to M.B. Ferrari.
Abbreviations used in this paper
- Bis I
bisindolylmaleimide I
- MLCK
myosin light chain kinase
- MLCKi
myosin light chain kinase inhibitory peptide
- pANT
antennapedia peptide
- PKC
protein kinase C
- PMA
phorbol 12-myristate, 13-acetate
- RLC
regulatory light chain
- RyR
ryanodine receptor Ca2+ release channel
Footnotes
Address all correspondence to Michael B. Ferrari, Ph.D., Department of Biology, University of California San Diego, La Jolla, CA 92093-0357. Tel.: (619) 534-2456. Fax: (619) 534-7309. E-mail: ferrari@biomail.ucsd.edu
K. Ribbeck's present address is Institut fuer Neurobiologie, Universitaet Heidelberg, Im Neuenheimer Feld 345, 69120, Heidelberg, Germany.
References
- Airey JA, Baring MD, Sutko JL. Ryanodine receptor protein is expressed during differentiation in the muscle cell lines BC3H1 and C2C12. Dev Biol. 1991;148:365–374. doi: 10.1016/0012-1606(91)90344-3. [DOI] [PubMed] [Google Scholar]
- Airey JA, Baring MD, Beck CF, Chelliah Y, Deerinck TJ, Ellisman MH, Houenou LJ, McKemy DD, Sutko JL, Talvenheimo J. Failure to make normal α ryanodine receptor is an early event associated with the crooked neck dwarf (cn) mutation in chicken. Dev Dynamics. 1993a;197:169–188. doi: 10.1002/aja.1001970303. [DOI] [PubMed] [Google Scholar]
- Airey JA, Deerinck TJ, Ellisman MH, Houenou LJ, Ivanenko A, Kenyon JL, McKemy DD, Sutko JL. Crooked neck dwarf (cn) mutant chicken skeletal muscle cells in low density primary cultures fail to express normal alpha ryanodine receptor and exhibit a partial mutant phenotype. Dev Dyn. 1993b;197:189–202. doi: 10.1002/aja.1001970304. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. The AM and FM of calcium signalling. Nature. 1997;386:759–760. doi: 10.1038/386759a0. [DOI] [PubMed] [Google Scholar]
- Bouche M, Goldfine SM, Fischman DA. Posttranslational incorporation of contractile proteins into myofibrils in a cell-free system. J Cell Biol. 1988;107:587–596. doi: 10.1083/jcb.107.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busa WB, Nuccitelli R. An elevated free cytosolic Ca2+ wave follows fertilization in eggs of the frog, Xenopus laevis. . J Cell Biol. 1985;100:1325–1329. doi: 10.1083/jcb.100.4.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronado R, Morrissette J, Sukhareva M, Vaughan DM. Structure and function of ryanodine receptors. Am J Physiol. 1994;266:1485–1504. doi: 10.1152/ajpcell.1994.266.6.C1485. [DOI] [PubMed] [Google Scholar]
- de Lanerolle P, Adelstein RS, Feramisco JR, Burridge K. Characterization of antibodies to smooth muscle myosin kinase and their use in localizing myosin kinase in nonmuscle cells. Proc Natl Acad Sci USA. 1981;78:4738–4742. doi: 10.1073/pnas.78.8.4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- Epstein HF, Fischman DA. Molecular analysis of protein assembly in muscle development. Science. 1991;251:1039–1044. doi: 10.1126/science.1998120. [DOI] [PubMed] [Google Scholar]
- Ferrari MB, Rohrbough JW, Spitzer NC. Spontaneous calcium transients regulate myofibrillogenesis in embryonic Xenopusmyocytes. Dev Biol. 1996;178:484–497. doi: 10.1006/dbio.1996.0233. [DOI] [PubMed] [Google Scholar]
- Fields RD, Eshete F, Stevens B, Itoh K. Action potential-dependent regulation of gene expression: temporal specificity in Ca2+, cAMP-responsive element binding proteins, and mitogen-activated protein kinase signaling. J Neurosci. 1997;17:7252–7266. doi: 10.1523/JNEUROSCI.17-19-07252.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flucher BE, Takekura H, Franzini-Armstrong C. Development of the excitation-contraction coupling apparatus in skeletal muscle: association of sarcoplasmic reticulum and transverse tubules with myofibrils. Dev Biol. 1993;160:135–147. doi: 10.1006/dbio.1993.1292. [DOI] [PubMed] [Google Scholar]
- Galione A, McDougall A, Busa WB, Willmott N, Gillot I, Whitaker M. Redundant mechanisms of calcium-induced calcium release underlying calcium waves during fertilization of sea urchin eggs. Science. 1993;261:348–352. doi: 10.1126/science.8392748. [DOI] [PubMed] [Google Scholar]
- Gallagher PJ, Garcia JG, Herring BP. Expression of a novel myosin light chain kinase in embryonic tissues and cultured cells. J Biol Chem. 1995;270:29090–29095. doi: 10.1074/jbc.270.49.29090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher PJ, Herring BP, Stull JT. Myosin light chain kinases. J Muscle Res Cell Motil. 1997;18:1–16. doi: 10.1023/a:1018616814417. [DOI] [PubMed] [Google Scholar]
- Gillot I, Whitaker M. Imaging calcium waves in eggs and embryos. J Exp Biol. 1993;184:213–219. [Google Scholar]
- Gomez TM, Snow DM, Letourneau PC. Characterization of spontaneous Ca2+transients in nerve growth cones and their effect on growth cone migration. Neuron. 1995;14:1233–1246. doi: 10.1016/0896-6273(95)90270-8. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca++indicators with greatly improved fluorescence properties. J BiolChem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gu X, Spitzer NC. Distinct aspects of neuronal differentiation encoded by frequency of spontaneous Ca2+transients. Nature. 1995;375:784–787. doi: 10.1038/375784a0. [DOI] [PubMed] [Google Scholar]
- Gu X, Spitzer NC. Breaking the code: regulation of neuronal differentiation by spontaneous calcium transients. Dev Neurosci. 1997;19:33–41. doi: 10.1159/000111183. [DOI] [PubMed] [Google Scholar]
- Gu X, Olson EC, Spitzer NC. Spontaneous neuronal Ca++spikes and waves during early differentiation. J Neurosci. 1994;14:6325–6335. doi: 10.1523/JNEUROSCI.14-11-06325.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero V, Jr, Rowley DR, Means AR. Production and characterization of an antibody to myosin light chain kinase and intracellular localization of the enzyme. Cell. 1981;27:449–458. doi: 10.1016/0092-8674(81)90386-x. [DOI] [PubMed] [Google Scholar]
- Herring BP, Stull JT, Gallagher PJ. Domain characterization of rabbit skeletal muscle myosin light chain kinase. J Biol Chem. 1990;265:1724–1730. [PMC free article] [PubMed] [Google Scholar]
- Huang CL-H, Hockaday AR. Development of myotomal cells in Xenopus laevislarvae. J Anat. 1988;159:129–136. [PMC free article] [PubMed] [Google Scholar]
- Ikebe M, Reardon S, Fay FS. Primary structure required for the inhibition of smooth muscle myosin light chain kinase. FEBS (Fed Eur Biochem Soc) Lett. 1992;312:245–248. doi: 10.1016/0014-5793(92)80944-c. [DOI] [PubMed] [Google Scholar]
- Jaffe LF. Calcium waves and development. Ciba Found Symp. 1995;188:4–12. doi: 10.1002/9780470514696.ch2. [DOI] [PubMed] [Google Scholar]
- Kater SB, Mattson MP, Cohan C, Connor J. Calcium regulation of the neuronal growth cone. Trends Neurosci. 1988;11:315–321. doi: 10.1016/0166-2236(88)90094-x. [DOI] [PubMed] [Google Scholar]
- Kato K, Gurdon JB. Single-cell transplantation determines the time when Xenopusmuscle precursor cells acquire a capacity for autonomous differentiation. Proc Natl Acad Sci USA. 1993;90:1310–1314. doi: 10.1073/pnas.90.4.1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidokoro Y, Saito M. Early cross-striation formation in twitching Xenopusmyocytes in culture. Proc Natl Acad Sci USA. 1988;85:1978–1982. doi: 10.1073/pnas.85.6.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidokoro Y, Anderson MJ, Gruener R. Changes in synaptic potential properties during acetylcholine receptor accumulation and neurospecific interactions in Xenopusnerve-muscle cell culture. Dev Biol. 1980;78:464–483. doi: 10.1016/0012-1606(80)90347-4. [DOI] [PubMed] [Google Scholar]
- Knighton DR, Pearson RB, Sowadski JM, Means AR, Ten LF, Eyck, Taylor SS, Kemp BE. Structural basis of the intrasteric regulation of myosin light chain kinases. Science. 1992;258:130–135. doi: 10.1126/science.1439761. [DOI] [PubMed] [Google Scholar]
- Labeit S, Kolmerer B. Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science. 1995;270:293–296. doi: 10.1126/science.270.5234.293. [DOI] [PubMed] [Google Scholar]
- Levine RJ, Kensler RW, Yang Z, Stull JT, Sweeney HL. Myosin light chain phosphorylation affects the structure of rabbit skeletal muscle thick filaments. Biophys J. 1996;71:898–907. doi: 10.1016/S0006-3495(96)79293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner G. Ryanodine activation and inhibition of the Ca2+release channel of sarcoplasmic reticulum. J Biol Chem. 1986;261:6300–6306. [PubMed] [Google Scholar]
- Muto A, Kume S, Inoue T, Okano H, Mikoshiba K. Calcium waves along the cleavage furrows in cleavage-stage Xenopusembryos and its inhibition by heparin. J Cell Biol. 1996;135:181–190. doi: 10.1083/jcb.135.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa M, Sellers JR, Adelstein RS, Hidaka H. Protein kinase C modulates in vitrophosphorylation of the smooth muscle heavy meromyosin by myosin light chain kinase. J Biol Chem. 1984;259:8808–8814. [PubMed] [Google Scholar]
- Prochiantz A. Getting hydrophilic compounds into cells: lessons from homeopeptides. Curr Opin Neurobiol. 1996;6:629–634. doi: 10.1016/s0959-4388(96)80095-x. [DOI] [PubMed] [Google Scholar]
- Rees DA, Charlton J, Ataliotis P, Woods A, Stones AJ, Bayley SA. Myosin regulation and calcium transients in fibroblast shape change, attachment, and patching. Cell Motil Cytoskeleton. 1989;13:112–122. doi: 10.1002/cm.970130206. [DOI] [PubMed] [Google Scholar]
- Reinhard E, Yokoe H, Niebling KR, Allbritton NL, Kuhn MA, Meyer T. Localized calcium signals in early zebrafish development. Dev Biol. 1995;170:50–61. doi: 10.1006/dbio.1995.1194. [DOI] [PubMed] [Google Scholar]
- Rhee D, Sanger JM, Sanger JW. The premyofibril: evidence for its role in myofibrillogenesis. Cell Motil Cytoskeleton. 1994;28:1–24. doi: 10.1002/cm.970280102. [DOI] [PubMed] [Google Scholar]
- Scholey JM, Taylor KA, Kendrick-Jones J. Regulation of non-muscle myosin assembly by calmodulin-dependent light chain kinase. Nature. 1980;287:233–235. doi: 10.1038/287233a0. [DOI] [PubMed] [Google Scholar]
- Sheng M, McFadden G, Greenberg ME. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron. 1990;4:571–582. doi: 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- Silver R B. Calcium, BOBs, QEDs, microdomains and a cellular decision: control of mitotic cell division in sand dollar blastomeres. Cell Calcium. 1996;20:161–179. doi: 10.1016/s0143-4160(96)90105-0. [DOI] [PubMed] [Google Scholar]
- Sobieszek A, Babiychuk EB, Ortner B, Borkowski J. Purification and characterization of a kinase-associated, myofibrillar smooth muscle myosin light chain phosphatase possessing a calmodulin-targeting subunit. J Biol Chem. 1997;272:7027–7033. doi: 10.1074/jbc.272.11.7027. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. [Published erratum appears in Nature. 1994. 372:812.] . Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- Spitzer NC, Lamborghini JE. The development of the action potential mechanism of amphibian neurons isolated in culture. Proc Natl Acad Sci USA. 1976;73:1641–1645. doi: 10.1073/pnas.73.5.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer NC, Sejnowski TJ. Biological information processing: bits of progress. Science. 1997;277:1060–1061. doi: 10.1126/science.277.5329.1060. [DOI] [PubMed] [Google Scholar]
- Spruce AE, Moody WJ. Developmental sequence of expression of voltage-dependent currents in embryonic Xenopus laevismyocytes. Dev Biol. 1992;154:11–22. doi: 10.1016/0012-1606(92)90043-g. [DOI] [PubMed] [Google Scholar]
- Takekura H, Sun X, Franzini-Armstrong C. Development of the excitation-contraction coupling apparatus in skeletal muscle: peripheral and internal calcium release units are formed sequentially. J Muscl Res Cell Mot. 1994;15:102–118. doi: 10.1007/BF00130422. [DOI] [PubMed] [Google Scholar]
- Takekura H, Nishi M, Noda T, Takeshima H, Franzini-Armstrong C. Abnormal junctions between surface membrane and sarcoplasmic reticulum in skeletal muscle with a mutation targeted to the ryanodine receptor. Proc Natl Acad Sci USA. 1995;92:3381–3385. doi: 10.1073/pnas.92.8.3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodore L, Derossi D, Chassaing G, Llirbat B, Kubes M, Jordan P, Chneiweiss H, Godement P, Prochiantz A. Intraneuronal delivery of protein kinase C pseudosubstrate leads to growth cone collapse. J Neurosci. 1995;15:7158–7167. doi: 10.1523/JNEUROSCI.15-11-07158.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trybus KM. Role of myosin light chains. J Muscle Res Cell Motil. 1994;15:587–594. doi: 10.1007/BF00121066. [DOI] [PubMed] [Google Scholar]
- Tsien RY. New calcium indicators and buffers with high selectivity against magnesium and protons. Biochemistry. 1980;19:2396–2404. doi: 10.1021/bi00552a018. [DOI] [PubMed] [Google Scholar]
- Turbedsky K, Pollard TD, Bresnick AR. A subset of protein kinase C phosphorylation sites on the myosin II regulatory light chain inhibits phosphorylation by myosin light chain kinase. Biochemistry. 1997;36:2063–2067. doi: 10.1021/bi9624651. [DOI] [PubMed] [Google Scholar]
- Webb SE, Lee KW, Karplus E, Miller AL. Localized calcium transients accompany furrow positioning, propagation, and deepening during the early cleavage period of zebrafish embryos. Dev Biol. 1997;192:78–92. doi: 10.1006/dbio.1997.8724. [DOI] [PubMed] [Google Scholar]